

Abstract
Maintenance and regulation of proper mitochondrial dynamics and functions are necessary for cellular homeostasis. Numerous diseases, including neurodegeneration and muscle myopathies, and overall cellular aging are marked by declining mitochondrial function and subsequent loss of multiple other cellular functions. For these reasons, optimized protocols are needed for visualization and quantification of mitochondria and their function and fitness. In budding yeast, mitochondria are intimately associated with the actin cytoskeleton and utilize actin for their movement and inheritance. This chapter describes optimal approaches for labeling mitochondria and the actin cytoskeleton in living budding yeast cells, for imaging the labeled cells, and for analyzing the resulting images.
Keywords: yeast, mitochondria, fluorescent proteins, vital staining, microscopy, live-cell imaging, deconvolution, ratio imaging, actin, cytoskeleton
1. Introduction
Live-cell imaging is the optimal approach to visualize dynamic structures, and several imaging techniques have been developed to efficiently visualize both actin and mitochondria in living yeast cells. These techniques enable visualization of events that cannot be seen in fixed cells, including mitochondrial motility, fusion, and fission, and retrograde actin cable flow. Some vital dyes can also be used to assess the function and quality of mitochondria, including mitochondrial membrane potential (Δψ), mitochondrial DNA (mtDNA) content, and mitochondrial redox state.
Here, we describe methods to tag yeast genes at their chromosomal loci, determine functionality of proteins with fluorescent tags, visualize tagged gene products expressed either from endogenous or other promoters, and carry out quantitative analysis of mitochondrial and actin dynamics. We also describe methods to measure mitochondrial quality using probes engineered to quantitatively measure mitochondrial Δψ, redox state, and mtDNA content.
1.1. Detection of cellular structures using targeted fluorescent proteins
Both mitochondria and the actin cytoskeleton can be visualized in living yeast cells using fluorescent proteins (FPs). Mitochondria can be visualized by tagging a protein native to mitochondria, or by adding a short mitochondrial targeting sequence to a fluorescent protein.
1.1.1. Choosing a fluorescent protein
Green fluorescent protein (GFP), discovered and cloned from the jellyfish Aequorea victoria, revolutionized live-cell imaging in cell biology. Now, GFP is one of a large and growing palette of FPs with different colors and molecular properties. Identification of novel FPs, such as mCherry derived from Discosoma sp. coral and Teal from Clavularia sp. coral, combined with laboratory mutagenesis, has yielded FPs with a variety of colors as well as improved brightness, faster folding, and decreased oligomerization (Bubnell et al., 2013).
To perform well in a live-cell experiment, an FP must be photostable enough to withstand repeated imaging. It also must be bright enough to allow the structure of interest to be seen over background fluorescence and detector noise, and spectrally distinct from any other labels being used. Finally, it must not be toxic or disrupt the behavior of the cell or the protein to which it is fused. The effective brightness of an FP is determined by its intrinsic brightness (the product of extinction coefficient and quantum yield), and the behavior of the FP upon ectopic expression (rate of folding and stability). The amount of this signal that is excited and detected depends on properties of the imaging system (light sources, lenses, filters and detectors). As an aid to selecting fluorescent proteins, Tally Lambert and Kurt Thorn have prepared an extensive database of physical properties of FPs, available at http://nic.ucsf.edu/FPvisualization/.
1.1.2. Tagging endogenous proteins
For chromosomal tagging of proteins in yeast, a double-stranded linear DNA that encodes the tag of interest and a selection marker is inserted into a target site in the genome by homologous recombination. This insertion cassette is most commonly produced by PCR using a tagging vector as a template. Tagging vectors have been developed for a variety of FPs (e.g. GFP, mCherry, mCitrine), epitopes (e.g. HA, myc), and affinity tags (e.g. GST, TAP, His) and have been linked to a variety of selection markers conferring drug resistance or rescue of auxotrophy. Families of tagging vectors have been constructed that share PCR-priming sequences. With these, a single set of primers can be used to insert any tag into a given target gene. Vectors are also available for expression of tagged genes from endogenous promoters, constitutively active promoters (e.g. AHD1, GPD1), and regulatable promoters (e.g. GAL1). FP and epitope tags can also be used for biochemical techniques including affinity purification, immunoprecipitation, western blot analysis, and immunofluorescence.
Some tagging cassettes, including the pOM family, allow removal of the selectable marker after tagging (Gauss et al., 2005). In these cassettes, the selection marker is flanked by LoxP sites and thus can be removed by bacteriophage Cre recombinase that is conditionally expressed from a plasmid (Gueldener et al., 2002). This technology is useful in several situations: 1) for N-terminal tagging without separation of the tagged protein from its endogenous promoter; 2) for inserting a tag internally within the coding region of the gene of interest; 3) for multiple rounds of tagging at the same locus; and 4) for use in yeast strains with a limited number of selectable markers. Some readily available tagging vectors are shown in Table 1.
Table 1:
Yeast tagging cassette vectors
| Plasmid family | Tag position | Promoter | Tags | Markers |
|---|---|---|---|---|
| pFA6a 1 | C terminal | endogenous | GFP(S65T) 3xHA 13xMyc GST |
TRP1
kanMX6 HIS3MX6 |
| pFA6a-PGAL1 1 | N terminal or internal | GAL1 | GFP(S65T) 3xHA GST |
TRP1
kanMX6 HIS3MX6 |
| pUR 2 | C terminal | endogenous | DsRed |
HIS3
URA3(K.l.) |
| pYM 3 | C terminal | endogenous | yEGFP EGFP EBFP ECFP EYFP DsRed, DsRedI RedStar, RedStar2 eqFP611 FlAsH 1xHA, 3xHA, 6xHA 3xMyc, 9xMyc 1xMyc+7xHis TAP Protein A |
kanMX4
hphNT1 natNT2 HIS3MX6 klTRP1 |
| pKT 4 | C terminal | endogenous | yEGFP yECFP yEVenus yECitrine yESapphire yEmCFP5 yEmCitrine tdimer26 yECitrine+3xHA yECitrine+13xMyc yECFP+3xHA yECFP+13xMyc |
KanMX
SpHIS5 CaURA3 |
| pOM 7 | N terminal or internal | endogenous8 | yEGFP 6xHA 9xMyc Protein A TEV-ProteinA TEV-GST-6xHis TEV-ProteinA-7xHis |
kanMX6
URA3(K.l.) LEU2(K.l.) |
| pCY 9 | C-terminal | endogenous | Cerulean yECFP yEmCFP yEGFP Venus yEVenus yECitrine yEmCitrine mCherry mEos2 yEc-MYC yEHA yEFlAsH |
HygromycinB
Zeocin |
monomeric version
tandem dimer of DsRed
After Cre-mediated removal of auxotrophic marker
When a tagging vector is used, primers should be designed with sequences to hybridize with both the tagging vector and the target site for homologous recombination within the yeast chromosome. An insertion cassette is then produced by PCR using the tagging vector as a template. The amplified DNA is transformed into yeast using a standard protocol (Gietz et al., 1995). The selection marker used in the insertion cassette is used to identify recombinants that carry the inserted tag, and PCR-based screening can be used to ensure that the homologous recombination occurred at the correct site.
1.1.3. Targeting FPs to mitochondria using a signal sequence
Mitochondria contain two membranes, the inner and outer membranes, and two soluble compartments, the intermembrane space and the matrix. Other submitochondrial compartments include the mtDNA nucleoids and contact sites between inner and outer membranes (Simbeni et al., 1991, Meeusen & Nunnari, 2003). Over 95% of mitochondrial proteins are encoded in nuclear DNA, synthesized in the cytoplasm, and imported into the organelle. Targeted import depends on signal sequences, which can exist at the amino terminus or within nuclear-encoded mitochondrial proteins (Koehler, 2004). Therefore, nuclear-encoded mitochondrial proteins are typically tagged at their C-terminus to preserve signal sequence function. A full-length protein containing a mitochondrial targeting sequence, or simply the signal sequence alone, can be used to target FPs to specific compartments within mitochondria. It is also possible to localize the FPs to specific mitochondrial components by tagging endogenous proteins that are imported to sites of interest, such as Tom70p (Translocation through the Outer Mitochondrial membrane) for outer membrane labeling and Cit1p (Citrate synthase 1) for matrix targeting (Fig. 1A). Expression of FP-tagged Tom70p or Cit1p has no obvious effect on mitochondrial morphology, motility, or respiratory activity. They are excellent tools for investigating mitochondrial distribution and morphology (Fig. 1B). Plasmid-borne targeted FPs can also be used to label yeast mitochondria and produce a robust, fluorescent signal that is specific to the organelle (Table 2). However, as a result of cell-to-cell variability in plasmid copy number, fluorescence intensity is variable using plasmid-borne mitochondria-targeted FPs.
Fig. 1. Visualization of mitochondrial morphology and distribution.
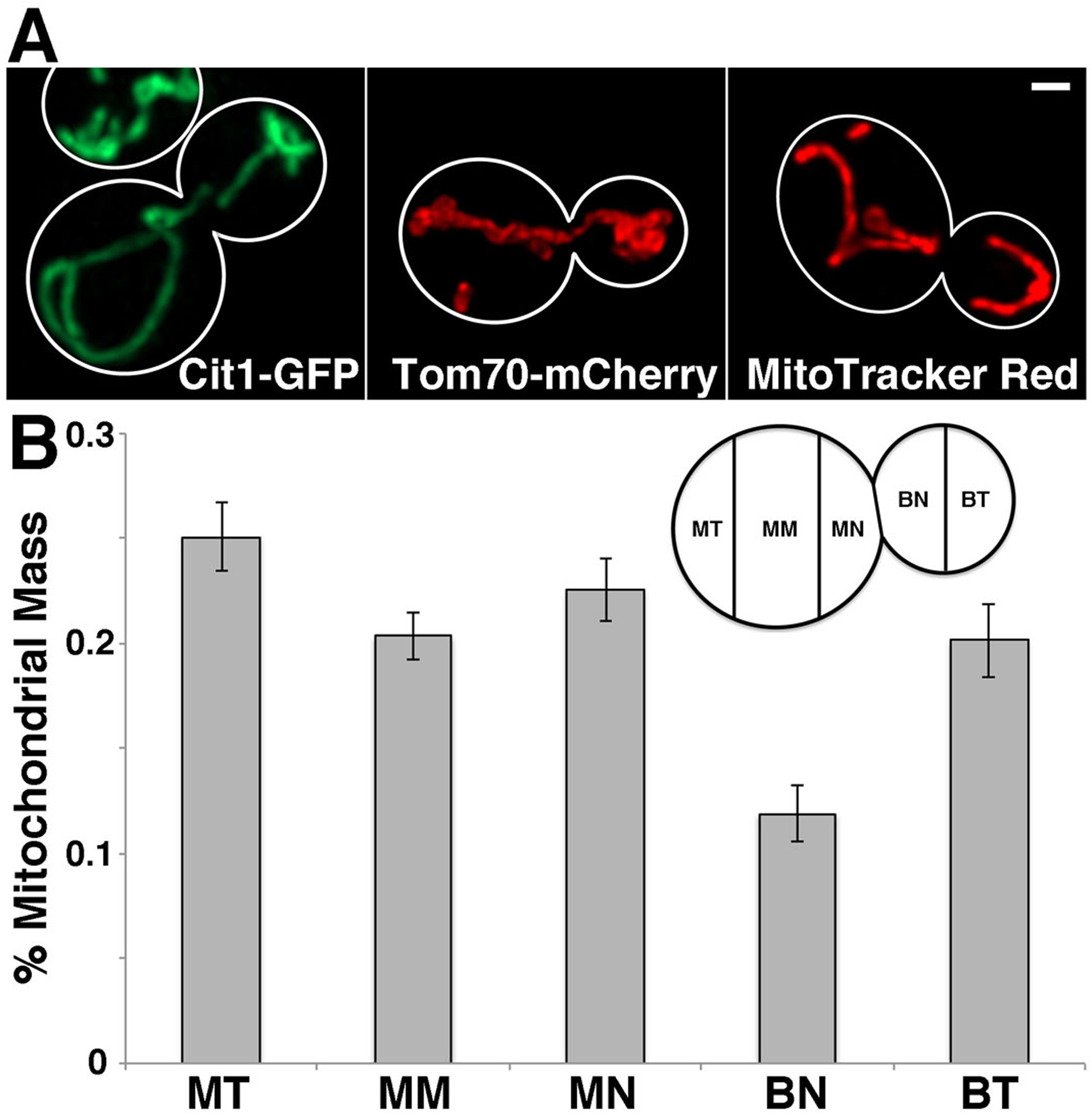
A) Mitochondria were visualized by tagging Cit1p, a mitochondrial matrix protein, with GFP (left panel); Tom70p, a mitochondrial outer membrane protein, with mCherry (middle panel); and using the lipophilic dye, MitoTracker Red (right panel). Scale bar = 1 μM. Images were acquired using a standard GFP filter with 100 ms exposure time for GFP and a standard DsRed filter with 200 ms exposure time for mCherry and MitoTracker Red. B) Mitochondrial distribution was measured by separating the yeast cell into 5 different compartments as shown in inset: tip of the bud distal to the mother (bud tip), tip of the bud adjacent to the mother (bud neck), tip of the mother cell adjacent to the bud (mother neck), middle of the mother cell (mother middle), and tip of the mother cell distal to the bud (mother tip). MT = mother tip; MM = mother middle; MN = mother neck; BN = bud neck; BT = bud tip. Mitochondrial content in each region was assessed by measuring the integrated intensity of Cit1-GFP fluorescence in yeast cells bearing a large bud (0.60 to 0.80 ratio of bud to mother size). Error bars indicate SEM. n = 40. Data is representative of 3 experiments.
Table 2:
Mitochondria-targeted signal sequence-FP fusion proteins
| Site | 4.1.1.1. Targeting | Promoter | Vector | FP | Reference |
|---|---|---|---|---|---|
| Matrix | OLI12 signal sequence | ADH1 | 2μ pRS426 derivative | HcRed | (Fehrenbacher et al., 2004) |
| CIT13 signal sequence | CIT1 | CEN-URA3 | bGFP (F99S, M153T, V163A) | (Okamoto et al., 1998) | |
| CIT1 signal sequence | GAL1 | CEN-URA3 | bGFP | (Okamoto et al., 1998) | |
| OLI1 | GAL1 | CEN-URA3 | DsRed | (Mozdy et al., 2000) | |
| OM | TOM6 signal sequence | GAL1 | CEN-URA3 | bGFP | (Okamoto et al., 1998) |
| IM | YTA10 | GAL1 | CEN-URA3 | bGFP | (Okamoto et al., 1998) |
| mtDNA | ABF2 | GAL1 | CEN-URA3 | bGFP | (Okamoto et al., 1998) |
Mitochondria are highly dynamic structures that undergo constant anterograde movement (toward the bud) and retrograde movement (towards the tip of the mother cell distal to the bud). They also undergo cycles of fusion and fission (Westermann, 2010, Higuchi-Sanabria et al., 2014, Vevea et al., 2014). Additionally, mitochondria are anchored at the bud tip and in the cortex of the mother cell by Mmr1p and Num1p, respectively (Swayne et al., 2011, Lackner et al., 2013) (Klecker et al., J Cell Sci, 2013). Mitochondrial dynamics, including velocity of mitochondrial motility, total mitochondrial motility, anchorage, and fusion and fission can be monitored by time-lapse imaging of mitochondria-targeted FPs, such as GFP-tagged Cit1p (Fig. 2).
Fig. 2. Visualization of mitochondrial dynamics in budding yeast.
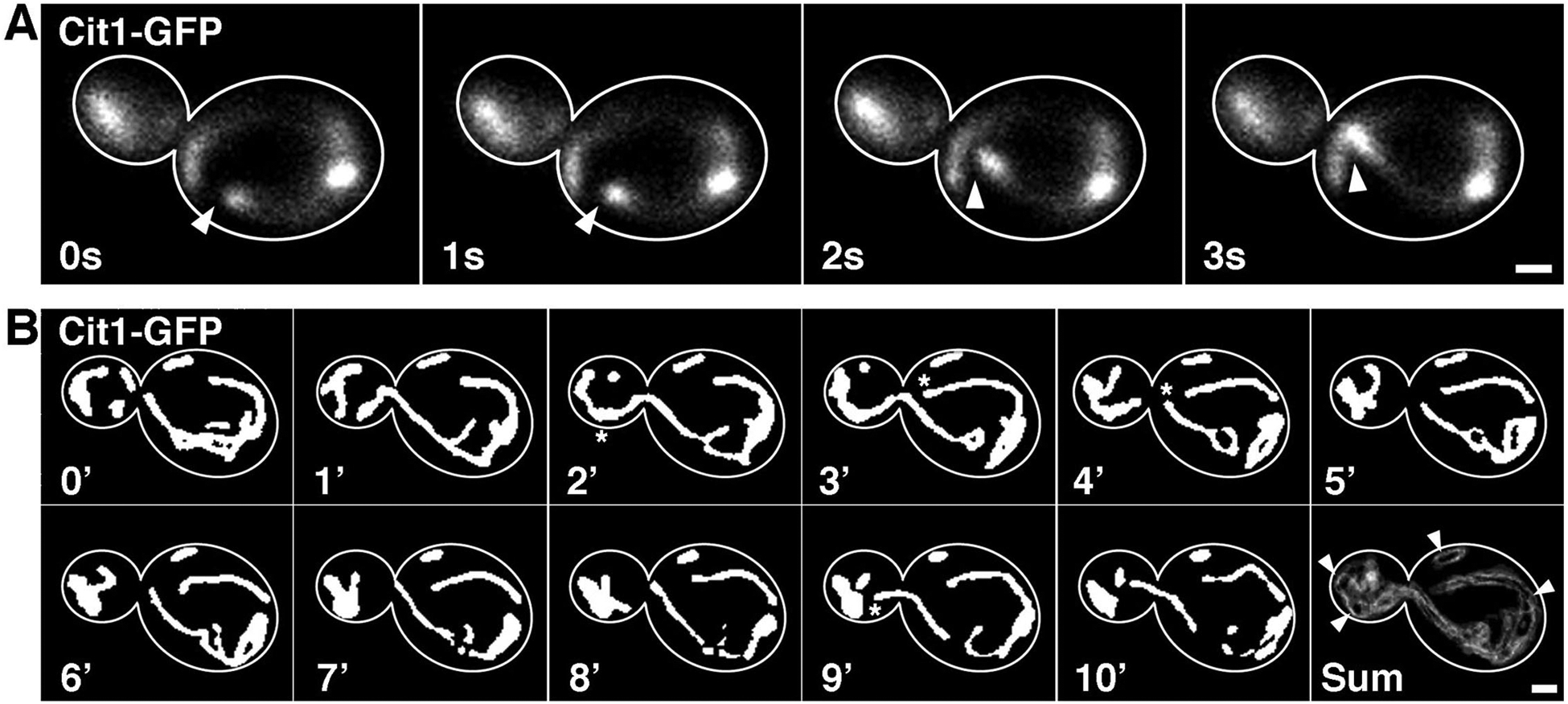
A) Velocity of mitochondrial motility was determined by taking single-plane images of Cit1-GFP every 1 s for 30 s. Images were captured using standard GFP filters and 100 ms exposure time. The optimal imaging plane is slightly above the center of the cell. Arrowheads follow the tip of a tubular mitochondrion moving in an anterograde direction. Scale bar = 1 μM. B) Total motility of mitochondria was determined by taking z-stacks of Cit1-GFP every 30 s for 10 min. Images were captured using a standard GFP filter, a 50% neutral density filters, and 100 ms exposure time. Thresholded maximum-intensity projections at 1-min intervals are shown. Stars indicate the largest movement events that were visualized during the 10 minute period. The “Sum” image (bottom-right corner) is the sum of the difference images calculated by the Total Motility plugin. This image depicts all motility events where white indicates movement and black mitochondrial centers indicate lack of movement. Edges of mitochondria commonly appear grey due to small non-directed movements. Arrowheads mark mitochondrial anchorage sites in the bud tip, mother tip, and mother cortex where mitochondrial movement is completely lacking. Scale bar = 1 μM.
1.1.4. Detecting mitochondrial DNA with DNA-binding dyes
In animal cells and fungi, mitochondria are the only extranuclear organelles containing DNA. Cytoplasmic DNA staining is therefore a reliable marker for mitochondria. In addition, the quantity and distribution of mitochondrial DNA (mtDNA) can illuminate mitochondrial structural integrity and fitness. In yeast and other eukaryotes, mtDNA associates with a complex of proteins involved in organization, replication, and expression of genes of mtDNA (Meeusen & Nunnari, 2003). This complex of proteins and mtDNA assemble into punctate structures localized to the inner leaflet of the inner mitochondrial membrane. Tagging of these proteins can be used to monitor nucleoid assembly and distribution. In yeast, a species in which mtDNA is dispensable for viability, DNA-binding dyes can also be used to determine if a strain lacks mtDNA (rho0).
Many DNA binding dyes do not readily cross the yeast cell wall. However, DAPI (4’,6’-diamidino-2-phenylindole), which stains nuclear and mtDNA in S. cerevisae, has become the most commonly used DNA-binding dye in yeast (Table 3). Its fluorescence increases greatly when bound to DNA in comparison to the unbound form or DAPI bound with RNA, making DAPI a reliable and robust nuclear and mtDNA marker with little cytoplasmic background. Additionally, DAPI staining of mtDNA is not dependent upon the metabolic state of the organelle. Finally, DAPI can be used in both live and fixed cells, producing a robust and persistent fluorescent signal.
Table 3:
Vital dyes for yeast mitochondria
| Dye | 4.1.1.1.1 IUPAC name | λ ex | λ em |
|---|---|---|---|
| DiOC6 (Riezman et al., 1983) | 3,3’-dihexyloxacarbocyanine iodide | 482 | 504 |
| DASPMI | 4-(4-(dimethylamino)styryl)-N-methylpyridinium iodide (4-Di-1-ASP) | 475 | 6051 |
| Rhodamine 123 | 2-(6-Amino-3-imino-3H-xanthen-9-yl)benzoic acid methyl ester | 505 | 5341 |
| MitoTracker | various | various | Various |
| DAPI | 4’,6’diamidino-2-phenylindole | 358 | 461 |
| DHE2 | dihydroethidium | 485 | 530 |
Broad emission range; not recommended for green/red double-label studies.
DHE is not a mitochondrial vital dye, but has been included in this list as it can reliably be used for measurements of mitochondrial ROS levels (see Fig. 3).
One disadvantage of DAPI is that due to its strong nuclear staining, mtDNA nucleoids close to the nucleus are not well resolved. In live cells, DAPI shows a preference for mtDNA over nuclear DNA, but nuclear staining does still occur, making accurate quantification of mtDNA using DAPI difficult. In addition, the UV illumination (350–400 nm) required to visualize DAPI is damaging. Therefore, it is not suitable for time-lapse or long-term live-cell imaging. In addition, because DAPI binds directly to DNA, it can reduce cell viability.
1.2. Visualization of organelle function: mitochondrial membrane potential and redox state
1.2.1. Measuring mitochondrial membrane potential
Positively charged lipophilic fluorophores can be utilized to measure mitochondrial Δψ because functioning mitochondria have the most negative Δψ in the cell, causing the fluorophores to accumulate in the organelle. Unhealthy or damaged mitochondria having low Δψ, such as those treated with the proton ionophore carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) or lacking mtDNA, fail to import these vital dyes into the organelle, making these dyes a useful tool in monitoring mitochondrial function in live cells.
DiOC6, the styryl dye DASPMI, the cationic rhodamine derivative rhodamine 123, and the fixable stains of the MitoTracker family work particularly well in yeast. DiOC6 and MitoTrackers are particularly useful for double-label experiments due to their narrow excitation and emission spectra, preventing bleed-through in more quantitative experiments. In addition, MitoTracker Orange, Red, and Deep Red are the only dyes that persist in mitochondria after aldehyde fixation and permeabilization by acetone or Triton X-100, making them the only membrane-potential dyes that can be used with immunofluorescence.
DiOC6 can be used to monitor mitochondrial Δψ as a quantitative measurement of overall mitochondrial quality (Hughes & Gottschling, 2012). In this assay, cells are exposed to DiOC6, and the total level of imported dye is normalized to total mitochondrial mass measured by a Δψ-independent mitochondrial marker, such as Tom70p that is tagged at its chromosomal locus with mCherry (Fig. 3A). This normalization is crucial to correct for strains or conditions that may cause increased mitochondrial quantity, which may overestimate DiOC6 measurements as an indicator for Δψ. Treatment with a proton ionophore will cause collapse of mitochondrial Δψ by uncoupling the proton gradient from ATP synthesis, while growth in non-fermentable carbon sources, such as glycerol, will result in increased Δψ due to increased respiration. These treatments can be used to determine the dynamic range of this quantitative tool (Fig. 3B).
Fig. 3. Characterization of mitochondrial quality.
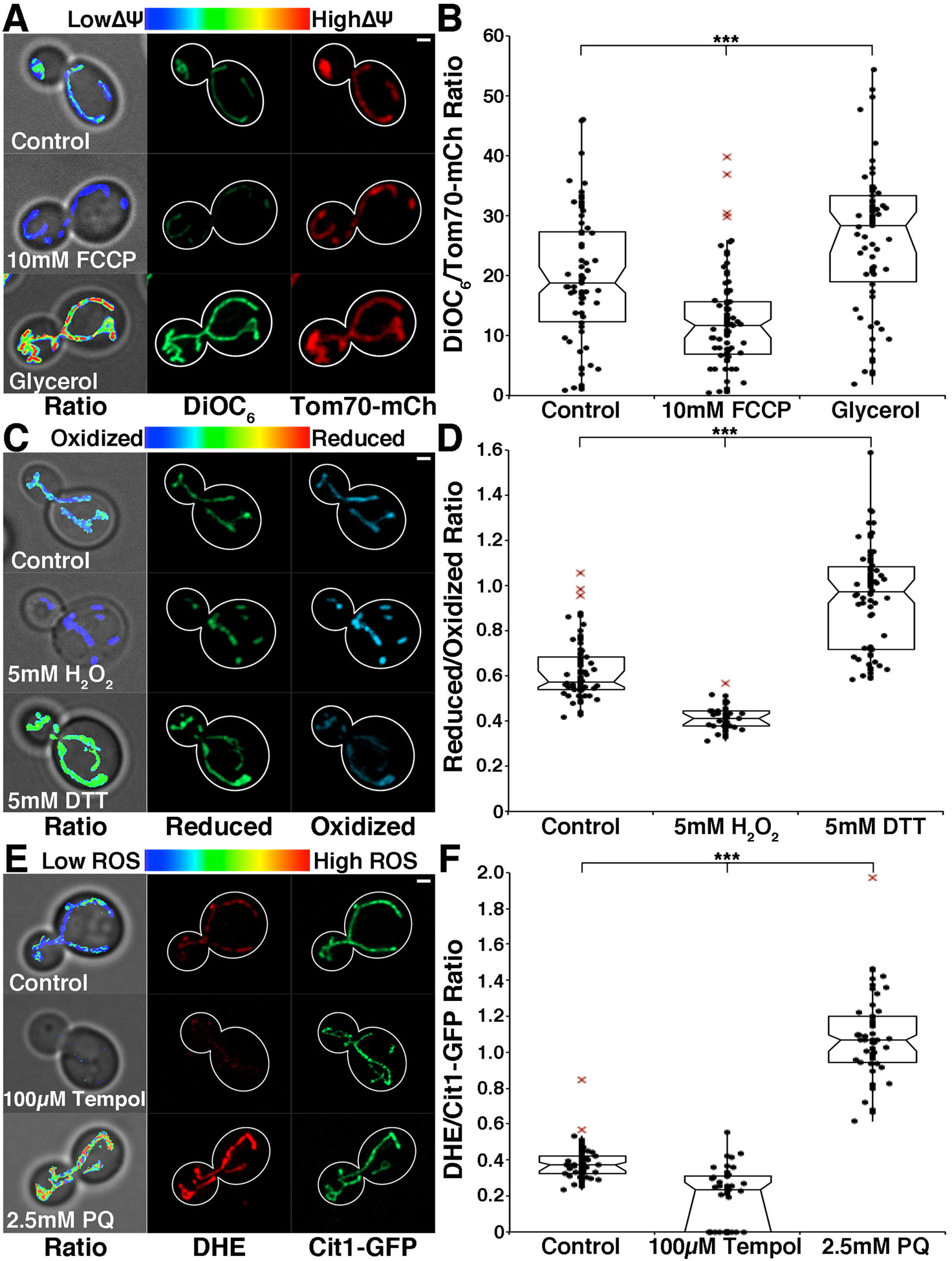
A) DiOC6 was used to visualize mitochondrial membrane potential in wild-type cells. Images were acquired using a 470 nm LED at 100% power, 50 ms exposure time and standard GFP filters for DiOC6 and 200 ms exposure time and metal-halide lamp with standard DsRed filters for Tom70-mCherry. Left panels: DiOC6:Tom70-mCherry ratios overlaid on bright-field images. Color scale indicates ratio values; higher numbers and warmer colors indicate higher membrane potential. Middle panels: DiOC6 images. Right panels: Tom70-mCherry images. Cell outlines were drawn from brightfield images. Scale bar = 1 μM. B) Notched dot box plot of the average DiOC6:Tom70-mCherry ratio in wild-type cells grown in glucose, 10 mM FCCP, and glycerol. Cells were treated with FCCP for 15 minutes prior to addition of DiOC6 or grown in YPG overnight and allowed to recover in SC with glucose for 3 hrs. n = 50 cells for each strain. Data is representative of 3 experiments. *** = p-value < 0.001 using non-parametric Kruskal-Wallis testing with pairwise Bonferroni correction. C) Mito-roGFP was used to visualize the redox environment of mitochondria in wild-type cells. Left panels: reduced:oxidized roGFP ratios overlaid on phase images. Images were acquired using a standard GFP filter with excitation filter removed, and LED excitation at 365 nm (25% power) and 100 ms exposure for the oxidized channel and 470 nm (100% power) and 100 ms exposure for the reduced channel. Color scale indicates ratio values; higher numbers and warmer colors indicate more reducing mitochondria. Middle panels: reduced roGFP. Right panels: oxidized roGFP. Cell outlines were drawn from bright-field images. Scale bar = 1 μM. D) Notched dot box plot of the average reduced:oxidized mito-roGFP1 ratio in wild-type cells untreated and treated with 5 mM H2O2 and 5 mM DTT. Cells were treated with H2O2 or DTT for 30 min prior to imaging. n = 50 cells for each strain. Data is representative of 3 experiments. *** = p-value < 0.001 using non-parametric Kruskal-Wallis testing with pairwise Bonferroni correction. E) DHE was used to visualize mitochondrial ROS levels in wild-type cells. Left panels: DHE:Cit1-GFP ratios overlaid on bright-field images. Cells were stained with DHE at 40 μM for 30 min and imaged using a standard rhodamine filter and 100 ms exposure for DHE and a standard GFP filter and 100 ms exposure for Cit1-GFP. Color scale indicates ratio values; higher numbers and warmer colors indicate lower ROS levels. Middle panels: DHE. Right panels: Cit1-GFP. Cell outlines were drawn from bright-field images. Scale bar = 1 μM. F) Notched dot box plot of the average DHE:Cit1-GFP ratio in wild-type cells untreated and treated with 2.5 mM paraquat (PQ) and 100 μM Tempol. Cells were treated with PQ or Tempol for 30 min prior to addition of DHE. n = 50 cells for each strain. Data is representative of 2 experiments. *** = p-value < 0.001 using non-parametric Kruskal-Wallis testing with pairwise Bonferroni correction.
1.2.2. Measuring mitochondrial redox state
The components of the electron transport chain in mitochondria are key contributors to reactive oxygen species (ROS) production in cells. ROS can be detoxified in by catalases and peroxidases both within the mitochondria and in the cytosol. However, accumulation of ROS and oxidative stress due to both age-dependent and age-independent factors contributes to decreased fitness and quality of the mitochondria overall. ROS and redox-sensing probes allow quantitative monitoring of these important aspects of mitochondrial function.
roGFP, a redox-sensitive GFP variant, reveals the mitochondrial redox state (Vevea et al., 2013). This ratiometric probe is constructed with cysteine molecules exposed on the surface of the GFP molecule. Oxidation of these cysteine residues results in a conformational change, shifting the optimal excitation wavelength to ~400nm. Reduction of these cysteine residues favors excitation at ~480nm. Thus, the ratio of GFP emission upon excitation of roGFP at 480nm and 400nm provides a relative readout of the redox state of the fluorophore’s environment (Fig 3C) (Hanson et al., 2004). roGFP can be targeted to the mitochondrial matrix by fusion to a mitochondrial localization sequence and ultimately used to determine redox fitness of mitochondria. Because oxidizing ROS molecules are a primary source of mitochondrial damage, reducing environments indicate healthier mitochondria and oxidizing environments indicate damaged or dysfunctional mitochondria. Treatment with H2O2 will result in a highly oxidizing environment, while treatment with dithiothreitol (DTT) will result in a highly reducing environment. These agents can be used to determine the dynamic range of roGFP (Fig 3D).
Dihydroethidium (DHE) is used to directly measure levels of mitochondrial superoxide. DHE fluoresces blue; however, when it is oxidized by superoxide anion, it emits red fluorescence. Since mitochondria are the main endogenous source of superoxide, DHE an ideal probe to specifically measure mitochondrial ROS levels. MitoSOX Red (Life Technologies) is a cationic derivative of DHE that has been used to detect mitochondrial ROS. Mitochondrial ROS levels can be quantified with DHE by normalizing to a bulk mitochondrial label such as GFP-tagged Cit1p (Fig. 3E) (McFaline-Figueroa et al., 2011). Paraquat, a drug that specifically increases mitochondrial superoxide formation, and TEMPOL, an agent that promotes elimination of superoxide, can be used to determine the dynamic range of DHE as an indicator for ROS (Fig. 3F). DHE is best used for short-term measurements of acute ROS, because its oxidized form can intercalate with DNA, causing it to accumulate in the nucleus after ~15 min.
1.3. Visualization of the actin cytoskeleton with fluorescent proteins
Actin patches and actin cables are two F-actin-containing structures that persist throughout the cell cycle in budding yeast. Actin patches are endosomes that are coated with F-actin filaments and localize to the bud and the mother-bud neck in polarized cells. Actin cables are dynamic bundles of F-actin that align along the mother-bud axis and continuously flow in a bud-to-mother direction via retrograde actin cable flow (RACF) (Yang & Pon, 2002). These cables serve as tracks for the movement of cellular cargo including secretory vesicles, mRNA, spindle alignment elements, mitochondria, Golgi and vacuoles. In addition, the actin cytoskeleton plays a major role in regulation of quality control of mitochondria (Higuchi et al., 2013).
Early studies revealed that tagging the actin-encoding ACT1 gene of yeast with GFP compromised function of the protein. Specifically, ACT1-GFP expressed on a plasmid did not rescue loss of the endogenous ACT1 gene (Doyle & Botstein, 1996). Presumably, GFP tagging results in loss of function because every surface of the actin protein is involved in protein-protein interactions in microfilament nucleation and polymerization, interaction with motor proteins and other force generators, microfilament assembly, capping, cross-linking, and severing. As an alternative, actin-binding proteins are often more tolerant of tagging with FPs. GFP tagging of Abp140p and Abp1p produces a fluorescent signal that localizes to actin cables and actin patches, respectively (Fig. 4A) (Yang & Pon, 2002, Huckaba et al., 2006). Live-cell imaging of the actin cytoskeleton allows quantification of velocity of RACF and the visualization of cargo trafficking, such as the movement of mitochondria along actin cables (Fig. 4B) (Fehrenbacher et al., 2004). A drawback of Abp140-GFP is low signal, which can be boosted by culturing cells in non-fermentable carbon sources as actin cables are thicker in these culture conditions (see Note 1). As an alternative to tagging the full-length protein, the first 17 amino acids of Abp140p are sufficient to mediate actin localization of fluorescent proteins without altering actin cable dynamics. This protein, termed LifeAct, provides reliable and robust staining of the actin cytoskeleton (Riedl et al., 2008). Table 4 lists genes that have been tagged with FPs to serve as probes for actin patches and actin cables in budding yeast.
Fig. 4. Colocalization of the actin cytoskeleton with cargo.
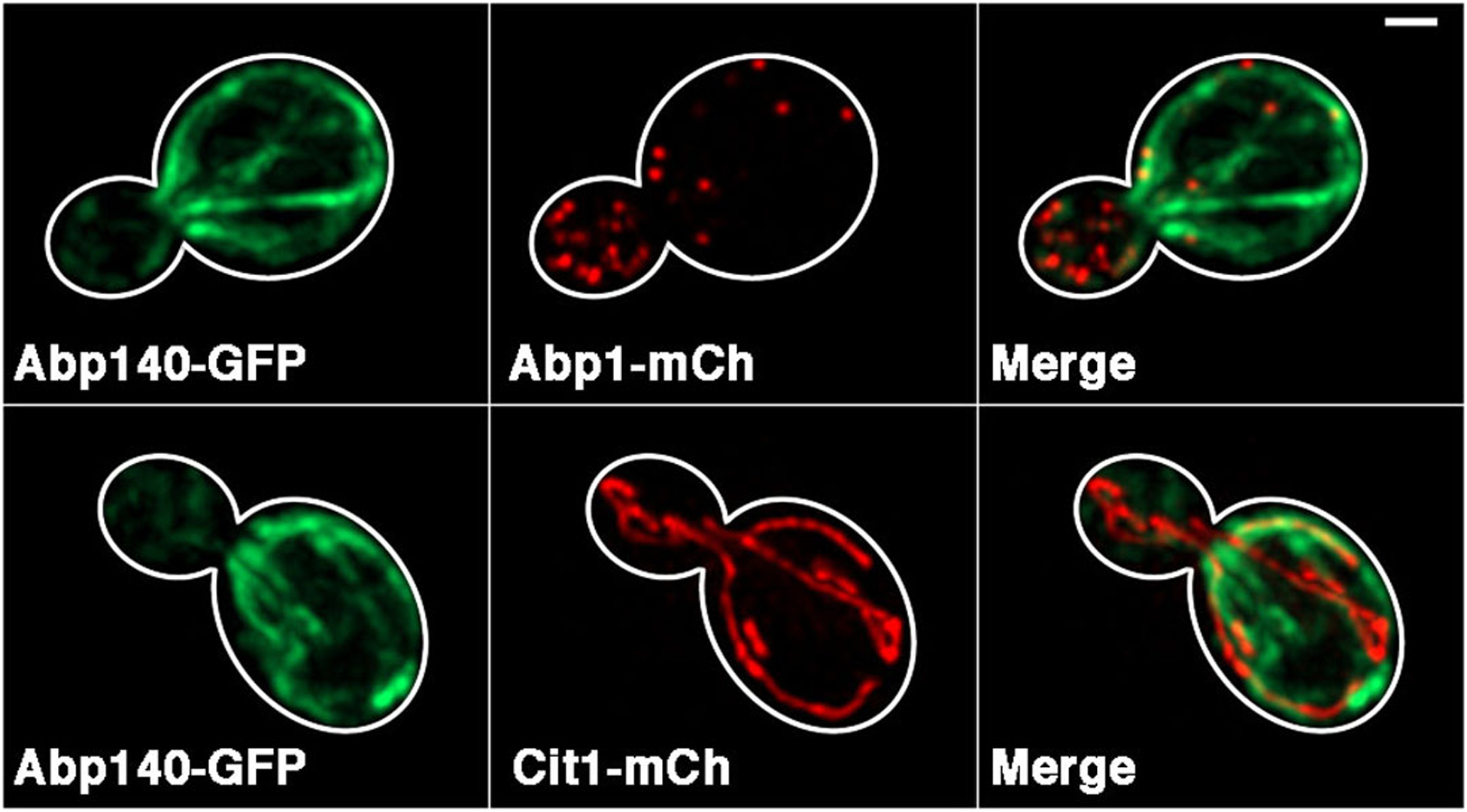
Yeast cells were grown in YPG and washed in SC medium prior to imaging. Actin cables were visualized with Abp140-GFP (left panels). Two cellular cargo structures, actin patches (top) and mitochondria (bottom), were visualized by tagging Abp1p and Cit1p with mCherry, respectively. Images were captured using standard GFP filters and 300 ms exposure time for GFP and standard DsRed filters and 200 ms exposure time for mCherry. Right panels: merged images indicating colocalization of actin patches and mitochondria along the actin cytoskeleton. Cell outlines were drawn from bright-field images. Scale bar = 1 μM.
Table 4:
GFP-tagged actin proteins in budding yeast
2. Materials
2.1. Yeast growth media
Amino acid supplements: For synthetic complete medium, supplement with all of the following. For dropout medium, omit one or more components to select for the desired strains: 10 ml adenine (2 mg/ml stock in 0.05 M HCl), 10 ml uracil (2 mg/ml stock in 0.5% NaHCO3), 10 ml arginine 10 ml (1 mg/ml stock in H2O), 10 ml histidine (1 mg/ml stock in H2O), 10 ml leucine (10 mg/ml stock in H2O), 10 ml lysine (3 mg/ml stock in H2O), 10 ml methionine (2 mg/ml stock in H2O), 10 ml phenylalanine (5 mg/ml stock in H2O), 10 ml tryptophan (2 mg/ml stock in H2O), 10 ml tyrosine (3 mg/ml stock in 0.05 M HCl).
Synthetic complete medium (SC): Dissolve 6.7 g yeast nitrogen base without amino acids, 20 g glucose, and amino acid supplements as needed in 800 mL distilled H2O. Adjust pH to 5.5 with NaHCO3 and bring volume to 1 L with distilled H2O. Sterilize by autoclaving.
Yeast peptone-dextrose medium (YPD): For 1 liter, dissolve 10 g yeast extract, 20 g bacto-peptone, and 20 g glucose in distilled H2O and bring volume to 1 L with distilled H2O. Sterilize by autoclaving.
Lactate medium: For 1 liter, combine 3.0 g yeast extract, 0.5 g glucose, 0.5 g CaCl2, 0.5 g NaCl, 0.5 g MgCl2, 1 g KH2PO4, 22 mL 90% lactic acid, and 7.5 g NaOH pellets. Dissolve ingredients in 800mL distilled H2O. Adjust pH to 5.5 with NaOH. Bring volume to 1 L with distilled H2O.
Synthetic raffinose medium: For 1 liter, combine 6.7 g of yeast nitrogen base without amino acids and amino acid supplements as needed. Dissolve ingredients in 600 mL distilled H2O. Adjust pH to 5.5 with NaHCO3. Bring volume to 900 mL with distilled H2O. Supplement after autoclaving with 100 mL filter-sterilized 20% raffinose.
Synthetic glycerol medium (SG): For 1 liter, combine 6.7 g of yeast nitrogen base without amino acids, 30 mL of glycerol, 0.5 g glucose, and amino acid supplements as needed. Dissolve ingredients in 800 mL distilled H2O. Adjust pH to 5.5 with NaHCO3.
Synthetic galactose medium: For 1 liter, combine 6.7 g of yeast nitrogen base without amino acids and amino acid supplements as needed. Dissolve ingredients in 600 mL distilled H2O. Adjust pH to 5.5 with NaHCO3. Bring volume to 950 mL with distilled H2O. Supplement after autoclaving with 50 mL filter-sterilized 40% galactose.
2.2. Reagents for PCR amplification
PCR buffer:100 mM Tris-HCl, pH 8.3 at 25°C; 500 mM KCl; 15 mM MgCl2; 0.01% gelatin (Sigma, P2912)
MgCl2 stock solution (25 mM)
dNTP stock solutions (10 mM each)
Forward and reverse primers (10 μM each)
Distilled H2O
2.3. Reagents for yeast transformation and marker excision
50% w/v Polyethylene glycol 3350 in H2O
1 M lithium acetate in H2O
0.1M lithium acetate in H2O
Carrier DNA: 2 mg/ml single-stranded calf thymus DNA in H2O. Store at −20°C. Boil for 5 min immediately before use. Do not subject to more than 2 freeze-thaw cycles.
DNA for transformation (1 PCR reaction as described in protocol, or 1 μg)
Sterile distilled H2O for resuspending cells before plating
Selective plates for growth of cells containing plasmid or integrated DNA
2.4. Materials for imaging living cells
Agarose pad: Add 0.1 g low-melting agarose to a 50mL conical-bottom tube and bring to a total volume of 5 mL with desired medium (final concentration will be 2% agarose). Dissolve agarose in medium by boiling the tube in a water bath. Dispense into 200-μl or smaller aliquots in microfuge tubes and store at room temperature in the dark. Avoid repeated boiling.
VALAP: combine equal weights of petrolatum (Vaseline), lanolin and paraffin (hard). Submerge mixture in a 70°C water bath to melt and aliquot into 60 × 15 mm glass Petri dishes. Store at room temperature.
2.5. Materials for measuring mitochondrial membrane potential using DiOC6
HEPES: For 1 liter, combine 2.5 g HEPES, 50 g glucose. Dissolve ingredients in 800 mL distilled H2O. Adjust pH to 7.6 using 10 N NaOH. Bring volume to 1 L with distilled H2O. Filter-sterilize; do not autoclave.
DiOC6: Dissolve DiOC6 in 100% ethanol to a stock concentration of 17.5 mM. Store at −20°C for up to two years. Dilute this stock to 17.5 μM in 100% ethanol as a working solution (Note 2). Store working solution at −20°C for up to two weeks.
FCCP: Dissolve FCCP in 100% DMSO to a stock concentration of 2 M. Store at −20°C.
2.6. Materials for measuring redox environment/ROS levels
Tempol: Dissolve Tempol in distilled H2O to a stock concentration of 100 mM. Store at −80°C.
Paraquat: Dissolve Paraquat in distilled H2O to a stock concentration of 100 mM. Store at −80°C.
H2O2: Dilute H2O2 to a working concentration of 1 M in distilled H2O2 prior to use (Note 3).
3. Methods
3.1. Modification of yeast genes at their chromosomal locus
3.1.1. Primer design
Primers will generally be 60 or more bases long. The 5’ end of each primer should contain at least 40 bases of perfect homology to the target site. For example, for C-terminal tagging with a FP, the 5’ end of the forward primer should contain the 40–45 bases directly upstream of the stop codon, while the 5’ end of the reverse primer should contain the reverse complement of the 40–45 bases directly downstream of the stop codon. The 3’ ends of the primers should contain 18–25 bases complementary to the sequences that will be inserted, which include the open reading frame of the FP, the transcription termination site, and the selectable marker (see Note 4).
The target gene and the FP tag are typically separated by a short linker, which is designed to allow proper folding and function of both the fluorescent tag and the protein of interest. For example, GFP fluorescence can often be optimized by increasing the length of the linker between the target gene and GFP molecule (we suggest starting with five alanines). The length and amino acid composition of this linker may need to be adjusted for different target proteins. Proteins with processive enzymatic functions, such as polymerases and telomerases, will benefit from poly-glycine linkers (Sabourin et al., 2007). In C-terminal tagging, the linker is encoded in the in the forward primer by including DNA encoding the appropriate amino acids between the 40 bases of DNA homologous to the target gene and the 20 bases corresponding to the template. For N-terminal tagging, a similar strategy can be used in the reverse primer.
3.1.2. Amplification of insertion cassette from tagging vector
Prepare the PCR reaction: PCR buffer, 1.5–5 mM MgCl2, dNTP mix (2.5 mM each dNTP), 0.1–1.0 μM forward and reverse primers, template DNA (see Note 5), 1.0–5.0 units of polymerase (see Note 6), distilled H2O up to 50 μL. A single 50 μL PCR reaction will provide sufficient DNA for a single yeast transformation (minimally 1 μg of DNA) if a high-fidelity, high-yield polymerase is used. For lower-yield polymerases, multiple PCR reactions can be pooled.
Amplify the cassette using the following thermocycler conditions: Initial denaturation cycle at 95°C for 2–5 minutes; 35 cycles of: 98°C for 20 s, 60–75°C (See Note 7) for 15 s, and 72°C for 60 s/kb; final extension cycle at 72°C for 1–5 minutes.
Use the PCR reaction directly in the lithium acetate transformation protocol described below. Alternatively, amplified DNA can be purified using any commercially available PCR purification kit (but see Note 8).
3.2. Lithium acetate transformation of yeast
The lithium acetate transformation protocol is the most commonly used method for yeast transformation (Gietz et al., 1995). The following protocol is for one transformation reaction. A negative control containing no DNA should always be carried out in parallel.
Grow yeast to mid-log phase.
Transfer 107 cells from the culture to a 1.5 mL microfuge tube.
Pellet cells (30 s, 7000 × g) and wash with 500 μL 0.1 M lithium acetate. Resuspend in 240 μL of 50% w/v polyethylene glycol 3350.
Add 36 μL 1.0 M lithium acetate, 25 μL of carrier DNA, and 1 μg of DNA to be transformed (PCR-amplified insertion cassette or plasmid DNA) or 50 μL of H2O for negative control.
Vortex vigorously and incubate in a water bath at 30°C for 30 min.
Heat shock in a water bath at 42°C for a minimum of 20 min and a maximum of 3 hrs.
Concentrate cells by mild centrifugation (30 s, 7000 × g).
For transformations using auxotrophic markers, resuspend cell pellet gently in 100 μL of sterile distilled H2O and plate on appropriate selective media.
For transformations using drug resistance markers (e.g. KanMX6), resuspend cell pellet gently in 500 μL of YPD or synthetic complete medium with dropouts as needed to maintain plasmids. Allow cells to recover at 30°C for 2–4 hrs. Concentrate and plate cells as described in steps 7–8 on appropriate selective medium containing drug.
3.3. Marker excision by Cre recombinase
If a loxP-containing vector such as the pOM family has been used to tag the gene of interest, the selectable marker can be removed by the following method.
Verify tagging by PCR and sequencing, as described below.
Using methods described above, transform the tagged strain with a plasmid that encodes Cre recombinase under control of a galactose-inducible promoter (Cheng et al., 2000). Select transformants on appropriate medium selecting for the Cre plasmid.
Grow cells to mid-log phase in 5 mL liquid medium in a 50-mL conical-bottom tube. Use medium that selects for the Cre plasmid.
Pellet cells (5 min, 4000 × g) and resuspend in 5 mL galactose medium.
Incubate cells in galactose medium in a shaking incubator at 30°C for 12–16 hrs.
Plate an aliquot of cells on non-selective (YPD or SC) medium.
Screen colonies for loss of the tagging cassette marker by replica plating on selective plates.
To induce dropout of the Cre plasmid, grow cells in non-selective liquid medium for 24–48 hrs, plate on non-selective medium, and screen for loss of the plasmid by replica plating on selective plates (see Note 9).
3.4. Validating and characterizing FP-tagged cytoskeletal proteins
3.4.1. PCR screening and sequencing
PCR screening should be used to validate the insertion of the tag into the target locus. This can be accomplished by isolating genomic DNA from transformants and using that as a template for a PCR reaction using a pair of primers that hybridize upstream and downstream of the tagging locus. Strains confirmed to have the correct tag at the target location should be validated by sequencing to confirm that the tagged genes do not carry any mutations.
3.4.2. Verification of protein expression and function
The first step to verify successful expression of tagged proteins is visual inspection of transformed cells. Cells should be prepared for short-term imaging as described below. Screen several colonies to find bright, consistent FP expression.
Western blotting can also be used to monitor protein expression. This can reveal 1) expression below the threshold of detection of microscopy; 2) the presence of degradation products that may not localize or function properly; or 3) differences in expression level relative to the native protein.
Tagged constructs should also be evaluated for correct localization and function if knowledge of the gene of interest is available. FP tags can change the function and/or localization of proteins, and some are prone to oligomerization (which can produce artifacts due to protein-protein interactions).
If the localization of the target protein is known, correct localization of the FP tagged protein suggests that this aspect of its function is preserved. For example, Cit1p is a mitochondrial matrix protein, and Cit1-GFP localization to the mitochondria will suggest that the tag does not perturb localization. Mitochondria can be counterstained by vital dyes or visualized using another tagged mitochondrial protein, such as Tom70p. If there is any question about the correct localization of the FP-tagged protein, it may be necessary to localize the target protein with a smaller tag such as myc or HA followed by fixation and immunostaining. After the correct localization is verified, the FP-tagged version can be used for live-cell analysis.
To check for proper function of a tagged protein, evaluate cell growth rate and any phenotypes characteristic of loss of function of the gene of interest. For example, if tagging of a non-essential gene compromises function of the protein, then the tagged strain may show a phenotype resembling that of cells mutated in that gene. Cell growth rate should be comparable to that of untagged strains. To demonstrate full wild-type function, a plasmid-borne version of the FP-tagged protein should rescue the wild-type phenotype in yeast cells with a deletion of the genomic copy of the gene of interest (see Note 10).
3.4.3. Characterization of mitochondria-targeted FPs
Mitochondrial targeting depends on signal sequences, which may be at the N terminus or internal to the protein, and the protein including the tag may need to traverse one or more membranes to be properly localized. FP-tagged proteins may mislocalize if targeting information is masked or if insertion of the fusion protein into the membrane is inhibited. For example, N-terminal tagging of Mdm33p inhibits mitochondrial localization, as the mitochondrial signal sequence of Mdm33p is at the N terminus. In contrast, GFP fusion to the C terminus of Mdm33p impairs the function of the protein, but does not alter mitochondrial localization. This problem is circumvented by internal tagging of Mdm33p between the signal sequence and the mature protein product (Messerschmitt et al., 2003), yielding a protein that is fully functional and correctly localized. Proper localization of mitochondria-targeted FPs can be assessed by visual inspection by counterstaining mitochondria with vital dyes or by biochemical methods for mitochondrial fractionation.
Adverse effects of fusion proteins on mitochondrial morphology or respiratory function can be assessed by visual inspection of mitochondria and by analysis of growth rates on non-fermentable carbon sources, such as glycerol.
3.5. Staining yeast mitochondria with vital dyes
Vital dyes that are used to detect mitochondria in living cells, and conditions used for staining are listed in Table 5.
Table 5:
Conditions for staining yeast mitochondria with vital dyes
| Dye | [Stock]1 | Staining conditions | |
|---|---|---|---|
| [Dye] | Incubation | ||
| DiOC6(Riezman et al., 1983)2 | 17.5 mM | 17.5 nM | 15 min RT |
| DASPMI3 | 1 mg/ml in ethanol | 10–100 μg/ml | 30 min RT |
| Rhodamine 1234 | 10 mg/ml in DMSO5 | 5–10 μg/ml | 15–30 min RT |
| MitoTracker6 | 1 mM in DMSO5 | 100 nM | 30 min RT |
| DAPI | 1 mg/ml in H2O | 0.1 μg/ml | 15 min RT |
All stock solutions should be stored in the dark.
(Skowronek et al., 1990); Broad emission range; not recommended for green/red double-label studies.
DMSO can be toxic and inhibits partitioning of the dye into the aqueous environment of the cells. If DMSO is the solvent, use a stock concentration that is at least 100X.
Grow yeast cells to mid-log phase in 5 mL liquid medium in a 50 mL conical tube (Note 11).
Concentrate 107 cells by gentle centrifugation at ~8,000 × g for 30 s if necessary (see Note 12).
Add appropriate volume of dye directly to cell culture medium and mix thoroughly (Note 13).
Incubate in the dark, shaking, at 30°C for the desired amount of time.
Remove excess dye by 3 washes with growth medium.
Concentrate cells by gentle centrifugation at ~8,000 × g for 30s.
Mount cells for short-term or long-term observation, as described below.
3.6. Wide-field imaging of living cells
3.6.1. Equipment for wide-field imaging
Budding yeast are among the smallest eukaryotic cells: buds are typically 2–3 μm and mother cells 4–6 μm in diameter. Resolving intracellular structures in such small cells requires a microscope equipped with a high-magnification objective lens for good spatial resolution, and a sensitive camera to capture the low signal levels emitted by small structures.
When imaging living cells, overall light throughput and sensitivity of the imaging system are especially important. High sensitivity supports lower-intensity illumination and shorter exposure times, which greatly reduce phototoxicity and photobleaching. In addition, shorter exposure time allows better time resolution when following rapid dynamics. Further improvements in time resolution are possible with a trigger connection between the computer and hardware components such as the camera and the focus drive, and by maximizing the speed and random access memory (RAM) of the computer.
An imaging system that works well for imaging yeast is based on a motorized inverted epifluorescence microscope (Zeiss AxioObserver.Z1) equipped with a metal halide lamp and Colibri LEDs for excitation; an EC Plan-NeoFluar 100x/1.3 NA or Plan-Apochromat 100x/1.4 NA objective lens; and an Orca ER cooled CCD camera with 1280 × 1024 pixel resolution. A software package such as ZEN (Zeiss) is used to control the camera and associated hardware, capture images at defined focus and time intervals, and export them for further analysis.
3.6.2. Equipment for 2-color imaging
Historically, multicolor imaging was first performed with dual-emission filters and color cameras. However, monochrome cameras offer superior dynamic range, resolution, and speed of detection. Therefore, for optimal multi-color imaging, each fluorophore should be imaged separately, and the images pseudocolored (e.g. green for EGFP and red for mCherry) and merged. Sequential imaging of multiple fluorophores is achieved by rapidly switching filters or light sources. Alternatively, a splitting device such as the Dual-View (Photometrics) can be used to image multiple colors simultaneously on subregions of the camera chip.
In our preferred imaging system there are two possible strategies for sequential imaging: 1) changing excitation and emission filters and 2) LED excitation switching. For example, for imaging of actin cables using Abp140-GFP and mitochondria using Cit1-mCherry, the sample is illuminated using a broad-spectrum light source (e.g. a metal halide lamp) and an image is taken first with the green excitation and emission filter sets, then with the red. In a motorized microscope stand, this is typically accomplished by rotating the filter turret. In a non-motorized stand, or for somewhat faster performance, external motorized filter wheels can be inserted in the excitation and/or emission path while a multi-line dichroic beamsplitter is placed in the filter turret. Changing the entire filter cube provides the best signal, because the single-band filters have high throughput and can be tailored precisely to the fluorophore of interest. However, because filter switching requires large mechanical movements, the time interval between acquisition of channels, at least 35 ms, may be too long for highly dynamic events. In addition, the rotation of the filter wheel may generate vibrations, necessitating a pause in the imaging that slows the actual time interval between channels to ~200 ms.
A faster, more stable approach is to switch the excitation light through the light source. This can be accomplished with lasers or narrow-spectrum, high intensity light-emitting diodes (LEDs), such as those in the Colibri 2 system (Zeiss). This illuminator holds up to 4 interchangeable LEDs that match the spectrum of many commonly used FPs, and are controlled individually by electrical signals with virtually no time lag. This system used in conjunction with a multi-line dichroic/emission filter cube allows rapid changing of the excitation light without any mechanical motion. This method is preferred for dual-color imaging of dynamic structures and ratio imaging.
Table 6 provides suggested imaging conditions for methods described in this chapter.
Table 6:
Suggested imaging conditions for methods described in this chapter
| FP/Dye | Excitation/Emission | Light source | Exposure time2 |
|---|---|---|---|
| Cit1-GFP | 488/507 | LED 470 nm (100%) or Metal-Halide Lamp + standard GFP Filter | 100 ms |
| Tom70-mCherry | 587/610 | Metal-halide lamp + standard DsRed filter | 200 ms |
| MitoTracker Red (100 nM for 30 min) | 581/644 | Metal-halide lamp + standard DsRed filter | 200 ms |
| DiOC6 | 482/504 | LED 470 nm (100%) | 50 ms |
| roGFP (Oxidized) | 365/507 | LED 365 nm (25%) | 100 ms |
| roGFP (Reduced) | 470/507 | LED 470 nm (100%) | 100 ms |
| DHE (40 μM for 30 min) | 485/530 | Metal-halide lamp + standard DsRed filter | 100 ms |
| Abp140-GFP | 488/507 | LED 470 nm (100%) or Metal-Halide Lamp + standard GFP Filter | 300 ms |
| Abp1-mCherry | 587/610 | Metal-halide lamp + standard DsRed filter | 200 ms |
| Cit1-mCherry | 587/610 | Metal-halide lamp + standard DsRed filter | 200 ms |
These are recommended concentrations, and can be adjusted as needed
These are recommended exposure times and can be adjusted as needed
3.6.3. Preparing cells for short-term imaging
For short-term visualization, concentrated cells in culture medium or staining buffers are added directly to a glass microscope slide and imaged immediately for maximally 10 min (see Note 14). After 10 min, cells will start to experience significant decrease in viability and may begin to exhibit signs of stress. The following protocol can be used for imaging actin cables labeled with Abp140-GFP together with mitochondria-targeted mCherry, as well as other cells expressing FPs and/or labeled with vital dyes as described above.
Inoculate cells from a colony into 5 mL of appropriate medium in a 50-mL conical-bottom tube and incubate overnight at 30° in a shaking incubator.
In the morning, dilute cultures to early log phase (OD600 = 0.01–0.1) and incubate under growth conditions at least 2 hrs, until cells are mid-log phase (OD600 = 0.2–0.6)
Transfer 1 mL of culture to a 1.5-mL microfuge tube and concentrate cells by centrifugation at ~8,000 × g for 30 s (see Note 15).
Without disturbing the pellet, remove almost all of the supernatant, leaving a volume of supernatant in the tube approximately twice the volume of the pellet.
Resuspend pellet in residual medium and transfer ~1.5 μL of the cell suspension to a microscope slide that has been cleaned with 70% ethanol and wiped dry with a Kimwipe.
Apply a #1.5 (170 μm thick) coverslip, taking care to avoid creating bubbles between slide and coverslip (see Note 16).
Acquire images. Prepare a fresh slide from the concentrated cell suspension after 10 min.
3.6.4. Preparing cells for long-term imaging
Cells can be immobilized on medium containing low-melting-point agarose, which will support cell growth at wild-type levels for up to 5 hrs. These agarose pads have minimal autofluorescence and remain transparent and thin enough for observation with oil-immersion lenses. Note that agarose pads take roughly 5 min to prepare and should not be left unused for extended periods of time as they may dry out. For time-sensitive studies, take care to prepare pads about 5 min before imaging.
Grow yeast cells to mid-log phase and concentrate in liquid medium or staining buffer as described above for short-term imaging (3.6.3 steps 1–3).
Melt an aliquot of agarose bed material in a boiling water bath, approximately 2 min for a 200-μL aliquot.
Pipet 35 μL of agarose bed material onto a glass microscope slide that has been cleaned with 70% ethanol and wiped dry with a Kimwipe (see Note 17), and cool for ~5 s (see Note 18).
Place a second cleaned microscope slide on top of the agarose and apply light pressure to spread the agarose bed to the diameter of a standard cover slip. Note that the agarose pad may adhere to either the top or bottom slide, so if using slides with charged surfaces, ensure that both slides have the charged side facing the agarose pad.
Let pad harden between microscope slides for ~2 min.
Gently remove top slide by rotating and sliding it past the bottom slide. If the pad is wrinkled or torn at this step, discard it and make a new one.
Pipet 1.5 μL of concentrated cells onto the center of the agarose pad and cover with a 22 × 22 mm coverslip.
Seal coverslip edges with VALAP: Using a metal spatula, pick up a chunk of solid VALAP about the size of a grain of rice. Melt the VALAP on the spatula in a Bunsen burner flame and drag the spatula along one edge of the coverslip to dispense a thin line of VALAP at the interface with the slide. Repeat on all edges of the coverslip. For imaging longer than 2 hr or at temperatures > 25°C, add a layer of nail polish over the VALAP for proper sealing.
3.7. Ratio imaging methods
3.7.1. Ratio imaging of Δψ with DiOC6
Grow yeast cells containing a red mitochondrial marker (we recommend Tom70p-mCherry) to mid-log phase in 5 mL of liquid medium in a 50-mL conical-bottom tube.
Transfer 3 × 107 cells to a 1.5 mL microfuge tube and concentrate by centrifugation at ~8,000 × g for 30s.
Resuspend cells in 1 mL HEPES and add DiOC6 to a final concentration of 17.5 nM. For drug treatment (e.g. FCCP), add drug to cells in HEPES prior to addition of DiOC6 (see Note 19).
Incubate cells in the dark at room temperature for 15 min.
Wash three times with HEPES.
Concentrate cells by centrifugation at ~8,000 × g for 30s.
Mount 1.5 μL cells for short-term imaging (Note 20).
Acquire images. Refer to Table 6 for suggested imaging conditions.
3.7.2. Ratio imaging of redox state using roGFP
Grow yeast cells with mito-roGFP plasmid to mid-log phase in 5 mL of liquid medium in a 50-mL conical-bottom tube.
Transfer 3 × 107 cells to a 1.5 mL microfuge tube and concentrate by gentle centrifugation at ~8,000 × g for 30s.
Mount 1.5 μL cells for short-term imaging.
Acquire images. Refer to Table 6 for suggested imaging conditions.
3.7.3. Ratio imaging of ROS using DHE
Grow yeast cells containing a green mitochondrial marker (we recommend Cit1-GFP) to mid-log phase in 5 mL of liquid medium in a 50-mL conical-bottom tube.
Transfer 3 × 107 cells to a 1.5 mL microfuge tube and concentrate by centrifugation at ~8,000 × g for 30s.
Resuspend cells in 1 mL SC medium and add DHE to a final concentration of 40 μM. For drug treatment, add drug to cells in SC prior to addition of DHE.
Incubate cells in the dark at room temperature for 30 min.
Wash three times with SC.
Concentrate cells by centrifugation at ~8,000 × g for 30s.
Mount 1.5 μL cells for short-term imaging.
Acquire images. Refer to Table 6 for suggested imaging conditions.
3.7.4. Optimizing imaging conditions and preventing toxicity
During live-cell imaging, illumination of cells for extended periods of time can lead to phototoxicity by two primary mechanisms: 1) photons at wavelengths close to the UV can cause direct cell damage; and 2) photons can react with cellular molecules to produce free radicals and reactive oxygen species. The following criteria can be used to check for photodamage to organelle and cytoskeletal function.
Cytoskeletal and mitochondrial structures: intense or long excitation can cause actin cable depolymerization and mitochondrial fragmentation.
Dynamics: While behavior of photodamaged mitochondria and cytoskeletal components has not been characterized, photodamage can be suspected if mitochondria or cytoskeletal elements change their velocity or other motility parameters in wild-type cells.
Staining with potential-sensitive dyes, such as DiOC6 throughout the mitochondria in wild-type cells. Loss of membrane potential indicates mitochondria are not functioning properly.
Even if cells remain healthy throughout imaging, excessive light exposure can cause photobleaching of fluorophores. To reduce phototoxicity and photobleaching, any or all of the following strategies can be used.
Reduce excitation intensity. With LEDs and many lasers, the intensity can be varied through the control software. With conventional light sources, neutral density filters can be used.
Reduce exposure time. In general, longer exposure time and lower intensity gives a comparable quality image with less photodamage.
Reduce spatial or time resolution. For example, increase the time interval and/or the z-section interval.
To increase signal-to-noise ratio without increasing light exposure:
Apply binning in the camera. This will result in decreased spatial resolution, but this can be alleviated by adding a projection tube before the camera.
Increase camera gain.
Reduce camera readout speed, if possible.
Use a filter set with a broader spectral window or more efficient coatings to increase throughput.
3.7.5. Special considerations for quantitative analysis
For quantitative fluorescence imaging, two criteria must be met: 1) emitted fluorescence must be proportional to the number of fluorescent molecules present; and 2) recorded pixel intensities must be proportional to the amount of light emitted by the sample. Under well-controlled conditions, these criteria are met at sufficient levels to measure many biological factors.
Absolute quantification of fluorescent molecules by imaging is a serious challenge due to many variables that complicate the linearity of fluorescence emission and pixel intensities (Pawley, 2000). However, proper controls and normalization can allow reliable measurements of relative changes in the volume and intensity of fluorescent structures, providing a powerful tool for hypothesis testing. At minimum, the following variables must be controlled in any wide-field fluorescence imaging experiment where intensities are compared:
fluorescent probe concentration and age, if an exogenous probe, such a dye, is used
sample age, unless photobleaching is known to be negligible
objective lens
illumination spectrum and intensity, including filters, field aperture setting, and light source alignment
camera model, binning, exposure time, gain, offset, readout speed, bit depth
Imaging parameters must be adjusted to give pixel values significantly above the background level of the detector and sample, and below the saturation level of the detector. Enough cells should be imaged to ascertain the variability within the population. Whenever possible, experimental design should be validated by positive and negative controls. For dyes, specifically those that are quantitative, such as DiOC6 for membrane potential or DHE for ROS, it is particularly useful to perform controls to determine the dynamic range of the dye (see Fig. 3). We also recommend normalizing to counterstains and producing ratio channels to compensate for local differences in mitochondrial mass or variability in thresholding (see Note 21).
3.8. Deconvolution of wide-field fluorescence images
Deconvolution is a computational method for removing out-of-focus light from wide-field fluorescence images. The result is a sharper image with a higher signal:noise ratio and enhanced three-dimensional information. Various algorithms are available, only some of which allow quantitation after processing (Wallace et al., 2001). The constrained iterative restoration algorithm (Volocity, Perkin-Elmer) preserves quantitative information.
For best results with deconvolution, z-series of images should be acquired at focus intervals of 0.2 μm. When studying fast dynamic processes such as mitochondrial motility, the z interval can be increased to 0.5 μm, resulting in some loss of spatial resolution but a doubled time resolution.
Acquire z-series images using short-term or long-term imaging conditions.
Load datasets in Volocity and verify that the x, y, and z scale are correctly set.
For each channel, generate a new calculated psf. When prompted, enter the numerical aperture of the objective lens and the maximum wavelength of fluorescence emission (e.g. 507 nm for GFP and 620 nm emission wavelength for mCherry and MitoTracker Red).
Using the calculated PSF for each channel, perform iterative deconvolution with 60 iterations, 100% confidence limit (see Note 22).
3.9. Quantification of fluorescent signals
To understand the mechanisms and consequences of actin-mediated mitochondrial trafficking and subsequent anchorage, it is helpful to quantify the amount of actin or mitochondria within a cell or subregions of a cell. There are two common measurements of fluorescence intensity: mean and integrated intensity. Mean fluorescence intensity is the average pixel value. It approximates the concentration of fluorescent probes in a given area. It is also proportional to the total number of fluorescent molecules present when measuring areas of similar sizes. However, the mean is not appropriate for measurements of areas of different sizes, such as comparing the amount of mitochondria or F-actin in yeast buds of different sizes. A small bud and a large bud may have the same number of labeled actin structures, but the higher density of fluorescence in the small bud would produce a higher mean intensity than in the large bud where the molecules are more spread out. For scenarios where ROI sizes vary greatly, it is best to indicate the total amount of fluorescent probes via measurements of integrated density or integrated intensity. This is the sum of all pixel intensities in the area. For scenarios where structures may be superimposed and thus cannot be resolved by fluorescence imaging, as in the cases of actin cables or highly aggregated structures, the integrated intensity is also a more accurate assessment than area in indicating the total amount of a labeled structure. Here, we show how to obtain integrated intensity of mitochondria using Volocity and ImageJ.
To make measurements using Volocity:
From the “Measurements” tab, create a protocol by selecting the following steps in order: Find Objects Using Intensity, Exclude Objects by Size, Clip Objects to ROIs, Measure Objects.
For images with multiple channels, make sure that the correct channel is selected under the “Find Objects Using Intensity” tab.
Under “Exclude Objects by Size,” select a size criterion that will exclude background pixels or other artifacts from image capture or deconvolution. This value will depend on imaging conditions and must be determined empirically.
Under the “Measure Objects” tab, click the gear icon to select which measurements you wish to use. Check “Intensity and Volume Measurements.”
Define the ROI you wish to measure. Volocity allows rectangular, circular, and freehand ROIs. Use the Zoom function to make it easier to outline the desired area.
Adjust the threshold under the “Find Objects Using Intensity” tab by manipulating the lower and upper limits. We suggest maximizing the upper limit to include all pixels that have high intensity values. The lower limit should be adjusted to remove enough background to only include signal that is found within the cellular structure of interest.
All measurements will appear in a tab below the image. Individual objects will be separated (for example, if mitochondrial fluorescence intensity is being measured and there are 3 mitochondrial structures, three measurements will be provided, each color-coded to depict which structure they represent). These measurements can be copied into a spreadsheet application such as Microsoft Excel or saved within Volocity by clicking Make Measurements under the Measurement tab.
The Sum value is the integrated intensity (total fluorescence of all the pixels within the object).
To make measurements using ImageJ:
From the Analyze menu, choose Set Measurements. Check the following: Area, Mean Gray Value, Integrated Density, Display Label.
Using the ROI tool, define the ROI you wish to measure.
From the Analyze menu, choose Measure (or press M).
Draw an ROI in the background (outside of cells) and measure background.
Copy or save the measurements that appear in the Results window.
Subtract the background integrated density from the integrated density of each measured ROI. The background integrated intensity for an ROI is the product of the background mean gray value and the area of the measured ROI.
3.10. Analysis of intracellular movement
Motility analysis can be used to test hypotheses about the mechanism and regulation of intracellular movements. The methods described here employ ImageJ (Schneider et al., 2012) and Volocity (Perkin-Elmer) software as the tools of choice in our own laboratory. Similar functions are available in most software packages for image analysis and processing.
Two strategies are presented here for quantifying motility: tracking and total motility measurement. The most direct and comprehensive way to measure motility is tracking analysis. Tracking can provide several quantitative measurements including velocity, distance traveled, frequency of movement, persistence/processivity of movement, and direction of movement. Here, we show how to calculate velocity and distance traveled.
Tracking involves marking the position of an object at successive timepoints. For punctate or roughly circular structures, the position is usually defined as the center of the structure. For structures with elongated, irregular or dynamic shapes, a point can be defined on the object of interest and be used as a reference point for velocity measurements. For example, to track mitochondria, which are highly tubular and dynamic, the leading tip can be used. In the case of Abp140-GFP-labeled actin, heterogeneity of Abp140 binding produces bright dots along the actin cable, and these can serve as fiduciary marks to track movement (Yang & Pon, 2002).
The second method, measurement of total motility, finds differences between successive image frames to quantify movement. A structure that does not change position will disappear in the subtracted image; any portion that does move will appear in the difference image. Measuring the area that changes relative to the total provides a quantitative indicator of the degree of motility. This method is more easily automated than tracking, especially for irregular structures. In addition, the regions of the cell where movement occurs are visible. This is helpful for qualitatively assessing anchorage, e.g. of mitochondria.
3.10.1. Tracking of intracellular movement
To make measurements in Volocity
Import image into Volocity. Make sure spatial and time scales are correctly set.
View the movie to identify structures meeting motility criteria (see Note 23).
Go to the first timepoint in which the movement occurs. Use the point tool to mark the center or leading edge of the structure of interest. For example, place a point at the leading edge of a mitochondrial tip.
Go to the next time point and mark the new position of the structure of interest.
Export point measurements into a spreadsheet or statistical application such as Microsoft Excel. Use the Pythagorean theorem to determine the distance moved and divide this by the time elapsed to calculate velocity.
To make measurements in ImageJ:
From the Analyze menu, choose Set Measurements. Check the following boxes: Display Label, Centroid.
View the movie to identify structures meeting motility criteria (see Note 23).
Select the Point Selection tool from the ImageJ toolbar and double-click the tool icon to set parameters: Check Auto-Measure and Auto-Next Slice.
Go to the first time point in which the movement occurs and click the structure.
Continue marking the motile structure for the duration of observable movement.
Copy or save the measurements that appear in the results window.
Calculate distance traveled between each set of successive timepoints t1 and t2 using the Pythagorean Theorem, where d is distance and the positions are (x1,y1) and (x2, y2) respectively:
Calculate velocity as distance traveled divided by time elapsed. To find mean velocity, calculate the average of all incremental velocities.
3.10.2. Qualitative and quantitative measurement of total motility
In this protocol, 4D image capturing is crucial to include movement in the z-axis. This analysis is done in ImageJ using a plugin published by Kurt De Vos (De Vos & Sheetz, 2007). Total motility for mitochondria is described below to provide an example of how this measurement can be made (Fig. 2).
Collect time-lapse z series. For mitochondria, we recommend using the long-term imaging preparation to collect a z-stack every 30 s for 10 min, with each z stack covering 6 μm of depth at an interval of 0.5 μm. We recommend imaging mitochondria using Cit1-GFP due to the high intensity and reliability of this mitochondria-targeted FP.
Deconvolve images in Volocity to remove out-of-focus light.
Open the deconvolved dataset as a hyperstack in ImageJ.
Optional: Contrast can be automatically enhanced using Process > Enhance Contrast. Any cell movement or drift can be corrected using the StackReg plugin in ImageJ (Note 24) (Thevenaz et al., 1998).
Generate a maximum-intensity projection for each timepoint: Image > Stacks > Z project; choose Projection Type: Max Intensity; check the box indicating All Time Frames.
Use Image > Adjust > Threshold to manually select a threshold for the projected stack. The thresholded areas should follow the outlines of mitochondria. Do not click Apply; rather, convert the stacks into binary images using Process > Binary > Make Binary.
Run the Total Motility plugin. The output will include a Results window and a new stack containing the difference images for each timepoint.
For quantitative analysis, each line of the Results window shows the percentage of mitochondrial area that was motile at a given timepoint.
For qualitative analysis, generate a sum projection of the new stack: Image > Stacks > Z project; choose Projection Type: Sum Slices. Here, the projection will summarize movement over the entire timecourse. This image depicts all motility events where white indicates movement and black indicates lack of movement.
3.10.3. Quantitative ratiometric measurements for measuring mitochondrial quality
Many tools used to measure mitochondrial quality are dyes that are imported into the mitochondria, such as the membrane potential-dependent dye, DiOC6. Measurements of total or mean intensity of these dyes are unreliable because they are skewed by the total mitochondrial mass. To correct for this, we suggest normalizing the dye intensities to a mitochondrial marker by calculating the ratio. The following protocol for quantitative ratiometric measurements using Volocity and ImageJ can be used for relative ratiometric measurements of any 2 fluorophores, including DiOC6 for mitochondrial membrane potential, roGFP for mitochondrial redox state, and DHE for mitochondrial ROS levels.
To produce a ratio channel in Volocity:
Draw an ROI around a field where there is zero fluorescence. This area will be used for background subtraction.
Under the Tools tab, select Ratio…
For Channel A (the numerator) select the dye to be quantified, such as DiOC6 or DHE. For Channel B (the denominator) select what you are normalizing to, such as Tom70-mCherry or Cit1-GFP.
Under Subtract, click Get From ROI for both Channel A and Channel B to subtract the background from each channel.
Click Calculate to automatically threshold each channel. Threshold values can also be manually set (see Note 25).
Optional: A rainbow LUT (look-up table or color scale) can make differences more obvious in the ratio channel, with warmer, redder colors indicating higher dye concentration (e.g. more DiOC6) and colder, bluer colors indicating lower dye concentration.
Click the Ratio button to produce the ratio channel.
To make measurements of your ratio channel in Volocity:
From the Measurements tab, create a protocol by selecting the following steps in order: Clip Objects to ROIs, Measure Objects.
Click on the gear icon in the Measure Objects tab and check Intensity measurements ignoring zero values. Ensure that the appropriate ratio channel is selected here.
Define the ROIs you wish to measure. You may select multiple ROIs for ratio measurements.
Under the Measurements tab, the measurements for each ROI are listed below the image and color-coded to match. These measurements can be copied into a spreadsheet application such as Microsoft Excel or saved within Volocity by clicking Make Measurements under the Measurement tab.
To compare values between strains, use the measurement item Mean (ignoring zero values).
To create and analyze ratiometric images in ImageJ:
Open deconvolved images and change type to 32 bit: Image>Type>32 bit.
Draw a region of interest (ROI) in an area where there are no cells. Calculate the mean intensity in this ROI: Analyze > Measure.
Subtract the calculated mean background from the stack: Process > Math > Subtract.
Using the subtracted z-stack, find the middle slice and threshold on mitochondria: Image > Adjust > Threshold and click Apply on the Threshold window. Apply to all slices in the stack. Check Set background pixels to NaN.
Create the ratio z stack: Process > Image Calculator. For the numerator, use the functional probe (DHE or DiOC6) or the reduced roGFP signal. For the denominator, use the mitochondrial marker or the oxidized roGFP signal.
Draw an ROI around the area of interest. Analyze > Tools > ROI Manager, and click Add to record the ROI. Multiple regions may be stored in the manager. In ROI Manager, select all ROIs, then choose More > Multi-Measure to measure all regions in stack slices. Export data to a spreadsheet for analysis.
4. Notes
Abp140-GFP signal is significantly higher in non-fermentable carbon sources. Lactate medium is the preferred choice as it is not autofluorescent and it provides more nutrients than synthetic complete medium with glycerol (SG). However, there may be some mitochondrial fragmentation in lactate medium. If this is a problem, use glycerol-based media, such as SG.
DiOC6 stock solutions are most stable at higher concentrations (~1000x working concentration). For best results, make working solutions fresh each day. Stock solutions may precipitate, so take care to resuspend prior to use.
H2O2 is unstable when diluted in water and diluted solutions should be prepared fresh and discarded after use for optimal results. DTT should be purchased in solution isolated in glass ampules for optimal results as reducing capacity can be lost upon contact with oxygen.
The sequences for the 3’ ends are often provided by the designers of the tagging plasmid. In many cases, the forward primer will include the sequence of the polylinker upstream of the FP tag while the reverse primer will include the reverse complement of the last few nucleotides of the selection marker, or some sequence downstream of the selection marker.
At least 250 ng of genomic DNA and 25 ng of plasmid DNA should be used.
We suggest using a high-fidelity polymerase for the amplification to prevent point mutations, especially when amplifying larger insertion cassettes.
The second step of the 35-cycle procedure is the primer annealing phase. The temperature here should be matched to the melting temperature of the DNA. For primers that have secondary structures, or when primer sequences cannot be manipulated to make the melting temperatures comparable, betaine, DMSO, or both can be added to the PCR reaction to a final concentration of 1 M and 5%, respectively (10 μL and 5 μL, respectively for a 50 μL reaction).
For most PCR reactions, we find that PCR purification is unnecessary. While purification will eliminate some contaminants from the PCR reaction, the accompanying loss of DNA from the process often outweighs the benefits of performing the purification. We specifically find that when using the KAPA HiFi HotStart or Platinum Taq DNA Polymerase High Fidelity PCR reaction kits, purification to eliminate contaminants is unnecessary.
The galactose-inducible Cre recombinase plasmids are CEN plasmids, which can often be dropped out by continuous growth on non-selective medium. Alternatively, pSH47 can be used as this galactose-inducible Cre recombinase expression plasmid contains the URA3 selectable marker and allows curing cells of the plasmid by counter-selection on 5-fluoroorotic acid (5-FOA) plates after marker selection.
When performing rescue/plasmid complementation assays, it is important to express the FP-tagged protein under the endogenous promoter of the gene. This will avoid unexpected errors, such as overexpression of a partially functional protein rescuing wild-type phenotypes.
For live-cell imaging, avoid using medium containing yeast extract (e.g. YPD, YPG) as the yeast extract is autofluorescent and will require repeated washing prior to imaging. Instead, use synthetic complete or lactate medium.
Generally, mid-log phase cultures (OD600 = 0.2–0.6) will have the optimal amount of cells for staining and do not require concentrating unless a higher number of cells is necessary for the experiment. A culture with OD600 = 1.0 contains ~107 cells/ml.
The dye concentrations suggested in Table 7 are a good starting point. The specificity of these dyes is dependent on mitochondrial membrane potential and dye concentration. For example, some dyes can accumulate in other organelles, such as ER and vacuoles, when concentrations are too high. Therefore, dye concentration and incubation times should be titrated when working with new strains. High concentrations of dyes should be avoided to prevent excessive accumulation of dye in mitochondria resulting in organelle swelling and respiratory defects.
Slides should be used for even less time for more sensitive applications. For example, DiOC6 slides should only be kept for 2–3 min, and slides for visualization of actin cables should be used within 4 min due to fading of Abp140-GFP signal.
Excessive/extended centrifugation can cause actin cable depolymerization and subsequent mitochondrial fragmentation.
The volume is important since excess volume can cause cells to float and move during image acquisition, while insufficient volume can compress cells or cause uneven spreading. Slight pressure should be applied to the edges of the coverslip to stably trap cells between the slide and coverslip and avoid movement of cells during image acquisition. Take care not to put too much pressure or cells may burst.
The agarose pad material should be placed slightly off center as it is likely to move when removing the top slide in a subsequent step.
Excess cooling will cause the agarose pad to harden before it is properly spread. 5 s cooling of agarose pad is appropriate for rooms at 25°C or higher. For colder rooms, no cooling is necessary.
It is not advisable to wash out the drug when staining with DiOC6 unless it is known that the drug will react with DiOC6 and make the dye unreliable. Because the incubation time is 15 min, the cells may recover or adapt during this time. If it is not desirable to keep cells in a specific drug for an additional 15 min, DiOC6 can be added 15 min prior to the completion of drug treatment. For example, if a 30-min treatment is desired, the drug can be added to cells in HEPES, incubated for 15 min, then DiOC6 can be added and incubation can continue for another 15 min.
DiOC6 imaging can only be performed for a very short time (maximally 2–3 min). After 2–3 min of imaging, the dye begins to leak out of the cell and stain slides, making visualization and quantification unreliable.
For measurements of fluorescence intensity, thresholding can affect quantitative values greatly. When normalizing and contrasting to a stable and valid counterstain, thresholding can be more appropriately controlled. For example, when measuring fluorescence intensity of a membrane potential-dependent dye, such as DiOC6, thresholding by normalizing to or contrasting signal intensities to Tom70-mCherry will help reduce subjective errors.
Artifacts occasionally appear in deconvolved images. These may include halo-like structures or the disappearance of structures that are visible in the raw data. If structures are lost, or halos appear, repeat the deconvolution with fewer iterations. If artifacts do not appear, but the image is still blurry or faint, repeat with more iterations. If deconvolution fails to exceed the 95% confidence limit, verify that optics are clean, light source is aligned, and cells are mounted under a 1.5 coverslip. Also make sure that images are acquired with pixel values at least in the middle of the detector range, and not saturating the detector.
For characterizing directed movement in mitochondria, we generally make velocity measurements on mitochondrial tubules that have made three or more consecutive movements in the same direction. For tracking of actin cables, we generally make measurements on fiduciary marks that have made 4 or more consecutive movements in the same direction. It is important to determine a selection criterion prior to measurement to decrease subjectivity of measurements.
The StackReg plugin is available from http://bigwww.epfl.ch/thevenaz/stackreg/.
Thresholding is very important in getting accurate measurements. The mitochondria-targeted FP should be used to threshold the ratio channel. Care must be taken to match the image of the ratio channel as close to the mitochondrial structure as possible (Vevea et al., 2013). In addition, it is important to keep the ratio between the two thresholds identical for each image to avoid introducing errors. For example, if the calculated thresholds are 220 and 200 for Channel A and Channel B respectively, they may be changed to 440 and 400.
References Cited
- Boldogh IR, Yang HC, Nowakowski WD, Karmon SL, Hays LG, Yates JR 3rd & Pon LA (2001) Arp2/3 complex and actin dynamics are required for actin-based mitochondrial motility in yeast. Proceedings of the National Academy of Sciences of the United States of America 98: 3162–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubnell J, Pfister P, Sapar ML, Rogers ME & Feinstein P (2013) beta2 adrenergic receptor fluorescent protein fusions traffic to the plasma membrane and retain functionality. PloS one 8: e74941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng TH, Chang CR, Joy P, Yablok S & Gartenberg MR (2000) Controlling gene expression in yeast by inducible site-specific recombination. Nucleic acids research 28: E108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ & Sheetz MP (2007) Visualization and quantification of mitochondrial dynamics in living animal cells. Methods in cell biology 80: 627–682. [DOI] [PubMed] [Google Scholar]
- Doyle T & Botstein D (1996) Movement of yeast cortical actin cytoskeleton visualized in vivo. Proceedings of the National Academy of Sciences of the United States of America 93: 3886–3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelista M, Klebl BM, Tong AH, Webb BA, Leeuw T, Leberer E, Whiteway M, Thomas DY & Boone C (2000) A role for myosin-I in actin assembly through interactions with Vrp1p, Bee1p, and the Arp2/3 complex. The Journal of cell biology 148: 353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehrenbacher KL, Yang HC, Gay AC, Huckaba TM & Pon LA (2004) Live cell imaging of mitochondrial movement along actin cables in budding yeast. Current biology : CB 14: 1996–2004. [DOI] [PubMed] [Google Scholar]
- Gauss R, Trautwein M, Sommer T & Spang A (2005) New modules for the repeated internal and N-terminal epitope tagging of genes in Saccharomyces cerevisiae. Yeast 22: 1–12. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH, Willems AR & Woods RA (1995) Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11: 355–360. [DOI] [PubMed] [Google Scholar]
- Gueldener U, Heinisch J, Koehler GJ, Voss D & Hegemann JH (2002) A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic acids research 30: e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson GT, Aggeler R, Oglesbee D, Cannon M, Capaldi RA, Tsien RY & Remington SJ (2004) Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. The Journal of biological chemistry 279: 13044–13053. [DOI] [PubMed] [Google Scholar]
- Higuchi R, Vevea JD, Swayne TC, Chojnowski R, Hill V, Boldogh IR & Pon LA (2013) Actin dynamics affect mitochondrial quality control and aging in budding yeast. Current biology : CB 23: 2417–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi-Sanabria R, Pernice WM, Vevea JD, Alessi Wolken DM, Boldogh IR & Pon LA (2014) Role of asymmetric cell division in lifespan control in Saccharomyces cerevisiae. FEMS yeast research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huckaba TM, Lipkin T & Pon LA (2006) Roles of type II myosin and a tropomyosin isoform in retrograde actin flow in budding yeast. The Journal of cell biology 175: 957–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huckaba TM, Gay AC, Pantalena LF, Yang HC & Pon LA (2004) Live cell imaging of the assembly, disassembly, and actin cable-dependent movement of endosomes and actin patches in the budding yeast, Saccharomyces cerevisiae. The Journal of cell biology 167: 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL & Gottschling DE (2012) An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast. Nature 492: 261–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, Magiera MM, Rathfelder N, et al. (2004) A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21: 947–962. [DOI] [PubMed] [Google Scholar]
- Jonsdottir GA & Li R (2004) Dynamics of yeast Myosin I: evidence for a possible role in scission of endocytic vesicles. Current biology : CB 14: 1604–1609. [DOI] [PubMed] [Google Scholar]
- Kaksonen M, Sun Y & Drubin DG (2003) A pathway for association of receptors, adaptors, and actin during endocytic internalization. Cell 115: 475–487. [DOI] [PubMed] [Google Scholar]
- Karpova TS, Reck-Peterson SL, Elkind NB, Mooseker MS, Novick PJ & Cooper JA (2000) Role of actin and Myo2p in polarized secretion and growth of Saccharomyces cerevisiae. Molecular biology of the cell 11: 1727–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler CM (2004) New developments in mitochondrial assembly. Annual review of cell and developmental biology 20: 309–335. [DOI] [PubMed] [Google Scholar]
- Lackner LL, Ping H, Graef M, Murley A & Nunnari J (2013) Endoplasmic reticulum-associated mitochondria-cortex tether functions in the distribution and inheritance of mitochondria. Proceedings of the National Academy of Sciences of the United States of America 110: E458–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P & Pringle JR (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953–961. [DOI] [PubMed] [Google Scholar]
- Madania A, Dumoulin P, Grava S, Kitamoto H, Scharer-Brodbeck C, Soulard A, Moreau V & Winsor B (1999) The Saccharomyces cerevisiae homologue of human Wiskott-Aldrich syndrome protein Las17p interacts with the Arp2/3 complex. Molecular biology of the cell 10: 3521–3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell SJ, Stewart LC, Talin A & Yaffe MP (1990) Temperature-sensitive yeast mutants defective in mitochondrial inheritance. The Journal of cell biology 111: 967–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFaline-Figueroa JR, Vevea J, Swayne TC, Zhou C, Liu C, Leung G, Boldogh IR & Pon LA (2011) Mitochondrial quality control during inheritance is associated with lifespan and mother-daughter age asymmetry in budding yeast. Aging cell 10: 885–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeusen S & Nunnari J (2003) Evidence for a two membrane-spanning autonomous mitochondrial DNA replisome. The Journal of cell biology 163: 503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messerschmitt M, Jakobs S, Vogel F, Fritz S, Dimmer KS, Neupert W & Westermann B (2003) The inner membrane protein Mdm33 controls mitochondrial morphology in yeast. The Journal of cell biology 160: 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miliaras NB, Park JH & Wendland B (2004) The function of the endocytic scaffold protein Pan1p depends on multiple domains. Traffic 5: 963–978. [DOI] [PubMed] [Google Scholar]
- Morishita M & Engebrecht J (2005) End3p-mediated endocytosis is required for spore wall formation in Saccharomyces cerevisiae. Genetics 170: 1561–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozdy AD, McCaffery JM & Shaw JM (2000) Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. The Journal of cell biology 151: 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J, Marshall WF, Straight A, Murray A, Sedat JW & Walter P (1997) Mitochondrial transmission during mating in Saccharomyces cerevisiae is determined by mitochondrial fusion and fission and the intramitochondrial segregation of mitochondrial DNA. Molecular biology of the cell 8: 1233–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Perlman PS & Butow RA (1998) The sorting of mitochondrial DNA and mitochondrial proteins in zygotes: preferential transmission of mitochondrial DNA to the medial bud. The Journal of cell biology 142: 613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okreglak V & Drubin DG (2007) Cofilin recruitment and function during actin-mediated endocytosis dictated by actin nucleotide state. The Journal of cell biology 178: 1251–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawley J (2000) The 39 steps: a cautionary tale of quantitative 3-D fluorescence microscopy. BioTechniques 28: 884–886, 888. [DOI] [PubMed] [Google Scholar]
- Riedl J, Crevenna AH, Kessenbrock K, et al. (2008) Lifeact: a versatile marker to visualize F-actin. Nature methods 5: 605–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riezman H, Hase T, van Loon AP, Grivell LA, Suda K & Schatz G (1983) Import of proteins into mitochondria: a 70 kilodalton outer membrane protein with a large carboxy-terminal deletion is still transported to the outer membrane. The EMBO journal 2: 2161–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues F, van Hemert M, Steensma HY, Corte-Real M & Leao C (2001) Red fluorescent protein (DsRed) as a reporter in Saccharomyces cerevisiae. Journal of bacteriology 183: 3791–3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabourin M, Tuzon CT, Fisher TS & Zakian VA (2007) A flexible protein linker improves the function of epitope-tagged proteins in Saccharomyces cerevisiae. Yeast 24: 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS & Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nature methods 9: 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiya-Kawasaki M, Groen AC, Cope MJ, et al. (2003) Dynamic phosphoregulation of the cortical actin cytoskeleton and endocytic machinery revealed by real-time chemical genetic analysis. The Journal of cell biology 162: 765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheff MA & Thorn KS (2004) Optimized cassettes for fluorescent protein tagging in Saccharomyces cerevisiae. Yeast 21: 661–670. [DOI] [PubMed] [Google Scholar]
- Simbeni R, Pon L, Zinser E, Paltauf F & Daum G (1991) Mitochondrial membrane contact sites of yeast. Characterization of lipid components and possible involvement in intramitochondrial translocation of phospholipids. The Journal of biological chemistry 266: 10047–10049. [PubMed] [Google Scholar]
- Skowronek P, Krummeck G, Haferkamp O & Rodel G (1990) Flow cytometry as a tool to discriminate respiratory-competent and respiratory-deficient yeast cells. Current genetics 18: 265–267. [DOI] [PubMed] [Google Scholar]
- Smith MG, Swamy SR & Pon LA (2001) The life cycle of actin patches in mating yeast. Journal of cell science 114: 1505–1513. [DOI] [PubMed] [Google Scholar]
- Sun Y, Martin AC & Drubin DG (2006) Endocytic internalization in budding yeast requires coordinated actin nucleation and myosin motor activity. Developmental cell 11: 33–46. [DOI] [PubMed] [Google Scholar]
- Sun Y, Carroll S, Kaksonen M, Toshima JY & Drubin DG (2007) PtdIns(4,5)P2 turnover is required for multiple stages during clathrin- and actin-dependent endocytic internalization. The Journal of cell biology 177: 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swayne TC, Zhou C, Boldogh IR, et al. (2011) Role for cER and Mmr1p in anchorage of mitochondria at sites of polarized surface growth in budding yeast. Current biology : CB 21: 1994–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevenaz P, Ruttimann UE & Unser M (1998) A pyramid approach to subpixel registration based on intensity. IEEE transactions on image processing : a publication of the IEEE Signal Processing Society 7: 27–41. [DOI] [PubMed] [Google Scholar]
- Vaduva G, Martin NC & Hopper AK (1997) Actin-binding verprolin is a polarity development protein required for the morphogenesis and function of the yeast actin cytoskeleton. The Journal of cell biology 139: 1821–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vevea JD, Swayne TC, Boldogh IR & Pon LA (2014) Inheritance of the fittest mitochondria in yeast. Trends in cell biology 24: 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vevea JD, Wolken DM, Swayne TC, White AB & Pon LA (2013) Ratiometric biosensors that measure mitochondrial redox state and ATP in living yeast cells. Journal of visualized experiments : JoVE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddle JA, Karpova TS, Waterston RH & Cooper JA (1996) Movement of cortical actin patches in yeast. The Journal of cell biology 132: 861–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace W, Schaefer LH & Swedlow JR (2001) A workingperson’s guide to deconvolution in light microscopy. BioTechniques 31: 1076–1078, 1080, 1082 passim. [DOI] [PubMed] [Google Scholar]
- Warren DT, Andrews PD, Gourlay CW & Ayscough KR (2002) Sla1p couples the yeast endocytic machinery to proteins regulating actin dynamics. Journal of cell science 115: 1703–1715. [DOI] [PubMed] [Google Scholar]
- Westermann B (2010) Mitochondrial fusion and fission in cell life and death. Nature reviews Molecular cell biology 11: 872–884. [DOI] [PubMed] [Google Scholar]
- Winder SJ, Jess T & Ayscough KR (2003) SCP1 encodes an actin-bundling protein in yeast. The Biochemical journal 375: 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HC & Pon LA (2002) Actin cable dynamics in budding yeast. Proceedings of the National Academy of Sciences of the United States of America 99: 751–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young CL, Raden DL, Caplan JL, Czymmek KJ & Robinson AS (2012) Cassette series designed for live-cell imaging of proteins and high-resolution techniques in yeast. Yeast 29: 119–136. [DOI] [PMC free article] [PubMed] [Google Scholar]