

Abstract
The c-Jun NH2-terminal kinase (JNK) group of mitogen-activated protein kinases (MAPKs) is activated in response to the treatment of cells with inflammatory cytokines and by exposure to environmental stress. JNK activation is mediated by a protein kinase cascade composed of a MAPK kinase and a MAPK kinase kinase. Here we describe the molecular cloning of a putative molecular scaffold protein, JIP3, that binds the protein kinase components of a JNK signaling module and facilitates JNK activation in cultured cells. JIP3 is expressed in the brain and at lower levels in the heart and other tissues. Immunofluorescence analysis demonstrated that JIP3 was present in the cytoplasm and accumulated in the growth cones of developing neurites. JIP3 is a member of a novel class of putative MAPK scaffold proteins that may regulate signal transduction by the JNK pathway.
Mitogen-activated protein kinase (MAPK) signal transduction pathways are evolutionarily conserved in eukaryotic cells and have been identified in plants, yeast, insects, nematodes, and mammals. Genetic studies have established that MAPK signaling pathways are critical mediators of the response of cells to changes in their environment. Thus, MAPK pathways are essential for complex physiological processes (for example, embryonic development and the immune response) and regulate cell survival, apoptosis, proliferation, and migration (24, 44, 49, 62). MAPKs are activated by conserved signaling modules that function as a protein kinase cascade. Dual phosphorylation on threonine and tyrosine residues within a Thr-Xaa-Tyr motif located in protein kinase subdomain VIII causes MAPK activation. This phosphorylation is mediated by a dual-specificity protein kinase (MAPK kinase [MAPKK]) which, in turn, is activated by phosphorylation mediated by a serine/threonine protein kinase (MAPKK kinase [MAPKKK]).
In the yeast Saccharomyces cerevisiae, five MAPK pathways that regulate mating, sporulation, filamentation, osmoregulation, and cell wall biosynthesis have been described (2, 32). In mammals, three major groups of MAPKs have been identified by molecular cloning (24, 44). These include the extracellular signal-regulated protein kinases (ERK), the p38 MAPKs, and the c-Jun NH2-terminal kinases (JNK). The physiological role of each of these groups of MAPKs has not been fully elucidated. However, each group of mammalian MAPKs is activated by a distinct signaling module and appears to be coupled to different biological responses.
The JNK group of MAPKs is activated by treatment of cells with inflammatory cytokines and by exposure to environmental stress. Biochemical studies demonstrate that the JNK protein kinases phosphorylate the NH2-terminal activation domain of components of the AP-1 transcription factor, including c-Jun and ATF2 (24). Phosphorylation by JNK increases AP-1 transcription activity. Thus, the JNK signaling pathway contributes to the regulation of AP-1 activity. This conclusion is supported by genetic evidence that Jun mediates some effects of the JNK signaling pathway (21, 41, 52) and by the observation that mice with targeted disruptions of genes that encode components of the JNK signaling pathway exhibit defects in stress-induced AP-1 transcription activity (67–69).
Recent studies of model genetic organisms have established physiological roles for the JNK signaling pathway. In the insect Drosophila melanogaster, JNK is required for early embryonic morphogenesis. JNK-deficient animals fail to initiate dorsal closure, a morphogenetic process in which the lateral epidermal cells spread (elongate and migrate) to cover the dorsal surface of the embryo (42, 53). JNK is also required for some forms of developmental apoptosis in Drosophila (1). In the nematode Caenorhabditis elegans, JNK is required for the normal function of type D GABAergic (GABA; γ-aminobutyric acid) motor neurons (26). The physiological role of the mammalian JNK signaling pathway has also been studied. Three genes encode the JNK protein kinase in mammals. The Jnk1 and Jnk2 genes are expressed ubiquitously, while the Jnk3 gene is expressed primarily in the brain (8, 16, 25, 28, 51). The JNK signaling pathway contributes to neuronal apoptosis in response to stress (4, 10, 64, 69) and during development (27). JNK is also required for apoptosis of CD4+ CD8+ double-positive thymocytes caused by anti-CD3 in vivo (43, 45). The JNK signaling pathway can also contribute to proliferative responses (24). In addition, the JNK signaling pathway is critical for immune cell function because Jnk1−/− (11) and Jnk2−/− (45, 68) mice exhibit defective immune responses. Further studies are required to fully establish the physiological role of the JNK signaling pathway. However, the studies outlined above demonstrate that the JNK pathway provides a mechanism that cells employ to mount an appropriate response to extracellular stimulation (24).
The JNK protein kinases are activated by evolutionarily conserved signaling modules that include MKK4 and MKK7 (24). Molecular cloning studies have identified MKK4 in mammals (9, 30, 47) and Drosophila (18). Similarly, the MKK7 protein kinase has been identified in mammals and Drosophila (15, 20, 35, 58, 59). Genetic analysis of Drosophila and gene targeting studies in mice demonstrate that MKK4 and MKK7 serve nonredundant roles during development (14, 15, 36, 37, 56, 67). The MKK4 and MKK7 protein kinases are phosphorylated and activated in response to extracellular stimulation by several MAPKKKs, including ASK1, TAK1, TPL2, and members of the MLK and MEKK groups (62).
The JNK signaling cascade can be reconstituted in vitro with purified JNK, MKK4 or MKK7, and a MAPKKK. However, it is likely that the JNK signaling cascade may be organized into defined modules in vivo (61). Thus, the MAPKKK MEKK1 binds to JNK, MKK4, and the Ste20p-related protein kinase NIK (54, 63, 65). Transmission of signals from MEKK1 to JNK may be facilitated by the formation of this complex in vivo (61). Functional signaling modules could also be created by the interaction of JNK signaling pathway components with other proteins (61). Recent studies have identified JIP1 (60) and JIP2 (70) as putative scaffold proteins that interact with multiple components of a JNK signaling module and facilitate JNK activation in vivo (61). A second example of a putative mammalian scaffold protein, MP1, was found to function within the ERK MAPK pathway (48). It is likely that such scaffold complexes contribute to the regulation of MAPK activation in vivo because previous studies of MAPK signaling in yeast have established that the activation and function of the mating MAPK pathway requires the scaffold protein Ste5p (6, 34, 39). A scaffolding function for Pbs2p in the yeast osmoregulatory MAPK pathway has also been reported (38). Thus, scaffold proteins are established, at least in some MAPK pathways, to be critical for physiological control of signal transduction. However, the number and function of other possible scaffold proteins that interact with MAPK signaling modules remain to be elucidated.
The purpose of the study described in this report was to identify a novel mammalian scaffold protein that interacts with a MAPK signaling module. We demonstrate that the JIP3 protein interacts with components of a JNK signaling module and facilitates JNK activation in vivo. JIP3 is structurally distinct from the previously identified JIP proteins and represents the founding member of a new class of putative MAPK scaffold proteins.
MATERIALS AND METHODS
Molecular cloning of JIP3.
Partial JIP3 cDNA clones were isolated from a mouse embryo cDNA library by the two-hybrid method using S. cerevisiae L40 (7, 10). The bait plasmid (pLexA-JNK1) was constructed by the insertion of the JNK1 cDNA in the polylinker of plasmid pBTM116 (7, 10). A full-length JIP3a cDNA clone was isolated from a mouse heart Uni-ZAPXR library (Stratagene Inc.), and a full-length JIP3b cDNA clone was isolated from a mouse brain λZAPII library (Stratagene) by plaque hybridization using a JIP3 cDNA fragment as a probe. The largest clones obtained (5,442 and 5,562 bp) included the complete open reading frame of mouse JIP3a and JIP3b. The sequences of these JIP3 cDNA clones were determined with an Applied Biosystems 373A machine.
Plasmids.
Expression vectors for JIP3 were constructed from plasmids pCDNA3 (Invitrogen Inc.), pEBG (47) and pGEX (Amersham Pharmacia Biotech Inc.) by subcloning PCR fragments of JIP3. Expression vectors for MAPK, MAPKK, MAPKKK, JIP1, and JIP2 were described previously (60, 70).
Antibodies.
The antibodies to the Flag epitope tag (M2; Sigma), the hemagglutinin (HA) epitope tag (12CA5; Boehringer Mannheim), the T7-Tag epitope tag (Novagen Inc.), glutathione S-transferase (GST; Pharmacia-LKB Biotechnology Inc.), and tubulin (Sigma Chemical Co.) were purchased from the indicated supplier. Antibodies to JIP1 and JIP2 were described previously (70). Antibodies to JIP3 were prepared by immunizing a rabbit with purified bacterially expressed JIP3b (residues 141 to 241), using standard techniques (19).
Cell culture.
COS7 cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum in an incubator with humidified air (5% CO2) at 37°C. The COS7 cells were transfected with plasmid expression vectors by the Lipofectamine procedure (Life Technologies Inc.). PC12 cells were cultured on collagen-coated plastic dishes in DMEM supplemented with 0.5 mM glutamine, 10% horse serum, and 5% fetal bovine serum (Life Technologies Inc.) in a humidified incubator (10% CO2) at 37°C (64). The cells were differentiated by incubation in DMEM supplemented with 1% horse serum, 0.5 mM glutamine, and 50 ng of nerve growth factor (NGF) per ml (64). The effect of NGF withdrawal was examined in cultures with neutralizing NGF antibody and without NGF (64).
Biochemical assays.
Cultured cells and mouse brain were solubilized in lysis buffer (20 mM Tris-Cl [pH 7.4], 137 mM NaCl, 2 mM EDTA, 25 mM β-glycerophosphate, 2 mM pyrophosphate, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 10 μg of leupeptin per ml, 10% glycerol, 1% Triton X-100). GST fusion proteins were isolated by incubation with glutathione-agarose (Amersham Pharmacia Biotech Inc.) beads (20 μl) for 3 h at 4°C. Proteins were immunoprecipitated by incubation for 3 h at 4°C with antibodies bound to protein G-Sepharose (Amersham Pharmacia Biotech Inc.). Immunoblot analysis was performed by enhanced chemiluminescence detection (Kirkegaard & Perry Inc.). Binding assays using purified proteins were performed by methods described previously (60, 70). The methods used for immune complex kinase assays, phosphoamino acid analysis, and phosphopeptide mapping have also been described elsewhere (17). The effect of scaffold proteins on MLK3-stimulated JNK activity was examined in COS7 cell transfection assays using the in-gel method with 0.25 mg of substrate (GST–c-Jun) per ml polymerized in the gel (60, 70). Proteolytic digestion experiments were performed with purified recombinant caspases (provided by S. Kharbanda, Dana-Farber Cancer Institute, Boston, Mass.) and [35S]methionine-labeled in vitro-translated poly(ADP-ribose) polymerase (PARP) and JIP3b by incubation at 37°C for 30 min. The products of the proteolytic digestion were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and detected by autoradiography.
Immunofluorescence analysis.
PC12 cells were grown on glass coverslips coated with laminin plus poly-d-lysine and differentiated in DMEM supplemented with 1% horse serum, 0.5 mM glutamine, and 50 ng of NGF per ml (64). The cells were fixed by incubation (10 min) with 4% paraformaldehyde in phosphate-buffered saline (PBS), washed with 10 mM glycine in PBS, and permeabilized with 0.2% Triton X-100 in PBS. After incubation (15 min) with 3% (wt/vol) bovine serum albumin (BSA) in PBS, the coverslips were incubated (1 h) with primary antibodies in PBS with 3% BSA. The primary antibodies were a rabbit polyclonal antibody to JIP3, a rabbit polyclonal antibody to neuron-specific enolase (Oncogene Research Products), and a mouse monoclonal antibody to tubulin (Sigma Chemical Co.). Immunocomplexes were detected with Texas red-conjugated anti-rabbit immunoglobulin and fluorescein-conjugated anti-mouse immunoglobulin secondary antibodies (Jackson ImmunoResearch Inc.) in 3% BSA in PBS. The coverslips were mounted in Vectashield (Vector Laboratories Inc.). Fluorescence microscopy was performed with a Zeiss Axioplan microscope.
Nucleotide sequence accession numbers.
The sequences of JIP3a and JIP3b have been deposited in GenBank with accession no. AF178636 and AF178637, respectively.
RESULTS
Molecular cloning of JIP3.
To identify novel proteins that interact with JNK, we screened a mouse embryo cDNA library by the two-hybrid method with JNK1 as the bait (7, 10). Two clones isolated from the library that interacted with JNK1, but not with ERK2, in the two-hybrid assay were found by sequence analysis to represent overlapping fragments corresponding to a partial cDNA clone. We refer to the protein encoded by this cDNA as JNK-interacting protein 3 (JIP3).
Two cDNA libraries were screened by plaque hybridization to isolate full-length clones (Fig. 1). The largest clone isolated from a mouse heart library (5,442 bp; JIP3a) and the largest clone isolated from a mouse brain library (5,512 bp; JIP3b) were each found to contain a single long open reading frame. An in-frame termination codon was identified in the predicted 5′ untranslated sequence, and a termination codon was identified prior to the predicted 3′ untranslated region. Sequence analysis demonstrated substantial identity between the JIP3a and JIP3b cDNA clones, but there were significant differences (Fig. 1b). JIP3b contains two insertions (18 and 27 bp) and a single base pair change at codon 376. JIP3b is therefore larger than JIP3a because of the two insertions (six and nine amino acids). In addition, Phe376 (TTT) of JIP3a is replaced by Leu (TTG) in JIP3b. It is likely that JIP3a and JIP3b are derived by alternative splicing of transcripts derived from a single gene.
FIG. 1.



Structure and expression of JIP3. (A) Structure of JIP3 illustrated schematically. (B) Primary sequence of mouse JIP3a and JIP3b deduced from the sequence of cDNA clones, presented in single-letter code. Numbering is based on the sequence of JIP3b. Residues identical to those in JIP3a (.), deletions (-), and termination codons (#) are indicated. (C) Expression of JIP3 mRNA, examined by Northern blot analysis of 2 μg of poly(A)+ mRNA isolated from different murine tissues (Clontech Inc.), using a 1.3-kb EcoRI/XbaI fragment of the JIP3a cDNA as a probe (inset). RNA size markers (in kilobases) are indicated on the left. Expression of JIP3 mRNA in different mouse tissues was also examined by dot blot analysis of 5 μg of total RNA hybridized to the JIP3a probe and was quantitated with a PhosphorImager (Molecular Dynamics Inc.). The data are presented graphically as the amount of expression relative to whole brain (100%). Sk, skeletal; Sm, smooth; Submax., submaxillary. (D) Expression of JIP3 protein, examined by Western blot analysis of 75 μg of total protein isolated from different murine tissues and brain subregions (Geno Technology Inc.), using a polyclonal antibody to JIP3. Protein size markers are indicated on the left.
Computer-assisted sequence analysis of the JIP3 proteins demonstrated the presence of an extended predicted coiled coil domain in the NH2-terminal region (5). This region also contained a predicted leucine zipper (29). These structural features were conserved in JIP3a and JIP3b (Fig. 1). Analysis of the GenBank database indicated four entries describing sequences similar to JIP3. One partial human cDNA (accession no. AB028989) encodes a predicted protein that is very similar to the COOH-terminal region of murine JIP3 and may represent a fragment of human JIP3 (55). A second partial human cDNA (accession no. AB011088) also encodes a protein with similarity to the COOH-terminal region of JIP3, but this protein is the product of a distinct gene (55) and may be a related member of the JIP3 group (JIP3β). A more distantly related human sequence with similarity to JIP3 (accession no. X91879) may correspond to a third member of a JIP3 gene family (JIP3γ) (50). A fourth sequence related to JIP3 in the database corresponds to a predicted protein encoded by the C. elegans genome and may be a nematode member of the JIP3 group of proteins (hypothetical protein ZK1098.10). No functional analysis of these JIP3-related sequences has been reported.
Expression of JIP3 in murine tissues.
We examined the expression of JIP3 in different murine tissues by Northern blot analysis (Fig. 1C). The largest amount of JIP3 mRNA (approximately 6 kb) expression was detected in the brain, with lower levels in the heart and other tissues. To confirm this pattern of JIP3 expression, we prepared a rabbit polyclonal antibody by using recombinant JIP3 as an antigen and examined expression of the JIP3 protein in different tissues by Western blot analysis (Fig. 1D). A high level of JIP3 expression in the brain was detected. In addition, expression of the JIP3 protein was found in the heart and lung. A low level of JIP3 expression was detected in other tissues.
The highest amount of JIP3 expression was detected in the brain (Fig. 1C and D). To test whether the expression of JIP3 in the brain was restricted to a subregion of the brain, we examined the expression of JIP3 by immunoblot analysis of protein extracts prepared from subregions of mouse brain. This analysis demonstrated that the JIP3 protein was widely expressed throughout many regions of the murine brain (Fig. 1D).
JIP3 selectively binds to the JNK group of MAPKs.
JIP3 was isolated in a two-hybrid screen using JNK as the bait. This result suggests that JIP3 may bind JNK. To test this hypothesis, we expressed JIP3 and JNK in COS7 cells and examined the interaction of these proteins by coprecipitation analysis (Fig. 2A). We found that JNK coprecipitated with JIP3 (Fig. 2A) and that JIP3 coprecipitated with JNK (Fig. 2B). However, no coprecipitation of JIP3 with ERK or p38 MAPKs was detected (Fig. 2A).
FIG. 2.

JIP3 binds to the JNK group of MAPKs. (A) Interaction of JIP3 with MAPKs, examined by expression of GST-JIP3a and HA epitope-tagged MAPKs in COS7 cells. GST-JIP3a was isolated from cell lysates by incubation with glutathione-agarose beads and was detected by immunoblot analysis using an antibody that binds GST. The binding to MAPKs was examined by immunoblot analysis using an antibody that binds the HA epitope tag. The interaction of JIP3 with JNK1α1, JNK2α2, JNK3α2, p38α, and ERK2 was investigated. Control experiments were performed by transfection of an empty expression vector instead of the MAPK expression vectors. (B) Epitope-tagged T7-JIP3a and HA-JNK1 were expressed in COS7 cells. Lysates were prepared, and the amount of JIP3 and JNK1 was examined by immunoblot analysis using monoclonal antibodies to the T7 and HA epitopes. HA-JNK1 was immunoprecipitated with the HA antibody, and T7-JIP3a in the immunoprecipitates was detected by immunoblot analysis with an antibody to the T7 epitope tag. (C) Comparison of the binding of 10 JNK isoforms to JIP3. GST and GST-JIP3a were expressed in COS7 cells and immobilized on glutathione-agarose beads. The JNK MAPKs were prepared by in vitro translation in the presence of [35S]methionine and incubated with immobilized GST and GST-JIP3a. No interaction of JNK with GST was detected. However, the JNK protein kinases bound to GST-JIP3a. The bound JNK was detected by SDS-PAGE and autoradiography. The radioactivity was quantitated by PhosphorImager analysis (Molecular Dynamics) and is presented graphically as relative binding. (D) Deletion analysis of JIP3. A series of GST-JIP3b fragments were expressed in bacteria, purified, and immobilized on glutathione-agarose beads. The interaction of these JIP3b fragments with JNK was examined by incubation of the immobilized JIP3b proteins with lysates prepared from COS7 cells expressing Flag epitope-tagged JNK1α1. Bound JNK was detected by immunoblot analysis using antibody M2, which binds the Flag epitope tag. (E) Mutational analysis of the JNK binding domain of JIP3. The function of the JNK binding domain was examined in binding assays using immobilized GST (lane 1) or GST-JIP3b (residues 141 to 241) (lanes 2 to 13) and Flag epitope-tagged JNK1α1 (lane 14). Competition assays were performed by including in the binding assay a synthetic peptide corresponding to the JNK binding domain (10 μg/ml) (lane 3). The effect of replacement of residues in the JNK binding domain with Gly was examined (lanes 4 to 13). The binding of JNK to the immobilized GST-JIP3b is presented. (F) Primary sequences of the JNK binding domains of JIP3 and JIP1 and the consensus sequence.
Interestingly, we found that the extent of coprecipitation of JIP3 was greater in assays using JNK3 than in assays using JNK1 or JNK2. This difference suggests that JIP3 may selectively bind JNK isoforms. We therefore tested the interaction of all 10 known JNK protein kinase isoforms with JIP3 in vitro (Fig. 2C). No binding of JNK to recombinant GST was observed. However, JNK binding to recombinant GST-JIP3 was detected. Quantitation of the extent of binding demonstrated that increased binding was observed for JNK1β and JNK3α isoforms compared to other JNK isoforms (Fig. 2C). These data indicate that while JIP3 does exhibit some selectivity in binding to JNK isoforms, JIP3 is able to bind all JNK isoforms.
Comparison of the two partial cDNA clones isolated from the two-hybrid screen indicated an overlapping region that encoded JIP3 residues 141 to 241. To test whether this region binds to JNK, we expressed this fragment of JIP3 as a GST fusion protein in bacteria. The GST-JIP3 protein was purified, immobilized on glutathione-agarose beads, and incubated with COS7 cell extracts. The agarose beads were washed, and the bound JNK was detected by immunoblot analysis. It was found that this JIP3 fragment bound JNK (Fig. 2D). To further define the region that is required for interaction with JNK, we constructed a deletion series by removal of NH2- and COOH-terminal amino acid residues. Binding assays demonstrated that the region surrounding JIP3 residues 207 to 216 was required for interaction with JNK (Fig. 2D).
To further define the JNK binding domain of JIP3, we examined the effect of point mutations within the predicted JNK binding domain. These experiments were performed by competition analysis (10) using synthetic peptides corresponding to the JIP3 predicted JNK binding domain. Binding assays were performed with JNK and immobilized recombinant GST-JIP3 (Fig. 2E). Addition of the synthetic peptide Arg202-Lys-Glu-Arg-Pro-Thr-Ser-Leu-Asn-Val-Phe-Pro213 caused a dose-dependent inhibition of JNK binding to GST-JIP3 (data not shown). Complete inhibition of JNK binding was observed in the presence of 10 μg of synthetic peptide per ml (Fig. 2E). Analysis of competitive binding assays using mutated synthetic peptides containing a Gly substitution indicated that JIP3 residues Arg205, Pro206, Thr207, Ser208, and Leu209 were important for interaction with JNK. Interestingly, this region of JIP3 shows primary sequence identity to the JNK binding domain of JIP1 (10). Alignment of the JNK binding domains of JIP3 and JIP1 indicated significant sequence identity (Fig. 2F).
JIP3 is phosphorylated by JNK in vitro and in vivo.
The interaction between JNK and JIP3 suggests that JIP3 may be a JNK substrate. To test this hypothesis, we performed in vitro protein kinase assays using purified bacterially expressed JIP3 (Fig. 3A). These experiments demonstrated that JIP3 was phosphorylated by JNK. Control experiments demonstrated that JIP3 was not phosphorylated by kinase-inactive JNK (data not shown). The extent of JIP3 phosphorylation by JNK was similar to that of a known JNK substrate, ATF2 (17, 31). Control experiments demonstrated that JIP3 was not phosphorylated by the related MAPKs ERK2 and p38α.
FIG. 3.

JIP3 is phosphorylated by JNK in vivo and in vitro. (A) JIP3 is phosphorylated by JNK in vitro. Epitope-tagged JNK1α1, p38α, and ERK2 MAPKs were expressed in COS-7 cells. The JNK and p38 MAPKs were activated by treatment of the cells without (−) and with (+) UV-C radiation (80 J/m2). The ERK MAPK was activated by treatment of the cells without (−) and with (+) 100 nM phorbol myristate acetate. The MAPKs were isolated by immunoprecipitation using a monoclonal antibody to the HA epitope tag, and immunocomplex protein kinase assays were performed with [γ-32P]ATP and GST-JIP3b (residues 190 to 380) as the substrate. Control experiments were performed with known substrates for JNK (GST-ATF2), p38 (GST-ATF2), and ERK (GST–c-Myc). Phosphorylation of the substrate proteins was detected following SDS-PAGE by autoradiography. Phosphorylation of JIP3b by JNK, but not by p38 or ERK, was observed. (B and C) Mutational analysis of JIP3b phosphorylation by JNK. Three potential JNK phosphorylation sites (Ser/Thr-Pro) were identified by sequence analysis (Thr266, Thr276, and Thr287). These potential phosphorylation sites were replaced with Ala residues, and the wild-type and mutated JIP3b proteins were examined as substrates for JNK in vitro. The phosphorylation of these JIP3b proteins was detected following SDS-PAGE by autoradiography (B) and was also examined by phosphoamino acid analysis (C). (D) JNK activation in vivo decreases the electrophoretic mobility of JIP3. Flag epitope-tagged JIP3b was expressed in COS7 cells and was detected by immunoblot analysis using antibody M2. The cells were treated without (−) and with (+) UV-C radiation (80 J/m2) to activate JNK. The effect of replacement of the JNK phosphorylation sites Thr266, Thr276, and Thr287 with Ala (Thr/Ala) was examined. (E) COS7 cells expressing epitope-tagged wild-type and mutated [Ala266, Ala276, Ala287] JIP3b were metabolically labeled with [32P]phosphate. The phosphorylated JIP3b proteins were detected following immunoprecipitation and SDS-PAGE by autoradiography (left) and were examined by phosphoamino acid analysis (right). (F) Analysis of JIP3 phosphorylation by phosphopeptide mapping. Wild-type and mutated (Thr/Ala) [Ala266, Ala276, Ala287] JIP3b phosphorylated in vivo were investigated by phosphopeptide mapping. Maps of JIP3 phosphorylated by JNK1 in vitro were also examined. Comparative phosphopeptide maps were prepared by mixing equal amounts of radioactivity derived from wild-type JIP3 phosphorylated in vivo and in vitro. The origin (o) is indicated on the right. The horizontal and vertical dimensions are electrophoresis and chromatography, respectively. The major [32P]phosphopeptide present in maps of in vivo phosphorylated wild-type JIP3b and absent in maps of mutated (Thr/Ala) JIP3b is indicated with an arrow.
Examination of the primary sequence of JIP3 indicates three potential JNK phosphorylation sites (Ser/Thr-Pro): Thr266, Thr276, and Thr287. To test whether these residues are sites of JNK phosphorylation, we investigated the effect of point mutations at each of these sites (Fig. 3B). Replacement of each Thr residue with Ala did not markedly decrease the phosphorylation of JIP3 by JNK. However, the simultaneous replacement of all three Thr residues with Ala caused a large decrease in phosphorylation. Phosphoamino acid analysis confirmed that JNK caused Thr phosphorylation of JIP3 (Fig. 3C). This phosphorylation on Thr was markedly decreased when Thr266, Thr276, and Thr287 were replaced with Ala. These data indicate that JNK phosphorylated JIP3 on Thr266, Thr276, and Thr287 in vitro.
To test whether JIP3 was phosphorylated by JNK in vivo, we investigated the effect of JNK activation on the electrophoretic mobility of JIP3 by Western blot analysis (Fig. 3D). Exposure of cells to UV-C radiation caused a marked decrease in JIP3 mobility. In contrast, no decrease in JIP3 mobility following exposure to UV radiation was detected when the three sites of in vitro phosphorylation by JNK (Thr266, Thr276, and Thr287) were replaced with Ala. The effect of these point mutations to eliminate the UV radiation-stimulated electrophoretic mobility shift is consistent with the hypothesis that JNK phosphorylates JIP3 in vivo.
To examine JIP3 phosphorylation by more direct biochemical methods, we isolated JIP3 from cells metabolically labeled with [32P]phosphate (Fig. 3E). This metabolic labeling procedure causes JNK activation, most likely as a consequence of the exposure of the cells to high levels of ionizing radiation. Phosphoamino acid analysis demonstrated that JIP3 was phosphorylated extensively on Ser residues. No Tyr phosphorylation was detected, but JIP3 phosphorylation on Thr was observed. Replacement of Thr266, Thr276, and Thr287 with Ala residues caused decreased Thr phosphorylation of JIP3 in vivo. Comparative tryptic phosphopeptide mapping demonstrated that wild-type JIP3 contained a phosphopeptide that was absent in maps of the mutated JIP3 protein (Fig. 3F). This phosphopeptide comigrated with a major phosphopeptide observed in maps of JIP3 phosphorylated by JNK in vitro. Together, these data establish that JIP3 is phosphorylated in vivo on sites phosphorylated by JNK in vitro.
JIP3 binds to the MAPKK MKK7 and the MAPKKK MLK3.
We have reported that JIP1 and JIP2 interact with several components of the JNK signaling pathway, including the JNK protein kinases (60, 70). Since JIP3 binds to JNK (Fig. 2), we tested whether JIP3, like the JIP1 and JIP2 proteins, might also interact with other components of the JNK signaling pathway.
The interaction of JIP3 with MAPKK was investigated by coprecipitation analysis (Fig. 4A). These experiments demonstrated that the JNK activator MKK7 coprecipitated with JIP3 (Fig. 4A) and that JIP3 coprecipitated with MKK7 (Fig. 4B). In contrast, no interaction between JIP3 and the JNK activator MKK4 was detected. Similarly, no binding of JIP3 to the p38 MAPK activators MKK3 and MKK6 or the ERK activator MEK1 was observed. These data indicate that JIP3 selectively interacts with MKK7 and not with other members of the MAPKK group of protein kinases (Fig. 4A). Binding assays performed with purified recombinant proteins demonstrated that MKK7 directly interacts with JIP3 (Fig. 4C). Deletion analysis of JIP3 demonstrated that the central region of the molecule (residues 410 to 815) bound strongly to MKK7 (Fig. 4D). A weak interaction between the NH2-terminal region of JIP3 (residues 1 to 442) and MKK7 was also detected (Fig. 4D). The central region of JIP3 that strongly binds MKK7 (residues 410 to 815) is distinct from the JNK binding domain located within the NH2-terminal region of JIP3 (residues 205 to 209 [Fig. 2]). The primary site of JIP3 interaction with MKK7 is therefore different from the site of JIP3 interaction with JNK.
FIG. 4.

JIP3 binds to the MAPKK MKK7. (A) JIP3a was expressed as a GST fusion protein in COS7 cells together with epitope-tagged MEK1, MKK3, MKK4, MKK6, and MKK7. Control experiments were performed with an empty vector instead of the MAPKK expression vector. The expression of JIP3a and MAPKK was examined by immunoblot analysis of cell lysates. GST-JIP3a was isolated on glutathione-agarose beads, and the bound MAPKKs were detected by immunoblot analysis. (B) Epitope-tagged T7-JIP3a and Flag-MKK7 were expressed in COS-7 cells. Lysates were prepared, and the amount of JIP3 and MKK7 was examined by immunoblot analysis using monoclonal antibodies to the T7 and Flag epitopes. The Flag-MKK7 was immunoprecipitated with antibody M2, and T7-JIP3a in the immunoprecipitates was detected by immunoblot analysis with an antibody to the T7 epitope tag. (C) Purified recombinant GST and GST-JIP3a were immobilized on glutathione-agarose and incubated with purified recombinant Flag-tagged MKK7. Bound MKK7 was detected by immunoblot analysis using an antibody that binds the Flag epitope tag. (D) Deletion analysis of JIP3. To define the MKK7 binding region of JIP3, in vitro-translated fragments of JIP3a (residues 1 to 442, 410 to 815, and 800 to 1337) were prepared in the presence of [35S]methionine. Control experiments were performed with in vitro-translated luciferase. These proteins were incubated with GST or GST-MKK7 immobilized on glutathione-agarose. The binding of JIP3 was detected following SDS-PAGE by autoradiography. Binding of in vitro-translated JIP3 to GST-MKK7, but not to GST, was detected.
Coprecipitation assays were performed to examine the interaction of MAPKKK with JIP3. No interaction of JIP3 with c-Raf, MEKK1, MEKK4, DLK, MLK2, or ASK1 was observed (Fig. 5A). However, MLK3 was found to coprecipitate with JIP3 (Fig. 5A), and JIP3 was found to coprecipitate with MLK3 (Fig. 5B). Thus, JIP3 appears to interact selectively with MLK3 and not with other members of the MAPKKK group of protein kinases. Binding assays performed with purified recombinant proteins demonstrated that MLK3 directly interacts with JIP3 (Fig. 5C). Deletion analysis of JIP3 indicated that the COOH-terminal region JIP3 (residues 800 to 1337) did not bind MLK3 (Fig. 5D). However, MLK3 binding to the NH2-terminal region of JIP3 was observed (residues 1 to 815). Deletions within this NH2-terminal region indicated strong binding to JIP3 residues 1 to 442, but weak binding of MLK3 was also detected in experiments using JIP3 residues 420 to 815.
FIG. 5.

Interaction of JIP3 with MAPKKK. (A) Epitope-tagged MAPKKKs and GST-JIP3a were expressed in COS7 cells. Control experiments were performed with empty vector instead of the MAPKKK expression vectors. GST-JIP3a protein was isolated on glutathione-agarose beads. The binding of MAPKKK to JIP3 was examined by immunoblot analysis using an antibody that binds the epitope tag. (B) Epitope-tagged T7-JIP3a and HA-MLK3 were expressed in COS7 cells. Lysates were prepared, and the amount of JIP3a and MLK3 was examined by immunoblot analysis using monoclonal antibodies to the T7 and HA epitopes. HA-MLK3 was immunoprecipitated with the HA antibody, and T7-JIP3a in the immunoprecipitates was detected by immunoblot analysis with an antibody to the T7 epitope tag. (C) Interaction of purified recombinant MLK3 with JIP3a. Epitope-tagged HA-MLK3 was isolated by immunoprecipitation and was eluted by incubation with HA synthetic peptide (20 μg/ml) for 2 h at 4°C. The purified soluble MLK3 was incubated with GST or GST-JIP3a immobilized on glutathione-agarose. The amount of MLK3, GST, and GST-JIP3a was examined by immunoblot analysis using HA or GST antibodies. The agarose beads were washed, and bound MLK3 was detected by immunoblot analysis with an antibody to the HA epitope tag. (D) Deletion analysis of JIP3. To define the MLK3 binding region of JIP3, fragments of JIP3a (residues 1 to 1337, 1 to 815, 1 to 442, 420 to 815, and 800 to 1337) fused to GST were immobilized on glutathione-agarose. Control experiments were performed with immobilized GST. These immobilized proteins were incubated with MLK3 (top) or luciferase (bottom) prepared by in vitro translation in the presence of [35S]methionine. Binding to the immobilized proteins was examined following SDS-PAGE by autoradiography.
Together, these data demonstrate that JIP3 interacts with proteins that can form a MAPK signaling module, including JNK, MKK7, and MLK3.
JNK activation increases complex formation with JIP3.
JNK binding to JIP3 was detected in experiments using purified recombinant proteins and in transfection assays using overexpressed recombinant proteins (Fig. 2). These observations suggested that endogenous JNK and JIP3 may form complexes in cells. To test this hypothesis, we prepared soluble extracts from mouse brain and performed coimmunoprecipitation analysis. This analysis confirmed that endogenous JIP3 forms complexes with JNK and MKK7 (Fig. 6A).
FIG. 6.

Identification of JIP3 complexes. (A) The interaction of JIP3 with components of the JNK signaling pathway was examined by coimmunoprecipitation analysis. Soluble extracts prepared from mouse brain were immunoprecipitated with a nonimmune antibody (Control) and with antibodies to JNK, MKK7, JIP1, and JIP2. The immunoprecipitates were examined by immunoblot analysis with an antibody to JIP3. (B) Complex formation with JIP3 is increased by JNK activation. COS cells were transfected with an empty expression vector (Control) or with an expression vector for Flag-tagged JIP3b. The effect of replacement of the three JNK phosphorylation sites (Thr266, Thr276, and Thr287) with Ala was examined. The cells were exposed without (−) and with (+) UV-C radiation (80 J/m2) and incubated for 1 h. JIP3b was isolated by immunoprecipitation with monoclonal antibody M2. The presence of JNK in the immunoprecipitates was examined by immunoblot analysis by probing with a rabbit polyclonal antibody to JNK.
The complexes formed between JNK and JIP3 might be regulated by activation of the JNK signaling pathway. We therefore examined the effect of JNK activation (caused by UV-C radiation) on the coimmunoprecipitation of JNK and JIP3 (Fig. 6B). JNK activation was found to markedly increase the coimmunoprecipitation of JIP3 with endogenous JNK. These data indicated that JNK activation is associated with increased formation of JIP3 complexes with JNK. Since JIP3 is a JNK substrate, it was possible that this increased complex formation was related to changes in JIP3 phosphorylation. To test this hypothesis, we examined the effect of replacement of the three sites of JNK phosphorylation on JIP3 (Thr266, Thr276, and Thr287) with Ala. The wild-type and phosphorylation-defective JIP3 proteins were found to form similar complexes with JNK (Fig. 6B). The phosphorylation of JIP3 by JNK therefore does not contribute to the regulated formation of JNK complexes with JIP3.
Together, these data demonstrate that JNK activation augments the formation of JNK complexes with JIP3.
JIP3 increases MLK3-stimulated JNK activity.
The interaction of JIP3 with JNK, MKK7, and MLK3 protein kinases suggested that JIP3 may act as a molecular scaffold for a JNK signaling module. If JIP3 serves this function, we would expect the expression of JIP3 to increase the activation of JNK caused by MLK3. To test this hypothesis, we performed transfection assays using COS7 cells, which do not express JIP3 (data not shown) or the putative scaffold proteins JIP1 and JIP2 (60, 70). JNK activity was measured with c-Jun as the substrate (Fig. 7). Expression of JIP3 did not cause JNK activation. However, JIP3 increased the activation of JNK caused by MLK3. Similar results were obtained in experiments using JNK1, JNK2, and JNK3. The effect of JIP3 to potentiate MLK3-stimulated JNK activation was similar to that caused by the putative scaffold proteins JIP1 and JIP2 (Fig. 7). It was also found that both JIP1a and JIP1b caused potentiation of MLK3-stimulated JNK activation (data not shown). These data indicated that JIP3 might be a mammalian MAPK scaffold protein for a JNK signaling module.
FIG. 7.

JIP3 potentiates MLK3-stimulated JNK activity. The effect of JIP3 on MLK3-stimulated JNK activity was examined in cotransfection assays using HA epitope-tagged JNK1α1 (A), JNK2α2 (B), and JNK3α2 (C). The effect of expression of MLK3 and JIP3a was examined. Control experiments were performed with the JIP1 and JIP2 scaffold proteins. The expression of MLK3, JIP1, JIP2, JIP3, and JNK was investigated by immunoblot analysis. JNK was immunoprecipitated with an antibody that binds the HA epitope tag, and protein kinase activity was measured with [γ-32P]ATP and c-Jun as substrates. The phosphorylated c-Jun was detected following SDS-PAGE by autoradiography and was quantitated by PhosphorImager (Molecular Dynamics) analysis. The data presented were derived from one experiment. Similar data were obtained in seven independent experiments.
To examine the role of JIP3 phosphorylation during MLK3-stimulated JNK activation, we compared the effect of wild-type JIP3b and the phosphorylation-defective [Ala266, Ala276, Ala287] JIP3b protein that lacks the three sites of phosphorylation by JNK. We found that the wild-type and mutated JIP3b proteins caused similar potentiation of MLK3-stimulated JNK activation (data not shown). The phosphorylation of JIP3 by JNK therefore does not appear to contribute to JNK regulation.
JIP3 selectively interacts with JNK MAPK scaffold proteins.
The JIP1 and JIP2 scaffold proteins appear to function as oligomeric complexes formed from dimers or higher-order aggregates (70). We therefore examined whether JIP3 might also form oligomeric complexes (Fig. 8). T7 epitope-tagged and GST-tagged JIP3a proteins were expressed in COS7 cells. GST-JIP3 was isolated with glutathione-agarose, and the binding of T7-tagged JIP3 was examined by immunoblot analysis. The JIP3 proteins were found to coprecipitate (Fig. 8A). These data indicated that JIP3 forms oligomeric complexes (either dimers or higher-order oligomeric complexes). Deletion analysis indicated that the NH2-terminal region (residues 1 to 442) of JIP3 was sufficient for this interaction (data not shown). This region includes the extended predicted coiled coil domain, which may therefore mediate JIP3-JIP3 association.
FIG. 8.

JIP3 selectively interacts with JIP scaffold proteins. (A) Epitope (T7)-tagged JIP3a was coexpressed with GST-tagged JIP3a in COS7 cells and incubated with glutathione-agarose. Bound proteins were detected by immunoblot analysis with an antibody to the T7 epitope. Expression of T7-JIP3 and GST-JIP3 proteins in the cell lysates was examined by immunoblot analysis. (B) Epitope (T7)-tagged JIP1 and JIP2 were expressed in COS7 cells. JIP3a was also expressed as a GST fusion protein in COS7 cells and immobilized on glutathione-agarose beads. Bound proteins were detected by immunoblot analysis with an antibody to the T7 epitope. Expression of T7-JIP1, T7-JIP2, and GST-JIP3a proteins in the cell lysates was monitored by immunoblot analysis. (C) Epitope-tagged T7-JIP3a and Flag-JIP2 were expressed in COS-7 cells. Lysates were prepared, and the amount of JIP3 and JIP2 was examined by immunoblot analysis using monoclonal antibodies to the T7 and Flag epitopes. The Flag-JIP2 was immunoprecipitated with antibody M2, and T7-JIP3 in the immunoprecipitates was detected by immunoblot analysis with an antibody to the T7 epitope tag. (D) Deletion analysis of JIP3. To define the JIP2 binding region of JIP3a, fragments of JIP3a (residues 1 to 1337, 1 to 815, 1 to 442, 420 to 815, and 800 to 1337) fused to GST were immobilized on glutathione-agarose. Control experiments were performed with immobilized GST. These immobilized proteins were incubated with JIP2 prepared by in vitro translation in the presence of [35S]methionine. Binding of JIP2 to the immobilized proteins was examined following SDS-PAGE by autoradiography. (E) Deletion analysis of JIP2. To define the JIP3 binding region of JIP2, fragments of JIP2 fused to GST were immobilized on glutathione-agarose. Control experiments were performed with GST. These immobilized proteins were incubated with JIP3a prepared by in vitro translation in the presence of [35S]methionine. The binding of JIP3 to an NH2-terminal fragment of JIP2 (residues 1 to 229) and a COOH-terminal fragment of JIP2 (residues 557 to 824) was examined following SDS-PAGE by autoradiography.
We also tested whether JIP3 interacted with the JIP1 and JIP2 scaffold complexes. We expressed T7-tagged JIP1, T7-tagged JIP2, and GST-tagged JIP3a in COS7 cells. The GST-JIP3 was isolated on glutathione-agarose, and the binding of JIP1 and JIP2 proteins to JIP3 was examined by immunoblot analysis. We found that JIP2 (but not JIP1) coprecipitated with JIP3 (Fig. 8B) and that JIP3 coprecipitated with JIP2 (Fig. 8C). Deletion analysis demonstrated that the central region of JIP3 (residues 420 to 815) (Fig. 8D) and the COOH-terminal region of JIP2 (residues 557 to 824) (Fig. 8E) were required for this interaction. These data demonstrate that JIP3 can form complexes with the JIP2 scaffold protein. However, complex formation with JIP2 was not required for the effect of JIP3 to increase MLK3-stimulated JNK activity (Fig. 7) since COS7 cells do not express JIP2 (70).
To test whether complexes are formed between endogenous JIP3 and JIP2, we performed coimmunoprecipitation assays using mouse brain. We found that JIP2 coimmunoprecipitated with JIP3 (Fig. 6A). In addition, a low level of JIP1 was found to coprecipitate with JIP3. Since recombinant JIP1 and JIP3 did not interact (Fig. 8B) and heterodimeric complexes are formed between JIP1 and JIP2 (70), the low level of coprecipitation of endogenous JIP1 with JIP3 (Fig. 6A) may reflect the interaction of JIP3 with a JIP1-JIP2 heterodimer.
These data demonstrate that different complexes can be formed by the association of various members of the JIP group of scaffold proteins.
NGF regulates the expression of JIP3.
The observation that a greater amount of JIP3 was detected in the brain than in other tissues (Fig. 1) suggested that JIP3 expression might be induced during neuronal differentiation. To test this hypothesis, we investigated the expression of JIP3 in the PC12 cell model of neuronal differentiation in response to NGF (Fig. 9A). Western blot analysis demonstrated that JIP3 was not detected in proliferating PC12 cells, but JIP3 expression was observed in differentiated PC12 cells. Time course analysis indicated that JIP3 expression was not an early event following NGF treatment (Fig. 9B). However, JIP3 expression was detected at 5 days following initiation of NGF-stimulated differentiation.
FIG. 9.

NGF regulates the expression of JIP3. (A) JIP3 expression was induced by treatment of PC12 cells with NGF. PC12 cells were differentiated to a neuron-like phenotype by culture in the presence of NGF for 12 days. The expression of JIP3 was examined by immunoblot analysis using preimmune and immune sera prepared from a rabbit immunized with recombinant JIP3. Control experiments were performed with COS7 cells transfected with an empty expression vector or with a JIP3 expression vector. (B) Expression of JIP3 by PC12 cells treated with NGF for various times was examined by immunoblot analysis. Extracts prepared from COS cells transfected with a JIP3b expression vector were examined in control experiments (JIP3). (C) Differentiated PC12 cells (+ NGF) were deprived of NGF (− NGF) for 24 h in the presence and absence of the caspase inhibitor zVAD (0.05 mM). Both the adherent (Adher.) and nonadherent (Non-Adher.) populations of cells were collected. The expression of JIP3 was examined by immunoblot analysis. (D) JIP3b (residues 1 to 781) was prepared by in vitro translation in the presence of [35S]methionine and incubated in the absence (−) or presence (+) of recombinant active caspase 1 (Casp-1) and caspase 3 (Casp-3). The effect of caspase digestion was examined following SDS-PAGE by autoradiography. Control experiments were performed with in vitro-translated PARP, which is a substrate of caspase 3. (E) In vitro-translated JIP3b (residues 1 to 781) was incubated without (−) and with (+) caspase 3 in the presence of the caspase inhibitors zVAD and DEVD (0.1 and 0.01 mM). The effect of caspase digestion was examined following SDS-PAGE by autoradiography. (F) Mutational analysis of the caspase 3 consensus site in JIP3. In vitro-translated JIP3b (residues 1 to 781) was incubated without (−) and with (+) caspase 1 and caspase 3. The effect of the replacement of Asp-344 with Glu in the predicted caspase 3 cleavage site of JIP3 was investigated. The products of caspase digestion were examined following SDS-PAGE by autoradiography. Sizes in panels C to F are indicated in kilodaltons.
The observation that NGF induced the expression of JIP3 raises questions about what might happen to JIP3 when NGF is withdrawn from NGF-differentiated PC12 cells. Western blot analysis demonstrated that the amount of JIP3 protein was markedly decreased when NGF was withdrawn from differentiated PC12 cells (Fig. 9C). Interestingly, NGF withdrawal caused an increase in the amount of lower-molecular-weight immunoreactive JIP3-related proteins. A plausible hypothesis was that NGF withdrawal, which causes caspase activation and apoptosis, results in the proteolytic cleavage of JIP3. To test this hypothesis, we examined the effect of the caspase inhibitor zVAD following NGF withdrawal. This analysis demonstrated that caspase inhibition prevented the loss of JIP3 protein caused by NGF withdrawal (Fig. 9C).
To investigate whether JIP3 was a caspase substrate, we performed in vitro assays using purified caspases (Fig. 9D). These experiments demonstrated that JIP3 was a substrate for caspase 3, but not caspase 1. Control experiments were performed using PARP, a known caspase 3 substrate (57). Addition of either the general caspase inhibitor zVAD or the more selective caspase 3 inhibitor DEVD to the in vitro assay blocked the cleavage of JIP3 caused by caspase 3 (Fig. 9E). Previous studies have established a consensus sequence (Asp-Xaa-Xaa-Asp) for substrate cleavage by caspase 3 (46). Examination of the primary sequence of JIP3 indicated three potential caspase 3 cleavage sites. Two of these sites were located very close to the COOH terminus of JIP3. Cleavage at either of these sites would cause a small decrease in the mass of JIP3 but would not account for the observed fragmentation of JIP3 observed in vivo (Fig. 9C) or in vitro (Fig. 9D). In contrast, a third site located in the central region of JIP3 may contribute to the observed proteolysis. To test this hypothesis, we replaced Asp344 with Glu to remove this potential site of caspase 3 cleavage. This mutation caused a marked decrease in the proteolysis of JIP3 caused by purified caspase 3 in vitro (Fig. 9F).
Together, these data indicate that the expression of JIP3 is induced during NGF-stimulated neuronal differentiation and that JIP3 is down-regulated, in part, by caspase-mediated cleavage during NGF withdrawal-induced apoptosis.
Subcellular localization of JIP3.
We investigated the subcellular distribution of JIP3 in NGF-differentiated PC12 cells by immunofluorescence analysis (Fig. 10). The specificity of the immunofluorescence observed was confirmed in control experiments which demonstrated that the intensity of JIP3 staining was reduced when recombinant JIP3 was included as a competitor during the incubation with primary antibodies. Two regions of the PC12 cells stained with antibodies to JIP3. First, we observed JIP3 immunofluorescence in the soma, with strong staining of the cytoplasmic compartment. Second, JIP3 immunofluorescence was detected at the end of the developing neurites that projected from the soma. Double-label immunofluorescence analysis with tubulin antibodies demonstrated that JIP3 was localized in the growth cone of the neurites. This localization might represent a preferential accumulation of JIP3 in the growth cone, but it was also possible that the apparent growth cone localization was an indirect consequence of cytoplasmic volume rather than specific localization. We therefore compared the immunofluorescence pattern of JIP3 with that of neuron-specific enolase. In contrast to JIP3 antibodies, which stain the soma and the growth cones, neuron-specific enolase antibodies were found to stain both the soma and the neurites. These data indicate that the growth cone localization of JIP3 represents compartmentalization rather than a nonspecific consequence of cytoplasmic volume.
FIG. 10.
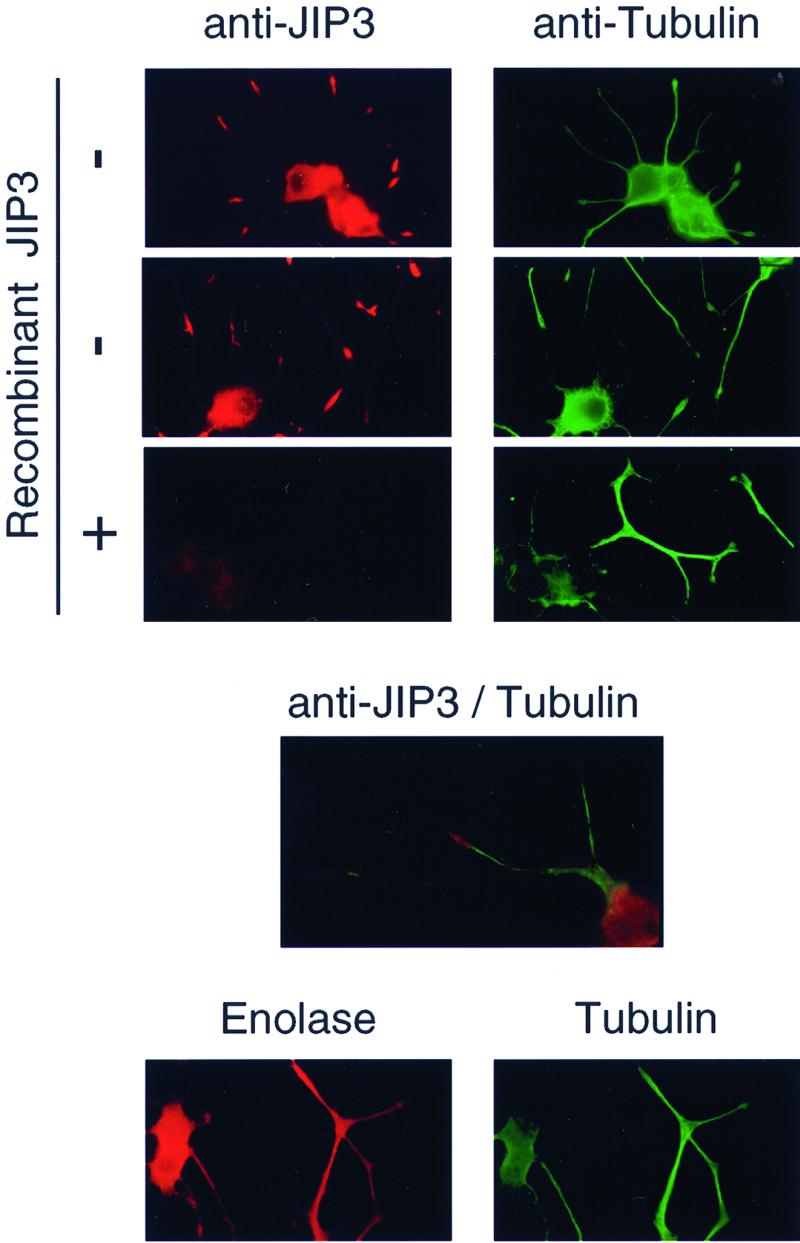
Subcellular localization of JIP3. PC12 cells differentiated in the presence of NGF for 12 days were fixed and processed for dual-label indirect immunofluorescence microscopy. The cells were stained with an antibody to JIP3 (red), neuron-specific enolase (red), and tubulin (green). Competition experiments were performed by including recombinant JIP3 (10 μg/ml) in the incubation with the primary antibody.
The localization of JIP3 in the soma and the growth cones of differentiated PC12 cells suggests that these cell compartments may represent sites where JIP3 exerts its physiological functions within the cell.
DISCUSSION
MAPKinase scaffold complexes.
MAPK signaling modules function to regulate MAPK activation. The minimal elements of a module include a MAPKKK, a MAPKK, and a MAPK. These three protein kinases function together to form a signaling cascade. The interactions between these protein kinases may involve sequential binary complexes between MAPKK and MAPKKK or MAPK (3, 63). It is also possible that larger complexes involving components of the MAPK module function to transmit signals through the protein kinase cascade (61). It is likely that both of these mechanisms are utilized by MAPK signaling pathways in response to extracellular stimulation. The mechanism that is used most likely depends upon the specific MAPK pathway and the nature of the extracellular stimulus. Two types of multicomponent complexes involving a MAPK signaling module have been identified.
(i) These complexes may be coordinated by a component of the MAPK module. For example, the MAPKK Pbs2p in yeast can create an osmoregulatory MAPK module through interactions with both the MAPKKK Ste11p and the MAPK Hog1p (38). A second example is provided by the observation that the mammalian MAPKKK MEKK1 binds to JNK, MKK4, and the Ste20-related protein kinase NIK (54, 63, 65). These interactions may participate in the transmission of signals from MEKK1 to JNK by the creation of a specific signaling module in vivo (61).
(ii) Complex formation by a MAPK module can also be coordinated by the interactions of the components of a MAPK module with other proteins that serve as a scaffold for the assembly of a functional signaling module. The best example of such a scaffold protein is Ste5p (12). The Ste5p scaffold protein is required for the activation of the mating MAPKinase pathway in yeast and appears to bind multiple components of the signaling module, including the MAPK Fus3p, the MAPKK Ste7p, and the MAPKKK Ste11p (6, 34, 39). Detailed genetic and biochemical studies establish that Ste5p organizes the MAPK signaling module that regulates mating (12). Scaffold proteins that are structurally similar to Ste5p have not been identified in mammals. However, putative scaffold proteins that may function to regulate mammalian MAPK signaling modules have been reported. Studies of the ERK MAPK pathway have led to the molecular cloning of MP1, a putative scaffold protein that binds both the MAPK ERK1 and the MAPKK MEK1 (48). Transfection assays indicate that MP1 potentiates the activation of ERK1 by MEK1 and may function as a scaffold protein in vivo. A second example of a putative mammalian scaffold protein was identified by studies of the JNK signal transduction pathway. The putative scaffold protein JIP1 (10) binds the MAPK JNK, the MAPKK MKK7, members of the MLK group of MAPKKK, and the Ste20-related protein kinase HPK1 (60). The JIP1 scaffold potentiates the activation of JNK caused by members of the MLK group of MAPKKK but does not participate in signaling by the MEKK group of MAPKKK (60). The JIP group of putative scaffold proteins includes the structurally related proteins JIP1 and JIP2 (70).
The JIP3 group of mammalian scaffold proteins.
We have identified a new class of scaffold proteins that may function to regulate MAPK signaling in mammals. The founding members of this group include the JIP3a and JIP3b proteins, which appear to represent the protein products of alternatively spliced transcripts derived from the JIP3 gene. The JIP3 group of proteins includes JIP3β (accession no. AB11088) and JIP3γ (accession no. X91879), which are structurally related to JIP3 (50, 55). Both JIP3 (Fig. 1) and JIP3β (55) are expressed in the brain, while JIP3γ appears to be most highly expressed in testis (50).
The JIP3 proteins interact with the MAPK JNK (Fig. 2), the MAPKK MKK7 (Fig. 4), and the MAPKKK MLK3 (Fig. 5). These binding interactions are similar to those of JIP1 and JIP2, which also bind JNK, MKK7, and MLK3. However, unlike JIP1 and JIP2, which bind several members of the mixed-lineage protein kinase group of MAPKKKs (including DLK, MLK2, and MLK3), the JIP3 proteins appear to selectively interact with MLK3. Transfection studies demonstrate that JIP3 potentiates the activation of JNK caused by MLK3 (Fig. 7). Thus, the JIP3 proteins may function as molecular scaffolds for the JNK signaling pathway.
JIP3 scaffold proteins may function as oligomers.
Coprecipitation assays demonstrated that JIP3 proteins can form oligomeric complexes, which may be dimers or higher-order aggregates (Fig. 8). Deletion analysis indicated that the NH2-terminal region of JIP3, which includes an extended coiled coil domain, was sufficient for the interaction of JIP3 molecules. This oligomeric structure of JIP3 is interesting because similar oligomeric complexes have been detected for other scaffold proteins, including the JIP1 and JIP2 proteins (70) and Ste5p (13, 23, 66). Studies of yeast demonstrate that the interaction between Ste5p molecules facilitates MAPK activation. Furthermore, interallelic complementation assays indicate that signal transmission through the MAPK module may occur in trans between protein kinases tethered to different Ste5p molecules (22). Thus, the function of scaffold proteins may be to aggregate components of the MAPK module to facilitate activation. Whether the oligomeric structure of the mammalian JIP scaffold proteins is essential for their function to potentiate JNK activation is not known. Testing this hypothesis will require the construction of mutated JIP proteins that are unable to oligomerize but retain the ability to bind MLK3, MKK7, and JNK.
The JIP1 and JIP2 proteins are known to form both homo-oligomeric and hetero-oligomeric complexes (70). This observation suggested that JIP3 proteins might interact with JIP1 and JIP2. Coprecipitation assays provided no evidence for the interaction of JIP1 with JIP3. However, JIP2 was found to interact with JIP3 (Fig. 8). Interestingly both JIP3 (Fig. 1) and JIP2 (70) are selectively expressed in the brain. Further studies are required to test the hypothesis that the interaction of the JIP3 and JIP2 scaffolds is relevant to the physiological function of these proteins. Expression of JIP proteins in COS cells (which do not express endogenous JIP1, JIP2, or JIP3) demonstrates that each of these proteins can potentiate JNK activation (Fig. 7). Thus, hetero-oligomeric complex formation by these putative scaffolds is not required for JNK activation. However, the observation of hetero-oligomeric complexes of JIP2 and JIP3 scaffold proteins suggests a mechanism by which combinatorial specificity of regulation may be conferred by the formation of specific scaffold assemblies in vivo. This speculative hypothesis warrants further study.
Subcellular location of MAPK scaffold proteins.
Studies of PC12 cells demonstrated that JIP3 expression was induced during neuron-like differentiation caused by NGF (Fig. 9). Withdrawal of NGF from differentiated PC12 cells caused a marked reduction of JIP3 expression which was mediated, in part, by caspase activation during apoptosis and the proteolysis of JIP3 (Fig. 9). Immunofluorescence analysis demonstrated that JIP3 was located in the cytoplasm and was accumulated in the growth cones of the developing neurites (Fig. 10). This distribution of JIP3 is similar to that detected for the JIP1 and JIP2 scaffold proteins, which also accumulate in growth cones (70). It is tempting to speculate that the growth cone location of the JIP proteins may indicate that these putative scaffolds contribute to JNK activation in these subcellular structures. However, it is also possible that the immunofluorescence images of fixed cells are a misleading representation of a more dynamic localization in vivo. Indeed, recent studies of Ste5p indicate that this yeast scaffold protein is recruited to pheromone-induced cell projections, where it activates the mating MAPK module (40) by a mechanism that requires the trafficking of Ste5p through the nucleus (33). Similar studies of the dynamics of scaffold protein localization in mammalian cells is required.
Conclusions.
The JIP3, JIP3β, and JIP3γ proteins represent a group of potential scaffold proteins that may function to regulate the activation of the JNK MAPK module. We present evidence that JIP3 binds components of a JNK signaling module (MLK3, MKK7, and JNK) and functions to potentiate JNK activation. Biochemical analysis of JIP3β and JIP3γ proteins will be required to establish whether these proteins have similar properties. These putative JIP3 scaffold complexes may aggregate components of a signaling module that activates JNK in a discrete cellular compartment. These JIP3 signaling complexes may function in a manner that is physiologically nonredundant with other mechanisms of JNK activation, including complexes with JIP1 and JIP2 proteins (60, 70) or MEKK proteins (54, 63, 65) and non-scaffold-mediated JNK activation (61). Genetic dissection of these alternative mechanisms of JNK activation will be required to identify the physiological role of each pathway.
ACKNOWLEDGMENTS
The first two authors contributed equally to this study.
We thank S. Kharbanda for providing purified recombinant caspases, A. Quail and T. Barrett for DNA sequence analysis, J. Cavanagh for technical assistance, and K. Gemme for administrative contributions to this research.
R.J.D. is an investigator of the Howard Hughes Medical Institute. This study was supported by a grant from the National Cancer Institute.
REFERENCES
- 1.Adachi-Yamada T, Fujimura-Kamada K, Nishida Y, Matsumoto K. Distortion of proximodistal information causes JNK-dependent apoptosis in Drosophila wing. Nature. 1999;400:166–169. doi: 10.1038/22112. [DOI] [PubMed] [Google Scholar]
- 2.Ammerer G. Sex, stress and integrity: the importance of MAP kinases in yeast. Curr Opin Cell Biol. 1994;4:90–95. doi: 10.1016/0959-437x(94)90096-5. [DOI] [PubMed] [Google Scholar]
- 3.Bardwell L, Thorner J. A conserved motif at the amino termini of MEKs might mediate high-affinity interaction with the cognate MAPKs. Trends Biochem Sci. 1996;21:373–374. [PubMed] [Google Scholar]
- 4.Behrens A, Sibilia M, Wagner E F. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet. 1999;21:326–329. doi: 10.1038/6854. [DOI] [PubMed] [Google Scholar]
- 5.Berger B, Wilson D B, Wolf E, Tonchev T, Milla M, Kim P S. Predicting coiled coils by use of pairwise residue correlations. Proc Natl Acad Sci USA. 1995;92:8259–8263. doi: 10.1073/pnas.92.18.8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi K Y, Satterberg B, Lyons D M, Elion E A. Ste5 tethers multiple protein kinases in the MAP kinase cascade required for mating in S. cerevisiae. Cell. 1994;78:499–512. doi: 10.1016/0092-8674(94)90427-8. [DOI] [PubMed] [Google Scholar]
- 7.Chow C-W, Rincon M, Cavanagh J, Dickens M, Davis R J. Nuclear accumulation of NFAT4 opposed by the JNK signal transduction pathway. Science. 1997;278:1638–1641. doi: 10.1126/science.278.5343.1638. [DOI] [PubMed] [Google Scholar]
- 8.Dérijard B, Hibi M, Wu I-H, Barrett T, Su B, Deng T, Karin M, Davis R J. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 9.Dérijard B, Raingeaud J, Barrett T, Wu I-H, Han J, Ulevitch R J, Davis R J. Independent human MAP kinase signal transduction pathways defined by MEK and MKK isoforms. Science. 1995;267:682–685. doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- 10.Dickens M, Rogers J S, Cavanagh J, Raitano A, Xia Z, Halpern J R, Greenberg M E, Sawyers C L, Davis R J. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science. 1997;277:693–696. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- 11.Dong C, Yang D D, Wysk M, Whitmarsh A J, Davis R J, Flavell R A. Defective T cell differentiation in the absence of Jnk1. Science. 1998;282:2092–2095. doi: 10.1126/science.282.5396.2092. [DOI] [PubMed] [Google Scholar]
- 12.Elion E A. Routing MAP kinase cascades. Science. 1998;281:1625–1626. doi: 10.1126/science.281.5383.1625. [DOI] [PubMed] [Google Scholar]
- 13.Feng Y, Song L Y, Kincaid E, Mahanty S K, Elion E A. Functional binding between Gbeta and the LIM domain of Ste5 is required to activate the MEKK Ste11. Curr Biol. 1998;8:267–278. doi: 10.1016/s0960-9822(98)70108-3. [DOI] [PubMed] [Google Scholar]
- 14.Ganiatsas S, Kwee L, Fujiwara Y, Perkins A, Ikeda T, Labow M A, Zon L I. SEK1 deficiency reveals mitogen-activated protein kinase cascade crossregulation and leads to abnormal hepatogenesis. Proc Natl Acad Sci USA. 1998;95:6881–6886. doi: 10.1073/pnas.95.12.6881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glise B, Bourbon H, Noselli S. hemipterous encodes a novel Drosophila MAP kinase kinase, required for epithelial cell sheet movement. Cell. 1995;83:451–461. doi: 10.1016/0092-8674(95)90123-x. [DOI] [PubMed] [Google Scholar]
- 16.Gupta S, Barrett T, Whitmarsh A J, Cavanagh J, Sluss H K, Derijard B, Davis R J. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996;15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- 17.Gupta S, Campbell D, Dérijard B, Davis R J. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science. 1995;267:389–393. doi: 10.1126/science.7824938. [DOI] [PubMed] [Google Scholar]
- 18.Han Z S, Enslen H, Hu X, Meng X, Wu I-H, Barrett T, Davis R J, Ip Y T. A conserved p38 mitogen-activated protein kinase pathway regulates Drosophila immunity gene expression. Mol Cell Biol. 1998;18:3527–3539. doi: 10.1128/mcb.18.6.3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harlow E, Lane D. Antibodies: a laboratory manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Press; 1988. [Google Scholar]
- 20.Holland P M, Suzanne M, Campbell J S, Noselli S, Cooper J A. MKK7 is a stress-activated mitogen-activated protein kinase kinase functionally related to hemipterous. J Biol Chem. 1997;272:24994–24998. doi: 10.1074/jbc.272.40.24994. [DOI] [PubMed] [Google Scholar]
- 21.Hou X S, Goldstein E S, Perrimon N. Drosophila Jun relays the Jun amino-terminal kinase signal transduction pathway to the Decapentaplegic signal transduction pathway in regulating epithelial cell sheet movement. Genes Dev. 1997;11:1728–1737. doi: 10.1101/gad.11.13.1728. [DOI] [PubMed] [Google Scholar]
- 22.Inouye C, Dhillon N, Durfee T, Zambryski P C, Thorner J. Mutational analysis of STE5 in the yeast Saccharomyces cerevisiae: application of a differential interaction trap assay for examining protein-protein interactions. Genetics. 1997;147:479–492. doi: 10.1093/genetics/147.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inouye C, Dhillon N, Thorner J. Ste5 RING-H2 domain: role in Ste4-promoted oligomerization for yeast pheromone signaling. Science. 1997;278:103–106. doi: 10.1126/science.278.5335.103. [DOI] [PubMed] [Google Scholar]
- 24.Ip Y T, Davis R J. Signal tansduction by the c-Jun NH2-terminal kinase (JNK)-from inflammation to development. Curr Opin Cell Biol. 1998;10:205–219. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- 25.Kallunki T, Su B, Tsigelny I, Sluss H K, Derijard B, Moore G, Davis R J, Karin M. JNK2 contains a specificity-determining region responsible for efficient c-Jun binding and phosphorylation. Genes Dev. 1994;8:2996–3007. doi: 10.1101/gad.8.24.2996. [DOI] [PubMed] [Google Scholar]
- 26.Kawasaki M, Hisamoto N, Iino Y, Yamamoto M, Ninomiya-Tsuji J, Matsumoto K. A Caenorhabditis elegans JNK signal transduction pathway regulates coordinated movement via type-D GABAergic motor neurons. EMBO J. 1999;18:3604–3615. doi: 10.1093/emboj/18.13.3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuan C-Y, Yang D, Roy D R S, Davis R J, Rakic P, Flavell R A. The Jnk1 and Jnk2 protein kinases regulate regional specific apoptosis during early brain development. Neuron. 1999;22:667–676. doi: 10.1016/s0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- 28.Kyriakis J M, Banerjee P, Nikolakaki E, Dai T, Rubie E A, Ahmad M F, Avruch J, Woodgett J R. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 29.Landschulz W H, Johnson P F, McKnight S L. The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science. 1988;240:1759–1764. doi: 10.1126/science.3289117. [DOI] [PubMed] [Google Scholar]
- 30.Lin A, Minden A, Martinetto H, Claret F X, Lange-Carter C, Mercurio F, Johnson G L, Karin M. Identification of a dual specificity kinase that activates the Jun kinases and p38-Mpk2. Science. 1995;268:286–290. doi: 10.1126/science.7716521. [DOI] [PubMed] [Google Scholar]
- 31.Livingstone C, Patel G, Jones N. ATF-2 contains a phosphorylation-dependent transcriptional activation domain. EMBO J. 1995;14:1785–1797. doi: 10.1002/j.1460-2075.1995.tb07167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Madhani H D, Fink G R. The control of filamentous differentiation and virulence in fungi. Trends Cell Biol. 1998;8:348–353. doi: 10.1016/s0962-8924(98)01298-7. [DOI] [PubMed] [Google Scholar]
- 33.Mahanty S K, Wang Y, Farley F W, Elion E A. Nuclear shuttling of yeast scaffold Ste5 is required for its recruitment to the plasma membrane and activation of the mating MAPK cascade. Cell. 1999;98:501–512. doi: 10.1016/s0092-8674(00)81978-9. [DOI] [PubMed] [Google Scholar]
- 34.Marcus S, Polverino A, Barr M, Wigler M. Complexes between STE5 and components of the pheromone-responsive mitogen-activated protein kinase module. Proc Natl Acad Sci USA. 1994;91:7762–7766. doi: 10.1073/pnas.91.16.7762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moriguchi T, Toyoshima F, Masuyama N, Hanafusa H, Gotoh Y, Nishida E. A novel SAPK/JNK kinase, MKK7, stimulated by TNFα and cellular stresses. EMBO J. 1997;16:7045–7053. doi: 10.1093/emboj/16.23.7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishina H, Fischer K D, Radvanyl L, Shahinian A, Hakem R, Ruble E A, Bernstein A, Mak T W, Woodgett J R, Penninger J M. Stress-signaling kinase Sek1 protects thymocytes from apoptosis mediated by CD95 and CD3. Nature. 1997;385:350–353. doi: 10.1038/385350a0. [DOI] [PubMed] [Google Scholar]
- 37.Nishina H, Vaz C, Billia P, Nghiem M, Sasaki T, la Pompa J L, Furlonger K, Paige C, Hui C, Fischer K D, Kishimoto H, Iwatsubo T, Katada T, Woodgett J R, Penninger J M. Defective liver formation and liver cell apoptosis in mice lacking the stress signaling kinase SEK1/MKK4. Development. 1999;126:505–516. doi: 10.1242/dev.126.3.505. [DOI] [PubMed] [Google Scholar]
- 38.Posas F, Saito H. Osmotic activation of the HOG MAPK pathway via Ste11p MAPKKK: scaffold role of Pbs2p MAPKK. Science. 1997;276:1702–1705. doi: 10.1126/science.276.5319.1702. [DOI] [PubMed] [Google Scholar]
- 39.Printen J A, Sprague G F., Jr Protein-protein interactions in the yeast pheromone response pathway: Ste5p interacts with all members of the MAP kinase cascade. Genetics. 1994;138:609–619. doi: 10.1093/genetics/138.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pryciak P M, Huntress F A. Membrane recruitment of the kinase cascade scaffold protein Ste5 by the Gβγ complex underlies activation of the yeast pheromone response pathway. Genes Dev. 1998;12:2684–2697. doi: 10.1101/gad.12.17.2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riesgo-Escovar J R, Hafen E. Drosophila Jun kinase regulates expression of decapentaplegic via the ETS-domain protein Aop and the AP-1 transcription factor DJun during dorsal closure. Genes Dev. 1997;11:1717–1727. doi: 10.1101/gad.11.13.1717. [DOI] [PubMed] [Google Scholar]
- 42.Riesgo-Escovar J R, Jenni M, Fritz A, Hafen E. The Drosophila Jun-N-terminal kinase is required for cell morphogenesis but not for DJun-dependent cell fate specification in the eye. Genes Dev. 1996;10:2759–2768. doi: 10.1101/gad.10.21.2759. [DOI] [PubMed] [Google Scholar]
- 43.Rincon M, Whitmarsh A, Yang D D, Weiss L, Derijard B, Jayaraj P, Davis R J, Flavell R A. The JNK pathway regulates the in vivo deletion of immature CD4(+)CD8(+) thymocytes. J Exp Med. 1998;188:1817–1830. doi: 10.1084/jem.188.10.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robinson M J, Cobb M H. Mitogen-activated protein kinase pathways. Curr Opin Cell Biol. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- 45.Sabapathy K, Hu Y, Kallunki T, Schreiber M, David J P, Jochum W, Wagner E F, Karin M. JNK2 is required for efficient T-cell activation and apoptosis but not for normal lymphocyte development. Curr Biol. 1999;9:116–125. doi: 10.1016/s0960-9822(99)80065-7. [DOI] [PubMed] [Google Scholar]
- 46.Salvesen G S, Dixit V M. Caspases: intracellular signaling by proteolysis. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- 47.Sanchez I, Hughes R T, Mayer B J, Yee K, Woodgett J R, Avruch J, Kyriakis J M, Zon L I. Role of SAPK/ERK kinase-1 in the stress-activated pathway regulating transcription factor c-Jun. Nature. 1994;372:794–798. doi: 10.1038/372794a0. [DOI] [PubMed] [Google Scholar]
- 48.Schaeffer H J, Catling A D, Eblen S T, Collier L S, Krauss A, Weber M J. MP1: a MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science. 1998;281:1668–1671. doi: 10.1126/science.281.5383.1668. [DOI] [PubMed] [Google Scholar]
- 49.Schaeffer H J, Weber M J. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol. 1999;19:2435–2444. doi: 10.1128/mcb.19.4.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shankar S, Mohapatra B, Suri A. Cloning of a novel human testis mRNA specifically expressed in testicular haploid germ cells, having unique palindromic sequences and encoding a leucine zipper dimerization motif. Biochem Biophys Res Commun. 1998;243:561–565. doi: 10.1006/bbrc.1997.7943. [DOI] [PubMed] [Google Scholar]
- 51.Sluss H K, Barrett T, Dérijard B, Davis R J. Signal transduction by tumor necrosis factor mediated by JNK protein kinases. Mol Cell Biol. 1994;14:8376–8384. doi: 10.1128/mcb.14.12.8376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sluss H K, Davis R J. Embryonic morphogenesis signaling pathway mediated by JNK targets the transcription factor JUN and the TGF-beta homologue decapentaplegic. J Cell Biochem. 1997;67:1–12. [PubMed] [Google Scholar]
- 53.Sluss H K, Han Z, Barrett T, Davis R J, Ip Y T. A JNK signal transduction pathway that mediates morphogenesis and an immune response in Drosophila. Genes Dev. 1996;10:2745–2758. doi: 10.1101/gad.10.21.2745. [DOI] [PubMed] [Google Scholar]
- 54.Su Y C, Han J, Xu S, Cobb M, Skolnik E Y. NIK is a new Ste20-related kinase that binds NCK and MEKK1 and activates the SAPK/JNK cascade via a conserved regulatory domain. EMBO J. 1997;16:1279–1290. doi: 10.1093/emboj/16.6.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Suyama M, Nagase T, Ohara O. HUGE: a database for human large proteins identified by Kazusa cDNA sequencing project. Nucleic Acids Res. 1999;27:338–339. doi: 10.1093/nar/27.1.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Swat W, Fujikawa K, Ganiatsas S, Yang D, Xavier R J, Harris N L, Davidson L, Ferrini R, Davis R J, Labow M A, Flavell R A, Zon L I, Alt F W. SEK1/MKK4 is required for maintenance of a normal peripheral lymphoid compartment but not for lymphocyte development. Immunity. 1998;8:625–634. doi: 10.1016/s1074-7613(00)80567-1. [DOI] [PubMed] [Google Scholar]
- 57.Tewari M, Quan L T, O'Rourke K, Desnoyers S, Zeng Z, Beidler D R, Poirier G G, Salvesen G S, Dixit V M. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 58.Tournier C, Whitmarsh A J, Cavanagh J, Barrett T, Davis R J. MAP kinase kinase 7 is an activator of the c-Jun NH2-terminal kinase. Proc Natl Acad Sci USA. 1997;94:7337–7342. doi: 10.1073/pnas.94.14.7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tournier C, Whitmarsh A J, Cavanagh J, Barrett T, Davis R J. The MKK7 gene encodes a group of c-Jun NH2-terminal kinase kinases. Mol Cell Biol. 1999;19:1569–1581. doi: 10.1128/mcb.19.2.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Whitmarsh A J, Cavanagh J, Tournier C, Yasuda J, Davis R J. A mammalian scaffold complex that selectively mediates MAP kinase activation. Science. 1998;281:1671–1674. doi: 10.1126/science.281.5383.1671. [DOI] [PubMed] [Google Scholar]
- 61.Whitmarsh A J, Davis R J. Structural organization of MAP kinase signaling modules in yeast and mammals. Trends Biochem Sci. 1998;23:481–485. doi: 10.1016/s0968-0004(98)01309-7. [DOI] [PubMed] [Google Scholar]
- 62.Widmann C, Gibson S, Jarpe M B, Johnson G L. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- 63.Xia Y, Wu Z, Su B, Murray B, Karin M. JNKK1 organizes a MAP kinase module through specific and sequential interactions with upstream and downstream components mediated by its amino-terminal extension. Genes Dev. 1998;12:3369–3381. doi: 10.1101/gad.12.21.3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xia Z, Dickens M, Raingeaud J, Davis R J, Greenberg M E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 65.Xu S, Cobb M H. MEKK1 binds directly to the c-Jun N-terminal kinases/stress-activated protein kinases. J Biol Chem. 1997;272:32056–32060. doi: 10.1074/jbc.272.51.32056. [DOI] [PubMed] [Google Scholar]
- 66.Yablonski D, Marbach I, Levitzki A. Dimerization of Ste5, a mitogen-activated protein kinase cascade scaffold protein, is required for signal transduction. Proc Natl Acad Sci USA. 1996;93:13864–13869. doi: 10.1073/pnas.93.24.13864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang D, Tournier C, Wysk M, Lu H-T, Xu J, Davis R J, Flavell R A. Targeted disruption of the MKK4 gene causes embryonic death, inhibition of c-Jun NH2-terminal kinase activation and defects in AP-1 transcriptional activity. Proc Natl Acad Sci USA. 1997;94:3004–3009. doi: 10.1073/pnas.94.7.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang D D, Conze D, Whitmarsh A J, Barrett T, Davis R J, Rincon M, Flavell R A. Differentiation of CD4+ T cells to Th1 cells requires MAP kinase JNK2. Immunity. 1998;9:575–585. doi: 10.1016/s1074-7613(00)80640-8. [DOI] [PubMed] [Google Scholar]
- 69.Yang D D, Kuan C-Y, Whitmarsh A J, Rincon M, Zheng T S, Davis R J, Rakic P, Flavell R A. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the JNK3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- 70.Yasuda J, Whitmarsh A J, Cavanagh J, Sharma M, Davis R J. The JIP group of mitogen-activated protein kinase scaffold proteins. Mol Cell Biol. 1999;19:7245–7254. doi: 10.1128/mcb.19.10.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]