

Abstract
TGFβ expression acts as a biomarker of poor prognosis in prostate cancer. It plays a dual functional role in prostate cancer. In the early stages of the tumor, it acts as a tumor suppressor while at the later stages of tumor development, it promotes metastasis. The molecular mechanisms of action of TGFβ are largely understood through the canonical and non-canonical signal transduction pathways. Our understanding of the mechanisms that establish transient TGFβ stimulation into stable gene expression patterns remains incomplete. Epigenetic marks like histone H3 modifications are directly linked with gene expression and they play an important role in tumorigenesis. In this report, we performed chromatin immunoprecipitation-sequencing (ChIP-Seq) to identify the genome-wide regions that undergo changes in histone H3 Lysine 4 trimethylation (H3K4me3) occupancy in response to TGFβ stimulation. We also show that TGFβ stimulation can induce acute epigenetic changes through the modulation of H3K4me3 signals at genes belonging to special functional categories in prostate cancer. TGFβ induces the H3K4me3 on its own ligands like TGFβ, GDF1, INHBB, GDF3, GDF6, BMP5 suggesting a positive feedback loop. The majority of genes were found to be involved in the positive regulation of transcription from the RNA polymerase II promoter in response to TGFβ. Other functional categories were intracellular protein transport, brain development, EMT, angiogenesis, antigen processing, antigen presentation via MHC class II, lipid transport, embryo development, histone H4 acetylation, positive regulation of cell cycle arrest, and genes involved in mitotic G2 DNA damage checkpoints. Our results link TGFβ stimulation to acute changes in gene expression through an epigenetic mechanism. These findings have broader implications on epigenetic bases of acute gene expression changes caused by growth factor stimulation.
Keywords: TGFβ, H3K4me3, epigenetic, histone modification, ChIP-sequencing
Introduction
Prostate cancer is the major cause of death in males, and it correlates with advanced age. However, the incidence of prostate cancer in young men is increasing and it thus poses a higher risk for metastatic disease and increased mortality in increasingly young subjects. 1 High levels of transforming growth factor-β (TGFβ) correlate with poor prognosis for cancer patients, especially in cases of prostate cancer. 2 TGFβ plays a dual functional role in many cancer types including prostate cancer. In the early stages of tumorigenesis, it acts as a tumor suppressor while at the later stages of tumor progression, it promotes metastasis. 3 TGFβ is a multifunctional cytokine that regulates proliferation, differentiation, development, angiogenesis, wound healing, and many other different processes in different cell types. 4 The known 32 members of TGFβ superfamily of ligands are grouped into TGFβ and BMP subfamilies based on the sequence similarity and functions. TGFβ subfamily consists of TGFβ ligands (TGFβ1, TGFβ2, TGFβ3), 2 activins (A and B), Nodal, growth, and differentiation factor GDF1, GDF3, GDF8, GDF9, and GDF11. The BMP subfamily consists of 10 BMPs, several GDFs, and anti-Mullerian hormones. The antagonistic ligands are Lefty and Inhibin. 5
TGFβ binds to the receptors (TGFβRI and TGFβRII) and then initiates a cascade of phosphorylation which subsequently activates different intracellular signaling pathways. 6 Overexpression of TGFβ and its receptors has been described in various types of human cancer, which correlates with tumor progression, metastasis, angiogenesis, and poor prognostic outcome. 7 TGFβ expression may be useful in clinic pathological features for prostate cancer. 8
In the canonical TGFβ signaling pathway, TGFβ binds to the TGFβR1 and TGFβR2 leading to the phosphorylation of the receptor Smads (Smad 2/3). The receptor Smads co-operate with Smad4 and translocate to the nucleus to regulate the expression of diverse TGFβ target genes. 9 TGFβ also signals through a non-canonical/non-Smad signaling pathway by activating ERK, p38, PI3K, Akt, and other kinases performing different functions. These non-Smad pathways work independently or together with Smad complexes to regulate the functions of TGFβ. For example, the activation of Akt signaling by TGFβ has been shown to promote cell proliferation. 10 TGFβ receptors activate MMPs, p38 MAPK, and Zinc finger E-box-binding homeobox 1 (ZEB1), ZEB2, Snail and Slug, leading to Epithelial to Mesenchymal transition (EMT), which is required for cancer cell invasion and metastasis. TGFβ can also regulate target gene expression through the MAPK signaling pathway. 11 The type III TGFβ receptor (TGFβRIII, also known as beta glycan) is a coreceptor that is not involved directly in TGFβ signal transduction; rather, it mediates TGFβ superfamily signaling to both Smad and non-Smad signaling pathways. 12
Epigenetic modifications like DNA methylation, histone acetylation or methylation, and non-coding RNAs play an important role in abnormal cell proliferation and cancer development. Global disruptions in the epigenetic landscape are a key feature of cancer. 13 Histone methylation is an essential part of histone modification involved in various biological processes including chromatin organization, transcriptional regulation, and DNA damage repair. Altered histone methylation plays an important role in tumor development and progression. Histone methylation is very complex and can lead to activation/repression of a gene, depending on the position of the lysine residue that is methylated.13,14 Histone H3 lysine 4 (H3K4) methylation leads to the activation of genes while the methylation at lysine 9 of histone H3 (H3K9) leads to a silencing of the target genes. H3K4 trimethylation is the major chromatin modification in eukaryotes and it plays an important role in cancer. 15 Some studies have shown the association of H3K4 methylation and cancer survival and many have provided conflicting results. 16 Another level of complexity is that Arginine can be mono- or di-methylated while lysine can be mono-, di-, and tri-methylated. Lysine residues can also be modified by mono-, di-, or tri-methylation of H3K4, H3K9, H3K27, H3K36, H3K79, and H4K20 residues. Histone methylation is controlled by site-specific histone methyltransferases (HMTs) and can be reversed by specific demethylases. Numerous experimental studies have suggested that the regulation of gene expression by HMTs and demethylases are closely associated with cancer development. 15 SET/MLL family and SMYD family are the most well-studied histone methylases for lysine 4 of histone H3. 17
A limited number of H3K4 methyltransferases have been studied in prostate cancer. It has been shown that MLL1 complex binds directly to the androgen receptor (AR) and is upregulated in castration-resistant prostate cancer. Upregulated MLL1 expression correlates with low overall survival in individuals diagnosed with prostate cancer. 18 MLL2 has been shown to activate the PI3K/EMT pathway and induce DNA damage in prostate cancer. 19 SMYD3 depletion inhibits prostate cancer progression through a mechanism that blocks the transcription of AR or cyclin D2.19,20 SET7/9 plays a proliferative role in prostate cancer. A recent study suggested that SETD1A can be used as an important marker for predicting the proliferation and prognosis of metastatic castrate-resistant prostate cancer. 21
There have been some studies that address the link between TGFβ signaling and epigenetic modifications like histone methylation. It has been shown that R-Smads forms a complex with histone methyltransferase in a TGFβ signaling-dependent manner. 22 Smad1/5 also binds to H3K9 methyltransferase Suv39h, leading to the silencing of the target genes. 23 Investigations on the epigenetic reprograming during the TGFβ induced EMT have found a global reduction in the heterochromatin mark H3 Lys9 dimethylation (H3K9me2), an increase in the euchromatin mark H3 Lys4 trimethylation (H3K4me3) and an increase in the transcriptional mark H3 Lys36 trimethylation (H3K36me3). 24 Comparison of the epigenetic switches and gene expression switches between normal primary and cancer cells indicated an H3K4me3/H3K27me3 epigenetic signature in prostate carcinogenesis. 25
However, the role of H3K4me3 in TGFβ-induced target genes in prostate cancer has not been explored. Thus, we wanted to understand the functional role of H3K4me3 in regulating TGFβ target genes and to identify the genes regulated by TGFβ in an H3K4me3-dependent manner. In the present work, we focus on a global search for the H3K4me3 associated regions in response to TGFβ. Chromatin Immunoprecipitation-sequencing (ChIP-Seq) was performed in Prostate cancer cells (PC3). Interestingly, the number of H3K4me3 associated regions were increased in response to TGFβ. Distribution of H3K4me3 occupancy at introns and distal intergenic regions showed increases in response to TGFβ stimulation for 6 and 24 hours. H3K4me3-associated regions were divided based on the molecular functions and biological processes they are involved in. Our results suggest a function-specific gene expression regulation by acute TGFβ stimulation through epigenetic mechanisms involving H3K4me3.
Results
Verification of TGFβ stimulation and summary of ChIP-sequencing-phred scores(Q), percentage-alignment, and peak calling
Our experimental strategy was to stimulate PC3 cells with TGFβ to characterize changes in H3K4me3 occupancy using ChIP-Seq. We first needed to validate the efficacy of TGFβ stimulation in our cellular system using established markers. Cells were stimulated with TGFβ for 6 or 24 hours. The RNA from these cells was extracted at time-points that mimic the points at which cells were to be harvested for ChIP-Seq. qRT-PCR was performed for 2 of the known TGFβ target genes in prostate cancer (PC3) cells, p21 26 and N-cadherin 27 to verify the TGFβ stimulation in these cells (Supplemental Figure 1). The qRT-PCR established that the TGFβ stimulation was effective in producing marker gene expression changes as expected.
At the same time points, cells were harvested for ChIP (see methods for details). The ChIP-DNA were subjected to sequencing using the Illumina HiSeq X platform. Paired-end ChIP-Sequencing was performed without TGFβ as well as with TGFβ stimulation for 6 and 24 hours. Table 1 details the basic parameters of the ChIP-Seq libraries and sequencing output including Phred quality score distributions. The percentage GC content of the aligned reads output of the Control (H3K4me3-Control) and TGFβ treated samples, 6 hours (H3K4me3-6h) and 24 hours (H3K4me3-24h) showed an interesting difference. H3K4me3-Control sample showed an average of 40% GC content while H3K4me3-6h and H3K4me3-24 hours showed an average of 50% GC content. Percentage Alignment for H3K4me3-Control was calculated and found to be 93.67% while it was 98.99% and 98.82% for H3K4me3-6h and H3K4me3-24h respectively. We used MACS2 callpeak to call the peaks in the aligned reads (Table 1). We found 26 917, 71 173, and 73 629 peaks (q-value <0.05) in H3K4me3-Control, H3K4me3-6h and H3K4me3-24h samples respectively. The increase in the number of peaks reflected the influence of TGFβ stimulation. Average Peak pile-up was found to be 54.30, 117.84, and 116.57 for H3K4me3-Control, H3K4me3-6h and H3K4me3-24 hours samples respectively (Table 1). Overall, we obtained good quality scores and substantial alignment with the 3 samples and we used these data for further analysis.
Table 1.
Details of the sequencing run. Each sample was subjected to paired-end sequencing on Illumina HiSeq X. More than 90% of reads were aligned to the Reference Human Genome which were used for further analysis.
| Sample | Read orientation | Mean read quality (Phred score) | Number of reads | %GC | % Reads with Q > 30 | Number of bases (MB) | Mean read length (bp) | # Reads aligned (% alignment) | # Peaks identified (q-value < .05) | Average peak pileup | Average peak length |
|---|---|---|---|---|---|---|---|---|---|---|---|
| H3K4me3-Control | R1 | 39.19 | 54673290 | 40.2 | 95.07 | 8255.67 | 151 | 98,609,122 (93.67) | 26918 | 54.30 | 1448.31 |
| R2 | 37.93 | 54673290 | 40.56 | 90.45 | 8255.67 | 151 | |||||
| H3K4me3-6 h | R1 | 39.05 | 68686449 | 50.53 | 94.68 | 10371.65 | 151 | 131,223,721 (98.99) | 71174 | 117.84 | 1327.1 |
| R2 | 37.3 | 68686449 | 50.83 | 88.34 | 10371.65 | 151 | |||||
| H3K4me3-24 h | R1 | 39.05 | 69591691 | 50.36 | 94.66 | 10508.35 | 151 | 132,970,431 (98.82) | 73630 | 116.57 | 1299.18 |
| R2 | 37.41 | 69591691 | 50.63 | 88.76 | 10508.35 | 151 |
H3K4me3 occupancy increases upon TGFβ stimulation
We analyzed the proportions of the total regions called among H3K4me3-Control and treated samples H3K4me3-6h and H3K4me3-24h. Total regions (q-value <0.05) associated with H3K4me3 were called using MACS2 callpeak function, wherein 23% regions were associated with Control, 38% were associated with H3K4me3-6h and 39% were associated with H3K4me3-24h (Figure 1a). The total peaks obtained across all samples were considered as 100. The gene list is mentioned in Supplemental File 1. It brings out the fact that the number of regions associated with H3K4me3 increases in response to TGFβ induction, elevating the transcriptional activity associated with H3K4me3. All the regions called were annotated using ChIPseeker, an R/Bioconductor package. 28 All the annotated genes were categorized based on their association to the time-point of TGFβ induction, that is, 0, 6, and 24 hours. Twenty-four percent of all annotated genes were present in Control, 38% were associated with 6 hours of TGFβ stimulation and 38% were associated with 24 hours of TGFβ stimulation (Figure 1b). The annotated gene list is mentioned in Supplemental File 2.
Figure 1.

Peaks obtained from MACS2 callpeak function were identified using the ChIPseeker R/Bioconductor package. The total number of peaks across all samples was taken as 100%. (a) Pie-Chart distribution of the number of regions that are associated with H3K4me3 modification in Control-0h (H3K4me3-Control), 6 hours (H3K4me3-6h) and 24 hours (H3K4me3-24h) induction of TGFβ1. (b) Pie-Chart distribution of Annotated genes obtained after removal of Pseudogenes and unidentified transcripts from total regions associated with H3K4me3.
A Venn diagram (Figure 2) demonstrates the overlapping and non-overlapping regions among the 3 datasets, that is, H3K4me3-Control (A), H3K4me3-6 hours (B) and H3K4me3-24h (C). From the Venn diagram, we observed that the majority of regions (Samples ABC; 14762 regions) were common to all 3 datasets. In samples BC the 9298 regions were common between H3K4me3-6h and H3K4me3-24h, but were absent in H3K4me3-Control. These were the regions potentially associated specifically with TGFβ induction. Sample C (1472) consisted of regions possibly associated only with relatively long-term (24 hours) induction of TGFβ. Regions exclusive to sample B (1182) were the regions associated with relative short-term induction (6 hours) with TGFβ. Exclusively present in sample A (266) were the regions which were not associated with TGFβ induction (Figure 2). Further analysis of regions in Sample BC can elucidate genes that are associated with H3K4me3 and are transcriptionally affected upon TGFβ stimulation. It is also possible that further analysis of these genes brings out key players in the process of EMT in cancer, some of which might be relevant to research on new therapeutic targets in cancer. Enriched regions from Sample B and Sample C hold the key to many time-specific activation of gene transcription. Regions enriched in Sample A interestingly could point to genes associated with H3K4me3 which are transcriptionally repressed in presence of TGFβ. It would be interesting to further find out if certain candidate genes from this category could be knocked out to potentially reverse the EMT process induced by TGFβ. The list of genes in each category is mentioned in Supplemental File 3.
Figure 2.

Venn diagram of comparison of 3 gene lists – H3K4me3-Control-A, H3K4me3-6h-B and H3K4me3-24h-C obtained by annotation of MACS2 peaks using ChIPseeker package. The comparative list was constructed by identifying common and unique genes using conditional formatting in MS Excel for visualization of common regions and unique regions with respect to each sample dataset.
The percentage of promoter regions associated with H3K4me3 decreases with the TGFβ stimulation, while the percentage of distal intergenic regions increases
After calling peaks using MACS2 callpeak from the aligned sequences, the list of H3K4me3 bound regions was obtained. We investigated the genomic distribution of H3K4me3 in response to TGFβ. The locations of the peaks were annotated in terms of genomic features. Annotate Peak function from the ChIPseeker package was used to assign the genomic annotation. We classified the peak annotations as promoters, 5′UTR, 3′UTR, first exon, other exons, first intron, other introns, downstream or distal intergenic. In the Control sample (Figure 3a), 31.54% of regions were located in the promoters, 34.57% were located in introns and 25.41% of regions were located in distal intergenic regions.
Figure 3.
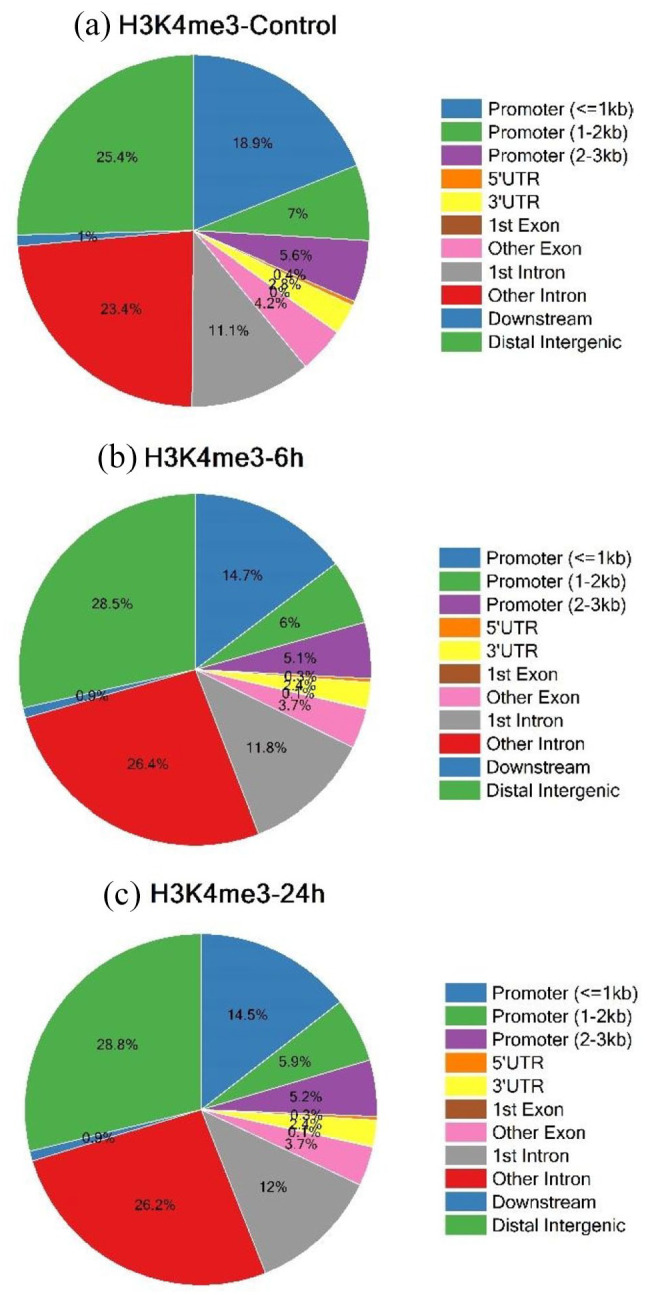
Visualization of Genomic annotations using annotatePeak function of ChIPseeker R/Bioconductor package of H3K4me3 occupied regions with 0 hours (H3K4me3-Control) (a), 6h-H3K4me3-6h (b) and 24h-H3K4me3-24h (c) of TGFβ induction. The categories used were Promoter(⩽1 kb), Promoter (1-2 kb), Promoter (2-3 kb), 5′UTR, 3′UTR, first exon, other exons, first intron, other introns, downstream (⩽300), and distal intergenic.
In samples with 6 hours of TGFβ induction (Figure 3b), 25.82% of regions were located in the promoters, 38.21% were located in introns and 28.53% regions were located in the distal intergenic regions. In samples with 24 hours of TGFβ induction (Figure 3c), 25.62% of regions were located in the promoters, 38.19% were located in introns and 28.82% regions were located in the distal intergenic regions. It was observed that the percentage distribution of distal intergenic regions increased in the treated samples − 28.53% in 6 hours and 28.82% in 24 hours of TGFβ stimulation respectively as compared to the Control sample with only 25.41%. This pointed to the role of TGFβ in increasing the association between the intergenic regions and H3K4me3. It was also interesting to note that the percentage distribution of promoters decreased from 31.54% in Control to 25.82% in the 6 hours and 25.62% in the 24 hours samples (Figure 3a–c). This implied the effect of TGFβ on the decrement of the extent of association of promoter regions of genes with H3K4me3.
Most of the genes associated with H3K4me3 in response to TGFβ belong to the functional category “ATP binding”
A Gene Ontology (GO) term analysis was performed using DAVID (https://david.ncifcrf.gov) for genes that were present in H3K4me3-6h and H3K4me3-24h, but were absent in H3K4me3-Control. Figure 4a shows a distribution of the different significantly detected GO Biological Process terms against the genes count for each term. One Hundred seventy-seven genes were observed to be associated with GO:0045944 (positive regulation of transcription from RNA-polymerase II promoter) indicating that these genes are involved in transcriptional activation or increasing the frequency of rate or extent of transcription from RNA polymerase II promoter upon TGFβ induction at the H3K4me3-occupied region. A set of 25 genes associated with GO:0051091 indicate the involvement of TGFβ signaling in increasing or activating the extent of various transcription factor bindings at the H3K4me3-occupied regions. A list of all the genes with their biological process is mentioned in Supplemental File 5. Enrichment of GO:0001837 (Epithelial to mesenchymal transition) points to the involvement of H3K4me3 in mediating the TGFβ-induced EMT. Enrichment of GO:0043967 (Histone H4 acetylation) interestingly suggests the coordination between TGFβ mediated H3K4 trimethylation and acetylation of Histone H4 (Figure 4a). This may also suggest possible crosstalk between the well-conserved H4K16ac and H3K4me3 marks in driving tumorigenesis.
Figure 4.

The annotated genes identified in H3K4me3-6h and H3K4me3-24h but absent in H3K4me3-control were used to identify enriched GO: biological processes (a) and GO: molecular function (b) terms associated with TGFβ stimulation using David Functional enrichment (v 6.8). qRT-PCR of randomly selected genes associated with ATP binding molecular function (c).
Figure 4b represents the graph of GO Molecular Function terms against the genes counts for each term. GO:0005524 (ATP binding) showed the highest gene count of 265 pointing out the importance of ATP as an enzyme regulator in TGFβ signaling. Genes associated with GO:0003379 (Actin binding) can play an important role in TGFβ in actin cytoskeleton reorganization during the invasion or migration of cancer cells. Other significant GO “Molecular Function” categories associated with H3K4me3 during TGFβ induction were serine-endopeptidase activity (52 genes), protein complex binding (49 genes), carbohydrate-binding (41 genes), protein tyrosine phosphatase activity (24 genes), core promoter binding (17 genes), tRNA binding (14 genes), TGFβ receptor binding (13 genes), aminopeptidase activity (9 genes), substrate-specific transmembrane transporter activity (6 genes), actinin binding (6 genes), dynactin binding (5 genes), and ion channel inhibitor activity (5 genes) (Figure 4b). A list of all the genes with their molecular functions is mentioned in Supplemental File 4. As H3K4me3 is linked with the transcriptional activation of genes, we performed RT-PCR to quantify the expression of some genes belonging to the ATP binding category as mentioned in Supplemental File 4. The expression of TGFβR1, KRAS, RUVBL1, NUBP1, and CHD7 was increased in response to TGFβ stimulation for both 6 hours and 24 hours, confirming the role of H3K4me3 in transcriptional activation (Figure 4c).
Minor overlap between H3K4me3 occupied genes in TGFβ stimulated prostate cancer cells (PC3) and TGFβ conditioned normal monocyte-derived dendritic cells (moDCs)
A list of genes identified only in 6 and 24 hours of TGFβ stimulation and not in the Control sample without TGFβ was prepared. We focused on the genes where H3K4me3 occupancy increases with the time of TGFβ stimulation. Alongside, for comparison with a non-cancerous cell line dataset, we extracted a list of genes that came up in TGFβ conditioned monocyte-derived Dendritic Cells (moDCs) bound to H3K4me3 with a log2 expression change greater than 1.2. 29 Only 72 common genes were found after comparing the 2 gene lists. The 72 genes were subjected to analysis for identification of enriched GO annotations using the DAVID annotation tool (Supplemental File 6). We found that 24 GO Biological Processes categories were enriched in these 72 genes (Figure 5a). Enrichment of biological processes such as Protein Phosphorylation including peptidyl serine and threonine phosphorylation and autophosphorylation indicated the association of phosphorylation event with H3K4me3 mark and TGFβ signaling. The analysis also revealed significant enrichment of genes associated with signal transduction, GTPase activity, cell adhesion and cell migration. Molecular Function categories such as protein kinase activity, specifically transmembrane receptor protein serine/threonine kinase activity were significantly enriched (Figure 5b). Consistent with our results (Figure 4b), the ATP binding function was also significantly enriched. Other enriched molecular functions included SMAD binding, cadherin binding in cell-cell adhesion and binding of metal ions and phospholipids.
Figure 5.

David functional enrichment analysis (a) biological process and (b) molecular function of enriched genes associated with H3K4me3 mark common between TGFβ conditioned moDCs and PC3 cells stimulated with TGFβ for 6 hours and 24 hours.
Changes in H3K4me3 occupancy upon TGFβ stimulation show a PC3 cell type specificity as compared to normal moDCs
Next, we prepared a list of genes that were absent in our Control sample and showed increasing fold-enrichment in the 6 and 24 hours sample and were also absent in TGFβ conditioned moDCs dataset (Supplemental File 7). Using the David Functional enrichment tool, 138 GO Biological Process categories were enriched (Figure 6a). We observed some processes that were associated with carcinogenesis such as positive regulation of GTPase activity, positive regulation of cell migration, Wnt signaling pathway, epithelial to mesenchymal transition, negative regulation of cell adhesion and response to hypoxia which indicated that TGFβ promotes carcinogenesis, EMT and metastases through active H3K4me3 mark. Alongside Biological Processes, 57 Molecular Functions were also identified (Figure 6b). Consistent with our earlier result and comparison with moDCs dataset, ATP binding function was significantly enriched indicating its role in the association of H3K4me3 mark with TGFβ signaling in a cancer-derived as well as a non-cancer-derived cell line. Enrichment of Histone Acetyltransferase binding also hinted toward possible crosstalk between active H3K4me3 mark and acetylation of another histone mark under the influence of TGFβ signaling. Other significantly enriched functions included protein kinase binding, actin-binding, chromatin binding, and SH3 domain binding.
Figure 6.

David Functional Enrichment of Gene list unique to H3K4me3-6h and H3K4me3-24h mark in TGFβ stimulated PC3 cells, and not found in TGFβ conditioned moDCs: (a) biological process and (b) molecular function.
H3K4me3 plays a major role in TGFβ induced EMT and malignant phenotype
A recent study by Ke and colleagues 30 has identified genes associated with H3K4me3 during epithelial to mesenchymal transition in prostate cells. Therefore, in addition to comparing the gene list associated with H3K4me3 mark in TGFβ stimulated PC3 cells with a non-cancerous cell line-moDCs, we also compared the genes obtained in our dataset for 6 and 24 hours of TGFβ stimulation associated with H3K4me3 with the genes identified by Ke and colleagues.
To specifically identify genes associated with EMT, we prepared a list wherein the genes showed the increasing intensity of H3K4me3 in terms of log2 ratio across EP156T (benign epithelial phenotype), EPT1 (benign mesenchymal phenotype) and EPT2 (malignant phenotype). The list was compared to the list of genes that had higher fold-enrichment in PC3 cells treated with TGFβ for 24 hours as compared to 6 hours but were absent in control cells. A panel of 1271 common genes were identified and were later subjected to David Functional enrichment to identify the categories of Biological Processes and Molecular Functions associated with the process of EMT and TGFβ stimulation enriched with H3K4me3 mark (Supplemental File 8). Biological Processes categories showed significant enrichment of positive regulation of GTPase activity, positive regulation of cell migration, Wnt signaling pathway, positive regulation of NF-kappa B transcription factor activity, positive regulation of peptidyl-serine phosphorylation, epidermal growth factor receptor signaling pathway, positive regulation of epithelial to mesenchymal transition, positive regulation of CREB transcription factor activity, hypoxia-inducible factor-1alpha signaling pathway. Enrichment of these processes indicates the activation of TGFβ signaling in promoting tumorigenesis and EMT through transcription activating H3K4me3 mark. Potentially, targeting the methylation of H3K4 can be a beneficial therapeutic measure. Another interesting result was the enrichment of the histone H4 acetylation process (Figure 7a). Consistent with our earlier comparison with moDCs dataset, acetylation of histone mark was positively associated with H3K4me3 indicating possible crosstalk between methylation and acetylation on histones in promoting tumorigenesis.
Figure 7.

David Functional enrichment of genes identified common in benign prostate epithelial cells undergoing EMT and malignant transformation and TGFβ stimulation in Prostate cancer cells moDCs: (a) biological process and (b) molecular function.
Consistent with our previous comparison with moDCs dataset, ATP binding function was again found to be significantly enriched in the process of EMT, indicating its active role in TGFβ signaling mediated downstream processes. Protein kinase activity was enriched indicating the association of phosphorylation with EMT (Figure 7b). GTPase-related molecular functions identified included GTPase activator activity, Rab GTPase binding and GTPase regulator activity which pointed out the possible role Rab GTPases and other GTPases in cancer progression.
The motifs found to be associated with H3K4me3 in response to TGFβ belong to HXC10, CPEB1, and PRDM6 genes
To discover novel DNA-binding motifs we extracted 500 bp sequences around the summits of the peaks using MEME-ChIP (https://meme-suite.org/meme/tools/meme-chip) with the sequence from H3K4me3-6h and H3K4me3-24h sample as primary Sequence and sequence from H3K4me3-Control as control Sequence (discriminative mode). The output of MEME-ChIP was imported into Tomtom to analyze the similarity of the motif we obtained against known binding motifs from the HOCOMOCO v11 database. There were 82 common motifs between H3K4me3-6h and H3K4me3-24h. We shortlisted 14 motifs whose p-value was less than 0.001. Out of the 14, the top 3 known motif hits are shown in Figure 8, remaining motif hits can be found in Supplemental File 9. The 3 known motifs that came up against our submitted motifs were binding sites of HXC10, CPEB1, and PRDM6 (Figure 8). Further analysis needs to be carried to study as to how these motifs play a role in the regulation of histone modification and TGFβ induced EMT. A list of all 14 motifs with their P-values is mentioned in Supplemental File 9.
Figure 8.

Discovery and identification of novel Nucleotide Motifs located in central regions of the Promoter Regions using MEME-ChIP (v5.3.3). The Motifs were discovered and identified by importing a 500bp-length sequence surrounding the summit of MACS peaks into the MEME-ChIP program (v5.3.3). The motifs were then compared against HOCOMOCO v11 Database using Tomtom-Motif Comparison Tool to identify similar known motifs and Top 3 hits with E-value < 0.05 were (a) HOXC10, (b) CPEB1, and (c) PRDM6.
Discussion
ChIP-Seq was performed to find out the changes in regions associated with H3K4me3 in prostate cancer cells in response to TGFβ for 6 and 24 hours. 28 The previous study on AML12 mouse hepatocytes reported a global increase in H3K4me3 levels and a decrease in H3K9me3 levels in response to TGFβ during the process of EMT. 24 Our results also confirmed a similar global increase in H3K4me3-occupied regions in response to TGFβ for 6 and 24 hours. The H3K4me3-associated regions increased by 15% in cells stimulated with TGFβ for 6 and 24 hours as compared to the control without TGFβ. We compared the number of H3K4me3 associated regions in control (without TGFβ) and TGFβ stimulated cells for 6 and 24 hours. The number of regions common between TGFβ stimulated samples were much higher than the ones compared with the control without TGFβ. It would be very interesting to study the regions associated with H3K4me3 unique to 6 or 24 hours of TGFβ stimulation in comparison to control.
Although all types of H3K4 methylations have a strong correlation with active transcription, the distribution of H3K4 methylation types on chromosomes is distinctly different. H3K4me1 is rich in active and priming enhancer regions whereas H3K4me2 is an enhancer marker, although it is concentrated predominantly toward the 5’ ends of actively transcribing genes. In contrast, H3K4me3 has been a hallmark of promoters and is predominantly localized near transcription start sites (TSSs) where it is responsible for RNA Pol II recruitment for the activation of gene expression.24,31 Our results show that 31.5% of the promoters, 23% of the intronic regions and 25.4% of distal intergenic regions were occupied by H3K4me3. Surprisingly, in response to TGFβ stimulation, the percentage of promoter regions decreased to 25.8% while the percentage occupancy at intronic and distal intergenic regions increased to 26.4% and 28.5%, respectively. The functional significance of H3K4me3 occupancy at intronic and intergenic regions is yet to be explored.
We analyzed the data and separated the genes found only in the TGFβ stimulated samples for both 6 and 24 hours as compared to the control without TGFβ. Next, we divided them based on their GO molecular function and biological process categories. We found that 177 genes were found to be involved in the positive regulation of transcription from RNA Pol II promoter in response to TGFβ stimulation for 6 and 24 hours. Other functional categories were intracellular protein transport, brain development, EMT, angiogenesis, antigen processing, antigen presentation via MHC class II, lipid transport, embryo development, H4 acetylation, a positive regulator of cell cycle arrest, genes involved in mitotic G2 DNA damage checkpoints and various other functions. Interestingly, 13 genes coding for proteins binding to the TGFβ receptor were found to be enriched with H3K4me3 in response to TGFβ for 6 and 24 hours. Surprisingly, TGFβ induced the H3K4 trimethylation on genes encoding its own ligands creating a possible feedback loop. Only 13 ligands were found to have this epigenetic change in response to TGFβ. The mechanism of how TGFβ regulates these genes has not been explored. This is the first report which shows that TGFβ can selectively regulate the H3K4me3 occupancy on specific genes with particular functions and thus regulate their expression. This enrichment was not found on all the TGFβ ligands, but it was only specific for these 11 genes enriched in H3K4me3 in response to TGFβ: GDF10, TGFβ2, LEFTY1, TGFβ, GDF1, INHBB, GDF3, GDF6, BMP5, TGFβR3, BMP2, LRG1, LEFTY2.
Based on the arrangement of cysteines, TGFβ ligands are divided into 4 categories. 32 Our results have specific genes from all 3 categories except from the third one which has only 6 cysteine residues instead of 7. Surprisingly, the antagonist ligands like Lefty and Inhibin were found to have H3K4me3 in response to TGFβ, suggesting the role of H3K4me3 in regulating both the positive and negative feedback loops in TGFβ signaling. In addition to the TGFβ ligands, we also found 2 other genes that were enriched in H3K4me3 methylation: leucine-rich alpha-2-glycoprotein-1 (LRG1) and TGFβR3. LRG1 has been shown to modulate TGFβ signaling via TGFβR2/Alk1/Smad1, Smad5, and Smad8 and plays an important role in TGFβ-induced angiogenesis. 33 LRG1 was also shown to inhibit the ESCC cell metastasis by reducing TGFβ/Smad signaling-induced EMT.33,34 Our results show an enrichment of H3K4me3 on LRG1 in response to TGFβ suggesting a positive regulation of LRG1 in response to TGFβ.
The other gene found was the TGFβR3. TGFβR3 is a membrane proteoglycan that often acts as a coreceptor with other TGFβ receptor superfamilies. As a co-receptor TGFβR3 plays an important role in mediating ligand-dependent TGFβ superfamily signaling and possesses importance for ligand-independent functions. TGFβR3 has been identified as a tumor suppressor gene in prostate cancer. 35 Less TGFβR3 was found in prostate cancer cells than in benign tissues, in metastatic versus primary tumors, and advanced clinical stage tumors or in tumors with higher PSA recurrence. Interestingly, a loss of heterozygosity in the TGFβR3 genomic locus was found in prostate cancer cells. 36 Restoration of TGFβR3 expression in human PCa cells may lead to inhibition of migration and invasion, independently of the ligand TGFβ. In a human PCa xenograft model, restoring TGFβR3 function decreased tumor growth, suggesting its tumor suppressor role. 36 LRG1 and TGFβR3 have been shown to regulate the TGFβ signaling but our results suggest that TGFβ can regulate H3K4me3 levels at these genes and thus affect their expression. It would be interesting to understand the mechanism and the functional importance of H3K4me3 on LRG1 and TGFβR3 in response to TGFβ in prostate cancer.
Several genes regulating the TGFβ-induced EMT like S100A4, HGF, hnRNP, HMGA2 were found to be occupied by H3K4me3 in response to TGFβ suggesting an important role of H3K4me3 in TGFβ-induced EMT. TGFβ has been shown to induce the expression of fibroblast-specific protein S100A4 and silencing of this gene can inhibit the TGFβ induced EMT. 37 TGFβ has been shown to regulate hepatocyte growth factor (HGF)-induced cell migration and HGF receptor MET expression in breast cancer. 38 TGFβ and HGF both can phosphorylate the linker regions of the Smads 39 and coordinate the process of EMT. HGF has also been shown to upregulate BMP7 in prostate cancer cells and lead to the development of bone metastasis. 40 Phosphorylation of heterogeneous nuclear ribonucleoprotein E1 (hnRNP E1) is required for the TGFβ induced EMT in NMuMG cells. 41 TGFβ can also induce the expression of HMGA2 and it has also been shown that TGFβ-induced Smad4 binds to the HMGA2 promoter. 42 Our results suggest the important role of H3K4me3 in regulating the TGFβ induced EMT.
One of the unique categories of genes found in our analysis was the genes regulating H4 acetylation like MSL3P1, IRF4, MORF4L2, LEF1, RUVBL1, EP400, EP300, CHD5, EPC1, ELP3. Most of the genes found in this category belong to the NuA4 HAT complex. NuA4 complex has been shown to play an important role in chromatin remodeling, cell cycle, and DNA repair. 43
MORF4L2 is a component of the NuA4 histone acetyltransferase complex which is involved in transcriptional activation of select genes principally by acetylation of nucleosome histone H4 and H2A. This modification may alter nucleosomes as well as promote interaction of the modified histones with other proteins which positively regulate transcription. 44 The ATPase and helicase activity of NuA4 seems to be in part contributed by the association of RUVBL1, RUVBL2 with EP400. 45 EPC1 is also a part of NuA4 acetyltransferase complex and can act as a transcriptional activator as well as a repressor. 46 ELP3 primarily acts as a tRNA acetyltransferase subunit of the RNA polymerase II elongator complex and is involved in transcriptional elongation. 47 MSL3P1 is a pseudogene and is thought to play an important role in chromatin remodeling and histone acetylation. 48
The association of TGFβ with the NuA4 complex has not been investigated. Following our findings reported here, it will be interesting to investigate this further and understand the functional role of H3K4me3 on the components of the NuA4 complex, in response to TGFβ.
The protein encoded by the gene IRF4, belongs to the IRF (interferon regulatory factor) family of transcription factors, characterized by a unique tryptophan pentad repeat DNA-binding domain. The IRFs are important in the regulation of interferons in response to infection by virus, and in the regulation of interferon-inducible genes. 49 IRF4 interacts with Smad2/3 and it cooperatively activates IL9 promoter.49,50 Our results suggest crosstalk between the TGFβ and interferon signaling pathway at the epigenetic level.
To verify the correlation of H3K4me3 with the transcriptional activation, RT-PCR were performed on some candidate ATP binding genes that were identified in our ChIP-Seq experiment. We have thus confirmed the upregulation of TGFBR1, KRAS, RUVBL1, NUBP1 and CHD7 in response to TGFβ for 6 and 24 hours. The upregulation of TGFBR1 suggests the role of H3K4me3 in regulating a positive feedback loop in TGFβ signaling. Studies have shown that CHD7 is required to maintain open, accessible chromatin, thus fine-tuning the transcriptional regulation of certain classes of genes. 51 CHD7 expression positively correlates with a small subset of classical oncogenes, notably NRAS, in breast cancer. The chromodomains of CHD7 have a unique specificity for the monomethylated H3K4 mark.51,52 To the best of our knowledge, no reports are suggesting a functional link between TGFβ and CHD7 to date. Our results report that TGFβ can induce the expression of CHD7 by modulating the H3K4me3 levels in response to TGFβ for 6 and 24 hours.
A comparison of H3K4me3 ChIP-Seq data in PC3 cells treated with TGFβ with normal dendritic cells treated with TGFβ, reinforces a significant role of TGFβ in the tumorigenesis. Only 72 regions were found to be common between the 2 data sets and a significant increase in the number of H3K4me3 occupied regions in PC3 cells treated with TGFβ was observed as compared to normal dendritic cells stimulated with TGFβ.
In humans, there are 4 types of TSS (classified as A, B, C, or D) depending on the GC- and AT-rich regions upstream or downstream of TSS. A and B are both GC rich in the upstream region while C and D are both AT-rich in the upstream region. The difference between A and B lies in the downstream region such that A is GC rich while B is AT-rich. The difference between C and D also lies in the downstream sequence, C is GC rich and D is AT-rich. Majority of the promoters belong to the A category with maximum CpG island content while B, C, and D are relatively rare. 53
Surprisingly in our dataset, we identified 3 motifs in the promoter region belonging to the rare category D. The 3 top hits with E-value <0.05 were motifs belonging to HXC10, CPEB1 and PRDM6 both in the 6 as well as 24 hours of TGFβ stimulation. Recently it has been shown that silencing HXC10 inhibited the expression of interleukin-6, TNF-α, TGFβ, and epidermal growth factor, and overexpressing HXC10 induced their expression both in vitro and in vivo. In PD gastric cancer cells, silencing of HXC10 suppressed proliferation, migration and invasion of these cells. 54 Cytoplasmic polyadenylation element-binding (CPEB) proteins are key mRNA-binding proteins that control the translation of several mRNAs. CPEB1 and CPEB4 knockdowns also attenuated TGFβ-activated Smad-dependent and -independent signaling cascades by reducing the levels of TAK1, P38, ERK, JNK, and phosphorylated Smad 2 and Smad 1/5/8 in fibroblasts. 55 The protein encoded by PRDM6 (putative histone-lysine N-methyltransferase) is a transcriptional repressor and a member of the PRDM family. The PRDM family members contain a PR domain and multiple zinc-finger domains. The encoded protein is involved in the regulation of vascular smooth muscle cells (VSMC) contractile proteins. 52 No research work has been reported on PRDM6 in the context of its regulation by TGFβ so it would be very interesting to understand the connection between these 2. Interestingly, all 3 hits belong to the category of transcriptional repressors and it would be very valuable to understand the functional role of these motifs in relation to TGFβ.
Material and Methods
In vitro cultivation and maintenance of prostate cancer cells
Prostate cancer (PC3) cells were cultured in RPMI-1640 Medium (HiMedia Labs, India) supplemented with 10% Fetal Bovine Serum (FBS) (Gibco, U.S.A) and 1% Antibiotic and Antimycotic solution (HiMedia Labs, India).
TGFβ treatment
PC3 cells were cultured in T-75 flask until 70% to 80% confluency. The cells were then serum starved for 24 hours in serum-free medium. Post serum starvation, TGFβ (Abcam, USA) was added to the cells to a final concentration of 5 ng/mL. TGFβ induction was given for 0 hours (Control), 6 and 24 hours.
qRT-PCR
PC3 cells were cultured in T-75 flask until 70% to 80% confluency. The cells were then Serum starved for 24 hours in Serum-free Medium. Post serum starvation, TGFβ (Abcam, USA) was added to the cells to a final concentration of 5 ng/mL. TGFβ induction was given for 0 hours (Control), 6 and 24 hours. The total RNA was extracted using RNeasy Mini Kit (Qiagen, Germany) according to the manufacturer’s protocol. DNA was removed from the samples by the RNAse free DNAse set (Qiagen, Germany). The mRNA was then reverse transcribed to cDNA (Eppendorf Mastercycler) with the help of iScript cDNA Synthesis kit (Biorad, USA). Primers specific for N-cadherin, p21, NUBP1, RUVBL1, CHD7, TGFBR1, and KRAS with the following sequences were used:
N-cadherin: For, 5′-TCTTTGTGCCCATCTCCCAT-3′ and Rev, 5′-AAGTCCCCAATGTCTCCAGG-3′; p21: For, 5′-AAGACCATGTGGACCTGTCA-3′ and Rev, 5′-GGCGTTTGGAGTGGTAGAAA-3′; NUBP1: For, 5′-AAAGCACATTCAGCGCCC-3′ and Rev, 5′-TCTCCCCAGTCCACATCTC-3′; RUVBL1: For, 5′-AGCCAAAGAAGACAGAAATCAC-3′ and Rev, 5′-ACCTCATCAACAAACAGCAC-3′; CHD7: For, 5′-GACGCTATAAACGCCAACTC-3′ and Rev, 5′-TCTGGGCTTTCACCTTCTTT-3′; TGFBR1: For, 5′-AGGATTTCTTTGGACCCAGG-3′ and Rev, 5′-CAGCACAGCAGAGTTACCTA-3′; KRAS: For, 5′-ACAGAGAGTGGAGGATGCTTT-3′ and Rev, 5′-TTTCACACAGCCAGGAGTCTT-3′ and GAPDH: For, 5′-ATGTTCGTCATGGGTGTGAA-3′ and Rev, 5′-TGTGGTCATGAGTCCTTCCA-3′. cDNA was amplified in Optical 8-tube strip (Applied Biosystems) and quantified in Applied Biosystems QuantStudio 5 qRT-PCR system using iTaq Universal SYBR Green Supermix (Biorad, U.S.A). The following were the cycling conditions: 30 seconds at 95°C, followed by 40 Cycles of 15 seconds at 95°C, and 1 minute at 60°C. Data was collected and analyzed using QuantStudio Design and Analysis Software v1.5.1 with GAPDH as reference gene.
Cross-linking of cells
Histone proteins were crosslinked to DNA using 37% Formaldehyde solution (Sigma-Aldrich, USA) to final concentration of 1% and cells were incubated for 10 minutes at 37°C in CO2 Incubator. The cross-linking reaction was terminated by quenching the formaldehyde by adding Glycine to a final concentration of 125 mM and incubated with shaking at Room Temperature. Post-Quenching, cells were washed with ice-cold Phosphate Buffered Saline (PBS) (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4) containing Protease Inhibitor Cocktail (Sigma-Aldrich, USA), scraped and pelleted by centrifugation. Pellet was flash-freezed using Liquid-Nitrogen and stored at −80°C until further use.
Cell Lysis, chromatin shearing, and Immunoprecipitation
Cell pellet was resuspended in SDS Lysis Buffer (1% SDS, 10 mM EDTA and 50 mM Tris, pH 8.1) with Protease inhibitors and incubated for 10 minutes on ice. The lysate was sonicated using Diagenode Bioruptor. The samples were centrifuged, and supernatant was collected. The Supernatant was diluted 10-fold in ChIP-Dilution Buffer (0.01% SDS, 1.1% Triton X- 100, 1.2 mM EDTA, 16.7 mM Tris-HCl, 167 mM NaCl, pH 8.1). Immunoprecipitation antibody specific for H3K4me3 (Abcam, USA) was added followed by addition of Protein A Sepharose beads (Abcam, USA) and were incubated overnight at 4°C. A No-Antibody sample to be used as Input-DNA was kept aside for each sample. The Histone-DNA-Antibody complex was washed with Low Salt Immune Complex Wash Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 150 mM NaCl), High Salt Immune Complex Wash Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 500 mM NaCl), LiCl Immune Complex Wash Buffer (0.25 M LiCl, 1% IGEPAL CA630, 1% deoxycholic acid (sodium salt), 1 mM EDTA, 10 mM Tris, pH 8.1), and 1X TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA) in that order. The DNA was eluted using Elution Buffer (1% SDS, 0.1M NaHCO3) by vortexing and incubating at Room Temperature for 15 minutes with rotation. The mixture was centrifuged and supernatant (eluate) was collected. Reverse crosslinking was carried out using 5 M NaCl by heating at 65°C for 4 hours followed by overnight RNase treatment at 60°C and Proteinase K treatment at 45°C for 1 hour. The DNA was recovered and purified using a DNA Purification kit.
Library preparation and sequencing
Libraries for ChIP sequencing were prepared using NEBNext® Ultra II DNA Library preparation kit. In brief, ChIP DNA and the Input DNA were subjected to various enzymatic steps for repairing the ends and tailing with dA-tail followed by ligation of adapter sequences. These adapter-ligated fragments were then size selected using SPRI beads. Next, the size selected fragments were indexed during limited cycle PCR to generate final libraries for paired-end sequencing. The resulting libraries were quantified before getting sequenced on Illumina HiSeq X system to generate 2 × 150 bp sequence reads. Prepared libraries were sequenced on Illumina HiSeqX to generate 100 M, 2 × 150 bp reads/samples. Sequenced data was processed to generate a FASTQ file.
Bioinformatic analysis
Read quality check: We checked the following parameters from fastq file (a) Base quality score distribution (b) Sequence quality score distribution (c) Average base content per read (d) GC distribution in the reads (e) PCR amplification issue (f) Check for overrepresented sequences.
Adapter trimming: Based on quality reports of fastq files, sequence reads were trimmed where necessary to only retain high-quality sequences for further analysis. In addition, the low-quality sequence reads were excluded from the analysis. The adapter trimming was performed using Trimmomatic (Ver-0.36). 56
Read alignment: The paired-end reads were aligned to the Human reference genome (hg19) Feb. 2009 release downloaded from UCSC database (GRCh37/hg19). The chromosome fasta file was downloaded from the following website (http://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/chromFa.tar.gz) and the Alignment was performed using BWA MEM (Ver-0.7.12).
Peak Calling and annotation: Model-based Analysis of ChIP-Seq (MACS 2.1.3 version) was used for peak calling analysis). 57 ChIPseeker, an R Bioconductor package 28 was used for the annotation of the enriched peaks obtained from the MACS2 callpeak function.
Functional annotation: Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.8 was used to identify enriched biological themes, particularly GO terms, discover enriched functional-related gene groups and cluster redundant annotation terms.
Functional analysis of gene lists
Annotated genes that were present in H3K4me3-6h and H3K4me3-24h but absent in H3K4me3-Control were used for the DAVID Functional Analysis. The gene list was imported to the David Analysis Wizard. “OFFICIAL_GENE_SYMBOL” was selected as the Identifier with List type set to “Gene List.” The list was analyzed with default parameters on DAVID platform. From the DAVID Functional annotation tools, Functional annotation Chart was downloaded and analyzed for enriched GO terms.58,59
MEME-ChIP motif analysis
The 500 bp Sequences around the peak summits from the Promoter Region were extracted. We imported the sequences into the MEME-ChIP motif discovery tool (v5.3.3). 60 Motif Discovery was performed in the Discriminative Mode by using Sequences obtained from treated samples as Primary Sequence and Sequences from Control Sample as Control sequences in the MEME-ChIP Program. Default Parameters were used for the discovery of motifs. HOCOMOCO Human (v11)60,61 was used as a Known Motif Database. Motifs discovered for H3K4me3-6h and H3K4me3-24h through the MEME-ChIP tool were compared against the HOCOMOCO Human(v11) motifs database using Tomtom-a Motif Comparison tool-using default settings. 62
Supplemental Material
Supplemental material, sj-xlsx-1-GAE-10.1177_25168657211051755 for Global Histone H3 Lysine 4 Trimethylation (H3K4me3) Landscape Changes in Response to TGFβ by Ankit Naik, Nidhi Dalpatraj and Noopur Thakur in Epigenetics Insights
Supplemental material, sj-xlsx-2-GAE-10.1177_25168657211051755 for Global Histone H3 Lysine 4 Trimethylation (H3K4me3) Landscape Changes in Response to TGFβ by Ankit Naik, Nidhi Dalpatraj and Noopur Thakur in Epigenetics Insights
Supplemental material, sj-xlsx-3-GAE-10.1177_25168657211051755 for Global Histone H3 Lysine 4 Trimethylation (H3K4me3) Landscape Changes in Response to TGFβ by Ankit Naik, Nidhi Dalpatraj and Noopur Thakur in Epigenetics Insights
Supplemental material, sj-xlsx-4-GAE-10.1177_25168657211051755 for Global Histone H3 Lysine 4 Trimethylation (H3K4me3) Landscape Changes in Response to TGFβ by Ankit Naik, Nidhi Dalpatraj and Noopur Thakur in Epigenetics Insights
Supplemental material, sj-xlsx-5-GAE-10.1177_25168657211051755 for Global Histone H3 Lysine 4 Trimethylation (H3K4me3) Landscape Changes in Response to TGFβ by Ankit Naik, Nidhi Dalpatraj and Noopur Thakur in Epigenetics Insights
Supplemental material, sj-xlsx-6-GAE-10.1177_25168657211051755 for Global Histone H3 Lysine 4 Trimethylation (H3K4me3) Landscape Changes in Response to TGFβ by Ankit Naik, Nidhi Dalpatraj and Noopur Thakur in Epigenetics Insights
Supplemental material, sj-xlsx-7-GAE-10.1177_25168657211051755 for Global Histone H3 Lysine 4 Trimethylation (H3K4me3) Landscape Changes in Response to TGFβ by Ankit Naik, Nidhi Dalpatraj and Noopur Thakur in Epigenetics Insights
Supplemental material, sj-xlsx-8-GAE-10.1177_25168657211051755 for Global Histone H3 Lysine 4 Trimethylation (H3K4me3) Landscape Changes in Response to TGFβ by Ankit Naik, Nidhi Dalpatraj and Noopur Thakur in Epigenetics Insights
Supplemental material, sj-xlsx-9-GAE-10.1177_25168657211051755 for Global Histone H3 Lysine 4 Trimethylation (H3K4me3) Landscape Changes in Response to TGFβ by Ankit Naik, Nidhi Dalpatraj and Noopur Thakur in Epigenetics Insights
Acknowledgments
Authors acknowledge the Department of Science and Technology (DST), Government of India for Ankit Naik’s INSPIRE-fellowship and Human Resource Development Group, Council of Scientific and Industrial Research (CSIR-HRDG), Government of India for Nidhi D’s fellowship. We also acknowledge MedGenome Labs for providing the sequencing facility.
Footnotes
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by funding from the Science and Engineering Research Board (SERB), Government of India (EMR/2017/001987) to Dr. Noopur Thakur.
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ Contributions: AN and NT designed the experiment. AN performed the ChIP-Sequencing experiment and bioinformatic analysis of the data. ND performed and analyzed the qPCR experiment. AN, ND, NT prepared the manuscript. NT supervised the study. All authors read and approved the manuscript.
Dataset Availability: The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus 63 and are accessible through GEO Series accession number GSE171058 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE171058). 64
ORCID iDs: Ankit Naik  https://orcid.org/0000-0002-0836-6934
https://orcid.org/0000-0002-0836-6934
Noopur Thakur
https://orcid.org/0000-0002-5531-7182
Supplemental Material: Supplemental material for this article is available online.
References
- 1. Torrealba N, Rodríguez-Berriguete G, Fraile B, et al. Expression of several cytokines in prostate cancer: correlation with clinical variables of patients. Relationship with biochemical progression of the malignance. Cytokine. 2017;89:105-115. [DOI] [PubMed] [Google Scholar]
- 2. Reis ST, Pontes-Júnior J, Antunes AA, et al. Tgf-β1 expression as a biomarker of poor prognosis in prostate cancer. Clinics. 2011;66:1143-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu S, Chen S, Zeng J. TGF-β signaling: a complex role in tumorigenesis (review). Mol Med Rep. 2018;17:699-704. [DOI] [PubMed] [Google Scholar]
- 4. Feng X-H, Derynck R. Specificity and versatility in TGF-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659-693. [DOI] [PubMed] [Google Scholar]
- 5. Heldin C-H, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465-471. [DOI] [PubMed] [Google Scholar]
- 6. Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685-700. [DOI] [PubMed] [Google Scholar]
- 7. Derynck R, Turley SJ, Akhurst RJ. TGFβ biology in cancer progression and immunotherapy. Nat Rev Clin Oncol. 2021;18:9-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tu WH, Thomas TZ, Masumori N, et al. The loss of TGF-β signaling promotes prostate cancer metastasis. Neoplasia. 2003;5:267-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weiss A, Attisano L. The TGFbeta Superfamily signaling pathway. Wiley Interdiscip Rev Dev Biol. 2013;2:47-63. [DOI] [PubMed] [Google Scholar]
- 10. Finnson KW, Almadani Y, Philip A. Non-canonical (non-SMAD2/3) TGF-β signaling in fibrosis: mechanisms and targets. Semin Cell Dev Biol. 2020;101:115-122. [DOI] [PubMed] [Google Scholar]
- 11. Tripathi V, Shin J-H, Stuelten CH, Zhang YE. TGF-β-induced alternative splicing of TAK1 promotes EMT and drug resistance. Oncogene. 2019;38:3185-3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu X-L, Xiao K, Xue B, et al. Dual role of TGFBR3 in bladder cancer. Oncol Rep. 2013;30:1301-1308. [DOI] [PubMed] [Google Scholar]
- 13. Meyers RA. Epigenetic Regulation and Epigenomics. John Wiley & Sons; 2012. [Google Scholar]
- 14. Berger SL, Nakanishi O, Haendler B. The Histone Code and Beyond: New Approaches to Cancer Therapy. Springer Science & Business Media; 2007. [Google Scholar]
- 15. Wang R, Xin M, Li Y, Zhang P, Zhang M. The functions of histone modification enzymes in cancer. Curr Protein Pept Sci. 2016;17:438-445. [DOI] [PubMed] [Google Scholar]
- 16. Li S, Shen L, Chen K-N. Association between H3K4 methylation and cancer prognosis: a meta-analysis. Thorac Cancer. 2018;9:794-799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rugo HS, Jacobs I, Sharma S, et al. The promise for histone methyltransferase inhibitors for epigenetic therapy in clinical oncology: a narrative review. Adv Ther. 2020;37:3059-3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Malik R, Khan AP, Asangani IA, et al. Targeting the MLL complex in castration-resistant prostate cancer. Nat Med. 2015;21:344-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lv S, Ji L, Chen B, et al. Histone methyltransferase KMT2D sustains prostate carcinogenesis and metastasis via epigenetically activating LIFR and KLF4. Oncogene. 2018;37:1354-1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bottino C, Peserico A, Simone C, Caretti G. SMYD3: an oncogenic driver targeting epigenetic regulation and signaling pathways. Cancers. 2020;12:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang L, Jin M, Jeong KW. Histone H3K4 methyltransferases as targets for drug-resistant cancers. Biology. 2021;10:581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Du D, Katsuno Y, Meyer D, et al. Smad3-mediated recruitment of the methyltransferase SETDB1/ESET controls snail1 expression and epithelial-mesenchymal transition. EMBO Rep. 2018;19:135-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wakabayashi Y, Tamiya T, Takada I, et al. Histone 3 lysine 9 (H3K9) methyltransferase recruitment to the interleukin-2 (IL-2) promoter is a mechanism of suppression of IL-2 transcription by the transforming growth factor-β-Smad pathway. J Biol Chem. 2011;286:35456-35465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McDonald OG, Wu H, Timp W, Doi A, Feinberg AP. Genome-scale epigenetic reprogramming during epithelial to mesenchymal transition. Nat Struct Mol Biol. 2011;18:867-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ke X-S, Qu Y, Rostad K, et al. Genome-wide profiling of histone H3 lysine 4 and lysine 27 trimethylation reveals an epigenetic signature in prostate carcinogenesis. PLoS One. 2009;4:e4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thakur N, Gudey SK, Marcusson A, et al. TGFβ-induced invasion of prostate cancer cells is promoted by c-Jun-dependent transcriptional activation of Snail1. Cell Cycle. 2014;13:2400-2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zheng Y, Wang Z, Bie W, et al. PTK6 activation at the membrane regulates epithelial–mesenchymal transition in prostate cancer. Cancer Research. 2013;73:5426-5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu G, Wang L-G, He QY. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382-2383. [DOI] [PubMed] [Google Scholar]
- 29. Huang Y, Min S, Lui Y, et al. Global mapping of H3K4me3 and H3K27me3 reveals chromatin state-based regulation of human monocyte-derived dendritic cells in different environments. Genes Immun. 2012;13:311-320. [DOI] [PubMed] [Google Scholar]
- 30. Ke X-S, Qu Y, Cheng Y, et al. Global profiling of histone and DNA methylation reveals epigenetic-based regulation of gene expression during epithelial to mesenchymal transition in prostate cells. BMC Genomics. 2010;11:669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Soares LM, He PC, Chun Y, Suh H, Kim T, Buratowski S. Determinants of Histone H3K4 methylation patterns. Mol Cell. 2017;68:773-785.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Levine AJ, Brivanlou AH. GDF3 at the crossroads of TGF-beta signaling. Cell Cycle. 2006;5:1069-1073. [DOI] [PubMed] [Google Scholar]
- 33. Wang X, Abraham S, McKenzie JAG, et al. LRG1 promotes angiogenesis by modulating endothelial TGF-β signalling. Nature. 2013;499:306-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang N, Ren Y, Wang Y, et al. LRG1 suppresses migration and invasion of esophageal squamous cell carcinoma by modulating epithelial to mesenchymal transition. J Cancer. 2020;11:1486-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jakowlew SB. Transforming Growth Factor-Beta in Cancer Therapy, Volume II: Cancer Treatment and Therapy. Springer Science & Business Media; 2010. [Google Scholar]
- 36. Turley RS, Finger EC, Hempel N, How T, Fields TA, Blobe GC. The type III transforming growth factor-beta receptor as a novel tumor suppressor gene in prostate cancer. Cancer Res. 2007;67:1090-1098. [DOI] [PubMed] [Google Scholar]
- 37. Ning Q, Li F, Wang L, et al. S100A4 amplifies TGF-β-induced epithelial–mesenchymal transition in a pleural mesothelial cell line. J Investig Med. 2017;66:334-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Breunig C, Erdem N, Bott A, et al. TGFβ1 regulates HGF-induced cell migration and hepatocyte growth factor receptor MET expression via C-ets-1 and miR-128-3p in basal-like breast cancer. Mol Oncol. 2018;12:1447-1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mori S, Matsuzaki K, Yoshida K, et al. TGF-β and HGF transmit the signals through JNK-dependent Smad2/3 phosphorylation at the linker regions. Oncogene. 2004;23:7416-7429. [DOI] [PubMed] [Google Scholar]
- 40. Ye L, Lewis-Russell JM, Davies G, Sanders AJ, Kynaston H, Jiang WG. Hepatocyte growth factor up-regulates the expression of the bone morphogenetic protein (BMP) receptors, BMPR-IB and BMPR-II, in human prostate cancer cells. Internet J Oncol. 2007;30:521-529. [PubMed] [Google Scholar]
- 41. Chaudhury A, Hussey GS, Ray PS, Jin G, Fox PL, Howe PH. TGF-beta-mediated phosphorylation of hnRNP E1 induces EMT via transcript-selective translational induction of Dab2 and ILEI. Nat Cell Biol. 2010;12:286-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang S, Mo Q, Wang X. Oncological role of HMGA2 (review). Internet J Oncol. 2019;55:775-788. [DOI] [PubMed] [Google Scholar]
- 43. Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev. 2000;64:435-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Scheibe M, Arnoult N, Kappei D, et al. Quantitative interaction screen of telomeric repeat-containing RNA reveals novel TERRA regulators. Genome Res. 2013;23:2149-2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dauden MI, López-Perrote A, Llorca O. RUVBL1-RUVBL2 AAA-ATPase: a versatile scaffold for multiple complexes and functions. Curr Opin Struct Biol. 2021;67:78-85. [DOI] [PubMed] [Google Scholar]
- 46. Doyon Y, Selleck W, Lane WS, Tan S, Côté J. Structural and functional conservation of the NuA4 Histone acetyltransferase complex from yeast to humans. Mol Cell Biol. 2004;24:1884-1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lin T-Y, Abbassi NEH, Zakrzewski K, et al. The elongator subunit Elp3 is a non-canonical tRNA acetyltransferase. Nat Commun. 2019;10:625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen B, Wang C, Zhang J, Zhou Y, Hu W, Guo T. New insights into long noncoding RNAs and pseudogenes in prognosis of renal cell carcinoma. Cancer Cell Int. 2018;18:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gualco G, Weiss LM, Bacchi CE. Mum1/Irf4. Appl Immunohistochem Mol Morphol. 2010;18:301-310. [DOI] [PubMed] [Google Scholar]
- 50. Tamiya T, Ichiyama K, Kotani H, et al. Smad2/3 and IRF4 play a cooperative role in IL-9-producing T cell induction. J Immunol. 2013;191:2360-2371. [DOI] [PubMed] [Google Scholar]
- 51. Chu X, Guo X, Jiang Y, et al. Genotranscriptomic meta-analysis of the CHD family chromatin remodelers in human cancers – initial evidence of an oncogenic role for CHD7. Mol Oncol. 2017;11:1348-1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Davis CA, Haberland M, Arnold MA, et al. PRISM/PRDM6, a transcriptional repressor that promotes the proliferative gene program in smooth muscle cells. Mol Cell Biol. 2006;26:2626-2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bajic VB, Tan SL, Christoffels A, et al. Mice and men: their promoter properties. PLoS Genet. 2006;2:e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li J, Tong G, Huang C, et al. HOXC10 promotes cell migration, invasion, and tumor growth in gastric carcinoma cells through upregulating proinflammatory cytokines. J Cell Physiol. 2020;235:3579-3591. [DOI] [PubMed] [Google Scholar]
- 55. Cui HS, Joo SY, Cho YS, Kim J-B, Seo CH. CPEB1 or CPEB4 knockdown suppresses the TAK1 and Smad signalings in THP-1 macrophage-like cells and dermal fibroblasts. Arch Biochem Biophys. 2020;683:108322. [DOI] [PubMed] [Google Scholar]
- 56. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 2014;30:2114-2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44-57. [DOI] [PubMed] [Google Scholar]
- 59. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ma W, Noble WS, Bailey TL. Motif-based analysis of large nucleotide data sets using MEME-ChIP. Nat Protoc. 2014;9:1428-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kulakovskiy IV, Vorontsov IE, Yevshin IS, et al. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-seq analysis. Nucleic Acids Res. 2018;46:D252-D259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS. Quantifying similarity between motifs. Genome Biol. 2007;8:R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets – update. Nucleic Acids Res. 2013;41:D991-D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-xlsx-1-GAE-10.1177_25168657211051755 for Global Histone H3 Lysine 4 Trimethylation (H3K4me3) Landscape Changes in Response to TGFβ by Ankit Naik, Nidhi Dalpatraj and Noopur Thakur in Epigenetics Insights
Supplemental material, sj-xlsx-2-GAE-10.1177_25168657211051755 for Global Histone H3 Lysine 4 Trimethylation (H3K4me3) Landscape Changes in Response to TGFβ by Ankit Naik, Nidhi Dalpatraj and Noopur Thakur in Epigenetics Insights
Supplemental material, sj-xlsx-3-GAE-10.1177_25168657211051755 for Global Histone H3 Lysine 4 Trimethylation (H3K4me3) Landscape Changes in Response to TGFβ by Ankit Naik, Nidhi Dalpatraj and Noopur Thakur in Epigenetics Insights
Supplemental material, sj-xlsx-4-GAE-10.1177_25168657211051755 for Global Histone H3 Lysine 4 Trimethylation (H3K4me3) Landscape Changes in Response to TGFβ by Ankit Naik, Nidhi Dalpatraj and Noopur Thakur in Epigenetics Insights
Supplemental material, sj-xlsx-5-GAE-10.1177_25168657211051755 for Global Histone H3 Lysine 4 Trimethylation (H3K4me3) Landscape Changes in Response to TGFβ by Ankit Naik, Nidhi Dalpatraj and Noopur Thakur in Epigenetics Insights
Supplemental material, sj-xlsx-6-GAE-10.1177_25168657211051755 for Global Histone H3 Lysine 4 Trimethylation (H3K4me3) Landscape Changes in Response to TGFβ by Ankit Naik, Nidhi Dalpatraj and Noopur Thakur in Epigenetics Insights
Supplemental material, sj-xlsx-7-GAE-10.1177_25168657211051755 for Global Histone H3 Lysine 4 Trimethylation (H3K4me3) Landscape Changes in Response to TGFβ by Ankit Naik, Nidhi Dalpatraj and Noopur Thakur in Epigenetics Insights
Supplemental material, sj-xlsx-8-GAE-10.1177_25168657211051755 for Global Histone H3 Lysine 4 Trimethylation (H3K4me3) Landscape Changes in Response to TGFβ by Ankit Naik, Nidhi Dalpatraj and Noopur Thakur in Epigenetics Insights
Supplemental material, sj-xlsx-9-GAE-10.1177_25168657211051755 for Global Histone H3 Lysine 4 Trimethylation (H3K4me3) Landscape Changes in Response to TGFβ by Ankit Naik, Nidhi Dalpatraj and Noopur Thakur in Epigenetics Insights