

Abstract
Cancer metastasis remains a major clinical challenge for cancer treatment. It is therefore crucial to understand how cancer cells establish and maintain their metastatic traits. However, metastasis-specific genetic mutations have not been identified in most exome or genome sequencing studies. Emerging evidence suggests that key steps of metastasis are controlled by reversible epigenetic mechanisms, which can be targeted to prevent and treat the metastatic disease. A variety of epigenetic mechanisms were identified to regulate metastasis, including the well-studied DNA methylation and histone modifications. In the past few years, large scale chromatin structure alterations including reprogramming of the enhancers and chromatin accessibility to the transcription factors were shown to be potential driving force of cancer metastasis. To dissect the molecular mechanisms and functional output of these epigenetic changes, it is critical to use advanced techniques and alternative animal models for interdisciplinary and translational research on this topic. Here we summarize our current understanding of epigenetic aberrations in cancer progression and metastasis, and their implications in developing new effective metastasis-specific therapies.
1. Introduction
Over 90% of cancer death is due to metastasis1. Although most patients with just localized diseases can often be cured with surgery, chemotherapy or radiation therapy, those with metastatic diseases are incurable in most cases, despite the development of new treatment options including targeted therapy and immunotherapy2.
Metastasis is a complex and surprisingly inefficient process3. During metastatic cascade, cancer cells acquire the ability to disseminate from the primary tumor, locally invade the surrounding tissue, intravasate into the bloodstream, survive in the circulation, exit the bloodstream, co-opt the foreign microenvironment, and eventually colonize and outgrow at distant organs4 (Figure 1). Only approximately 0.01% of the circulated tumor cells are able to overcome all the upfront physical and biological barriers to form metastases5. These barriers force/encourage the transformed cancer cells to gain many additional abilities in a timely manner. As the result, metastatic cells often become too malignant to target.
Figure 1. The metastatic cascade.
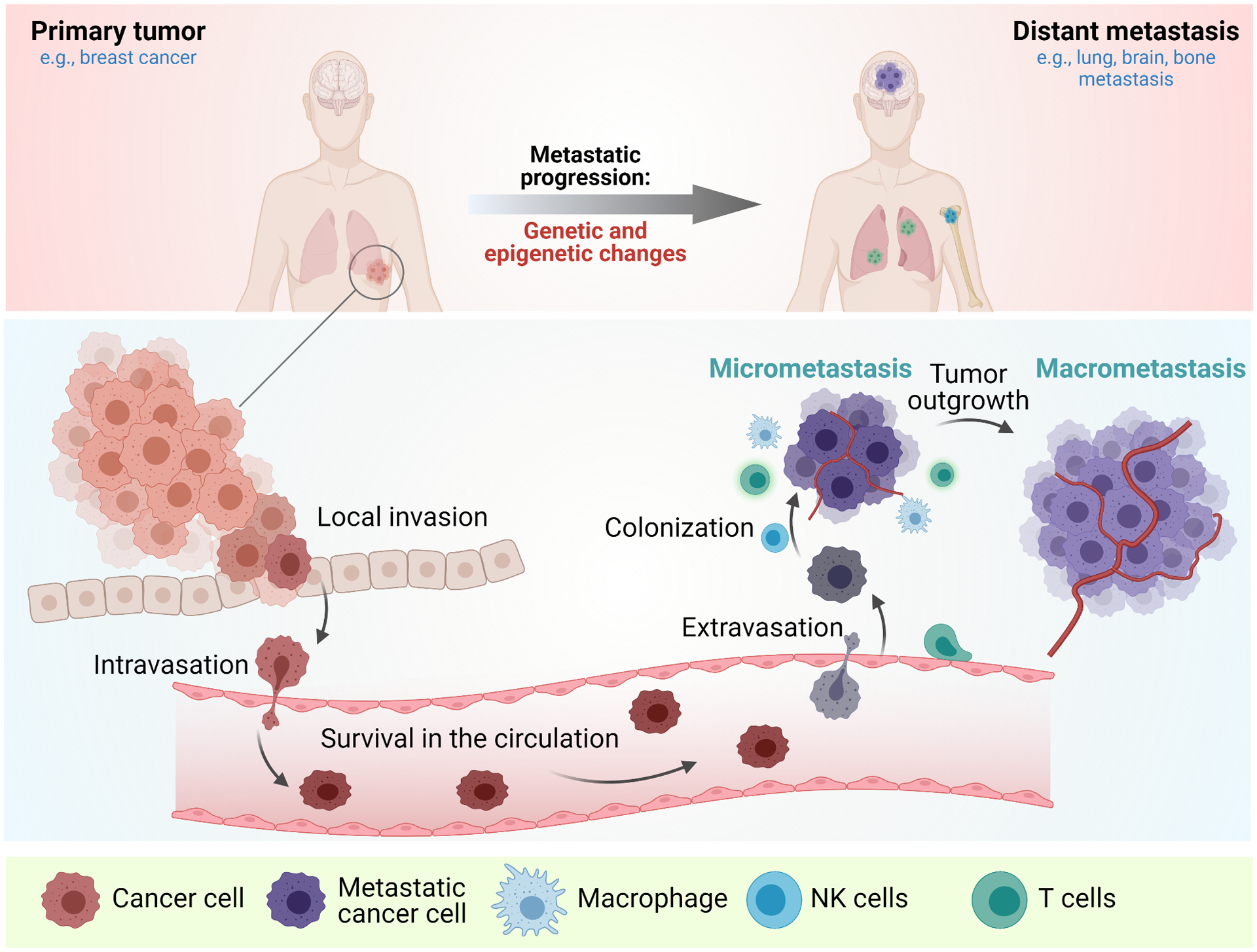
Top-During the metastatic progression, primary tumor (for example (e.g.,) breast cancer undergoes epigenetic changes to acquire the metastatic traits to form distant metastases (e.g., lung, brain, and bone metastasis). Bottom-The cancer cells at the primary site acquire the ability to invade and migrate through the blood vessels, survive in the circulation, extravasate the blood vessels, settle down at the distant microenvironment, and proliferate to form metastases.
Identifying the keys that contribute to the metastatic path is therefore extremely crucial, which could lead to strategies to treat metastatic diseases. Through intensive studies using experimental systems, many genes that mediate these steps have been identified6–9. For example, the transcription factors TWIST, SNAIL1, and SNAIL2 mediate epithelial-mesenchymal transition (EMT) at the initial step of metastatic cascade10. The lysyl oxidase and cytokine angiopoietin-like 4 enhance cancer cell invasion through crosstalk with the tumor microenvironment11,12. However, these mechanisms that contribute to metastasis may be context-dependent. It remains an open question how the upstream regulators control the initiation and progression of metastasis.
Tumor cells progress through acquiring driver mutations that confer the growth advantage over time13. In line with this notion, metastasis progression should be driven by additional genetic mutations. However, even with extensive characterizing of primary tumor cells and metastatic lesions using advanced sequencing methods, recurrent metastasis-specific mutations that drive metastasis are not identified in most cases13–16. In fact, many oncogenic mouse models that developed cancer lesions did not establish distant metastasis17. In addition, the study from Jacob et al. have shown that the genetic alterations that confer the metastatic potential already exist in the primary tumors or the parental cancer cell lines that is functionally less metastatic18. Thus, if genetic mutations are not the driver, what could be the alternative mechanisms that contribute to the process?
“Epigenetics” are defined as non-genetic regulation of heritable traits including gene expression. It is well accepted that epigenetic regulators are a powerful class of proteins that creates the selective advantage of tumor cells by exerting dynamic and rapid changes19. The main epigenetic mechanisms are histone modifications, DNA methylations, chromatin remodeling and non-coding RNA regulation20,21. These mechanisms alter regional chromosome structure into a “opened” or “closed” state, allowing upregulation or downregulation of gene expression, respectively (Figure 2A). There are four groups of proteins known to act in concert for mediating a specific modification-writers, erasers, readers, and chromatin remodeler proteins (Figure 2B). Because of the complexity and interplay of epigenetic regulation in normal state, these proteins must be tightly regulated. Therefore, aberrant expression or functional alteration in a single component could lead to severe diseases including metastasis. Because epigenetic changes are reversible, which allow the functional recovery of the affected genes, targeting epigenetic changes provides great potential in current therapeutic development.
Figure 2. The epigenetic landscape and epigenetic control.
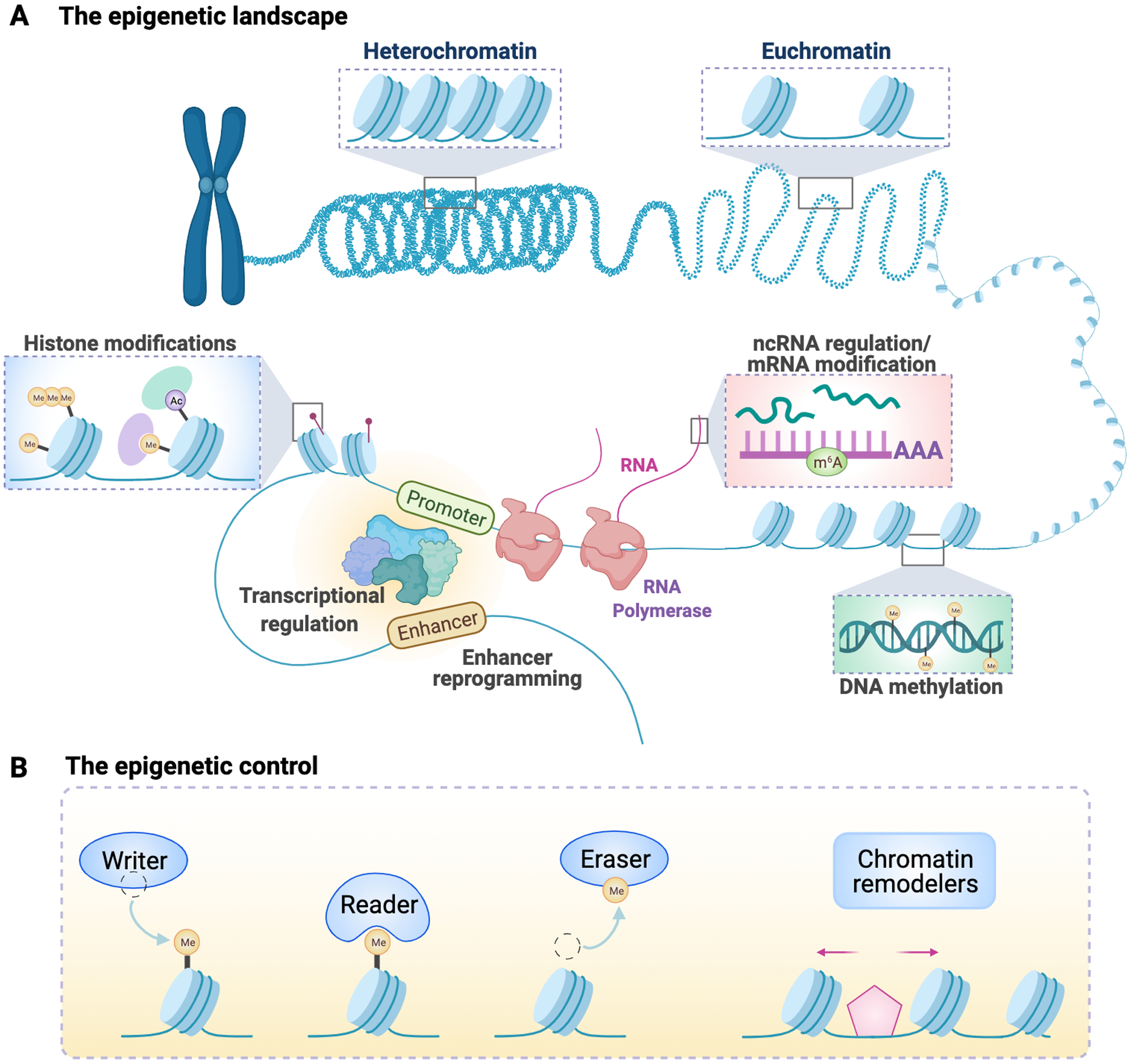
(A) Chromatin states can be switched between “opened” (Euchromatin) and “closed” (Heterochromatin) states to maintain gene activation or gene silencing, respectively. Many types of epigenetic regulations act in concert to maintain certain chromatin state, including histone modifications, transcriptional regulation, enhancer reprogramming, DNA methylation, non-coding RNA regulation, and RNA modifications. The opened chromatin state allows the binding of transcription factors to the promoter and enhancer regions of a genome locus to drive gene expression. (B) The epigenetic control is reversible. Each type of modification can be added to the modification site through a “writer” protein, removed by an “eraser” protein, and can be recognized by a “reader” protein through a specific interacting domain. Chromatin remodelers are a class of proteins that are able to restructure chromatin packaging.
Emerging evidence suggested epigenetic regulation may be the key machinery to achieve metastatic properties22–25. Because cancer cells need to adapt to the changing environmental cues, having cellular plasticity is extremely critical in those transition states. This adaptation is sometimes reversible. A well-studied example is the process of EMT and mesenchymal-epithelial transition (MET). Although this process may not be necessary in all cancer types, regaining the epithelial characteristic lost during local invasion is required for metastatic outgrowth at least in some contexts26. This is evident that the process could be driven in a transient and reversible manner, suggesting the process is epigenetically regulated. In fact, during cancer progression, the promoter of E-cadherin has been shown to become hypermethylated, leading to loss of E-cadherin gene expression27. Additionally, several transcription factors that are linked to the EMT process, SNAIL, SLUG, TWIST, and ZEB 1/2, could also recruit DNA/histone modification complexes to repress E-cadherin expression10.
In this review, we summarize the recent findings of epigenetic regulatory mechanisms in cancer metastasis with the emphasis on histone modifications, enhancer reprogramming, chromosome accessibility, and transcription regulation; evaluate the potential therapeutic implications; and discuss the current strategy and future directions.
2. Regulation of cancer metastasis by chromatin opening and transcription factor binding
Chromatin architecture and accessibility to transcription factor binding is by far the most comprehensive epigenomic characteristic that determines the biological output of specific gene locus. Multiple layers of regulation are interconnected to alter chromatin structure and complete the entire gene expression process. Firstly, many enzymes such as histone modifiers or chromatin remodelers are able to unpack the condensed nucleosome. Secondly, the binding of transcription factors to its target enhancers or promoters can further drive gene expression. In fact, transcription factors are the central factors that control the rate of gene expression due to their ability in DNA binding and the tight regulation of these factors. Thus, through mechanisms including activation or altered expression, specific gene expression program can be turned on rapidly in response to the environmental cues.
Several studies have shown transcription factors or altered chromatin accessibility as the key driver of metastasis progression28–32. An interesting study from Siersbæk et al. identified the transcriptional mechanism underlying IL6/STAT3-signaling controlled estrogen receptor (ER)+ breast cancer metastasis. Upon IL6 induction, the activated STAT3 establishes and hijacks the enhancers shared with ER and FOXA1 in ER+ breast cancer cells33. STAT3 therefore drives the transcriptional program to promote the expression of genes involved in tumor invasion and metastasis instead of the genes targeted by ER/FOXA1 that controls cell proliferation33 (Figure 3A). The shift of gene expression programs renders the cancer cells insensitive to the standard ER-targeted endocrine therapy. Instead, targeting STAT3 with JAK inhibitor, ruxolitinib, decreases breast cancer cell invasion in an in vivo model33. These findings suggest that STAT3 and ER/FOXA1, though sharing the regulatory regions, are driving functionally independent oncogenic pathways. Therefore, targeting specific transcriptional pathway would be an alternative for treating the metastatic diseases.
Figure 3. Transcription factors regulate chromatin opening in cancer metastasis.
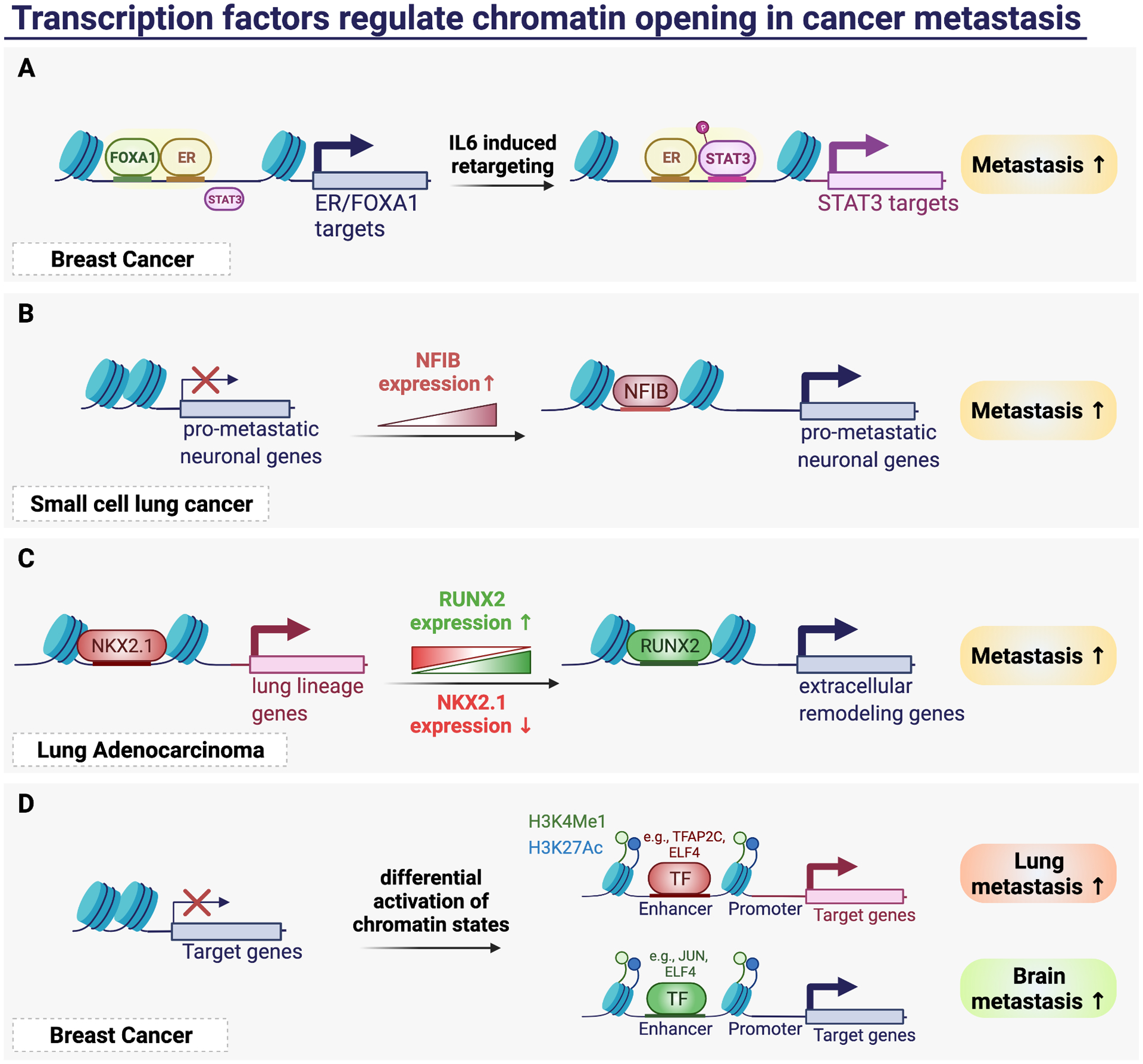
(A) IL6 induction activates transcription factor STAT3 through hijacking the enhancer element of FOXA1 and ER, thereby drives the expression of STAT3 targets and metastasis. (B) Upregulation of NFIB drives the chromatin accessibility in small cell lung cancer to activate pro-metastatic neuronal genes and metastasis. (C) Alteration of chromatin accessibility driven by downregulation of NKX2.1 and upregulation of RUNX2 switches the transcription program to extracellular remodeling gene program and activates lung cancer metastasis. (D) Differential activation of chromatin states at specific locus leads to binding of specific transcription factors that regulate organotropic metastasis.
In small cell lung cancer, Denny et al. discovered dramatic remodeling of its chromatin state which is again driven by a single transcription factor nuclear factor 1B (NFIB). Using ATAC-seq, the authors first identified global increase in chromatin accessibility in liver metastases samples compared to that of primary tumors34. They further discovered the differential accessible regions are highly enriched for NFIB binding motifs, and that NFIB is highly expressed in invasive primary tumors and metastases34 (Figure 3B). Manipulating NFIB expression with either shRNA knockdown or ectopically expressed, they showed that NFIB is both necessary and sufficient to maintain a subset of open chromatin regions to drive the pro-metastatic neuronal gene expression program, thereby promoting several requisite steps of the metastatic cascade34.
Recent development which combined the technology of single-cell RNA sequencing with the ATAC-seq becomes a powerful tool to profile epigenomics at the single-cell level. To discover the potential heterogeneity of TF activity in tumor, LaFave et al. has thus leveraged scATAC-seq to characterize chromatin accessibility and transcriptional changes within the progression of lung adenocarcinoma (LUAD) from tumor initiation to metastasis in the genetically engineering mouse Kras/p53 model35. Their analyses showed that primary tumors are more heterogeneous than metastases, which has a small fraction of cells resembling the regulatory states of cells isolated from metastatic sites35. Importantly, they discovered the epigenomic state transition during LUAD progression, where cells are altering lineage restriction and cell identity by losing the TF motif accessibility of lung lineage factor NKX2.1 and progressive gaining the activity of transcription factor RUNX2. The master regulator RUNX2 initiated the expression of extra-cellular matrix proteins to induce EMT35 (Figure 3C). They further showed RUNX2 knockout cells developed fewer lung metastases and increased survival in the mice injected intravenously35. Still, in other cancer types, many transcription factors have been shown to mediate metastasis through altering cell differentiation or cell fate or driving EMT gene program28,36. However, the stimulating signals which initiate such epigenomic transition remains to be elucidated.
Transcriptional mechanisms which control metastasis varies across different cancer types. In fact, such gene expression program could also vary by which secondary organ the cancer cells are targeting to. For example, in triple negative breast cancer model, Peluffo et al. identified engrailed 1 (EN1) as a transcription factor that associated with brain metastasis37. EN1 targets genes involved in WNT, hedgehog signaling, and neural-related functions, which allows the cells to disseminate and survive specifically in the brain tissue37. This idea prompted us to identify unique chromatin states and transcription factors which could explain the metastatic organotropism of breast cancer cells. Specifically, we integrated RNA-seq, ChIP-seq, and ATAC-seq to profile the transcriptome and epigenome of the triple negative breast cancer cell line MDA-MB-231 and its metastatic brain and lung organotropic derivatives38. We identified increased activation of both promoters and enhancers in the metastatic sub-populations38. we found that the activated promoters and enhancers are enriched for regulators of vasculature development in the lung metastatic derivatives, while the activated promoters are enriched for homophilic cell adhesion in the brain metastasis derivatives. Using ATAC-seq, we determined differential accessible regions and specific transcription factor motifs that they harbor38. We uncovered several transcription factors that are associated specifically to the open chromatin landscape of either lung or brain metastasis and their expression levels correlate with poor outcome38(Figure 3D). These lines of evidence suggest transcription factors as potential biomarkers for cancer metastasis, which are likely the key to maintain specific chromatin landscape for organotropic metastasis.
3. Control of cancer metastasis by histone modifications
Post-translational modifications (PTMs) on histones are a major category of changes on chromatin that could alter chromatin structure. There are a large number of different histone PTMs identified to date including the two major classes, histone methylation and histone acetylation. The site and degree of histone methylation correlate with the states of gene expression21. In general, tri- and di-methylation of lysine 4 on histone H3 (H3K4me3/2) are associated with gene activation, while methylation of lysine 9 or lysine 27 on histone H3 correlates with gene repression21. The methylation of histone lysine or arginine residues is highly dynamic. It is mediated by opposing action of histone methyltransferases and histone demethylases to add or remove the methylation marks, respectively. In addition, lysine methylation can interact with reader proteins that contain either PHDs, chromodomains, tudor domains, or PWWP domains, which mediate the function of these marks39. Similar to the dynamic regulation of histone methylation, histone acetylation marks can be added by histone acetyltransferases (HATs), removed by histone deacetylases (HDACs), and can be read by bromodomain and YEATS domain proteins39.
There is accumulating evidence that the alteration of histone modifications plays a major role in cancer metastasis20,24. In fact, several histone methyltransferases have been linked to metastatic progression40,41. A well-studied example is enhancer of zeste homolog 2 (EZH2), the catalytic subunit of the PRC2 methyltransferase complex that mediate H3K27 methylation. EZH2 is highly expressed in many cancer types including advanced and metastatic prostate cancer42,43. Chen et al. first showed its causal role in driving prostate cancer metastasis through epigenetic suppression of a metastasis-suppressor DAB2IP (AIP1)44. It was further shown that silencing of DAB2IP led to activation of NF-kB pathway and induced EMT to drive metastasis43,45 (Figure 4A). These studies have therefore underscored the potential of targeting EZH2 to treat metastasis. In fact, EZH2 has been reported to promote metastasis of many cancers42,46–48. Notably, EZH2 was even found to be able to mediate breast cancer secondary metastasis stimulated by the bone microenvironment, which underscores the important role of EZH2 in metastatic spread49. On the other hand, the mechanisms for increased EZH2 in many of the cases is likely through post-translational modification50. Recently, it was found that asymmetrically di-methylation of EZH2 by protein arginine methyltransferase 1 (PRMT1) inhibits EZH2 phosphorylation at nearby residues, and subsequent ubiquitination and degradation in breast cancer51. The stabilization of EZH2 by PRMT1 decreases its target gene expression including DAB2IP thereby promotes metastasis51. Additional study has identified that SET-domain containing protein 2 (SETD2), a canonical H3K36 methyltransferase, mediates EZH2-K735 mono-methylation and promotes its degradation to suppress metastasis in prostate cancer52. The mutation at R1523 in SETD2 disrupts its interaction with EZH2, which results in unmethylated EZH2 and promotes metastasis52. This demonstrates an antagonistic relationship between the two epigenetic writers within metastasis progression, and demonstrates the functional role of SETD2 modification on the non-histone proteins. In addition to the metastasis-promoting role of EZH2, PRC2 activity has also been shown to suppress metastasis. In the model of VHL-HIF pathway-driven ccRCC, loss of the repressive H3K27 trimethylation can liberate the expression of chemokine CXCR4, which increases the metastatic fitness through supporting chemotactic cell invasion53 (Figure 4B). Thus, depending on the context of tumor cells, PRC2 activity can either promote or inhibit metastatic progression.
Figure 4. Control of cancer metastasis by histone modifiers.
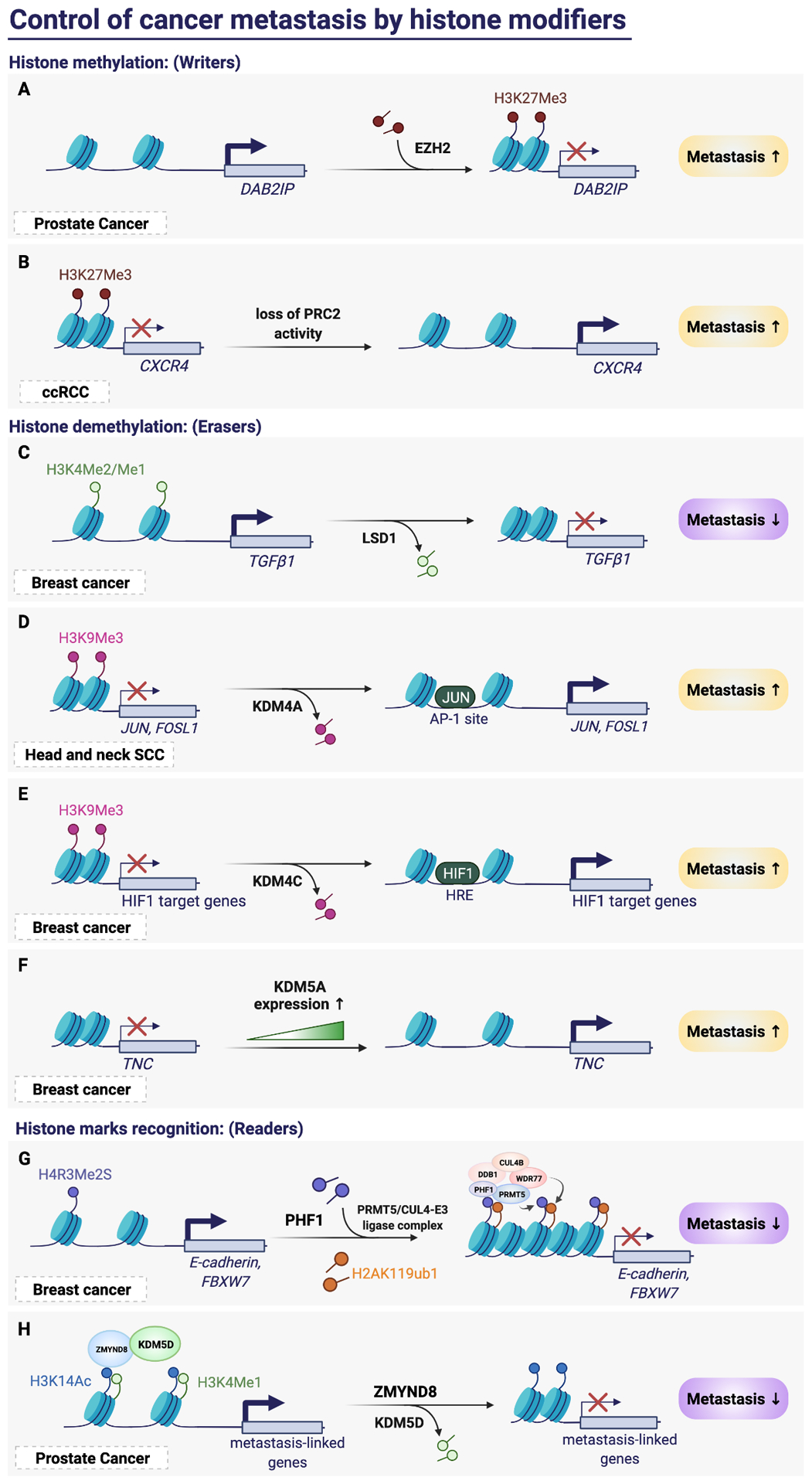
(A) EZH2 promotes cancer metastasis in prostate cancer by silencing DAB2IP with the addition of H3K27me3 on the nucleosome. (B) Loss of PRC2 complex de-represses its target CXCR4, thereby promotes metastasis in ccRCC. (C) LSD1 suppresses breast cancer metastasis by removing H3K4 methylation to silence TGFβ1. (D) KDM4A promotes head and neck SCC metastasis by activating the expression of JUN and FOSL1 through removing the H3K9me3 mark. The activation of JUN further enhanced its transcription through binding to its promoter AP-1 site. (E) KDM4C promotes breast cancer metastasis by removing H3K9me3 thereby activating the transcription of HIF1 target genes. The activation of HIF1 further enhanced its transcription through binding to the HIF-response element. (F) In breast cancer, the expression of KDM5A correlates with metastasis progression, and KDM5A activates its target TNC in a demethylase-independent manner. (G) The H4R3me2S modification can be read by PHF1, which recruits writer proteins PRMT5 and Cul4B-ubiquitin E3 ligase complex to silence its target E-cadherin and FBXW7, thereby suppresses metastasis. (H) ZMYD3 recruits KDM5D and recognizes the dual histone mark H3K14ac and H3K4me1. KDM5D then erases the methylation from histone to suppress metastasis-linked genes and metastasis progression.
After the first lysine demethylase, lysine-specific demethylase (LSD1), was identified, the knowledge of epigenetics in cancer and metastasis has been expanded. The study from Wang et al. has found that LSD1 expression is downregulated in breast cancer and it is able to inhibit the invasion and breast cancer metastasis54. Mechanistically, LSD1 was found a subunit of the nucleosome remodeling and histone deacetylase (NuRD) complex, which erases the H3K4 methylation at the promoter of TGFβ1 to suppress TGFβ1 gene expression, thereby suppress EMT and metastasis54 (Figure 4C). This study not only demonstrates the functional role of a demethylase in cancer metastasis but also indicates the potential interdependence of histone deacetylation function of the NuRD complex and the demethylase function of LSD1 in chromatin remodeling. In addition, KDM5B (JARID1B/PLU-1) was later found as another component of the LSD1/NuRD complex, which suppresses the angiogenesis and metastasis of breast cancer cells through repressing the expression of CC chemokine ligand 14, an epithelial derived chemokine55. This study provides a functional connection between the H3K4me2/1-specific demethylase LSD1, and the H3K4me3/2-specific demethylase KDM5B. To bring the target genes to a complete silenced state, it is likely that the KDM5B recognizes the H3K4me3 first and then the methylation mark will be removed sequentially by the two physically associated demethylases55.
Similar to KDM5B, many other demethylases within the jumonji (JmjC) domain-containing hydroxylase family have been reported to participate in caner metastasis24. KDM4A, a H3K9me3 and H3K36me3 demethylase, was identified as a promoting factor in head and neck SCC invasion and metastasis through the induction of AP1 transcription and AP1 positive feedback loop56. The authors showed with chromatin immunoprecipitation (ChIP) assays that demethylation of H3K9me3 by KDM4A increases the chromatin accessibility, thereby allowing the recruitment of AP1 to the promoter encoding JUN and FOSL156 (Figure 4D). In breast cancer, KDM4C is recruited by HIF1 to the hypoxia response element (HRE) of the HIF1 target gene, which enhances HIF1 binding to the HRE through decreasing H3K9me357. KDM4C therefore facilitates the expression of HIF1 targeted genes that involved in metabolic reprogramming and lung metastasis in breast cancer57 (Figure 4E). Our study identified KDM5A (also known as RBP2/JARID1A) as a critical driver of breast cancer metastasis58. We found a strong association of KDM5A expression with breast cancer metastasis in patient data set and cell lines58. Using two in vivo models, we showed that knockdown of KDM5A or deletion of KDM5A inhibites breast cancer metastasis to the lungs, while did not affect bone metastasis58. These results indicate KDM5A likely regulates metastasis in a tissue-specific manner. Intriguingly, our gene-expression analysis revealed that KDM5A positively regulates a set of lung-metastasis genes including TNC independent of its demethylase activity (Figure 4F), unlike the study by Teng et al. which showed KDM5A promotes the expression of integrin-β1 to regulate lung cancer metastasis through a demethylase-dependent manner58,59. These findings demonstrated that KDM5A is a context-dependent pleiotropic regulator and a potential target for tumor progression and metastasis.
In addition to the writers and erasers of histone modifications, the readers of histone marks are important epigenetic effectors. For example, PHD finger protein 1 (PHF1) is identified a novel reader for the histone H4R3 symmetric di-methylation catalyzed by PRMT5/WDR77 complex. The recognition is through the N-terminal of PHD domain in PHF1, while its additional C-terminal PHD domain is able to recruit the writer cullin 4B-RING E3 ligase complex to promote H2AK119ub160. The coordination between the reader PHF1 and the two writer complexes creates the repressive histone code and silences their target genes including E-cadherin and FBXW7, which then promotes the proliferation and invasion in breast cancer cells60 (Figure 4G). Like PHF1, ZMYND8 is another reader protein that has dual module cassettes to read two histone marks. The study revealed the dual histone mark H3K4me1-H3K14ac, which usually denotes active genes, can be recognized by the PHD-bromo domains in ZMYND861. This interaction may bridge the demethylase KDM5D (JARID1D) to their co-targeted sites, resulting in the repressive H3K4me0-H3K14ac and downregulation of metastasis-linked genes61 (Figure 4H). The results provide the mechanism of how the reader and eraser proteins could act coordinately on top of pre-existing histone marks, thereby switching the histone mark from active to inactive to regulate metastasis.
Lastly, in the model of highly lethal pancreatic ductal adenocarcinoma, McDonald et al. indeed detected a large-scale reprogramming of histone H3K9 and DNA methylation at the large heterochromatin domains during the evolution of distant metastasis62. They further found that such chromatin modification is controlled by increased dependence of the distant metastatic subclones to the oxidative branch of the pentose phosphate pathway62. These findings uncovered how the metastatic subclones adapt to the foreign environment through the altered metabolism machinery that confer increased fitness to the cells. However, in this case, the specific epigenetic modifiers were not revealed.
4. Enhancer reprogramming drives cancer metastasis
Enhancers are distal cis-regulatory element that harbor clusters of transcription factor (TF) binding sites within the DNA sequence63. Upon the binding of transcription factors, enhancers can promote transcription independent of their orientation or distance63. Mechanistically, transcription factors will recruit chromatin modifiers to their bound enhancers, establishing a stereotypical chromatin modification pattern including the histone modifications H3K4me1 and H3K27ac, which open up the regional chromatin63. Notably, H3K27ac specifically distinguishes active enhancers from poised enhancers that only contain H3K4me164. Leveraging the advanced techniques of genome-wide sequencing including H3K27ac and H3K4me1 enrichment using ChIP-sequencing (ChIP-seq) or identifying chromatin accessibility using the assay for transposase-accessible chromatin using sequencing (ATAC-seq), researchers were able to annotate poised and activated enhancer landscapes in a specific cellular state.
The regulatory events and modifications on enhancer elements control specific gene expression program, therefore contributing to a variety of biological events such as cell differentiation during the developmental process63. In fact, emerging studies have linked the role of enhancer to cancer progression65. For example, mice lacking a Myc enhancer becomes resistant to the development of intestinal tumors66. In addition, pharmacologically targeting bromodomain containing 4 (BRD4) and mediators that associated at the super-enhancers impact the expression of key genes in cancer including MYC67,68. These lines of evidence therefore raised the interests in studying the functional roles of enhancers in metastasis and whether enhancers could be effective targets for the metastatic diseases.
In fact, several recent studies highlighted the importance of enhancer activation in metastasis. In advanced prostate cancer, Takeda et al. identified a somatically acquired enhancer of the androgen receptor (AR) that drives the disease progression69 (Figure 5A). This study provides a deeper understanding of the transcriptional regulation in addition to the promoter of its key target, AR gene, in advanced prostate cancer. CRISPRi targeting the functional relevant enhancer element suppresses AR signaling and cell proliferation69. Conversely, cell clones carrying an extra copy of AR enhancer have increased AR gene expression, which was sufficient to drive an AR antagonist-resistant phenotype69. Although the mechanism of how cancer cells acquire the enhancer function is unknown, the observations have expanded the range of potentials targets in the advanced prostate cancer.
Figure 5. Enhancer reprogramming drives cancer metastasis.
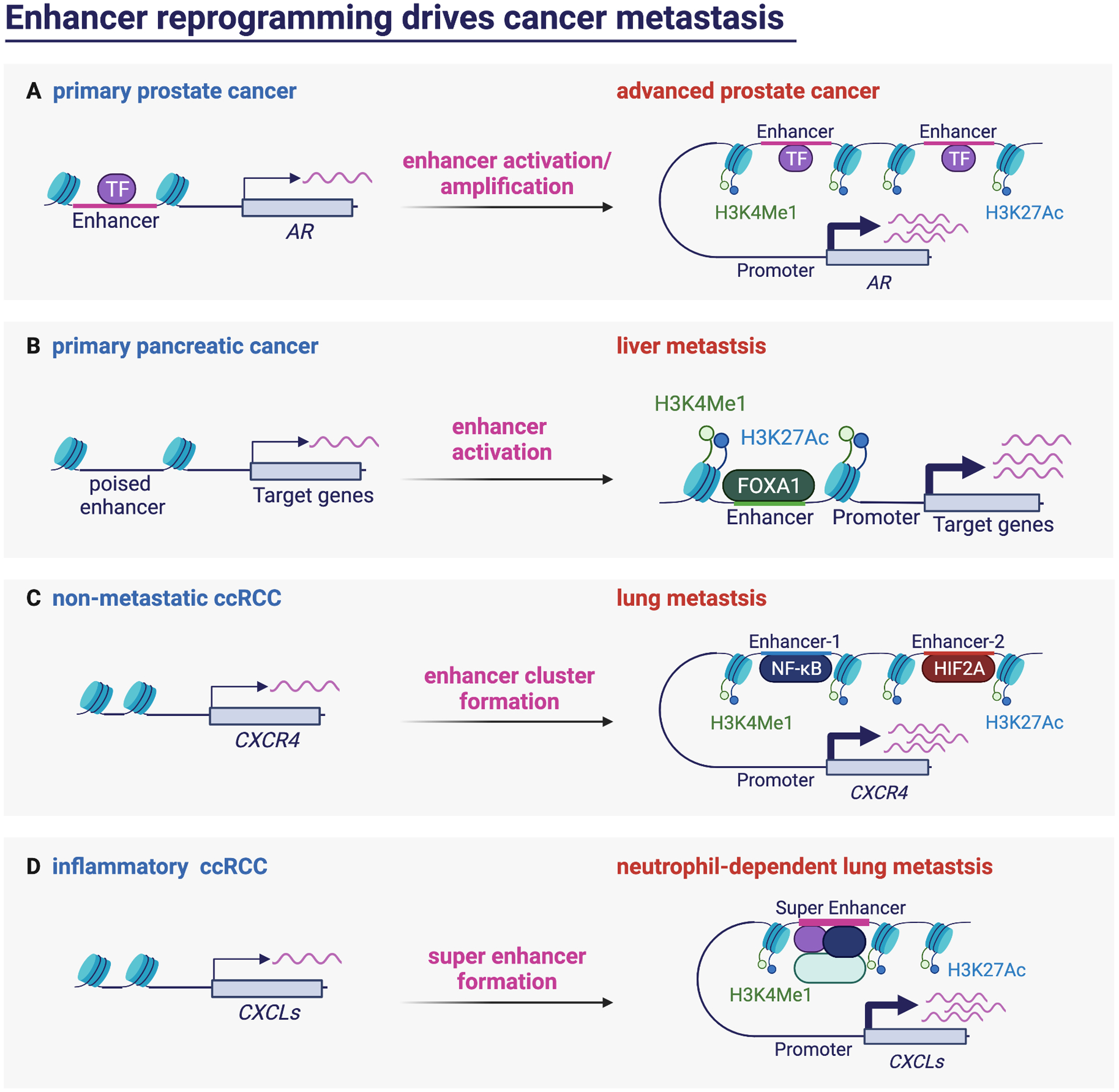
(A) AR enhancer activation and amplification drives the progression of primary prostate cancer to advanced stage. (B) In pancreatic cancer, FOXA1 activates enhancers of its target to drive liver metastasis. (C) The formation of super enhancer allows the binding of NF-κB and HIF2A to drive the expression of their common target CXCR4, thereby promotes lung metastasis in ccRCC. (D) The formation of super enhancer in inflammatory ccRCC activates the expression of CXCLs to promote neutrophil infiltration thereby promotes lung metastasis.
Similar enhancer reprogramming was identified in pancreatic cancer when Roe et al. aimed to identify the underlying changes in chromatin landscape associated with the metastatic phenotype70. They utilized paired tumor- and metastasis-derived organoids to profile genome-wide enrichment of H3K27ac as an indication of active enhancer landscape. Interestingly, the authors not only identified large-scale enhancer reprogramming during the progression of primary tumors to metastatic lesions, but also found that this reprogramming is directed by the transcription factor forkhead box protein A1 (FOXA1)70 (Figure 5B). Furthermore, aberrant upregulation of FOXA1 activates genes involved in embryonic foregut endoderm development, suggesting that the metastatic cancer cells are able to acquire metastatic capabilities by resetting their transcriptional program to a more primitive and developmental plastic state70.
Likewise, the activation of certain enhancers was identified to promote metastasis in renal cancer. As mentioned in the previous section, chemokine receptor CXCR4 supports metastatic progression through increasing cell survival and chemotactic migration, where its expression is epigenetic silenced by PRC2 complex in the non-metastatic state53. On top of this finding, Rodrigues et al. showed that the expression of CXCR4 is controlled by combined action of distant enhancer cluster, which are activated by two transcription factors, HIF2A and NF-κB71 (Figure 5C). The authors further found that the abovementioned enhancers and most of the identified metastasis-associated enhancers are co-localized with normal enhancers. These analyses indicate that cancer cells may co-opt lineage-specific enhancer states to acquire metastasis phenotype. More recently, Nishida et al. discovered the underlying mechanisms of how ccRCC cells reshape their intrinsic inflammation response and immune landscape to facilitate neutrophil-dependent metastasis progression72. They identified the expression of those inflammation-related genes are activated partially through super-enhancer formation72 (Figure 5D). Super-enhancers are clusters of enhancers occupied by high densities of the transcription machinery, which allows the cancer cells to exhibit sharp transitions of gene expression profile, thereby altering their immune response in this case.
To target these enhancer changes, several groups turned to BRD4, a member of the bromodomain and extraterminal (BET) family. BRD4 recognizes the acetylated chromatin and promotes transcriptional activation67,73. Additionally, BRD4 and MED1 are identified as transcriptional coactivators that form phase separation condensates at the super-enhancers, which concentrates the transcription apparatus for essential gene expression control74,75. Nishida et al. therefore evaluated the potential of targeting BRD4 to suppress the metastasis phenotype in ccRCC72. Treatment of ccRCC cells with BET inhibitor JQ1 decreased BRD4 binding and the transcription of CXC chemokines, thereby suppressing ccRCC metastasis and neutrophil infiltration72. A similar strategy was used in the study of Morrow et al. to manage osteosarcoma lung metastasis76. In addition to BET inhibition, they have also depleted the expression of enhancer-associated transcription factor AP-1 or inhibited individual genes activated by the metastasis-associated enhancers to suppress metastasis progression successfully76. These lines of evidence demonstrated that enhancer dysregulation is a main driver for metastasis and could be utilized to develop targeted therapies to treat metastatic diseases.
5. Other epigenetic mechanisms of metastasis
Chromatin remodeling in metastasis
Chromatin remodelers are a group of the ATP-dependent chromatin remodeling complexes that determine how DNA is packaged. Such regulation can be done through activating nucleosome sliding along the DNA, assembling or removing histones, or facilitating the exchange of histone variants77. In physiological conditions, chromatin remodelers are critical in many biological processes including, DNA replication, chromosome assembly, and transcription. The SWI/SNF complexes are the most-studied chromatin remodeling complexes. Notably, more than 20% of cancers carry mutations of the SWI/SNF complex genes, which underscores its role in tumor initiation78. However, the contribution of the SWI/SNF complex to cancer metastasis is less well known. In breast cancer, Wang et al. uncovered the role of the SWI/SNF core subunit BAF155 in promoting tumor progression and metastasis through arginine methylation. Arginine methylation of BAF155 by protein arginine methyltransferase 4 (PRMT4) redirects it to unique genomic locations including c-Myc pathway genes and genes involved in metastasis79 (Figure 6A). Restoring the expression of WT BAF155 but not unmethylable mutant in the BAF155 knockdown cells is able to rescue cell migration activity in vitro and lung metastasis ability in vivo79.
Figure 6. Other epigenetic mechanisms of metastasis.
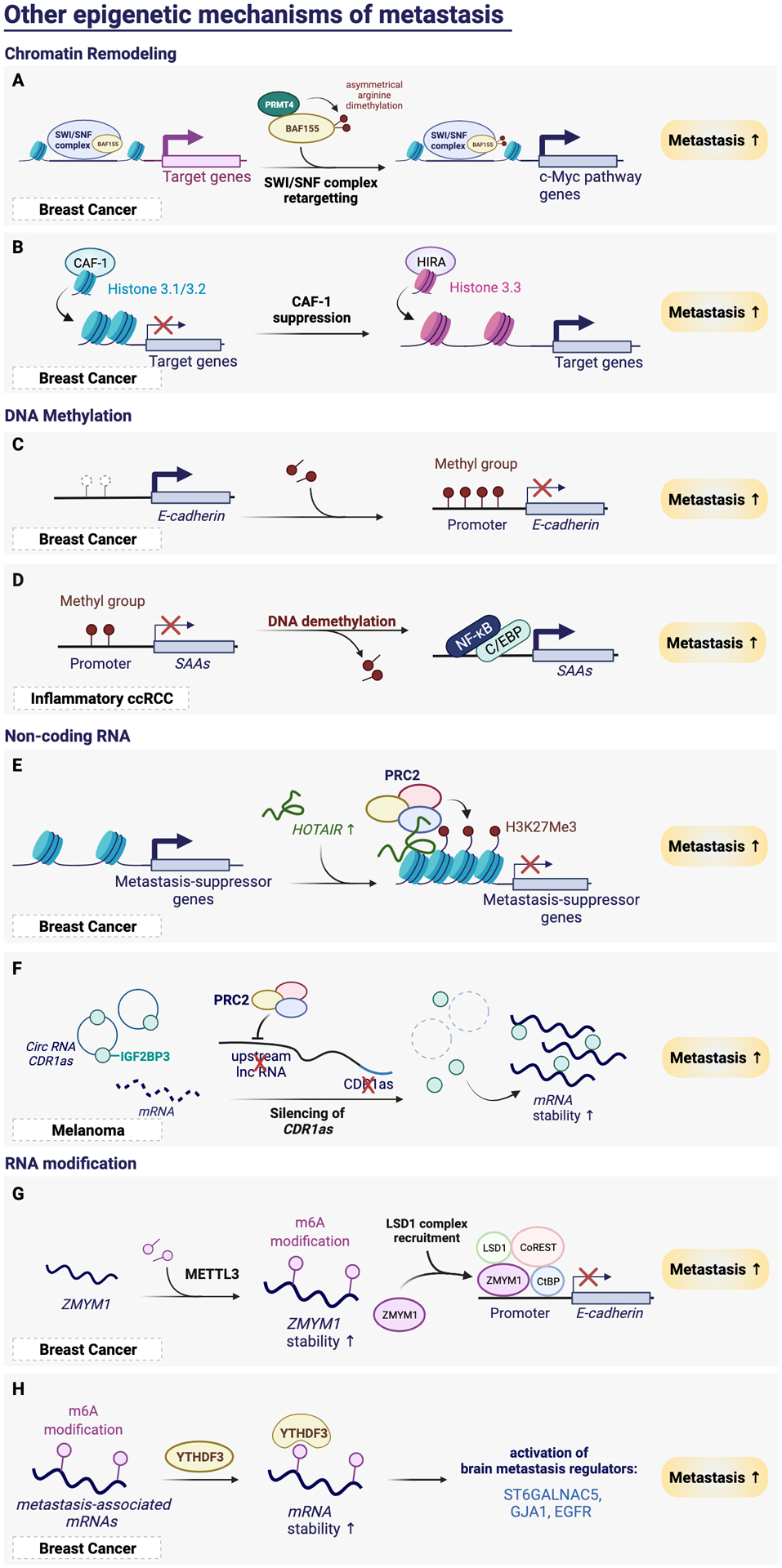
(A) Asymmetrical arginine demethylation of BAF155 by PRMT4 retargets the SWI/SNF complex to the c-Myc pathway genes to drive breast cancer metastasis. (B) CAF-1 suppression reduces the incorporation of canonical histone H3.1/3.2, and increases the incorporation of H3.3 variant to increase chromatin accessibility and activates the target gene expression and metastasis. (C) E-cadherin 5’ CpG island hypermethylation silences its expression and promotes metastasis in breast cancer. (D) Promoter demethylation at SAA genes allows for the binding of NF-κB and C/EBP to activate SAAs to promote metastasis. (E) Upregulation of HOTAIR recruits PRC2 complex to silence metastasis-suppressor genes, thereby promotes metastasis. (F) PRC2 silencing of the lncRNA upstream of circRNA CDR1as decreases the expression of CDR1as, which releases its binding partner IGF2BP2 to stabilize its mRNA target, thereby promotes metastasis. (G) m6A modification increases the stabilization of ZMYM1. Increased expression of ZMYM1 recruits the LSD1 complex to silence E-cadherin, thereby promotes metastasis. (H) YTHDF3 reads m6A marks on brain metastasis-associated mRNAs, which increases the mRNA stability and activates brain metastasis phenotypes.
In addition to the SWI/SNF complex activity, Gomes et al. recently identified the essential role of increased incorporation of histone H3 variant into the chromatin in metastasis colonization using a breast cancer model80. CAF-1 complex is a histone chaperone which deposits the canonical histone H3 into chromatin. Metastasis signaling leads to repressed CAF-1 expression and induced expression of histone cell cycle regulator (HIRA), which mediates H3.3 incorporation80. The replacement of histone variant at promoters and enhancers therefore increases chromatin accessibility and elicits an aggressive transcriptional program, resulting in tumor progression and metastasis formation80 (Figure 6B). These results suggest that histone chaperon proteins are critical regulators of chromatin structures which may act as a cell fate determinant and serve as valuable target for invasive cancers.
DNA methylation
DNA cytosine methylation is perhaps the most-characterized epigenetic mechanism, which is usually associated with transcriptional silencing81. Alteration in DNA methylation at CpG islands of specific gene loci can influence tumor progression. Several oncogenes are highly expressed through promoter hypomethylation82. In contrast, tumor suppressor genes are frequently silenced through DNA hypermethylation, such as VHL in ccRCC and p16 in lung cancer83. Furthermore, DNA methylation is able to regulate metastasis through silencing metastasis-suppressing genes. For example, E-cadherin 5’ CpG island was found hypermethylated in metastatic breast cancers27 (Figure 6C). In ccRCC, DNA demethylation at the serum amyloid A (SAA) genes was shown to contribute to neutrophil-dependent metastasis progression72 (Figure 6D). Thus, DNA methylation serves as important driver of many stages during cancer development, which has also been reviewed extensively previously23.
Profiling DNA methylation status now becomes a powerful analytical tool to improve cancer diagnosis. The study from Orizco et al. profiled DNA methylome of brain metastases (BM) developed from three primary cancer types: melanoma, breast, and lung cancer84. They discovered the DNA methylation profile from BM is distinct from that of primary CNS neoplasm, and that brain metastases from different origins harbor specific methylation patterns84. These findings implicate that the DNA methylation landscape of BM serve as an indication of tissue of origin, which not only aids the current diagnostic algorithm, but also guides therapeutic decisions in BM patients84. Genome-wide profiling of DNA methylome can also be applied to detect patient plasma sample or isolated circulating tumor cells from liquid biopsies85,86. These technologies not only provide comprehensive understanding of epigenome alteration during the transition stage from primary tumor to metastasis progression, but could also similarly facilitate the diagnosis and prediction of patient outcome in a less invasive way.
Non-coding RNA
In addition to protein-coding genes, non-coding RNAs including long non-coding RNAs (lncRNAs) and microRNAs have emerged as another important class of epigenetic regulators in various biological processes. Gupta et al. has demonstrated strong effect of lncRNA HOTAIR in promoting breast cancer metastasis. They discovered HOTAIR expression is upregulated in metastatic breast cancers, which is associated with patient prognosis87. Increased HOTAIR expression promotes selective re-targeting of the PRC2 complex genome-wide, leading to epigenetically silencing of metastasis-suppressor genes and increasing invasive ability of cancer cells87 (Figure 6E). Targeting HOTAIR or inhibiting the interaction between HOTAIR and PRC2 could thus be potential strategies of cancer therapy.
A list of microRNAs has been identified as metastasis-regulatory miRNAs88. In many cases, the upstream transcription factors which induce or repress the expression of these effector miRNAs have been identified. Notably, there are multiple miRNAs-regulated mechanisms that were identified in a single cancer type88. For example, in breast cancer, miR-9, activated by MYC/MYCN, is able to inhibit E-cadherin to promote migration and invasion, while miR-10b, activated by TWIST, is also able to promote metastasis through inhibiting HOXD10/RHOC axis88–90. These lines of evidence demonstrates that multiple layers of miRNA regulation may act in concert during cancer metastasis. Recently, the study from Hanniford et al. identified the role of circular RNAs (circRNA) in melanoma metastasis. Both the expression of circRNA CDR1as and its originated lncRNA were lost upon EZH2/PRC2-mediated-epigenetically silencing, thereby releasing their interactor, IGF2BP391. The released IGF2BP3 thus stabilizes their RNA targets to promotes melanoma progression and metastasis (Figure 6F)91. Consistently, low CDR1as level is associated with poor patient outcomes91. These findings have thus expanded the prognostic and therapeutic application potential to a rarer class of non-coding RNAs in cancer metastasis.
RNA modifications
The array of possible epigenetic changes mentioned above allows cancer cells to gain metastatic competence. Intriguingly, like the epigenome, the “epitranscriptome” generated by chemical modification of internal RNA has drawn increasing attention on its role in physiological and pathological conditions. The most abundant form of mRNA modification, N6-Methyladenosine (m6A) has been implicated in cellular processes including cancer, which is often resulting from the altered regulation in proteins that write, remove, or read the RNA modification marks92,93.
The study from Yue et al. recently discovered the critical role of m6A methyltransferase, METTL3 in metastasis of gastric cancer. METTL3 increased the stability of its target ZMYM1, which encodes a zinc finger protein that bound to and repressed the expression of E-cadherin to promote EMT and metastasis94 (Figure 6G). YTHDF3, one of the m6A reader, was identified by Chang et al. as a poor prognostic marker for breast cancer brain metastasis95. Knockdown of YTHDF3 significantly affects multiple critical steps of brain metastasis formation, including extravasation, overcome the blood-brain barrier, cell adhesion to astrocytes, and angiogenesis. They further identified that YTHDF3 specifically binds through its m6A recognition motif to multiple brain metastasis-associated mRNAs, such as ST6GALNAC5, GJA1, and EGFR. Such binding increases the translation of these key brain metastasis genes, thereby promoting breast cancer brain metastasis95 (Figure 6H). These findings demonstrate the potential of epitranscriptomic modifiers as metastasis biomarkers and specific targets for fighting against the metastasis diseases.
6. Targeting epigenetics in metastatic diseases
Regardless of the advancement of traditional cancer therapies, targeted therapies, and the recent developed immunotherapies, cancer may still relapse due to conditions such as drug resistance and cancer metastasis. Since the treatment options are limited when these situations occur, identifying alternative cancer targets are critical for the development of therapeutic intervention to overcome the deadly diseases.
In the previous sections, we have highlighted multiple critical findings on the role of epigenetics as the driver of cancer metastasis. Because many of these epigenetic alterations are induced upon the metastasis signaling, novel epigenetic therapeutic approaches would be specific for targeting metastatic diseases. Despite until now, there is no approved epigenetic drug specifically targeting the metastatic diseases, several types of these medications have been approved by the FDA for cancer treatment96. There are several advantages of targeting epigenetic modifiers. Cancer cells are more dependent on the specific epigenetic changes they acquired during cancer progression than the normal cells, and the reversible feature of epigenetic modifications allow cancer cells to be reprogrammed back to normal phenotype.
Currently, DNA methyltransferase inhibitors, HDAC inhibitors and EZH2 inhibitor are the only approved epigenetic anticancer drugs for specific disease conditions including myelodysplastic syndrome, T-cell lymphoma (CTCL/PTCL), or multiple myeloma97–101 (Table 1). However, it is worth noticing that many other epigenetic-targeted therapies have been investigated in multiple on-going clinical trials either alone in solid tumors or in combination with chemotherapy, targeted therapy or immunotherapy for synergistical effects, suggesting the great potential of developing epigenetic-targeted therapy for cancer patients20,97.
Table 1.
Epigenetic drugs approved by US or China FDA for cancer treatment.
| Category | Target | Compound | Brand name | Condition | Approved year |
|---|---|---|---|---|---|
| DNMT inhibitor | DNA methylation writers | Azacitidine | Vidaza | Myelodysplastic syndromes | U.S. FDA (2004) |
| Decitabine | Dacogen | Myelodysplastic syndromes | U.S. FDA (2006) | ||
| HDAC inhibitor | Histone acetylation erasers | Vorinostat | Zolinza | Cutaneous T-cell lymphoma | U.S. FDA (2006) |
| Romidepsin | Istodax | Cutaneous T-cell lymphoma | U.S. FDA (2009) | ||
| Belinostat | Beleodaq | Peripheral T-cell lymphoma | U.S. FDA (2014) | ||
| Panobinostat | Farydak | Multiple myeloma | U.S. FDA (2015) | ||
| Chidamide | Epidaza | Peripheral T-cell lymphoma | China FDA (2015) | ||
| EZH2 inhibitor | Histone methylation writer | Tazemetostat | Tazverik | Epithelioid sarcoma | U.S. FDA (2020) |
With accumulating studies that uncovered large-scale epigenetic changes in metastatic progression, more and more chemical inhibitors have been designed accordingly to target the epigenetic changes20,93. Among them, multiple inhibitors were able to suppress metastasis progression by targeting the step of EMT (Figure 7). Specifically, EZH2 methyltransferase inhibitor, GSK126, is able to reverse the repression of forkhead box C1 (FOXC1)-driven transcriptional program mediated by EZH2 in luminal B breast cancer102. Targeting EZH2 could therefore de-repressed the EMT marker, FOXC1, and reactivate a list of anti-metastatic genes102. Inhibiting WD repeat domain 5 (WDR5), the core subunit of histone H3K4 methyltransferase complex, with OICR-9429, is able to reverse EMT through repressing its target gene TGFβ1103. Furthermore, WDR5 inhibition sensitizes breast cancer cells to chemotherapy, paclitaxel treatment, which could be a promising approach to overcome resistance to standard treatment103. Moreover, histone deacetylase inhibitors, Pracinostat and SAHA, are able to suppress breast cancer metastasis and growth through inactivating the IL-6/STAT3 signaling104. Lastly, the BET family contains proteins are readers of the acetylated histone lysine, which accumulates on hyperacetylated chromatin regions as active promoters or enhancers. Therefore, targeting these BET proteins could disrupt the scaffolds and displace the transcription factors binding to repress transcription. Findings using the BET inhibitor tool compound JQ1 has promoted further development of many BET family inhibitors for cancer therapy105. For instance, JQ1 treatment on gastric cancer suppresses metastasis through inducing MET106. These lines of evidence, though not limited to, have shown numerous ways of targeting epigenetics during metastasis progression in different context. Further investigation of expanding the treatment of a single inhibitor to other cancer types or analyzing the effect of combining different inhibitors could be beneficial to elevate the epigenetic-targeted therapies to the next stage.
Figure 7. Strategies for targeting epigenetics in metastatic diseases.
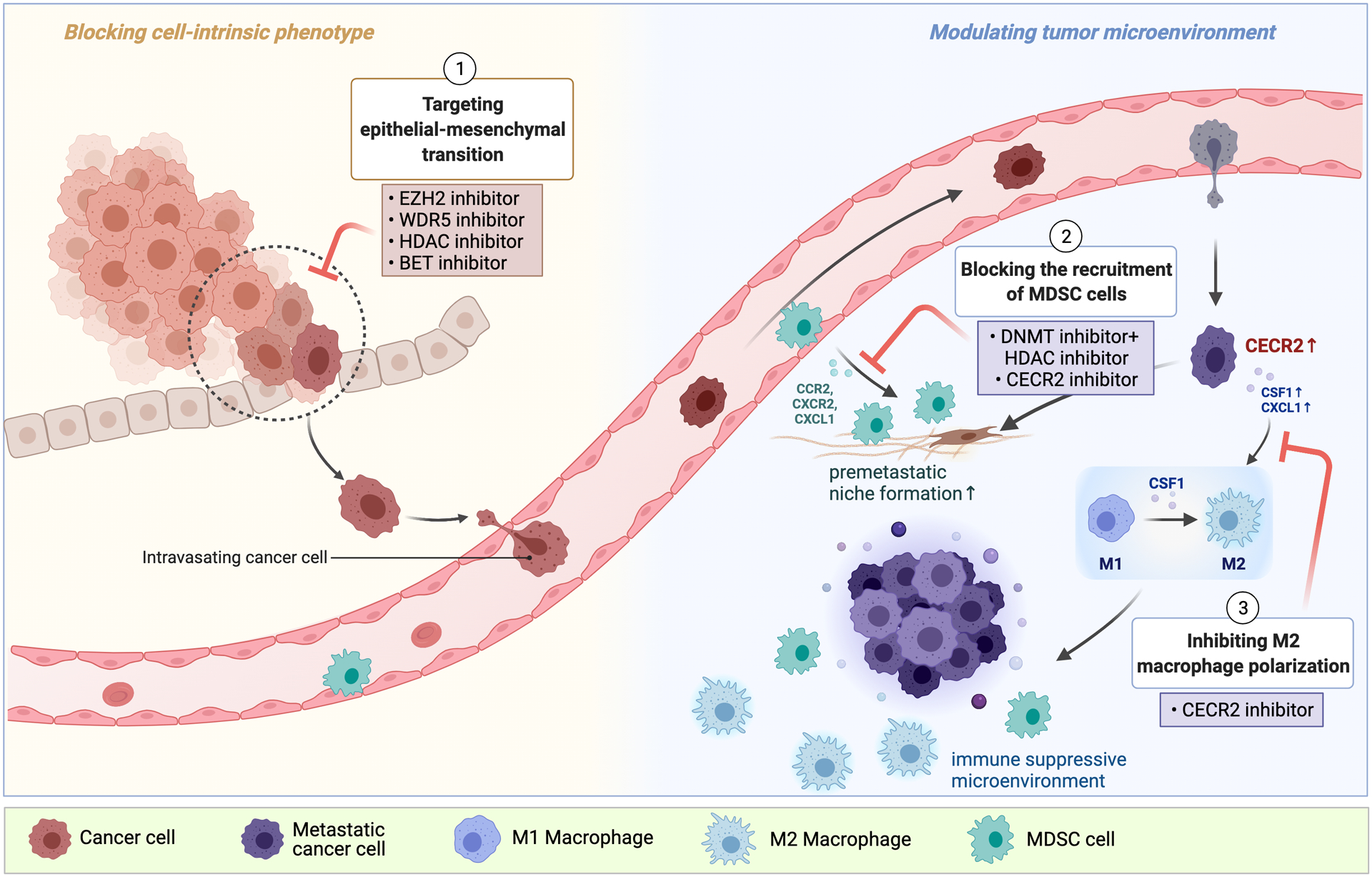
Multiple epigenetic regulators participate in cancer metastasis. These epigenetic changes are able to alter cell-intrinsic phenotypes or the tumor microenvironment, which could be targeted by specific inhibitors. (1) The step of epithelial-mesenchymal transition could be reversed by EZH2 inhibitor, WDR5 inhibitor, HDAC inhibitors, or BET inhibitor. These drugs suppress metastatic progression by decreasing the expression of genes regulating migratory capacity, cell invasiveness, and decrease the production of ECM components of cancer cells. (2) Modulating tumor microenvironment unfavored by the cancer cells could discourage metastasis seeding. Low dose DNMT inhibitor and HDAC inhibitor or CECR2 inhibitor could inhibit the production of several chemokines that attract MDSC cells, thereby suppresses premetastatic niche formation. This treatment approach could prevent the occurrence of metastatic diseases. (3) Activating the immune system could be effective in suppressing cancer growth. In metastatic cancer cells, upregulation of CECR2 promotes M2 macrophage polarization. Targeting M2 macrophage polarization with CECR2 inhibitor is able to reverse the immune suppressive microenvironment, thereby suppresses metastasis outgrowth.
The tumor microenvironments of distant organs are another critical factor to determine the success of metastasis colonization, which could be selectively modified by the primary tumor before metastasis occurs. Therefore, in addition to reverse the cell-intrinsic phenotype to block cancer metastasis, researchers have expanded the knowledge to assess the effect of leveraging inhibitors to diminish the interactions of cancer and stromal cells. The myeloid cells derived from bone marrow is one of the stromal components that contribute to the premetastatic microenvironment. Importantly, Lu et al. has provided strong evidence showing the use of low-dose DNA methyltransferase inhibitor, 5-azacytidine, and HDAC inhibitor, entinostat, in lung, breast, and oesophageal cancer models, which could disrupt the premetastatic niche formation and further inhibit lung metastasis after the removal of primary tumor107 (Figure 7). Specifically, low-dose epigenetic therapy not only impedes the migration of myeloid-derived suppressor cells (MDSCs) to the premetastatic microenvironment by downregulating expression of CCR2 and CXCR2, but also skew monocytic MDSCs differentiation107. In three mouse models, these treatments successfully prolonged the overall survival, which could be especially beneficial to the patients who received primary tumor resection as adjuvant therapy to avoid recurrence and inhibit further metastasis diseases to occur107.
Macrophage is another critical immune regulator within the tumor microenvironment. Recently, we have identified a targetable epigenetic factor, Cat eye syndrome critical region protein 2 (CECR2), which is able to drive breast cancer metastasis by promoting M2 macrophage polarization to suppress the anti-tumor immune responses108. Mechanistically, CECR2 is an acetyl-lysine reader, which can be recruited by acetylated NF-κB family member to activate macrophage-mediated immunosuppressor, CSF1 and CXCL1108. Since the pharmacological inhibitors of CECR2 bromodomain have been developed, we have tested a CECR2 inhibitor NVS-CECR2–1 in vitro and in a syngeneic mouse model. NVS-CECR2–1 treatment significantly inhibits the ability of breast cancer cells to metastasize to the lung108. CECR2 inhibition would thus be a promising therapeutic strategy to fight against metastasis (Figure 7).
7. Conclusion and future perspective
In the past decade, more and more studies have revealed that cancer cells use epigenetic mechanisms to reprogram cells to acquire metastatic traits. Due to the fact that metastasis is a multi-step process, the epigenetic reprograming could occur at any step or more than one step within the metastatic cascade in which different types of epigenetic modifications could act in concert to exert a more pronounced metastatic phenotype. Nowadays, the advancement of experimental and computational techniques such as ChIP-seq and ATAC-seq assists researchers to uncover a more comprehensive picture of the global changes in transcriptome and epigenome profiles during metastasis. Moreover, the expansion of paired sample collection from different experimental models and patients have provided a fruitful of information of the altered epigenetic status between primary tumor and metastases. These findings show great potential in developing metastasis-specific targeted therapy for cancer patients. However, it would still require huge efforts to further confirm the specificity, effectiveness, and the optimal timing of applying drug treatment to targeting certain epigenetic changes in the preclinical stage.
Because of the complexity of cancer, many open questions remain in the field. First, albeit many epigenetic regulators and transcription factors are identified as “drivers” for the metastasis progression, these drivers can influence multiple aspects of cancer, resulting in a combined net promoting effect in cancer progression. In addition, studies have suggested that acquiring genetic mutation might be an alternative mechanism by cancer cells during metastasis progression. For example, ESR1 mutation, the ligand-binding domain mutations of estrogen receptor, is primarily detected in metastatic breast cancer patients but very rare in primary tumors109,110. Therefore, researchers would need to be cautious in determining the therapeutic window and potential side effects when targeting a particular driver in cancer. In addition, the paired primary tumor and metastases samples collected by researchers are snapshots within the entire metastasis progression, which may not capture the initial change which drives metastasis, or it could lose the contribution of other epigenetic changes that occurred over time. Thus, lineage-tracing experiments or single-cell analyses may provide more comprehensive observations of the epigenetic changes over time. In addition, these studies could discover the specific population of cells leading the entire process during the metastasis transitions from the primary cancer lesions.
Secondly, the tumor microenvironment, including immune compartments, is critical for metastasis seeding and outgrowth. However, the limitations of current studies are that most of the in vivo studies are using xenograft models that lack the entire immune system. Thus, establishing additional syngeneic models or humanized mouse models to dissect the effect of epigenetic reprogramming in immune modulation would be much needed to capture the role of such regulators in late-stage metastasis. Many large-scale epigenome sequencings are done using metastatic derivative cell lines due to the requirement of collecting many starting materials, which do not capture the cancer cell and stromal cell interaction. The improved sequencing methods such as Omni-ATAC allow for the generation of chromatin accessibility profiles from frozen tissue samples111. Such method can be combined with spatial genomics platforms such as DBiT-seq to interrogate the epigenomic profiles of the tumor microenvironment112,113.
Lastly, since many signaling pathways in cancers are context-dependent, it would be worth studying whether a specific epigenetic regulation only happened in one cancer type or across multiple cancer types, potentially benefiting more patients. Further, there are also many different epigenetic mechanisms being identified in a single cancer type. These findings are likely generated using multiple model systems, thus identifying various dominant factors. It would be interesting to investigate which epigenetic regulator is essential and whether the combination of targeting multiple mechanisms could lead to a synergistic effect.
Metastatic diseases are hard to treat not only because of the complexity and malignancy of cancer cells but because they could occur years after the detection or removal of primary lesions or during malignant dormancy even before the detection of primary cancer114. Frequently, patients already progressed to advanced-stage diseases when they are diagnosed with cancer. Thus, a better understanding of metastatic tumor dormancy could be beneficial in metastasis prevention. Future studies could explore epigenetic mechanisms that maintain cell survival and dormancy and identify the critical factor in awakening dormant malignant cells at the distant site. Mechanistically, in addition to various types of epigenetic regulation identified in metastasis, epitranscriptomic regulation is likely another major player. Therefore, even with extensive studies in the epigenetic field, the picture of the epigenetic profile remains incomplete, and it can be more complicated in the future. It is crucial to assign the function to epigenetic changes and identify the causal relationship between epigenetics and metastasis. Until then, we will be able to revert the phenotype by applying specific modulators to target metastasis.
Acknowledgements
This work was partly supported by National Institutes of Health Awards F31CA243295 (to JFC) and R01CA237586 (to QY), and the Department of Defense Breast Cancer Research Program Award W81XWH-21-1-0411 (to QY). The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript. All the figures are adapted from images created with BioRender.com.
Abbreviations
- AP1
activator protein 1
- AR
androgen receptor
- ATAC-seq
transposase-accessible chromatin using sequencing
- BET
bromodomain and extraterminal
- BM
brain metastases
- BRD4
bromodomain containing 4
- CAF-1
chromatin assembly factor 1
- CCL14
C-C Motif chemokine ligand 14
- ccRCC
clear cell renal cell carcinoma
- CECR2
cat eye syndrome critical region protein 2
- ChIP
chromatin immunoprecipitation
- circRNA
circular RNAs
- CRISPR
clustered regularly interspaced short palindromic repeats
- CTCL
cutaneous T-cell lymphoma
- CXCR4
C-X-C Motif chemokine receptor 4
- DAB2IP
DAB2 interacting protein
- EGFR
epidermal growth factor receptor
- EMT
epithelial-mesenchymal transition
- EN1
engrailed 1
- ER
estrogen receptor
- EZH2
enhancer of zeste homolog 2
- FOSL1
fos-like antigen 1
- FOXA1
forkhead box protein A1
- GJA1
gap junction alpha-1 protein
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- HIF1
hypoxia-inducible factor 1
- HIF2A
hypoxia-inducible factor 2 alpha
- HIRA
histone cell cycle regulator
- HOXD10
homeobox D10
- HRE
hypoxia response element
- IGF2BP3
Insulin-like growth factor 2 mRNA-binding protein 3
- IL6
interleukin 6
- JAK
janus kinase
- KDM4A
histone demethylase 4A
- KDM4C
histone demethylase 4C
- KDM5A
histone demethylase 5A
- KDM5B
histone demethylase 5B
- LSD1
lysine-specific demethylase
- LUAD
lung adenocarcinoma
- m6A
N6-methyladenosine
- MDSC
myeloid-derived suppressor cells
- METTL3
methyltransferase Like 3
- NFIB
nuclear factor 1B
- NF-kB
nuclear factor kappa B
- NKX2.1
NK2 homeobox 1
- NuRD
nucleosome remodeling and histone deacetylase
- PHD
plant homeodomain
- PHF1
PHD finger protein 1
- PRC2
polycomb repressive complex 2
- PRMT1
protein arginine methyltransferase 1
- PRMT4
protein arginine methyltransferase 4
- PRMT5
protein arginine methyltransferase 5
- PTCL
peripheral T-cell lymphoma
- PTMs
post-translational modifications
- RHOC
Ras homolog gene family, member C
- RUNX2
RUNX Family Transcription Factor 2
- SAA
serum amyloid A
- SCC
squamous cell carcinoma
- SETD2
SET-domain containing protein 2
- ST6GALNAC5
alpha-N-acetylgalactosaminide alpha-2,6-sialyltransferase 5
- STAT3
signal transducer and activator of transcription 3
- SWI/SNF
switch/sucrose non-fermentable
- TF
transcription factor
- TGFβ1
transforming growth factor β1
- TNC
tenascin C
- TWIST
twist-related protein
- WDR5
WD repeat domain 5
- WDR77
WD Repeat Domain 77
- YTHDF3
YTH N6-methyladenosine RNA binding protein 3
- ZEB 1/2
zinc finger E-box-binding protein 1/2
- ZMYM1
zinc finger MYM-type containing 1
- ZMYND8
zinc finger MYND-type containing 8
Footnotes
Competing Interests
The authors declare no competing interests.
References:
- 1.Wittekind C, Neid M. Cancer invasion and metastasis. Oncology 2005;69 Suppl 1:14–6. [DOI] [PubMed] [Google Scholar]
- 2.Ganesh K, Massagué J. Targeting metastatic cancer. Nature Medicine 2021;27:34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science (New York, NY) 2011;331:1559–64. [DOI] [PubMed] [Google Scholar]
- 4.Nguyen DX, Bos PD, Massagué J. Metastasis: from dissemination to organ-specific colonization. Nature reviews Cancer 2009;9:274–84. [DOI] [PubMed] [Google Scholar]
- 5.Fidler IJ. Metastasis: quantitative analysis of distribution and fate of tumor emboli labeled with 125 I-5-iodo-2’-deoxyuridine. Journal of the National Cancer Institute 1970;45:773–82. [PubMed] [Google Scholar]
- 6.Bos PD, Zhang XH, Nadal C, et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009;459:1005–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Minn AJ, Gupta GP, Siegel PM, et al. Genes that mediate breast cancer metastasis to lung. Nature 2005;436:518–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nguyen DX, Bos PD, Massagué J. Metastasis: from dissemination to organ-specific colonization. Nature Reviews Cancer 2009;9:274–84. [DOI] [PubMed] [Google Scholar]
- 9.Robinson DR, Wu Y-M, Lonigro RJ, et al. Integrative clinical genomics of metastatic cancer. Nature 2017;548:297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 2008;14:818–29. [DOI] [PubMed] [Google Scholar]
- 11.Padua D, Zhang XH, Wang Q, et al. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 2008;133:66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erler JT, Bennewith KL, Cox TR, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 2009;15:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr., Kinzler KW. Cancer genome landscapes. Science (New York, NY) 2013;339:1546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casasent AK, Schalck A, Gao R, et al. Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing. Cell 2018;172:205–17.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Makohon-Moore AP, Zhang M, Reiter JG, et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat Genet 2017;49:358–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vanharanta S, Massagué J. Origins of metastatic traits. Cancer Cell 2013;24:410–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Minna JD, Kurie JM, Jacks T. A big step in the study of small cell lung cancer. Cancer Cell 2003;4:163–6. [DOI] [PubMed] [Google Scholar]
- 18.Jacob LS, Vanharanta S, Obenauf AC, et al. Metastatic Competence Can Emerge with Selection of Preexisting Oncogenic Alleles without a Need of New Mutations. Cancer research 2015;75:3713–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feinberg AP, Koldobskiy MA, Göndör A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet 2016;17:284–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahuja N, Sharma AR, Baylin SB. Epigenetic Therapeutics: A New Weapon in the War Against Cancer. Annu Rev Med 2016;67:73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blair LP, Yan Q. Epigenetic mechanisms in commonly occurring cancers. DNA Cell Biol 2012;31 Suppl 1:S49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patel SA, Vanharanta S. Epigenetic determinants of metastasis. Mol Oncol 2017;11:79–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chatterjee A, Rodger EJ, Eccles MR. Epigenetic drivers of tumourigenesis and cancer metastasis. Semin Cancer Biol 2017. [DOI] [PubMed] [Google Scholar]
- 24.Cao J, Yan Q. Histone demethylases set the stage for cancer metastasis. Science signaling 2013;6:pe15, 1–2. [DOI] [PubMed] [Google Scholar]
- 25.Cock-Rada A, Weitzman JB. The methylation landscape of tumour metastasis. Biol Cell 2013;105:73–90. [DOI] [PubMed] [Google Scholar]
- 26.Bakir B, Chiarella AM, Pitarresi JR, Rustgi AK. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol 2020;30:764–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nass SJ, Herman JG, Gabrielson E, et al. Aberrant methylation of the estrogen receptor and E-cadherin 5’ CpG islands increases with malignant progression in human breast cancer. Cancer research 2000;60:4346–8. [PubMed] [Google Scholar]
- 28.Tiwari N, Tiwari VK, Waldmeier L, et al. Sox4 is a master regulator of epithelial-mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell 2013;23:768–83. [DOI] [PubMed] [Google Scholar]
- 29.Luo C, Lim JH, Lee Y, et al. A PGC1alpha-mediated transcriptional axis suppresses melanoma metastasis. Nature 2016;537:422–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han HJ, Russo J, Kohwi Y, Kohwi-Shigematsu T. SATB1 reprogrammes gene expression to promote breast tumour growth and metastasis. Nature 2008;452:187–93. [DOI] [PubMed] [Google Scholar]
- 31.Pomerantz MM, Qiu X, Zhu Y, et al. Prostate cancer reactivates developmental epigenomic programs during metastatic progression. Nat Genet 2020;52:790–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu W, Huang W, Yang Y, et al. GATA3 recruits UTX for gene transcriptional activation to suppress metastasis of breast cancer. Cell Death Dis 2019;10:832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siersbæk R, Scabia V, Nagarajan S, et al. IL6/STAT3 Signaling Hijacks Estrogen Receptor α Enhancers to Drive Breast Cancer Metastasis. Cancer Cell 2020;38:412–23.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Denny SK, Yang D, Chuang CH, et al. Nfib Promotes Metastasis through a Widespread Increase in Chromatin Accessibility. Cell 2016;166:328–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.LaFave LM, Kartha VK, Ma S, et al. Epigenomic State Transitions Characterize Tumor Progression in Mouse Lung Adenocarcinoma. Cancer Cell 2020;38:212–28 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheung WK, Zhao M, Liu Z, et al. Control of alveolar differentiation by the lineage transcription factors GATA6 and HOPX inhibits lung adenocarcinoma metastasis. Cancer Cell 2013;23:725–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peluffo G, Subedee A, Harper NW, et al. EN1 Is a Transcriptional Dependency in Triple-Negative Breast Cancer Associated with Brain Metastasis. Cancer research 2019;79:4173–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cai WL, Greer CB, Chen JF, et al. Specific chromatin landscapes and transcription factors couple breast cancer subtype with metastatic relapse to lung or brain. BMC Med Genomics 2020;13:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yun M, Wu J, Workman JL, Li B. Readers of histone modifications. Cell Res 2011;21:564–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu PC, Lu JW, Yang JY, et al. H3K9 histone methyltransferase, KMT1E/SETDB1, cooperates with the SMAD2/3 pathway to suppress lung cancer metastasis. Cancer research 2014;74:7333–43. [DOI] [PubMed] [Google Scholar]
- 41.Salz T, Deng C, Pampo C, et al. Histone Methyltransferase hSETD1A Is a Novel Regulator of Metastasis in Breast Cancer. Mol Cancer Res 2015;13:461–9. [DOI] [PubMed] [Google Scholar]
- 42.Yi X, Guo J, Guo J, et al. EZH2-mediated epigenetic silencing of TIMP2 promotes ovarian cancer migration and invasion. Sci Rep 2017;7:3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Min J, Zaslavsky A, Fedele G, et al. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat Med 2010;16:286–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen H, Tu SW, Hsieh JT. Down-regulation of human DAB2IP gene expression mediated by polycomb Ezh2 complex and histone deacetylase in prostate cancer. The Journal of biological chemistry 2005;280:22437–44. [DOI] [PubMed] [Google Scholar]
- 45.Xie D, Gore C, Liu J, et al. Role of DAB2IP in modulating epithelial-to-mesenchymal transition and prostate cancer metastasis. Proceedings of the National Academy of Sciences of the United States of America 2010;107:2485–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mahmoud F, Shields B, Makhoul I, Hutchins LF, Shalin SC, Tackett AJ. Role of EZH2 histone methyltrasferase in melanoma progression and metastasis. Cancer biology & therapy 2016;17:579–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chu W, Zhang X, Qi L, et al. The EZH2-PHACTR2-AS1-Ribosome Axis induces Genomic Instability and Promotes Growth and Metastasis in Breast Cancer. Cancer research 2020;80:2737–50. [DOI] [PubMed] [Google Scholar]
- 48.Zhao Y, Ding L, Wang D, et al. EZH2 cooperates with gain-of-function p53 mutants to promote cancer growth and metastasis. Embo j 2019;38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang W, Bado IL, Hu J, et al. The bone microenvironment invigorates metastatic seeds for further dissemination. Cell 2021;184:2471–86.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Z, Li M, Wang D, et al. Post-translational modifications of EZH2 in cancer. Cell Biosci 2020;10:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Z, Wang D, Lu J, et al. Methylation of EZH2 by PRMT1 regulates its stability and promotes breast cancer metastasis. Cell Death Differ 2020;27:3226–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yuan H, Han Y, Wang X, et al. SETD2 Restricts Prostate Cancer Metastasis by Integrating EZH2 and AMPK Signaling Pathways. Cancer Cell 2020;38:350–65 e7. [DOI] [PubMed] [Google Scholar]
- 53.Vanharanta S, Shu W, Brenet F, et al. Epigenetic expansion of VHL-HIF signal output drives multiorgan metastasis in renal cancer. Nat Med 2013;19:50–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Y, Zhang H, Chen Y, et al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 2009;138:660–72. [DOI] [PubMed] [Google Scholar]
- 55.Li Q, Shi L, Gui B, et al. Binding of the JmjC demethylase JARID1B to LSD1/NuRD suppresses angiogenesis and metastasis in breast cancer cells by repressing chemokine CCL14. Cancer research 2011;71:6899–908. [DOI] [PubMed] [Google Scholar]
- 56.Ding X, Pan H, Li J, et al. Epigenetic activation of AP1 promotes squamous cell carcinoma metastasis. Sci Signal 2013;6:ra28.1–13, S0–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Luo W, Chang R, Zhong J, Pandey A, Semenza GL. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proceedings of the National Academy of Sciences of the United States of America 2012;109:E3367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cao J, Liu Z, Cheung WK, et al. Histone demethylase RBP2 is critical for breast cancer progression and metastasis. Cell Rep 2014;6:868–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Teng YC, Lee CF, Li YS, et al. Histone demethylase RBP2 promotes lung tumorigenesis and cancer metastasis. Cancer research 2013;73:4711–21. [DOI] [PubMed] [Google Scholar]
- 60.Liu R, Gao J, Yang Y, et al. PHD finger protein 1 (PHF1) is a novel reader for histone H4R3 symmetric dimethylation and coordinates with PRMT5-WDR77/CRL4B complex to promote tumorigenesis. Nucleic Acids Res 2018;46:6608–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li N, Li Y, Lv J, et al. ZMYND8 Reads the Dual Histone Mark H3K4me1-H3K14ac to Antagonize the Expression of Metastasis-Linked Genes. Mol Cell 2016;63:470–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McDonald OG, Li X, Saunders T, et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet 2017;49:367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ong CT, Corces VG. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat Rev Genet 2011;12:283–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Creyghton MP, Cheng AW, Welstead GG, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences of the United States of America 2010;107:21931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fagnocchi L, Poli V, Zippo A. Enhancer reprogramming in tumor progression: a new route towards cancer cell plasticity. Cell Mol Life Sci 2018;75:2537–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sur IK, Hallikas O, Vähärautio A, et al. Mice lacking a Myc enhancer that includes human SNP rs6983267 are resistant to intestinal tumors. Science (New York, NY) 2012;338:1360–3. [DOI] [PubMed] [Google Scholar]
- 67.Yin M, Guo Y, Hu R, et al. Potent BRD4 inhibitor suppresses cancer cell-macrophage interaction. Nat Commun 2020;11:1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lovén J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013;153:320–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Takeda DY, Spisak S, Seo JH, et al. A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell 2018;174:422–32 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roe JS, Hwang CI, Somerville TDD, et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 2017;170:875–88 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rodrigues P, Patel SA, Harewood L, et al. NF-kappaB-Dependent Lymphoid Enhancer Co-option Promotes Renal Carcinoma Metastasis. Cancer Discov 2018;8:850–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nishida J, Momoi Y, Miyakuni K, et al. Epigenetic remodelling shapes inflammatory renal cancer and neutrophil-dependent metastasis. Nat Cell Biol 2020;22:465–75. [DOI] [PubMed] [Google Scholar]
- 73.Wu SY, Chiang CM. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. The Journal of biological chemistry 2007;282:13141–5. [DOI] [PubMed] [Google Scholar]
- 74.Sabari BR, Dall’Agnese A, Boija A, et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science (New York, NY) 2018;361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ahn JH, Davis ES, Daugird TA, et al. Phase separation drives aberrant chromatin looping and cancer development. Nature 2021;595:591–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Morrow JJ, Bayles I, Funnell APW, et al. Positively selected enhancer elements endow osteosarcoma cells with metastatic competence. Nat Med 2018;24:176–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nature reviews Cancer 2011;11:481–92. [DOI] [PubMed] [Google Scholar]
- 78.Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci Adv 2015;1:e1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang L, Zhao Z, Meyer MB, et al. CARM1 methylates chromatin remodeling factor BAF155 to enhance tumor progression and metastasis. Cancer Cell 2014;25:21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gomes AP, Ilter D, Low V, et al. Dynamic Incorporation of Histone H3 Variants into Chromatin Is Essential for Acquisition of Aggressive Traits and Metastatic Colonization. Cancer Cell 2019;36:402–17 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schübeler D Function and information content of DNA methylation. Nature 2015;517:321–6. [DOI] [PubMed] [Google Scholar]
- 82.Feinberg AP, Vogelstein B. Hypomethylation of ras oncogenes in primary human cancers. Biochem Biophys Res Commun 1983;111:47–54. [DOI] [PubMed] [Google Scholar]
- 83.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. The New England journal of medicine 2003;349:2042–54. [DOI] [PubMed] [Google Scholar]
- 84.Orozco JIJ, Knijnenburg TA, Manughian-Peter AO, et al. Epigenetic profiling for the molecular classification of metastatic brain tumors. Nat Commun 2018;9:4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gkountela S, Castro-Giner F, Szczerba BM, et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019;176:98–112 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wu A, Cremaschi P, Wetterskog D, et al. Genome-wide plasma DNA methylation features of metastatic prostate cancer. J Clin Invest 2020;130:1991–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gupta RA, Shah N, Wang KC, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010;464:1071–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pencheva N, Tavazoie SF. Control of metastatic progression by microRNA regulatory networks. Nat Cell Biol 2013;15:546–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ma L, Young J, Prabhala H, et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol 2010;12:247–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 2007;449:682–8. [DOI] [PubMed] [Google Scholar]
- 91.Hanniford D, Ulloa-Morales A, Karz A, et al. Epigenetic Silencing of CDR1as Drives IGF2BP3-Mediated Melanoma Invasion and Metastasis. Cancer Cell 2020;37:55–70 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017;169:1187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jonkhout N, Tran J, Smith MA, Schonrock N, Mattick JS, Novoa EM. The RNA modification landscape in human disease. Rna 2017;23:1754–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yue B, Song C, Yang L, et al. METTL3-mediated N6-methyladenosine modification is critical for epithelial-mesenchymal transition and metastasis of gastric cancer. Mol Cancer 2019;18:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chang G, Shi L, Ye Y, et al. YTHDF3 Induces the Translation of m(6)A-Enriched Gene Transcripts to Promote Breast Cancer Brain Metastasis. Cancer Cell 2020;38:857–71 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Park JW, Han JW. Targeting epigenetics for cancer therapy. Arch Pharm Res 2019;42:159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cheng Y, He C, Wang M, et al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct Target Ther 2019;4:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Agrawal K, Das V, Vyas P, Hajdúch M. Nucleosidic DNA demethylating epigenetic drugs - A comprehensive review from discovery to clinic. Pharmacol Ther 2018;188:45–79. [DOI] [PubMed] [Google Scholar]
- 99.McClure JJ, Li X, Chou CJ. Advances and Challenges of HDAC Inhibitors in Cancer Therapeutics. Adv Cancer Res 2018;138:183–211. [DOI] [PubMed] [Google Scholar]
- 100.Chan TS, Tse E, Kwong YL. Chidamide in the treatment of peripheral T-cell lymphoma. OncoTargets and therapy 2017;10:347–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.First EZH2 Inhibitor Approved-for Rare Sarcoma. Cancer Discov 2020;10:333–4. [DOI] [PubMed] [Google Scholar]
- 102.Hirukawa A, Smith HW, Zuo D, et al. Targeting EZH2 reactivates a breast cancer subtype-specific anti-metastatic transcriptional program. Nat Commun 2018;9:2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Punzi S, Balestrieri C, D’Alesio C, et al. WDR5 inhibition halts metastasis dissemination by repressing the mesenchymal phenotype of breast cancer cells. Breast Cancer Res 2019;21:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen J, Li N, Liu B, et al. Pracinostat (SB939), a histone deacetylase inhibitor, suppresses breast cancer metastasis and growth by inactivating the IL-6/STAT3 signalling pathways. Life Sci 2020;248:117469. [DOI] [PubMed] [Google Scholar]
- 105.Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature 2010;468:1067–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhou S, Zhang S, Wang L, et al. BET protein inhibitor JQ1 downregulates chromatin accessibility and suppresses metastasis of gastric cancer via inactivating RUNX2/NID1 signaling. Oncogenesis 2020;9:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lu Z, Zou J, Li S, et al. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature 2020;579:284–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang M, Liu ZZ, Aoshima K, et al. CECR2 Drives Breast Cancer Metastasis by Suppressing Macrophage Inflammatory Responses. bioRxiv 2020:2020.09.10.291799. [Google Scholar]
- 109.Bertucci F, Ng CKY, Patsouris A, et al. Genomic characterization of metastatic breast cancers. Nature 2019;569:560–4. [DOI] [PubMed] [Google Scholar]
- 110.Robinson DR, Wu YM, Vats P, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet 2013;45:1446–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Corces MR, Trevino AE, Hamilton EG, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 2017;14:959–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu Y, Yang M, Deng Y, et al. High-Spatial-Resolution Multi-Omics Sequencing via Deterministic Barcoding in Tissue. Cell 2020;183:1665–81.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Deng Y, Bartosovic M, Ma S, et al. Spatial-ATAC-seq: spatially resolved chromatin accessibility profiling of tissues at genome scale and cellular level. bioRxiv 2021:2021.06.06.447244. [Google Scholar]
- 114.Manjili MH. Tumor Dormancy and Relapse: From a Natural Byproduct of Evolution to a Disease State. Cancer research 2017;77:2564–9. [DOI] [PMC free article] [PubMed] [Google Scholar]