

Abstract
Prefrontal cortex (PFC) serves as the chief executive officer of the brain, controlling the highest level cognitive and emotional processes. Its local circuits among glutamatergic principal neurons and GABAergic interneurons, as well as its long-range connections with other brain regions, have been functionally linked to specific behaviors, ranging from working memory to reward seeking. The efficacy of synaptic signaling in PFC network is profundedly influenced by monoaminergic inputs via the activation of dopamine, adrenergic or serotonin receptors. Stress hormones and neuropeptides also exert complex effects on the synaptic structure and function of PFC neurons. Dysregulation of PFC synaptic transmission is strongly linked to the social deficits, affective disturbance, and memory loss in brain disorders including autism, schizophrenia, depression and Alzheimer’s disease. Critical neural circuits, biological pathways, and molecular players that go awry in these mental illnesses have been revealed by integrated electrophysiological, optogenetic, biochemical, and transcriptomic studies of PFC. Novel epigenetic mechanism-based strategies are proposed as potential avenues of therapeutic intervention for PFC-involved diseases. This review provides an overview on PFC network organization and synaptic modulation, as well as the mechanisms linking PFC dysfunction to the pathophysiology of neurodevelopmental, neuropsychiatric and neurodegenerative diseases. Insights from the preclinical studies offer the potential for discovering new medical treatment of human patients with these brain disorders.
Keywords: prefrontal cortex, synaptic transmission, glutamate, GABA, monoamine, stress, epigenetics, autism, schizophrenia, depression, Alzheimer’s disease
Prefrontal cortex (PFC) is the central hub for high-level executive functions critically involved in mental health and diseases. In this review, we will provide an overview on how synaptic transmission in PFC is regulated in normal and pathological conditions. We will first summarize PFC functional organization by describing the local and long-range neuronal circuits and the behavioral correlates to these top-down and bottom-up synaptic connections. Next, we will summarize PFC modulation by describing the influence of monoaminergic systems, stress hormones and neuropeptides on the synaptic structure and function of PFC neurons. Finally, we will summarize the PFC synaptic dysregulation in a variety of brain disorders and potential treatment strategies. Because of the broad scope of this review, only selective neuronal circuits, signaling molecules and disease mechanisms are included. Interested readers are encouraged to find more comprehensive details on specific aspects of the topic in the orignial papers and review articles cited here.
PFC Organization and Function
PFC network organization and functional implications
PFC is the cortical region located at the anterior part of the frontal lobe. PFC of humans is delineated into two functionally, morphologically, and evolutionarily different regions: ventromedial PFC and lateral PFC. PFC is the last portion of the brain to fully develop. In monkey PFC neurons, the rapid phase of synaptogenesis reaches the plateau at 2 months old, and maintains the consistently high synaptic density from early adolescence through puberty (2 months to 3 years), the formative time when learning experiences are most intense 1.
Early studies of macaque monkeys have revealed PFC as a crucial hub necessary for successful maintenance of working memory (WM), a cognitive process involving information holding and manipulation, which serves as the fundamental backbone for executive functions 2,3. The key findings is that in a delayed reaching task, some primate dorsolateral PFC neurons exhibit the increased firing during the delay period when response-related information needs to be held in mind to guide the subsequent execution of correct responses 4,5. The delayed and persistent spiking activity that encodes spatial “working memory” signals arises from recurrent excitation among PFC pyramidal neurons 5,6. Cortical network models predict that NMDA receptors subserve the recurrent synaptic excitation and persist firing of PFC pyramidal neurons during working memory 7,8. In parallel with these neurophysiological studies, neuroimaging studies also implicate the human PFC in higher-order cognitive processing including working memory 9 and sustained attention 10.
Based on anatomical, functional and connectivity homologies, it is thought that rodent PFC is composed of three cytoarchitecturally defined parts: the prelimbic (PL), infralimbic (IL), and anterior cingulate cortex (ACC) 11,12. Electro-physiological and pharmacological evidence further supports the existence of a frontal cortical area in the rat that exhibits increased firing during the delay period in the absence of sensory stimulus that instruct the orienting response, as in primate PFC 13. Recordings of neuronal ensembles in rats during new task learning further find that the dynamic changes in PFC neuronal activity parallel with behavioral decoding, suggesting that working memory can be mediated by dynamic activation of different neural populations in PFC 14.
PFC projects to distributed subcortical autonomic, motor, and limbic regions (Fig. 1). Tracing studies have found that deep layer PFC pyramidal neurons project to mediodorsal thalamus, lateral hypothalamus, ventral and dorsal striatum, and basolateral amygdala (BLA) 15. Prefrontal circuitry can dynamically control reward-seeking behaviour through projection-specific cue encoding 16: stimulation of PFC to nucleus accumbens (NAc) neurons promotes conditioned reward-seeking behaviour, while activation of PFC to thalamic neurons suppresses both the acquisition and expression of conditioned reward seeking. A specific subset of mPFC projections to NAc neurons encodes the decision to initiate or suppress reward seeking when faced with risk of punishment 17.
Figure 1.
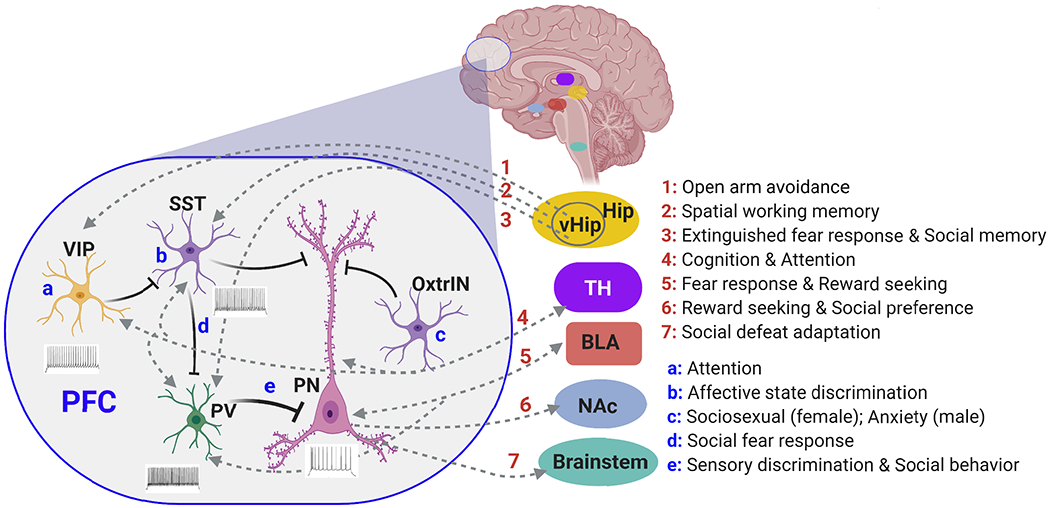
Plot of PFC organization highlighting the functional mapping of some long-range and local circuits of PFC. VIP+ interneurons (VIP) in PFC sends inhibitory inputs to SST+ interneurons (SST), which causes disinbition of PV+ interneurons (PV), resulting in the strong somatic inhibition and weak dendritic inhibition of pyramidal neurons (PN). PFC interneurons and PN are innervated by hippocampus (HIP) or ventral HIP (vHIP), thalamus (TH) and basolateral amygdala (BLA), while PN send output to TH, BLA, nucleus accumbens (NAc) and brainstem. Some behavioral correlates of these neuronal circuits are listed, along with the functional role of VIP+ or SST+ interneurons, as well as a subtype of SST+ interneurons expressing oxytocin receptor (OxtrIN).
Dorsomedial PFC exerts the top-down control of motor cortex ensembles to inhibit inappropriate responses during a delayed-response task 18. The neural projection from medial PFC (mPFC) to a brainstem area has been found to control behavioral adaptation to social defeat 19: social defeat weakens functional connectivity of these two areas, while selective inhibition of this pathway recaptures behavioral effects of social defeat. A spatiotemporal dynamic network, which originates in PFC and ventral striatum, relays through the amygdala and ventral tegmental area (VTA), and converges in ventral hippocampus (vHIP), is found to encode depression vulnerability in mice subjected to social defeat stress 20.
PFC also receives inputs from diverse regions, including hippocampus, thalamus, and BLA (Fig. 1). The direct hippocampal to prefrontal afferents in mice are found to be critical for encoding of spatial cues during working memory, and successful encoding of task-related information is mediated by gamma-frequency synchrony between hippocampus and PFC 21. Proper vHIP-mPFC signaling is necessary to recall social memories 22. Reciprocal interactions between PFC and thalamus play a critical role in high-level cognitive function 23. Thalamic input amplifies PFC connectivity, sustaining attentional control 24 and enabling sensory selection in divided attention 25. The reciprocal connection between PFC and BLA is implicated in reward-seeking and fear-related responses, and optogenetic studies have revealed that BLA to PL circuit is critical in governing the selection of behavioral responses amid conflicting cues of reward and punishment 26.
PFC cellular components and synaptic circuits
PFC is composed of pyramidal neurons with wide arbor axons, wide spikes, low discharge rates, and interneurons, many of which having horizontal axonal arbors, narrow action potentials (APs) and high evoked firing rates 27. PFC fast spiking (FS) interneurons (presumably expressing parvalbumin, PV+) form contacts on the perisomatic domains of pyramidal neurons, while non-FS interneurons (presumably expressing somatostatin, SST+) contact peridendritic domains 28, enabling PV+ and SST+ interneurons to specialize in the control of the output and input of principal neurons, respectively. PV+ interneurons, which could fire in millisecond synchrony, exert fast and powerful inhibition on principal neuron firing, whereas SST+ interneurons exert weak and more variable inhibitory effects on firing output in behaving mice 29. PFC in awake mice also has a basic disinhibitory circuit module in which activation of VIP+ interneurons transiently suppresses primarily SST+ and a fraction of PV+ interneurons 30. Such disinhibitory control may serve to amplify local processing and modulate information gating. A subclass of VIP neurons expressing ChAT, ChAT-VIP, directly excites neurons throughout cortical layers via fast synaptic transmission of acetylcholine in mPFC, providing a local source to control attention 31.
Opotogenetics, in vivo electrophysiological recording and single-cell microendoscopic Ca2+ imaging of PFC neurons in behaving animals have revealed the cell-type functional response diversity that contributes to diverse behavioral correlates (Fig. 1). PL pyramidal neurons are found to contain anatomically and molecularly distinct subpopulations that target three downstream regions implicated in social behavior: NAc, BLA, and VTA. Activation of NAc-projecting PL neurons, but not the other subpopulations, decreases the preference for a social target 32. PV+ interneurons in mPFC also play a key role in social behaviors: activation of mPFC PV+ neurons rescues social memory impairment caused by vHIP inhibition 33. Dampening thalamic activity causes significant reductions of GABA signaling in the mPFC and concomitant abnormalities in cognition and social interaction, which is ameliorated by selectively activating mPFC PV+ interneurons 34. BLA inputs also make strong connections onto PFC PV+ and SST+ interneurons, driving feedforward inhibition of PFC pyramidal neurons 35,36.
In mice performing a reward foraging task, SST+ interneurons in ACC are found to selectively respond at reward approach, whereas PV+ interneurons respond at reward leaving 29. In mice performing a PFC-dependent sensory discrimination task, PV+ interneurons are responsive to sensory cues, motor action, and trial outcomes, while SST+ and VIP+ interneurons respond more selectively to motor action or trial outcomes, respectively, and pyramidal neurons show much greater functional heterogeneity with varied responses across cortical layers 37. This cell type-specific encoding of task-related signals may be crucial for local computation within the PFC microcircuit to guide goal-directed behavior. Furthermore, SST+ interneurons in mPFC of mice are found to play a primary role in orchestrating the ability to discriminate positive and negative affective states 38: inhibition of mPFC SST+ interneurons abolishes affective state discrimination, while an increased synchronous activity of PFC SST+ interneurons, which guides the inhibition of PFC pyramidal neurons, is associated with this social cognitive function.
More recent studies have further revealed the functional role of PFC interneurons in working memory. During a spatial WM task, optogenetically inhibiting PFC SST+, but not PV+, interneurons decreases the long-range synchrony between hippocampus and PFC and impairs working memory accuracy 39, suggesting that SST+ interneurons are uniquely involved in facilitating hippocampal-prefrontal synchrony and prefrontal spatial encoding. Inhibiting PFC pyramidal neurons by optogenetically activating SST+ or PV+ interneurons during the delay impairs working memory performance, while activating VIP+ interneurons in PFC improves memory retention 40, which has provided a circuit mechanism underlying memory-guided behavior. PV+ and SST+ interneurons in mPFC are also found to have distinct roles in WM: PV interneurons show weak target-dependent delay-period activity and are strongly inhibited by reward; while SST+ interneurons show strong target-dependent delay-period activity, and only a subtype of them is inhibited by reward 41.
Moreover, in a social fear conditioning paradigm, SST+ interneurons suppress PV+ interneurons and disinhibit pyramidal cells, which consequently enhances PFC output to mediate social fear responses 42. This disinhibitory microcircuit in PFC through interactions between interneuron subtypes provides an important circuit mechanism in gating social fear behavior. The relapse of extinguished fear is found to be mediated by a feedforward inhibitory pathway from ventral hippocampus to IL cortex, which involves the recruitment of PV+ interneurons in IL 43. In the avoidance behavior of elevated plus maze, PFC VIP+ interneurons enable cortical circuits to integrate hippocampal inputs, therefore effectively gating the transmission of signals related to open arm avoidance across hippocampal-prefrontal network 44 (Fig. 1).
Regulation of PFC Synaptic Function
Monoaminergic regulation of PFC synaptic transmission
PFC is very sensitive to neurochemical environment (Fig. 2), and monoaminergic modulation of the efficacy of synaptic connections in PFC circuits has profound effects on PFC-mediated executive function, including working memory, regulation of attention, inhibition of inappropriate behaviors, and impulsivity control 45. Reduced catecholamine transmission has been linked to ADHD, while overamplified noradrenergic transmission is associated with PTSD 46. The therapeutic actions of psychostimulant methylphenidate for ADHD involve the preferential increase of catecholamine neurotransmission within PFC 47 and dose-dependent, bi-directional regulation of PFC glutamate receptors 48.
Figure 2.
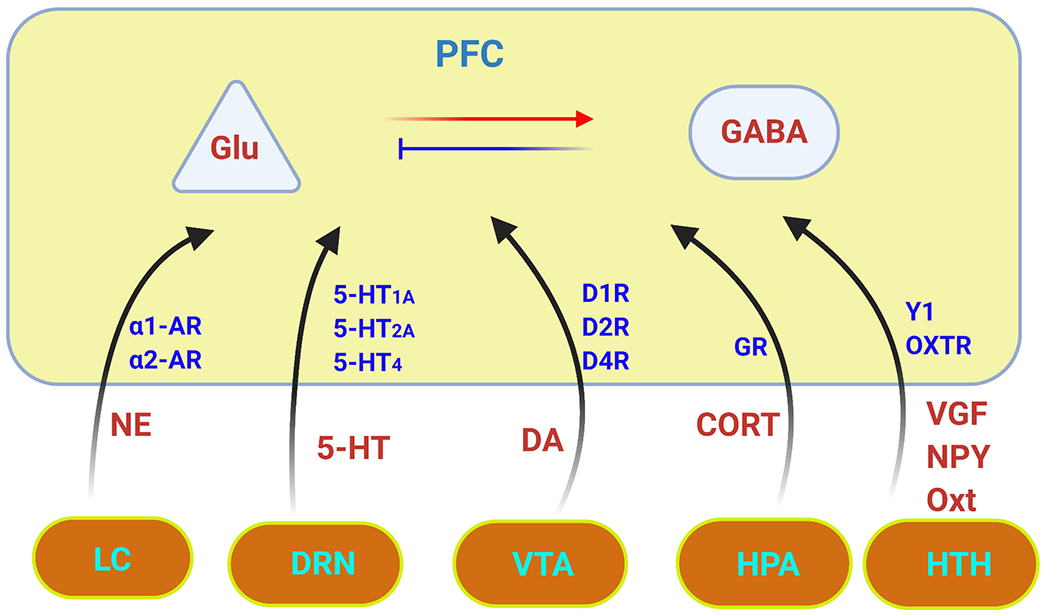
Plot of PFC regulation illustrating some neurochemical influences on glutamatergic (Glu) excitation and GABAergic (GABA) inhibition in PFC neurons. The major modulators include dopamine (DA) from ventral tegmental area (VTA), serotonin (5-HT) from dorsal raphe (DRN), norepinephrine (NE) from locus ceruleus (LC), corticosterone (CORT) stress hormone from hypothalamic–pituitary–adrenal (HPA) axis, and neuropeptides, such as VGF nerve growth factor (VGF), Neuropeptide Y (NPY), and oxytocin (OT), from hypothalamus (HTH). These monoaminergic and homornal modulations are achieved by activing corresponding receptors. The complex intracellular signaling pathways initiated by each neuromodulator can be found in relevant papers or previous reviews.
Dopamine
Serial section electron microscopy studies suggest that the majority of dopamine synapses in all PFC layers are on pyramidal neurons, but a significant fraction are on GABAergic interneurons 49. Electrophysiological studies revealed that dopamine selectively modulates PFC excitatory and inhibitory microcircuits. DA depressed inhibitory transmission between FS interneurons and pyramidal neurons, but enhanced inhibition between non-FS interneurons and pyramidal cells 28. Dopamine also reduces recurrent excitation in layer V pyramidal neurons 50, which may involve target-specific expression of presynaptic and postsynaptic D1Rs 50,51. Dopamine has no effect on excitatory transmission from pyramidal neurons to FS interneurons, but increases the excitability of FS interneurons 52. In an experimental condition mimicking NMDA receptor hypofunction and GABAA receptor deficiency in schizophrenia, dopamine, by acting on D2 receptors, promoted burst firing in a subset of PFC pyramidal neurons 53.
Stimulation of D1R produces an inverted-U dose response on PFC neuronal firing and cognitive performance during working memory tasks 46. High levels of D1R activation of cAMP signaling reduces PFC neuronal firing and impairs working memory via opening of HCN channels at excitatory synapses 54. It suggests that optimal levels of dopamine are essential for PFC function, while excessive dopamine stimulation could be detrimental.
Optogenetic stimulation of VTA neurons to trigger the synaptically released dopamine has revealed a marked and prolonged enhancement of the excitability of PFC PV+ interneurons and a modest and short-lived enhancement of the excitability of PFC principal neurons 55. Blocking dopamine receptor activation in PFC shortens the VTA excitation of PFC PV+ interneurons and prolongs the VTA excitation of PFC principal neurons, and pharmacological evidence suggests that the dopaminergic effect on PFC principal neurons is through influencing the inhibitory transmission system 55. These results have unmasked a role of dopamine in regulating the temporal dynamics of excitation/inhibition balance in VTA-PFC circuit.
Dopamine D4 receptor, which is largely restricted to PFC neurons, is highly implicated in ADHD and schizophrenia 56,57. It has been revealed that D4 receptors serve as a homeostatic synaptic factor to stabilize cortical excitability by dynamically regulating CaMKII 58,59 and the downstream NMDA and AMPA receptor channels 60–62. In contrast to the activity-dependent, bi-directional effect of D4R stimulation on AMPARs in PFC pyramidal neurons 62,63, D4 receptors suppress AMPARs in PFC GABAergic interneurons via the unique calcineurin/Slingshot/cofilin signaling mechanism 64. Moreover, D4R stimulation elicits distinct effects on synaptic-driven action potential firing in PFC projection neurons versus fast-spiking interneurons, which are differentially altered in neuropsychiatric disorder-related conditions 65. Stress exposure in D4R knockout mice induces schizophrenia-like behaviors via disruption of GABAergic transmission in PFC 66. These results have provided a framework for understanding how D4R signaling is involved in the regulation of PFC functions.
D4R is one of the most variable proteins (and genes) in humans. A unique primate-specific feature of D4R is the additional 2 to 11 proline-rich repeats (most commonly 4) located in the third intracellular loop, which allows more complex simultaneous interactions with other proteins containing the SH3 domain. Interestingly, genetic studies have linked novelty-seeking behavior and ADHD to long-repeat D4R allele (hD4.7) 57,67. Using D4R knockout mice with PFC expression of human D4R variants containing different repeats (hD4.4 and hD4.7), researchers have revealed the impact of hD4 variants on synaptic transmission and network activity in PFC neurons, the proteins interacting with hD4 variants and the signaling pathways activated by hD4 variants, as well as the behavioral changes caused by hD4 variants 68,69. These studies provide novel insights into the functional roles of the remarkable polymorphism of human D4 receptors, and shed light on the development of new therapeutic agents for diseases associated with dopaminergic dysfunction (Fig. 2).
Noradrenaline
The noradrenergic system in PFC is involved in many physiological and psychological processes, including working memory and mood control 46. Endogenous norepinephrine (NE, also called noradrenaline), by acting on α1-ARs, exerts complex effects in PFC: NE increases the firing of some PFC pyramidal neurons via presynaptic excitation of glutamate release under basal conditions, but when high levels of α1-AR stimulation occurs, such as with stress exposure, NE suppresses PFC neuronal firing through opening of K+ channels on spines 70. Stimulation of α2A-ARs enhances the spatially-tuned, delay-related firing of PFC neurons and strengthens working memory networks through inhibition of cAMP and closing HCN (Hyperpolarization-activated Cyclic Nucleotide-gated) channels 71. The interaction of HCN channels and cAMP regulating proteins in dendritic spines of PFC is considered to be potential substrate of working memory 72.
Activation of α1-ARs or α2-ARs suppresses NMDAR currents in PFC pyramidal neurons by distinct mechanisms: the α1-AR effect depends on the PLC/IP3/Ca2+ pathway, whereas the α2-AR effect depends on PKA and the microtubule-based transport of NMDARs that is regulated by ERK signaling. Moreover, the effects of α1-ARs and α2-ARs are differentially modified by RGS4 73, a member of the RGS family protein that plays an important role in modulating GPCR signaling 74 and an identified schizophrenia susceptibility gene 75,76 (Fig. 2).
Serotonin
PFC receives a major serotonergic projection, which is dysfunctional in individuals who show impulsive aggression and violence 77. Aberrant serotonergic neurotransmission has long been implicated in the pathogenesis of neuropsychiatric disorders associated with PFC dysfunction, including schizophrenia, depression and anxiety 78. Most clinically effective antipsychotic drugs, antidepressants and anxiolytics all exert potent effects on the serotonin system. The pleiotropic functions of serotonin are afforded by the concerted actions of multiple serotonin receptor subtypes 79.
A series of studies have revealed the synaptic functions of serotonin receptors in PFC and how dysfunction of the serotonin system contributes to mental illnesses 80–89. One of the key targets of serotonin receptor signaling is the glutamatergic system. It has been found that 5-HT1A receptors in PFC pyramidal neurons suppress NMDAR function by disrupting the microtubule/kinesin-based dendritic transport of NMDA receptors 83, while this effect of 5-HT1A is opposed by 5-HT2A/C signaling 82. In the human PFC, serotonin suppresses monosynaptic excitatory connections from pyramidal cells to interneurons 90. At thalamocortical synapses, 5-HT2A activation enhances NMDA transmission, gates the induction of NMDAR-mediated temporal plasticity, and facilitates associated cognitive functions 91.
Another main target of serotonin receptor signaling is the GABAergic system. 5-HT2 receptors inhibit GABAAR-mediated currents in PFC pyramidal neurons through phosphorylation of GABAA receptors by the activation of anchored PKC 87, while 5-HT4 receptors modulate GABAergic signaling bi-directionally, depending on the basal PKA activation levels that are determined by neuronal activity 80. Serotonergic regulation of GABA transmission in PFC is subject to the alteration by corticotropin-releasing factor (CRF) in response to stressful stimuli 89 or chronic antidepressant treatment 86. In addition to pyramidal neurons, serotonin also affects GABAergic fast-spiking interneurons in PFC by enhancing their excitability and gamma frequency temporal integration via 5-HT2A suppression of an inward-rectifying potassium conductance 92. These studies have provided unique and complex mechanisms for serotonin receptors to dynamically regulate synaptic transmission and neuronal excitability in the PFC network in physiological and pathophysiological conditions (Fig. 2).
Hormonal regulation of PFC synaptic transmission
Stress hormones
Stress hormones exert complex and profound effects on the synaptic structure and function in PFC neurons (Fig. 2). Glucocorticoid action in the PFC shows a marked functional heterogeneity. Glucocorticoid receptor (GR) knockdown in IL induces compromised stress adaptation and depression-like behavior, while GR knockdown in PL increases HPA axis responses to acute stress but does not affect stress sensitization or helplessness behavior 93, highlighting the region-specific impact of PFC on stress coping and emotional control.
Exposing animals to an acute stressor, such as forced swim, foot shock or restraint, produces significantly enhanced glutamatergic transmission in PFC circuitry 94–97. One mechanism is related to the enhancement of readily releasable pool of glutamate vesicles via a non-genomic mechanism mediated by membrane receptors 95. Acute stress, via acting on GR, also induces a delayed potentiation of NMDAR-and AMPAR-mediated synaptic currents, which is correlated with the increased level of synaptic NMDAR and AMPAR subunits 96,98. Serum-and glucocorticoid-inducible kinases (SGKs), an immediate early gene activated by stress hormone, is shown to be involved by enhancing Rab4-mediated trafficking of glutamate receptors from early endosomes to plasma membrane in PFC neurons 98,99. The GR-induced upregulation of SGK and glutamatergic signaling in acutely stressed animals is blocked by inhibition of HDAC6, a unique member of the HDAC family that directly regulates the GR chaperone protein, heat shock protein 90 (HSP90) 97. Interestingly, reduced SGK expression is found in postmortem brains of PTSD patients, and inhibition of SGK in rat PFC produces helplessness- and anhedonia-like phenotypes 100.
Adhesion molecules that anchor glutamate receptors at the synaptic surface are also implicated in the acute stress-induced enhancement of GluR2 membrane trafficking and memory facilitation 101. In addition, acute footshock stress increases spine density and induces dendritic remodeling in medial PFC, which can be partially blocked by chronic treatment with the antidepressant desipramine 102,103. This effect of stress and antidepressant is linked to the alteration of key genes involved in synaptic plasticity and spine structure 104.
In contrast to the positive effects of acute stress, chronic stress (e.g. 21-day restraint stress) produces impaired dendritic branching, atrophy and spine loss in PFC pyramidal neurons 102,105, and such structural reorganization is reversed after 3-week cessation of stress 106. Chronic stress impairs synaptic plasticity by reducing LTP induction in the hippocampus-PFC connection 107, and repeated social stress during mid-adolescence significantly decreases synaptic activity and intrinsic excitability of mPFC neurons 108. Chronic exposure to corticosterone also causes the reduction of NR2B and GluR2/3 subunit expression in ventromedial PFC 109.
A prominent loss of GluR1 and NR1 subunit expression has been found in PFC pyramidal neurons from repeatedly stressed animals, which leads to the depression of AMPAR- and NMDAR-mediated synaptic currents 110. This stress-induced loss of glutamate receptor expression is attributable to the increased ubiquitin/proteasome-mediated degradation of GluR1 and NR1 that is controlled by E3 ubiquitin ligases Nedd4 and Fbx2, and inhibition of proteasomes or knockdown of these E3 ligases in PFC prevents the loss of glutamatergic responses and recognition memory in stressed animals 110. The transcription of Nedd4 is upregulated by repeated stress via an epigenetic mechanism involving the elevated histone deacetylase 2 (HDAC2) and the ensuing suppression of histone methyltransferase Ehmt2 (also known as G9a) that catalyzes the repressive mark H3K9me2 111. The impairment of glutamate receptors and excitatory transmission in PFC of chronically stressed animals is blocked by HDAC2 inhibitors 111.
There are many additional molecules that have been implicated in governing the effects of chronic stress in PFC. One key player is the mTORC (mammalian target of rapamycin complex) signaling pathway. Decreased levels of mTORC are found in PFC of humans with major depressive disorder 112, and the fast-acting antidepressant ketamine increases mTORC signaling in rat PFC 113. REDD1, an endogenous inhibitor of mTOR, shows the elevated expression in PFC of animals exposed to chronic unpredictable stress and PFC of post-partum depressed humans 114. REDD1 knockdown renders animals to have greater resilience to the chronic stress-induced spine shrinkage and AMPAR current reduction 114.
Neuropeptides
The neuropeptide precursor VGF plays a critical role in depression and antidepressant efficacy 115. Reduced levels of VGF have been found in PFC of MDD patients and PFC of chronically stressed mice 116. VGF in PFC regulates susceptibility to stress and the antidepressant response to ketamine, and infusion of VGF C-terminal peptide TLQP-62 to PFC produces sustained antidepressant responses 116.
Neuropeptide Y (NPY), a stress modulatory transmitter, has been associated with PTSD. NPY in IL cortex, acting on NPY Y1 receptors, increases inhibitory synaptic transmission onto IL projection neurons and dampens their excitability, resulting in the significant impairment of fear extinction memory 117.
The “prosocial” hormone oxytocin (OT) plays a key role in regulating social and emotional behaviors through a population of SST+ interneurons that express the oxytocin receptor (OxtrIN) in mPFC. Silencing OxtrIN of female mice or deleting Oxtr gene in mPFC results in sociosexual deficits 118, while OxtrIN in male mice regulate anxiety-related behaviors by interacting with the co-expressed corticotropin-releasing-hormone-binding protein (CRHBP), an antagonist of the stress hormone CRH 119 (Fig. 2).
PFC Dysregulation and Diseases
Transcriptomic meta-analyses of human cerebral cortex across major neuropsychiatric disorders, including autism (ASD), schizophrenia (SZ), bipolar disorder (BD) and depression (MDD), have found the shared transcriptional dysregulation underlying convergent molecular neuropathology, such as a gradient of synaptic gene down-regulation and astroglial gene up-regulation in ASD, SZ and BD (ASD > SZ = BD), and dysregulation of HPA-axis and hormonal signaling in MDD 120. Synaptic plasticity deficits in PFC of animal models have been linked to clinical findings from human patients with frontal cortical dysfunction, including the decreased functional connectivity in prefrontal-limbic circuits, executive function and memory deficits, inflexible and maladaptive behaviors 121. In what follows, we will summarize mechanisms of synaptic transmission dysregulation in the PFC and pathophysiological implications in a few neurodevelopmental, neuropsychiatric and neurodegenerative diseases linked to the impairment of social, emotional, and cognitive processes.
Autism
Autism is a prevalent neurodevelopmental disorder characterized by core symptoms like social deficits and stereotypic behaviors. The importance of PFC in autism is demonstrated by structural, functional, and genomic studies. Postmortem studies have found focal patches of abnormal laminar cytoarchitecture and cortical disorganization of neurons, but not glia, in prefrontal and temporal cortical tissue from a majority of young children with autism, with the clearest signs of abnormal expression of excitatory neuronal markers in deep cortical layers 4 and 5 122. Many of the high-level executive functions controlled by PFC are impaired in autism. Transcriptomic analysis of autistic brains has found 444 genes showing significant expression changes in PFC, but only 2 in cerebellum 123.
Haploinsufficiency of the Shank3 gene, which encodes a scaffolding protein at postsynaptic density of glutamatergic synapses, has been causally linked to autism 124,125. High-resolution MRI has found that loss of Shank3 in mice results in the disrupted local and long-range prefrontal and frontostriatal functional connectivity, which is predictive of social communication deficits 126. In PFC pyramidal neurons of Shank3-deficient mice, NMDAR function and synaptic distribution are significantly diminished, which is caused by a marked loss of actin filaments at glutamatergic synapses due to the altered Rac1/PAK/cofilin signaling 127. Inhibiting cofilin or activating Rac1 rescues the social deficits and NMDAR hypofunction in Shank3-deficient mice 127 (Fig. 3), suggesting that targeting actin regulators to restore actin-based NMDAR synaptic delivery could be a therapeutic option for autism 128. Chemogenetic activation of PFC pyramidal neurons in Shank3-deficient mice also restores social preference behaviors and elevates glutamatergic synaptic function 129. Moreover, the reduced synaptic plasticity in hippocampus-mPFC pathway and impaired social recognition memory and attention in Shank3-deficient rats are attenuated by oxytocin treatment 130.
Figure 3.
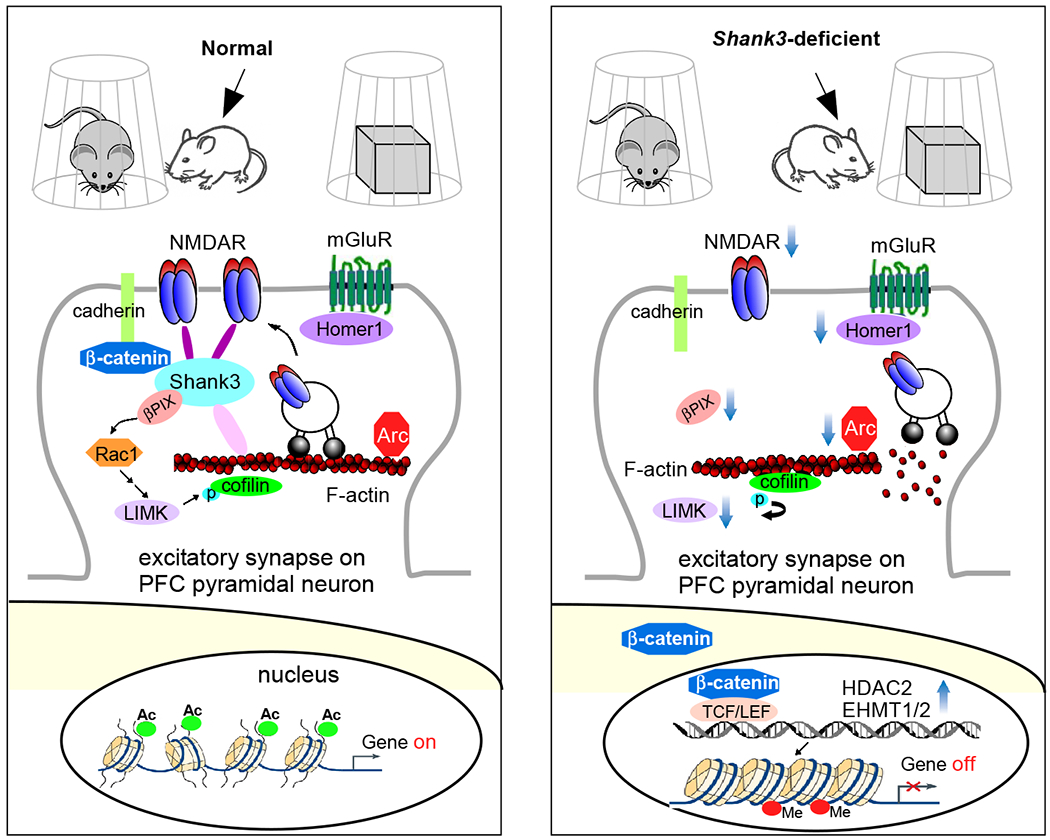
Schematic diagram illustrating a potential mechanism underlying the social deficits in Shank3 autism models. Normally, Shank3 crosslinks NMDARs to actin cytoskeleton, and binds to the adhesive junction-associated protein β-catenin. Loss of Shank3 leads to the translocation of β-catenin from synapses to nucleus, inducing the upregulation of histone modifiers HDAC2 and EHMT1/2. The ensuing transcriptional suppression of actin regulators, such as βPIX (Rac1 activator) and LIMK, and synaptic plasticity genes, such as Arc and Homer1 (mGluR anchor), results in the disruption of actin filaments (via cofilin-mediated depolymerization) and the diminished actin-based synaptic delivery of NMDARs in PFC pyramidal neurons. Consequently, the autism-like social preference deficits are manifested. Treatment with HDAC or EHMT inhibitors restores or elevates many target genes, which collectively leads to the normalization of NMDAR synaptic function in PFC and the rescue of social deficits127,145,146.
Another prominent genetic link to autism spectrum disorders and intellectual disability is the microdeletion or microduplication of the human 16p11.2 gene locus 131,132. A 16p11.2 deletion mouse model, 16p11+/−, exhibits NMDAR hypofunction in PFC pyramidal neurons 133. Chemogenetic activation of PFC pyramidal neurons leads to the amelioration of cognitive and social impairments and the restoration of NMDAR function in 16p11+/− mice 133. On the other hand, a 16p11.2 duplication mouse model, 16p11dp/+, exhibits deficient GABAergic synaptic transmission and elevated excitability in PFC pyramidal neurons 134. Restoring the expression of Npas4, a key regulator of GABA synapses 135,136, ameliorates the social and cognitive deficits, as well as GABAergic synaptic impairment and neuronal hyperexcitability in 16p11dp/+ mice 134. Synaptic dysfunction and excitation/inhibition imbalance is identified as a common pathophysiological feature of multiple ASD models 137–139.
Large scale genetic screenings have revealed that many of the identified top-ranking autism risk factors are genes implicated in synaptic homeostasis, transcriptional regulation and chromatin remodeling pathways 140–142. Epigenomic studies show that PFC neurons from subjects with autism exhibit the altered H3K4me3 peaks at numerous genes regulating neuronal connectivity, social behaviors, and cognition, often in conjunction with altered expression of the corresponding transcripts 143. A recent series of studies have shown that targeting epigenetic enzymes to adjust gene expression and ameliorate synaptic defects in PFC is a potential strategy to normalize behavioral phenotypes associated with autism 144–147.
In PFC of Shank3-deficient mice, the loss of histone acetylation and upregulation of HDAC2 have been identified 145. Systemic administration of the class I HDAC inhibitor romidepsin or MS-275 leads to the robust and persistent alleviation of social deficits, as well as the restoration of actin regulators and NMDAR function in PFC 144,145. A histone acetylome-wide association study (H3K27ac ChIP-seq) of human ASD postmortem samples also reveals common “epimutations” on genes involved in synaptic transmission, ion transport, and histone deacetylation 148. Moreover, the repressive histone mark H3K9me2 and its catalyzing enzymes EHMT1/2 are selectively increased in the PFC of Shank3-deficient mice and autistic human postmortem brains 146. Inhibition of EHMT1/2 rescues social deficits and restores PFC NMDAR function in Shank3-deficient mice via restoring the expression of plasticity gene Arc, which encodes the activity-regulated cytoskeleton-associated protein Arc 146. The nucleocytoplasmic shuttle protein involved in both cell adhesion and transcriptional regulation, β-catenin, is found to link the loss of Shank3 in the synapse to the upregulation of HDAC2 and EHMT1/2 transcription in the nucleus 145,146 (Fig. 3).
Another identified high-risk factor for autism is the Cullin 3 (Cul3) gene 141, which encodes a core component of the E3 ubiquitin ligase complex that mediates proteasomal degradation 149. Loss of Cul3 in forebrain or PFC leads to social deficits and NMDAR hypofunction, while loss of Cul3 in striatum leads to stereotypic behaviors and cell type-specific alteration of neuronal excitability in striatal circuits 147. One of the misregulated proteins resulting from forebrain Cul3-deficiency is Smyd3, a histone methyltransferase involved in gene transcription. Inhibition or knockdown of Smyd3 ameliorates social deficits and restores PFC NMDAR function in forebrain Cul3-deficient mice 147. These studies have demonstrated how aberrations in molecular and epigenetic pathways implicated in autism are interconnected.
Schizophrenia
Schizophrenia (SZ) is a chronic psychiatric disorder that is characterized by delusions, hallucinations, disorganized speech and behavior, and negative symptoms 150. SZ has been linked to both aberrant PFC development during embryonic stages and disrupted PFC maturation in later adolescence driven by environmental stressors, making SZ a unique disorder with two critical susceptibility periods 151. SZ patients exhibit reduced gray matter in frontal cortex, evidenced by MRI studies 152,153. It has been suggested that SZ is driven by disrupted cortical synaptic pruning 154 because its symptoms tend to appear late in adolescence corresponding with the developmental period in which PFC connectivity undergoes maturation 155. One hypothesis is that psychosocial stress in late development drives microglial overactivation, leading to the increased synaptic pruning in PFC 156. In accordance with these theories, SZ patients display significant reductions in dendritic spine density in PFC 157, and lower density of perineuronal nets (PNN) - extracellular structures that stabilize synapses, also suggesting synaptic loss or destabilization 158. SZ patients show reduced blood flow in PFC during task performance 159 and reduced functional connectivity between PFC and other brain regions when performing a cognitive task 160.
SZ etiology has a strong genetic component, and many high-risk SZ genes identified by GWAS studies are highly enriched in mid-frontal lobe 161, form protein interactions in fetal dorsolateral (DL) and ventrolateral PFC 162, and/or are functionally implicated in synaptic transmission 162,163. Proteomic analysis of SZ DLPFC samples identified 15 differentially expressed proteins, 7 of which were synaptic 164. These studies suggest synaptic dysfunction in PFC as a core mechanism in SZ.
The synaptic pathology of SZ in the PFC is complex, involving disrupted glutamatergic, GABAergic, dopaminergic, and serotonergic transmission (Fig. 4A). There is strong support for NMDA hypofunction in SZ, as treatment with NMDAR antagonists, such as MK-801 or phencyclidine, can induce SZ-like symptoms in both human subjects 165 and in animals 166. NMDAR-mediated connectivity between PFC and hippocampus is impaired in SZ, which is suggested as a mechanism underlying working memory deficits 167. Blocking NMDARs with MK-801 injection leads to the impairment of mPFC-dependent cognitive flexibility and hippocampus-mPFC pathway-dependent spatial working memory 168.
Figure 4.
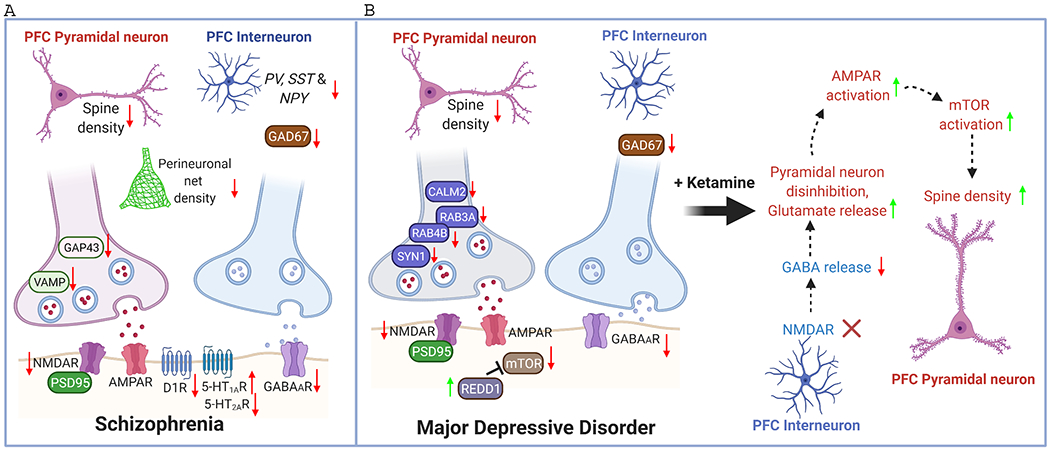
Schematic diagram illustrating the synaptic changes in PFC of schizophrenia (SZ) and major depressive disorder (MDD). NMDAR hypofunction, GABA deficiency, dopamine hypoactivity, serotonin dysregulation, and dendritic spine loss are major aberrations in SZ PFC. The reduced density of perineuronal nets (PNN) may contriubute to the destabilization of synapses. Similar reduction on NMDAR, GABA system and spine density is found in MDD PFC. Some presynaptic molecules involved in transmitter release, including CALM2, SYN1, RAB3A, RAB4B, are also diminished in MDD PFC. In addition, mTOR signaling is decreased in MDD PFC because of the elevated expression of the endogenous inhibitor REDD1. The fast-acting antidepressant ketamine primarily blocks NMDARs on PFC interneurons, leading to the disinhibition of PFC pyramidal neurons and the increase of mTORC signaling, resulting in the restoration of dendritic spines in MDD PFC.
Paradoxically, MK-801 drives the increased release of glutamate in the mPFC, though the antipsychotic medications clozapine and haloperidol are both able to block MK-801-induced glutamate release in PFC of rats 169. NMDAR antagonists may elicit this paradoxical response by primarily blocking NMDA receptors on GABAergic interneurons, thereby promoting disinhibition of pyramidal neurons 170. It has thus been postulated that the NMDA hypofunction in SZ mainly occurs in cortical GABAergic interneurons 165. Postmortem schizophrenia studies have revealed supporting evidence 171, including the reduced density of NR2A-expressing PV interneurons in cortical layers 3 and 4 172 and the reduced density of GAD67- and NR2A-coexpressing neurons in layers 2 and 5 of anterior cingulate cortex 173.
In rats treated with NMDAR antagonists, PFC neurons display the elevated and irregular spiking activity and the reduced coordinated burst firing, suggesting that NMDA hypofunction in PFC may drive disorganized network activity 174. In agreement with this, monkeys performing a SZ-relevant cognitive task exhibit the reduced synchronous firing and disrupted connectivity in PFC neurons after the administration of an NMDAR antagonist 175. PFC-specific inhibition of either NMDA or GABA transmission produces working memory deficits in rats, suggesting that either system could contribute to working memory deficits in SZ 176. Several biochemical indicators of NMDA dysfunction are reported in SZ PFC, including the reduced expression of several synaptic plasticity-related genes 177, and the significantly lower postsynaptic expression of the NMDAR subunit NR1 and PSD95 178. Reduced expression of the presynaptic vesicle protein VAMP has also been reported in SZ PFC 179, while other presynaptic markers appear unchanged 177,179.
Several well-replicated cellular indications of disrupted GABAergic synaptic transmission have been reported in PFC of SZ patients, including the decreased GAD67 expression and reduced gamma oscillations, suggesting deficient GABA synthesis and GABAergic synaptic transmission, respectively 180. While no change in the number of PV+ cells 181 or the density of GABAergic synaptic inputs on DLPFC neurons 182 is observed in SZ patients, the reduced PV expression is reported both at the individual cellular level 181 and specifically within presynaptic boutons of PV+ basket cells 182. Furthermore, an expression analysis of DLPFC from SZ patients indicated the reduced expression of several GABA-related genes, including neuropeptides and both presynaptic and postsynaptic markers 183. Inhibiting GABAergic transmission in PFC of rats produces several behavioral phenotypes mirroring SZ symptomology 184. The evidence implicating GABAergic deficits in PFC as a core mechanism in SZ-related cognitive dysfunction has been reviewed previously 185.
Dopamine modulation of PFC, which also plays a major role in working memory 186, is thought to contribute to PFC dysfunction in SZ. In a mouse model carrying a genetic risk factor for neuropsychiatric disorders, adolescent isolation stress induces DNA hypermethylation of the tyrosine hydroxylase (TH) gene in mesocortical dopaminergic neurons, causing the reduced TH expression and cortical dopamine release, leading to SZ-associated behavioral abnormalities 187. D2R activation leads to long-lasting NMDAR hypofunction, resulting in the profound disruption of functional connectivity between hippocampus and PFC 167. D2R overexpression in mouse striatum causes the reduced inhibitory synaptic input to PFC pyramidal neurons, suggesting that the deficit in PFC GABAergic function in schizophrenia could be secondary to alterations in the striatal dopamine system 188. On the other hand, D1R activation in PFC facilitates the maintenance of long-term potentiation (LTP) of glutamatergic transmission, as well as the induction of long-term depression (LTD) of glutamatergic transmission, indicating its bidirectional modulation of synaptic plasticity in mPFC 189. Computational models predict that an imbalance of D1R/D2R activation in the PFC may underlie the associated cognitive, positive and negative symptoms of SZ 190. Positron emission tomography studies indicate that radioligand binding to D1R is reduced in PFC of SZ patients, and the extent of the reduction correlates directly with the severity of SZ-related negative symptoms 191. Further postmortem studies have confirmed the reduced D1R expression in PFC of SZ patients 192.
In addition to dopamine, serotonin dysfunction in PFC is also implicated in SZ, as 5-HT receptors are the major targets of many atypical antipsychotics used to treat SZ, such as clozapine and olanzapine. A systematic review of literatures reported the increased expression of 5-HT1A and the reduced expression of 5-HT2A receptors in PFC of SZ patients 193. PET studies in SZ patients have similarly shown the reduced 5-HT2A availability in frontal cortex 194. The novel antipsychotic drug lurasidone restores the diminished NMDAR function in PFC pyramidal neurons from the phencyclidine (PCP) model of schizophrenia via antagonizing 5-HT7 receptors 195, which may underlie its effect on reversing cognitive impairment in SZ. The exact mechanisms by which serotonin dysfunction in PFC contributes to SZ pathophysiology remains to be further elucidated. Collectively, the existing evidence suggests a model whereby the disruption of NMDA and GABA transmission in PFC, which is partially driven by aberrant dopamine and serotonin neuromodulation, produces excitation/inhibition imbalance and network dysconnectivity in SZ (Fig. 4A).
Emerging studies have revealed the epigenetic alterations in SZ 196. ATAC-seq analysis of SZ DLPFC shows that genetic regions with high chromatin accessibility are highly enriched in heritable genetic risk factors for SZ 197. In agreement with this, two histone markers associated with transcriptional activation (H3K4me3 and H3K27ac) are enriched at SZ genetic risk variants 198. Broadly disrupted DNA methylation profiles are also reported in SZ PFC 199. Epigenetic dysregulation of 5-HT2A receptor has been demonstrated in human frontal cortex 200. In a rat model of SZ, abnormal epigenetic repression of the NMDAR subunit Grin2b in PFC drives glutamatergic synaptic deficits 201. Upregulation of HDAC1 is found in PFC of human SZ patients and Hdac1 overexpression in PFC is associated with SZ-like phenotypes in mice 202. These findings suggest that epigenetic dysregulation of selective genes may play an important role in SZ.
Depression
Substantial evidence indicates that mPFC dysfunction plays a critical role in the pathology of major depressive disorder (MDD) 203. MDD patients display the reduced glucose metabolism in DLPFC 204, and the significantly reduced mPFC volume spanning across dorsomedial PFC, ventromedial (VM) PFC and ACC 205. While both DLPFC and VMPFC are involved in MDD, they play different roles: damage to VMPFC is associated with low levels of depression, and damage to DLPFC is associated with high levels of depression 206. The PFC has also emerged as a hub for MDD treatment, as repeated transcranial magnetic stimulation (rTMS) therapy targeting the left PFC appears effective in mitigating depression symptoms 207.
MDD is associated with aberrant synaptic transmission in PFC, affecting both GABA and glutamate systems (Fig. 4B). A meta-analysis of GWAS studies identified 102 genetic risk variants in MDD patients, many of which display high expression in frontal cortex and are enriched in biological pathways related to synaptic transmission 208. Diminished expression of several synaptic genes, including CALM2, SYN1, RAB3A, RAB4B and TUBB4, is observed in PFC of MDD patients, along with reduced spine density in layers II/III of DLPFC 209. Protein expression levels of the NMDA subunits NR2A/NR2B and PSD95 are also reduced in PFC of MDD patients 210, and RNA-sequencing analyses of postmortem DLPFC from MDD patients similarly revealed the downregulation of genes related to glutamatergic transmission 211. Moreover, an optogenetic study reveals that activating D1R-, but not D2R-, expressing PFC pyramidal neurons produces rapid and long-lasting antidepressant and anxiolytic responses, and disruption of D1R activity blocks the rapid antidepressant effects of ketamine 212.
In addition to the disrupted glutamatergic transmission, there is also evidence for GABAergic impairment in MDD. Microarray analyses of PFC tissue from MDD patients indicate the downregulation of genes related to GABAergic signaling, including multiple GABAA receptor subunits 213. Additionally, protein levels of GAD67, the primary enzyme responsible for GABA synthesis, are reduced in MDD PFC, and PET imaging studies reveal lower GABA levels in PFC of MDD patients 214. RNA-seq of postmortem DLPFC tissue from individuals who died by suicide also revealed the downregulation of GABAAR γ2 subunit 215. Interestingly, mice carrying heterozygous deficiency of the GABAAR γ2 subunit in forebrain display depression-like behavioral phenotypes 216 and the reduced surface expression of NMDA and AMPA receptor subunits in mPFC that could be restored via ketamine treatment 217, suggesting that GABAergic impairment could affect glutamatergic signaling in MDD PFC. Studies in mice indicate that scopolamine, an effective antidepressant medication acting on muscarinic acetylcholine receptors, enacts its antidepressant effects via targeting somatostatin-expressing GABAergic interneurons in mPFC 218, further implicating the prefrontal cortical GABA system in MDD. The density of PNNs is also reduced in PFC of stressed mice 219 and rats 220, suggesting that PNNs may be disrupted in MDD, similar to SZ.
The NMDAR antagonist ketamine produces rapid antidepressant effects in those with treatment-resistant depression 221, and its therapeutic efficacy appears to be dependent upon its effects on PFC, along with its connecting regions. In rodents, low doses of ketamine have been shown to drive the increased glutamate release 222 and glutamate cycling 223 in PFC. Electrophysiological recordings similarly indicate that ketamine elevates the frequency of excitatory postsynaptic currents (EPSC) in mouse PFC pyramidal neurons 224. In human MDD patients, ketamine administration similarly increases glutamate/glutamine cycling in PFC 225. The paradoxical increase in glutamate activity driven by ketamine initially appeared to be mediated by the loss of NMDA-mediated excitation of GABA interneurons, resulting in disinhibition of pyramidal neurons 170 (Fig. 4B). However, deletion of the NMDAR subunit GluN2B from mPFC pyramidal neurons in vivo produces antidepressant-like effects, while also preventing ketamine’s effects on behavior, EPSC and the induction of mTOR activation in mouse mPFC 224, suggesting that ketamine enacts direct effects on pyramidal neurons via GluN2B antagonism.
The ketamine-induced elevation in PFC glutamate release promotes AMPA receptor activation, which is necessary for ketamine’s antidepressant effects; pre-administration of the AMPAR receptor antagonist NBQX prevents ketamine’s antidepressant-like behavioral effects in mice 226. NBQX treatment also prevents ketamine’s induction of mTOR signaling, suggesting that mTOR activation is mediated by AMPA receptor activation 227. mTOR inhibition is sufficient to prevent the ketamine-dependent induction of synaptogenesis in PFC and antidepressant-like behavioral effects in rats 113. The mTOR activation of BDNF and synaptogenesis in PFC appear to be involved in Ketamine’s therapeutic function, because transgenic mice carrying a mutant form of BDNF that cannot translocate to dendrites do not display ketamine-dependent synaptogenesis nor antidepressant-like behavioral responses 228. Notably, the significantly reduced protein expression level of mTOR and several downstream signaling molecules is reported in postmortem PFC tissue from depressed patients 112. Mice expressing a non-phosphorylatable mutant copy of the signaling molecule eIF4E (eukaryotic initiation factor 4E), which is activated downstream of mTORC1, display depression-like behaviors 229, further implicating the mTOR signaling pathway in MDD. The ketamine metabolite hydroxynorketamine similarly produces antidepressant effects in mice when administered exclusively in the PFC, and this effect is dependent upon the activation of BDNF, and downstream TrkB and mTORC1 signaling 230. Furthermore, direct activation of the mTORC1 pathway via NV-5138 produces antidepressant effects in rats, suggesting that mTORC1 activation alone may be sufficient to alleviate depressive symptoms 231.
Collectively, these studies suggest that in MDD patients, the PFC is characterized by diminished expression of glutamatergic and GABAergic receptors, accompanied by a reduction in dendritic spine density. Ketamine treatment thus enacts its therapeutic effects by transiently promoting glutamate release in PFC to activate AMPA receptors, which in turn activates the downstream mTOR signaling pathway and promotes BDNF expression, leading to synaptogenesis and elevated glutamatergic transmission (Fig. 4B).
In addition to the involvement of PFC synaptic dysregulation in depression, epigenetic changes have been described in PFC of individuals with MDD. Twenty genes are found to be differentially methylated in PFC of MDD patients from a genome-wide methylation analysis, including hypermethylation of Grin2a 232. HDAC inhibitor produces antidepressant-like effects in mice 233. DNA methyltransferases have also been implicated in depression 234. Elevated DNA methylation is observed in PFC of rats subjected to learned helplessness stress, which is reversed by antidepressant imipramine treatment 235. Moreover, systemic administration of DNA methyltransferase inhibitors induces antidepressant-like effects in rats 236. These findings suggest that pharmacological targeting of epigenetic enzymes may be a novel treatment strategy for depressive symptoms.
Alzheimer’s Disease
PFC is one of the first regions to be affected in clinical AD, which may explain the early occurrence of the impairment of PFC-mediated cognitive processes, such as working memory and attention. Aging is the leading risk factor for developing AD, so many studies have focused on revealing neurobiological changes that affect synaptic integrity with aging and why aged brain is vulnerable to AD. In aged PFC, the cAMP/PKA pathway is disinhibited, and PKA inhibition provides an effective approach for treating age-related cognitive decline 237. The marked loss of PFC persistent firing in aged monkeys is also partially restored by inhibiting cAMP signaling or blocking HCN or KCNQ channels 238. Age-related increase in PKA phosphorylation of tau at serine 214 in monkey PFC is thought to confer risk for degeneration of glutamatergic synapses 239. From aging monkey studies, it is found that PFC is particularly vulnerable to calcium dysregulation and tau phosphorylation 240. The loss of thin spines in monkey PFC, which are particularly plastic and linked to learning, is selectively correlated with age-related cognitive impairment 241. Aβ infused into the monkey brain induces accelerated cortical aging by triggering the specific loss of dendritic thin spines in the PFC 242.
Cholinergic system is crucial for cognitive processes, and deficient acetylcholine (ACh) function has been implicated in AD. A major AD therapy is acetylcholinesterase (AChE) inhibitors, which act to enhance cholinergic function by prolonging the action of endogenously released ACh. AChE inhibitors produce a strong and persistent reduction of synaptic NMDAR responses in PFC pyramidal neurons via nicotinic ACh receptors 243. Activation of muscarinic or nicotinic ACh receptors in PFC pyramidal neurons also enhances inhibitory synaptic transmission through PKC-dependent mechanisms 244,245. The cholinergic regulation of NMDA and GABA function is impaired in transgenic mice overexpressing mutant β-amyloid precursor protein (APP) 243,245,246. Aβ-induced loss of RACK1 (an anchoring protein for activated PKC) distribution in the membrane fraction of cortical neurons is found to underlie the impairment of muscarinic regulation of PKC 247. In aged monkeys with naturally occurring cholinergic depletion, low-dose muscarinic M1R stimulation enhances delay firing in dorsolateral PFC neurons and improves working memory via a mechanism involving KCNQ channels 248. These studies suggest that restoring cholinergic regulation of PFC synaptic activity and neuronal excitability might help to treat AD-associated cognitive disorders.
The brain noradrenergic system is also critical for cognition and noradrengergic dysfunction is a critical early step in AD progression 249. A recent study has found that β-amyloid oligomers bind to an allosteric site on α2-ARs to redirect norepinephrine signaling to activate the pathogenic GSK3β/tau cascade 250. Guanfacine, a specific α2-AR antagonist, has been used to treat cognitive disorders linked to PFC dysfunction 251.
Systematic search for global gene expression changes in PFC during the course of AD has found a temporally orchestrated increase of genes involved in synaptic activity during the very early pre-symptomatic stage, probably representing a coping mechanism against increased β-amyloid levels, and the decreased expression of these synaptic genes in later Braak stages, suggesting a reduction in synaptic activity that coincides with AD neuropathology and cognitive impairment 252. Studies of postmortem human brains have found the significant association between cognitive decline and the loss of synaptic proteins in PFC, such as SNAP25, SNAP47, SV2C, SYT2 and GRIA3 253,254, suggesting that these PFC synaptic markers are predictive molecular fingerprint for neurodegenerative diseases, and understanding the basis of synaptic impairment is the most effective route for early intervention and prevention of cognitive decline.
Both AD patients and symptomatic AD mouse models show impaired synaptic plasticity in PFC, which is related to the impaired NMDAR function 255. The diminished transcription of NMDA and AMPA receptors in late-stage AD mouse models has been linked to the EHMT1/2-mediated repressive histone methylation at their promoters 256. Treatment of AD models with specific EHMT1/2 inhibitors reverses the increased H3K9me2 enrichment at genes involved in neuronal signaling (e.g. Grin2b encoding NR2B subunit and Gria2 encoding GluR2 subunit), restores glutamate receptor expression and excitatory synaptic function in PFC, and rescues the impaired recognition memory, working memory, and spatial memory 256 (Fig. 5).
Figure 5.
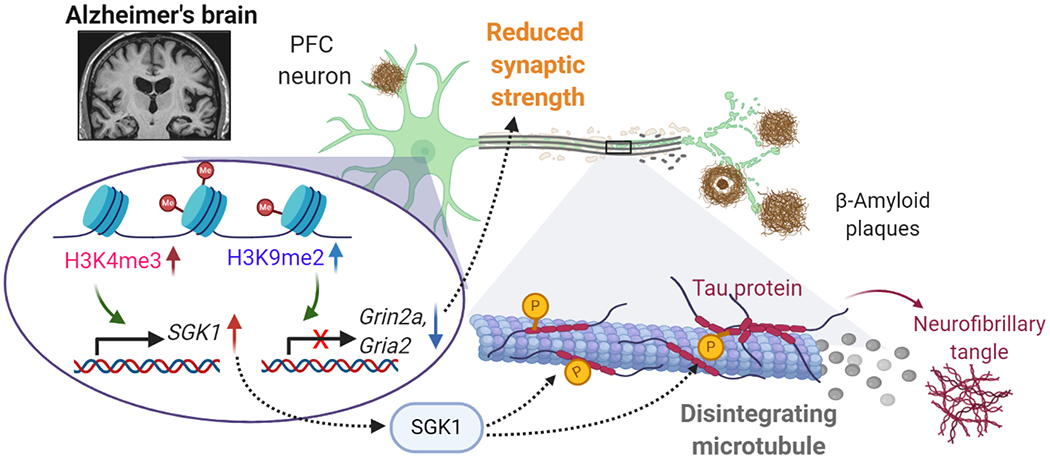
Schematic diagram illustrating the potential epigenetic mechanisms underlying gene dysregulation and synaptic deficits in PFC of AD. The elevation of repressive histone mark H3K9me2 in AD leads to the downregulation of genes involved in synaptic transmission, such as AMPAR subunit Gria2 and NMDAR subunit Grin2a, resulting in the reduction of synaptic strength256. On the other hand, the elevation of permissive histone mark H3K4me3 in AD leads to the upregulation of genes involved in cell stress, such as serum- and glucocorticoid-inducible kinase 1 (SGK1), resulting in hyperphosphorylation of tau, disintegration of microtubules and disruption of vital protein transport260. Targeting histone methytransferases to normalize histone modification or key target genes is the potential new therapeutic strategy for treating synaptic and cognitive deficits in AD.
In addition to the downregulation of genes involved in synaptic transmission and synapse organization, there is also prominent upregulation of genes involved in cell stress, immune response activation and cell death in PFC of AD patients 257–259. A recent studie has found that H3K4 trimethylation (H3K4me3), a histone mark for gene activation, is significantly elevated in PFC of human AD postmortem tissues and AD mouse models, and inhibiting H3K4-specific methyltransferases, including MLL1-4 (also named as KMT2A-D) and SETD1a/b, leads to the substantial recovery of glutamatergic synaptic function in PFC pyramidal neurons, and the significant improvement of memory-related behaviors in mutant Tau transgenic mice 260. One of the top-ranking genes elevated by the abnormally high H3K4me3 in PFC of AD patients and mouse models is SGK1, which encodes the stress-responsive kinase SGK1. A short treatment with the specific SGK1 inhibitor results in the significant reduction of hyper-phosphorylated tau (a pathological hallmark of AD) and the recovery of synaptic receptor expression and glutamatergic transmission in PFC, as well as the restoration of recognition and spatial memories, in mutant Tau transgenic mice 260 (Fig. 5). These results suggest that targeting histone methylation enzymes may represent a novel therapeutic avenue for normalizing the aberrant epigenetic regulation of gene transcription in PFC that underlies cognitive deficits in AD.
Concluding Remarks
Overwhelming evidence has indicated that PFC is a uniquely important brain region for the highest level of excecutive function, which drives self-regulatory and goal-directed behaviors 261–263. Dysregulation of PFC synaptic transmission directly contributes to the impairment of social, affective, motivational, and cognitive function associated with brain disorders including autism, schizophrenia, depression and Alzheimer’s disease. Studies in animal models have revealed critical neural circuits, biological pathways, and molecular players that converge to control the excitatory and inhibitory signals in PFC networks. Novel epigenetic mechanism-based strategies have been proposed as potential avenues of therapeutic intervention for mental diseases linked to PFC dysfunction.
There are many open questions that await to be further explored in future studies, and a few examples are listed as follows:
What makes PFC so special in terms of the organizational and cellular function? Circuit mapping with modern technologies have revealed the links of specific pathways and cell types to particular behavioral aspects. How specific and robust are such links? Is it likely that complex cognitive and emotional behaviors usually need to recruit multiple circuits and neuronal populations? What are the differential roles of PFC and its connecting regions in the control of these behaviors?
How does PFC integrate the influences of monoaminergic systems, stress hormones and neuropeptides in different conditions? What effects of these neuromodulators are unique to PFC synaptic function? How much does the aberrant modulation of PFC contribute to the pathophysiology of neurodevelopmental, neuropsychiatric and neurodegenerative diseases?
What are the convergent and divergent factors causing PFC synaptic dysfunction in autism, schizophrenia, depression, and Alzheimer’s disease? Is PFC synaptic dysregulation directly linked to behavioral abnormalities in these illnesses?
How are the synaptic, epigenetic and transcriptomic alterations mechanistically connected in each of PFC-involved brain disorder? What is the translational potential of the epigenetics-based treatment strategies found in preclinical models for human patients with these disorders?
Acklowdgements:
We are grateful to former and current members in Dr. Yan’s laboratory for their contributions to the original findings and NIH grants (MH108842, MH112237, DA037618, AG056060, AG064656 to Z.Y.) for their support of some of the work reviewed here.
Footnotes
Conflict of Interests:
The authors declare no conflict of interest.
References:
- 1.Bourgeois JP, Goldman-Rakic PS & Rakic P Synaptogenesis in the prefrontal cortex of rhesus monkeys. Cerebral cortex (New York, N.Y. : 1991) 4, 78–96 (1994). [DOI] [PubMed] [Google Scholar]
- 2.Diamond A Executive functions. Annu Rev Psychol 64, 135–168 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stokes MG ‘Activity-silent’ working memory in prefrontal cortex: a dynamic coding framework. Trends Cogn Sci 19, 394–405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fuster JM & Alexander GE Neuron activity related to short-term memory. Science 173, 652–654 (1971). [DOI] [PubMed] [Google Scholar]
- 5.Goldman-Rakic PS Cellular basis of working memory. Neuron 14, 477–485 (1995). [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, et al. Heterogeneity in the pyramidal network of the medial prefrontal cortex. Nature neuroscience 9, 534–542 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Compte A, Brunel N, Goldman-Rakic PS & Wang XJ Synaptic mechanisms and network dynamics underlying spatial working memory in a cortical network model. Cerebral cortex (New York, N.Y. : 1991) 10, 910–923 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Wang M, et al. NMDA receptors subserve persistent neuronal firing during working memory in dorsolateral prefrontal cortex. Neuron 77, 736–749 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCarthy G, et al. Functional magnetic resonance imaging of human prefrontal cortex activation during a spatial working memory task. Proceedings of the National Academy of Sciences of the United States of America 91, 8690–8694 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pardo JV, Fox PT & Raichle ME Localization of a human system for sustained attention by positron emission tomography. Nature 349, 61–64 (1991). [DOI] [PubMed] [Google Scholar]
- 11.Uylings HB, Groenewegen HJ & Kolb B Do rats have a prefrontal cortex? Behavioural brain research 146, 3–17 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Ongür D & Price JL The organization of networks within the orbital and medial prefrontal cortex of rats, monkeys and humans. Cerebral cortex (New York, N.Y. : 1991) 10, 206–219 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Erlich JC, Bialek M & Brody CD A cortical substrate for memory-guided orienting in the rat. Neuron 72, 330–343 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baeg EH, et al. Dynamics of population code for working memory in the prefrontal cortex. Neuron 40, 177–188 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Gabbott PL, Warner TA, Jays PR, Salway P & Busby SJ Prefrontal cortex in the rat: projections to subcortical autonomic, motor, and limbic centers. The Journal of comparative neurology 492, 145–177 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Otis JM, et al. Prefrontal cortex output circuits guide reward seeking through divergent cue encoding. Nature 543, 103–107 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim CK, et al. Molecular and Circuit-Dynamical Identification of Top-Down Neural Mechanisms for Restraint of Reward Seeking. Cell 170, 1013–1027.e1014 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Narayanan NS & Laubach M Top-down control of motor cortex ensembles by dorsomedial prefrontal cortex. Neuron 52, 921–931 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Franklin TB, et al. Prefrontal cortical control of a brainstem social behavior circuit. Nature neuroscience 20, 260–270 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hultman R, et al. Brain-wide Electrical Spatiotemporal Dynamics Encode Depression Vulnerability. Cell 173, 166–180.e114 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spellman T, et al. Hippocampal-prefrontal input supports spatial encoding in working memory. Nature 522, 309–314 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phillips ML, Robinson HA & Pozzo-Miller L Ventral hippocampal projections to the medial prefrontal cortex regulate social memory. Elife 8(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Collins DP, Anastasiades PG, Marlin JJ & Carter AG Reciprocal Circuits Linking the Prefrontal Cortex with Dorsal and Ventral Thalamic Nuclei. Neuron 98, 366–379.e364 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmitt LI, et al. Thalamic amplification of cortical connectivity sustains attentional control. Nature 545, 219–223 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wimmer RD, et al. Thalamic control of sensory selection in divided attention. Nature 526, 705–709 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burgos-Robles A, et al. Amygdala inputs to prefrontal cortex guide behavior amid conflicting cues of reward and punishment. Nature neuroscience 20, 824–835 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krimer LS & Goldman-Rakic PS Prefrontal microcircuits: membrane properties and excitatory input of local, medium, and wide arbor interneurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 21, 3788–3796 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao WJ, Wang Y & Goldman-Rakic PS Dopamine modulation of perisomatic and peridendritic inhibition in prefrontal cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 23, 1622–1630 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kvitsiani D, et al. Distinct behavioural and network correlates of two interneuron types in prefrontal cortex. Nature 498, 363–366 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pi HJ, et al. Cortical interneurons that specialize in disinhibitory control. Nature 503, 521–524 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Obermayer J, et al. Prefrontal cortical ChAT-VIP interneurons provide local excitation by cholinergic synaptic transmission and control attention. Nature communications 10, 5280 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murugan M, et al. Combined Social and Spatial Coding in a Descending Projection from the Prefrontal Cortex. Cell 171, 1663–1677.e1616 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun Q, et al. Ventral Hippocampal-Prefrontal Interaction Affects Social Behavior via Parvalbumin Positive Neurons in the Medial Prefrontal Cortex. iScience 23, 100894 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferguson BR & Gao WJ Thalamic Control of Cognition and Social Behavior Via Regulation of Gamma-Aminobutyric Acidergic Signaling and Excitation/Inhibition Balance in the Medial Prefrontal Cortex. Biol Psychiatry 83, 657–669 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McGarry LM & Carter AG Inhibitory Gating of Basolateral Amygdala Inputs to the Prefrontal Cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 36, 9391–9406 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dilgen J, Tejeda HA & O’Donnell P Amygdala inputs drive feedforward inhibition in the medial prefrontal cortex. J Neurophysiol 110, 221–229 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pinto L & Dan Y Cell-Type-Specific Activity in Prefrontal Cortex during Goal-Directed Behavior. Neuron 87, 437–450 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scheggia D, et al. Somatostatin interneurons in the prefrontal cortex control affective state discrimination in mice. Nature neuroscience 23, 47–60 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Abbas AI, et al. Somatostatin Interneurons Facilitate Hippocampal-Prefrontal Synchrony and Prefrontal Spatial Encoding. Neuron 100, 926–939.e923 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kamigaki T & Dan Y Delay activity of specific prefrontal interneuron subtypes modulates memory-guided behavior. Nature neuroscience 20, 854–863 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim D, et al. Distinct Roles of Parvalbumin- and Somatostatin-Expressing Interneurons in Working Memory. Neuron 92, 902–915 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Xu H, et al. A Disinhibitory Microcircuit Mediates Conditioned Social Fear in the Prefrontal Cortex. Neuron 102, 668–682.e665 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Marek R, et al. Hippocampus-driven feed-forward inhibition of the prefrontal cortex mediates relapse of extinguished fear. Nature neuroscience 21, 384–392 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee AT, et al. VIP Interneurons Contribute to Avoidance Behavior by Regulating Information Flow across Hippocampal-Prefrontal Networks. Neuron 102, 1223–1234.e1224 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robbins TW & Arnsten AF The neuropsychopharmacology of fronto-executive function: monoaminergic modulation. Annual review of neuroscience 32, 267–287 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arnsten AF Catecholamine influences on dorsolateral prefrontal cortical networks. Biol Psychiatry 69, e89–99 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Berridge CW, et al. Methylphenidate preferentially increases catecholamine neurotransmission within the prefrontal cortex at low doses that enhance cognitive function. Biol Psychiatry 60, 1111–1120 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Cheng J, et al. Methylphenidate exerts dose-dependent effects on glutamate receptors and behaviors. Biol Psychiatry 76, 953–962 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smiley JF & Goldman-Rakic PS Heterogeneous targets of dopamine synapses in monkey prefrontal cortex demonstrated by serial section electron microscopy: a laminar analysis using the silver-enhanced diaminobenzidine sulfide (SEDS) immunolabeling technique. Cerebral cortex (New York, N.Y. : 1991) 3, 223–238 (1993). [DOI] [PubMed] [Google Scholar]
- 50.Gao WJ, Krimer LS & Goldman-Rakic PS Presynaptic regulation of recurrent excitation by D1 receptors in prefrontal circuits. Proceedings of the National Academy of Sciences of the United States of America 98, 295–300 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paspalas CD & Goldman-Rakic PS Presynaptic D1 dopamine receptors in primate prefrontal cortex: target-specific expression in the glutamatergic synapse. The Journal of neuroscience : the official journal of the Society for Neuroscience 25, 1260–1267 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao WJ & Goldman-Rakic PS Selective modulation of excitatory and inhibitory microcircuits by dopamine. Proceedings of the National Academy of Sciences of the United States of America 100, 2836–2841 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y & Goldman-Rakic PS D2 receptor regulation of synaptic burst firing in prefrontal cortical pyramidal neurons. Proceedings of the National Academy of Sciences of the United States of America 101, 5093–5098 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gamo NJ, et al. Stress Impairs Prefrontal Cortical Function via D1 Dopamine Receptor Interactions With Hyperpolarization-Activated Cyclic Nucleotide-Gated Channels. Biol Psychiatry 78, 860–870 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhong P, Qin L & Yan Z Dopamine Differentially Regulates Response Dynamics of Prefrontal Cortical Principal Neurons and Interneurons to Optogenetic Stimulation of Inputs from Ventral Tegmental Area. Cerebral cortex (New York, N.Y. : 1991) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seeman P, Guan HC & Van Tol HH Dopamine D4 receptors elevated in schizophrenia. Nature 365, 441–445 (1993). [DOI] [PubMed] [Google Scholar]
- 57.LaHoste GJ, et al. Dopamine D4 receptor gene polymorphism is associated with attention deficit hyperactivity disorder. Mol Psychiatry 1, 121–124 (1996). [PubMed] [Google Scholar]
- 58.Gu Z, Jiang Q, Yuen EY & Yan Z Activation of dopamine D4 receptors induces synaptic translocation of Ca2+/calmodulin-dependent protein kinase II in cultured prefrontal cortical neurons. Molecular pharmacology 69, 813–822 (2006). [DOI] [PubMed] [Google Scholar]
- 59.Gu Z & Yan Z Bidirectional regulation of Ca2+/calmodulin-dependent protein kinase II activity by dopamine D4 receptors in prefrontal cortex. Molecular pharmacology 66, 948–955 (2004). [DOI] [PubMed] [Google Scholar]
- 60.Wang X, Zhong P, Gu Z & Yan Z Regulation of NMDA receptors by dopamine D4 signaling in prefrontal cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 23, 9852–9861 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang X, et al. Aberrant regulation of NMDA receptors by dopamine D4 signaling in rats after phencyclidine exposure. Mol Cell Neurosci 31, 15–25 (2006). [DOI] [PubMed] [Google Scholar]
- 62.Yuen EY, Zhong P & Yan Z Homeostatic regulation of glutamatergic transmission by dopamine D4 receptors. Proceedings of the National Academy of Sciences of the United States of America 107, 22308–22313 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yuen EY & Yan Z Cellular mechanisms for dopamine D4 receptor-induced homeostatic regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. The Journal of biological chemistry 286, 24957–24965 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yuen EY & Yan Z Dopamine D4 receptors regulate AMPA receptor trafficking and glutamatergic transmission in GABAergic interneurons of prefrontal cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 29, 550–562 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhong P & Yan Z Distinct Physiological Effects of Dopamine D4 Receptors on Prefrontal Cortical Pyramidal Neurons and Fast-Spiking Interneurons. Cerebral cortex (New York, N.Y. : 1991) 26, 180–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tan T, et al. Stress Exposure in Dopamine D4 Receptor Knockout Mice Induces Schizophrenia-Like Behaviors via Disruption of GABAergic Transmission. Schizophr Bull 45, 1012–1023 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ebstein RP, et al. Dopamine D4 receptor (D4DR) exon III polymorphism associated with the human personality trait of Novelty Seeking. Nat Genet 12, 78–80 (1996). [DOI] [PubMed] [Google Scholar]
- 68.Qin L, et al. The ADHD-linked human dopamine D4 receptor variant D4.7 induces over-suppression of NMDA receptor function in prefrontal cortex. Neurobiology of disease 95, 194–203 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhong P, Liu W & Yan Z Aberrant regulation of synchronous network activity by the attention-deficit/hyperactivity disorder-associated human dopamine D4 receptor variant D4.7 in the prefrontal cortex. The Journal of physiology 594, 135–147 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Datta D, et al. Noradrenergic α1-Adrenoceptor Actions in the Primate Dorsolateral Prefrontal Cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 39, 2722–2734 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang M, et al. Alpha2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell 129, 397–410 (2007). [DOI] [PubMed] [Google Scholar]
- 72.Paspalas CD, Wang M & Arnsten AF Constellation of HCN channels and cAMP regulating proteins in dendritic spines of the primate prefrontal cortex: potential substrate for working memory deficits in schizophrenia. Cerebral cortex (New York, N.Y. : 1991) 23, 1643–1654 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu W, et al. Adrenergic modulation of NMDA receptors in prefrontal cortex is differentially regulated by RGS proteins and spinophilin. Proceedings of the National Academy of Sciences of the United States of America 103, 18338–18343 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.De Vries L, Zheng B, Fischer T, Elenko E & Farquhar MG The regulator of G protein signaling family. Annu Rev Pharmacol Toxicol 40, 235–271 (2000). [DOI] [PubMed] [Google Scholar]
- 75.Chowdari KV, et al. Association and linkage analyses of RGS4 polymorphisms in schizophrenia. Hum Mol Genet 11, 1373–1380 (2002). [DOI] [PubMed] [Google Scholar]
- 76.Williams NM, et al. Support for RGS4 as a susceptibility gene for schizophrenia. Biol Psychiatry 55, 192–195 (2004). [DOI] [PubMed] [Google Scholar]
- 77.Davidson RJ, Putnam KM & Larson CL Dysfunction in the neural circuitry of emotion regulation--a possible prelude to violence. Science 289, 591–594 (2000). [DOI] [PubMed] [Google Scholar]
- 78.Leonardo ED & Hen R Genetics of affective and anxiety disorders. Annu Rev Psychol 57, 117–137 (2006). [DOI] [PubMed] [Google Scholar]
- 79.Hoyer D, Hannon JP & Martin GR Molecular, pharmacological and functional diversity of 5-HT receptors. Pharmacol Biochem Behav 71, 533–554 (2002). [DOI] [PubMed] [Google Scholar]
- 80.Cai X, Flores-Hernandez J, Feng J & Yan Z Activity-dependent bidirectional regulation of GABA(A) receptor channels by the 5-HT(4) receptor-mediated signalling in rat prefrontal cortical pyramidal neurons. The Journal of physiology 540, 743–759 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yan Z Regulation of GABAergic inhibition by serotonin signaling in prefrontal cortex: molecular mechanisms and functional implications. Molecular neurobiology 26, 203–216 (2002). [DOI] [PubMed] [Google Scholar]
- 82.Yuen EY, Jiang Q, Chen P, Feng J & Yan Z Activation of 5-HT2A/C receptors counteracts 5-HT1A regulation of n-methyl-D-aspartate receptor channels in pyramidal neurons of prefrontal cortex. The Journal of biological chemistry 283, 17194–17204 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yuen EY, et al. Serotonin 5-HT1A receptors regulate NMDA receptor channels through a microtubule-dependent mechanism. The Journal of neuroscience : the official journal of the Society for Neuroscience 25, 5488–5501 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yuen EY, et al. Synergistic regulation of glutamatergic transmission by serotonin and norepinephrine reuptake inhibitors in prefrontal cortical neurons. The Journal of biological chemistry 289, 25177–25185 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhong P, Liu W, Gu Z & Yan Z Serotonin facilitates long-term depression induction in prefrontal cortex via p38 MAPK/Rab5-mediated enhancement of AMPA receptor internalization. The Journal of physiology 586, 4465–4479 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhong P & Yan Z Chronic antidepressant treatment alters serotonergic regulation of GABA transmission in prefrontal cortical pyramidal neurons. Neuroscience 129, 65–73 (2004). [DOI] [PubMed] [Google Scholar]
- 87.Feng J, Cai X, Zhao J & Yan Z Serotonin receptors modulate GABA(A) receptor channels through activation of anchored protein kinase C in prefrontal cortical neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 21, 6502–6511 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gu Z, Jiang Q & Yan Z RGS4 modulates serotonin signaling in prefrontal cortex and links to serotonin dysfunction in a rat model of schizophrenia. Molecular pharmacology 71, 1030–1039 (2007). [DOI] [PubMed] [Google Scholar]
- 89.Tan H, Zhong P & Yan Z Corticotropin-releasing factor and acute stress prolongs serotonergic regulation of GABA transmission in prefrontal cortical pyramidal neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 24, 5000–5008 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Komlósi G, et al. Fluoxetine (prozac) and serotonin act on excitatory synaptic transmission to suppress single layer 2/3 pyramidal neuron-triggered cell assemblies in the human prefrontal cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 32, 16369–16378 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barre A, et al. Presynaptic serotonin 2A receptors modulate thalamocortical plasticity and associative learning. Proceedings of the National Academy of Sciences of the United States of America 113, E1382–1391 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Athilingam JC, Ben-Shalom R, Keeshen CM, Sohal VS & Bender KJ Serotonin enhances excitability and gamma frequency temporal integration in mouse prefrontal fast-spiking interneurons. Elife 6(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McKlveen JM, et al. Role of prefrontal cortex glucocorticoid receptors in stress and emotion. Biol Psychiatry 74, 672–679 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang M, Perova Z, Arenkiel BR & Li B Synaptic modifications in the medial prefrontal cortex in susceptibility and resilience to stress. The Journal of neuroscience : the official journal of the Society for Neuroscience 34, 7485–7492 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Treccani G, et al. Stress and corticosterone increase the readily releasable pool of glutamate vesicles in synaptic terminals of prefrontal and frontal cortex. Mol Psychiatry 19, 433–443 (2014). [DOI] [PubMed] [Google Scholar]
- 96.Yuen EY, et al. Acute stress enhances glutamatergic transmission in prefrontal cortex and facilitates working memory. Proceedings of the National Academy of Sciences of the United States of America 106, 14075–14079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee JB, et al. Histone deacetylase 6 gates the synaptic action of acute stress in prefrontal cortex. The Journal of physiology 590, 1535–1546 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yuen EY, et al. Mechanisms for acute stress-induced enhancement of glutamatergic transmission and working memory. Mol Psychiatry 16, 156–170 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu W, Yuen EY & Yan Z The stress hormone corticosterone increases synaptic alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors via serum- and glucocorticoid-inducible kinase (SGK) regulation of the GDI-Rab4 complex. The Journal of biological chemistry 285, 6101–6108 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Licznerski P, et al. Decreased SGK1 Expression and Function Contributes to Behavioral Deficits Induced by Traumatic Stress. PLoS Biol 13, e1002282 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Conboy L & Sandi C Stress at learning facilitates memory formation by regulating AMPA receptor trafficking through a glucocorticoid action. Neuropsychopharmacology 35, 674–685 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Musazzi L, Treccani G & Popoli M Functional and structural remodeling of glutamate synapses in prefrontal and frontal cortex induced by behavioral stress. Front Psychiatry 6, 60 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nava N, et al. Chronic desipramine prevents acute stress-induced reorganization of medial prefrontal cortex architecture by blocking glutamate vesicle accumulation and excitatory synapse increase. Int J Neuropsychopharmacol 18(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nava N, et al. The expression of plasticity-related genes in an acute model of stress is modulated by chronic desipramine in a time-dependent manner within medial prefrontal cortex. Eur Neuropsychopharmacol 27, 19–28 (2017). [DOI] [PubMed] [Google Scholar]
- 105.Radley JJ, et al. Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cerebral cortex (New York, N.Y. : 1991) 16, 313–320 (2006). [DOI] [PubMed] [Google Scholar]
- 106.Radley JJ, et al. Reversibility of apical dendritic retraction in the rat medial prefrontal cortex following repeated stress. Exp Neurol 196, 199–203 (2005). [DOI] [PubMed] [Google Scholar]
- 107.Cerqueira JJ, Mailliet F, Almeida OF, Jay TM & Sousa N The prefrontal cortex as a key target of the maladaptive response to stress. The Journal of neuroscience : the official journal of the Society for Neuroscience 27, 2781–2787 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Urban KR & Valentino RJ Age- and Sex-Dependent Impact of Repeated Social Stress on Intrinsic and Synaptic Excitability of the Rat Prefrontal Cortex. Cerebral cortex (New York, N.Y. : 1991) 27, 244–253 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gourley SL, Kedves AT, Olausson P & Taylor JR A history of corticosterone exposure regulates fear extinction and cortical NR2B, GluR2/3, and BDNF. Neuropsychopharmacology 34, 707–716 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yuen EY, et al. Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron 73, 962–977 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wei J, et al. Histone Modification of Nedd4 Ubiquitin Ligase Controls the Loss of AMPA Receptors and Cognitive Impairment Induced by Repeated Stress. The Journal of neuroscience : the official journal of the Society for Neuroscience 36, 2119–2130 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jernigan CS, et al. The mTOR signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Progress in neuro-psychopharmacology & biological psychiatry 35, 1774–1779 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li N, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329, 959–964 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ota KT, et al. REDD1 is essential for stress-induced synaptic loss and depressive behavior. Nat Med 20, 531–535 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jiang C, et al. VGF function in depression and antidepressant efficacy. Mol Psychiatry 23, 1632–1642 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jiang C, et al. VGF and its C-terminal peptide TLQP-62 in ventromedial prefrontal cortex regulate depression-related behaviors and the response to ketamine. Neuropsychopharmacology 44, 971–981 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vollmer LL, et al. Neuropeptide Y Impairs Retrieval of Extinguished Fear and Modulates Excitability of Neurons in the Infralimbic Prefrontal Cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 36, 1306–1315 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nakajima M, Görlich A & Heintz N Oxytocin modulates female sociosexual behavior through a specific class of prefrontal cortical interneurons. Cell 159, 295–305 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li K, Nakajima M, Ibañez-Tallon I & Heintz N A Cortical Circuit for Sexually Dimorphic Oxytocin-Dependent Anxiety Behaviors. Cell 167, 60–72.e11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gandal MJ, et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 359, 693–697 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Price RB & Duman R Neuroplasticity in cognitive and psychological mechanisms of depression: an integrative model. Mol Psychiatry 25, 530–543 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stoner R, et al. Patches of disorganization in the neocortex of children with autism. N Engl J Med 370, 1209–1219 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Voineagu I, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bonaglia MC, et al. Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am J Hum Genet 69, 261–268 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Betancur C & Buxbaum JD SHANK3 haploinsufficiency: a “common” but underdiagnosed highly penetrant monogenic cause of autism spectrum disorders. Mol Autism 4, 17 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pagani M, et al. Deletion of Autism Risk Gene Shank3 Disrupts Prefrontal Connectivity. The Journal of neuroscience : the official journal of the Society for Neuroscience 39, 5299–5310 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Duffney LJ, et al. Autism-like Deficits in Shank3-Deficient Mice Are Rescued by Targeting Actin Regulators. Cell Rep 11, 1400–1413 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yan Z, Kim E, Datta D, Lewis DA & Soderling SH Synaptic Actin Dysregulation, a Convergent Mechanism of Mental Disorders? The Journal of neuroscience : the official journal of the Society for Neuroscience 36, 11411–11417 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Qin L, Ma K & Yan Z Chemogenetic Activation of Prefrontal Cortex in Shank3-Deficient Mice Ameliorates Social Deficits, NMDAR Hypofunction, and Sgk2 Downregulation. iScience 17, 24–35 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Harony-Nicolas H, et al. Oxytocin improves behavioral and electrophysiological deficits in a novel Shank3-deficient rat. Elife 6(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Weiss LA, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med 358, 667–675 (2008). [DOI] [PubMed] [Google Scholar]
- 132.Rein B & Yan Z 16p11.2 Copy Number Variations and Neurodevelopmental Disorders. Trends Neurosci 43, 886–901 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang W, et al. Chemogenetic Activation of Prefrontal Cortex Rescues Synaptic and Behavioral Deficits in a Mouse Model of 16p11.2 Deletion Syndrome. The Journal of neuroscience : the official journal of the Society for Neuroscience 38, 5939–5948 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rein B, et al. Reversal of synaptic and behavioral deficits in a 16p11.2 duplication mouse model via restoration of the GABA synapse regulator Npas4. Mol Psychiatry (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bloodgood BL, Sharma N, Browne HA, Trepman AZ & Greenberg ME The activity-dependent transcription factor NPAS4 regulates domain-specific inhibition. Nature 503, 121–125 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lin Y, et al. Activity-dependent regulation of inhibitory synapse development by Npas4. Nature 455, 1198–1204 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lee E, Lee J & Kim E Excitation/Inhibition Imbalance in Animal Models of Autism Spectrum Disorders. Biol Psychiatry 81, 838–847 (2017). [DOI] [PubMed] [Google Scholar]
- 138.Spratt PWE, et al. The Autism-Associated Gene Scn2a Contributes to Dendritic Excitability and Synaptic Function in the Prefrontal Cortex. Neuron 103, 673–685.e675 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lazaro MT, et al. Reduced Prefrontal Synaptic Connectivity and Disturbed Oscillatory Population Dynamics in the CNTNAP2 Model of Autism. Cell Rep 27, 2567–2578.e2566 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Satterstrom FK, et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 180, 568–584.e523 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.De Rubeis S, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Coe BP, et al. Neurodevelopmental disease genes implicated by de novo mutation and copy number variation morbidity. Nat Genet 51, 106–116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Shulha HP, et al. Epigenetic Signatures of Autism: Trimethylated H3K4 Landscapes in Prefrontal Neurons. Archives of General Psychiatry 69, 314–324 (2012). [DOI] [PubMed] [Google Scholar]
- 144.Ma K, et al. Histone deacetylase inhibitor MS-275 restores social and synaptic function in a Shank3-deficient mouse model of autism. Neuropsychopharmacology 43, 1779–1788 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Qin L, et al. Social deficits in Shank3-deficient mouse models of autism are rescued by histone deacetylase (HDAC) inhibition. Nature neuroscience 21, 564–575 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wang ZJ, et al. Amelioration of autism-like social deficits by targeting histone methyltransferases EHMT1/2 in Shank3-deficient mice. Mol Psychiatry (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rapanelli M, et al. Behavioral, circuitry, and molecular aberrations by region-specific deficiency of the high-risk autism gene Cul3. Mol Psychiatry (2019). [DOI] [PubMed] [Google Scholar]
- 148.Sun W, et al. Histone Acetylome-wide Association Study of Autism Spectrum Disorder. Cell 167, 1385–1397.e1311 (2016). [DOI] [PubMed] [Google Scholar]
- 149.Genschik P, Sumara I & Lechner E The emerging family of CULLIN3-RING ubiquitin ligases (CRL3s): cellular functions and disease implications. EMBO J 32, 2307–2320 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.American Psychiatric Asociation. Diagnostic and Statistical Manual of Mental Disorders: DSM-5. 5th ed., (Arlington: American Psychiatric Association, 2013). [Google Scholar]
- 151.Selemon LD & Zecevic N Schizophrenia: a tale of two critical periods for prefrontal cortical development. Translational psychiatry 5, e623 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sanfilipo M, et al. Volumetric measure of the frontal and temporal lobe regions in schizophrenia: relationship to negative symptoms. Arch Gen Psychiatry 57, 471–480 (2000). [DOI] [PubMed] [Google Scholar]
- 153.Goldstein JM, et al. Cortical abnormalities in schizophrenia identified by structural magnetic resonance imaging. Arch Gen Psychiatry 56, 537–547 (1999). [DOI] [PubMed] [Google Scholar]
- 154.Feinberg I Cortical pruning and the development of schizophrenia. Schizophr Bull 16, 567–570 (1990). [DOI] [PubMed] [Google Scholar]
- 155.Lewis DA Development of the prefrontal cortex during adolescence: insights into vulnerable neural circuits in schizophrenia. Neuropsychopharmacology 16, 385–398 (1997). [DOI] [PubMed] [Google Scholar]
- 156.Howes OD & McCutcheon R Inflammation and the neural diathesis-stress hypothesis of schizophrenia: a reconceptualization. Translational psychiatry 7, e1024 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Glausier JR & Lewis DA Dendritic spine pathology in schizophrenia. Neuroscience 251, 90–107 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mauney SA, et al. Developmental pattern of perineuronal nets in the human prefrontal cortex and their deficit in schizophrenia. Biol Psychiatry 74, 427–435 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Goldman-Rakic PS & Selemon LD Functional and anatomical aspects of prefrontal pathology in schizophrenia. Schizophr Bull 23, 437–458 (1997). [DOI] [PubMed] [Google Scholar]
- 160.Fornito A, Yücel M, Dean B, Wood SJ & Pantelis C Anatomical abnormalities of the anterior cingulate cortex in schizophrenia: bridging the gap between neuroimaging and neuropathology. Schizophr Bull 35, 973–993 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Schizophrenia Working Group of the Psychiatric Genomics, C. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gulsuner S, et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 154, 518–529 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Jaffe AE, et al. Developmental and genetic regulation of the human cortex transcriptome illuminate schizophrenia pathogenesis. Nature neuroscience 21, 1117–1125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pennington K, et al. Prominent synaptic and metabolic abnormalities revealed by proteomic analysis of the dorsolateral prefrontal cortex in schizophrenia and bipolar disorder. Mol Psychiatry 13, 1102–1117 (2008). [DOI] [PubMed] [Google Scholar]
- 165.Nakazawa K, Jeevakumar V & Nakao K Spatial and temporal boundaries of NMDA receptor hypofunction leading to schizophrenia. NPJ Schizophr 3, 7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kehrer C, Maziashvili N, Dugladze T & Gloveli T Altered Excitatory-Inhibitory Balance in the NMDA-Hypofunction Model of Schizophrenia. Front Mol Neurosci 1, 6 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Banks PJ, et al. Disruption of hippocampal-prefrontal cortex activity by dopamine D2R-dependent LTD of NMDAR transmission. Proceedings of the National Academy of Sciences of the United States of America 112, 11096–11101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Blot K, et al. Modulation of hippocampus-prefrontal cortex synaptic transmission and disruption of executive cognitive functions by MK-801. Cerebral cortex (New York, N.Y. : 1991) 25, 1348–1361 (2015). [DOI] [PubMed] [Google Scholar]
- 169.López-Gil X, et al. Clozapine and haloperidol differently suppress the MK-801-increased glutamatergic and serotonergic transmission in the medial prefrontal cortex of the rat. Neuropsychopharmacology 32, 2087–2097 (2007). [DOI] [PubMed] [Google Scholar]
- 170.Homayoun H & Moghaddam B NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 27, 11496–11500 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Cohen SM, Tsien RW, Goff DC & Halassa MM The impact of NMDA receptor hypofunction on GABAergic neurons in the pathophysiology of schizophrenia. Schizophr Res 167, 98–107 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bitanihirwe BK, Lim MP, Kelley JF, Kaneko T & Woo TU Glutamatergic deficits and parvalbumin-containing inhibitory neurons in the prefrontal cortex in schizophrenia. BMC Psychiatry 9, 71 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Woo TU, Walsh JP & Benes FM Density of glutamic acid decarboxylase 67 messenger RNA-containing neurons that express the N-methyl-D-aspartate receptor subunit NR2A in the anterior cingulate cortex in schizophrenia and bipolar disorder. Arch Gen Psychiatry 61, 649–657 (2004). [DOI] [PubMed] [Google Scholar]
- 174.Jackson ME, Homayoun H & Moghaddam B NMDA receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proceedings of the National Academy of Sciences of the United States of America 101, 8467–8472 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zick JL, et al. Blocking NMDAR Disrupts Spike Timing and Decouples Monkey Prefrontal Circuits: Implications for Activity-Dependent Disconnection in Schizophrenia. Neuron 98, 1243–1255.e1245 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Auger ML & Floresco SB Prefrontal cortical GABAergic and NMDA glutamatergic regulation of delayed responding. Neuropharmacology 113, 10–20 (2017). [DOI] [PubMed] [Google Scholar]
- 177.Fung SJ, Sivagnanasundaram S & Weickert CS Lack of change in markers of presynaptic terminal abundance alongside subtle reductions in markers of presynaptic terminal plasticity in prefrontal cortex of schizophrenia patients. Biol Psychiatry 69, 71–79 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Catts VS, Derminio DS, Hahn CG & Weickert CS Postsynaptic density levels of the NMDA receptor NR1 subunit and PSD-95 protein in prefrontal cortex from people with schizophrenia. NPJ Schizophr 1, 15037 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Halim ND, et al. Presynaptic proteins in the prefrontal cortex of patients with schizophrenia and rats with abnormal prefrontal development. Mol Psychiatry 8, 797–810 (2003). [DOI] [PubMed] [Google Scholar]
- 180.Lewis DA, Fish KN, Arion D & Gonzalez-Burgos G Perisomatic inhibition and cortical circuit dysfunction in schizophrenia. Current opinion in neurobiology 21, 866–872 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hashimoto T, et al. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. The Journal of neuroscience : the official journal of the Society for Neuroscience 23, 6315–6326 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Glausier JR, Fish KN & Lewis DA Altered parvalbumin basket cell inputs in the dorsolateral prefrontal cortex of schizophrenia subjects. Mol Psychiatry 19, 30–36 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hashimoto T, et al. Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol Psychiatry 13, 147–161 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Enomoto T, Tse MT & Floresco SB Reducing prefrontal gamma-aminobutyric acid activity induces cognitive, behavioral, and dopaminergic abnormalities that resemble schizophrenia. Biol Psychiatry 69, 432–441 (2011). [DOI] [PubMed] [Google Scholar]
- 185.Tse MT, Piantadosi PT & Floresco SB Prefrontal cortical gamma-aminobutyric acid transmission and cognitive function: drawing links to schizophrenia from preclinical research. Biol Psychiatry 77, 929–939 (2015). [DOI] [PubMed] [Google Scholar]
- 186.Seamans JK & Yang CR The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Progress in neurobiology 74, 1–58 (2004). [DOI] [PubMed] [Google Scholar]
- 187.Niwa M, et al. Adolescent stress-induced epigenetic control of dopaminergic neurons via glucocorticoids. Science 339, 335–339 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Li YC, Kellendonk C, Simpson EH, Kandel ER & Gao WJ D2 receptor overexpression in the striatum leads to a deficit in inhibitory transmission and dopamine sensitivity in mouse prefrontal cortex. Proceedings of the National Academy of Sciences of the United States of America 108, 12107–12112 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Huang YY, Simpson E, Kellendonk C & Kandel ER Genetic evidence for the bidirectional modulation of synaptic plasticity in the prefrontal cortex by D1 receptors. Proceedings of the National Academy of Sciences of the United States of America 101, 3236–3241 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Durstewitz D & Seamans JK The dual-state theory of prefrontal cortex dopamine function with relevance to catechol-o-methyltransferase genotypes and schizophrenia. Biol Psychiatry 64, 739–749 (2008). [DOI] [PubMed] [Google Scholar]
- 191.Okubo Y, et al. Decreased prefrontal dopamine D1 receptors in schizophrenia revealed by PET. Nature 385, 634–636 (1997). [DOI] [PubMed] [Google Scholar]
- 192.Kaalund SS, et al. Contrasting changes in DRD1 and DRD2 splice variant expression in schizophrenia and affective disorders, and associations with SNPs in postmortem brain. Mol Psychiatry 19, 1258–1266 (2014). [DOI] [PubMed] [Google Scholar]
- 193.Selvaraj S, Arnone D, Cappai A & Howes O Alterations in the serotonin system in schizophrenia: a systematic review and meta-analysis of postmortem and molecular imaging studies. Neurosci Biobehav Rev 45, 233–245 (2014). [DOI] [PubMed] [Google Scholar]
- 194.Takano H Cognitive Function and Monoamine Neurotransmission in Schizophrenia: Evidence From Positron Emission Tomography Studies. Front Psychiatry 9, 228 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Yuen EY, et al. The novel antipsychotic drug lurasidone enhances N-methyl-D-aspartate receptor-mediated synaptic responses. Molecular pharmacology 81, 113–119 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Rajarajan P, Jiang Y, Kassim BS & Akbarian S Chromosomal Conformations and Epigenomic Regulation in Schizophrenia. Prog Mol Biol Transl Sci 157, 21–40 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Bryois J, et al. Evaluation of chromatin accessibility in prefrontal cortex of individuals with schizophrenia. Nature communications 9, 3121 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Girdhar K, et al. Cell-specific histone modification maps in the human frontal lobe link schizophrenia risk to the neuronal epigenome. Nature neuroscience 21, 1126–1136 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Lee SA & Huang KC Epigenetic profiling of human brain differential DNA methylation networks in schizophrenia. BMC Med Genomics 9, 68 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Abdolmaleky HM, et al. Epigenetic dysregulation of HTR2A in the brain of patients with schizophrenia and bipolar disorder. Schizophr Res 129, 183–190 (2011). [DOI] [PubMed] [Google Scholar]
- 201.Gulchina Y, Xu SJ, Snyder MA, Elefant F & Gao WJ Epigenetic mechanisms underlying NMDA receptor hypofunction in the prefrontal cortex of juvenile animals in the MAM model for schizophrenia. J Neurochem 143, 320–333 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Bahari-Javan S, et al. HDAC1 links early life stress to schizophrenia-like phenotypes. Proceedings of the National Academy of Sciences of the United States of America 114, E4686–E4694 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Koenigs M & Grafman J The functional neuroanatomy of depression: distinct roles for ventromedial and dorsolateral prefrontal cortex. Behavioural brain research 201, 239–243 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Baxter LR Jr., et al. Reduction of prefrontal cortex glucose metabolism common to three types of depression. Arch Gen Psychiatry 46, 243–250 (1989). [DOI] [PubMed] [Google Scholar]
- 205.Belleau EL, Treadway MT & Pizzagalli DA The Impact of Stress and Major Depressive Disorder on Hippocampal and Medial Prefrontal Cortex Morphology. Biol Psychiatry 85, 443–453 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Koenigs M, et al. Distinct regions of prefrontal cortex mediate resistance and vulnerability to depression. The Journal of neuroscience : the official journal of the Society for Neuroscience 28, 12341–12348 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Perera T, et al. The Clinical TMS Society Consensus Review and Treatment Recommendations for TMS Therapy for Major Depressive Disorder. Brain Stimul 9, 336–346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Howard DM, et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nature neuroscience 22, 343–352 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kang HJ, et al. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat Med 18, 1413–1417 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Feyissa AM, Chandran A, Stockmeier CA & Karolewicz B Reduced levels of NR2A and NR2B subunits of NMDA receptor and PSD-95 in the prefrontal cortex in major depression. Progress in neuro-psychopharmacology & biological psychiatry 33, 70–75 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Pantazatos SP, et al. Whole-transcriptome brain expression and exon-usage profiling in major depression and suicide: evidence for altered glial, endothelial and ATPase activity. Mol Psychiatry 22, 760–773 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Hare BD, et al. Optogenetic stimulation of medial prefrontal cortex Drd1 neurons produces rapid and long-lasting antidepressant effects. Nature communications 10, 223 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Klempan TA, et al. Altered expression of genes involved in ATP biosynthesis and GABAergic neurotransmission in the ventral prefrontal cortex of suicides with and without major depression. Mol Psychiatry 14, 175–189 (2009). [DOI] [PubMed] [Google Scholar]
- 214.Fogaca MV & Duman RS Cortical GABAergic Dysfunction in Stress and Depression: New Insights for Therapeutic Interventions. Front Cell Neurosci 13, 87 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Yin H, et al. A pilot integrative genomics study of GABA and glutamate neurotransmitter systems in suicide, suicidal behavior, and major depressive disorder. Am J Med Genet B Neuropsychiatr Genet 171B, 414–426 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Earnheart JC, et al. GABAergic control of adult hippocampal neurogenesis in relation to behavior indicative of trait anxiety and depression states. The Journal of neuroscience : the official journal of the Society for Neuroscience 27, 3845–3854 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Ren Z, et al. Bidirectional Homeostatic Regulation of a Depression-Related Brain State by Gamma-Aminobutyric Acidergic Deficits and Ketamine Treatment. Biol Psychiatry 80, 457–468 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Wohleb ES, et al. GABA interneurons mediate the rapid antidepressant-like effects of scopolamine. The Journal of clinical investigation 126, 2482–2494 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Ueno H, et al. Juvenile stress induces behavioral change and affects perineuronal net formation in juvenile mice. BMC Neurosci 19, 41 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Yu Z, et al. Decreased Density of Perineuronal Net in Prelimbic Cortex Is Linked to Depressive-Like Behavior in Young-Aged Rats. Front Mol Neurosci 13, 4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Abdallah CG, Sanacora G, Duman RS & Krystal JH Ketamine and rapid-acting antidepressants: a window into a new neurobiology for mood disorder therapeutics. Annual review of medicine 66, 509–523 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Moghaddam B, Adams B, Verma A & Daly D Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 17, 2921–2927 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Chowdhury GM, et al. Transiently increased glutamate cycling in rat PFC is associated with rapid onset of antidepressant-like effects. Mol Psychiatry 22, 120–126 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Miller OH, et al. GluN2B-containing NMDA receptors regulate depression-like behavior and are critical for the rapid antidepressant actions of ketamine. Elife 3, e03581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Abdallah CG, et al. The effects of ketamine on prefrontal glutamate neurotransmission in healthy and depressed subjects. Neuropsychopharmacology 43, 2154–2160 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Maeng S, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry 63, 349–352 (2008). [DOI] [PubMed] [Google Scholar]
- 227.Zhou W, et al. Ketamine-induced antidepressant effects are associated with AMPA receptors-mediated upregulation of mTOR and BDNF in rat hippocampus and prefrontal cortex. Eur Psychiatry 29, 419–423 (2014). [DOI] [PubMed] [Google Scholar]
- 228.Liu RJ, et al. Brain-derived neurotrophic factor Val66Met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol Psychiatry 71, 996–1005 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Aguilar-Valles A, et al. Translational control of depression-like behavior via phosphorylation of eukaryotic translation initiation factor 4E. Nature communications 9, 2459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Fukumoto K, et al. Activity-dependent brain-derived neurotrophic factor signaling is required for the antidepressant actions of (2R,6R)-hydroxynorketamine. Proceedings of the National Academy of Sciences of the United States of America 116, 297–302 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Hasegawa Y, Zhu X & Kamiya A NV-5138 as a fast-acting antidepressant via direct activation of mTORC1 signaling. The Journal of clinical investigation 129, 2207–2209 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Kaut O, et al. Aberrant NMDA receptor DNA methylation detected by epigenome-wide analysis of hippocampus and prefrontal cortex in major depression. Eur Arch Psychiatry Clin Neurosci 265, 331–341 (2015). [DOI] [PubMed] [Google Scholar]
- 233.Schroeder FA, Lin CL, Crusio WE & Akbarian S Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry 62, 55–64 (2007). [DOI] [PubMed] [Google Scholar]
- 234.Duan Z & Lu J DNA Methyltransferases in Depression: An Update. Front Psychiatry 11, 538683 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Sales AJ & Joca SRL Antidepressant administration modulates stress-induced DNA methylation and DNA methyltransferase expression in rat prefrontal cortex and hippocampus. Behavioural brain research 343, 8–15 (2018). [DOI] [PubMed] [Google Scholar]
- 236.Sales AJ, et al. Antidepressant-like effect induced by systemic and intra-hippocampal administration of DNA methylation inhibitors. Br J Pharmacol 164, 1711–1721 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Ramos BP, et al. Dysregulation of protein kinase a signaling in the aged prefrontal cortex: new strategy for treating age-related cognitive decline. Neuron 40, 835–845 (2003). [DOI] [PubMed] [Google Scholar]
- 238.Wang M, et al. Neuronal basis of age-related working memory decline. Nature 476, 210–213 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Carlyle BC, et al. cAMP-PKA phosphorylation of tau confers risk for degeneration in aging association cortex. Proceedings of the National Academy of Sciences of the United States of America 111, 5036–5041 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Arnsten AFT, et al. Alzheimer’s-like pathology in aging rhesus macaques: Unique opportunity to study the etiology and treatment of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America 116, 26230–26238 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Dumitriu D, et al. Selective changes in thin spine density and morphology in monkey prefrontal cortex correlate with aging-related cognitive impairment. The Journal of neuroscience : the official journal of the Society for Neuroscience 30, 7507–7515 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Beckman D, et al. Oligomeric Aβ in the monkey brain impacts synaptic integrity and induces accelerated cortical aging. Proceedings of the National Academy of Sciences of the United States of America 116, 26239–26246 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Chen G, et al. Regulation of the NMDA receptor-mediated synaptic response by acetylcholinesterase inhibitors and its impairment in an animal model of Alzheimer’s disease. Neurobiology of aging 29, 1795–1804 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Ma XH, Zhong P, Gu Z, Feng J & Yan Z Muscarinic potentiation of GABA(A) receptor currents is gated by insulin signaling in the prefrontal cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 23, 1159–1168 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Chen GJ, Xiong Z & Yan Z Aβ impairs nicotinic regulation of inhibitory synaptic transmission and interneuron excitability in prefrontal cortex. Molecular neurodegeneration 8, 3 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Zhong P, et al. Impaired modulation of GABAergic transmission by muscarinic receptors in a mouse transgenic model of Alzheimer’s disease. The Journal of biological chemistry 278, 26888–26896 (2003). [DOI] [PubMed] [Google Scholar]
- 247.Liu W, Dou F, Feng J & Yan Z RACK1 is involved in н╡-amyloid impairment of muscarinic regulation of GABAergic transmission. Neurobiology of aging 32, 1818–1826 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Galvin VC, et al. Muscarinic M1 Receptors Modulate Working Memory Performance and Activity via KCNQ Potassium Channels in the Primate Prefrontal Cortex. Neuron 106, 649–661.e644 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Rorabaugh JM, et al. Chemogenetic locus coeruleus activation restores reversal learning in a rat model of Alzheimer’s disease. Brain 140, 3023–3038 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Zhang F, et al. β-amyloid redirects norepinephrine signaling to activate the pathogenic GSK3β/tau cascade. Sci Transl Med 12(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Arnsten AF & Wang M Targeting Prefrontal Cortical Systems for Drug Development: Potential Therapies for Cognitive Disorders. Annu Rev Pharmacol Toxicol 56, 339–360 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Bossers K, et al. Concerted changes in transcripts in the prefrontal cortex precede neuropathology in Alzheimer’s disease. Brain 133, 3699–3723 (2010). [DOI] [PubMed] [Google Scholar]
- 253.Bereczki E, et al. Synaptic markers of cognitive decline in neurodegenerative diseases: a proteomic approach. Brain 141, 582–595 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Bereczki E, et al. Synaptic proteins predict cognitive decline in Alzheimer’s disease and Lewy body dementia. Alzheimer’s & dementia : the journal of the Alzheimer’s Association 12, 1149–1158 (2016). [DOI] [PubMed] [Google Scholar]
- 255.Battaglia F, et al. Cortical plasticity in Alzheimer’s disease in humans and rodents. Biol Psychiatry 62, 1405–1412 (2007). [DOI] [PubMed] [Google Scholar]
- 256.Zheng Y, et al. Inhibition of EHMT1/2 rescues synaptic and cognitive functions for Alzheimer’s disease. Brain 142, 787–807 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Mathys H, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Zhang B, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 707–720 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Zhou Y, et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med 26, 131–142 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Cao Q, et al. Targeting histone K4 trimethylation for treatment of cognitive and synaptic deficits in mouse models of Alzheimer’s disease. Sci Adv 6(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.McEwen BS & Morrison JH The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron 79, 16–29 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Popoli M, Yan Z, McEwen BS & Sanacora G The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci 13, 22–37 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Arnsten AF Stress weakens prefrontal networks: molecular insults to higher cognition. Nature neuroscience 18, 1376–1385 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]