

Abstract
In this full article, a detailed study of a distal alkenyl C–H arylation and alkylation through the palladium/norbornene (NBE) cooperative catalysis is described. Both aminopyridine- and oxime ether-type directing groups have been found effective for this transformation, allowing functionalization of diverse allyl amines and homoallyl alcohols. In addition, the C5,C6-substititued NBEs show optimal reactivity and selectivity. Various cis-olefins can be transformed to the corresponding arylated or alkylated trisubstituted alkenes with excellent regio- and stereoselectivity. Preliminary mechanistic studies support the Catellani pathway instead of the Heck pathway.
Keywords: Palladium/norbornene, C–H activation, Alkenes, Distal C–H functionalization, “exo”-type directing group
1. Introduction
Transition metal-catalyzed directed alkene C–H activation provides a distinct and efficient approach to install functional groups and increase complexity to olefin substrates [1]. In particular, it offers a straightforward, regio- and stereoselective strategy to convert widely available disubstituted alkenes into trisubstituted ones that are commonly found in natural products, pharmaceutical agents, and functional materials (Fig. 1) [2]. The choices of directing groups (DGs) and metal catalysts have been known to greatly affect the reaction efficiency and selectivity [3]. To date, two major directing modes have been reported for alkenyl C–H activation (Scheme 1) [1]. One involves vicinal activation, in which the DG and the target C–H bond are at vicinal positions of the olefin (Scheme 1a). With this activation mode, an endo-metallocycle is formed after C–H activation, which leads to alkene functionalization through transforming the resulting metal–C bond [4]. Various functional groups such as ester [4a–b], amide [4c–f], imine [4g], oxime ether [4h], and free alcohol [4i] have been demonstrated to be effective DGs for vicinal C–H activation. The other directing mode involves geminal activation to form an exo-metallocycle [5,6](Scheme 1b). This mode has been much less developed; To date, only several examples have been reported under this category, including using cyclohexenyl carboxylic acids [5a–b] and linear cis-olefins with a tethered 8-aminoquinoline [5c–e] or a free alcohol [5f] as DGs. In both cases, the site-selectivity of the C–H functionalization reactions [7] is controlled by spatial proximity between the DGs and potential reactive positions; the reactions are also driven by forming relatively stable five- or six-membered metallocycles. Hence, the activation of alkene C–H bonds distal to existing functional groups remains a challenging task through the conventional DG approach.
Fig. 1.

Trisubstituted alkenes in natural products, drug molecules and synthetic intermediates.
Scheme 1.

Functionalization of sp2 C–H bonds.
As an alternative approach to directed C–H activation, the palladium/norbornene (Pd/NBE) cooperative catalysis, also known as the Catellani-type reactions, has merged as a useful tool for site-selective arene functionalization [8,9]. For example, the Yu group [10a] and our group [10b] have independently reported the Pd/NBE-catalyzed arene meta C–H activation in 2015, which was initiated by an ortho-palladation process (Scheme 1c). While wide success has been achieved on arene functionalization [8], the use of Pd/NBE catalysis for functionalizing regular alkene substrates was not reported until recently [11] (Scheme 1d). The choice of structurally modified NBEs [12] was found to be critical for the success of these reactions. Stimulated by the challenge of activating alkene C–H bonds distal to existing functional groups, we questioned whether the concept of directed meta C–H activation and the geminal directing mode could be merged with the alkenyl Catellani reactions. In this article we describe a full story of developing distal C–H arylation and alkylation of cis-olefins using the Pd/NBE cooperative catalysis (Scheme 1e) [13].
We envisioned that, upon the directed C–H palladation through the geminal activation, the NBE is expected to relay the Pd to the distal C–H bond via migratory addition, followed by another C–H palladation. The resulting alkenyl-norbornyl palladacycle II (ANP) serves as the key intermediate to react with an electrophile to install the distal substituent. The subsequent β-carbon elimination and proto-depalladation process regenerates the NBE and the Pd(II) catalyst (Scheme 1e). While the alkene distal functionalization is formally similar to the arene meta functionalization, substantial differences and challenges can still be envisaged. First, compared to aromatic compounds, simple alkenes are much less stable and more reactive. For example, a 3-exo-trig cyclization has been reported when reacting an alkenyl bromide, Pd(0) and NBE [14]. In addition, the added electrophile could also directly react with the alkenes through regular pathways. Moreover, compared to rigid arene scaffolds, alkene molecules are generally more flexible, which could introduce additional difficulties for the C–H palladation process. To address these challenges, we anticipated that judicious choices of the DGs and the NBE cofactors would likely be the key for the success of the distal alkene functionalization, as it is critical to match the reaction rates among the ANP formation, the reaction between the ANP and the electrophile, and the Pd–C(sp2) protonation to minimize undesired pathways such as the direct electrophile coupling with the alkene and the four-membered-ring formation via direct reductive elimination of the ANP.
2. Results and discussion
2.1. Using pyridine-type DGs
In our first model study, a cyclic alkene with an ether-tethered pyridine DG (1a) [15] was employed as the substrate. To our delight, the initial reaction with methyl 2-iodobenzoate 2a as the electrophile delivered the desired arylated product (3a) in 18% yield with 20% 5-CF3-2-pyridone [15] (L1) as the concerted metalation deprotonation (CMD) promotor (Scheme 2). Given that a seven-membered palladacycle needs to be formed in the first C–H palladation process, it was questioned that whether DGs with a more rigid and shorter linker would be beneficial for this reaction. Towards the end, substrate 1b with several different protecting groups were tested under similar conditions (Table 1, entries 1–3). While the use of pivalate protecting group afforded no desire product (entry 1), the carbamate-type substrates proved to be much more reactive with 68% and 77% arylated products obtained with Boc and Cbz as the protecting group, respectively (entries 2 and 3). As a large excess of regular NBE (N1) was employed in these reactions, further optimization focused on reducing the NBE loading through examining the NBE effect. While several substituted NBEs were found still effective with 50 mol% loading, the C5–CN substituted N6 [16] proved to be optimal (entries 4–11). Notably, in the absence of the NBE cofactor, no desired arylated product was detected (entry 12).
Scheme 2.

Initial discovery of the distal alkenyl C–H arylation.
Table 1.
Selected optimization of alkenes with aminopyridine DGs.

| |||
|---|---|---|---|
| entry | PG | NBE | Yield % (3b/1b) |
| 1 | Pivb | N1 b | 0/50 |
| 2 | Bocb | N1 b | 68/18 |
| 3 | Cbzb | N1 b | 77/6 |
| 4 | Cbz | N1 | 69/4 |
| 5 | Cbz | N2 | 68/4 |
| 6 | Cbz | N3 | 72/4 |
| 7 | Cbz | N4 | 34/25 |
| 8 | Cbz | N5 | 25/33 |
| 9 | Cbz | N6 | 74/2 |
| 10 | Cbz | N7 | 19/27 |
| 11 | Cbz | N8 | 34/23 |
| 12 | Cbz | none | 0/48 |

Standard conditions are 1b (0.1 mmol, 1.0 equiv), 2a (1.5 equiv), Pd(OAc)2 (10 mol %), 5-CF3-2-pyridone (20 mol %), NBE (50 mol %), AgOAc (1.5 equiv), MTBE (0.5 mL, 0.2 M), air, 90 °C, 24 hours. Yields were determined by 1H NMR analysis using CH2Br2 as the internal standard.
with 3.0 equiv 2a at 80 °C intead.
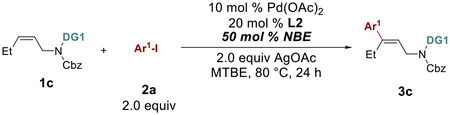
Next, the more challenging acyclic alkene substrate (1c) was explored. With the rigid ring system, one can imagine that forming the ANP intermediate would be significantly more difficult. Under the very similar conditions, the desired distal C–H arylation product can indeed be obtained albeit only in 30% yield (Scheme 3). The alkene geometry in the product matches with what is predicted with the Pd/NBE catalysis pathway, rather than the Heck pathway. The major side-product (3c’), however, was the one derived from the Catellani-type self-arylation of 2a followed by the Heck-quench.
Scheme 3.

Distal C–H Arylation of acyclic allylic amines.
With the promising preliminary result in hand, a number of different CMD promotors were examined to facilitate ANP formation (Table 2). The substituents on 2-pyridone ligands had a notable effect on reaction yields and selectivity. It was found that 3-trifluoromethyl-2-pyridone L2 was more reactive than the previously optimal CMD promotor L1, affording 44% 3c with only 6% 3c’ formation (entry 2). More electron-deficient NO2-substituted L3 led to low reactivity (entry 3); in contrast, bidentate 2-acetamide substituted L4 favored the Heck pathway and inhibited 3c formation (entry 4). Simple pyridone L5 gave low conversion and low selectivity towards the desired product.
Table 2.
Optimization of CMD promotors with linear alkene 1c.

| ||
|---|---|---|
| Entry | CMD promotor | Yield % (3c/3c’/lc) |
| 1 | L1 | 28/4/39 |
| 2 | L2 | 44/6/43 |
| 3 | L3 | trace/0/67 |
| 4 | L4 | trace/44/24 |
| 5 | L5 | 7/26/55 |
A survey of substituted NBEs was next conducted setting L2 as the optimal CMD reagent (Table 3). Consistent with the cyclic substrate, N6 still proved to be the best, affording the desired product 3c in 80% yield with forming only 4% side product 3c’ (entry 4). Notably, the C2-substituted NBEs N7 and N8 resulted in low yields of the desired product and poor mass balance due to forming a complex reaction mixture.
Table 3.
NBE effect with linear alkene 1c.
| ||
|---|---|---|
| Entry | NBE | Yield % (3c/3c’/1c) |
| 1 | N1 | 44/6/43 |
| 2 | N2 | 45/7/41 |
| 3 | N3 | 54/6/20 |
| 4 | N6 | 80/4/trace |
| 5 | N9 | 65/5/5 |
| 6 | N10 | 69/6/10 |
| 7 | N7 | 7/trace/trace |
| 8 | N8 | 16/2/22 |
With these optimized conditions in hand, the relationship between the NBE equivalency and the E/Z selectivity of the arylated products were studied (Table 4). As aforementioned, forming the other alkene isomer was likely due to the undesired Heck reaction pathway, which is supported by the fact that such Heck-pdt 3c” was formed in 70% in the absence of the NBE (entry 1). However, even with 20 mol% of NBE, the selectivity was largely shifted favoring the Catellani product (entry 2). Both the yield and the selectivity for the desired product increased dramatically when the increasing loading of NBE N6. When 1.0 equiv N6 was added, the product (3c) was formed exclusively (entry 4), indicating that the NBE cofactor can indeed suppress the unproductive Heck pathway.
Table 4.
Effect of NBE equivalency on the selectivity.

| ||||
|---|---|---|---|---|
| Entry | X (mol %) | Yield % (3c) | Yield % (3c”) | ratio(3c/3c”) |
| 1 | 0 | 0 | 70 | -- |
| 2 | 20 | 59 | 5.4 | 11:1 |
| 3 | 50 | 74 | 2.4 | 31:1 |
| 4 | 100 | 84 | trace | >50:1 |
While silver salts are commonly used in Pd-catalyzed C–H activation reactions and often give high efficiency, one intriguing question is whether they are necessary in this distal alkene functionalization reaction. Using alkene 1b as the model substrate, a silver-free protocol was investigated (Table 5). While replacing silver with lithium was found not effective at all, the use of sodium or potassium salts instead can indeed provide the desired product (entries 1–4). Given this trend, the use of larger cesium cations was tested, which gave 48% yield for the Boc-protected substrate (entry 5). It is likely that, with similar size to iodide, the soft cesium cation can effectively interact with the iodide in the reaction system; however, the solubility factor cannot be ignored as cesium salts are typically more soluble. Although CsOPiv gave comparable results to CsOAc (entry 6), the less basic CsTFA and CsOMs were much less reactive. Finally, employing Cbz as the protecting group and NBE N6 as the NBE cofactor, the desired product was obtained in 71% yield (entry 9).
Table 5.
Survey of non-silver salts for alkene 1b.

| |||
|---|---|---|---|
| Entry | PG | Salts | Yield %(3b/1b) |
| 1 | Boc | LiTFA | 0/45 |
| 2 | Boc | Li(acac) | 0/81 |
| 3 | Boc | NaOAc | 4/65 |
| 4 | Boc | KOAc | 6/29 |
| 5 | Boc | CsOAc | 48/22 |
| 6 | Boc | CsOPiv | 45/36 |
| 7 | Boc | CsTFA | 0/80 |
| 8 | Boc | CsOMs | 3/69 |
| 9a | Cbz | CsOAc | 71/11 |
100 mol % N6 was used.
A preliminary alkene scope with the aminopyridine-type DG was illustrated in Table 6. These data suggest that both cyclic and acyclic amines are suitable substrates. In addition, the alkene derived from a homo allyl amine reacted as well, with 50% 3d formation and >20:1 E/Z selectivity. The reduced efficiency was likely due to the need of forming a 7-membered palladacycle in the first C–H activation step. While the aminopyridine DGs proved to be efficient for this transformation, they are difficult to remove under mild conditions. Thus, the exploration of other DGs were carried out to seek a broader substrate scope.
Table 6.
Preliminary alkene scope with the aminopyridine DGs.

|
2.2. Oxime ethers as the DG
2.2.1. C–H arylation
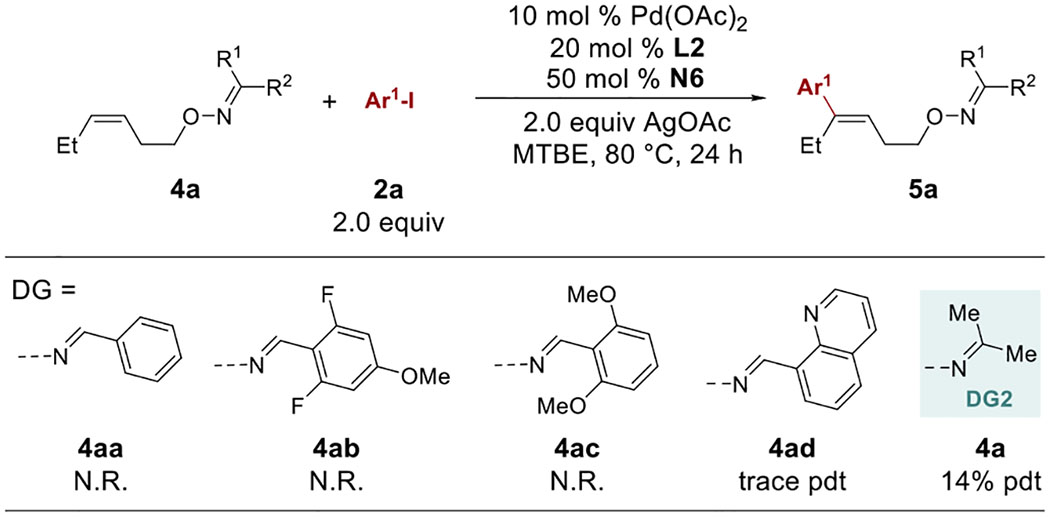
From the synthetic utility viewpoint, it would be attractive to use a DG that can be easily installed and removed. Considering our ongoing interest in using oxime ethers as exo-type DGs for sp3 and sp2 C–H activation [17], they were next investigated as DGs for the distal alkene functionalization. Such DGs can be easily prepared from the corresponding alcohols and the O–N bond can be readily cleaved under reductive conditions (vide infra). First, a number of cis-alkenes tethered with several different types of oxime ethers were synthesized and tested under the distal arylation conditions developed earlier (Table 7). Surprisingly, no reactivity was observed for almost all aldehyde-derived DGs (4aa-4ad); however, the one derived from simple acetone (4a) afforded the desired product 5a in 14% yield.
Table 7.
Survey of oxime ether DGs in the C–H arylation.
|
With this preliminary result in hand, we continued to optimize other reaction parameters. First, a variety of CMD promotors were tested with substrate 4a (Table 8). When the prior best pyridone (L1) was used at slightly elevated temperature, 12% desired product 5a was obtained, together with a significant amount of Heck side-products 5a′ and 5a” formation (entry 1). Different CMD promotors including different pyridines and carboxylic acids appear to have some impact on the reaction selectivity, though the yields were low in all cases (entries 2–8). It is worthy to note that L2 gave a cleaner reaction with only a trace amount of Heck side-products.
Table 8.
Optimization of CMD promotors with alkene 4a.

| ||
|---|---|---|
| Entry | CMD promotor | Yield % (5a/5a’/5a”/4a) |
| 1 | L1 | 12/24/38/38 |
| 2 | L2 | 14/trace/trace/74 |
| 3 | L3 | 9/6/35/35 |
| 4 | L4 | 11/24/38/34 |
| 5 | L6 | 14/trace/10/62 |
| 6 | L7 | 5/trace/9/74 |
| 7 | L8 | trace/3/23/24 |
| 8 | L9 | 3/3/32/19 |

Standard conditions are 4a (0.1 mmol, 1.0 equiv), 2a (1.5 equiv), Pd(OAc)2 (10 mol %), CMD promoter (20 mol %), NBE N6 (50 mol %), AgOAc (2.0 equiv), MTBE (0.5 mL, 0.2 M), under air, 100 °C, 24 hours. Yields were determined by 1H NMR analysis using CH2Br2 as the internal standard.
With the optimal CMD promotor L2 in hand, different NBE co-catalysts were next surveyed (Table 9). Simple NBE N1 completely shut down the reactivity (entry 1), with no Heck side products formation either, though the exact reason is unclear. The C5–CN-substituted N6 and the C5,C6-imide-substituted N9 gave similar results to the C5-amide substituted N3 (entries 2–4). Interestingly, the C5,C6-diester substituted N10 afforded the desired product with notably improved yield (20%, entry 5). Again, the C2-substituted NBEs (N7 and N8) showed poor reactivity (entries 6 and 7). In the absence of NBE, the Heck side pathway dominated to form 5a’ in 41% yield (entry 8), which is similar to the observation with the aminopyridine-based substrates (vide supra, Table 4).
Table 9.
The NBE effect with alkene 4a.

| ||
|---|---|---|
| Entry | NBE | Yield % (5a/5a’/5a”/4a) |
| 1 | N1 | trace/trace/trace/92 |
| 2 | N3 | 12/trace/trace/76 |
| 3 | N6 | 14/trace/trace/74 |
| 4 | N9 | 10/trace/trace/71 |
| 5 | N10 | 20/trace/trace/65 |
| 6 | N7 | 5/17/trace/55 |
| 7 | N8 | 6/trace/trace/88 |
| 8 | none | 0/41/0/23 |
The solvent effect was also evaluated (Table 10). Halogenated solvents, such as DCE and CHCl3, proved to be more effective than MTBE, and better mass balance was observed with CHCl3 (entry 3). Lower selectivity was obtained with ether solvents (entries 4 and 5) and toluene (entry 6). While using mixed solvent of DCE and MTBE gave slightly improved yields, albeit with worse selectivity (vs the Heck side products) (entries 8–10). Therefore, CHCl3 was chosen as the optimal reaction solvent for further studies. Considering the relative low conversion with these reactions, prolonging the reaction time was found beneficial (Table 11). Finally, the desired product (5a) was obtained in 59% yield after 42h (entry 4).
Table 10.
Optimization of solvents with alkene 4a.

| ||
|---|---|---|
| Entry | solvent | Yield % (5a/5a”/4a) |
| 1 | MTBE | 20/trace/65 |
| 2 | DCE | 38/10/24 |
| 3 | CHCl3 | 35/3/36 |
| 4 | THF | 19/11/52 |
| 5 | 1,4-dioxane | 25/5/57 |
| 6 | toluene | 11/4/52 |
| 7 | DMF | 0/20/0 |
| 8 | DCE/MTBE(10:1) | 43/8/20 |
| 9 | DCE/MTBE(5:1) | 44/7/23 |
| 10 | DCE/MTBE(2:1) | 43/6/26 |
Table 11.
Optimization of the reaction time with alkene 4a.

| ||
|---|---|---|
| Entry | Xhrs | Yield % (5a/5a”/4a) |
| 1 | 6 | 22/2/61 |
| 2 | 18 | 42/3/33 |
| 3 | 24 | 48/5/25 |
| 4 | 42 | 59/5/7 |
Following the reaction optimization, the alkene scope with oxime ether DGs was next investigated (Table 12). Besides linear alkyl alkenes (5a), other olefins, such as conjugated (5b) and cyclic ones (5c, 5d), all worked well. In addition to primary alcohol-derived substrates, those based on secondary alcohols were found to be slightly better substrates. It is likely that the additional steric hindrance promotes the first CMD process by positively influencing the orientation of the DG. A range of styrenyl alkenes underwent the distal C–H arylation smoothly (5e-5j), with both electron-deficient (5f) and -rich (5g) substituents tolerated. Notably, no phenyl C–H arylation product was observed when substrate 4i was applied, indicating that the chelation of the cis-olefin to the metal center might inhibit the undesired meta-arylation. Alkenes bearing methyl (5k), ethyl (5l), cyclopropyl (5o), and protected alcohol (5m, 5n) substituents all generated the desired trisubstituted olefins with good yields and moderate to excellent diastereoselectivity.
Table 12.
Scope of the alkenes with an oxime ether DGa.

|
Standard conditions are 4 (0.2 mmol, 1.0 equiv), 2a (2.0 equiv), Pd(OAc)2 (10 mol %), L2 (20 mol %), N9 (30 mol %), AgOAc (2.0 equiv), CHCl3 (1.0 mL, 0.2 M), under air, 100 °C, 12 hours.
N10 was used instead of N9, 42 hours.
24 hours.
When terminal alkene 4p was used as the substrate, mono-arylated product 5p was obtained in 47% yield as a single E isomer and the diarylation product 5p′ was isolated in 15% yield. However, the same products with a similar ratio were formed in the absence of NBE cocatalyst. Due to the lack of success with terminal alkenes in the Catellani-type reactions [11], this result indicates that these arylation products were likely formed via the direct Heck mechanism. On the other hand, when 1,1-disubstituted alkene 4q with the proximal position blocked was subjected to the standard reaction conditions, the desired distal alkene functionalization indeed cannot take place; instead, the Catellani arylation-then-Heck product 5q’ was formed as the major product in 48% yield, along with 15% Heck product 5q.
The electrophile scope was next explored (Table 13). Aryl iodides with ortho electron-withdrawing groups (EWGs) proved to be most reactive, and this is consistent with the typically Catellani-type arylation [18]. Aryl iodide (6a), bromide (6e) and chloride (6c) were all tolerated, which allows for further elaboration of the alkene products. Interestingly, alkenylation with methyl 1,4-diiodo benzoate occurred exclusively at the position ortho to the EWG (6a). Beside benzoates, 2-nitro phenyl iodides also reacted, with both electronic-rich (6f) and -deficient (6e) substituents well tolerated. Heteroarenes such as thiophene (6g) and pyridine (6h) were competent substrates, despite that diminished selectivity was observed for thiophene. Other ortho EWGs such as ketone (6i), amide (6j), and Weinreb amide (6k) also delivered the arylated products in moderate to good yield. Simple phenyl iodide and aryl halides with only para or meta EWGs afforded no desired products, instead giving Heck side products predominantly.
Table 13.
Scope of aryl iodides.a.

|
Standard conditions are 4e (0.2 mmol, 1.0 equiv), 2 (2.0 equiv), Pd(OAc)2 (10 mol %), L2 (20 mol %), N9 (30 mol %), AgOAc (2.0 equiv), CHCl3 (1.0 mL, 0.2 M), under air, 100 °C, 12 hours.
110 °C.
24 hours.
The utility of this transformation has also been examined. Firstly, the Pd catalyst loading can be reduced to 5 mol % on a larger scale (2 mmol), with 68% desired product isolated (Scheme 4a). The oxime ether DGs can be easily removed under mild conditions (with Zn/HOAc) to release the free alcohol 8 in 81% yield (Scheme 4b).
Scheme 4.

Synthetic utility.
2.2.2. C–H alkylation
Besides arylation, alkylation of the distal alkene C–H bonds via the Pd/NBE catalysis has also been studied. Compared to arylation, the C–H alkylation [9] is anticipated to be more challenging as it involves more electrophilic species that can lead to background reactions, and regarding the reaction with ANP, the sp2 and sp3 reductive elimination is generally more difficult than the one only with sp2 carbons. Styrenyl substrate 4e was first chosen as a model substrate to investigate the distal C–H methylation. As shown in Table 14, the desired methylation product (7a) was obtained in 7% yield (entry 1) under the previously optimized conditions with pyridone L2 as the CMD promotor. A survey of pyridine-type ligands revealed that simple pyridine (L13) and electron-rich pyridine (L14) did not afford any methylation product, but the electron-deficient ones were more effective (L10–12, L15). Among the ligands screened, methyl picolinate (L10) proved to be optimal with 14% yield of the desired product. Interestingly, the yield can be further increased to 19% when L2 was added together with L10 (entry 9). This indicates that pyridone L2 acted as a CMD promotor while picolinate L10 acting as a ligand to facilitate the reaction between the ANP intermediate and methyl iodide.
Table 14.
Survey of pyridine ligands for C–H methylationa.

| ||
|---|---|---|
| Entry | Ligand | Yield %(7/4e) |
| 1 | L2 | 7/70 |
| 2 | L10 | 14/70 |
| 3 | L11 | 14/71 |
| 4 | L12 | 10/71 |
| 5 | L13 | 0/100 |
| 6 | L14 | 0/95 |
| 7 | L15 | 8/92 |
| 8 | L16 | 6/94 |
| 9 b | L2 + L10 | 19/67 |

Standard conditions are 4e (0.2 mmol, 1.0 equiv), Mel (3.0 equiv), Pd(OAc)2 (10 mol %), ligand (20 mol %), N9 (30 mol %), AgOAc (3.0 equiv), CHCl3 (1.0 mL, 0.2 M), under air, 100 °C, 24 hours.
20 mol % of L10 and 10 mol % L2 added.
Inspired by this result, a number of other CMD promotors were surveyed with methyl picolinate L10 as the dative ligand (Table 15). Although electron-deficient NO2-substituted pyridones (L3, L17) gave slightly lower yield than L2, the CF3-substituted ones were more effective. For example, methylated product 7a was obtained in 20% and 27% yield when L1 and L6 were used respectively (entries 1 and 5). It is worthy to note that the mass balance in these reactions are excellent.
Table 15.
Effect of the CMD promotors for the C–H methylation.

| ||
|---|---|---|
| Entry | CMD promotor | Yield % (7a/4e) |
| 1 | L1 | 20/73 |
| 2 | L2 | 19/67 |
| 3 | L3 | 17/71 |
| 4 | L4 | 21/73 |
| 5 | L6 | 27/72 |
| 6 | L17 | 14/84 |
With L6 and L10 as the CMD/ligand combination, several structurally modified NBEs were next examined (Table 16). The C2-substituted NBE (N7) was still less reactive than other NBEs (entry 2); the C5,C6-disubstituted ones generally gave better conversions (entries 3–5). The imide-derived NBE (N9) remained be the optimal cofactor (entry 3).
Table 16.
Survey of NBEs for C–H methylation.

| ||
|---|---|---|
| Entry | NBE | Yield % (7a/4e) |
| 1 | N6 | 13/83 |
| 2 | N7 | 6/83 |
| 3 | N9 | 27/72 |
| 4 | N10 | 15/85 |
| 5 | N11 | 17/77 |
The reaction conditions were further tweaked by choosing different additives and varying the reaction set-up (Table 17). First, when 20 mol% of the more soluble ammonium iodide or benzoate salts were added, the reaction was completely shut down (entries 2 and 3). It is likely that a large amount of anions can inhibit the Pd(II) activity by forming the corresponding “ate” complexes. One major issue for the low conversion under the standard conditions (entry 1) was due to formation of Pd/Ag black. Thus, additional oxidants, such as BQ or Cu(II) salts, were added to prevent Pd(0) formation (entries 4–6), which were unfortunately unfruitful. Interestingly, when the reaction was run in a test tube with heating in an oil bath, consistent yield (30%) was obtained (entry 7), likely due to better stirring and more stable reaction temperature. Notably, CsOAc can again serve as an effective halide scavenger other than AgOAc, providing a comparable result (entry 8).
Table 17.
Survey of additives/oxidants/bases for the C–H methylation.

| ||
|---|---|---|
| Entry | Additive/Variants | Yield % (7a/4e) |
| 1 | None | 27/61 |
| 2 | 20% (nBu)4I | 0/100 |
| 3 | 20% (nBu)4OBz | 0/100 |
| 4 | 50% BQ | 5/88 |
| 5 | 50% CuCl2 | 15/28 |
| 6 | 50% Cu(OAc)2 | 29/10 |
| 7a | run in a test tube | 30/70 |
| 8a | CsOAc instead of AgOAc | 25/72 |
| 9a | KOAc instead of AgOAc | 16/84 |
| 10a | LiOAc instead of AgOAc | 8/90 |
The reactions were run in test tubes instead of vials and heated in an oil bath instead of with pi-blocks.
While further optimization of the distal C–H methylation remains challenging and is a topic of our ongoing investigations, a preliminary substrate scope was nevertheless investigated (Table 18). Besides methyl iodide, α-halo carbonyls were competent substrates as well [19]. Notably, methyl bromoacetate as the electrophile afforded the desired alkylation product in 65% yield (7b). Other carbonyl-based electrophiles, such as amide- and ketone-derived ones (7c and 7d), were much less reactive. In contrast, simply alkyl iodides, bromides, and sulfonates were not sufficiently reactive to serve as good electrophiles in this reaction.
Table 18.
Preliminary C–H alkylation scope.

|
2.3. Proposed catalytic cycle
Deuterium-labelling studies and control experiments were next carried out to acquire some mechanistic understanding of this distal alkene C–H functionalization (Scheme 5). When CD3CO2D was used as an additive to replace the electrophile in the reaction system, moderate deuterium incorporation was observed at both the distal and proximal alkene positions in the recycled alkene substrate (Scheme 5a). This result suggests that the germinal C–H palladation and the ANP formation are likely reversible (vide supra, Scheme 1e). In addition, under the standard conditions, a trans-olefin (E-4b) gave no desired product; there was also no E/Z isomerization observed when subjecting the Heck product (Z-5b) to the standard conditions (Scheme 5b). These control experiments could exclude other possible pathways, such as first alkene-isomerization-then-Heck or Heck-then-isomerization.
Scheme 5.

Mechanistic studies and the proposed catalytic cycle.
While some mechanistic details remain unclear and will continue to be investigated, the current data allowed us to propose a simplified catalytic cycle of the distal alkene C–H functionalization (Scheme 5c). The reaction starts with the oxime ether-directed germinal C–H palladation, which forms the six-membered palladacycle (I). Followed by the NBE migratory insertion and the second reversible C–H activation, the ANP intermediate (II) is generated. Analogous to the reactivity of the aryl ANP, it is anticipated that oxidative addition with aryl or alkyl iodides can take place next with II to give a Pd(IV) intermediate (III). Subsequent C–C reductive elimination delivers intermediate IV. Finally, NBE extrusion and protonation of the resulting alkenyl Pd species regenerate the active Pd(II) catalyst and NBE cofactor.
3. Conclusion
In conclusion, we have described a detailed study of discovering a distal alkenyl C–H arylation and alkylation via the Pd/NBE cooperative catalysis. This method provides a distinct strategy to access tri-carbon-substituted alkenes in a regio- and stereoselective manner from readily available disubstituted ones. Aminopyrdines and oxime ethers have been identified as suitable DGs, which allows use of amines and alcohols as handles for functionalization. The C5/C6-disubstituted NBEs were most effective for this type of transformations. The scope of cis alkenes is generally broad, tolerating both linear and cyclic ones as well as conjugated and nonconjugated ones. Further mechanistic exploration supports the proposed Pd/NBE catalysis pathway instead of the Heck pathway. Ongoing investigations involve further improving the efficiency for the distal alkylation, expanding the aryl halide scope for the arylation reaction, and exploring new classes of electrophiles for this type of reactions.
4. Experimental section
4.1. General information
Unless noted otherwise, all solvents were dried by filtration through a Pure-Solv MD-5 Solvent Purification System (Innovative Technology). Chloroform was dried and stored with K2CO3 under air. Sensitive reagents and solvents were transferred under nitrogen into a nitrogen-filled glovebox with standard techniques. Palladium (II) acetate Pd(OAc)2 and silver acetate AgOAc were purchased from Sigma-Aldrich and used as received. Pyrdione ligands were purchased from Combi-blocks and used as received. Unless otherwise noted, the reactions were run in flame-dried 4 mL vials under air. Analytical thin-layer chromatography (TLC) was carried out using 0.2 mm commercial silica gel plates (silica gel 60, F254, EMD chemical). Vials (15 × 45 mm 1 dram (4 mL)/17 × 60 mm 3 dram (7.5 mL) with PTFE lined cap attached) were purchased from Qorpak and flame-dried or put in an oven overnight and cooled in a desiccator. Mass spectra were recorded on an Agilent 6530 LC Q-TOF mass spectrometer using electrospray ionization with fragmentation voltage set at 115 V and processed with an Agilent MassHunter Operating System. Infrared spectra were recorded on a Nicolet 380 FTIR using neat thin film technique. Nuclear magnetic resonance spectra (1H NMR and 13C NMR) were recorded with Bruker Model DMX 500 (500 MHz, 1H at 500 MHz, 13C at 126 MHz) or 400 (400 MHz, 1H at 400 MHz, 13C at 101 MHz). Unless otherwise noted, all spectrums were acquired in CDCl3. Chemical shifts are reported in parts per million (ppm, δ), downfield from tetramethylsilane (TMS, δ = 0.00 ppm) and are referenced to residual solvent (CDCl3, δ = 7.26 ppm (1H) and 77.16 ppm (13C)). Coupling constants were reported in Hertz (Hz). Data for 1H NMR spectra were reported as follows: chemical shift (ppm, referenced to protium, s = singlet, d = doublet, t = triplet, q = quartet, quin = quintet, dd = doublet of doublets, td = triplet of doublets, ddd = doublet of doublet of doublets, m = multiplet, coupling constant (Hz), and integration). All other materials were obtained from Aldrich Chemical Company or Combi-blocks and were used as received.
4.2. Substrate synthesis
Unless otherwise noted, olefin substrates are prepared according to the literature [13] and aryl halides are commercially available or known compounds [13].
4.2.1. Preparation of substrate 1a
To a dry 100 mL round-bottom flask were charged a stir bar, 0.88 g NaH (22 mmol, 2.2 equiv) and 20 mL THF. The reaction flask was then cooled to 0 °C (ice bath), followed by the addition of 1.97 g cyclohex-2-en-1-ol (20 mmol, 2.0 equiv). The resulting mixture was then stirred at 0 °C for 35 min before 1.78 g 2-(chloromethyl)-3-methylpyridine hydrochloride (10 mmol, 1.0 equiv) was added. Afterwards, the reaction was heated up to 80 °C and allowed to stir at the same temperature overnight. The crude was quenched by the addition of water and extracted with EtOAc three times. The combining organic layers then were washed by water, brine, then dried over MgSO4. After concentrated in vacuo, the crude was purified by flash column chromatography on silica gel to afford 1a in 75% yield. Rf (hexanes: EtOAc = 2 : 1) 0.3; dH (400 MHz, CDCl3) 8.38 (ddd, J = 4.9, 1.7, 0.8 Hz, 1H), 7.46 (ddd, J = 7.7, 1.7, 0.8 Hz, 1H), 7.13 (dd, J = 7.6, 4.8 Hz, 1H), 5.99–5.63 (m, 2H), 4.74 (d, J = 10.7 Hz, 1H), 4.67 (d, J = 10.7 Hz, 1H), 4.05–3.95 (m, 1H), 2.42 (s, 3H), 2.13–1.90 (m, 2H), 1.88–1.71 (m, 3H), 1.65–1.48 (m, 1H). dc (101 MHz, CDCl3) 156.25, 146.51, 138.30, 133.31, 131.21, 127.75, 123.17, 72.81, 71.24, 28.31, 25.36, 19.35, 18.30. HRMS (ESI-TOF) m/z: [M + H+] calculated for C13H17NO, 204.1383; found, 204.1388.
4.2.2. Preparation of substrates 4aa-4ad and 4p-q
To a dry 100 mL round-bottom flask were charged a stir bar, the alcohol substrate (10 mmol, 1.0 equiv), 1.80 g N-hydroxylphthalimide (12 mmol, 1.1 equiv), 5.90 g PPh3 (11 mmol, 1.1 equiv) and 40 mL THF. After the reaction mixture was cooled to 0 °C (ice bath), 2.2 mL DIAD (11 mmol, 1.1 equiv) was added dropwise. The resulting mixture was allowed to stir at the room temperature for 3 h followed by addition of 0.9 mL NH2NH2 H2O (12 mmol, 1.2 equiv). After being stirred for another 30 min, corresponding aldehyde or ketone (12 mmol, 1.2 equiv) was added to the reaction mixture, which was then allowed to stir overnight. The reaction was diluted by Et2O, followed by filtration through Celite. The filtrate was then concentrated in vacuo and purified by flash column chromatography on silica gel to afford the desired product.
4.2.2.1. (E)-benzaldehyde O-((Z)-hex-3-en-1-yl) oxime (4aa).
Prepared according to the above described procedure as a colorless liquid in 85% yield. Rf (hexanes: EtOAc = 20 : 1) 0.65; dH (400 MHz, CDCl3) 8.08 (s, 1H), 7.69–7.53 (m, 2H), 7.41–7.33 (m, 3H), 5.56–5.46 (m, 1H), 5.45–5.34 (m, 1H), 4.17 (t, J = 6.9 Hz, 2H), 2.54–2.43 (m, 2H), 2.16–2.01 (m, 2H), 1.86 (d, J = 9.3 Hz, 1H), 1.57 (s, 0H), 0.97 (t, J = 7.5 Hz, 3H).dc (101 MHz, CDCl3): 148.59, 134.21, 132.57, 129.82, 128.80, 127.11, 124.50, 73.91, 27.48, 20.78, 14.40. IR (KBr, cm−1) 3008, 2963, 2832, 2873, 1447, 1371, 1340, 1212, 1046, 945, 755, 692; HRMS (ESI-TOF) m/z: [M + H+] calculated for C13H17NO, 204.1383; found, 204.1385.
4.2.2.2. (E)-2,6-difluoro-4-methoxybenzaldehyde O-((Z)-hex-3-en-1-yl) oxime (4 ab).
Prepared according to the above described procedure as a colorless liquid in 78% yield. Rf (hexanes: EtOAc = 20 : 1) 0.55; dH (400 MHz, CDCl3) 8.18 (s, 1H), 6.50 (d, J = 2.4 Hz, 1H), 6.47 (d, J = 2.4 Hz, 1H), 5.57–5.46 (m, 1H), 5.44–5.34 (m, 1H), 4.18 (t, J = 6.9 Hz, 2H), 3.81 (s, 3H), 2.53–2.42 (m, 2H), 2.14–2.01 (m, 2H), 0.97 (t, J = 7.5 Hz, 3H). dc (101 MHz, CDCl3) 162.99, 139.72, 134.24, 124.46, 108.72, 98.72 (d, J = 2.4 Hz), 98.45 (d, J = 20.77, = 1.9 Hz), 74.07, 56.06, 27.41, 14.40. IR (KBr, cm−1) 3011, 2965, 2934, 2874, 1635, 1574, 1443, 1348, 1200, 1149, 1043, 838; HRMS (ESI-TOF) m/z: [M + H+] calculated for C14H17F2NO2, 270.1300; found, 270.1305.
4.2.2.3. (E)-2,6-dimethoxybenzaldehyde O-((Z)-hex-3-en-1-yl) oxime (4ac).
Prepared according to the above described procedure as a colorless liquid in 82% yield. Rf (hexanes: EtOAc = 20 : 1) 0.45; dH (500 MHz, CDCl3) 8.45 (s, 1H), 7.28 (t, J = 8.4 Hz, 1H), 6.57 (d, J = 8.4 Hz, 2H), 5.54–5.47 (m, 1H), 5.45–5.38 (m, 1H), 4.23 (t, J = 7.0 Hz, 2H), 3.86 (s, 6H), 2.56–2.43 (m, 2H), 2.16–2.02 (m, 2H), 0.97 (t, J = 7.5 Hz, 3H). dc (101 MHz, CDCl3) 159.12, 144.20, 133.98, 130.84, 124.79, 109.67, 104.18, 73.49, 56.18, 27.55, 20.75, 14.41. IR (KBr, cm−1) 3006, 2962, 2934, 2872, 1596, 1471, 1432, 1257, 1208, 1114, 936, 778, 721; HRMS (ESI-TOF) m/z: [M + H+] calculated for C15H21NO3, 264.1594; found, 264.1596.
4.2.2.4. (E)-quinoline-8-carbaldehyde O-((Z)-hex-3-en-1-yl) oxime (4ad).
Prepared according to the above described procedure as a colorless liquid in 87% yield. Rf (hexanes: EtOAc = 20 : 1) 0.45; dH (500 MHz, CDCl3) 9.40 (s, 1H), 9.08–8.90 (m, 1H), 8.28 (dd, J = 7.3, 1.5 Hz, 1H), 8.20 (dd, J = 8.3, 1.8 Hz, 1H), 7.87 (dd, J = 8.1, 1.5 Hz, 1H), 5.56–5.48 (m, 7.58 (t, J = 7.7 Hz, 1H), 7.46 (dd, J = 8.3, 4.2 Hz, 1H), 1H), 5.47–5.40 (m, 1H), 4.26 (t, J = 7.0 Hz, 2H), 2.53 (qd, J = 7.1, 1.5 Hz, 2H), 2.10 (td, J = 7.4, 2H), 1.4 Hz, 0.98 (t, J = 7.5 Hz, 3H). dc (101 MHz, CDCl3) 150.10, 146.29, 146.07, 136.30, 134.12, 130.42, 129.49, 128.40, 126.51, 126.27, 124.64, 121.51, 73.99, 27.59, 20.81, 14.43. IR (KBr, cm−1) 3009, 2962, 2931, 2872, 1587, 1496, 1367, 1328, 1166, 1052, 828, 792; HRMS (ESI-TOF) m/z: [M + H+] calculated for C16H18N2O, 255.1492; found, 255.1499.
4.2.2.5. Propan-2-one O-pent-4-en-2-yl oxime (4p).
Prepared according to the above described procedure as a colorless liquid in 80% yield. Rf (hexanes: EtOAc = 20 : 1) 0.6; dH (400 MHz, CDCl3) 1H NMR (400 MHz, CDCl3) δ 5.81 (ddt, J = 17.2, 10.2, 7.1 Hz, 1H), 5.12–5.00 (m, 2H), 4.18 (td, J = 6.5, 5.7 Hz, 1H), 2.42 (dddt, J = 13.9, 6.9, 5.6, 1.4 Hz, 1H), 2.31–2.17 (m, 1H), 1.86 (s, 3H), 1.84 (s, 3H), 1.20 (d, J = 6.3 Hz, 3H). dc (101 MHz, CDCl3) 154.30, 135.08, 116.86, 76.84, 40.36, 22.08, 19.40, 15.77. IR (KBr, cm−1) 3078, 2978, 2919, 1642, 1439, 1374, 1335, 1272, 1068, 963, 913; HRMS (ESI-TOF) m/z: [M + H+] calculated for C8H15NO, 142.1226; found, 142.1230.
4.2.2.6. Propan-2-one O-(3-methylbut-3-en-1-yl) oxime (4q).
Prepared according to the above described procedure as a colorless liquid in 50% yield. Rf (hexanes: EtOAc = 20 : 1) 0.65; dH (400 MHz, CDCl3) 4.76 (tt, J = 2.3,1.1 Hz, 1H), 4.72 (dq, J = 2.1,1.1 Hz, 1H), 4.11 (t, J = 6.9 Hz, 2H), 2.42–2.31 (m, 2H), 1.86 (s, 3H), 1.83 (s, 3H), 1.76 (t, J = 1.2 Hz, 3H). dc (101 MHz, CDCl3) 13C NMR (101 MHz, CDCl3) δ 154.74, 142.99, 111.57, 71.89, 37.41, 22.96, 21.99, 15.71. IR (KBr, cm−1) 3076, 2921, 2876, 1650, 1441, 1369, 1272, 1067, 1047, 891; HRMS (ESI-TOF) m/z: [M + H+] calculated for C8H15NO, 142.1226; found, 142.1225.
4.3. General procedure A
To a flame-dried 4 mL vial were added 4.4 mg Pd(OAc)2 (0.02 mmol, 10 mol %), 6.6 mg 3-CF3-2-pyridone (0.04 mmol, 20 mol %), 11.9 mg N6 (0.1 mmol, 50 mol %), 68.0 mg AgOAc (0.4 mmol, 2.0 equiv), alkene 1 (0.2 mmol, 1.0 equiv), aryl iodide 2 (0.4 mmol, 2.0 equiv) and 1.0 mL MTBE under air atmosphere. The vial was heated up to 100 °C and allowed to stir for 24 h. The crude reaction mixture was diluted with DCM, followed by filtration through Celite, concentrated in vacuo and then purified by flash column chromatography on silica gel to afford the desired product.
4.3.1. Methyl 5’-((3-methylpyridin-2-yl)methoxy)-2′,3′,4′,5′-tetrahydro-[1,1′-biphenyl]-2-carboxylate (3a)
Prepared according to the General procedure with 20 mol % L1 and 300 mol % N1 in 18% NMR yield; Rf (hexanes: EtOAc = 1 : 1) 0.2; dH (500 MHz, CDCl3) 8.47–8.25 (m, 1H), 7.87–7.65 (m, 1H), 7.57–7.42 (m, 2H), 7.34–7.30 (m, 1H), 7.21–7.07 (m, 2H), 5.83–5.58 (m, 1H), 4.86–4.64 (m, 2H), 4.16 (m, 1H), 3.82 (s, 3H), 2.43 (s, 3H), 2.28–2.18 (m, 2H), 2.01–1.88 (m, 3H), 1.87–1.68 (m, 2H).
4.3.2. Methyl 5’-(((benzyloxy)carbonyl)(3-methylpyridin-2-yl) amino)-2′,3′,4′,5′-tetra hydro-[1,1′-biphenyl]-2-carboxylate (3b)
Prepared according to the General procedure A in 83% isolated yield as a colorless liquid; Rf (hexanes: EtOAc = 2 : 1) 0.6; dH (500 MHz, CDCl3, at 55 °C) 8.37 (dd, J = 4.9, 1.9 Hz, 1H), 7.77 (dd, J = 7.7, 1.5 Hz, 1H), 7.62–7.49 (m, 1H), 7.39–7.31 (m, 1H), 7.31–7.19 (m, 6H), 7.14 (dd, J = 7.5, 4.7 Hz, 1H), 7.03–6.56 (m, 1H), 5.81–5.35 (m, 1H), 5.15 (s, 2H), 5.07–4.95 (m, 1H), 3.78 (s, 3H), 2.18 (s, 3H), 2.16–2.08 (m, 3H), 1.95–1.70 (m, 3H); dc (126 MHz, CDCl3, at 55 °C) 168.14, 154.91, 153.03, 146.45, 145.06, 139.28, 136.89, 132.58, 131.73, 130.09, 129.79, 129.54, 128.47, 127.94, 127.86, 126.88, 122.90, 67.27, 56.37, 51.93, 30.28, 28.10, 22.22, 17.94; IR (KBr, cm−1) 3036, 2947, 2866, 1707, 1574, 1447, 1431, 1303, 1254, 1128, 754, 734, 698; HRMS (ESI-TOF) m/z: [M + H+] calculated for C28H28N2O4, 457.2122; found, 457.2130.
4.3.3. Methyl (E)-2-(1-(((benzyloxy)carbonyl)(3-methylpyridin-2-yl)amino)pent-2-en-3-yl)benzoate (3c)
Prepared according to the General procedure A in 84% isolated yield as a colorless liquid; Rf (hexanes: EtOAc = 2 : 1) 0.6; dH (500 MHz, CDCl3, at 55 °C) 8.36 (dd, J = 4.8, 1.9 Hz, 1H), 7.76 (dd, J = 7.7, 1.5 Hz, 1H), 7.59–7.50 (m, 1H), 7.36 (td, J = 7.5, 1.4 Hz, 1H), 7.31–7.22 (m, 6H), 7.12 (dd, J = 7.5, 4.7 Hz, 1H), 6.99 (dd, J = 7.7, 1.4 Hz, 1H), 5.33 (t, J = 7.0 Hz, 1H), 5.18 (s, 2H), 4.71–4.57 (m, 2H), 3.71 (s, 3H), 2.34 (q, J = 7.6 Hz, 2H), 2.17 (s, 3H), 0.73 (t, J = 7.6 Hz, 3H); dc (126 MHz, CDCl3, at 55 °C) 168.23, 154.91, 153.52, 146.65, 146.31, 144.76, 139.58, 136.78, 132.03, 131.28, 130.37, 130.11, 130.06, 128.54, 128.07, 128.01, 126.88, 123.15, 122.74, 67.57, 51.82, 46.82, 25.24, 17.70, 12.83; IR (KBr, cm−1) 3034, 2966, 2936, 1709, 1576, 1448, 1400, 1290, 1254, 1123, 764, 699; HRMS (ESI-TOF) m/z: [M + H+] calculated for C27H28N2O4, 445.2122; found, 445.2129.
4.3.4. Methyl (E)-2-(6-(((benzyloxy)carbonyl)(3-methylpyridin-2-yl)amino)hex-3-en-3-yl)benzoate (3d)
Prepared according to the General procedure A in 50% isolated yield as a colorless liquid; Rf (hexanes: EtOAc = 2 : 1) 0.6; dH (500 MHz, CDCl3, at 55 °C) 8.45–8.29 (m, 1H), 7.75 (dd, J = 7.7, 1.5 Hz, 1H), 7.55 (d, J = 7.5 Hz, 1H), 7.33–7.18 (m, 7H), 7.17–7.12 (m, 1H), 7.09 (d, J = 7.7 Hz, 1H), 5.22–5.12 (m, 3H), 3.86 (m, 2H), 3.78 (s, 3H), 2.50 (m, 2H), 2.35 (q, J = 7.6 Hz, 2H), 2.19 (s, 3H), 0.84 (t, J = 7.5 Hz, 3H); dc (126 MHz, CDCl3, at 55 °C) 168.68, 155.02, 153.68, 146.68, 145.31, 145.22, 139.70, 136.75, 131.57, 131.16, 130.44, 130.35, 129.93, 128.56, 128.09, 128.00, 126.65, 124.32, 122.75, 67.54, 51.85, 49.02, 27.84, 25.31, 17.62, 12.95; IR (KBr, cm−1) 3036, 2965, 2934, 2876, 1710, 1576, 1448, 1402, 1292, 1252, 1184, 914, 765, 699; HRMS (ESI-TOF) m/z: [M + H+] calculated for C28H30N2O4, 459.2278; found, 459.2284.
4.4. General procedure B
To a flame-dried 4 mL vial were added 4.4 mg Pd(OAc)2 (0.02 mmol, 10 mol %), 6.6 mg 3-CF3-2-pyridone (0.04 mmol, 20 mol %), 14.2 mg N9 (0.06 mmol, 30 mol %), 68.0 mg AgOAc (0.4 mmol, 2.0 equiv), alkene 4 (0.2 mmol, 1.0 equiv), aryl iodide 2 (0.4 mmol, 2.0 equiv) and 1.0 mL CHCl3 under air atmosphere. The vial was heated up to 100 °C and allowed to stir for 12 h. The crude reaction mixture was diluted with DCM, followed by filtration through Celite, concentrated in vacuo and then purified by flash column chromatography on silica gel to afford the desired product.
4.4.1. Methyl (E)-2-(6-((propan-2-ylideneamino)oxy)hex-3-en-3-yl)benzoate (5a)
Prepared according to the General procedure B with 50 mol % N10 for 42 h, in 59% isolated yield as a colorless liquid with >20:1 E/Z ratio; Rf (hexanes: EtOAc = 4 : 1) 0.5; dH (500 MHz, CDCl3) 7.76 (dd, J = 7.8, 1.5 Hz, 1H), 7.41 (td, J = 7.5, 1.5 Hz, 1H), 7.29 (td, J = 7.6, 1.4 Hz, 1H), 7.18 (dd, J = 7.8, 1.3 Hz, 1H), 5.26 (t, J = 7.3 Hz, 1H), 4.07 (t, J = 7.0 Hz, 2H), 3.81 (s, 3H), 2.53 (q, J = 7.1 Hz, 2H), 2.43 (q, J = 7.6, Hz, 2H), 1.88 (s, 3H), 1.86 (s, 3H), 0.90 (t, J = 7.6 Hz, 3H); dc (126 MHz, CDCl3) 168.95, 154.73, 145.25, 144.67, 131.19, 130.40, 130.35, 129.84, 126.64, 124.21, 72.85, 51.99, 28.54, 25.34, 22.01, 15.74, 13.15; IR (KBr, cm−1) 2968, 2927, 2872, 1731, 1598, 1446, 1433, 1289, 1251, 1124, 1074, 763, 714; HRMS (ESI-TOF) m/z: [M + H+] calculated for C17H23NO3, 290.1751; found, 290.1755.
4.4.2. Methyl (E)-2-(1-phenyl-4-((propan-2-ylideneamino)oxy) but-1-en-1-yl) benzoate (5b)
Prepared according to the General procedure B in 69% isolated yield as a colorless liquid in >20:1 E/Z ratio; Rf (hexanes: EtOAc = 4 : 1) 0.5; dH (400 MHz, CDCl3) 7.66–7.61 (m, 1H), 7.42 (td, J = 7.3, 1.5 Hz, 1H), 7.37–7.27 (m, 4H), 7.25–7.14 (m, 3H), 5.78 (t, J = 7.4 Hz, 1H), 4.10 (t, J = 6.6 Hz, 2H), 3.59 (s, 3H), 2.65 (q, J = 6.8 Hz, 2H), 1.85 (s, 3H), 1.84 (s, 3H); dc (101 MHz, CDCl3) 169.06, 154.90, 144.27, 142.76, 139.73, 131.72, 131.19, 131.03, 129.96, 129.48, 127.98, 127.87, 127.13, 127.07, 72.80, 52.00, 30.00, 22.01, 15.78; IR (KBr, cm−1) 3028, 2920, 2850,1729,1601,1443,1290,1253,1078, 761, 702; HRMS (ESI-TOF) m/z: [M + H+] calculated for C21H23NO3, 338.1751; found, 338.1756.
4.4.3. Methyl 5’-(((propan-2-ylideneamino)oxy)methyl)-2′,3′,4′,5′-tetrahydro-[1,1′-biphe-nyl]-2-carboxylate (5c)
Prepared according to the General procedure B in 75% isolated yield as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.5; dH (500 MHz, CDCl3) 7.74 (dd, J = 7.8,1.5 Hz, 1H), 7.42 (td, J = 7.6,1.5 Hz, 1H), 7.28 (td, J = 7.7,1.4 Hz, 1H), 7.20 (dd, J = 7.6,1.3 Hz, 1H), 5.49 (dt, J = 3.1, 1.8 Hz, 1H), 3.94 (dd, J = 7.2, 1.7 Hz, 2H), 3.83 (s, 3H), 2.72–2.60 (m, 1H), 2.29–2.19 (m, 2H), 1.87 (s, 3H), 1.85 (s, 3H), 1.85–1.80 (m, 1H), 1.76–1.65 (m, 2H), 1.46–1.36 (m, 1H); dc (126 MHz, CDCl3) 169.02, 154.74, 145.39, 140.86, 131.44, 130.16, 129.81, 129.55, 126.72, 125.96, 77.30, 52.04, 35.78, 30.68, 25.42, 21.99, 21.58, 15.72; IR (KBr, cm−1) 2989, 2929, 2859, 1728, 1598,1446, 1432, 1290, 1252, 1085, 758; HRMS (ESI-TOF) m/z: [M + H+] calculated for C18H23NO3, 302.1751; found, 302.1755.
4.4.4. Methyl (E)-2-(3-(((propan-2-ylideneamino)oxy)methyl) cyclooct-1-en-1-yl) benzoate (5d)
Prepared according to the General procedure B in 47% isolated yield as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.5; dH (400 MHz, CDCl3) 7.80 (dd, J = 7.7, 1.4 Hz, 1H), 7.42 (td, J = 7.5, 1.5 Hz, 1H), 7.29 (td, J = 7.6, 1.3 Hz, 1H), 7.22 (dd, J = 7.6, 1.3 Hz, 1H), 5.28 (d, J = 8.5 Hz, 1H), 4.10–3.93 (m, 2H), 3.81 (s, 3H), 2.99–2.87 (m, 1H), 2.83–2.68 (m, 1H), 2.20 (dt, J = 13.8, 3.8 Hz, 1H), 1.87 (s, 3H), 1.85 (s, 3H), 1.83–1.75 (m, 1H), 1.73–1.46 (m, 5H), 1.39–1.26 (m, 2H); dc (101 MHz, CDCl3) 168.63, 154.97, 146.08, 142.33, 131.55, 130.34, 130.15, 129.69, 129.48, 126.69, 77.94, 52.06, 37.94, 33.30, 32.55, 28.57, 26.91, 26.01, 22.00, 15.79; IR (KBr, cm−1) 2924, 2852, 1731, 1610, 1447, 1289, 1252, 1124, 1078, 762, 713; HRMS (ESI-TOF) m/z: [M + H+] calculated for C20H27NO3, 330.2064; found, 330.2074.
4.4.5. Methyl (E)-2-(1,6-diphenyl-4-((propan-2-ylideneamino) oxy)hex-1-en-1-yl) benzoate (5e)
Prepared according to the General procedure B in 79% isolated yeidl as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.5; dH (400 MHz, CDCl3) 7.64 (dd, J = 8.0, 1.5 Hz, 1H), 7.49–7.39 (m, 1H), 7.36–7.20 (m, 7H), 7.21–7.13 (m, 5H), 5.83 (t, J = 7.3 Hz, 1H), 4.19 (dq, J = 7.6, 5.6 Hz, 1H), 3.54 (s, 3H), 2.84–2.49 (m, 4H), 2.03–1.89 (m, 2H), 1.87 (s, 3H), 1.86 (s, 3H); dc (101 MHz, CDCl3) 169.14, 154.31, 144.45, 142.54, 142.46, 139.80, 131.60, 131.21, 131.05, 130.00, 129.48, 128.55, 128.39, 127.81, 127.78, 127.06, 126.99, 125.77, 81.30, 51.99, 35.24, 33.93, 31.84, 22.09, 15.84; IR (KBr, cm−1) 3025, 2948, 1729, 1598, 1495, 1443, 1432, 1290, 1254, 701; HRMS (ESI-TOF) m/z: [M + H+] calculated for C29H31NO3, 442.2377; found, 442.2381.
4.4.6. Methyl (E)-2-(6-phenyl-4-((propan-2-ylideneamino)oxy)-1-(4-(trifluoro-methyl)phenyl)hex-1-en-1-yl)benzoate (5f)
Prepared according to the General procedure B in 75% isolated yield as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.5; dH (500 MHz, CDCl3) 7.68 (dd, J = 7.7, 1.4 Hz, 1H), 7.54 (d, J = 8.1 Hz, 2H), 7.46 (td, J = 7.5, 1.4 Hz, 1H), 7.34 (td, J = 7.6, 1.3 Hz, 1H), 7.32–7.27 (m, 3H), 7.29–7.22 (m, 2H), 7.21–7.09 (m, 3H), 5.88 (t, J = 7.3 Hz, 1H), 4.18 (dq, J = 10.8, 5.7 Hz, 1H), 3.55 (s, 3H), 2.80–2.51 (m, 4H), 2.05–1.90 (m, 1H), 1.86 (s, 3H), 1.85 (s, 3H), 1.70–1.63 (m, 1H); dc (125 MHz, CDCl3) 168.54, 154.45, 143.88, 143.54, 142.32, 141.48, 131.36, 131.34, 134.31, 130.28, 129.83, 129.29, 128.97 (q, J = 32.2 Hz), 128.55, 128.45, 127.45, 125.87, 124.77 (q, J = 3.7 Hz), 124.37 (q, J = 271.8 Hz), 81.17, 52.02, 35.30, 34.08, 31.93, 22.06, 15.85; IR (KBr, cm−1) 3026, 2949, 2926, 1730, 1615,1433, 1325, 1125, 1066, 848, 700; HRMS (ESI-TOF) m/z: [M + H+] calculated for C30H30F3NO3, 510.2251; found, 510.2255.
4.4.7. Methyl (E)-2-(1-(4-methoxyphenyl)-6-phenyl-4-((propan-2-ylideneamino) oxy)hex-1-en-1-yl)benzoate (5g)
Prepared according to the General procedure B in 66% isolated yeidl as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.2; dH (400 MHz, CDCl3) 7.62 (ddd, J = 7.6, 1.5, 0.6 Hz, 1H), 7.42 (td, J = 7.5, 1.5 Hz, 1H), 7.34–7.27 (m, 2H), 7.28–7.21 (m, 2H), 7.19–7.13 (m, 3H), 7.09 (d, J = 8.7 Hz, 2H), 6.82 (d, J = 8.8 Hz, 2H), 5.75 (t, J = 7.3 Hz, 1H), 4.18 (dq, J = 7.6, 5.6 Hz, 1H), 3.79 (s, 3H), 3.54 (s, 3H), 2.88–2.47 (m, 4H), 2.04–1.90 (m, 1H), 1.87 (s, 3H), 1.85 (s, 3H), 1.69–1.52 (m, 1H); dc (101 MHz, CDCl3) 169.24, 158.49, 154.32, 144.78, 142.52, 142.11, 132.41, 131.65, 131.17, 131.13, 131.03, 129.45, 128.57, 128.40, 127.00, 126.96, 125.77, 113.19, 81.39, 55.32, 52.04, 35.27, 33.98, 31.86, 22.11, 15.86; IR (KBr, cm−1) 3031, 2948, 2921, 1728, 1607, 1510, 1291, 1248; HRMS (ESI-TOF) m/z: [M + H+] calculated for C30H33NO4, 472.2482; found, 472.2481.
4.4.8. Methyl (E)-2-(1-phenyl-4-((propan-2-ylideneamino)oxy) pent-1-en-1-yl) benzoate (5h)
Prepared according to the General procedure B in 67% isolated yeidl as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.4; dH (500 MHz, CDCl3) 7.63 (dd, J = 7.9,1.4 Hz, 1H), 7.42 (td, J = 7.5,1.4 Hz, 1H), 7.35–7.27 (m, 4H), 7.25–7.17 (m, 3H), 5.81 (t, J = 7.3 Hz, 1H), 4.26 (q, J = 6.3 Hz, 1H), 3.59 (s, 3H), 2.75–2.60 (m, 1H), 2.57–2.46 (m, 1H), 1.85 (s, 3H), 1.83 (s, 3H), 1.23 (d, J = 6.3 Hz, 3H); dc (125 MHz, CDCl3) 169.20, 154.29, 144.47, 142.51, 139.86, 131.67, 131.18, 131.02, 130.02, 129.46, 127.95, 127.82, 127.05, 126.97, 78.13, 52.02, 35.88, 22.08, 19.81, 15.84; IR (KBr, cm−1) 3022, 2949, 2918, 1730, 1570, 1443, 1289, 1255, 1125, 1083, 762, 703; HRMS (ESI-TOF) m/z: [M + H+] calculated for C22H25NO3, 352.1907; found, 352.1916.
4.4.9. (E)-2-(1,6-diphenyl-4-((propan-2-ylideneamino)oxy)hex-1-en-1-yl)-N-methoxy-N-methylbenzamide (5i)
Prepared according to the General procedure B in 63% isolated yeidl as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.5; dH (500 MHz, CDCl3) 7.62 (dd, J = 7.7,1.5 Hz, 1H), 7.41 (td, J = 7.5,1.5 Hz, 1H), 7.35–7.28 (m, 3H), 7.29–7.23 (m, 4H), 7.23–7.16 (m, 3H), 7.09–7.02 (m, 2H), 5.81 (t, J = 7.3 Hz, 1H), 5.17 (t, J = 6.6 Hz, 1H), 3.53 (s, 3H), 2.91 (dt, J = 14.7, 7.3 Hz, 1H), 2.77 (ddd, J = 14.9, 7.5, 5.9 Hz, 1H), 1.92 (s, 3H), 1.82 (s, 3H); dc (125 MHz, CDCl3) 169.05, 155.16, 144.37, 142.89, 142.49, 139.69, 131.57, 131.27, 131.09, 129.91, 129.52, 128.33, 127.77, 127.42, 127.34, 127.12, 126.99, 126.65, 84.09, 51.95, 36.91, 22.05, 16.08; IR (KBr, cm−1) 3028, 2920, 2850, 1728, 1597, 1290,1254,1126, 701; HRMS (ESI-TOF) m/z: [M + H+] calculated for C27H27NO3, 414.2064; found, 414.2065.
4.4.10. Methyl (E)-2-(4-(2,6-difluoro-4-methoxyphenyl)-1-phenyl-4-((propan-2-ylideneamino)oxy)but-1-en-1-yl)benzoate (5j)
Prepared according to the General procedure B in 60% isolated yeidl as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.2; dH (500 MHz, CDCl3) 7.62 (dd, J = 7.6,1.4 Hz, 1H), 7.42 (td, J = 7.6,1.5 Hz, 1H), 7.30 (td, J = 7.5, 1.3 Hz, 1H), 7.26–7.23 (m, 3H), 7.22–7.17 (m, 1H), 7.13–7.07 (m, 2H), 6.37 (s, 1H), 6.35 (s, 1H), 5.76 (t, J = 7.4 Hz, 1H), 5.47 (t, J = 7.4 Hz, 1H), 3.75 (s, 3H), 3.54 (s, 3H), 3.11 (dt, J = 14.5, 7.2 Hz, 1H), 2.88 (dt, J = 14.9, 7.6 Hz, 1H), 1.85 (s, 3H), 1.80 (s, 3H); dc (125 MHz, CDCl3) 168.98, 162.99 (d, J = 11.7 Hz), 161.02 (d, J = 12.1 Hz), 155.52, 144.32, 143.51, 139.53, 131.53, 131.26, 131.17, 129.90, 129.57, 127.75, 127.16, 127.01, 126.71, 109.90 (t, J = 16.8 Hz), 98.13 (d, J = 29.5 Hz), 75.63, 55.81, 51.93, 33.97, 22.04,15.82; IR (KBr, cm−1) 2948, 2921, 2847, 1728, 1638, 1586, 1442, 1290, 1143, 1043, 703; HRMS (ESI-TOF) m/z: [M + H+] calculated for C28H27F2NO4, 480.1981; found, 480.1984.
4.4.11. Methyl (E)-2-(7-phenyl-5-((propan-2-ylideneamino)oxy) hept-2-en-2-yl) benzoate (5k)
Prepared according to the General procedure B with 50 mol % N10 for 36 h, in 68% isolated yield as a colorless liquid with 9:1 E/Z ratio; Rf (hexanes: EtOAc = 4 : 1) 0.55; dH (400 MHz, CDCl3) 7.77 (dd, J = 7.7, 1.5 Hz, 1H), 7.44 (td, J = 7.6, 1.5 Hz, 1H), 7.37–7.12 (m, 7H), 5.42–5.36 (m, 1H), 4.21 (ddd, J = 12.1, 7.2, 5.0 Hz, 1H), 3.78 (s, 3H), 2.89–2.79 (m, 1H), 2.77–2.67 (m, 1H), 2.63–2.56 (m, 1H), 2.54–2.46 (m, 1H),1.99 (s, 3H),1.95 (m, 1H), 1.92 (s, 3H), 1.90 (s, 3H), 1.87 (m, 1H); dc (101 MHz, CDCl3) 168.85, 154.21, 146.85, 142.65, 138.25, 131.48, 130.27, 129.77, 129.71, 128.58, 128.41, 126.57, 125.77, 124.70, 81.32, 52.08, 35.14, 33.22, 32.11, 22.11, 18.57, 15.80; IR (KBr, cm−1) 3062, 3026, 2948, 2918, 1731, 1599, 1496, 1447, 1433, 1289, 1252, 1085, 950, 763, 700; HRMS (ESI-TOF) m/z: [M + H+] calculated for C24H29NO3, 380.2220; found, 380.2222.
4.4.12. Methyl (E)-2-(8-phenyl-6-((propan-2-ylideneamino)oxy) oct-3-en-3-yl) benzoate (5l)
Prepared according to the General procedure B with 50 mol % N10 for 24 h, in 74% isolated yield as a colorless liquid with 18:1 E/Z ratio; Rf (hexanes: EtOAc = 4 : 1) 0.5; dH (400 MHz, CDCl3) 7.76 (dd, J = 7.8, 1.4 Hz, 1H), 7.41 (td, J = 7.5, 1.5 Hz, 1H), 7.33–7.27 (m, 2H), 7.26–7.21 (m, 3H), 7.19–7.13 (m, 2H), 5.28 (t, J = 7.3 Hz, 1H), 4.17 (tt, J = 7.2, 5.1 Hz, 1H), 3.73 (s, 3H), 2.80 (ddd, J = 13.7, 9.9, 5.9 Hz, 1H), 2.68 (ddd, J = 13.8, 9.8, 6.8 Hz, 1H), 2.59 (ddd, J = 14.7, 7.1, 5.3 Hz, 1H), 2.53–2.45 (m, 1H), 2.44–2.33 (m, 2H), 2.01–1.91 (m, 2H), 1.89 (s, 3H), 1.87 (s, 3H), 0.88 (t, J = 7.6 Hz, 3H); dc (101 MHz, CDCl3) 168.86, 154.25, 145.44,144.40, 142.65, 131.23, 130.49, 130.18, 129.86, 128.59, 128.40, 126.59, 125.76, 124.06, 81.42, 52.01, 35.13, 32.84, 32.10, 25.43, 22.12, 15.83, 13.06; IR (KBr, cm−1) 3031, 2953, 2930, 1730, 1598, 1446, 1432, 1288, 1251, 1073, 762, 700; HRMS (ESI-TOF) m/z: [M + H+] calculated for C25H31NO3, 394.2377; found, 394.2373.
4.4.13. Methyl (Z)-2-(2,11,11,12,12-pentamethyl-5-phenethyl-4,10-dioxa-3-aza-11 -silatrideca-2,7-dien-8-yl)benzoate (5 m)
Prepared according to the General procedure B with 50 mol % N10 for 24 h, in 66% isolated yield as a colorless liquid with 15:1 Z/E ratio; Rf (hexanes: EtOAc = 4 : 1) 0.6; dH (500 MHz, CDCl3) 7.81 (dd, J = 7.8, 1.4 Hz, 1H), 7.41 (td, J = 7.5, 1.4 Hz, 1H), 7.34–7.19 (m, 6H), 7.20–7.14 (m, 1H), 5.42 (t, J = 7.4 Hz, 1H), 4.47 (s, 2H), 4.23–4.14 (m, 1H), 3.74 (s, 3H), 2.80 (ddd, J = 13.7, 10.1, 5.6 Hz, 1H), 2.68 (ddd, J = 13.8, 10.0, 6.6 Hz, 1H), 2.61 (ddd, J = 14.8, 7.2, 5.3 Hz, 1H), 2.57–2.48 (m, 1H), 1.99–1.91 (m, 2H), 1.89 (s, 3H), 1.87 (s, 3H), 0.72 (s, 9H), −0.15 (s, 6H); dc (126 MHz, CDCl3) 168.25, 154.38, 143.93, 142.81, 142.62, 131.77, 131.31, 129.98, 129.58, 128.61, 128.41, 126.70, 125.77, 125.42, 81.29, 61.72, 51.93, 35.12, 32.54, 32.09, 25.84, 22.13, 18.27, 15.85, −5.47; IR (KBr, cm−1) 2950, 2927, 2855, 1728, 1599, 1288, 1254, 1083, 837, 775, 699; HRMS (ESI-TOF) m/z: [M + H+] calculated for C30H43NO4Si, 510.3034; found, 510.3039.
4.4.14. Methyl (Z)-2-(1-(benzyloxy)-7-phenyl-5-((propan-2-ylide neamino)oxy) hept-2-en-2-yl) benzoate (5n)
Prepared according to the General procedure B with 50 mol % N10 for 24 h, in 48% isolated yield as a colorless liquid with 14:1 Z/E ratio; Rf (hexanes: EtOAc = 4 : 1) 0.3; dH (400 MHz, CDCl3) 7.85–7.80 (m, 1H), 7.49–7.42 (m, 1H), 7.35–7.29 (m, 2H), 7.30–7.22 (m, 5H), 7.22–7.17 (m, 4H), 7.17–7.12 (m, 1H), 5.61 (t, J = 7.4 Hz, 1H), 4.41 (s, 2H), 4.33 (s, 2H), 4.24–4.12 (m, 1H), 3.71 (s, 3H), 2.77 (ddd, J = 13.8, 10.2, 5.6 Hz, 1H), 2.70–2.62 (m, 1H), 2.62–2.49 (m, 2H), 2.01–1.89 (m, 2H), 1.87 (s, 3H), 1.85 (s, 3H); dc (101 MHz, CDCl3) 158.21, 154.00, 144.24, 142.56, 139.87, 138.55, 138.37, 131.72, 131.10, 129.94, 129.13, 128.61, 128.42, 128.37, 127.79, 127.55, 126.96, 125.79, 81.17, 72.36, 68.76, 52.03, 35.19, 32.94, 32.04, 22.12, 15.85; IR (KBr, cm−1) 3063, 3027, 2920, 2851, 1726, 1599, 1496, 1445, 1289, 1254, 1084, 948, 738, 699; HRMS (ESI-TOF) m/z: [M + H+] calculated for C31H35NO4, 486.2639; found, 486.2641.
4.4.15. Methyl (E)-2-(1-cyclopropyl-7-phenyl-5-((propan-2-ylideneamino)oxy) hept-2-en-2-yl)benzoate (5o)
Prepared according to the General procedure B with 50 mol % N10 for 36 h, in 58% isolated yield as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.5; dH (400 MHz, CDCl3) 7.78 (dd, J = 7.7, 1.4 Hz, 1H), 7.42 (td, J = 7.5, 1.5 Hz, 1H), 7.32–7.27 (m, 2H), 7.26–7.20 (m, 4H), 7.17 (td, J = 6.8, 1.7 Hz, 1H), 5.31 (t, J = 7.3 Hz, 1H), 4.16 (tt, J = 7.2, 5.2 Hz, 1H), 3.72 (s, 3H), 2.80 (ddd, J = 13.8, 9.7, 6.1 Hz, 1H), 2.73–2.65 (m, 1H), 2.64–2.55 (m, 1H), 2.48 (dt, J = 14.6, 7.2 Hz, 1H), 2.31 (qd, J = 14.5, 6.9 Hz, 2H), 2.02–1.91 (m, 1H), 1.89 (s, 3H), 1.87 (s, 3H), 0.69–0.55 (m, 1H), 0.34–0.22 (m, 2H), −0.03 to −0.13 (m, 2H); dc (101 MHz, CDCl3) 168.66, 154.22, 146.21, 142.99, 142.66, 131.28, 130.84, 129.84, 128.60, 128.40, 126.53, 125.76, 124.35, 81.47, 51.98, 36.98, 35.12, 33.01, 32.10, 22.13, 15.83, 10.28, 4.84, 4.74; IR (KBr, cm−1) 3025, 3001, 2948, 2925, 1730, 1598, 1496, 1446, 1290, 1251, 1080, 965, 700; HRMS (ESI-TOF) m/z: [M + H+] calculated for C27H33NO3, 420.2533; found, 420.2535.
4.4.16. Methyl (E)-2-(4-((propan-2-ylideneamino)oxy)pent-1-en-1-yl)benzoate (5p)
Prepared according to the General procedure B with 50 mol % N10 for 12 h, in 47% isolated yield as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.45; dH (500 MHz, CDCl3) 7.84 (dd, J = 7.8, 1.4 Hz, 1H), 7.54 (d, J = 7.8 Hz, 1H), 7.44 (td, J = 7.6, 1.5 Hz, 1H), 7.30–7.23 (m, 1H), 7.17 (d, J = 15.7 Hz, 1H), 6.13 (dt, J = 15.7, 7.3 Hz 1H), 4.31 (h, J = 6.2 Hz, 1H), 3.89 (s, 3H), 2.61 (dddd, J = 14.2, 7.1, 5.6, 1.5 Hz, 1H), 2.52–2.40 (m, 1H), 1.89 (s, 3H), 1.86 (s, 3H), 1.28 (d, J = 6.3 Hz, 3H).; dc (126 MHz, CDCl3) 168.17, 154.71, 139.61, 132.06, 130.80, 130.40, 129.83, 128.37, 127.44, 126.77, 77.94, 52.14, 39.74, 22.07, 19.56, 15.89; IR (KBr, cm−1) 2950, 2916, 1722, 1480, 1433, 1292, 1252, 1077, 965, 747; HRMS (ESI-TOF) m/z: [M + H+] calculated for C16H21NO3, 276.1594; found, 276.1596.
5p can also be obtained in 50% yield according to the General procedure B in the absence of NBE cocatalyst.
4.4.17. Dimethyl 2,2’-(4-((propan-2-ylideneamino)oxy)pent-1-ene-1,1-diyl)dibenzoate (5p′)
Prepared according to the General procedure B with 50 mol % N10 for 12 h, in 15% isolated yield as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.2; dH (400 MHz, CDCl3) d 7.83–7.72 (m, 1H), 7.55–7.42 (m, 3H), 7.41–7.30 (m, 3H), 7.26–7.20 (m, 1H), 5.86 (t, J = 7.3 Hz, 1H), 4.18 (h, J = 6.2 Hz, 1H), 3.68 (s, 3H), 3.68 (s, 3H), 2.35 (ddd, J = 14.7, 7.1, 5.6 Hz, 1H), 2.26 (ddd, J = 14.5, 7.8, 6.2 Hz, 1H), 1.85 (s, 3H), 1.83 (s, 3H), 1.16 (d, J = 6.3 Hz, 3H). dc (101 MHz, CDCl3) 170.10, 168.49, 154.45, 142.44, 140.19, 139.97, 132.69, 132.04, 131.70, 131.17, 131.13, 130.26, 129.65, 129.44, 128.69, 127.26, 126.75, 77.75, 52.20, 52.16, 35.98, 22.05, 19.68, 15.85; IR (KBr, cm−1) 2950, 2917, 1729, 1596, 1444, 1432, 1290, 1256, 1127, 1081, 957, 760; HRMS (ESI-TOF) m/z: [M + H+] calculated for C24H27NO5, 410.1962; found, 410.1956.
4.4.18. Methyl (E)-2-(2-methyl-4-((propan-2-ylideneamino)oxy) but-1-en-1-yl)benzoate (5q)
Prepared according to the General procedure B with 50 mol % N10 for 12 h, in 15% isolated yield (1.4:1 E/Z ratio) as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.4; dH (400 MHz, CDCl3) E isomer: 7.89 (dd, J = 3.1, 1.4 Hz, 1H), 7.45–7.40 (m, 1H), 7.33 (d, J = 7.6 Hz, 1H), 7.28 (dd, J = 7.8, 1.4 Hz, 2H), 6.70 (s, 1H), 4.23 (t, J = 6.9 Hz, 2H), 3.85 (s, 3H), 2.55 (td, J = 6.9, 1.2 Hz, 2H), 1.89 (s, 3H), 1.86 (s, 3H), 1.73 (d, J = 1.4 Hz, 3H). Z isomer: 7.91 (dd, J = 3.1, 1.4 Hz, 1H), 7.45–7.40 (m, 1H), 7.28 (dd, J = 7.8, 1.4 Hz, 2H), 7.24 (d, J = 7.4 Hz, 2H), 6.75 (s, 1H), 4.11 (t, J = 6.9 Hz, 2H), 3.85 (s, 3H), 2.46–2.38 (m, 2H), 1.96 (d, J = 1.5 Hz, 3H), 1.83 (s, 3H), 1.81 (s, 3H); dc (101 MHz, CDCl3) mixture of E/Z isomers: 168.04, 167.86, 154.87, 154.71, 139.88, 139.85, 135.36, 134.95, 131.68, 131.50, 131.33, 131.30, 130.40, 130.37, 129.60, 129.37, 127.22, 126.40, 126.34, 72.20, 71.54, 51.99, 51.97, 39.68, 32.42, 23.79, 22.02, 21.95, 18.12, 15.78.; IR (KBr, cm−1) 2950, 2919, 2875, 1726, 1598, 1568, 1480, 1434, 1294, 1254, 1077, 1044, 745; 710. HRMS (ESI-TOF) m/z: [M + H+] calculated for C16H21NO3, 276.1594; found, 276.1592.
5q can also be obtained in 52% yield (1:1 E/Z ratio) according to the General procedure B in the absence of NBE cocatalyst.
4.4.19. Dimethyl 2’-(2-methylene-4-((propan-2-ylideneamino)oxy) butyl)-[1,1’-biphenyl]-2,3’-dicarboxylate (5q′)
Prepared according to the General procedure B with 50 mol % N10 for 12 h, in 48% isolated yield as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.2; dH (500 MHz, CDCl3) 7.96 (dd, J = 7.7, 1.5 Hz, 1H), 7.83 (dd, J = 7.7, 1.6 Hz, 1H), 7.47 (td, J = 7.5, 1.6 Hz, 1H), 7.42 (td, J = 7.6, 1.4 Hz, 1H), 7.30 (t, J = 7.6 Hz, 1H), 7.25–7.19 (m, 2H), 4.70 (d, J = 2.2 Hz, 1H), 4.19 (d, J = 2.0 Hz, 1H), 3.87 (td, J = 7.1, 2.1 Hz, 2H), 3.83 (s, 3H), 3.73 (d, J = 16.8 Hz, 1H), 3.58 (s, 3H), 3.23 (d, J = 16.8 Hz, 1H), 2.19 (td, J = 7.2, 2.6 Hz, 2H), 1.83 (s, 3H), 1.76 (s, 3H); dc (126 MHz, CDCl3) 168.76, 167.55, 154.68, 146.32, 143.52, 141.97, 137.65, 132.79, 131.55, 131.46, 131.14, 130.28, 130.19, 129.62, 127.70, 125.54, 111.43, 71.80, 52.13, 51.96, 37.29, 36.87, 21.94, 15.71; IR (KBr, cm−1) 3072, 2950, 2923, 2875, 1727, 1646, 1599, 1434, 1289, 1256, 1137, 1092, 1061, 763; 714. HRMS (ESI-TOF) m/z: [M + H+] calculated for C24H27NO5, 410.1962; found, 410.1959.
4.4.20. Methyl (E)-2-(1,6-diphenyl-4-((propan-2-ylideneamino) oxy)hex-1-en-1-yl)-5-iodobenzoate (6a)
Prepared according to the General procedure B in 63% isolated yield as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.5; dH (400 MHz, CDCl3) 7.94 (d, J = 1.9 Hz, 1H), 7.73 (dd, J = 8.1,1.9 Hz, 1H), 7.32–7.20 (m, 5H), 7.18–7.10 (m, 5H), 7.02 (d, J = 8.1 Hz, 1H), 5.80 (t, J = 7.3 Hz, 1H), 4.15 (dq, J = 7.7, 5.6 Hz, 1H), 3.52 (s, 3H), 2.69–2.50 (m, 4H), 2.02–1.88 (m, 1H), 1.86 (s, 3H), 1.84 (s, 3H), 1.83–1.75 (m, 1H); dc (101 MHz, CDCl3) 167.55, 154.40, 143.97, 142.39, 141.58, 139.91, 139.26, 138.10,133.40, 132.90, 130.00, 128.56, 128.50, 128.42, 127.93, 127.22, 125.82, 91.93, 81.18, 52.25, 35.32, 34.02, 31.85, 22.10, 15.85; IR (KBr, cm−1) 3025, 2920, 2850,1733,1601,1495,1434,1282, 1242, 1085, 700; HRMS (ESI-TOF) m/z: [M + H+] calculated for C29H30INO3, 568.1343; found, 568.1347.
4.4.21. Dimethyl (E)-2-(1,6-diphenyl-4-((propan-2-ylideneamino) oxy) hex-1-en-1-yl) terephthalate (6b)
Prepared according to the General procedure B in 80% isolated yield as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.3; dH (400 MHz, CDCl3) 8.01–7.93 (m, 2H), 7.66 (d, J = 8.4 Hz, 1H), 7.35–7.19 (m, 5H), 7.20–7.13 (m, 5H), 5.87 (t, J = 7.3 Hz, 1H), 4.19 (dq, J = 7.7, 5.7 Hz, 1H), 3.92 (s, 3H), 3.55 (s, 3H), 2.79–2.51 (m, 4H), 2.05–1.93 (m, 1H), 1.87 (s, 3H), 1.87 (s, 3H), 1.86–1.80 (m, 1H); dc (101 MHz, CDCl3) 168.53, 166.44, 154.39, 144.51, 142.40, 141.61, 139.25, 135.71, 132.24,132.02, 130.04, 129.41, 128.93,128.56, 128.40, 128.08, 127.95, 127.23, 125.78, 81.18, 52.49, 52.24, 35.36, 34.09, 31.86, 22.08, 15.84; IR (KBr, cm−1) 3025, 2950, 2920, 2851, 1727, 1602, 1495, 1435, 1287, 1114, 758, 702; HRMS (ESI-TOF) m/z: [M + H+] calculated for C31H33NO5, 500.2431; found, 500.2441.
4.4.22. Methyl (E)-4-chloro-2-(1,6-diphenyl-4-((propan-2-ylidene amino)oxy)hex-1-en-1-yl)benzoate (6c)
Prepared according to the General procedure B in 81% isolated yield as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.4; dH (400 MHz, CDCl3) 7.64–7.55 (m, 1H), 7.35–7.27 (m, 5H), 7.25–7.22 (m, 2H), 7.19–7.12 (m, 5H), 5.83 (t, J = 7.3 Hz, 1H), 4.18 (dq, J = 7.8, 5.6 Hz, 1H), 3.53 (s, 3H), 2.75–2.51 (m, 4H), 1.99–1.90 (m, 1H), 1.88 (s, 3H), 1.86 (s, 3H), 1.84–1.77 (m, 1H); dc (101 MHz, CDCl3) 168.12, 154.37, 146.37, 142.37, 141.45, 139.14, 137.13, 131.19, 131.06, 129.95, 129.91, 128.84, 128.56, 128.43, 127.94, 127.24, 127.20, 125.82, 81.23, 52.12, 35.41, 34.12, 31.88, 22.11, 15.86; IR (KBr, cm−1) 3025, 2948, 2918, 1731, 1588, 1433, 1284, 1103, 777, 700; HRMS (ESI-TOF) m/z: [M + H+] calculated for C29H30ClNO3, 476.1987; found, 476.1988.
4.4.23. (E)-propan-2-one O-(6-(2-nitrophenyl)-1,6-diphenylhex-5-en-3-yl) oxime (6d)
Prepared according to the General procedure B in 74% isolated yield as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.3; dH (400 MHz, CDCl3) 7.75 (dd, J = 8.0, 1.4 Hz, 1H), 7.52 (td, J = 7.5, 1.4 Hz, 1H), 7.44–7.35 (m, 2H), 7.35–7.22 (m, 5H), 7.23–7.12 (m, 5H), 5.88 (t, J = 7.3 Hz, 1H), 4.19 (dq, J = 7.7, 5.6 Hz, 1H), 2.79–2.47 (m, 4H), 2.00–1.90 (m, 1H), 1.87 (s, 3H), 1.86 (s, 5H), 1.84–1.77 (m, 1H); dc (101 MHz, CDCl3) 154.47, 149.39, 142.37, 139.21, 138.87, 138.07, 132.39, 132.24,129.84, 129.33,128.56, 128.40, 128.09, 127.98, 127.51, 125.78, 124.12, 81.04, 35.19, 33.85, 31.83, 22.09, 15.85; IR (KBr, cm−1) 3026, 2919, 2851, 1604, 1528, 1495, 1443, 1363, 1067, 701; HRMS (ESI-TOF) m/z: [M + H+] calculated for C27H28N2O3, 429.2173; found, 429.2183.
4.4.24. (E)-propan-2-one O-(6-(4-bromo-2-nitrophenyl)-1,6-diph enylhex-5-en-3-yl) oxime (6e)
Prepared according to the General procedure B in 67% isolated yield as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.3; dH (400 MHz, CDCl3) 7.88 (d, J = 2.0 Hz, 1H), 7.63 (dd, J = 8.3, 2.1 Hz, 1H), 7.35–7.20 (m, 6H), 7.20–7.12 (m, 5H), 5.87 (t, J = 7.3 Hz, 1H), 4.18 (dq, J = 7.7, 5.6 Hz, 1H), 2.82–2.51 (m, 4H), 1.99–1.89 (m, 1H), 1.87 (s, 3H), 1.86 (s, 3H), 1.84–1.74 (m, 1H); dc (101 MHz, CDCl3) 154.51, 149.71, 142.29, 138.23, 137.76, 137.59, 135.23, 133.66, 130.09, 129.81, 128.55, 128.42, 128.22, 127.74, 127.05, 125.82, 120.96, 80.93, 35.25, 33.96, 31.84, 22.09, 15.84; IR (KBr, cm−1) 3026, 2920, 2853, 1601, 1554, 1533, 1495, 1354, 1093, 910, 701; HRMS (ESI-TOF) m/z: [M + H+] calculated for C27H27BrN2O3, 507.1278; found, 507.1274.
4.4.25. (E)-propan-2-one O-(6-(4-methoxy-2-nitrophenyl)-1,6-diphenylhex-5-en-3-yl) oxime (6f)
Prepared according to the General procedure B in 63% isolated yield as a yellow liquid; Rf (hexanes: EtOAc = 4 : 1) 0.15; dH (500 MHz, CDCl3) 7.32–7.21 (m, 7H), 7.21–7.12 (m, 5H), 7.05 (dd, J = 8.5, 2.7 Hz, 1H), 5.81 (t, J = 7.3 Hz, 1H), 4.18 (dq, J = 7.3, 5.6 Hz, 1H), 3.85 (s, 3H), 2.78–2.51 (m, 4H), 1.98–1.90 (m, 1H), 1.86 (s, 3H), 1.85 (s, 3H), 1.84–1.77 (m, 1H); dc (125 MHz, CDCl3) 158.99, 154.40, 149.83, 142.45, 138.94, 138.50, 133.28, 131.35, 129.81, 128.74, 128.58, 128.41, 128.05, 127.41, 125.78, 118.70, 108.93, 81.19, 55.99, 35.22, 33.89, 31.86, 22.06, 15.84; IR (KBr, cm−1) 3024, 2918, 2855, 1619, 1564, 1531, 1365, 1269, 1034, 700; HRMS (ESI-TOF) m/z: [M + H+] calculated for C28H30N2O4, 459.2278; found, 459.2283.
4.4.26. Methyl (E)-3-(1,6-diphenyl-4-((propan-2-ylideneamino) oxy) hex-1-en-1-yl) thiophene-2-carboxylate (6g)
Prepared according to the General procedure in B 61% isolated yield as a colorless liquid with 4:1 E/Z ratio; Rf (hexanes: EtOAc = 4 : 1) 0.45; dH (400 MHz, CDCl3) Major isomer (E): 7.39 (d, J = 5.1 Hz, 1H), 7.34–7.12 (m, 10H), 6.93 (d, J = 5.0 Hz, 1H), 5.89 (t, J = 7.3 Hz, 1H), 4.27–4.10 (m, 1H), 3.63 (s, 3H), 2.73–2.57 (m, 4H), 2.01–1.88 (m, 2H), 1.86 (s, 3H), 1.85 (s, 3H). Minor isomer (Z): 7.54 (d, J = 5.0 Hz, 1H), 7.34–7.12 (m, 10H), 6.92 (d, J = 5.0 Hz, 1H), 6.24 (t, J = 7.4 Hz, 1H), 4.27–4.10 (m, 1H), 3.60 (s, 3H), 2.41–2.25 (m, 4H), 2.01–1.88 (m, 2H), 1.87 (s, 3H), 1.86 (s, 3H); dc (101 MHz, CDCl3) Major isomer (E): 162.55, 154.35, 150.41, 142.50, 139.70, 137.05, 131.92, 129.69, 129.41, 129.26, 128.55, 128.38, 127.89, 126.92, 126.30, 125.76, 81.23, 51.87, 35.19, 33.75, 31.82, 22.09, 15.85. Minor isomer (Z): 162.55, 154.35, 150.41, 146.47, 142.45, 141.22, 137.29, 131.73, 130.70, 129.26, 128.52, 128.40, 128.26, 126.99, 126.88, 125.78, 81.23, 51.87, 34.61, 33.75, 31.90, 22.09, 15.85; IR (KBr, cm−1) 3026, 2947, 2919, 2851, 1722, 1702, 1601, 1526, 1494, 1436, 1406, 1244, 1072, 775; HRMS (ESI-TOF) m/z: [M + H+] calculated for C27H29NO3S, 448.1941; found, 448.1942.
4.4.27. Methyl (E)-3-(1,6-diphenyl-4-((propan-2-ylideneamino) oxy) hex-1-en-1-yl) picolinate (6h)
Prepared according to the General procedure B at 110 °C for 24 h, in 61% isolated yield as a light yellow liquid; Rf (hexanes: EtOAc = 4 : 1) 0.1; dH (400 MHz, CDCl3) 8.54 (dd, J = 4.7, 1.6 Hz, 1H), 7.60 (dd, J = 7.9, 1.5 Hz, 1H), 7.37–7.29 (m, 3H), 7.30–7.21 (m, 3H), 7.22–7.12 (m, 6H), 5.88 (t, J = 7.3 Hz, 1H), 4.16 (dq, J = 7.7, 5.6 Hz, 1H), 3.63 (s, 3H), 2.79–2.51 (m, 4H), 2.06–1.89 (m, 1H), 1.86 (s, 3H), 1.85 (s, 3H), 1.84–1.74 (m, 1H); dc (101 MHz, CDCl3) 167.19, 154.45, 149.27, 147.69, 142.32, 139.70, 139.53, 138.82, 138.79, 130.10, 129.97, 128.56, 128.41, 128.13, 127.47, 125.82, 125.07, 81.01, 52.60, 35.24, 34.07, 31.84, 22.09, 15.85; IR (KBr, cm−1) 3025, 2949, 2920, 2854, 1737, 1601, 1561, 1495, 1443, 1300, 1204, 1096, 702; HRMS (ESI-TOF) m/z: [M + H+] calculated for C28H30N2O3, 443.2329; found, 443.2319.
4.4.28. (E)-1-(2-(1,6-diphenyl-4-((propan-2-ylideneamino)oxy) hex-1-en-1-yl)phenyl) ethan-1-one (6i)
Prepared according to the General procedure B in 75% isolated yield as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.3; dH (500 MHz, CDCl3) 7.53–7.49 (m, 1H), 7.47–7.40 (m, 4H), 7.39–7.33 (m, 1H), 7.31–7.24 (m, 2H), 7.21–7.14 (m, 5H), 7.06–7.00 (m, 1H), 5.66–4.94 (brm, 1H), 4.92–4.77 (m, 1H), 2.83 (dd, J = 14.3, 10.5 Hz, 1H), 2.84–2.74 (m, 1H), 2.68 (ddd, J = 13.9, 10.1, 6.2 Hz, 1H), 2.56 (dd, J = 14.3, 3.2 Hz, 1H), 1.99–1.90 (m, 1H), 1.88–1.81 (m, 1H), 1.80 (s, 3H), 1.59 (s, 3H), 1.51 (s, 3H); dc (125 MHz, CDCl3) 153.87, 151.37, 146.33, 142.32, 142.28, 139.11, 135.19, 129.33, 128.54, 128.52, 128.48, 127.61, 127.49, 125.96, 125.84, 121.65, 119.86, 82.43, 80.08, 36.77, 32.65, 32.45, 25.08, 21.51, 15.76; IR (KBr, cm−1) 3025, 2922, 2851, 1735, 1602, 1495, 1457, 1399, 1364, 1094, 1068, 701; HRMS (ESI-TOF) m/z: [M + H+] calculated for C29H31NO2, 426.2428; found, 426.2430.
4.4.29. (E)-2-(1,6-diphenyl-4-((propan-2-ylideneamino)oxy)hex-1-en-1-yl)-N,N-diethyl benzamide (6j)
Prepared according to the General procedure B in 59% isolated yield as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.1; dH (500 MHz, CDCl3, at 55 °C) 7.30–7.27 (m, 2H), 7.25–7.17 (m, 8H), 7.16–7.11 (m, 4H), 5.92 (t, J = 7.4 Hz, 1H), 4.15 (dq, J = 7.5, 5.7 Hz, 1H), 3.32–3.18 (m, 2H), 3.03 (brs, 1H), 2.65 (ddd, J = 13.9, 10.0, 5.7 Hz, 1H), 2.57 (ddd, J = 13.9, 9.9, 6.4 Hz, 1H), 2.56–2.44 (m, 1H), 1.84 (s, 3H), 1.84 (s, 3H), 1.84–1.79 (m, 1H), 1.69–1.50 (m, 2H), 1.28 (s, 3H), 1.05 (t, J = 7.1 Hz, 3H), 0.96 (t, J = 7.1 Hz, 3H); dc (125 MHz, CDCl3, at 55 °C) 170.64, 142.66, 141.97, 141.71, 139.96, 136.91, 130.70, 130.20, 129.11, 128.63, 128.55, 128.41, 127.98, 127.03, 126.84, 126.59, 125.76, 81.47, 43.38, 39.08, 35.35, 34.19, 31.90, 29.86, 21.96, 15.76, 13.99, 13.11; IR (KBr, cm−1) 3025, 2923, 2851, 1634, 1494, 1455, 1427, 1366, 1290, 1222, 1072, 912, 702; HRMS (ESI-TOF) m/z: [M + H+] calculated for C32H38N2O2, 483.3006; found, 483.3008.
4.4.30. (E)-2-(1,6-diphenyl-4-((propan-2-ylideneamino)oxy)hex-1-en-1-yl)-N-methoxy -N-methylbenzamide (6k)
Prepared according to the General procedure B in 76% isolated yield as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.25; dH (500 MHz, CDCl3, at 55 °C) 7.32–7.26 (m, 4H), 7.26–7.22 (m, 6H), 7.20–7.12 (m, 4H), 5.95 (t, J = 7.4 Hz, 1H), 4.16 (dq, J = 7.5, 5.7 Hz, 1H), 3.39 (s, 3H), 3.00 (s, 3H), 2.73–2.49 (m, 4H), 2.00–1.93 (m, 1H), 1.89–1.86 (m, 1H), 1.85 (s, 6H); dc (125 MHz, CDCl3, at 55 °C) 153.96, 142.66, 142.40, 142.02, 140.18, 135.08, 130.36, 130.33, 130.19, 129.04, 128.63, 128.38, 127.99, 127.17, 127.08, 126.54, 125.74, 81.43, 60.76, 35.23, 34.27, 31.90, 21.96, 15.76; IR (KBr, cm−1) 3024, 2926, 2854, 1654, 1599, 1495, 1442, 1370, 991, 766, 702; HRMS (ESI-TOF) m/z: [M + H+] calculated for C30H34N2O3, 471.2642; found, 471.2644.
4.5. General procedure C
To a flame-dried 4 mL vial were added 2.2 mg Pd(OAc)2 (0.01 mmol, 10 mol %), 1.7 mg 4-CF3-2-pyridone (0.01 mmol, 10 mol %), 2.7 mg 2-methyl picolinate (0.02 mmol, 20 mol%), 7.1 mg N9 (0.03 mmol, 30 mol %), 34 mg AgOAc (0.2 mmol, 2.0 equiv), alkene (0.1 mmol, 1.0 equiv), and 0.5 mL CHCl3 were added to a flame-dried 8 mL tube under air atmosphere. 19 μL MeI (0.3 mmol, 3.0 equiv) or 28uL methyl bromoacetate (0.3 mmol, 3.0 equiv) was then added. The vial was heated up to 100 °C using oil bath and allowed to stir for 12 h. The reaction was diluted with DCM, followed by filtration through Celite, concentrated in vacuo. Then crude mixture was purified by flash column chromatography on silica gel to afford the desired product.
4.5.1. (E)-2-(1,6-diphenyl-4-((propan-2-ylideneamino)oxy) hex −1-en-1-yl)-N-methoxy -N-methylbenzamide (7a)
Prepared according to the General procedure C in 31% isolated yield as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.65; dH (400 MHz, CDCl3) 7.36–7.30 (m, 2H), 7.29–7.21 (m, 3H), 7.21–7.10 (m, 5H), 5.52 (t, J = 6.9 Hz, 1H), 4.08 (p, J = 6.0 Hz, 1H), 2.67–2.46 (m, 2H), 2.42–2.24 (m, 2H), 2.04 (s, 3H), 1.87 (s, 3H), 1.85 (s, 3H), 1.83–1.71 (m, 2H); dc (101 MHz, CDCl3) 154.91, 142.66, 142.24, 138.02, 128.53, 128.38, 128.18, 128.16, 126.60, 125.73, 123.39, 81.62, 35.08, 33.45, 31.75, 25.93, 22.11, 15.85; IR (KBr, cm−1) 3026, 2921, 2852, 1601, 1495, 1435, 1367, 1095, 913, 700; HRMS (ESI-TOF) m/z: [M + H+] calculated for C22H27NO, 322.2165; found, 322.2167.
4.5.2. Methyl (Z)-3,8-diphenyl-6-((propan-2-ylideneamino)oxy) oct-3-enoate (7b)
Prepared according to the General procedure C in 65% isolated yield as a colorless liquid; Rf (hexanes: EtOAc = 4 : 1) 0.3; dH (500 MHz, CDCl3) 7.37–7.31 (m, 2H), 7.29–7.21 (m, 3H), 7.21–7.17 (m, 2H), 7.16–7.10 (m, 3H), 5.69 (t, J = 7.2 Hz, 1H), 4.09 (dq, J = 7.5, 5.7 Hz, 1H), 3.58 (s, 3H), 3.37 (s, 2H), 2.68–2.47 (m, 2H), 2.38 (hept, J = 8.2, 7.3 Hz, 2H), 1.92–1.87 (m, 1H), 1.86 (s, 3H), 1.85 (s, 3H), 1.81–1.69 (m, 1H); dc (101 MHz, CDCl3) 172.12, 154.32, 142.49, 139.94, 135.07, 128.57, 128.51, 128.38, 128.27, 128.18, 127.08, 125.75, 81.10, 51.83, 44.64, 35.01, 33.46, 31.71, 22.08, 15.82; IR (KBr, cm−1) 3025, 2948, 2918, 2850, 1740, 1601, 1495, 1435, 1264, 1163, 1067, 701; HRMS (ESI-TOF) m/z: [M + H+] calculated for C24H29NO3, 380.2220; found, 380.2223.
4.5.3. Benzyl (3-methylclohex-2-en-1-yl) (3-methylpyridin-2-yl) carbamate (7f)
2.2 mg Pd(OAc)2 (0.01 mmol, 10 mol %), 3.3 mg 5-CF3-2-pyridone (0.02 mmol, 20 mol %), 2.7 mg 2-methyl picolinate (0.02 mmol, 20 mol%), 10.5 mg N10 (0.05 mmol, 50 mol %), 51 mg AgOAc (0.3 mmol, 3.0 equiv), 32 mg alkene (0.1 mmol, 1.0 equiv), and 0.5 mL DCE were added to a flame-dried 4 mL vial under air atmosphere. 19 μL MeI (0.3 mmol, 3.0 equiv) was then added. The vial was heated up to 90 °C and allowed to stir for 24 h. The reaction was diluted with DCM, followed by filtration through Celite, concentrated in vacuo. Then crude mixture was purified by flash column chromatography on silica gel to afford the desired product (7f) in 25% yield; Rf (hexanes: EtOAc = 4 : 1) 0.15; m.p. 86–88 °C; dH (500 MHz, CDCl3, 55 °C) 8.32 (dd, J = 4.9, 1.8 Hz, 1H), 7.51 (dd, J = 7.5, 1.9 Hz, 1H), 7.40–7.16 (m, 5H), 7.10 (dd, J = 7.6, 4.7 Hz, 1H), 5.44 (br, 1H), 5.16 (d, J = 12.5 Hz, 1H), 5.10 (d, J = 12.6 Hz, 1H), 4.84–4.74 (m, 1H), 2.19 (s, 3H), 2.07–1.97 (m, 1H), 1.92–1.69 (m, 4H), 1.64–1.50 (m, 4H); dc (126 MHz, CDCl3, 55 °C) 154.94, 153.17, 146.52, 139.17, 136.99, 132.41, 128.69, 128.44, 127.86, 127.78, 127.10, 122.79, 67.13, 56.45, 29.74, 27.87, 23.61, 21.72, 17.95; IR (KBr, cm−1) 3032, 2931, 2868, 1705, 1575, 1395, 1305, 698; HRMS (ESI-TOF) m/z: [M + H+] calculated C21H24N2O2, 337.1911; found, 337.1910.
4.6. Deprotection of oxime ether
To a flame-dried 4 mL vial were charged a stir bar, 44 mg 5e (0.1 mmol, 1.0 equiv), and solvent mixture (0.9 mL HOAc + 0.3 mL THF + 0.3 mL H2O). 65 mg activated Zn powder (1.0 mmol, 10 equiv) was then added in one portion at the room temperature. The reaction was then heated up to 80 °C and stirred for 12 h. The reaction crude was diluted with DCM, followed by filtration through Celite. The filtrate was then concentrated in vacuo and purified by flash column chromatography on silica gel to afford product as a colorless liquid in 81% yield.; Rf (hexanes: EtOAc = 4 : 1) 0.1; dH (500 MHz, CDCl3) 7.72 (dd, J = 7.8, 1.4 Hz, 1H), 7.42 (td, J = 7.6, 1.5 Hz, 1H), 7.34–7.28 (m, 4H), 7.25 (m, 5H), 7.19 (m, 3H), 5.70 (dd, J = 8.8, 6.5 Hz, 1H), 3.73 (tt, J = 8.4, 4.4 Hz, 1H), 3.69 (s, 3H), 2.79 (ddd, J = 14.7, 9.4, 5.8 Hz, 1H), 2.68 (ddd, J = 13.8, 9.3, 7.0 Hz, 1H), 2.55–2.40 (m, 2H), 1.86–1.73 (m, 2H); dc = (126 MHz, CDCl3) 169.17, 144.89, 144.03, 142.29, 139.49, 131.31, 130.93, 130.86, 129.93, 129.76, 128.61, 128.47, 128.06, 127.79, 127.25, 127.12, 125.88, 71.10, 52.33, 38.64, 37.62, 32.20; IR (KBr, cm−1) 3380, 2934, 2865, 1720, 1600, 1435, 1282, 755; HRMS (ESI-TOF) m/z: [M + H+] calculated C26H26O3, 387.1955; found, 387.1956.
Supplementary Material
Acknowledgments
The authors thank NIGMS (1R01GM124414-01A1) and the University of Chicago and for financial supports.
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Guangbin Dong reports financial support was provided by NIGMS.
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.tet.2021.132173.
References
- [1].For alkenyl C H activation reviews, see:.; a) Zhan J, Lu X, Shen C, Xu L, Ding L, Zhong G, Chem. Soc. Rev 50 (2021) 3263–3314; [DOI] [PubMed] [Google Scholar]; b) Liu B, Yang L, Li P, Wang F, Li X, Org. Chem. Front 8 (2021) 1085–1101; [Google Scholar]; c) Maraswami M, Loh T-P, Synthesis 51 (2019) 1049–1062. [Google Scholar]
- [2].a) Smith III AB, Qiu Y, Jones DR, Kobayshi K, J. Am. Chem. Soc 117 (1995) 12011–12012; [Google Scholar]; b) Villalobos A, Danishefsky SJ, J. Org. Chem 55 (1990) 2776–2786; [Google Scholar]; c) White JD, Carter RG, Sundermann KF, Wartmann M, J. Am. Chem. Soc 123 (23) (2001) 5407–5413. [DOI] [PubMed] [Google Scholar]
- [3].For directed C H activation reviews, see:.; a) Lyons TW, Sandford MS, Chem. Rev 110 (2010) 1147–1169; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) He J, Wasa M, Chan KL, Shao Q, Yu J-Q Chem. Rev 117 (2017) 8754–8786; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sambiagio C, Schonbauer D, Blieck R, Dao-Huy T, Pototschnig G, Schaaf P, Wiesinger T, Zia MF, Wencel-Delord J, Besset T, Maes BUW, Schnurch M, Chem. Soc. Rev 47 (2018) 6603–6743; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Rej S, Das A, Chatani N, Coord. Chem. Rev 431 (2021) 213683. [Google Scholar]
- [4].For selected examples, see:.; a) Trost BM, Imi K, Davies IW, J. Am. Chem. Soc 117 (1995) 5371–5372; [Google Scholar]; b) Hu XH, Zhang J, Yang XF, Xu YH, Loh T-P, J. Am. Chem. Soc 137 (2015) 3169–3172; [DOI] [PubMed] [Google Scholar]; c) Zhou H, Xu Y, Chung W, Loh T-P, Angew. Chem. Int. Ed 48 (2009) 5355–5357; [DOI] [PubMed] [Google Scholar]; d) Wang H, Beiring B, Yu D, Collins KD, Glorius F, Angew. Chem. Int. Ed 52 (2013) 12430–12434; [DOI] [PubMed] [Google Scholar]; e) Ilies L, Matsubara T, Ichikawa S, Asako S, Nakamura E, J. Am. Chem. Soc 136 (2014) 13126–13129; [DOI] [PubMed] [Google Scholar]; f) Monks BM, Fruchey ER, Cook SP, Angew. Chem. Int. Ed 53 (2014) 11065–11069; [DOI] [PubMed] [Google Scholar]; g) Ilies L, Ichikawa S, Matsubara T, Nakamura E, Adv. Synth. Catal 357 (2015) 2175–2179; [Google Scholar]; h) Tsai AS, Bergman RG, Ellan JA, J. Am. Chem. Soc 130 (2008) 6316–6317; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Hou W, Zhou B, Yang Y, Feng H, Li Y, Org. Lett 15 (2013) 1814–1817; [DOI] [PubMed] [Google Scholar]; j) Liang Q, Yang C, Meng F-F, Jiang B, Xu Y-H, Loh T-P, Angew. Chem. Int. Ed 56 (2017) 5091–5095. [DOI] [PubMed] [Google Scholar]
- [5].For selected examples, see:.; a) Tsai H-C, Huang Y-H, Chou C-M, Org. Lett 20 (2018), 1338–1332; [DOI] [PubMed] [Google Scholar]; b) Wang Y-C, Huang Y-H, Tsai H-C, Basha RS, Chou C-M, Org. Lett 22 (2020) 6765–6770; [DOI] [PubMed] [Google Scholar]; c) Liu M, Yang P, Karunananda MK, Wang Y, Liu P, Engle KM, J. Am. Chem. Soc 140 (2018) 5805–5813; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Schreib BS, Carreira EM, J. Am. Chem. Soc 141 (2019) 8758–8763; [DOI] [PubMed] [Google Scholar]; e) Schreib BS, Fadel M, Carreira EM, Angew. Chem. Int. Ed 59 (2020) 7818–7822; [DOI] [PubMed] [Google Scholar]; f) Meng K, Li T, Yu C, Shen G, Zhang J, Zhong G, Nat. Commun 10 (2019) 5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].For a review about “exo”-type directing groups, see; Xu Y, Dong G, Chem. Sci 9 (2018) 1424–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].For selected reviews, see:.; a) Huang Z, Dong G, Acc. Chem. Res 50 (2017) 465–471; [DOI] [PubMed] [Google Scholar]; b) Hartwig JF, Acc. Chem. Res 50 (2017) 549–555; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Meng G, Lam NYS, Lucas EL, Saint-Denis TG, Verma P, Chekshin N, Yu J-Q, J. Am. Chem. Soc 142 (2020) 10571–10591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].For selected reviews, see:.; a) Ye J, Lautens M, Nat. Chem 7 (2015) 863–870; [DOI] [PubMed] [Google Scholar]; b) Della Ca’ N, Fontana M, Motti E, Catellani M, Acc. Chem. Res 49 (2016) 1389–1400; [DOI] [PubMed] [Google Scholar]; c) Wang J, Dong G, Chem. Rev 119 (2019) 7478–7528; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Cheng H-G, Chen S, Chen R, Zhou Q, Angew. Chem. Int. Ed 58 (2019) 5832–5844. [DOI] [PubMed] [Google Scholar]
- [9].Catellani M, Frignani F, Rangoni A, Angew. Chem. Int. Ed 36 (1997) 119–122. [Google Scholar]
- [10].a) Wang X-C, Gong W, Fang L-Z, Zhu R-Y, Li S, Engle KM, Yu J-Q, Nature 519 (2015) 334–338; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dong Z, Wang J, Dong G, J. Am. Chem. Soc 137 (2015) 5887–5890. [DOI] [PubMed] [Google Scholar]
- [11].For the Catellani reaction with regular alkenes, see; Wang J, Dong Z, Yang C, Dong G, Nat. Chem 11 (2019) 1106–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].For a review about structurally modified NBEs, see; Li R, Dong G, J. Am. Chem. Soc 142 (2020) 17859–17875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].For a preliminary communication, see; Wu Z, Fatuzzo N, Dong G, J. Am. Soc. Chem 142 (2020) 2715–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Catellani M, Chiusoli GP, J. Organomet. Chem 233 (1982) C21–C24; [Google Scholar]; b) Khanna A, Premachandra IDUA, Sung PD, Van Vranken DL, Org. Lett 15 (2013) 3158–3161. [DOI] [PubMed] [Google Scholar]
- [15].Wang P, Farmer ME, Huo X, Jain P, Shen P-X, Ishoey M, Bradner JE, Wisniewski SR, Eastgate MD, Yu J-Q, J. Am. Chem. Soc 138 (2016) 9269–9276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chen S, Liu Z-S, Yang T, Hua Y, Zhou Z, Cheng H-G, Zhou Q, Angew. Chem. Int. Ed 57 (2018) 7161–7165. [DOI] [PubMed] [Google Scholar]
- [17].a) Ren Z, Mo F, Dong G, J. Am. Chem. Soc 134 (2012) 16991–16994; [DOI] [PubMed] [Google Scholar]; b) Ren Z, Schulz JE, Dong G, Org. Lett 17 (2015) 2696–2699. [DOI] [PubMed] [Google Scholar]
- [18].a) Motti E, Della Ca N’, Deledda S, Fava E, Panciroli F, Catellani M, Chem. Commun 46 (2010) 4291–4293; [DOI] [PubMed] [Google Scholar]; b) Martins A, Candito DA, Lautens M, Org. Lett 12 (2010) 5186–5188. [DOI] [PubMed] [Google Scholar]
- [19].Zhang S-Y, He G, Nack WA, Zhao Y, Li Q, Chen G, J. Am. Chem. Soc 135 (6) (2013) 2124–2127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.