

Abstract
A promoter is a region in the DNA sequence that defines where the transcription of a gene by RNA polymerase initiates, which is typically located proximal to the transcription start site (TSS). How to correctly identify the gene TSS and the core promoter is essential for our understanding of the transcriptional regulation of genes. As a complement to conventional experimental methods, computational techniques with easy-to-use platforms as essential bioinformatics tools can be effectively applied to annotate the functions and physiological roles of promoters. In this work, we propose a deep learning-based method termed Depicter (Deep learning for predicting promoter), for identifying three specific types of promoters, i.e. promoter sequences with the TATA-box (TATA model), promoter sequences without the TATA-box (non-TATA model), and indistinguishable promoters (TATA and non-TATA model). Depicter is developed based on an up-to-date, species-specific dataset which includes Homo sapiens, Mus musculus, Drosophila melanogaster and Arabidopsis thaliana promoters. A convolutional neural network coupled with capsule layers is proposed to train and optimize the prediction model of Depicter. Extensive benchmarking and independent tests demonstrate that Depicter achieves an improved predictive performance compared with several state-of-the-art methods. The webserver of Depicter is implemented and freely accessible at https://depicter.erc.monash.edu/.
Keywords: eukaryotic promoters, bioinformatics, sequence analysis, machine learning, deep learning
Introduction
Gene expression refers to the process of synthesizing functional gene products from genetic information, during which the initiation of transcription is of vital importance [1]. In eukaryotes, there are three types of RNA polymerases responsible for transcription of different subsets of genes: (i) RNA polymerase I (RNA pol I) transcribes genes encoding ribosomal RNA (rRNA); (ii) RNA polymerase II (RNA pol II) transcribes mRNA, miRNA, snRNA, and snoRNA genes; and (iii) RNA polymerase III (RNA pol III) transcribes genes encoding transfer RNA (tRNA) [2, 3]. According to the type of RNA polymerases, eukaryotic promoters are classified into three different categories: RNA pol I promoters, RNA pol II promoters and RNA pol III promoters, of which the RNA pol II promoters are more essential for transcribing all protein-coding and many noncoding genes [4, 5]. Promoter sequences are short, conserved, noncoding DNA sequences and usually located around the transcription start site (TSS), which define where transcription of a gene by RNA polymerase begins [5, 6]. TSS is embedded in the core promoter sequence encompassing ~40 bp upstream and ~40 bp downstream or ~50 bp upstream and ~100 bp downstream, which is deemed as the gateway to transcription and largely dependent on the core promoter elements or motifs [7, 8]. The location diagram of the TSS and core promoter is shown in Figure 1B. Inr (initiator), TATA box, BREu and BREd, TCT (polypyrimidine initiator), downstream promoter element (DPE), motif ten element (MTE), specificity protein 1 (Sp 1) and other core promoter elements (such as the X core promoter element 1 (XCPE1) and XCPE2 motif, the downstream core element, three downstream elements, termed GLE, DPE-L1, and DPE-L2, the downstream transcrsiption initiation element (DTIE)) are well-known sequence motifs discovered in several eukaryotes [1, 9]. In addition, there are some other promoter elements, such as ‘CpG island’ and ‘ATG desert’ in mammalian promoters. For the detailed information about these motifs, please refer to the reviews [10]. TATA box is the most distinctive core promoter element and also is regarded as the most ancient for it is present in organisms ranging from yeast to plants and metazoans [11]. For eukaryotes, promoters are usually classified into TATA-containing and TATA-less or non-TATA according to whether containing TATA-box in their sequences. In the work of Yella and Bansal [10], they showed that there are three obviously distinct structural properties, e.g. DNA duplex stability, bendability and curvature in TATA-containing and TATA-less promoters of six eukaryotes. Promoters have demonstrated essential roles in the regulation of gene expressions, such as alternative splicing [12], the stability of transcripts [13], mRNA localization [14] and translation. Therefore, the identification of promoters is essential not only for recognition of the complete structure of a gene, but also for further understanding the mechanisms of gene transcription and expression regulation [15].
Figure 1.
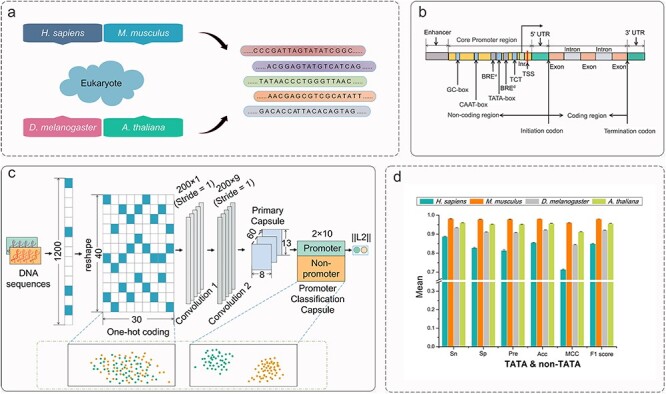
Depicter overview. (A) Depicter studies species including H. sapiens, M. musculus, D. melanogaster and A. thaliana. (B) Genetic structure of eukaryotic cells. (C) Overall architecture of Depicter. (D) Models evaluation and validation, and the part of the figure show the average performances of 5-fold cross-validation running five-time on the benchmark datasets for the TATA and non-TATA type.
A variety of computational methods based on traditional machine learning and deep learning techniques have been proposed to identify promoters in different species. A consensus of these studies is to analyze the data and extract relevant and useful information to make accurate decision and facilitate knowledge discovery [16, 17]. Here, we summarize and categorize the strategies of information extraction into three major types, which extract the information based on DNA structure, shape and sequence, respectively. DNA structural features such as flexibility/bendability, curvature, base stacking and duplex stability have been used to characterize promoter regions [18–20]. DNA shape features are more helpful for understanding and characterizing transcription factor binding sites, origins of replications and other genomic regions [21]. Sequence-based features are extracted from the genomic context of the promoter, such as the biological signal of core promoter elements (Inr, TATA-box), the statistical properties of k-mer composition and the characteristics of DNA secondary structure [22]. Using the extracted information, the majority of the existing approaches are developed based on conventional machine learning algorithms such as support vector machine (SVM) [23, 24], random forest (RF) [25], logistic regression (LR) [26] and gradient boosting decision tree (GBDT) [27], etc. Among these, iProEP is the most recently developed predictor [24], which employed the SVM algorithm to train the classifier for predicting promoter sequences of multiple species by utilizing pseudo k-tuple nucleotide composition and position-correlation scoring matrix to capture sequence information. However, it does not take into account the promoters in plant species, and requires domain knowledge-related features to get information from DNA sequences.
Deep learning is a cutting-edge machine learning technique which has proved to be extraordinarily powerful for mining valuable information through multiple layers of feature representations from the raw biological sequence data (i.e. DNA, RNA or protein sequences) without any domain knowledge in constructing feature vectors [28–32]. Due to its breakthrough performance in various machine learning applications, deep learning has recently been increasingly employed in the bioinformatics field [33]. To the best of our knowledge, there are three deep learning-based predictors for the promoters, namely CNNProm [34], DeeReCT-PromID [35], DeePromoter [36], respectively. Different from previous approaches, CNNPorm divided the promoters into two classes: TATA and non-TATA promoters [37] and then constructed the convolution neural network prediction model for H. sapiens, M. musculus and A. thaliana. DeePromoter [36] is developed based on the combination of CNN and bidirectional long short-term memory to further improve the recognition ability. Evaluated on their own independent test datasets, DeePromoter achieved an MCC of 0.88 for TATA promoters and 0.92 for non-TATA promoters of H. sapiens, an MCC of 0.87 for TATA promoters and 0.82 for non-TATA promoters of M. musculus, respectively. Rather than focusing on the high-precision of classification, DeeReCT-PromID [35] can be used to predict the exact locations of TSS in the genomic sequences by detecting each possible location. In contrast to the traditional machine learning algorithms that require complicated feature engineering and various feature selection schemes, the three deep learning methods only use the one-hot encoding scheme, and then directly input to the multiple convolutional neural networks.
Although extensive studies have been conducted on this topic, several issues remain to be addressed. Firstly, a large amount of new genomes has been recently annotated and collected; secondly, the species for which each promoter identification model was specifically designed are not uniform; thirdly, the predictive performances of some tools are still unsatisfactory, especially for identifying non-TATA promoters of M. musculus and A. thaliana; and lastly, three different models would need to be specifically designed for TATA promoters, non-TATA promoters and indistinguishable promoters (i.e.TATA and non-TATA promoters) according to cater for users’ requirements as someone may be not interested in the specific type of promoters. To address these problems, in this work we propose Depicter, which employs the connected architecture of CNN and capsule network, to identify TATA promoters and non-TATA promoters of H. sapiens, M. musculus, D. melanogaster and A. thaliana. To illustrate the effectiveness of this architecture, we visualized the analysis results using t-distributed Stochastic Neighbor Embedding (t-SNE). The performance of Depicter was compared with different promoter prediction models at the species-specific level and is shown to provide a superior performance compared with the existing methods on newly constructed independent test datasets. In order to facilitate community-wide research efforts, a user-friendly webserver of Depicter is developed and made freely used at https://depicter.erc.monash.edu/, with the source code and datasets freely available at https://github.com/zhuyaner/Depicter/.
Materials and methods
Overall framework
In this study, we propose a computational framework named Depicter to predict species-specific TATA promoters and non-TATA promoters. There are four major steps involved in the development of Depicter, including data collection and preprocessing (Figure 1A), one-hot encoding, training (Figure 1C) and model performance evaluation (Figure 1D). At the first step, we construct reliable and rigorous benchmark and independent test datasets for model training and performance validation. At the second step, each DNA sequence fragment is transformed to the input feature matrix using the one-hot encoding scheme. At the third step, we build a novel deep learning framework consisting of two one-dimensional convolutional layers and a one-dimensional convolutional capsule layer, and a fully connected layer to train the prediction model based on the input feature matrix (Figure 1C). Finally, we assess and evaluate the predictive performance of Depicter based on the independent test datasets. The schematic representation of the Depicter approach is shown in Figure 1.
Data collection and preprocessing
Constructing a rigorous and objective benchmark dataset is a fundamental step to establish a robust and forceful prediction model [38]. In this study, we collected promoter sequences for four different species, including Homo sapiens (H. sapiens), Mus musculus (M. musculus), Drosophila melanogaster (D. melanogaster) and Arabidopsis thaliana (A. thaliana) from the EPDNew database (http://epd.vital-it.ch, last update October 2019), which is an update version of the Eukaryotic Promoter Database (EPD) that collects experimentally validated eukaryotic promoters [39]. We developed prediction models for these four eukaryotic species as they have sufficient numbers of promoters to ensure statistical significance. All collected eukaryotic promoter sequences have a length of 300 bp which were extracted from 249 bp upstream to 50 bp downstream regions of TSS (TSS is regarded as the 0-th site). There are two reasons why we chose a promoter region 300 bp (−249 bp to +50 bp) for eukaryotes: (i) the core regulatory elements of transcription recognized by the polymerase in eukaryotes are located between −250 bp and + 50 bp of TSS [40]; (ii) the promoter region 300 bp (−249 bp to +50 bp) has been extensively used in [36, 41]. Therefore, we selected this region to make an objective comparison with these models. Subsequently, according to whether a promoter sequence contained a TATA-box at the position −28 (±3 bp) from the TSS, these collected sequences were labeled as TATA promoters and non-TATA promoters [42]. To construct the corresponding negative datasets, we collected exon sequences of H. sapiens, intron sequences of M. musculus, exon and intron sequences of A. thaliana from the Exon–Intron Database (EID) [6], exon sequences of D. melanogaster from FlyBase (release 6.3) (ftp://ftp.flybase.net/genomes/dmel/current/fasta/). In order to avoid the oversampling or downsampling process of negative samples, which may affect the predictor [43], we select the number of exon or intron sequences that match the positive samples as negative samples for the four species, respectively. The downloaded exon or intron sequences were treated as negative samples, whose length was also 300 bp, which was of the same length as the eukaryotic promoter samples.
The original sample sequences obtained from the databases should be preprocessed to remove noise samples and ensure the robustness of the prediction model [44]. For each species, we used CD-HIT-EST [45] with the cut-off value of 0.8 to exclude highly similar promoter sequences [46]. The negative samples were similarly treated and then the same number of negative samples as positive samples was randomly selected. A statistical summary of the negative samples is provided in the Supplementary Table S1. We then combined the positive and negative samples and randomly divided them into the training sets (including training and validation datasets) and independent test datasets with the dataset size ratio of 9:1 between the former and the latter. The statistical summary of the promoter and nonpromoter (the last column) datasets for four different species in our study are shown in Table 1. All datasets curated in this work can be freely downloaded at https://github.com/zhuyaner/Depicter/.
Table 1.
A statistical summary of the curated promoter datasets for four different species in this study
| Kingdom | Species | TATA | Original data | CD-HIT-EST (80%) | Promoter | Training dataset | Independent test dataset | Location | Non-promoter |
|---|---|---|---|---|---|---|---|---|---|
| Eukaryotes (300 bp) | H. sapiens | With | 3065 | 2927 | 2927 | 2634 | 293 | [−249,+50] | 2927 |
| Without | 26 533 | 25 460 | 25 460 | 22 914 | 2546 | [−249,+50] | 25 460 | ||
| M. musculus | With | 3305 | 3077 | 3077 | 2769 | 308 | [−249,+50] | 3077 | |
| Without | 21 805 | 21 040 | 21 040 | 18 936 | 2104 | [−249,+50] | 21 040 | ||
| D. melanogaster | With | 2598 | 2585 | 2585 | 2326 | 259 | [−249,+50] | 2585 | |
| Without | 14 372 | 14 035 | 14 035 | 12 631 | 1404 | [−249,+50] | 14 035 | ||
| A. thaliana | With | 6405 | 6323 | 6323 | 5691 | 632 | [−249,+50] | 6323 | |
| Without | 16 298 | 15 858 | 15 858 | 14 272 | 1586 | [−249,+50] | 15 858 |
Training the deep learning model
In this section, we describe the framework design of Depicter in detail. We use the processed gene sequences as the input, employ the one-hot encoding scheme to construct the sparse feature matrix, and then concatenate the convolutional neural network and capsule network. These major steps are introduced in the following sections in detail.
One-hot encoding of the input gene sequence
The one-hot encoding scheme has been widely applied in deep learning and has demonstrated its effectiveness in the research areas of computer science [47–49] and bioinformatics [50–53]. The one-hot encoding transforms each nucleotide to a 4-dimensional binary vector, that is, A is transformed to (1, 0, 0, 0), C is transformed to (0, 1, 0, 0), G is transformed to (0, 0, 1, 0) and T is transformed to (0, 0, 0, 1), respectively [54].
Architecture design of Depicter
A critical factor for promoter identification is to determine the specific locations in the promoter region where some promoter elements, such as the GC-box, TATA-box, CAAT-box and so on, are localized [35]. Although such positional information is important for promoter recognition, the average pooling layer or the maximum pooling layer used in CNN tends to deteriorate the positional information of the sequences to some extent [55]. A useful strategy to address this is to use the capsule network, which was proposed by Geoffrey Hinton in NIPS 2017 (Sabour, et al., 2017). Its improvement lies in the application of dynamic routing instead of the primitive pooling. Dynamic routing can deliver the information selectively through protocols between the lower layer and the higher layer, whereas the maximum pooling focuses only on the maximum value and the averaging pooling focuses on the average of the feature point in the neighborhood.
Therefore, we constructed the Depicter architecture based on two one-dimensional convolutional layers and a one-dimensional convolutional capsule layer, and a fully connected layer (i.e. the promoter classification capsule). The first two layers are the conventional convolutional layers, which serve to capture middle-level features from the one-hot encoding matrix. Then they feed into the following capsule network layers for representative feature extraction and more accurate classification. Depicter is implemented using the Keras 2.1.1 package with a Tensor Flow 1.3.0 backend in Python 3.7. Detailed parameter settings of these layers can be found in the Supplementary Information (File S1).
Deep capsule neural network training
In this section, we apply several strategies to avoid the over-fitting issue of the trained model and automatically tune parameter setting of the number of epochs. These strategies include: first, use the ReLU activation function [56] in the first two layers that may repair the vanishing gradient issue in the back-propagation training algorithm. Second, adopt the dropout mechanism [57] to randomly remove certain neurons when training neural networks. Third, the ‘early stopping’ [58–60] strategy is applied to halt the model training when the loss is no longer dropping or the extent of dropping is less than a specific threshold. By doing this, we can address the problem of manually setting the number of epochs and minimizing the overfitting risk of the neural network on the training datasets. The ‘Adam’ optimizer [61] with the separate margin loss function was used for training the model [62]. To examine the fitting effect of Depicter, we also draw the loss and accuracy curves of each model in Figure S1 that demonstrates the model training process.
Parameter optimization
Identifying the optimal parameters is one of most crucial aspects of establishing a valid prediction model because the selection of parameters will affect the predictive performance of the trained model significantly [63]. However, it is impossible to manually select and test each combination of parameters exhaustively. In this work, we adopt a two-step parameter adjustment strategy. We randomly partition the processed training datasets into 10 subsets with the ratio of 9:1 (nine subsets are merged as the training dataset and the remaining subset will be used as the validation dataset) for parameters search. First, the initial parameters are set according to a previous work [64] to roughly compare the prediction results. Then Bayesian optimization [65] is used to fine-tune the key parameters including learning rate, batch size, dropout rate, etc. Finally, the optimal parameters are selected according to the area under curve (AUC) value. The finally selected parameters of the models for each species and their corresponding AUC values are provided in Table S2.
Performance evaluation
In order to comprehensively assess the performance of promoter prediction, six generally applied statistical measures [66–70] are adopted in this work, including sensitivity (Sn), specificity (Sp), precision (Pre), accuracy (Acc), Matthew’s correlation coefficient (MCC) and F1 score. They are defined as follows:
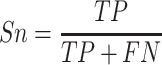 |
(1) |
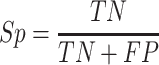 |
(2) |
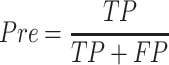 |
(3) |
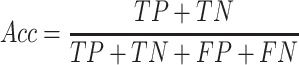 |
(4) |
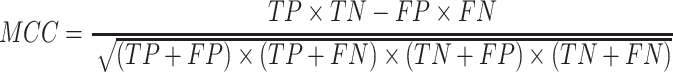 |
(5) |
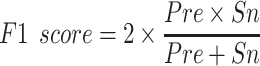 |
(6) |
where TP represents the number of promoters correctly classified, TN represents the number of nonpromoters correctly classified, FN represents the number of promoters incorrectly classified as nonpromoters, and FP represents the number of nonpromoters incorrectly classified as promoters, respectively. Accordingly, Sn (also called true positive rate) measures the percentage of promoters correctly classified; Sp calculates the percentage of nonpromoters correctly classified analogously; Pre indicates the ratio of true promoters that are classified as promoters by Depicter; F1 score comprehensively considers precision and recall; MCC represents the balance quality of the positive and negative data. In addition, receiver-operating characteristic (ROC) curves and the area under ROC curve (AUC) are also applied to assess the overall classification performance. The AUC value closer to 1 indicates a close-to-perfect prediction, for which the ROC curve would be localized closer to the upper left corner.
Results and discussion
Visualization of learning characteristics in different periods
When using traditional machine learning methods or deep learning techniques to train and optimize the prediction model, the intermediate process executed by hidden layers is often invisible, just like a ‘black box’. This leads to the difficulty to interpret the various components of the machine learning model and understand how the prediction decision is made [71, 72]. Accordingly, substantial efforts have been devoted to improving the interpretability of learning process and providing explanations for the predictions [73]. In this study, we employed a popular visualization algorithm termed t-distributed t-SNE to visualize the intermediate results [74]. T-SNE is a nonlinear dimensionality reduction algorithm aiming to embed high-dimensional data for visualization in a two-dimensional or three-dimensional space [75].
Using the t-SNE algorithm, we mapped the high-dimensional feature space to the two-dimensional space. The results are shown in Figure 2A, which provides a visual comparison of the feature representations of for the one-hot encoding and after the capsule layers of TATA models for H. sapiens. Mixing of the two types of dots in Figure 2A (a) indicates that it is difficult to distinguish promoters from nonpromoters. In contrast, the selected and processed features by the convolutional capsule layers could be clearly separated as shown in Figure 2A (b). Comparison of t-SNE visualizations of promoter predictions for all species is shown in Figures S2–S5. Taken together, we conclude that the Depicter framework can effectively learn the informative feature representations from the one-hot encoding mapped from the DNA sequences.
Figure 2.
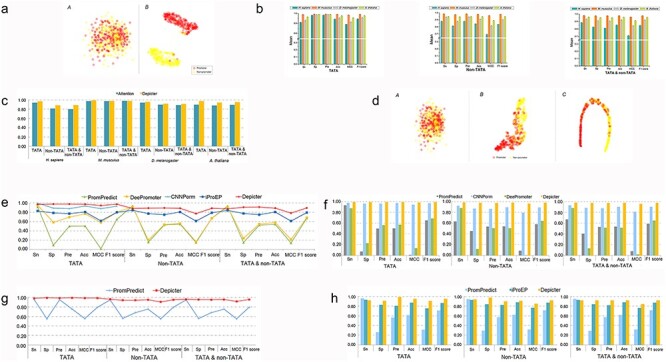
Using Depicter to explore comparative performance. (A) T-SNE plots for the one-hot encoding vector (a) and feature representation after capsule layer (b), in which red dots represent promoters and yellow dots represent nonpromoters, respectively. (B) The average performances of all four species on five times 5-fold cross-validation tests using the benchmark datasets. (C) Comparison of AUC values of attention model and Depicter on the benchmark datasets. (D) T-SNE visualization of the one-hot encoding feature representation (a), feature representation after the attention layer (b), and feature representation for the 2nd fully connected layer (c). (E–H) Performance comparison between Depicter and other existing methods on the independent test sets for H. sapiens (E), M. musculus (F), A. thaliana (G) and D. melanogaster (H).
Five-fold cross-validation test on the benchmark datasets
As there were a total of 12 models with a large amount of data in the experiments, we performed 5-fold cross-validation test by running five times to examine the robustness of Depicter, and then provided the corresponding prediction results in the Supplementary Table S3. Moreover, the average results of three models of each of the four species on five times 5-fold cross-validation tests are shown in Figure 2B. It can be seen from Table S3 and Figure 2B that our Depicter models achieved an outstanding predictive performance. More specifically, Depicter achieved the average Sn of 89.14%, Sp of 89.96%, Pre of 89.58%, Acc of 89.34%, MCC of 0.7889, and F1 score of 0.8924 for H. sapiens; the average Sn of 98.64%, Sp of 98.33%, Pre of 98.32%, Acc of 98.48%, MCC of 0.9696, F1 score of 0.9848 for M. musculus; the average Sn of 93.63%, Sp of 92.66%, Pre of 92.28%, Acc of 92.98%, MCC of 0.8613, and F1 score of 0.9286 for D. melanogaster; the average Sn of 95.58%, Sp of 96.35%, Pre of 96.36%, Acc of 95.95%, MCC of 0.9192, and F1 score of 0.9596 for A. thaliana, respectively.
Effective performance evaluation on the framework of Depicter
The attention mechanism in the deep neural network is also an important component. It has been extensively applied in many bioinformatics studies and achieved superior performances. Therefore, we attempted to combine the attention layer after the one-dimensional convolution neural network layer to examine the possibility of further improving the predictive performance. The output from the third hidden state of CNN is directly input into the attention mechanism, and its trans-position form is input into another attention mechanism analogously. Then the outputs of the two attention mechanisms are combined and input into the first connected neural network layer. The second fully connected layer is a single neural network layer with the softmax output. The same training set and one-hot encoding scheme were used by Depicter to train and calculate the variable indices. Refer Figure S6 for details. We compared the predictive performance of the attention model with Depicter on the benchmarking dataset for different species, and all the performance comparison results are listed in Table 2. The AUC values of the two frameworks are shown in Figure 2C. As can be seen for H. sapiens, D.melanogaster and A. thaliana, the average AUCs of Depicter were 0.062, 0.023 and 0.067, respectively higher than those of attention model on three types of models. On the other hand, it should be noted that for M. musculus, Depicter secured the best AUC values on TATA and non-TATA types of promoters, whereas the attention model achieved the best AUC value on TATA and non-TATA type of promoters. Figure 2D and Figures S7–S10 provide the t-SNE plots that include the mapped feature representations for the one-hot encoding, after the attention layer, and for the 2nd fully connected layer. From Figure 2A (b) and Figure 2D (c), we can clearly observe that Depicter could more correctly classify the promoter samples than the attention model in the final classification layer.
Table 2.
Performance comparison of the attention model and Depicter on the validation datasets
| Species | Types | Model | Sn (%) | Sp (%) | Pre (%) | Acc (%) | MCC | F1 score |
|---|---|---|---|---|---|---|---|---|
| H. sapiens | TATA | Attention | 96.75 | 92.17 | 91.54 | 94.31 | 0.8873 | 0.9407 |
| Depicter | 96.60 | 98.47 | 98.46 | 97.53 | 0.9508 | 0.9752 | ||
| Non-TATA | Attention | 85.47 | 79.07 | 77.53 | 82.00 | 0.6431 | 0.8131 | |
| Depicter | 88.61 | 89.14 | 89.46 | 88.87 | 0.7774 | 0.8903 | ||
| TATA and non-TATA | Attention | 78.63 | 82.54 | 83.81 | 80.45 | 0.6102 | 0.8114 | |
| Depicter | 88.22 | 89.95 | 90.25 | 89.06 | 0.7814 | 0.8922 | ||
| M. musculus | TATA | Attention | 98.56 | 97.11 | 97.15 | 97.83 | 0.9568 | 0.9785 |
| Depicter | 99.29 | 99.63 | 99.64 | 99.46 | 0.9892 | 0.9947 | ||
| Non-TATA | Attention | 99.41 | 96.74 | 96.70 | 98.05 | 0.9613 | 0.9804 | |
| Depicter | 99.20 | 97.13 | 97.12 | 98.15 | 0.9633 | 0.9815 | ||
| TATA and non-TATA | Attention | 99.15 | 97.83 | 97.78 | 98.48 | 0.9697 | 0.9846 | |
| Depicter | 98.38 | 98.17 | 98.15 | 98.27 | 0.9654 | 0.9826 | ||
| D. melanogaster | TATA | Attention | 92.21 | 96.85 | 96.98 | 94.42 | 0.8896 | 0.9454 |
| Depicter | 93.17 | 100.00 | 100.00 | 96.35 | 0.9295 | 0.9647 | ||
| Non-TATA | Attention | 90.33 | 89.75 | 89.16 | 90.03 | 0.8005 | 0.8974 | |
| Depicter | 94.10 | 91.12 | 90.37 | 92.52 | 0.8509 | 0.9220 | ||
| TATA and non-TATA | Attention | 89.58 | 88.98 | 88.30 | 89.27 | 0.7853 | 0.8893 | |
| Depicter | 92.09 | 91.42 | 90.90 | 91.74 | 0.8348 | 0.9149 | ||
| A. thaliana | TATA | Attention | 96.04 | 85.49 | 84.06 | 90.17 | 0.8101 | 0.8965 |
| Depicter | 97.09 | 98.38 | 98.44 | 97.72 | 0.9544 | 0.9776 | ||
| Non-TATA | Attention | 91.55 | 85.46 | 84.30 | 88.27 | 0.7677 | 0.8778 | |
| Depicter | 95.00 | 9456 | 94.53 | 94.78 | 0.8956 | 0.9477 | ||
| TATA and non-TATA | Attention | 91.83 | 87.48 | 86.76 | 89.53 | 0.7918 | 0.8922 | |
| Depicter | 96.06 | 95.38 | 95.34 | 95.72 | 0.9144 | 0.9570 |
Bold value in the table represents the maximum value for each performance metric.
Performance comparison on the independent test datasets
We uniformly and comprehensively compared the predictive performance of Depicter against other state-of-the-art prediction tools on the independent test datasets that were built for different types of promoters. There were three key points related to the performance comparison. The first point is that the data used in all other tools are not unified due to the continuous updating of the data; the second point is that several tools only focus on one species; the third point is that most approaches do not distinguish the specific types of TATA, non-TATA, and TATA and non-TATA. In this section, we compare the performance of Depicter with other state-of-the-art approaches across the four different species.
It should be noted that the prediction webserver of PromPredict required a minimum of 1000 nucleotides for making the prediction [18] whereas CNNPorm required that eukaryotic sequences had a length of 251 bp [34]. Therefore, we first processed our independent test sequences to keep the length consistent and then compared with PromPredict and CNNPorm. To meet the requirement of PromPredict, we extended the promoter sequences from 300 bp to 1001 bp according to the whole genome sequences. Whereas for the introns and exons, we retained those with the lengths of longer than 1001 bp, and then deleted the redundant sequences a the threshold of 0.8 by applying CD-HIT-EST. Lastly, we randomly selected the same number of negative samples as the selected positive samples to constitute the new negative test dataset. For CNNPorm, we extracted promoter sequences from the region of 200 bp upstream to 50 bp downstream regions of TSS, and extracted the negative samples with the length of 251 (1:251) bp from the corresponding original negative dataset.
For H. sapiens, Depicter was compared with iProEP, DeePromoter, PromPredict and CNNPorm, which are most recently reported prediction tools [18, 34, 36, 41]. However, as CNNPorm could only predict TATA promoters, we compared the performance of its Depicter for predicting the other types of promoters with that of the other three methods iProEP, DeePromoter and PromPredict. The detailed comparison results are listed in Table S4. We can see that among these five predictors, Depicter achieved the best performances with an average AUC of 0.940 over the three types of promoters across all the four species (Table S5). When compared with the second best iProEP, Depicter increased the AUC of 0.185 for predicting TATA promoters, 0.136 for non-TATA promoters, and 0.075 for TATA and non-TATA promoters, respectively. In addition to the AUC, Depicter also achieved the best performance in terms of all major measurements with the only exception of Sn for predicting the TATA type of promoters, which was 2.65% lower than that of CNNPorm (Figure 2E).
For M. musculus, Depicter was compared with CNNPorm, PromPredict and DeePromoter on the independent test dataset because the method iProEP did not consider this species. As a result, Depicter achieved an average AUC of 0.997 over the three types of promoters, which was 0.07 higher than that of CNNPorm (Table S5). More specifically, Depicter increased the AUC by 0.027 on TATA promoters, 0.098 on non-TATA promoters, and 0.085 on TATA and non-TATA promoters, respectively. In addition to the AUC, Depicter demonstrated the best performance in terms of all other six performance measurements, as shown in Figure 2F and Table S6.
For A. thaliana, Depicter was compared with PromPredict on the independent test datasets. The performance comparison results are provided in Table S7. Depicter attained the best performance with an average AUC of 0.983 on the three types of promoters (Table S5). Specifically, Depicter increased the AUC by 0.196 for TATA promoters, 0.166 for non-TATA promoters, and 0.182 for TATA and non-TATA promoters, respectively. Apart from the AUC score, Depicter demonstrated the best performance in terms of all performance metrics (Figure 2G).
For D. melanogaster, Depicter was compared with the other two methods iProEP and PromPredict, with the performance comparison results provided in Figure 2H and Table S8. As can be seen, Depicter also achieved a better predictive performance than iProEP and PromPredict in terms of Sp, Pre, Acc, MCC and F1 score, with the only exception of Sn for predicting promoters, which were slightly lower than those of PromPredict. Nevertheless, PromPredict achieved lower Sp than the other predictors. This was presumably due to the fact that PromPredict only considered the DNA duplex stability and did not consider the sequence statistics information. Moreover, Depicter achieved an average AUC of 0.980 on the three types of promoters, which was 0.096 higher than that of iProEP, and 0.267 higher than that of PromPredict, respectively (Table S5).
Overall, the empirical benchmarking tests indicate that Depicter provides improved predictive performances for predicting the three types promoters from H. sapiens, M. musculus, A. thaliana, and D. melanogaster, which is superior to the performance shown by the currently available models. Furthermore, we plotted and displayed the ROC curves of Depicter on the independent test datasets for all the four species in Figure 3. It can be seen that Depicter performed the best for prediction of three specific types of promoters for M. musculus than the other three species. It might be that the sequence-level signals of promoter sequences of M. musculus can be more easily captured by the machine learning than other species. Whereas for H. sapiens, an AUC score of 0.883 was achieved for TATA and non-TATA promoters, which was much lower than that for TATA promoters (AUC = 0.994) and non-TATA promoters AUC = 0.944. A possible reason might be that the one-hot encoding cannot extract sufficient information to discriminate TATA and non-TATA promoters from the data. Potentially useful strategies, such as adding the flexibility, base stacking, duplex stability and other structural features need to be developed to improve this respect in future work.
Figure 3.
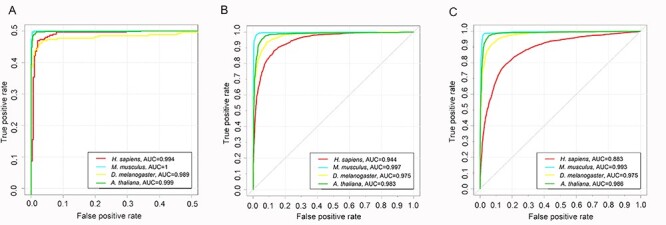
ROC curves of Depicter models for: (A) TATA types promoters; (B) non-TATA types promoters; and (C) TATA and non-TATA types promoters on the independent test datasets.
Webserver implementation
As an implementation of the proposed Depicter method, we have developed a user-friendly online webserver, which is freely available at https://depicter.erc.monash.edu/. To utilize the webserver, users need to upload the DNA sequences or paste them in the sequence window in the FASTA format at the prediction web page. The generated prediction results for all the submitted jobs will be presented in a table with detailed information about the sequence information and the predicted promoter type. In addition, the webserver also provides a probability score ranging from 0 to 1 to indicate the probability of the prediction results. The score close to 1 means the result is most reliable, whereas the score close to 0 means the result is least reliable. The prediction results can be copied directly or downloaded in the CSV, Excel or PDF formats. Moreover, the input of our webserver is not limited to a DNA sequence with a length of 300 bp. Users can also input multiple full-length DNA sequences. The webserver will automatically intercept in units of 300 bp and generate the prediction output for each full-length DNA sequence. Then, the user can move the mouse to a specific position with a higher score and the position of the predicted promoter will be will be highlighted. Step-by-step instructions for using the Depicter server can be found at the help page of the webserver. Figure 4 shows an example of the prediction webpage of the web server with the detailed prediction outputs.
Figure 4.
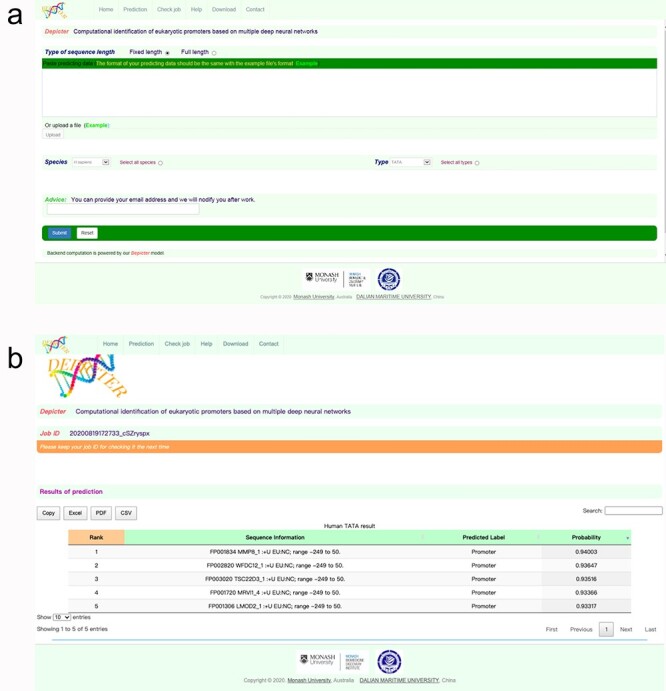
Screenshot of the webserver interface of Depicter. (A) The input interface of Depicter. (B) The output interface of Depicter, which shows the predicted results for the query sequences.
Conclusion
In this work, we have developed a novel approach called Depicter based on deep capsule neural networks for identifying specific types of promoters (including TATA, non-TATA, and TATA and non-TATA) across four eukaryote species that have most abundant promoter data. The up-to-date and reliable datasets for H. sapiens, M. musculus, D. melanogaster and A. thaliana were collected and processed. Then the training and independent datasets were rigorously established to train the models and evaluate the predictive performance. The concatenation of CNN and capsule network was utilized to build the framework of Depicter, which showed its superiority when compared to the connecting framework of CNN and the attention mechanism. In addition, we also provided a visual illustration to show that Depicter could effectively learn feature representations to accurately differentiate the different types of promoters. We performed empirical assessment of Depicter with several other state-of-the-art methods for the four species. Remarkably, in terms of seven performance measurements and three specific types of promoters across the four different species, there were a total of 84 performance results. Amongst these, Depicter achieved 78 better predictive performance results compared with the other existing methods. The user-friendly web server and the source code of Depicter are freely available at https://depicter.erc.monash.edu/. Both are designed to facilitate users to perform the prediction analysis and retrieve the results in an intuitive and accessible fashion. We expect that this deep capsule learning framework will be exploited as a powerful and useful alternative to address other sequence-based prediction tasks, such as prediction of enhancers and other functional elements from the DNA or RNA sequence data.
Key Points
Accurate identification of the gene transcription start site and the core promoter is essential for our understanding of the transcriptional regulation of genes.
We reviewed main existing methods for promoter recognition and categorized these methods into two major groups according to the operating algorithms.
A deep capsule neural network framework termed Depicter is designed to identify eukaryotic promoters across four species including Homo sapiens, Mus musculus, Drosophila melanogaster and Arabidopsis thaliana from the DNA sequences.
Comprehensive benchmarking tests demonstrate that Depicter outperforms several existing state-of-the-art methods.
The online webserver of Depicter is implemented and freely accessible at https://depicter.erc.monash.edu/.
Supplementary Material
Yan Zhu is currently pursuing the MS degree in the School of Science, Dalian Maritime University, China. Her research interests are bioinformatics, deep learning and machine learning.
Fuyi Li received his PhD in Bioinformatics from Monash University, Australia. He is currently a research fellow in the Peter Doherty Institute for Infection and Immunity, The University of Melbourne, Australia. His research interests are bioinformatics, computational biology, machine learning and data mining.
Dongxu Xiang received his BEng from Northwest A&F University, China. He is currently a research assistant in the Department of Biochemistry and Molecular Biology and Biomedicine Discovery Institute, Monash University, Australia. His research interests are bioinformatics, computational biology, machine learning and data mining.
Tatsuya Akutsu has been a professor in Bioinformatics Center, Institute for Chemical Research, Kyoto University since 2001. He obtained Dr Eng. degree from University of Tokyo in 1989. His research interests include bioinformatics, complex networks and discrete algorithms.
Jiangning Song is an associate professor and group leader in the Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, Australia. He is also affiliated with the Monash Centre of Data Science, Faculty of Information Technology, Monash University. His research interests include bioinformatics, computational biology, machine learning, data mining and pattern recognition.
Cangzhi Jia is an associate professor in the College of Science, Dalian Maritime University. She obtained her PhD degree in the School of Mathematical Sciences from Dalian University of Technology in 2007. Her major research interests include mathematical modeling in bioinformatics and machine learning.
Contributor Information
Yan Zhu, School of Science, Dalian Maritime University, China.
Fuyi Li, Peter Doherty Institute for Infection and Immunity, The University of Melbourne, Australia.
Dongxu Xiang, Northwest A&F University, China.
Tatsuya Akutsu, Bioinformatics Center, Institute for Chemical Research, Kyoto University.
Jiangning Song, Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, Australia.
Cangzhi Jia, College of Science, Dalian Maritime University.
Data availability
All the datasets generated for this study are either included in this article/Supplementary material or available at the ‘Depicter’ https://github.com/zhuyaner/Depicter/ mentioned in the Materials and methods section.
Conflict of interest
The authors declare that they have no conflict of interest.
Funding
National Natural Scientific Foundation of China (grant 62071079), Fundamental Research Funds for the Central Universities (3132020170, 3132019323), the National Natural Science Foundation of Liaoning Province (20180550223), double first-class construction special items (‘innovative project’) (345148012004). National Health and Medical Research Council of Australia (NHMRC) (1144652 and 1127948), the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01 AI111965), a Major Inter-Disciplinary Research (IDR) project awarded by Monash University, and the Collaborative Research Program of Institute for Chemical Research, Kyoto University (2019-32 and 2018-28).
References
- 1. Ngoc LV, Wang YL, Kassavetis GA, et al. The punctilious RNA polymerase II core promoter. Gene Dev 2017;31:1289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gangal R, Sharma P. Human POL II promoter prediction: time series descriptors and machine learning (vol 33, pg 1332, 2005). Nucleic Acids Res 2005;33:4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carter R, Drouin G. Structural differentiation of the three eukaryotic RNA polymerases. Genomics 2009;94:388–96. [DOI] [PubMed] [Google Scholar]
- 4. Gao ZL, Herrera-Carrillo E, Berkhout B. RNA polymerase II activity of type 3 pol III promoters. Mol Ther-Nucl Acids 2018;12:135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zeng W, Wang F, Ma Y, et al. Dysfunctional mechanism of liver cancer mediated by transcription factor and non-coding RNA. Curr Bioinform 2019;14:100–7. [Google Scholar]
- 6. Shepelev V, Fedorov A. Advances in the exon-intron database (EID). Brief Bioinform 2006;7:178–85. [DOI] [PubMed] [Google Scholar]
- 7. Kadonaga JT. The DPE, a core promoter element for transcription by RNA polymerase II. Exp Mol Med 2002;34:259–64. [DOI] [PubMed] [Google Scholar]
- 8. Ohler U. Identification of core promoter modules in drosophila and their application in accurate transcription start site prediction. Nucleic Acids Res 2006;34:5943–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Juven-Gershon T, Cheng S, Kadonaga JT. Rational design of a super core promoter that enhances gene expression. Nat Methods 2006;3:917–22. [DOI] [PubMed] [Google Scholar]
- 10. Yella VR, Bansal M. DNA structural features of eukaryotic TATA-containing and TATA-less promoters. Febs Open Bio 2017;7:324–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Deng WS. Roberts SGE. A core promoter element downstream of the TATA box that is recognized by TFIIB. Gene Dev 2005;19:2418–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cramer P, Pesce CG, Baralle FE, et al. Functional association between promoter structure and transcript alternative splicing. P Natl Acad Sci USA 1997;94:11456–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Slobodin B, Agami R. Transcription initiation determines its end. Mol Cell 2015;57:205–6. [DOI] [PubMed] [Google Scholar]
- 14. Giordano A, Eder PS, Mcgill DB, et al. Methods for Identifying Novel Nucleic Acid Regulatory Elements and Compounds that Affect the Regulation. US Patent 20100781308[P]. 2006. [Google Scholar]
- 15. Knudsen S. Promoter2.0: for the recognition of PolII promoter sequences. Bioinformatics 1999;15:356–61. [DOI] [PubMed] [Google Scholar]
- 16. Rahman MS, Aktar U, Jani MR, et al. iPro70-FMWin: identifying Sigma70 promoters using multiple windowing and minimal features. Mol Genet Genomics 2019;294:69–84. [DOI] [PubMed] [Google Scholar]
- 17. Ning C, You FQ. Optimization under uncertainty in the era of big data and deep learning: when machine learning meets mathematical programming. Comput Chem Eng 2019;125:434–48. [Google Scholar]
- 18. Yella VR, Kumar A, Bansal M. Identification of putative promoters in 48 eukaryotic genomes on the basis of DNA free energy. Sci Rep 2018;8:4520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Abeel T, Saeys Y, Bonnet E, et al. Generic eukaryotic core promoter prediction using structural features of DNA. Genome Res 2008;18:310–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kumar A, Bansal M. Characterization of structural and free energy properties of promoters associated with primary and operon TSS in helicobacter pylori genome and their orthologs. J Biosci 2012;37:423–31. [DOI] [PubMed] [Google Scholar]
- 21. Abe N, Dror I, Yang L, et al. Deconvolving the recognition of DNA shape from sequence. Cell 2015;161:307–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schaefer U, Kodzius R, Kai C, et al. High sensitivity TSS prediction: estimates of locations where TSS cannot occur. PLoS One 2010;5:e13934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang M, Li FY, Marquez-Lago TT, et al. MULTiPly: a novel multi-layer predictor for discovering general and specific types of promoters. Bioinformatics 2019;35:2957–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lai HY, Zhang ZY, Su ZD, et al. iProEP: a computational predictor for predicting promoter. Mol Ther-Nucl Acids 2019, 46;17:337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu B, Yang F, Huang DS, et al. iPromoter-2L: a two-layer predictor for identifying promoters and their types by multi-window-based PseKNC. Bioinformatics 2018;34:33–40. [DOI] [PubMed] [Google Scholar]
- 26. Chen Z, Zhao P, Fs L, et al. iLearn: an integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA, RNA and protein sequence data. Brief Bioinform 2020;21:1047–57. [DOI] [PubMed] [Google Scholar]
- 27. Li F, Chen J, Ge Z, et al. Computational prediction and interpretation of both general and specific types of promoters in Escherichia coli by exploiting a stacked ensemble-learning framework. Brief Bioinform 2020;1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu Q, Chen J, Wang Y, et al. DeepTorrent: a deep learning-based approach for predicting DNA N4-methylcytosine sites. Brief Bioinform 2020;10. doi: 1093/bib/bbaa124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu B, Zhang H, Lin L, et al. A similarity searching system for biological phenotype images using deep convolutional encoder-decoder architecture. Curr Bioinform 2019;14:628–39. [Google Scholar]
- 30. Yu L, Sun X, Tian SW, et al. Drug and nondrug classification based on deep learning with various feature selection strategies. Curr Bioinform 2018;13:253–9. [Google Scholar]
- 31. Chen Z, Zhao P, Li F, et al. Comprehensive review and assessment of computational methods for predicting RNA post-transcriptional modification sites from RNA sequences. Brief Bioinform 2020;21:1676–96. [DOI] [PubMed] [Google Scholar]
- 32. Li F, Chen J, Leier A, et al. DeepCleave: a deep learning predictor for caspase and matrix metalloprotease substrates and cleavage sites. Bioinformatics 2020;36:1057–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aristodemou L, Tietze F. The state-of-the-art on intellectual property analytics (IPA): a literature review on artificial intelligence, machine learning and deep learning methods for analysing intellectual property (IP) data. World Patent Info 2018;55:37–51. [Google Scholar]
- 34. Umarov RK, Solovyev VV. Recognition of prokaryotic and eukaryotic promoters using convolutional deep learning neural networks. PLoS ONE 2017;12:e0171410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Umarov R, Kuwahara H, Li Y, et al. Promoter analysis and prediction in the human genome using sequence-based deep learning models. Bioinformatics 2019;35:2730–7. [DOI] [PubMed] [Google Scholar]
- 36. Oubounyt M, Louadi Z, Tayara H, et al. DeePromoter: robust promoter predictor using deep learning. Front Genet 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zou Q, Wan S, Ju Y, et al. Pretata: predicting TATA binding proteins with novel features and dimensionality reduction strategy. BMC Syst Biol 2016;10:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lv H, Dao F-Y, Zhang D, et al. iDNA-MS: an integrated computational tool for detecting DNA modification sites in multiple genomes. iScience 2020;23:100991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dreos R, Ambrosini G, Perier RC, et al. The eukaryotic promoter database: expansion of EPDnew and new promoter analysis tools. Nucleic Acids Res 2015;43:D92–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xu MY, Gonzalez-Hurtado E, Martinez E. Core promoter-specific gene regulation: TATA box selectivity and initiator-dependent bi-directionality of serum response factor-activated transcription. Bba-Gene Regul Mech 2016;1859:553–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lai HY, Zhang ZY, Su ZD, et al. iProEP: a computational predictor for predicting promoter. Mol Ther Nucleic Acids 2019;17:337–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dreos R, Ambrosini G, Perier RC, et al. EPD and EPDnew, high-quality promoter resources in the next-generation sequencing era. Nucleic Acids Res 2013;41:D157–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhu Y, Jia C, Li F, et al. Inspector: a lysine succinylation predictor based on edited nearest-neighbor undersampling and adaptive synthetic oversampling. Anal Biochem 2020;593:113592. [DOI] [PubMed] [Google Scholar]
- 44. He W, Jia C. EnhancerPred2.0: predicting enhancers and their strength based on position-specific trinucleotide propensity and electron-ion interaction potential feature selection. Mol Biosyst 2017;13:767–74. [DOI] [PubMed] [Google Scholar]
- 45. Huang Y, Niu BF, Gao Y, et al. CD-HIT suite: a web server for clustering and comparing biological sequences. Bioinformatics 2010;26:680–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zou Q, Lin G, Jiang X, et al. Sequence clustering in bioinformatics: an empirical study. Brief Bioinform 2020;21:1–10. [DOI] [PubMed] [Google Scholar]
- 47. Yu ZY, Niu ZW, Tang WH, et al. Deep learning for daily peak load forecasting-a novel gated recurrent neural network combining dynamic time warping. Ieee Access 2019;7:17184–94. [Google Scholar]
- 48. Wei L, Su R, Wang B, et al. Integration of deep feature representations and handcrafted features to improve the prediction of N 6-methyladenosine sites. Neurocomputing 2019;324:3–9. [Google Scholar]
- 49. Uriarte-Arcia AV, Lopez-Yanez I, Yanez-Marquez C. One-hot vector hybrid associative classifier for medical data classification. Plos One 2014;9:e95715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lv ZB, Ao CY, Zou Q. Protein function prediction: from traditional classifier to deep learning. Proteomics 2019;19:2. [DOI] [PubMed] [Google Scholar]
- 51. Li F, Zhang Y, Purcell AW, et al. Positive-unlabelled learning of glycosylation sites in the human proteome. BMC Bioinformatics 2019;20:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen W, Li Y, Tsangaratos P, et al. Groundwater spring potential mapping using artificial intelligence approach based on kernel logistic regression, random Forest, and alternating decision tree models. Appl Sci-Basel 2020;10:425. [Google Scholar]
- 53. Li F, Li C, Wang M, et al. GlycoMine: a machine learning-based approach for predicting N-, C- and O-linked glycosylation in the human proteome. Bioinformatics 2015;31:1411–9. [DOI] [PubMed] [Google Scholar]
- 54. Chen Z, Zhao P, Li F, et al. iLearn: an integrated platform and meta-learner for feature engineering, machine learning analysis and modeling of DNA, RNA and protein sequence data. Brief Bioinform 2020;21:1047–57. [DOI] [PubMed] [Google Scholar]
- 55. Chen YS, Jiang HL, Li CY, et al. Deep feature extraction and classification of hyperspectral images based on convolutional neural networks. IEEE T Geosci Remote 2016;54:6232–51. [Google Scholar]
- 56. Petersen P, Voigtlaender F. Optimal approximation of piecewise smooth functions using deep ReLU neural networks. Neural Netw 2018;108:296–330. [DOI] [PubMed] [Google Scholar]
- 57. Sainath TN, Mohamed AR, Kingsbury B, et al. Deep convolutional neural networks for Lvcsr. Int Conf Acoust Spee 2013;8614–8. [Google Scholar]
- 58. Yao Y, Rosasco L, Caponnetto A. On early stopping in gradient descent learning. Constr Approx 2007;26:289–315. [Google Scholar]
- 59. Prechelt L. Early stopping - but when? Neural Networks: Tricks of the Trade 1998;1524:55–69. [Google Scholar]
- 60. Zhao YM, He NN, Chen Z, et al. Identification of protein lysine Crotonylation sites by a deep learning framework with convolutional neural networks. Ieee Access 2020;8:14244–52. [Google Scholar]
- 61. Kingma D, Ba J. Adam: A method for stochastic optimization. In: 3rd Int. Conf. Learn. Represent ICLR 2015 - Conf. Track Proc., 2015; 1–15. [Google Scholar]
- 62. Beykikhoshk A, Quinn TP, Lee SC, et al. DeepTRIAGE: interpretable and individualised biomarker scores using attention mechanism for the classification of breast cancer sub-types. BMC Med Genomics 2020;13, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhang SY, Zhang SW, Fan XN, et al. FunDMDeep-m6A: identification and prioritization of functional differential m6A methylation genes. Bioinformatics 2019;35:i90–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang D, Liang Y, Xu D. Capsule network for protein post-translational modification site prediction. Bioinformatics 2019;35:2386–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Snoek J, Larochelle H, Adams RP. Practical Bayesian optimization of machine learning algorithms. Adv Neural Inform Process Syst 2012;4:2951–9. [Google Scholar]
- 66. Jia C, Zhang M, Fan C, et al. Formator: predicting lysine formylation sites based on the most distant undersampling and safe-level synthetic minority oversampling. IEEE/ACM Trans Comput Biol Bioinform 2019. doi: 10.1109/TCBB.2019.2957758. [DOI] [PubMed] [Google Scholar]
- 67. Jia C, Bi Y, Chen J, et al. PASSION: an ensemble neural network approach for identifying the binding sites of RBPs on circRNAs. Bioinformatics 2020;36:4276–82. [DOI] [PubMed] [Google Scholar]
- 68. Li F, Leier A, Liu Q, et al. Procleave: predicting protease-specific substrate cleavage sites by combining sequence and structural information. Genomics Proteomics Bioinformatics 2020;18:52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Li F, Wang Y, Li C, et al. Twenty years of bioinformatics research for protease-specific substrate and cleavage site prediction: a comprehensive revisit and benchmarking of existing methods. Brief Bioinform 2019;20:2150–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Li F, Li C, Marquez-Lago TT, et al. Quokka: a comprehensive tool for rapid and accurate prediction of kinase family-specific phosphorylation sites in the human proteome. Bioinformatics 2018;34:4223–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zeiler MD, Fergus R. Visualizing and understanding convolutional networks. Computer Vision - Eccv 2014, Pt I2014;8689:818–33. [Google Scholar]
- 72. Zhang HJ, Wang FJ, Xi T, et al. A novel quality evaluation method for resistance spot welding based on the electrode displacement signal and the Chernoff faces technique. Mech Syst Signal Pr 2015;62-63:431–43. [Google Scholar]
- 73. Bau D, Zhou BL, Khosla A, et al. Network Dissection: Quantifying Interpretability of Deep Visual Representations, 30th Ieee Conference on Computer Vision and Pattern Recognition (Cvpr 2017) 2017; 10.1109/Cvpr.2017.354:3319-27. [DOI]
- 74. Maaten L, Hinton G. Visualizing data using t-SNE. J Mach Learn Res 2008;9:2579–605. [Google Scholar]
- 75. Husnain M, Missen MMS, Mumtaz S, et al. Visualization of high-dimensional data by pairwise fusion matrices using t-SNE. Symmetry-Basel 2019;11:107. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the datasets generated for this study are either included in this article/Supplementary material or available at the ‘Depicter’ https://github.com/zhuyaner/Depicter/ mentioned in the Materials and methods section.