

Abstract
Heart failure is prevalent in the elderly population. Inflammatory processes can contribute to the progression of heart failure by altering the balance of tissue healing and pathological remodeling during the injury response. New findings show that aging can alter immune cell phenotypes through the process of clonal hematopoiesis. This condition results from acquired somatic DNA mutations in specific driver genes that give rise to clonal expansions of mutant hematopoietic cells with overactive inflammatory properties. Recent clinical and experimental studies have shown that clonal hematopoiesis is prevalent in heart failure patients and associated with poor prognosis. In this review, we summarize current evidence that associates clonal hematopoiesis with the progression of heart failure. We further describe the mechanistic links between clonal hematopoiesis and the pro-inflammatory responses that can contribute to pathological outcomes in the heart. Finally, we provide perspectives on future research directions in the area of clonal hematopoiesis and heart failure.
Keywords: Clonal Hematopoiesis, Heart Failure, Inflammation
Introduction
Heart failure is a prevalent disease with poor prognosis that affects 6.2 million adults in the United States (1), and its predicted prevalence will increase ~30% over the next decade due to the ageing of the population (2). Growing evidence suggests that inflammation contributes to the pathogenesis of heart failure. Proinflammatory cytokine levels correlate with worse outcomes in patients with this condition (3–8). Similarly, the circulation, infiltration, and expansion of immune cells in myocardial tissue is correlated to poor outcomes in clinical studies of heart failure (9–12). Recent clinical trials indicate that targeting proinflammatory pathways can have beneficial effects in patients with heart failure (13–15). Immune cells have also been shown to contribute to pathological cardiac remodeling in mouse models of heart failure (12, 16–18). Collectively, these data suggest that alterations in the immune response towards tissue injury can lead to the progression of heart failure.
An elderly individual’s immune response can be altered through acquired somatic gene mutations in hematopoietic stem cells (HSC). When these mutations occur in a “driver” gene, the mutant HSC can gain a competitive growth advantage which leads to clonal expansion of that mutant cell (19). This process, referred to as “clonal hematopoiesis”, can lead to a condition in which a substantial fraction of an individual’s leukocytes is derived from one or more mutant HSC clones. The frequency of clonal hematopoiesis increases with age (20–22), and clonal hematopoiesis is associated with an increase in mortality due in large part to an increased risk of cardiovascular disease (20). Clonal hematopoiesis was initially associated with increased risk of inflammation-related atherosclerotic cardiovascular diseases including coronary heart disease and myocardial infarction (20, 23). However, more recent studies have also investigated the link between clonal hematopoiesis and heart failure, and how this may be mediated by alterations in immune system function.
In this review, we discuss clinical studies showing a correlation between clonal hematopoiesis and heart failure progression. We also discuss experimental models that reveal mechanistic links that involving altered immune system responses to cardiac injury.
Clinical studies associating clonal hematopoiesis with heart failure progression
Cohorts of heart failure patients have been analyzed to investigate the association between clonal hematopoiesis and long-term prognosis (24–28). These studies used chromosomal DNA from blood cells collected from chronic ischemic heart failure patients, and advanced targeted sequencing techniques to identify DNA mutations in clonal hematopoiesis driver genes. Mutations are identified as variants from the standard DNA sequence, and the percentage of that mutant variant sequence compared with all sequencing reads of the specific gene is defined as the Variant Allele Frequency (VAF). A key aspect of clonal hematopoiesis is a lack of absolute blood cell number expansion, which would be indicative of blood cancer. Instead, the mutant HSC clone expands and replaces non-mutant HSC in a clonal expansion event, resulting in a greater percentage of blood cells that arise from the mutant HSC and contain the mutation for the driver gene.
Clonal hematopoiesis in heart failure
In addition to atherosclerotic diseases, recent epidemiological studies have revealed an association between clonal hematopoiesis and heart failure. Dorsheimer et al. initially analyzed bone marrow-derived mononuclear cells from 200 patients with ischemic heart failure employing deep, error-corrected sequencing on 56 driver genes (24). Using the VAF threshold of 2%, they identified 38 patients carrying clonal hematopoiesis where the top two driver gene mutations were DNMT3A and TET2. In this cohort, the frequency of clonal hematopoiesis in patients with heart failure increased with age, reaching ~10% of patients 50-59 years old, ~21% in 60-69 years old, ~28% in 70-79 years old, and ~50% in >80 years old. These findings were recently corroborated by a study from Pascual-Figal et al. that investigated clonal hematopoiesis in patients with either ischemic and non-ischemic heart failure with reduced left ventricular ejection fraction (29). This study found clonal hematopoiesis driver gene mutations at VAF >2% in ~21% of patients 60-67 years old, ~41% of patients 68-75 years old, ~40% of patients 76-83 years old, and ~67% of patients >84 years old. These rates of clonal hematopoiesis are generally higher than that reported in other cohorts (20, 21), indicating that the incidence of clonal hematopoiesis is enriched in patients with heart failure (Figure 1). Both studies further investigated clinical outcomes of patients with the top 2 mutations, DNMT3A and TET2. Dorsheimer et al. (24) described that clonal hematopoiesis with either of these two mutations was associated with worse outcome with the hazard ratio of 2.09 compared to patients without clonal hematopoiesis, which is slightly higher than the increased all-cause mortality risk from clonal hematopoiesis that was reported in other populations. Intriguingly, when the VAF threshold was lowered to 0.5%, leading to the identification of 119 additional mutated clones (66 and 53 clones for DNMT3A-and TET2-mutation, respectively) affecting 69 patients, a dose-dependent relationship between clone size and the mortality was indicated. While mortality in the <1% VAF group was comparable to control group, the 1-2% VAF and VAF>2% groups showed increasing worse outcomes. Additionally, Pascual-Figal et al. found that clonal hematopoiesis associated with TET2 or DNMT3A mutations is predictive of worse outcomes in patients with heart failure, regardless of etiology, and this increased risk was also found in patients with only TET2 mutations or DNMT3A mutations compared to patients without clonal hematopoiesis (29).
Figure 1. Clonal hematopoiesis prevalence and mortality risk in various patient cohorts.
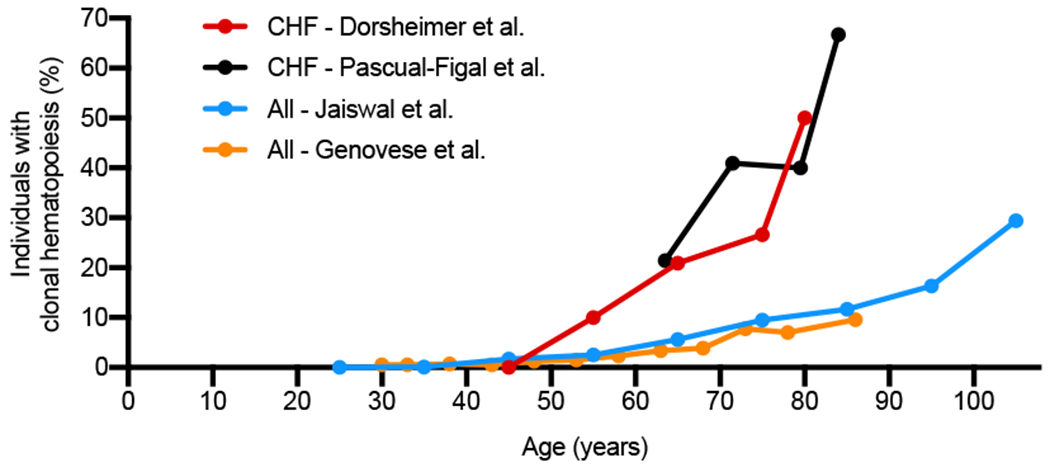
Prevalence of clonal hematopoiesis from mutations in known driver genes (>2% VAF) at different ages as presented in chronic heart failure (CHF) patient cohorts (CHF – Dorsheimer et al., CHF – Pascual-Figal et al.) compared to other patient cohorts or the general population (All - Jaiswal et al., All - Genovese et al.). Original clinical data from Dorsheimer et al. JAMA Cardiology 2018 (24), Pascual-Figal et al. JACC 2021 (29), Jaiswal et al. NEJM 2014 (20), and Genovese et al. NEJM 2014 (21).
An important question is raised by this finding: what is the threshold of VAF that affects cardiovascular outcomes? In the early epidemiological studies, clonal hematopoiesis of indeterminate potential (CHIP) was typically defined as VAF>2% in individuals without overt hematologic disorders. However, this threshold is somewhat arbitrary and influenced by technical limitations in the detection of low-abundance mutations rather than reflecting the biological or epidemiological findings. Advances in sequencing technologies have allowed researchers to identify clones with driver gene mutations as small as 0.03% VAF (30), revealing a greater prevalence of clonal hematopoiesis in healthy individuals (31). Thus, it is critical to clarify the biological significance of small VAF clones and assess their impact on human diseases. Recently, Zeiher’s group provided additional insights about this issue. In a study that employed error-corrected DNA sequencing, an analysis of 419 patients with chronic heart failure identified 154 patients with clonal hematopoiesis above newly defined VAF thresholds caused by mutations in 9 driver genes (26). Employing a receiver operating characteristic-curve analysis allowed the optimized VAF cut-off value to predict mortality for each of 9 driver genes (and 233 mutations in total): DNMT3A (1.15%), TET2 (0.73%), PHF6 (0.62%), SMC1A (0.62%), PPM1D (0.59%), EZH2 (0.58%), CEBPA (0.55%), SRSF2 (0.53%), and SETBP (0.5%). In another study, the same group focused on the patients with mutations in DNMT3A and/or TET2 in this cohort and reported that the patients with VAF larger than the aforementioned thresholds displayed significantly worse 5-year mortality compared to those with VAF values below the threshold (28, 32). A separate study also employed error-corrected DNA sequencing in 399 ischemic heart failure patients to identify the consequences of relatively rare clonal hematopoiesis driver gene mutations that are present at low VAF values (33). In this study, patients were excluded if they exhibited mutations TET2 or DNMT3A, or other clonal hematopoiesis mutations with a VAF > 2%. This analysis found that somatic mutations with low VAF in a set of genes (CBL, CEBPA, EZH2, GNB1, PHF6, SMC1A and SRSF2), are associated with mortality independently of the prevalent mutations in DNMT3A and TET2 (33). Furthermore, it has been shown that the sum of all VAF values derived from mutant genes within an individual also increased with mortality (26). Specifically, it was shown that a mutated gene VAF sum of greater than 3% is more predictive of a poor prognosis compared with individuals with total mutation burden less than 3%. These lines of evidence indicate that the pathology of heart failure may be impacted by clonal hematopoiesis, and that the association between VAF and the pathological phenotype may depend upon the identity of the driver gene mutation and the size of the mutant clone.
Increased chronic inflammation in heart failure patients with clonal hematopoiesis mutations
The pathophysiology of heart failure is associated with immune cell infiltration to cardiac tissue and dysregulated cytokine expression. In clonal hematopoiesis, the progeny leukocytes of the mutant HSC also harbor the mutation(s) such that the phenotypes of these circulating mutant immune cells can be altered and contribute inflammatory processes in the failing heart (34). The role of inflammation in clonal hematopoiesis-driven disease was highlighted by a study showing that a genetic mutation in IL6R, resulting in reduced IL6 signaling, is associated with reduced cardiovascular disease risk for individuals DNMT3A or TET2 clonal hematopoiesis (35). A study employing single cell RNA sequencing on mononuclear blood cells from individuals with heart failure revealed that patients with DNMT3A-induced clonal hematopoiesis have increased expression of pro-inflammatory genes IL1B, IL6R, NLRP3, and CD163 (25). It was also reported that TET2-mediated clonal hematopoiesis elevates CD14dim CD16+ nonclassical monocytes, whereas DNMT3A-mediated clonal hematopoiesis increases the Th17/Treg ratio, although within the normal blood count range (36). CD14dim CD16+ nonclassical monocytes can secrete pro-inflammatory cytokines TNF-a, IL-1B, and IL-6 (37), and Th17/Treg ratio is used to determine the pro-inflammatory profile of T-cells (38). These data indicate that mutation-specific alterations in the immune system could contribute to the pathophysiology of heart failure.
Mechanisms of clonal hematopoiesis-mediated heart failure progression
Experimental mouse models of clonal hematopoiesis have been generated to investigate the effects of different driver gene mutations on heart failure progression. Currently, three driver gene mutations have been tested in clonal hematopoiesis-mediated models of heart failure: Dnmt3a, Tet2, and JAK2V617F. In these models, the chromosomal DNA of HSC are altered to mimic the driver gene mutations that cause clonal hematopoiesis. Mutations in DNMT3A and TET2 that result in loss-of-function will lead to clonal hematopoiesis (39). Therefore, Dnmt3a or Tet2 mutant HSC have been generated through knockout approaches, including Cre-lox recombination or CRISPR/Cas9-mediated gene silencing (40–43). On the other hand, JAK2V617F is an activating driver mutation, and this mutation has been modeled through lentivirus-mediated transduction of HSC and the specific expression of the mutant gene in myeloid cells (44). In these studies, the genetically engineered HSC are transplanted into recipient mice to recapitulate the clinical presentation of clonal hematopoiesis. In some cases, this can be accomplished through a competitive bone marrow transplant method where mice are lethally irradiated and transplanted with a mixture of mutant and wild-type HSC to replenish the bone marrow. Alternatively, this can be accomplished through an adoptive transfer method where donor mutant HSC are administered intravenously to recipient in non-conditioned mice (45). The latter approach results in a low percentage of mutant HSC that engraft in the recipient and then can undergo a slow expansion process, while avoiding complications arising from unintended effects of radiation on the bone marrow stem cell niche and leukocyte composition (43, 45). These models of clonal hematopoiesis have then been combined with experimental models of heart failure to elucidate how clonal hematopoiesis affects the biology of injury-induced cardiac remodeling. As revealed by these studies and described in detail below, clonal hematopoiesis accelerates cardiac dysfunction largely through pro-inflammatory immune responses that promote greater cardiac remodeling (Figure 2).
Figure 2. Mechanisms of how clonal hematopoiesis promotes heart failure progression.
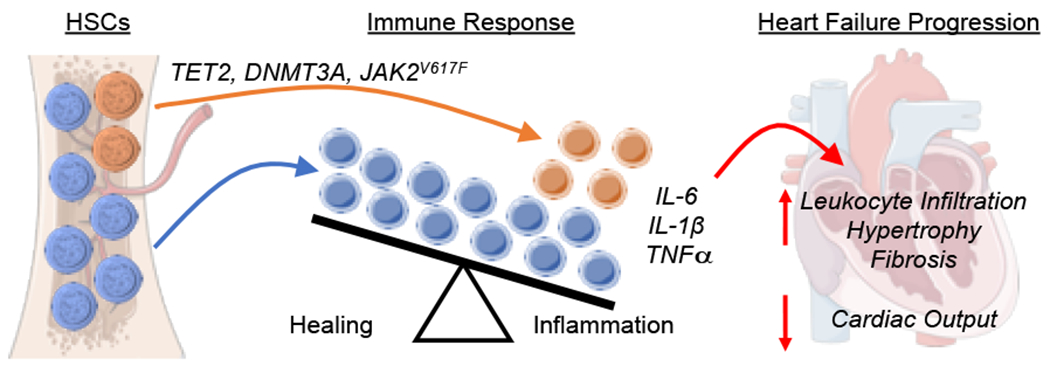
Driver gene mutations including TET2, DNMT3A, and JAK2V617F, will lead to aberrant clone expansions in hematopoietic stem cells (HSCs) that increasingly give rise to mutant progeny leukocytes. The mutant leukocytes display altered immune responses, shifting the balance away from a healing and towards an overt inflammatory response involving the increased expression of IL-6, IL-1β, and TNFα in addition to other immune cell perturbations. These mutant leukocytes promote myocardial cardiac dysfunction, hypertrophy and fibrosis, and accelerate the progression of heart failure.
Clonal hematopoiesis promotes heart failure in mouse models
Experimental mouse models of clonal hematopoiesis display greater cardiac dysfunction in aging mice or in young mice that are challenged by injury. To date, four heart failure models have been employed to assess the effects of clonal hematopoiesis in mice: infusion of angiotensin II, transverse aortic constriction (TAC) surgery, permanent left anterior descending (LAD) artery ligation, and age-associated cardiomyopathy in the absence of induced cardiac injury (46). These experimental systems have been applied to different models of clonal hematopoiesis that have focused on the driver genes Tet2, Dnmt3a, and JAK2V617F.
The initial study to investigate the effects of clonal hematopoiesis on heart failure examined the consequences of the Tet2 driver gene (47). In the experimental heart failure models of LAD artery ligation and TAC surgery, Tet2-mediated clonal hematopoiesis was modeled by competitive bone marrow transplantation, and this condition led to worsened cardiac remodeling in the later stages of heart failure (47). In both heart failure models, Tet2-mediated clonal hematopoiesis mice showed reduced cardiac output and increased cardiac fibrosis, with the LAD ligation model resulting in increased left ventricular systolic and diastolic volumes and the TAC model resulting in increased heart size and left ventricular posterior wall thickness.
In another study, the ablation of Tet2 or Dnmt3a in isolated HSC was achieved by lentiviral-mediated CRISPR/Cas9 gene editing, followed by bone marrow transplantation and angiotensin II infusion to induce heart failure (41). In this study, it was shown that employing CRISPR/Cas9 to edit Tet2 led to a similar reduction in cardiac function when compared with a conventional bone marrow transplantation model. This study was also the first to investigate Dnmt3a-mediated clonal hematopoiesis in cardiovascular disease model, showing that angiotensin II infusion induced greater cardiac remodeling and dysfunction in the Dnmt3a-deficient condition than in control mice (41). The lentivirus-mediated approach for transducing HSPC prior to transplantation was then applied to overexpress the JAK2V617F mutation in myeloid cells in the experimental heart failure models of LAD artery ligation and TAC surgery (44). After LAD artery ligation or TAC surgery, the JAK2V617F clonal hematopoeisis model displayed reduced cardiac output and worsened cardic remodeling, resembling the results from Tet2-mediated clonal hematopoeisis mice.
In addition to the experimental models of heart failure, the effects of ageing on cardiac function of mice with Tet2-mediated clonal hematopoiesis was also examined (45). This study avoided the negative effects of radiation treatment in the bone marrow transplantation procedure by implementing an adoptive transfer method for inducing clonal hematopoiesis. In this procedure, driver gene-manipulated, lineage-negative bone marrow cells are injected into the circulation of the mice and the HSC are allowed to naturally engraft into their niches and then slowly expand over time (45). Non-conditioned mice implanted with Tet2-deficient HSC displayed time-dependent cardiac dysfunction, with systolic dysfunction, hypertrophy and fibrosis occurring at an earlier age (18 months) in the Tet2 clonal hematopoiesis model than in control mice. Collectively, these data show that clonal hematopoiesis induced by multiple driver gene mutations can promote cardiac dysfunction in mouse models of heart failure and advanced age.
Clonal hematopoiesis contributes to experimental heart failure through pro-inflammatory mechanisms
Consistent with clinical findings of increased inflammation associated with clonal hematopoiesis, experimental models of clonal hematopoiesis and heart failure show greater levels of cardiac inflammation through increased leukocyte infiltration and increased cytokine expression from mutant leukocytes. After heart failure induction, mice with Dnmt3a-mediated clonal hematopoiesis showed enhanced cardiac infiltration of macrophages and increased myocardial inflammation with greater gene expression of markers for monocytes (Cd68) and T cells (Cd3e, Cd4, Cd8) in cardiac tissue (41). This pro-inflammatory signature is similar to what was subsequently observed in heart failure patients with DNMT3A-mediated clonal hematopoiesis, where single cell RNA sequencing of peripheral blood showed an increased inflammatory profile of monocytes and an increase in monocyte-T cell interactions in patients with DNMT3A mutations (48). In the lentivirus overexpression model of JAK2V617F-mediated clonal hematopoiesis, greater macrophage infiltration of the heart was observed following cardiac injury (44). Similarly, models of Tet2-mediated clonal hematopoiesis identified greater leukocyte infiltration in the heart following injury and greater myocardial cytokine production (47). In cell culture studies, the expression of pro-inflammatory cytokines is increased in myeloid cells with clonal hematopoiesis driver gene mutations. Myeloid cells lacking Dnmt3a or Tet2 showed greater induction of II6, II1b, CxcI1, CxcI2, and CcI5 genes after LPS stimulation (41). JAK2V617F mutant myeloid cells showed increased expression of II6, II1b, Tnfa, CcI2, and Aim2 after LPS stimulation (44).
In some of these models, the NLRP3 inflammasome was found to have a causal role in accelerated heart failure progression. The NLRP3 inflammasome consists of a group of proteins that promote inflammation by inducing the secretion of IL-1β in macrophages upon activation by various stimuli (49). Prior studies have shown that NLRP3 inflammasome activation has a role in heart failure (15). In experimental clonal hematopoiesis mediated by Tet2 mutations, NLRP3 inflammasome inhibition by treatment with the MCC950 small molecule inhibitor negated the detrimental effects of clonal hematopoiesis on cardiac parameters after TAC or LAD ligation surgeries (47). Similarly, inflammasome inhibition has been shown to reverse the adverse effects of Tet2-mediated clonal hematopoiesis in models of atherogenesis (40) and insulin resistance caused by diet-induced obesity (50). Taken together, these results highlight that acquired driver gene mutations in HSC will affect the biology of progeny leukocytes and promote myocardial inflammation in models of heart failure.
Mutations in TET2 or DNMT3A may promote hyper-active inflammatory reactions through epigenetic modifications in immune cells. DNMT3A promotes methylation of DNA to inhibit gene expression which can promote hematologic malignancies (51), whereas TET2 is a multifunctional epigenetic regulator that can affect gene expression through diverse mechanisms (52). While TET2 and DNMT3A have opposing roles on DNA methylation (53), multiple lines of evidence suggest that TET2 regulates IL-1β and the production of downstream cytokines, such as IL-6, through a histone deacetylation mechanism (54–56). Supporting an HDAC-mediated mechanism of IL-1β regulation by TET2, our lab has reported that TET2-deficient macrophages show greater histone H3 acetylation at the II1b promoter region and treatment of macrophages with an HDAC inhibitor increased IL-1b expression in macrophages and abolished expression differences between TET2-deficient and wild-type genotypes (40). Furthermore, a catalytically inactive form of TET2, unable to oxidize 5-methylcytosine in DNA to 5-hydroxymethylcytosine, was able to suppress IL-1b expression in macrophages as efficiently as wild-type TET2. Therefore, although DNMT3A and TET2 can promote opposing effects on DNA methylation, mechanistic studies on macrophage function and IL-1β production suggest that mutations in DNMT3A or TET2 promote hyper-active inflammatory reactions through different epigenetic regulatory mechanisms.
As noted above, a key question is what proportion of the mutant peripheral blood cells is sufficient to modify the pathophysiology of heart failure, particularly when a very low VAF can be associated with disease prognosis (26, 28). Thus, how can such a small population change lead to a physiologically relevant alteration in disease progression? Findings from experimental studies in a mouse atherosclerosis model may provide potential explanations (40). Clonal hematopoiesis resulting from Tet2-deficiency leads to overactivation of IL-1β in myeloid cells. This can activate vascular endothelial cells and further recruit monocytes to the plaque regardless of their Tet2 genotype (40). Further, since IL-1β can augment its own expression, a positive feedback loop can potentially be initiated by a small population of mutant myeloid cells. In future experimental studies on clonal hematopoiesis, it will be important to develop a better understanding of how the somatic mutations can potentially exert non-cell autonomous effects, that may provide insights about disease processes can be impacted by low VAF clones.
Finally, during heart failure progression, excessive fibrosis can interfere with cardiac contraction and worsens heart failure (57, 58). Immune system perturbations will promote myofibroblast extracellular matrix deposition (59), and it is reasonable to expect that immune cell perturbations by clonal hematopoiesis will trigger fibrotic responses. Supportive of this supposition, all of the experimental models of clonal hematopoiesis (Dnmt3a, Tet2, and JAK2V617F) have displayed greater levels of cardiac fibrosis (41, 44, 47). Additionally, elevation of renal fibrosis was noted in the angiotensin II infusion models of Tet2-and Dnmt3a-mediated clonal hematopoiesis (41). In some of these systems, it was shown that reducing inflammatory response through inhibition of the NLRP3 inflammasome can diminish the increases in fibrosis that are brought about by the clonal hematopoiesis model (47).
Future Directions
Recent clinical and experimental studies have provided ample evidence that heart failure can be accelerated by the condition of clonal hematopoiesis. However, many questions remain unanswered. First, the number of patients with heart failure in investigations for clonal hematopoiesis has been limited. Future studies increasing patient number could strengthen the association between clonal hematopoiesis and heart failure, particularly elderly patients. An analysis of inflammatory phenotype and patient outcome as a function of VAF would strengthen the hypothesis that aberrant inflammatory responses are a critical of the pathological mechanism. Additionally, numerous driver gene mutations have been discovered in patient populations, but the role of only a few of these mutations have been investigated in heart failure models to date. It is reasonable to assume that the mutations in different driver genes will exert functionally different effects on cardiac pathophysiology, but current studies have yet to elucidate this level of granularity in the data. It is also possible that as-yet-unidentified clonal hematopoiesis driver genes may play a role in the pathogenesis of heart failure. Currently, most studies have focused on mutations in driver genes that are recurrently mutated in hematologic disorders. However, an unbiased genome wide survey has suggested that as many as 80% of these aberrant clonal events in the hematopoietic system are caused by unknown drivers (22). Future studies that aim to discover novel clonal hematopoiesis driver mutations would be of interest and would advance the field.
Beyond age, the underlying causes of clonal hematopoiesis are relatively unknown. In addition to genotoxic stress (60) and smoking (22, 61–63), the roles of other environmental factors on this process require further attention. In addition, numerous genomic loci, in particular the TERT locus, have been associated with a propensity to develop clonal hematopoiesis (22, 61). However, the molecular connections between these heritable loci and the prevalence of clonal hematopoiesis are unknown.
With regard to heart failure, epidemiological studies have indicated that relatively small hematopoietic clone expansion can be indicative of poor prognosis. It is worth noting that standard next generation DNA sequencing has an inherent error rate that limits the detection of low-abundance DNA mutations (64, 65). However, a recent advance in error-corrected DNA sequencing, that incorporates internal DNA barcodes, can greatly reduce sequencing error and allow for accurate detection of mutations as low as 0.01% VAF (27, 28, 66). Although error-corrected sequencing is costly and typically only provides information about a select group of driver genes, these more advanced techniques will be helpful in assessing conditions that contribute to the genesis of clonal hematopoiesis in longitudinal studies. Furthermore, advanced sequencing techniques can identify patients with multiple driver gene mutations with low VAF values. In this regard, Cremer et al. (26), showed with error-corrected sequencing that the combined VAF of multiple driver gene mutations within a single patient can be predictive of poor outcome in heart failure. Thus, more sensitive methods to identify driver gene mutations may give a more accurate description of overall clonal hematopoiesis burden and its impacts on disease processes.
Additional research into the mechanisms by which inflammation is regulated by clonal hematopoiesis driver gene mutations can yield novel insights about this new disease mechanism. As many of the clonal hematopoiesis driver genes are epigenetic modifiers, additional studies of their effects on chromatin structure in hematopoietic cells could be fruitful. In this regard, epigenetic modulation of DNA methylation patterns are associated with an individual’s age (67), and epigenetic age is markedly increased in individuals that harbor with clonal expansions in their blood. Thus, clonal hematopoiesis driver genes may accelerate the biological aging process in part through the direct epigenetic alteration of chromatin (68). Thus, additional studies that employ Assay for Transposase Accessible Chromatin using Sequencing (ATAC-Seq), to reveal transcriptionally active regions in chromatin (69–71), may prove useful in understanding how clonal hematopoiesis driver gene mutations confer inflammatory phenotypes to hematopoietic cells.
Conclusions
Clonal hematopoiesis is prevalent in the elderly and prognostic of worse outcome in patients with heart failure. To the extent that it has been studied, the mutations that cause clonal hematopoiesis also confer pro-inflammatory phenotypes to leukocytes. Experimental studies have shown that clonal hematopoiesis elevates myocardial inflammation leading to cardiac dysfunction, hypertrophy, and fibrosis. Identifying clonal hematopoiesis in individuals with heart failure could provide diagnostic information as well as guidance for therapeutic strategies that target the immune system to treat this condition.
Acknowledgements
The authors were supported by grants to N.W.C. (NIH T32 HL007284) and K.W. (NIH R01 HL138014, R01 HL139819, R01 HL141256, and R01 HL142650).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation. 2020;141(9):e139–e596. [DOI] [PubMed] [Google Scholar]
- 2.Savarese G, Lund LH. Global Public Health Burden of Heart Failure. Card Fail Rev. 2017;3(1):7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323(4):236–41. [DOI] [PubMed] [Google Scholar]
- 4.Testa M, Yeh M, Lee P, Fanelli R, Loperfido F, Berman JW, et al. Circulating levels of cytokines and their endogenous modulators in patients with mild to severe congestive heart failure due to coronary artery disease or hypertension. J Am Coll Cardiol. 1996;28(4):964–71. [DOI] [PubMed] [Google Scholar]
- 5.Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation. 2001;103(16):2055–9. [DOI] [PubMed] [Google Scholar]
- 6.Kardys I, Knetsch AM, Bleumink GS, Deckers JW, Hofman A, Stricker BH, et al. C-reactive protein and risk of heart failure. The Rotterdam Study. Am Heart J. 2006;152(3):514–20. [DOI] [PubMed] [Google Scholar]
- 7.Kalogeropoulos A, Georgiopoulou V, Psaty BM, Rodondi N, Smith AL, Harrison DG, et al. Inflammatory markers and incident heart failure risk in older adults: the Health ABC (Health, Aging, and Body Composition) study. J Am Coll Cardiol. 2010;55(19):2129–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swirski FK, Nahrendorf M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat Rev Immunol. 2018;18(12):733–44. [DOI] [PubMed] [Google Scholar]
- 9.Engstrӧm G, Melander O, Hedblad B. Leukocyte count and incidence of hospitalizations due to heart failure. Circ Heart Fail. 2009;2(3):217–22. [DOI] [PubMed] [Google Scholar]
- 10.Tsujioka H, Imanishi T, Ikejima H, Kuroi A, Takarada S, Tanimoto T, et al. Impact of heterogeneity of human peripheral blood monocyte subsets on myocardial salvage in patients with primary acute myocardial infarction. J Am Coll Cardiol. 2009;54(2):130–8. [DOI] [PubMed] [Google Scholar]
- 11.Glezeva N, Voon V, Watson C, Horgan S, McDonald K, Ledwidge M, et al. Exaggerated inflammation and monocytosis associate with diastolic dysfunction in heart failure with preserved ejection fraction: evidence of M2 macrophage activation in disease pathogenesis. J Card Fail. 2015;21(2):167–77. [DOI] [PubMed] [Google Scholar]
- 12.Hulsmans M, Sager HB, Roh JD, Valero-Muñoz M, Houstis NE, Iwamoto Y, et al. Cardiac macrophages promote diastolic dysfunction. J Exp Med. 2018;215(2):423–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Everett BM, Cornel JH, Lainscak M, Anker SD, Abbate A, Thuren T, et al. Anti-Inflammatory Therapy With Canakinumab for the Prevention of Hospitalization for Heart Failure. Circulation. 2019;139(10):1289–99. [DOI] [PubMed] [Google Scholar]
- 14.Van Tassell BW, Canada J, Carbone S, Trankle C, Buckley L, Oddi Erdle C, et al. Interleukin-1 Blockade in Recently Decompensated Systolic Heart Failure: Results From REDHART (Recently Decompensated Heart Failure Anakinra Response Trial). Circ Heart Fail. 2017; 10(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abbate A, Toldo S, Marchetti C, Kron J, Van Tassell BW, Dinarello CA. Interleukin-1 and the Inflammasome as Therapeutic Targets in Cardiovascular Disease. Circ Res. 2020;126(9):1260–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sager HB, Hulsmans M, Lavine KJ, Moreira MB, Heidt T, Courties G, et al. Proliferation and Recruitment Contribute to Myocardial Macrophage Expansion in Chronic Heart Failure. Circ Res. 2016;119(7):853–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liao X, Shen Y, Zhang R, Sugi K, Vasudevan NT, Alaiti MA, et al. Distinct roles of resident and nonresident macrophages in nonischemic cardiomyopathy. Proc Natl Acad Sci U S A. 2018;115(20):E4661–E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Sano S, Oshima K, Sano M, Watanabe Y, Katanasaka Y, et al. Wnt5a-Mediated Neutrophil Recruitment Has an Obligatory Role in Pressure Overload-Induced Cardiac Dysfunction. Circulation. 2019;140(6):487–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forsberg LA, Gisselsson D, Dumanski JP. Mosaicism in health and disease - clones picking up speed. Nat Rev Genet. 2017;18(2):128–42. [DOI] [PubMed] [Google Scholar]
- 20.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zink F, Stacey SN, Norddahl GL, Frigge ML, Magnusson OT, Jonsdottir I, et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood. 2017;130(6):742–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med. 2017. ;377(2): 111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dorsheimer L, Assmus B, Rasper T, Ortmann CA, Ecke A, Abou-El-Ardat K, et al. Association of Mutations Contributing to Clonal Hematopoiesis With Prognosis in Chronic Ischemic Heart Failure. JAMA Cardiol. 2019;4(1):25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abplanalp WT, Mas-Peiro S, Cremer S, John D, Dimmeler S, Zeiher AM. Association of Clonal Hematopoiesis of Indeterminate Potential With Inflammatory Gene Expression in Patients With Severe Degenerative Aortic Valve Stenosis or Chronic Postischemic Heart Failure. JAMA Cardiol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cremer S, Kirschbaum K, Berkowitsch A, John D, Kiefer K, Dorsheimer L, et al. Multiple Somatic Mutations for Clonal Hematopoiesis Are Associated With Increased Mortality in Patients With Chronic Heart Failure. Circ Genom Precis Med. 2020;13(4):e003003. [DOI] [PubMed] [Google Scholar]
- 27.Dorsheimer L, Assmus B, Rasper T, Ortmann CA, Abou-El-Ardat K, Kiefer KC, et al. Hematopoietic alterations in chronic heart failure patients by somatic mutations leading to clonal hematopoiesis. Haematologica. 2020;105(7):e328–e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Assmus B, Cremer S, Kirschbaum K, Culmann D, Kiefer K, Dorsheimer L, et al. Clonal haematopoiesis in chronic ischaemic heart failure: prognostic role of clone size for DNMT3A-and TET2-driver gene mutations. Eur Heart J. 2021. ;42(3):257–65. [DOI] [PubMed] [Google Scholar]
- 29.Pascual-Figal DA, Bayes-Genis A, Diez-Dies M, Hernandez-Vincente A, Vazquez-Andres D, de la Barrera J, et al. Clonal Hematopoiesis and Risk of Progression of Heart Failure With Reduced Left Ventricular Ejection Fraction. JACC 2021. p. 1747–59. [DOI] [PubMed] [Google Scholar]
- 30.Watson CJ, Papula AL, Poon GYP, Wong WH, Young AL, Druley TE, et al. The evolutionary dynamics and fitness landscape of clonal hematopoiesis. Science. 2020;367(6485):1449–54. [DOI] [PubMed] [Google Scholar]
- 31.Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun. 2016;7:12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Evans MA, Sano S, Walsh K. Clonal haematopoiesis and cardiovascular disease: how low can you go? Eur Heart J. 2021;42(3):266–8. [DOI] [PubMed] [Google Scholar]
- 33.Kiefer KC, Cremer S, Pardali E, Assmus B, Abou-El-Ardat K, Kirschbaum K, et al. Full spectrum of clonal haematopoiesis-driver mutations in chronic heart failure and their associations with mortality. ESC Heart Fail. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yura Y, Sano S, Walsh K. Clonal Hematopoiesis: A New Step Linking Inflammation to Heart Failure. JACC Basic Transl Sci. 2020;5(2):196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bick AG, Pirruccello JP, Griffin GK, Gupta N, Gabriel S, Saleheen D, et al. Genetic Interleukin 6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation. 2020;141(2):124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mas-Peiro S, Hoffmann J, Fichtlscherer S, Dorsheimer L, Rieger MA, Dimmeler S, et al. Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. Eur Heart J. 2020;41(8):933–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kapellos TS, Bonaguro L, Gemünd I, Reusch N, Saglam A, Hinkley ER, et al. Human Monocyte Subsets and Phenotypes in Major Chronic Inflammatory Diseases. Front Immunol. 2019;10:2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Noack M, Miossec P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun Rev. 2014;13(6):668–77. [DOI] [PubMed] [Google Scholar]
- 39.Sato H, Wheat JC, Steidl U, Ito K. DNMT3A and TET2 in the Pre-Leukemic Phase of Hematopoietic Disorders. Front Oncol. 2016;6:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355(6327):842–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sano S, Oshima K, Wang Y, Katanasaka Y, Sano M, Walsh K. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ Res. 2018;123(3):335–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sano S, Wang Y, Evans MA, Yura Y, Sano M, Ogawa H, et al. Lentiviral CRISPR/Cas9-Mediated Genome Editing for the Study of Hematopoietic Cells in Disease Models. J Vis Exp. 2019(152). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park E, Evans MA, Doviak H, Horitani K, Ogawa H, Yura Y, et al. Bone Marrow Transplantation Procedures in Mice to Study Clonal Hematopoiesis. JoVE. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sano S, Wang Y, Yura Y, Sano M, Oshima K, Yang Y, et al. JAK2V617F-Mediated Clonal Hematopoiesis Accelerates Pathological Remodeling in Murine Heart Failure. JACC Basic Transl Sci. 2019;4(6):684–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y, Sano S, Yura Y, Ke Z, Sano M, Oshima K, et al. Tet2-mediated clonal hematopoiesis in nonconditioned mice accelerates age-associated cardiac dysfunction. JCI Insight. 2020;5(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Min KD, Kour A, Sano S, Walsh K. The role of clonal haematopoiesis in cardiovascular diseases: epidemiology and experimental studies. J Intern Med. 2020;288(5):507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1beta/NLRP3 Inflammasome. J Am Coll Cardiol. 2018;71(8):875–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abplanalp WT, Cremer S, John D, Hoffmann J, Schuhmacher B, Merten M, et al. Clonal Hematopoiesis-Driver DNMT3A Mutations Alter Immune Cells in Heart Failure. Circ Res. 2020. [DOI] [PubMed] [Google Scholar]
- 49.Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019;10(2):128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fuster JJ, Zuriaga MA, Zorita V, MacLauchlan S, Polackal MN, Viana-Huete V, et al. TET2-Loss-of-Function-Driven Clonal Hematopoiesis Exacerbates Experimental Insulin Resistance in Aging and Obesity. Cell Rep. 2020;33(4):108326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chaudry SF, Chevassut TJ. Epigenetic Guardian: A Review of the DNA Methyltransferase DNMT3A in Acute Myeloid Leukaemia and Clonal Haematopoiesis. Biomed Res Int. 2017;2017:5473197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol. 2013;14(6):341–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lyko F The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. 2018;19(2):81–92. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525(7569):389–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tanaka S, Ise W, Inoue T, Ito A, Ono C, Shima Y, et al. Tet2 and Tet3 in B cells are required to repress CD86 and prevent autoimmunity. Nat Immunol. 2020;21(8):950–61. [DOI] [PubMed] [Google Scholar]
- 56.Huang F, Sun J, Chen W, He X, Zhu Y, Dong H, et al. HDAC4 inhibition disrupts TET2 function in high-risk MDS and AML. Aging (Albany NY). 2020;12(17):16759–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.González A, Schelbert EB, Díez J, Butler J. Myocardial Interstitial Fibrosis in Heart Failure: Biological and Translational Perspectives. J Am Coll Cardiol. 2018;71(15):1696–706. [DOI] [PubMed] [Google Scholar]
- 58.Frangogiannis NG. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ Res. 2019;125(1):117–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kania G, Blyszczuk P, Eriksson U. Mechanisms of cardiac fibrosis in inflammatory heart disease. Trends Cardiovasc Med. 2009;19(8):247–52. [DOI] [PubMed] [Google Scholar]
- 60.Chen S, Gao R, Yao C, Kobayashi M, Liu SZ, Yoder MC, et al. Genotoxic stresses promote clonal expansion of hematopoietic stem cells expressing mutant p53. Leukemia. 2018;32(3):850–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bick AG, Weinstock JS, Nandakumar SK, Fulco CP, Bao EL, Zekavat SM, et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature. 2020;586(7831):763–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dawoud AAZ, Tapper WJ, Cross NCP. Clonal myelopoiesis in the UK Biobank cohort: ASXL1 mutations are strongly associated with smoking. Leukemia. 2020;34(10):2660–72. [DOI] [PubMed] [Google Scholar]
- 63.Bolton KL, Ptashkin RN, Gao T, Braunstein L, Devlin SM, Kelly D, et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet. 2020;52(11):1219–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456(7218):66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pfeiffer F, Gröber C, Blank M, Händler K, Beyer M, Schultze JL, et al. Systematic evaluation of error rates and causes in short samples in next-generation sequencing. Sci Rep. 2018;8(1):10950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Young AL, Wong TN, Hughes AE, Heath SE, Ley TJ, Link DC, et al. Quantifying ultra-rare pre-leukemic clones via targeted error-corrected sequencing. Leukemia. 2015;29(7):1608–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet. 2018;19(6):371–84. [DOI] [PubMed] [Google Scholar]
- 68.Robertson NA, Hillary RF, McCartney DL, Terradas-Terradas M, Higham J, Sproul D, et al. Age-related clonal haemopoiesis is associated with increased epigenetic age. Curr Biol. 2019;29(16):R786–R7. [DOI] [PubMed] [Google Scholar]
- 69.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10(12):1213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523(7561):486–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet. 2016;48(10):1193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]