

Summary
We developed Miscell, a self-supervised learning approach with deep neural network as latent feature encoder for mining information from single-cell transcriptomes. We demonstrated the capability of Miscell with canonical single-cell analysis tasks including delineation of single-cell clusters and identification of cluster-specific marker genes. We evaluated Miscell along with three state-of-the-art methods on three heterogeneous datasets. Miscell achieved at least comparable or better performance than the other methods by significant margin on a variety of clustering metrics such as adjusted rand index, normalized mutual information, and V-measure score. Miscell can identify cell-type specific markers by quantifying the influence of genes on cell clusters via deep learning approach.
Subject areas: Biological sciences, Neural networks, Transcriptomics
Graphical abstract
Highlights
-
•
We presented a deep learning approach Miscell to dissecting single-cell transcriptomes
-
•
Miscell achieved high performance on canonical single-cell analysis tasks
-
•
Miscell can transfer knowledge learned from single-cell transcriptomes to bulk tumors
Biological sciences; Neural networks; Transcriptomics
Introduction
Comprehensive analysis of single-cell RNA-seq (scRNA-seq) is helpful for dissecting tumor microenvironment, exploring developmental lineages, and characterizing dynamic evolution of immune checkpoint blockade (ICB) therapy response in multiple cancer types (Chalmers et al., 2017; Guo et al., 2018; Jerby-Arnon et al., 2018; Zhang et al., 2018; Zheng et al., 2017). Jerby and colleagues identified a T cell exclusion signature via analyzing scRNA-seq data of malignant melanoma (Jerby-Arnon et al., 2018). Meanwhile, the heterogeneity, clonal expansion and migration of T cells are reported to be key players in the efficacy of ICB therapy including exhaustion and dysfunction of tumor infiltrating lymphocytes (Li et al., 2019; Tirosh et al., 2016a). There are several scRNA-seq analysis methods such as Seurat (Butler et al., 2018; Kang et al., 2018), Scanpy (Wolf et al., 2018) and scVI (Lopez et al., 2018) that have been proposed to elucidate the complex cellular biological processes involved in tumor immune microenvironment and organ development (Asp et al., 2019; Guo et al., 2018; Liao et al., 2020; Zheng et al., 2017). However, the gene expression profile of single-cell is highly sparse, heterogeneous, noisy and subjected to unavoidable batch effect, which poses a tremendous challenge to learn cell-type invariant representation from scRNA-seq data.
The core procedure of scRNA-seq analysis methods is to learn feature representation of single-cells by embedding each cell into a lower and more compact feature space, where downstream tasks such as cell type clustering and annotation could be performed more efficiently. The aforementioned methods Seurat (Butler et al., 2018; Kang et al., 2018) and Scanpy (Wolf et al., 2018) rely on linear dimensional reduction such as principal component analysis of gene expression profiles to capture the relationships among genes and cells. However, the rationality of the linear assumption of gene expression data remained hypothetical and unproven (Lopez et al., 2018). Deep learning algorithms offer an alternative way to modeling nonlinear features of gene expression profiles with deep neural networks. For instance, scVI (Lopez et al., 2018) and MARS (Brbic et al., 2020) employed deep generative models to learn single-cell representation for variational inference and unknown cell-type detection, respectively. Deep generative models learn the latent representation space with a unified encoder and decoder by reconstructing the input and output data via minimizing reconstruction loss, which is often difficult to optimize and associated with high computational cost (Grill et al., 2020). Self-supervised learning is a new paradigm for learning the latent representation of high dimensional data by using itself as a label to predict any parts of its input from any observed parts (Liu et al., 2020). The self-supervised learning is rapidly approaching the performance of supervised learning in computer vision including image classification (Chen et al., 2020a, 2020b).
Here we presented Miscell, a self-supervised learning approach for Mining Information from Single-cell transcriptomes. Miscell is built upon contrastive learning in that it learns representation of the expression pattern by narrowing the gap between the original expression profile and its various augmented expression ones. Miscell is capable of delineating single-cell clusters in the latent feature representation space, identifying cluster-specific marker genes with an attribution method and transferring knowledge learned from single-cells to extract biologically meaningful features from bulk tumor expression data.
We systematically evaluated Miscell with three different datasets that were subjected to different sequencing protocols. The PMBC dataset consisted of 43,095 peripheral blood mononuclear cells (Kang et al., 2018) subjected to 10x sequencing. The Smart-seq dataset consisted of 71,494 single-cells subjected to smart-seq sequencing protocol that were aggregated from studies encompassing tumor cells, stroma cells, dendritic cells, CD4+ T cells, and CD8+ T cells from 7 cancer types. The Muris dataset includes 55,656 single-cells across 20 mouse organs. Compared with Seurat (Butler et al., 2018; Kang et al., 2018), Scanpy (Wolf et al., 2018) and scVI (Lopez et al., 2018), Miscell achieved at least comparable or better performance across these three datasets. Besides, the latent features learned by Miscell were found to be associated with genomic and transcriptomic alteration signatures, immune checkpoint blockade therapy response and prognosis.
Results
An overview of Miscell workflow
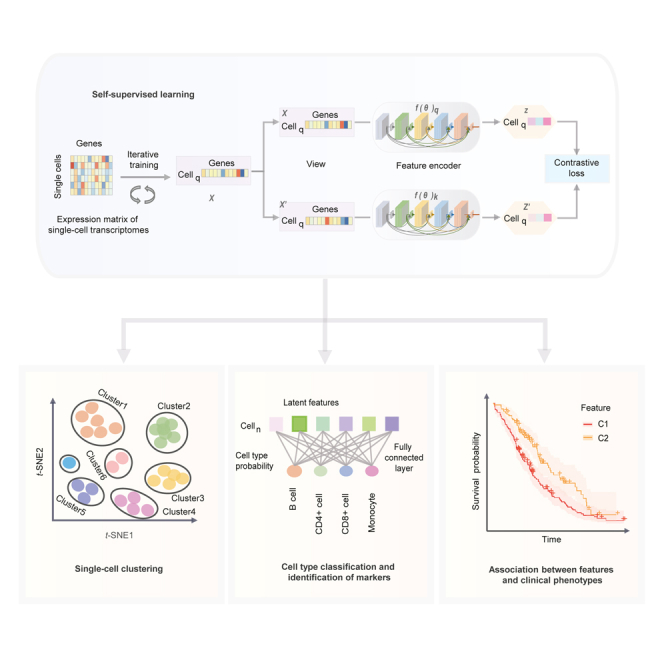
The analytical framework of Miscell (Figure 1) consists of developing a momentum contrast self-supervised learning framework (Chen et al., 2020b; He et al., 2019) for feature representation learning of gene expression (Figure 1A), application of the learned features in downstream tasks including delineation of single-cell clusters (Figure 1B), identification of cluster-specific genes (Figure 1C), classification of different cell types (Figure 1D), and examination of the learned single-cell features with clinical phenotypes in bulk tumors (Figure 1E). The feature encoder used in the self-supervised learning framework is a densely connected deep neural network (DenseNet) with 21 layers (Data S1). The DenseNet has the advantages of improving feature propagation, encouraging feature reuse and alleviating vanishing-gradient problems (Huang et al., 2016). The feature representation component in Miscell is trained end-to-end to minimize the contrastive loss function (Chen et al., 2020b) of the original gene expression pattern and its corresponding augmented version . Data augmentation for gene expression includes random zeroing out and shuffling 20% of genes. Parameters of the encoder and multiple layer perceptron (MLP) projection head are updated end-to-end by stochastic gradient descent, whereas parameters of momentum encoder and MLP are updated gradually in a moving-averaged manner (Figure 1A and see STAR Methods). The MLP maps latent features learned by the encoder to a new space that is beneficial to contrastive learning (Chen et al., 2020a). Miscell maintains a queue of negative examples in that the oldest one is discarded and the current one is enqueued. The momentum encoder can prevent solution collapsing (Chen et al., 2020b). The developed encoder can be viewed as a trainable feature extractor for condensing high-dimensional and sparse single-cell expression profiles into much lower and more compact features. The extracted latent features are used as input for downstream tasks including delineation of single-cell clusters and identification of cluster-specific marker genes. To detect cluster-specific markers, we added a 1-layer linear classifier at the end of the developed encoder and trained it end-to-end by freezing the parameters of the encoder. Subsequently, we applied an integrated gradient algorithm (Sundararajan et al., 2017) to quantify the impact of each gene on the decision made by the linear classifier as a surrogate statistic to identify marker genes (Figure 1D and STAR Methods). We examined the association of single-cell features with clinical phenotypes such as immune chokepoint blockade therapy response and prognosis in bulk tumors by applying the developed encoder to expression profiles of bulk tumors.
Figure 1.

The analytical framework of Miscell
(A–E)This framework consists of five components: (A) development of a feature encoder via momentum contrast self-supervised learning, (B) single-cell clustering and (C) identification of cluster-specific marker genes, (D) cell type classification and (E) examination of the learned single-cell features with clinical phenotypes in bulk tumors.
High clustering performance of Miscell
We evaluated the clustering performance of Miscell on three datasets including 43,095 peripheral blood mononuclear cells (PBMC) (Kang et al., 2018) subjected to 10x sequencing, 55,656 single-cells across 20 mouse organs in the Muris (Tabula Muris et al., 2018) and 71,494 single-cells subjected to smart-seq sequencing protocol (Smart-Seq) that were aggregated from 10 previous studies (Filbin et al., 2018; Guo et al., 2018; Jerby-Arnon et al., 2018; Puram et al., 2017; Tirosh et al., 2016a, 2016b; Venteicher et al., 2017; Zhang et al., 2018, 2020; Zheng et al., 2017) (Table S1), which encompass tumor cells, stroma cells, dendritic cells, CD4+ T cells, and CD8+ T cells from 7 cancer types. We set the queue size (q) = 1024 and momentum coefficient (θ) = 0.999 according to the parameter tuning results on the Smart-seq dataset (Table S2).
We systematically compared the clustering performance of Miscell to three state-of-the-art methods that are widely used for single-cell transcriptome analysis. These three methods include Scanpy (Wolf et al., 2018), Seurat (Butler et al., 2018; Kang et al., 2018) and scVI (Lopez et al., 2018). Miscell performed clustering in the latent feature space projected by the encoder. Scanpy and Seurat performed clustering in a lower dimensional space by applying principal component analysis to the original expression matrices, whereas scVI performed clustering in latent space projected by a deep generative model. The t-SNE embedded scatterplots shown in Figure 2, depicted single-cell clusters detected by Miscell, Scanpy (Wolf et al., 2018), Seurat (Butler et al., 2018; Kang et al., 2018) and scVI (Lopez et al., 2018). For each dataset, the cluster labels and true labels (i.e., cell types) are shown adjacently (Figures 2A–2D, 2F–2I, and 2K–2N). The true labels for Smart-seq dataset are only available for CD4+ and CD8+ T cells from Fluorescence-Activated Cell Sorting, which acounted for 30% (21,213/71,494) of total cells. In the PBMC dataset, Miscell is better than the other methods in identifying cell types of small numbers. For example, Miscell can detect distinct clusters of platelets and dendritic cells, whereas the other methods often intermingle these small number of cells into other cell clusters (Figure S1). Miscell achieved higher clustering metrics as measured by true labels or cluster compactness (Figure 2E). The qualitative values of these clustering metrics are provided in Table S3. In the Smart-seq dataset, Miscell is able to identify visually more compact and less admixed clusters of CD4+ and CD8+ T cells as compared with the other three methods (Figures 2F–2I). Miscell achieved significantly higher clustering performance among a variety of clustering metrics in the Smart-seq dataset (Figures 2J; Table S4). In the Muris dataset, Miscell is able to identify cell clusters (Table S5) across 20 mouse organs (Figure 2K). More specifically, in the PBMC dataset, Miscell achieved a gain of 33% for ARI, 6% for NMI, and 7% for V-measure score over the second-best method (Figure 2E). Meanwhile, Miscell achieved a gain of 120% for ARI, 66% for NMI, and 67% for V-measure score over the second-best method on the Smart-seq dataset (Figure 2J), and Miscell gained 10% for ARI and comparable NMI and V-measure as compared with the second-best method on the Muris dataset (Figure 2O; Table S6). In addition, a better clustering result (Figure S2) of Miscell was also observed when it was applied to the batch-corrected expression matrix of the PBMC dataset corrected by Harmony (Korsunsky et al., 2019).
Figure 2.

The clustering performances of Miscell versus Scanpy, scVI and Seurat
(A–N) t-SNE visualization of feature representations learned by four methods in the PBMC (A–D), Smart-seq (F–I) and Muris dataset (K–N). The corresponding scatterplots of true labels are presented on the right. The clustering metrics of Miscell versus Scanpy, scVI and Seurat on the PBMC (E), Smart-seq (J) and Muris (O) dataset, respectively. Data in the bar plots are represented as mean ± SEM.
In addition, Scanpy can also benefit from latent features learned by Miscell. Scanpy achieved significant improvement across these two datasets among all clustering metrics except ARI; however, its performance in this scenario is still inferior to Miscell (Figure S3).
Miscell enabled functional annotation of identified clusters
Miscell allows for identifying cluster-specific marker genes with an attribution method (see STAR Methods), which is to quantify the impact of a given gene on a specific cluster. We took the Smart-seq dataset as an exemplar to demonstrate the ability of Miscell in this scenario. Miscell detected 63 clusters (Figure 3A) on the Smart-seq dataset. CD4+ T cells are dispersed in cluster C29, C49, C54, and C55, whereas CD8+ T cells are dispersed in cluster C8, C47, C50, C51, C52, and C53 (Figure 3B). We calculated the attribution scores of each gene for each cluster. The attribution scores reflect the quantitative influence of genes on cell clusters and are used as a surrogate statistical value in downstream functional annotation. The result shows that Miscell is able to identify cell-type specific markers. For example, C29, a sub-cluster of CD4+ T cells, is characterized by high attribution score of SLAMF7 and PRF1, which are specific markers of cytotoxic CD4+ T cells (Della-Torre et al., 2018; Herndler-Brandstetter et al., 2018) (Figure 3B). C49 is a sub-cluster of CD4+ T cells, which is marked with genes associated with CD4+ Treg cells (Kavanagh et al., 2008) such as TIGIT and CCDC22, and enrichment of T cell dysfunction (Figure 3B). C8, a sub-cluster of CD8+ T cells, is marked with XCR1, SLC4A10, and CTLA4 (Figure 3B). Functional annotation indicated that C8 was associated with tumor evasion (Qin et al., 2019; Ribas and Wolchok, 2018). The C53 cluster is featured by overrepresentation of KLRG1, which is a marker of KLRG1+ effector CD8+ T cell and can induce proliferation of effector CD8+ T cells while receiving inflammatory signals (Herndler-Brandstetter et al., 2018).
Figure 3.

Functional annotation of identified cell clusters detected by Miscell in the Smart-seq dataset
(A) t-SNE plot representation of single-cell clusters. Cell clusters of CD4+ and CD8+ T cells are depicted by rectangles.
(B) Heatmap representation of cluster-specific markers in cell clusters with more than 600 cells highlighted by attribution score.
(C) Gene set enrichment analysis of cell clusters with more than 600 cells.
We compared the marker genes of CD4+ and CD8+ T cells identified by Miscell, Seurat and Scanpy from the Smart-seq dataset (Table S8). Miscell is able to identify traditional marker genes of CD4+ T cells (i.e., CD4 and CD40LG) and for CD8+ T cells (i.e., CD8A, CD8B, and KLRK1) that were also found by Scanpy and Seurat. In addition, Miscell identified CCR4 as the marker gene of CD4+ T cell (Sugiyama et al., 2013) and XCL1 as the marker gene of CD8+ T cell (Brewitz et al., 2017).
Miscell identified clinically significant latent features
Here, we demonstrated that Miscell is capable of transferring knowledge learned from single-cells to extract biologically meaningful features from bulk tumor expression data. We applied Miscell to extract latent features from an ICB clinical trial of 298 urothelial carcinoma patients subjected to bulk tumor RNA sequencing. Sixty-eight patients achieved CR/PR whereas 230 achieved SD/PD (see STAR Methods). Among 64 latent features, three of them are significantly associated with ICB therapy response (Wilcoxon test, adjusted p < 0.05, Figure 4A). For instance, patients with low score (i.e., less than 25% quantile value) of 31th feature was characterized by upregulation of genes related to activated immune environment including IFNG (Spranger et al., 2013), IL12B, CXCR3 (House et al., 2020), and CD19 (Yazawa et al., 2003) (Figure 4C). In addition, one of these three features exhibited significant association with prognosis (Log rank test, p = 0.004) whereas the other two also exhibited marginal significant association (Figure S4). Analogously, we examined the association between extracted latent features by Miscell from TCGA pan-cancer expression data and overall survival. We found that the 54th feature (Figure 4B) and the other 16 features are significantly associated with prognosis after controlling for Age, Sex, and TNM stage (Table S7 and Data S2). In addition, we found that the latent features learned by Miscell are correlated with clinical features, genomic and transcriptomic alteration signatures (Thorsson et al., 2018), Hallmarks of cancer and immune-related circuits (Liberzon et al., 2015) (Figure 4D). For instance, the 12th feature is positively correlated with Age, Proliferation, Wound healing, G2M checkpoint and tumor evasion (Figure 4D).
Figure 4.

Association between the latent features learned by Miscell versus clinical and genomic features
(A) Box plot representation of latent features associated with ICB therapy. SD: stable disease, PD: progression disease, CR: complete response, PR: partial response. Adjusted p values were shown above each boxplot.
(B) Kaplan–Meier survival curves representation of the association between the latent feature and prognosis in TCGA pan-cancer dataset.
(C) Marker genes of the 31th feature and clinical information across 298 urothelial carcinoma patients.
(D) Heatmap representation of the association between the latent features versus clinical features, genomic and transcriptomic alteration signatures, and Hallmarks of cancer and immune-related circuits. Only latent features significantly associated with ICB therapy response and overall survival were shown.
Cell type classifier built upon latent features of Miscell
We developed a cell type classifier based on the latent features of Miscell applied to the PBMC dataset with a single-layer linear classifier (STAR Methods). Miscell achieved high classification performance in detecting 7 cell types of PBMC with area under the curve ranging from 0.908 for CD8+ T cell to 0.997 for monocyte and accuracy from 92.6% to 99.8% (Figure S5).
Discussion
Miscell is a self-supervised learning approach, which is used to address the fundamental analytical tasks of single-cell transcriptomes including delineation of single-cell clusters and identification of cluster-specific marker genes. Noticeably, Miscell can transfer knowledge learned from single-cells to extract meaningful features from bulk tumors. Miscell outperforms state-of-the-art methods examined in this study by significant margin on multiple clustering metrics. This is probably because of better feature representation learning capability of Miscell enabled by the utilization of DenseNet as feature encoder and the contrastive momentum self-supervised learning. The DenseNet can improve information flow, enable feature reuse and substantially reduce the number of parameters; consequently, it is less prone to overfitting (Huang et al., 2016). Study showed that the surface of loss function of DenseNet is smoother than its plain and sequential counterpart; therefore, it is easier to minimize the loss function (Li et al., 2017). Apart from the advantage of using DenseNet as encoder in Miscell, momentum contrastive self-supervised learning is the key determinant leading to its superior performance. Momentum contrastive self-supervised learning achieved comparable performance in visual representation learning of images as compared with supervised representation learning (Chen et al., 2020b; He et al., 2019). As compared with a similar method proposed by Ciotran and colleagues (Ciortan and Defrance, 2021) used a network of 3 linear layers as feature encoder, we observed that Miscell achieved better performance (Figure S6). This is probably because of better representation capacity of the feature encoder used by Miscell.
The use of integrated gradient algorithm to identify cluster-specific markers by Miscell has unique advantage in that it defines markers in the context of complex interactions among all genes as compared with differential analysis such as t test and Wilcoxon rank-sum test adopted by Seurat and Scanpy, which assume independent expression of genes. Our analysis showed that Miscell is able to identify KLRG1 as markers of KLRG1+ effector CD8+ T cell and SLAMF7 and PRF1 as markers of cytotoxic CD4+ T cell, which justifies the feasibility of using the integrated gradient algorithm as marker detection. Besides, Miscell has the potential to discover new cell types. For example, C8, a subgroup of CD8+ T cells, is marked with overrepresentation of CTLA4 (Figure 3) and involved in tumor escape (Pardoll, 2012). C49, a subgroup of CD4+ Treg cells, is enriched for TIGIT and CCDC22 (Figures 3A and 3B) and may contribute to HIV persistence during antiretroviral therapy (Fromentin et al., 2016). Miscell is able to identify traditional marker genes for CD4+ and for CD8+ T cells and new marker genes that might be missed by Scanpy and Seurat such as CCR4 and XCL1. CCR4 is a chemokine receptor of CD4+ T cells whose upregulation could evoke antitumor immune response (Sugiyama et al., 2013). XCL1 was reported to be significantly associated with the number of tumor-infiltrating CD8+ T cells in squamous cell carcinoma (Tamura et al., 2020). Miscell would complement Scanpy and Seurat in identifying marker genes as it uses a different strategy. In this study, the identification of cluster-specific marker genes by Miscell is limited to the highly variable genes. However, cluster-specific marker genes may be missed in HVGs. Therefore, training Miscell on a full gene list would allow it to identify new marker genes.
The latent features learned by Miscell can improve the performance of Scanpy when we replace the principal component analysis outputs with latent features of Miscell. In this scenario, Scanpy achieved marginal improvement in the identification of single-cell clusters albeit it is still inferior to Miscell (Figure S3). This indicates Miscell is better at disentangling the complex expression patterns of single cells, therefore, beneficial for downstream clustering tasks. Although both Miscell and scVI are all based on deep learning algorithms, the former is based on self-supervised learning whereas the latter is based on deep generative approach. Miscell uses contrastive loss to minimize the similarity of the input data and its various augmented versions, whereas scVI needs original feature generation and may not be necessary for feature representation learning. As Miscell is built upon deep neural networks, it requires a large amount of data for training. Therefore, it would not be appropriate for small sample sizes. In addition, there are different batch-correction methods such as Harmony (Korsunsky et al., 2019), MNN (Haghverdi et al., 2018) and BBKNN (Polański et al., 2020). The selection of batch-correction often depends on the scientific issues to address; therefore, we did not include batch-correction in Miscell and leave it as a data preprocessing step.
Analogous to autoencoders, the trained encoder of Miscell can be used to extract latent features from bulk tumors. These latent features showed significant associations with cancer immunotherapy response (Figure 4A), overall survival (Figures 4B and S4), alteration of immune signatures and cancer hallmarks (Liberzon et al., 2015) (Figure 4D). This indicates that Miscell may be helpful to facilitate basic research findings into clinical utilities. The biological mechanisms underlying the latent features of Miscell trained on these single-cell datasets remain unknown and necessitate exploration in future study.
Conclusion
In summary, we present a new approach Miscell that is built upon contrastive momentum self-supervised learning to dissect single-cell transcriptomes with state-of-the-art performance. Miscell will facilitate basic research of single-cell transcriptomics into clinical utility.
Limitations of the study
Analogous to methods built upon deep neural networks, Miscell demands a large sample size to obtain good performance. Miscell does not internally include any batch-correction methods. Therefore, users are required to correct for batch effects if necessary before running Miscell. The clinically significant latent feature extracted by Miscell is lack of explainability and future works are required to address this issue.
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for resources and materials should be directed to and will be fulfilled by the lead contact, Xiangchun Li (lixiangchun2014@foxmail.com).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
We downloaded single-cell gene expression matrices (Filbin et al., 2018; Guo et al., 2018; Jerby-Arnon et al., 2018; Kang et al., 2018; Puram et al., 2017; Tirosh et al., 2016a, 2016b; Venteicher et al., 2017; Zhang et al., 2018, 2020; Zheng et al., 2017) of the PBMC and Smart-seq dataset from Gene Expression Omnibus repository. The Tabula Muris dataset was downloaded from Figshare (Tabula Muris et al., 2018). The PBMC dataset consists of 43,095 single cells from 3 systemic lupus erythematosus and 2 control samples (Kang et al., 2018) subjected to 10x sequencing. This PBMC dataset includes B cells, CD4+ T cells, CD8+ T cells, dendritic cells, platelets, monocytes and natural killer cells. The Smart-seq dataset consists of 71,494 single cells subjected to smart-seq sequencing aggregated from 10 studies (Filbin et al., 2018; Guo et al., 2018; Jerby-Arnon et al., 2018; Puram et al., 2017; Tirosh et al., 2016a, 2016b; Venteicher et al., 2017; Zhang et al., 2018, 2020; Zheng et al., 2017) encompassing tumour cells, stroma cells, dendritic cells, CD4+ and CD8+ T cells. The Tabula Muris dataset consists of 55,656 single-cells across 20 mouse organs, which were subjected to smart-seq sequencing. We used the cell types sorted by Fluorescence-Activated Cell Sorting as gold standard labels. We collected gene expression and clinical data from a dataset of 298 patients with urothelial carcinoma who received immune checkpoint blockade (ICB) therapy (Mariathasan et al., 2018).
Method details
Data preprocessing
We discarded genes that were expressed in less than 3 cells in each batch and identified the HVG using Scanpy (Wolf et al., 2018) toolkit. We denoted the gene expression matrix as M where rows are samples and columns are genes. The raw gene count expression data was transformed into counts per million with the cpm in R package edgeR (v3.20.8) followed by log2 transformation. We normalized the gene expression matrix M by columns via subtracting each column by its mean and dividing with standard deviation. The preprocessing steps for bulk tumors consisted of conversion of gene count to counts per million, log2 transformation and feature normalization. The expression matrix for the highly variable genes (HVGs) obtained from the Smart-Seq data was inputted to the model for feature extraction.
The architecture of Miscell
Miscell implements self-supervised feature representation learning with momentum contrast by minimizing the contrastive loss of the original single-cell and its corresponding augmented version. The feature encoder of Miscell is a DenseNet (Huang et al., 2016) with 21 layers consisted of 4 dense blocks. We replaced the convolutional layer in the original convolutional DenseNet with linear layer to make it able to process gene expression data. For each layer, all the outputs from preceding layers are used as input via feature concatenation, which could make feature transmission more efficient. The use of multi-layer perceptron (MLP) as project head was demonstrated to be beneficial for contrastive learning (Chen et al., 2020b).
Miscell trains a feature encoder of single-cell gene expression by matching an encoded query to a dictionary of encoded keys defined on-the-fly by a set of trained cells. The dictionary is built as a queue, and the contrastive loss function is used to iteratively optimize the model. The contrastive loss function is defined as (Chen et al., 2020b):
The is the feature of query cell, where is the feature of matching cell in the queue. are the features of negative examples that do not match cellq. is a hyper-parameter whose value was set to 0.2. are updated by back-propagation while were updated slowly via , where . m is a momentum coefficient. The strategy of self-supervised learning is to minimize the differences between the latent features of two augmented versions of each sample towards zero. Therefore, the self-supervised labels are all zeros.
We used stochastic gradient descent to train the network for 500 epochs with a weight decay of 0.0001, initial learning rate of 0.24 and batch size of 256. The learning rate was decayed by 0.1 at epoch 150 and 250. We use data augmentation to increase data diversity and mimic data variation. The data augmentation operations include random zeroing out and random shuffling of gene expression values. We conducted parameter tuning for queue size (q) and momentum coefficient (θ). We implemented Miscell with PyTorch framework (v1.6.0).
The developed feature encoder can be used as a nonlinear feature extractor , where F is the dimension of the extracted features. The extracted features can be used as input for downstream tasks.
Single-cell clustering
We applied dbscan algorithm (Ester et al., 1996) implemented in scikit-learn package to the embedded features of fast interpolation-based t-SNE (van der Maaten and Hinton, 2008) applied to the extracted features mapped by . In addition, the extracted features obtained from Miscell were used as input to the K-Nearest Neighbors (KNN) algorithm to construct KNN graph for subsequent clustering by Leiden (Traag et al., 2019) algorithm using Scanpy with default parameters named Miscell-KNN.
Identification of cluster-specific marker genes
We constructed a new deep neural network (denoted as ) by adding a single linear classifier at the end of the trained feature encoder to predict the identity of the cell clusters. We froze the parameters of the developed encoder and train only the newly added linear classifier. Identification of cluster-specific marker genes can be viewed as quantifying the impact of each input feature (i.e. gene expression) on the classification made by the classifier. We used the attribution score calculated by integrated gradient algorithm (Sundararajan et al., 2017) as surrogate metric for the impact of each gene on classification output. Genes exhibited high variation of attribution scores (i.e. variation coefficient >0.15) among different clusters are considered to be cluster-specific marker genes.
For each cell cluster, we applied integrating gradient algorithm to calculate the important score of the ith gene as the gradient of along the ith dimension, which is expanded as (Sundararajan et al., 2017):
The and are the actual and baseline expression levels, respectively. We used zero as the baseline expression.
Transferring single-cell features to bulk tumors
We applied the developed feature encoder to extract features from bulk tumors, i.e. , where is the gene expression matrix of bulk tumor and is the corresponding extracted feature matrix. N denoted the number of samples and G the number of genes. We then analyzed the associations of extracted features with clinical and biological features.
Benchmarking tools
We systematically compared the clustering performance of Miscell to Scanpy (Wolf et al., 2018), Seurat (Butler et al., 2018; Kang et al., 2018) and scVI (Lopez et al., 2018). Miscell, Scanpy and scVI use the same set of HVGs obtained from Scanpy while Seurat used its own routine to identify HVGs.
Scanpy is a scalable toolkit for analyzing single-cell gene expression data implemented in Python. It implements canonical single-cell analysis tasks such as clustering and differential expression testing etc. Scanpy selects highly variable genes (HVGs) by calculating the dispersion coefficient (defined as the ratio of variance to mean). It places genes into a given bin based on average expression. The HVGs are identified based on the condition that the expression values of genes are highly variable compared to genes with similar average expression within the bin. Scanpy captures the relationships among genes and cells relied on principal components of the expression profiles of HVGs, on which a neighborhood graph of cells is built using KNN algorithm. Subsequently, cell cluster cells are detected by Leiden algorithm (Traag et al., 2019). We filtered out cells with less than 200 genes expressed or more than 30% mitochondrial counts. The HVGs were selected using Scanpy with default parameter (i.e. min_mean = 0.0125 and max_mean = 3). Leiden community detection algorithm was applied to the first 50 principal components obtained from Scanpy with the default resolution (i.e. resolution = 1). In addition, we used rank_genes_groups from Scanpy to identify cluster-specific markers parameters.
Seurat is an R package designed for analysis and exploration of single-cell RNA-seq data. Seurat is able to identify and interpret sources of heterogeneity from single-cell transcriptomic measurements, and to integrate diverse-source single-cell data. Seurat constructs a Shared Nearest Neighbor (SNN) graph (Xu and Su, 2015) via the first 50 principal components of expression matrix for optimal modularity detection (Waltman and Van Eck, 2013) to determine cell clusters. The gene expression matrix was log-normalized with a scale factor of 1.0 × 10−4. HVGs were selected by FindVariableGenes with the number of HVGs set to 2000. We used the default 50 principal components to construct the Shared Nearest-Neighbor (SNN) graph and subsequently applied FindClusters to delineate cluster with default parameter.
scVI (single-cell variational inference) is a PyTorch-based package for probabilistic modeling of single-cell omics data. scVI uses deep neural network and stochastic optimization to aggregate information across similar cells and genes. The scVI supports fundamental analysis tasks of single-cell including clustering and differential expression. We used the default parameters for scVI. The number of neurons of the hidden layer is set to 128, the output neurons to 10 dimensions and dropout rate to 0.1. scVI was trained with a batch size of 256, learning rate of 0.001.
Functional annotation
We curated a number of gene sets to annotate the identified cell clusters. The function of these gene sets include markers natural killer cell, tumor cell, fibroblast, B cell, monocyte and macrophage, T cell and endothelial cell, follicular T cell, mitotic CD8+ T cell, mitotic macrophage, M1 macrophage, M2 macrophage, dendritic cell, monocyte, naïve T cell, co-simulator, T regulatory cell, cytotoxic CD8+ T cell, CD8+ T effector memory cell and CD8+ resident memory cell, antigen presentation, ligand, cell adhesion, receptor, and co-inhibitor, interferon associated immune checkpoint blockade therapy, dysfunction T cell, cytotoxic T lymphocyte and tumor evasion. Gene set enrichment analysis was conducted with in R package fgsea (v 1.6.0). The marker genes of the 31th feature were analyzed with R package limma (v 3.34.9), and the patients with urothelium carcinoma were stratified into two clusters (i.e. <25% quantile group versus >75% quantile group). A differential expressed marker gene was required to satisfy these criteria: absolute value of log-transformed fold-change > 0.15 and adjusted p value < 0.05. In addition, we annotated the marker genes with clinical information including Sex, Stage, Tumor mutation burden (TMB), Response Evaluation Criteria in Solid Tumours (RECIST), Cytotoxic Lymphocytes Infiltration (CTL) score and the expression of CTLA4, CD274 and PDCD1. The CTL score was calculated as the average gene expression level of CD8A, CD8B, GZMA, GZMB and PRF1.
Quantification and statistical analysis
Clustering metrics
We used V-measure score, normalized mutual information (NMI), adjusted rand index (ARI), Calinski-Harabaz index and Davies Bouldin index as measurement of clustering. We compared the performance of Miscell versus Scanpy (v1.6.0), scVI (v0.6.8), and Seurat (v3.1.5). To determine whether the latent feature learned by Miscell can be helpful for the other method, we replaced the principal component analysis results with the latent features of Miscell and proceed towards clustering scheme implemented by Scanpy. The clustering metrics were calculated with sklearn (v0.21.2) python package. The confidence interval of mean and standard deviation of these metrics were calculated via random permutation with 1000 times.
V-measure score is defined as the harmonic average value of homogeneity and compactness. The homogeneity score is defined as:
is the conditional entropy of category division under given cluster division conditions, which is calculated as:
is the conditional entropy of category division, which is defined as:
where is the number of instances. and are the number of instances in cluster and , respectively. is the number of instances in cluster which are classified into cluster .
The compactness score is defined as :
V-measure score is defined as:
Normalized Mutual Information (NMI). It is used to evaluate the similarity between two label assignments. Assuming that the clustering labels and actual labels of N cells are U and V, their entropy is defined as:
where is the probability that a cell picked at random from U falls into . In a similar manner, H(V) is defined as:
where . The mutual information (MI) between U and V is calculated by:
where is the probability that a cell picked at random falls into classes and . The normalized mutual information is thus defined as:
Adjusted Rand Index (ARI). The ARI metric is calculated according to the contingency table summarizing the clustering labels and actual labels. Rows of the contingency table are actual labels and columns are clustering labels. ARI is formulated as:
where denoted the numbers of cell in common between clustering labels and actual labels, the sum of row and the sum of column from the contingency table.
Calinski-Harabaz Index. It is also known as variance ratio criterion, and based on the concept of dense and well-separated cluster to comprehensively measure the dispersions for between-cluster and within-cluster. Assuming that a population of N cells can be partitioned into clusters, the Calinskin-Harabaz index is defined as the ratio of between-clusters dispersion and the within-cluster dispersion:
where is the within-cluster dispersion matrix and defined as:
is the between group dispersion matrix defined as:
where N is the total number of cells, the set of cells in cluster , the cell in center of cluster , the center of cluster , be the number of cells in cluster .
Davies Bouldin Index. Davies Bouldin index is defined as the average similarity between each cluster for and most similar one . Lower Davies Bouldin index sugggests better clustering results. The Davies Bouldin index is defined as:
is defined by:
is the average distance between each example in cluster i and the centroid of cluster i. is the distance between centroids of cluster i and j.
Evaluation of cell-type classifier
The area under the receiver operating curve (AUROC) was used to evaluate classification performance on cell type identification. The AUROC value was calculated by roc routine implemented in R package pROC (v1.10.0). We performed 10-fold cross-validation with sklearn (v0.21.2) to evaluate the classification performance.
Survival analysis
We performed the Kaplan-Meier survival analyses to examine the association between latent features learned by Miscell and prognosis using R package survival (v 3.1.12). We stratified patients into two clusters (i.e. C1/2) by determining whether the extracted feature is greater than zero. The survival curves of C1/2 were plotted by R package survminer and their difference was evaluated by log-rank test. p values were subjected for multiple hypothesis test. Two-sided test was used if not specified.
Additional resources
This study did not generate additional data.
Acknowledgments
We are grateful for researchers for their generosity to made their data publicly available. This work was supported by the Chinese National Key Research and Development Project (no. 2018YFC1315600), National Natural Science Foundation of China (no. 31801117 to X.L., no. 31900471 to M.Y. and 82073287 to Q.Z.), Tianjin Municipal Health Commission Foundation (grant no. RC20027 to Y.L) and IRT_14R40 from the Program for Changjiang Scholars and Innovative Research Team in University in China (K.C.).
Author contributions
Xiangchun Li and Kexin Chen designed and supervised the study; Hongru Shen and Xiangchun Li performed data collection, analysis, and wrote the manuscript; Chao Zhang, Dan Wu, Xilin Shen, Mengyao Feng, Jiani Hu, Jilei Liu, Yichen Yang, Yang Li, and Meng Yang collected data; Wei Wang and Qiang Zhang interpreted the data; Xiangchun Li, Kexin Chen, Jilong Yang and Hongru Shen revised the manuscript.
Declaration of interests
The authors declare that they have no conflict of interest.
Published: November 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103200.
Contributor Information
Kexin Chen, Email: chenkexin@tmu.edu.cn.
Xiangchun Li, Email: lixiangchun2014@foxmail.com.
Supplemental information
and S2
Data and code availability
-
•
Data analyzed in the manuscript are available in open access database. All data relevant to the study are included in the main text or supplemental information. The detailed information for the collected datasets was provided in the Key resources table and Table S1. The raw count matrix of the datasets was downloaded from the Gene Expression Omnibus including GEO: GSE96583, GSE103322, GSE89567, GSE108989, GSE98638, GSE102130, GSE146771, GSE99254, GSE72056, GSE115978, GSE70630. The Tabula Muris data were collected from Figshare Data: https://figshare.com/projects/Tabula_Muris_Senis/64982. The source code of Miscell is available at https://github.com/lixiangchun/Miscell.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Asp M., Giacomello S., Larsson L., Wu C.L., Furth D., Qian X.Y., Wardell E., Custodio J., Reimegard J., Salmen F. A spatiotemporal organ-wide gene expression and cell atlas of the developing human heart. Cell. 2019;179:1647–1660.e1619. doi: 10.1016/j.cell.2019.11.025. [DOI] [PubMed] [Google Scholar]
- Brbic M., Zitnik M., Wang S., Pisco A.O., Altman R.B., Darmanis S., Leskovec J. MARS: discovering novel cell types across heterogeneous single-cell experiments. Nat. Methods. 2020;1200-1206 doi: 10.1038/s41592-020-00979-3. [DOI] [PubMed] [Google Scholar]
- Brewitz A., Eickhoff S., Dahling S., Quast T., Bedoui S., Kroczek R.A., Kurts C., Garbi N., Barchet W., Iannacone M. CD8(+) T cells orchestrate pDC-XCR1(+) dendritic cell spatial and functional cooperativity to optimize priming. Immunity. 2017;46:205–219. doi: 10.1016/j.immuni.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers Z.R., Connelly C.F., Fabrizio D., Gay L., Ali S.M., Ennis R., Schrock A., Campbell B., Shlien A., Chmielecki J. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34. doi: 10.1186/s13073-017-0424-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T., Kornblith S., Norouzi M., Hinton G. A simple framework for contrastive learning of visual representations. (PMLR) arXiv. 2020:1597–1607. 2002.05709. [Google Scholar]
- Chen X., Fan H., Girshick R., He K. Improved baselines with momentum contrastive learning. arXiv. 2020 2003.04297. [Google Scholar]
- Ciortan M., Defrance M. Contrastive self-supervised clustering of scRNA-seq data. BMC Bioinformatics. 2021;22:280. doi: 10.1186/s12859-021-04210-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della-Torre E., Bozzalla-Cassione E., Sciorati C., Ruggiero E., Lanzillotta M., Bonfiglio S., Mattoo H., Perugino C.A., Bozzolo E., Rovati L. A CD8alpha-subset of CD4+SLAMF7+ cytotoxic T cells is expanded in patients with IgG4-related disease and decreases following glucocorticoid treatment. Arthritis Rheumatol. 2018;70:1133–1143. doi: 10.1002/art.40469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ester M., Kriegel H.-P., Sander J., Xu X. Proceedings of the second international conference on knowledge discovery and data mining. AAAI Press; 1996. A density-based algorithm for discovering clusters in large spatial databases with noise. [Google Scholar]
- Filbin M.G., Tirosh I., Hovestadt V., Shaw M.L., Escalante L.E., Mathewson N.D., Neftel C., Frank N., Pelton K., Hebert C.M. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science. 2018;360:331–335. doi: 10.1126/science.aao4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromentin R., Bakeman W., Lawani M.B., Khoury G., Hartogensis W., DaFonseca S., Killian M., Epling L., Hoh R., Sinclair E. CD4+ T cells expressing PD-1, TIGIT and LAG-3 contribute to HIV persistence during ART. Plos Pathog. 2016;12:e1005761. doi: 10.1371/journal.ppat.1005761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill J.-B., Strub F., Altché F., Tallec C., Richemond P.H., Buchatskaya E., Doersch C., Pires B.A., Guo Z.D., Azar M.G. Bootstrap your own latent: a new approach to self-supervised learning. arXiv. 2020 preprint arXiv:2006.07733. [Google Scholar]
- Guo X., Zhang Y., Zheng L., Zheng C., Song J., Zhang Q., Kang B., Liu Z., Jin L., Xing R. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018;24:978–985. doi: 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- Haghverdi L., Lun A.T., Morgan M.D., Marioni J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018;36:421–427. doi: 10.1038/nbt.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K., Fan H., Wu Y., Xie S., Girshick R. Momentum contrast for unsupervised visual representation learning. arXiv. 2019 1911.05722. [Google Scholar]
- Herndler-Brandstetter D., Ishigame H., Shinnakasu R., Plajer V., Stecher C., Zhao J., Lietzenmayer M., Kroehling L., Takumi A., Kometani K. KLRG1(+) effector CD8(+) T cells lose KLRG1, differentiate into all memory T cell lineages, and convey enhanced protective immunity. Immunity. 2018;48:716–729.e718. doi: 10.1016/j.immuni.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- House I.G., Savas P., Lai J., Chen A.X.Y., Oliver A.J., Teo Z.L., Todd K.L., Henderson M.A., Giuffrida L., Petley E.V. Macrophage-derived CXCL9 and CXCL10 are required for antitumor immune responses following immune checkpoint blockade. Clin. Cancer Res. 2020;26:487–504. doi: 10.1158/1078-0432.CCR-19-1868. [DOI] [PubMed] [Google Scholar]
- Huang G., Liu Z., van der Maaten L., Weinberger K.Q. Densely connected convolutional networks. arXiv. 2016 1608.06993. [Google Scholar]
- Jerby-Arnon L., Shah P., Cuoco M.S., Rodman C., Su M.J., Melms J.C., Leeson R., Kanodia A., Mei S., Lin J.R. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell. 2018;175:984–997.e924. doi: 10.1016/j.cell.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H.M., Subramaniam M., Targ S., Nguyen M., Maliskova L., McCarthy E., Wan E., Wong S., Byrnes L., Lanata C.M. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol. 2018;36:89–94. doi: 10.1038/nbt.4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanagh B., O'Brien S., Lee D., Hou Y., Weinberg V., Rini B., Allison J.P., Small E.J., Fong L. CTLA4 blockade expands FoxP3+ regulatory and activated effector CD4+ T cells in a dose-dependent fashion. Blood. 2008;112:1175–1183. doi: 10.1182/blood-2007-11-125435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Xu Z., Taylor G., Studer C., Goldstein T. Visualizing the loss landscape of neural nets. arXiv. 2017 1712.09913. [Google Scholar]
- Li H., van der Leun A.M., Yofe I., Lubling Y., Gelbard-Solodkin D., van Akkooi A.C.J., van den Braber M., Rozeman E.A., Haanen J., Blank C.U. Dysfunctional CD8 T cells Form a proliferative, dynamically regulated compartment within human melanoma. Cell. 2019;176:775–789.e718. doi: 10.1016/j.cell.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M.F., Liu Y., Yuan J., Wen Y.L., Xu G., Zhao J.J., Cheng L., Li J.X., Wang X., Wang F.X. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- Liberzon A., Birger C., Thorvaldsdottir H., Ghandi M., Mesirov J.P., Tamayo P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Zhang F., Hou Z., Wang Z., Mian L., Zhang J., Tang J. Self-supervised learning: generative or contrastive. arXiv. 2020 2006.08218. [Google Scholar]
- Lopez R., Regier J., Cole M.B., Jordan M.I., Yosef N. Deep generative modeling for single-cell transcriptomics. Nat. Methods. 2018;15:1053–1058. doi: 10.1038/s41592-018-0229-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten L., Hinton G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008;9:2579–2605. [Google Scholar]
- Mariathasan S., Turley S.J., Nickles D., Castiglioni A., Yuen K., Wang Y., Kadel E.E., III, Koeppen H., Astarita J.L., Cubas R. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polański K., Young M.D., Miao Z., Meyer K.B., Teichmann S.A., Park J.-E. BBKNN: fast batch alignment of single cell transcriptomes. Bioinformatics. 2020;36:964–965. doi: 10.1093/bioinformatics/btz625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puram S.V., Tirosh I., Parikh A.S., Patel A.P., Yizhak K., Gillespie S., Rodman C., Luo C.L., Mroz E.A., Emerick K.S. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell. 2017;171:1611–1624.e1624. doi: 10.1016/j.cell.2017.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S., Xu L., Yi M., Yu S., Wu K., Luo S. Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol. Cancer. 2019;18:155. doi: 10.1186/s12943-019-1091-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A., Wolchok J.D. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S., Spaapen R.M., Zha Y., Williams J., Meng Y., Ha T.T., Gajewski T.F. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl Med. 2013;5:200ra116. doi: 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama D., Nishikawa H., Maeda Y., Nishioka M., Tanemura A., Katayama I., Ezoe S., Kanakura Y., Sato E., Fukumori Y. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc. Natl. Acad. Sci. U S A. 2013;110:17945–17950. doi: 10.1073/pnas.1316796110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundararajan M., Taly A., Yan Q. Axiomatic attribution for deep networks. arXiv. 2017 1703.01365. [Google Scholar]
- Tabula Muris Consortium, Overall coordination, Logistical coordination, Organ collection and processing, Library preparation and sequencing, Computational data analysis, Cell type annotation, Writing group, Supplemental text writing group, Principal investigators Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature. 2018;562:367–372. doi: 10.1038/s41586-018-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura R., Yoshihara K., Nakaoka H., Yachida N., Yamaguchi M., Suda K., Ishiguro T., Nishino K., Ichikawa H., Homma K. XCL1 expression correlates with CD8-positive T cells infiltration and PD-L1 expression in squamous cell carcinoma arising from mature cystic teratoma of the ovary. Oncogene. 2020;39:3541–3554. doi: 10.1038/s41388-020-1237-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsson V., Gibbs D.L., Brown S.D., Wolf D., Bortone D.S., Ou Yang T.H., Porta-Pardo E., Gao G.F., Plaisier C.L., Eddy J.A. The immune landscape of cancer. Immunity. 2018;48:812–830.e814. doi: 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., 2nd, Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I., Venteicher A.S., Hebert C., Escalante L.E., Patel A.P., Yizhak K., Fisher J.M., Rodman C., Mount C., Filbin M.G. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016;539:309–313. doi: 10.1038/nature20123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traag V.A., Waltman L., van Eck N.J. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 2019;9:5233. doi: 10.1038/s41598-019-41695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venteicher A.S., Tirosh I., Hebert C., Yizhak K., Neftel C., Filbin M.G., Hovestadt V., Escalante L.E., Shaw M.L., Rodman C. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. 2017;355 doi: 10.1126/science.aai8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltman L., Van Eck N.J. A smart local moving algorithm for large-scale modularity-based community detection. Eur. Phys. J. B. 2013;86:1–14. [Google Scholar]
- Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C., Su Z. Identification of cell types from single-cell transcriptomes using a novel clustering method. Bioinformatics. 2015;31:1974–1980. doi: 10.1093/bioinformatics/btv088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazawa N., Fujimoto M., Sato S., Miyake K., Asano N., Nagai Y., Takeuchi O., Takeda K., Okochi H., Akira S. CD19 regulates innate immunity by the toll-like receptor RP105 signaling in B lymphocytes. Blood. 2003;102:1374–1380. doi: 10.1182/blood-2002-11-3573. [DOI] [PubMed] [Google Scholar]
- Zhang L., Yu X., Zheng L., Zhang Y., Li Y., Fang Q., Gao R., Kang B., Zhang Q., Huang J.Y. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. 2018;564:268–272. doi: 10.1038/s41586-018-0694-x. [DOI] [PubMed] [Google Scholar]
- Zhang L., Li Z., Skrzypczynska K.M., Fang Q., Zhang W., O'Brien S.A., He Y., Wang L., Zhang Q., Kim A. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell. 2020;181:442–459.e429. doi: 10.1016/j.cell.2020.03.048. [DOI] [PubMed] [Google Scholar]
- Zheng C., Zheng L., Yoo J.K., Guo H., Zhang Y., Guo X., Kang B., Hu R., Huang J.Y., Zhang Q. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell. 2017;169:1342–1356.e1316. doi: 10.1016/j.cell.2017.05.035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
and S2
Data Availability Statement
-
•
Data analyzed in the manuscript are available in open access database. All data relevant to the study are included in the main text or supplemental information. The detailed information for the collected datasets was provided in the Key resources table and Table S1. The raw count matrix of the datasets was downloaded from the Gene Expression Omnibus including GEO: GSE96583, GSE103322, GSE89567, GSE108989, GSE98638, GSE102130, GSE146771, GSE99254, GSE72056, GSE115978, GSE70630. The Tabula Muris data were collected from Figshare Data: https://figshare.com/projects/Tabula_Muris_Senis/64982. The source code of Miscell is available at https://github.com/lixiangchun/Miscell.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.