

Abstract
There is hope that host-directed therapy (HDT) for Tuberculosis (TB) can either shorten treatment duration, help cure drug resistant disease or limit the immunopathology. Many candidate HDT drugs have been proposed, however solid evidence only exists for a few select patient groups. The clinical presentation of TB is variable, with differences in severity, tissue pathology, and bacillary burden. TB clinical phenotypes likely determine the potential benefit of HDT. Underlying TB clinical phenotypes, there are TB “endotypes,” defined as distinct molecular profiles, with specific metabolic, epigenetic, transcriptional, and immune phenotypes. TB endotypes can be characterized by either immunodeficiency or pathologic excessive inflammation. Additional factors, like comorbidities (HIV, diabetes, helminth infection), structural lung disease or Mycobacterial virulence also drive TB endotypes. Precise disease phenotyping, combined with in-depth immunologic and molecular profiling and multimodal omics integration, can identify TB endotypes, guide endotype-specific HDT, and improve TB outcomes, similar to advances in cancer medicine.
Keywords: Tuberculosis, endotypes, immune correlates of protection
eTOC:
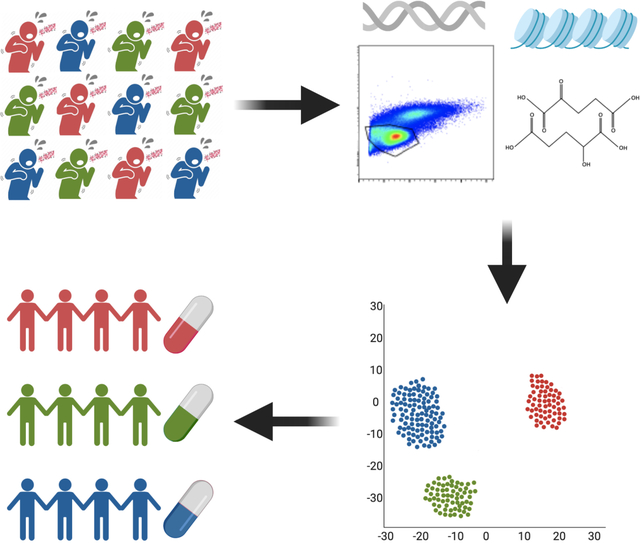
“DiNardo et al” discuss Tuberculosis endotypes. Cancer and asthma therapy depend on the disease endotype, the distinct molecular pathology driving disease. There is no single immune correlate of protection against TB, but multiple pathways that lead to TB. This article discusses the potential benefit of an endotype-specific HDT approach.
Graphical Abstract
Introduction:
With 10 million estimated annual cases, tuberculosis (TB) remains a global scourge. Following infection with Mycobacterium tuberculosis (Mtb), a minority of people progress to active TB disease, with a complex interaction of host, mycobacterial, and environmental factors determining the risk of disease progression. Active TB mostly presents as pulmonary disease, ranging from subclinical localized infiltrative disease, pleurisy, or hilar lymphadenopathy, to severe or widespread disease with extensive necrosis, lung cavities and fibrosis, disseminated miliary disease, or extrapulmonary disease including osteoarticular, abdominal, spinal, and cerebral involvement that may be life-threatening. Also, following treatment, some patients recover uneventfully, while others are slow to clear disease even with effective antimicrobial therapy or even experience paradoxical worsening of TB with progressive immunopathology(1). Such worsening has been described as immune reconstitution inflammatory syndrome (IRIS) in the context of HIV-infection but may also occur in HIV-negative patients. Finally, after successful completion of treatment, some patients experience disease recurrence. These variable presentations and outcomes of TB disease can be termed ‘phenotypes’.
While antibiotics target mycobacteria, there is enthusiasm that host-directed therapy (HDT), which acts by modulating host immunity or other molecular mechanisms, can improve TB outcomes. The potential benefit of HDT is likely to depend on TB disease phenotype and on timing of the use of HDT. Goals of HDT include clearance of Mtb (potentially shortening antimicrobial treatment or improving outcomes of multi-drug resistant TB) and/or limiting damaging inflammation and immunopathology. So far, although many candidate drugs have been proposed or trialed as HDT for TB, there is only evidence for a beneficial effect of corticosteroids for two specific narrow indications: TB meningitis in HIV-negative patients(2), and IRIS in the context of HIV co-infection(3, 4). In addition, in patients with HIV-associated TB, antiretroviral therapy restores host immunity, improves long-term outcomes, and can be considered an effective form of HDT. One reason behind the lack of consistent evidence for other drugs is the fact that their effect may depend on disease phenotype, or on underlying biological pathways, or endotypes.
While phenotype is the observable, clinical presentation, endotypes are the distinct immunologic and molecular pathophysiologic mechanisms that lead to disease(5, 6). Different endotypes can result in similar or dissimilar clinical phenotypes. For example, asthma has a relatively uniform phenotypic presentation, that can be due to eosinophilic or neutrophilic endotypes. Asthma treatment is supposed to target the specific endotype(5, 6). For example, asthma can be classified as “T2-high,” “T2-low,” obesity-related, smoking-related, or late-onset; “T2-high” asthma is characterized by increased IgE, IL-5, IL-25 and IL-33 and treated with inhaled monoclonal antibodies targeting IL-5(6, 7). In this perspective, we will review what is known about TB endotypes, in particular the specific immune profiles and patterns of inflammatory markers (endotypes) that are regulated by a complex interplay of cellular metabolism, host genetic background, epigenetics, and gene transcriptional regulation. Other factors, especially M. tuberculosis genotype and bacillary load, structural lung damage, and environmental factors such as co-medication or smoking are likely to influence host endotypes. In cancer medicine, large collaborative efforts, such as The Cancer Genome Atlas (TCGA) have helped to identify distinct endotypes leading to targeted endotype-specific therapy and improved clinical outcomes. Better characterization of distinct TB endotypes, and their underlying host, environmental, and pathogen- related factors holds promise to identify TB endotype-specific therapy and improved clinical outcomes.
Leveraging Host Immunity to combat Mtb
Defects in host immunity, immune polarization due to HIV or helminth co-infection, or rare inborn genetic defects drastically increase the risk of TB disease progression. Unlike bacterial pneumonia, which is treated for five to 14 days with antibiotics, TB therapy requires six months of antibiotics for drug-sensitive pulmonary disease, and nine to twenty-four months for drug-resistant disease. Each year, ~1.5 million of the 10 million TB patients die. In searching to define immune correlates of protection, the immune response that protects against TB disease progression, there is a hope that these efforts will also identify the means to harness host immunity to improve TB treatment outcomes.
From a therapeutic perspective, the major objective of treatment is to kill intracellular mycobacteria, without inflicting collateral damage to host organs(8–10). Dendritic cells (DCs), macrophages, and neutrophils are the main phagocytic cells and all can possess pro and anti-inflammatory characteristics under specific circumstances (Fig. 1)(11). Lymphocytes, both classic CD4+ and CD8+ T lymphocytes, as well as Natural killer (NK) cells, Innate Lymphoid Cells (ILCs), Mucosal associated invariant T cells (MAITs), and γδ T cells produce cytokines and implement cytotoxic killing. Cytokines can generally be classified as Th1 (such as TNF, IL-2, and IFN-γ for combatting intracellular pathogens), Th2 (IL4, IL-5, and IL-13 for combatting helminths and wound healing), and Th17 (IL-17, IL-21, and IL-22 for combatting extracellular bacteria and fungi). Lymphocytes, especially NK cells and CD8+ T cells, also have cytotoxic killing potential; host cells infected by Mtb are destroyed by injection of perforin and granzymes into their membranes(12–14). B cells, often ignored in mycobacterial immunity, aid in opsonophagocytosis and antibody-dependent cell-mediated cytotoxicity(15). Increasing knowledge of the complex interaction of multiple immune cell types and the delicate regulation of immune homeostasis has made it clear that there is not a single immune correlate of protection since there are multiple pathways by which an individual can progress to TB disease.
Figure 1:
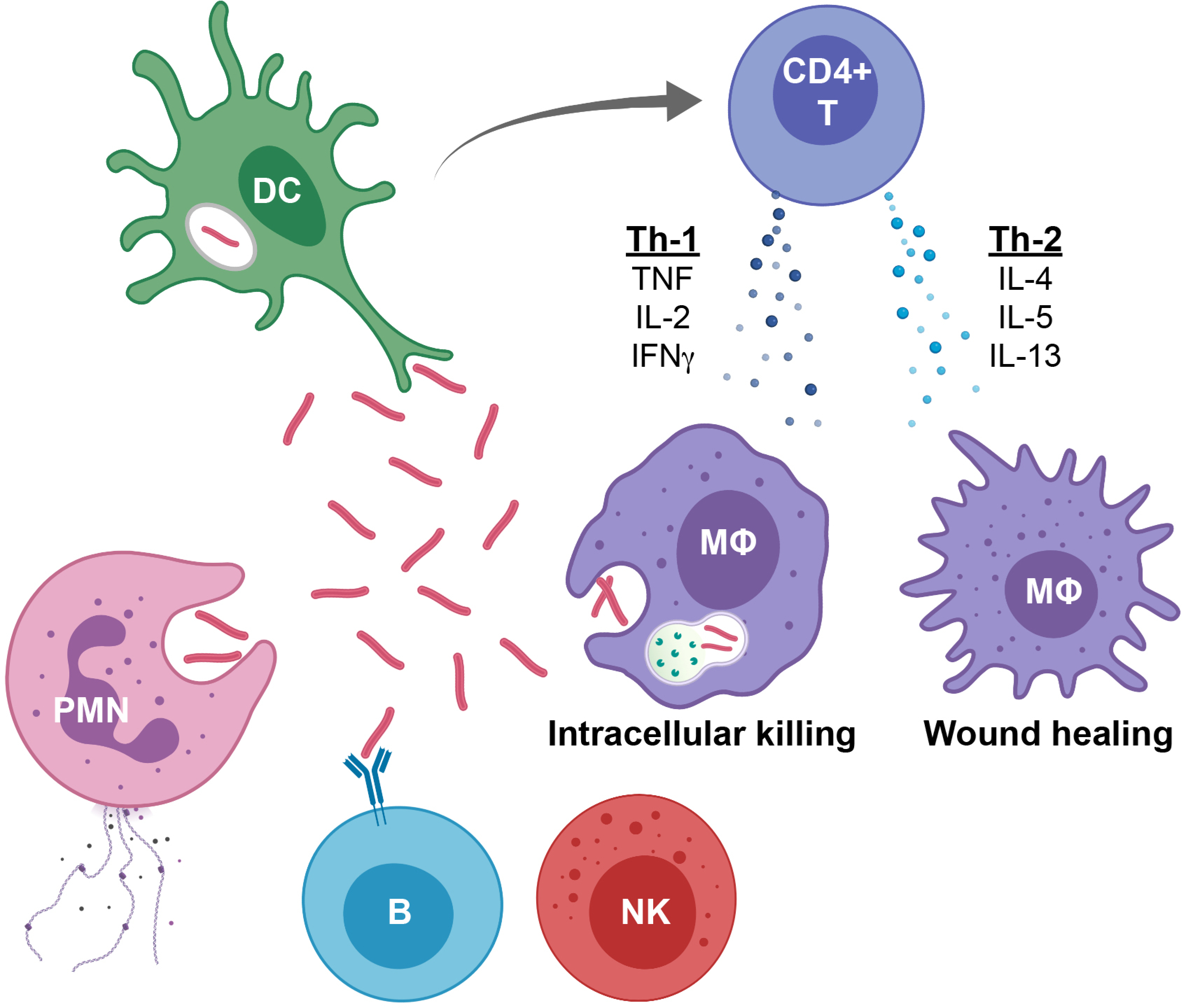
Overview of immune response to Mtb infection
1. Dendritic cells (DCs), polymorphonuclear neutrophils (PMNs), and macrophages phagocytose Mtb leading to either mycobacterial survival or death.
2. DCs are professional antigen presenting cells and bring Mtb antigens to the draining lymph node, where DCsimplement antigen presentation and activation the classic CD4+ T cell response.
3. Macrophages are the preferred intracellular niche for Mtb. When activated by TNF and IFN-γ, they upregulate ROS, lysosomal acidification and phagolysosome maturation and increase Mtb killing capacity (More M1-like macrophage). When activated by IL-10, IL-4, IL-5 or IL-13, they down-regulate these processes, temper intracellular killing and focus on wound healing (More M2-like macrophage). Tissue macrophages represent a spectrum that typically includes elements of both M1 and M2 phenotypic characteristics.
4. Neutrophils, phagocytose and kill Mtb, but also produce cytokines and when they die, they spew out an extracellular matrix of chromatin and proteins that alert other immune cells.
5. CD4+ T cells produce cytokines, such as Th-1 cytokines TNF, IL-2 and IFN-γ that stimulate macrophages for intracellular killing, or Th2 cytokines IL-4, IL-5 and IL-13 that promote macrophage wound healing.
6. Cytotoxic T cells, as well as Natural Killer (NK) cells, MAIT (mucosal associated invariant T cells) and Innate LymphoidCells (ILCs), in addition to producing activating or suppressive cytokines, can induce perforin and granzyme-mediate cytotoxic killing of Mtb-infected cells.
7. B cells produce antibodies that neutralize extracellular Mtb, mediate NK CD16 antibody dependent cytotoxicity, and mark Mtb for opsonophagocytic clearance.
Known TB Immune endotypes and the multiple immune requisites against Mtb
IL-12 - IFN-γ up and downstream immune deficient endotypes
Interferon-gamma (γ) is the immune biomarker most often evaluated as a potential immune correlate of TB protection; yet, an in-depth evaluation of IFN-γ signaling pathways highlights the heterogeneity of immune dysfunction that can lead to TB progression. The Mendelian susceptibility to mycobacterial diseases (MSMDs) are a collection of rare genetic defects that increase an individual’s risk for TB and other mycobacterial diseases(16, 17). A large number of these defects are related to IFN-γ signaling, either upstream (IL12B2, IL12RB1, NEMO, IRF8, SPPL2a) or downstream (IFNGR1, IFNGR2, STAT1, IRF8) of IFN-γ (17). IL12RB1 and IFNGR1 mutations, representative of IFN-γ upstream and downstream mutations highlight why a single immune correlate of protection is unlikely to be identified(16).
Mutations upstream in the IFN-γ pathway result in decreased IFN-γ production and decreased capacity to kill intracellular bacteria (Fig. 2A). The most common means to trigger the IFN-γ pathway starts with the IL12 receptor, a heterodimer consisting of IL12RB1 and IL12RB2. Aberrant IL12RB1 mutations affect IL12, IL-17 and IL23 signaling(18). Intracellular components of the IL12 receptor interact with tyrosine kinases (JAK2 and TYK2) to transmit the IL12 signal to STAT4. Phosphorylation of STAT4 and subsequent activation of other transcription factors, including IRF8 (interferon regulatory factor), increases expression of multiple genes involved in the host defense against intracellular pathogens (GBP1, GBP2, CXCL9, etc.), and in particular the production of IFN-γ (Fig. 2A)(19). Upstream defects in IFN-γ signaling lead to decreased lymphocyte proliferation, poor granuloma formation, decreased CD4 memory formation, immature NK cells and decreased intracellular killing capacity(20, 21). The major functional result of IL12RB1, and other upstream MSMD mutations, is a decrease in the abundance of IFN-γ produced (Fig. 2A). Production of IFN-γ results in cell biologic changes that upregulate ROS production, lysosomal acidification, and phagolysosome maturation, leading to increased killing of intracellular pathogens(8, 19). Treatment of upstream defects includes targeted antimicrobials augmented by adjuvant recombinant IFN-γ treatment.
Figure 2:
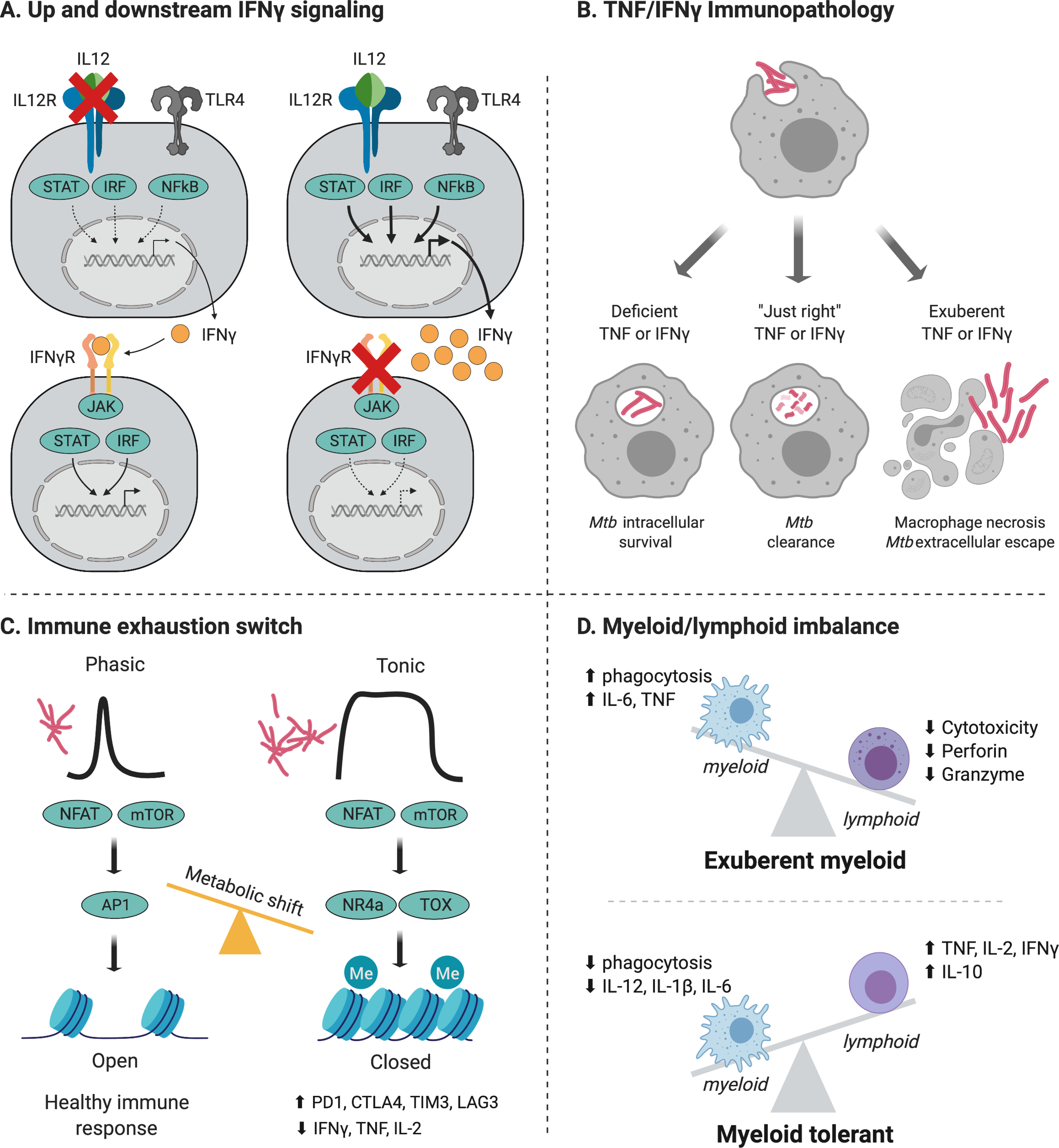
Endotypes are the distinct host molecular pathways by which an individual can progress to TB. While some endotypes are exclusive, other endotypes overlap. For example, defects in the IL-12- IFN-γ axis (A) result in immune deficiency and overlap with the IFN-γ deficient endotype (B). Similar, after tonic antigenic stimulation, immune exhaustion (C) leads to deficiencies in both TNF and IFN-γ.
A. IL-12- IFN-γ upstream defects in IL12, the IL12 receptor, IKKB or IRF8 result in decreased IFN-γ production and decreased mycobacterial killing capacity. In contrast, downstream defects in the IFN-γ receptor, STAT1 or IRF1 result in increased IFN-γ, but decreased IFN-γ signal transduction and decreased mycobacterial killing capacity.
B. Host immunity has a narrow therapeutic window: deficiencies in TNF and IFN-γ result in decreased capacity to kill intracellular Mtb. In contrast, exuberant TNF and IFN-γ lead to macrophage necrosis and viable Mtb escape into the extracellular space.
C. Short antigenic stimulation induces Warburg metabolism, increased glycolysis and glutaminolysis that triggers beneficial epigenetic immune changes. In contrast, chronic antigenic stimulation, either from TB itself, or from previous HIV, helminth or other chronic infection, results in tonic NFAT and mTOR activation resulting in metabolic and epigenetic mediated immune exhaustion. Immune exhaustion is characterized by decreased cytokine (TNF, IL-2 and IFN-γ) production, so this phenotype overlaps with above.
D. Mycobacterial immunity requires both intact and well-balanced myeloid and lymphoid immunity. Hemophagocytic lymphohistiocytosis (HLH) represents imbalance, with deficient cytotoxic T cell and NK cell immunity and myeloid driven immunopathology with excessive TNF, IL-6 and phagocytosis. Likely, this overlaps with the exuberant phenotype depicted in B.
In contrast, downstream defects are characterized by excess IFN-γ and a failure to transmit the signal. IFN-γ signals through the IFN-γ receptor, a hetero-dimer of IFNGR1 and IFNGR2. Extracellular activation of the receptor signals to the intracellular domains to activate the tyrosine kinases JAK1 and JAK2, which phosphorylate STAT1(16). In the nucleus, pSTAT1 induces epigenetic and transcriptional changes resulting in the cell biology changes requisite for control of intracellular pathogens such as mycobacteria, endemic mycosis and salmonellae(16, 22, 23). Classically, IFN-γ responsiveness was measured by stimulating cells with IFN-γ followed by measuring TNF production. Nevertheless, this readout is limited in scope since the IFN-γ response is cell-specific resulting in increased chemokine production (CXCL9, CXCL10), improved antigen presentation (TAP, HLADR), improved phagolysosome maturation (GBP1, GBP5), and lysosomal acidification (PFK, IDO, AIM, DUOX)(17, 19, 24). Clinically, and in contrast to individuals with upstream mutations, downstream mutations induce more severe disease, are not responsive to exogenous IFN-γ and have worse outcomes after bone marrow transplantation(16). Rarely individuals can develop auto-antibodies to IFN-γ that block downstream IFN-γ signaling, similar to a downstream defect. These individuals are at increased risk of intracellular fungal and mycobacterial pathogens(23).
TNF deficient endotype
Like IFN-γ, TNF is necessary, but not sufficient for protection against Mtb and TNF is often evaluated in parallel with IFN-γ as a biomarker(25–28). One study demonstrated that individuals with asymptomatic infection are more likely to have Mtb-specific “polyfunctional” CD4+ T cells that simultaneously produce TNF, IFN-γ and IL-2(25, 29). TB diseased individuals are more likely to have CD4+ T cells producing only antigen-specific TNF, with a decrease in IL-2 and IFN-γ (25). As TNF and IFN-γ act synergistically(30, 31), clarifying poly-functionality (simultaneous production of multiple cytokines by a single cell) had inspired hope for the identification of immune correlates of protection. There is, however, growing skepticism in this approach since IFN-γ is modified by both upstream and downstream signaling as described above, and as described below, TNF itself has a narrow therapeutic window.
Clinically, the critical role of TNF is evidenced by the multifold increased risk of progression to active TB among individuals treated with monoclonal antibodies antagonizing TNF(28, 32). TNF is produced by both innate and adaptive immune cells and induces profuse and pleiotropic anti-mycobacterial activity resulting in increased phagocytosis, increased granuloma formation, and increased Mtb killing(27). TNF induces the endothelial cell adhesion requisite for granuloma formation. In addition, it results in increased chemokine production thereby recruiting immune cell to the site of infection(33).
Intracellularly, TNF induces phagosome-lysosome maturation, acidification of the lysosome, and up-regulation of NO synthetase and ROS in order to augment intracellular Mtb killing(34). The TNF cellular effects are synergistic with IFN-γ with inhibition of TNF resulting in inhibition of IFN-γ-inducible phagosome-lysosome maturation and Mtb killing(35). Ideally, treatment of TB patients with TNF-deficient endotypes would remove the offending TNF inhibitory agent, and/or a drug perturbing TNF signaling. Immune exhaustion, discussed in detail below, inhibits TNF and therefore strategies to reverse immune exhaustion may restore TNF.
Narrow therapeutic window and need to avoid an Immunopathology endotype
Further complicating the identification of a singular immune correlate of protection against TB disease progression is the fact that many immune functional pathways and cytokines against Mtb have a narrow therapeutic windows (Fig. 2B). Host immunity must be robust enough to kill mycobacteria, but not so therapeutic window within which to achieve balanced killing of intracellular pathogens rather than inducing exuberant immunopathology. While IFN-γ deficiency increases the risk of TB, mouse studies demonstrate that excessive IFN-γ results in immune-induced pathology and increased mortality(36–39). The addition of IFN-γ to healthy macrophages ex vivo increases Mtb killing capacity(40, 41); however, in animal studies, when IFN-γ production was increased (either via PD1 inhibition or cell transfer), there was increased pulmonary necrosis, escape of viable Mtb into extracellular spaces, and death of the animals(36, 37). Results from these studies suggest that IFN-γ has a narrow therapeutic window with excessive IFN-γ inducing immunopathology. Further, the effect of IFN-γ is modulated by IL-4, IL-10, HIF1α, and other factors(42, 43). Therefore, despite IFN-γ increasing macrophage capacity to kill Mtb in vitro(40, 41), measuring this cytokine alone is not a predictive biomarker of sufficient anti-Mtb immunity; further, increases in IFN-γ are associated with increased risk of disease progression(44, 45).
Similar to IFN-γ, TNF has a narrow therapeutic window. Specifically, multiple studies have demonstrated the mechanisms by which Mtb is capable of evading an unbalanced immune response(2, 46). TNF is regulated by the arachidonic acid- leukotriene pathway with leukotriene A4 hydroxylase as a key regulator(47). The arachidonic acid pathway is capable of producing both the pro-inflammatory leukotriene B4 (LTB4) and/or the anti-inflammatory Lipoxin A4 (LXA4). In a zebrafish model, the heterozygous LTB4 genotype improved Mtb killing, while in contrast the homozygous CC LTA4H polymorphism decreased LTB4, decreased TNF, decreased intracellular ROS up-regulation, and decreased mycobacterial killing capacity(2, 46). Similarly, the homozygous TT LTA4H polymorphism has decreased mycobacterial killing capacity, however through an alternative mechanism: excessive TNF leads to RIP3-mediated necroptosis (organized cell necrosis) resulting in macrophage cell death with viable Mtb escaping into the extracellular space(46). Although the studies were initially performed in zebra fish, results correlated with the benefits of steroids for humans suffering from TB meningitis (TBM)(2, 46) where LTB4 high patients benefited from steroids, while LTB4 low patients did not(2). TNF is also regulated by PD1, and like IFN-γ, inhibiting PD1 boosts TNF production; but, this boosting can be excessive resulting in increased Mtb growth(48). Therefore, like IFN-γ, TNF has a narrow therapeutic window with excess inducing immunopathology. Clinically, exuberant IFN-γ and TNF TB endotypes could be treated with glucocorticoids, cyclooxygenase inhibitors (aspirin, ibuprofen, montelukast), calcineurin inhibitors (cyclosporin and tacrolimus), or mTOR inhibitors (rapamycin). Animal models fail to fully recapitulate human disease; most mouse strains have poorly formed granulomas and zebra fish only have innate immunity(2, 49, 50). However, the anecdotal evidence that TNF blockade can successfully be implemented in TB patients with steroid-refractory IRIS and that steroids are beneficial in some forms of TB lends support that exuberant immunity is detrimental in humans.
Additional evidence for the need for balanced host immunity and of the detrimental effect of exuberant immunity is hemophagocytic lymphohistiocytosis (HLH), a relatively rare manifestation of TB(51). HLH is a devastating disorder characterized by clinical and laboratory evidence of extreme inflammation characterized by defects in cytotoxic T cell and NK cell degranulation whereby the stimulating antigen is not cleared resulting in macrophage over-activation syndrome (MAS) with pathologic phagocytosis and pro-inflammatory cytokine secretion. Interleukin-1 plays a key role in the pathogenesis of HLH/MAS, and interleukin-1 receptor blockade has shown benefit in MAS and HLH due to rheumatologic disorders and sepsis(52). Studies in macaques(53) and anecdotal evidence also supports a role for interleukin-1 receptor blockade in selected TB patients with hyperinflammation (van Crevel, unpublished data), but this clearly needs more evidence. While HLH/MAS is an extreme and rare TB endotype, markers of sHLH/MAS like hyperferritinemia, coagulopathy and cytopenia should be checked in TB patients with unexplained hyperinflammation, and IL-1 receptor blockade or dexamethasone plus etoposide should be considered if sHLH/MAS is diagnosed(54).
Transition from robust to exhausted immune endotype
Even excluding individuals with pre-existing immune suppression, many previously immunocompetent individuals develop TB. However, there is evidence that after extensive antigenic stimulation, the immune system will not stay robust, but will transition to exhausted. Since 1984, LCMV infection has been the prototypical model for studying immune exhaustion(55). These studies provide guidelines how a healthy immune response evolves and transitions towards immune exhaustion in the setting of TB (Fig. 2C).
Immune exhaustion is defined by decreased cytokine production (TNF, IL-2 and IFN-γ), decreased proliferative capacity and increased immune checkpoint markers (PD1, TIM3, CTLA4)(56). Upon immune activation, intracellular signaling cascades are mediated by the influx of calcium, triggering the activation of multiple transcription factors, especially NFAT(57). The Phosphoinositide-3-kinase (PI3K) cascade then activates the AKT-mTOR pathway, resulting in activation of the transcription factors IRF4 and HIF1α(39, 58). Activation of mTOR, NFAT, IRF4 and HIF1α induces cellular metabolic shifts towards glycolysis and glutaminolysis and depletion of intracellular glutathione stores(56, 59–61). Initially these metabolic shifts promote immune activation, however, if persistent, increases in TCA metabolites alter the epigenetic enzymes inducing epigenetic-mediated exhaustion(61–63). As intracellular calcium stores become depleted, AP-1 transcription factors become dysregulated resulting in NFAT homodimers(64). Tonic activation of mTOR and NFAT induces NFAT, TOX and NR4A up-regulation of checkpoint inhibitors (PD1, TIM3, CTLA4)(65–68). The nucleosome remodeling deacetylase complex (NuRD) consisting of histone deacetylases, methyl binding domains proteins (MBDs), PAC1, DNA methyltransferases (DNMTs) and the polycomb repressive complex (PRC, including EZH2) work in concert to close chromatin, inhibit gene transcription and limit immune function(69).
We hypothesize that this is likely beneficial to prevent exuberant immune pathology, but human studies demonstrate that these inhibitory epigenetic marks persist even after removal of antigenic stimulation. For example, in human studies, despite successful antiretroviral therapy, HIV patients have DNA hyper-methylation of IL-2 for two years post aviremia(70). Children with schistosomiasis have DNA hyper-methylation that lasts at least 6 months after successful de-worming(71). In TB, DNA hyper-methylation marks are persistent for six months after completing successful anti-TB therapy(72). HIV, schistosomiasis and TB are chronic infections, with patients having antigenic stimulation for months. In mouse studies, immune exhaustion is reversible early, but becomes fixed when activation is persistent(73). Memory-like exhausted immune cells develop 1 week into continuous antigenic stimulation in mice. TB patients have a median of 87 days of symptoms before diagnosis(74) suggesting that individuals who initially have intact immunity will transition to an exhausted phenotype when diagnosis is delayed. If TB induced immune exhaustion follows mechanisms similar to LCMV induced immune exhaustion, then patients with exhausted TB endotypes could be treated with mTOR, NFAT, NR4A inhibitors, DNA hypomethylating agents, and/ or histone deacetylase inhibitors to block and even reverse TB induced immune exhaustion(56, 66, 75–78).
Other potential immune deficiency endotypes: Mal, GM-CSF, IL23, ORAI
In addition to TNF and IFN-γ, TIRAP (Toll/ interleukin-1 receptor domain containing adaptor protein) signaling may also have a narrow therapeutic immune window. TIRAP is involved in non-canonical signaling of IFN-γ and regulation of TLR2 and TLR4 activities by regulating MAPK activation(79). Interestingly, a heterozygous TIRAP mutation had improved Mtb killing capacity, despite the SS homozygous SNP having increased production of CXCL10 and TNF(80). Other critical components of anti-mycobacterial immunity include GM-CSF, IL-17, IL-21, IL-23 and STIM1. It is unclear how frequently a perturbation in these mechanisms leads to disease. A mycobacteria immune-deficient endotype characterized by auto-antibodies to GM-CSF is well described in humans(81) and the mechanisms of IL-17, IL21, IL23 and STIM1 TB endotypes have also been observed in mouse models(18, 82). Identifying the role these defects play in human TB requires larger studies with the capacity and resolution to identify rare endotypes. This is discussed in more detail below.
External drivers of host endotypes:
Susceptibility to and clinical presentation of TB is due to the interaction of host, environment, and pathogen. HIV infection is a classic example of an external factor driving host endotype and increasing the risk for TB. Additional risks include helminth co-infection, diabetes, and airway dysfunction from smoking and air pollution. In-depth reviews on HIV(83) and helminths(84, 85) perturbations on host immunity have been previously covered; therefore, below we review how diabetes, airway dysfunction, and Mtb features effect host endotypes and increase the risk of TB progression.
Diabetes is becoming one of the most common risk factors for TB and poor TB treatment outcomes(86, 87). Macrophages from people with diabetes show decreased phagocytosis, autophagy, and antigen presentation(88–90). In contrast to HLH where myeloid cells are exuberantly hyper-activated and lymphocytes are deficient, diabetes is characterized by hyper-activated T cells and hypo-activated myeloid cells(89, 91–93). Diabetics also have other factors like dysregulated fatty acids and lipid metabolism, vitamin D deficiency, glycosylation end products, diminished cell signaling molecule diacylglycerol (DAG), and diminished glutathione, a key mediator of ROS signaling, which may be involved in increasing the risk for TB disease progression or worsening TB outcomes(88, 92, 94–96). This remains an area of speculation, but experimental and epidemiologic data suggests that metformin, a regulator of mTOR and one of the most commonly used drugs for diabetes, may be a promising example of an endotype-specific HDT. Metformin ameliorates lung pathology and limits Mtb growth in mice(97), while in humans it improves phagocytosis, increases ROS production and inhibits TNFa and IL-1b production(98). In epidemiological studies, metformin decreases negative TB outcomes, including cavitation, death, and disease recurrence,(9, 99) and a trial evaluating metformin as HDT for TB in non-diabetic individuals is in preparation (Kornfeld, personal communication).
Cigarette smoking and indoor air pollution increase the risk for TB due to airway damage, mucociliary dysfunction, and impaired anti-mycobacterial immunity(100–102). Cigarette smoking substantially delays the time to culture conversion in patients who receive TB treatment(103); therefore, we hypothesize that the increased duration of antigenic stimulation increases immune exhaustion. Studies are needed to evaluate this hypothesis. TB patients have rates of recurrence higher than asymptomatic household contacts; and, this risk of relapse increases further with smoking(104). The deleterious effects of air pollution and smoking on anti-mycobacterial immunity have been reviewed; in short, they induce inflammation and impair innate (macrophage, dendritic cells and NK cell) and adaptive immunity (T cells, B cells, antibodies)(105, 106). Of note, bronchiectasis and other mechanical airway diseases that increase airway diameter result in permanent mucociliary dysfunction. Therefore, even if host immunity normalizes, an individual with structural lung disease is likely more prone to TB progression with a lower bacillary burden. Likely, the chronic antigenic stimulation from bronchiectasis is not its own endotype, but induces epigenetic-mediated immune exhaustion as described above. Future studies need to clarify if mechanical airway deficiencies induce a distinct immune endotype or overlap with other endotypes.
Mtb genotype and load are likely additional drivers of TB endotypes. Even individuals with robust, well-balanced host immunity and intact airway function can progress to disease if the bacillary burden is high enough. Considering the narrow therapeutic window of host immunity described above, full clarification and interpretation of the appropriateness of host immunity requires precise knowledge regarding the bacillary burden in patients which is not currently available with existing technology. In addition to bacillary burden, different Mtb strains produce different virulence factors(107, 108). There are nine distinct Mtb strains that differ in their mycobacterial cell wall components, their metabolic induction of host immune cells, their induction of host inflammation, their transmissibility, and their success in different regions of the world(109). Each Mtb lineage contains distinct glycolipid cell wall components (110) and we hypothesize this is one means strains may be differentially modulating host immunity. Evidence for this hypothesis may be supported by studies demonstrating that rabbits infected with CDC1551 (common Lineage 4 strain) induce less myeloperoxidase, less pulmonary necrosis, less STAT1 and NFKB signaling, and had lower bacillary loads than HN878 (common Lineage 2, Beijing strain)(111). Other studies suggest the HN878 strain is more likely to induce IL4 and IL-13 and less IL-12 and IFN-γ than the CDC1551 strain(112). Even within lineages, reports suggest significant differential immune induction between strains. When compared to a non-MDR Beijing strain, an MDR Beijing strain induced less TLR2-mediated AKT-mTOR glycolytic metabolism and IL1β up-regulation(113). Sequencing and molecular epidemiologic studies have demonstrated that strain-specific transmissibility and disease progression depend on environmental factors and host genotype(114). As integrating strain sequencing and host immunity becomes more feasible, the bi-directional relationship between Mtb strain and host endotypes should become more clarified.
Integration of multimodal omics to clarify TB endotypes
There is compelling evidence from case reports and animal studies for the existence of multiple distinct and contradictory TB endotypes. To determine the presence of TB endotypes, including rare endotypes, a large clinical cohort with uniformly collected data is required. This does not yet exist for immune phenotyping, metabolism or epigenetics, however multiple studies have systematically reviewed publicly available gene expression data(115, 116). We recently embarked on analyzing publicly available transcriptomics and implementing an unbiased bioinformatic clustering analysis to identify TB endotypes with distinct metabolic, epigenetic and immune gene expression patterns(117). The publicly available data has limited epidemiology, and therefore limited analysis of the external drivers of distinct TB endotypes. Further, resting gene expression data alone is not indicative of function. TB inhibits host immunity and multiple studies have demonstrated that TB patients have elevated baseline immune activation, but a failure to up-regulate upon stimulation(25, 72, 118, 119). Immune function depends on metabolic activation upon stimulation and epigenetic confirmation, and therefore paired metabolic, epigenetic and functional immune phenotyping studies are needed to fully characterize preliminary TB endotypes based on gene expression.
While simultaneous evaluation of all these factors may seem a daunting obstacle, many fields have developed integrative multimodal omics approaches that have advanced clinical care (Fig. 3). Endotypes can be clarified by integrating these data sources with rich and longitudinal clinical information and by leveraging approaches similar to The Cancer Genome Atlas (TCGA). Initiated in 2006, the TCGA include publicly available data on >40 tumor types for more than 30,000 patients(120). Publicly available data include paired gene expression transcriptomics, copy number variation, nucleotide polymorphisms, genome-wide DNA methylation, microRNA, and protein expression. Robust and uniformly collected epidemiology and clinical outcomes are linked to each sample. By mining the TCGA, researchers determined that breast cancer consists of three subtypes with different dysregulated molecular pathways that can be differentially treated(121). Similarly, data from the TCGA identified mechanisms by which cancers escape immune checkpoint therapy, thereby developing improved combination therapies(122). A TB equivalent of the TCGA could first clarify the relationship between metabolic, genetic, epigenetic, and immune phenotypes that characterize distinct TB endotypes. To clarify external drivers of specific endotypes, the data should be properly linked with epidemiology, clinical outcomes, and description of Mtb strain, similar to the CRyPTIC consortium(123). TB can present as paucibacillary pulmonary TB, miliary TB, cavitary TB, gastrointestinal TB, TB meningitis, and in many other forms of extrapulmonary TB. A TB equivalent of the TCGA, that combines robust clinical and immune information, is likely to identify immune endotype- clinical phenotype correlations that could make implementing endotype-specific HDT easier. Clinically relevant outcomes, such as relapse-free cure must be included to determine which endotypes require additional clinical monitoring and interventions. To date, there have been over twenty clinical trials evaluating host directed therapies and more clinical trials evaluating new antibiotic regimens and shorter treatment courses. If biologic samples from these studies are contributed to a TB equivalent of the TCGA, within this decade, the field should be able to identify endotype-specific treatment regimens and endotype-specific antibiotic and HDT treatment durations.
Figure 3:
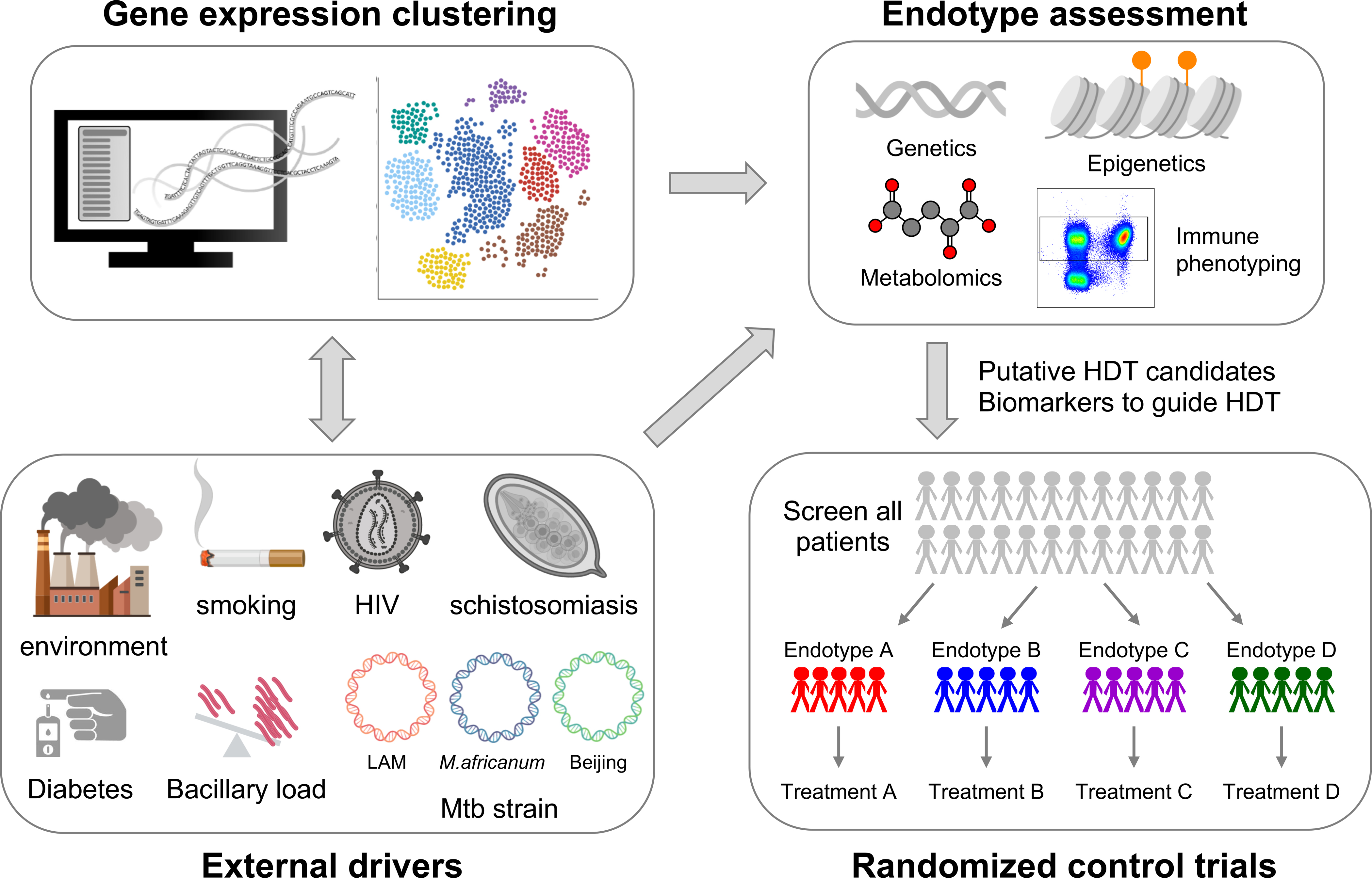
Unbiased clustering of publicly available data allows for identification of gene expression derived clusters. Applying multimodal integration techniques, endotypes can be discovered and characterized based on the their metabolic, epigenetic, genetic and immune phenotype. Similarly, multimodal integration would clarify which epidemiologic factors are likely driving specific endotypes. Multimodal integration will identify the constellation of clinical epidemiology and biomarkers best suitable for treatment with putative HDT candidates that should be prospectively evaluated in umbrella and basket clinical trials.
CONCLUSION:
In summary, a complex interplay between host, pathogen, and environment contributes to susceptibility and clinical presentation of TB. Distinct and overlapping immunological or molecular pathophysiologic mechanism or ‘endotypes’ likely affect TB phenotypes. We propose at least three mutually non-exclusive endotypes, including one caused by IL-12-IFN-γ signaling defects, one characterized by exuberant hyperinflammation, and one by immune exhaustion. Additional factors that drive these endotypes, like co-morbidities, structural lung damage, and Mtb lineage should also be considered.
Powerful bio-informatic approaches, that effectively integrate large epidemiologic and multi-modal omics datasets, promise to clarify these distinct TB endotypes and help identify endotype-specific HDTs. At times, endotype-specific HDT will temper exuberant immunity, while at other times it will augment immunity to improve mycobacterial clearance. Previous trials failed to identify a benefit for HDTs such as exogenous IFN-γ,(124) but these trials were generically implemented in all TB patients without designating their endotype or clinical phenotype. A recent trial demonstrated that Vitamin D combined with the histone deacetylase inhibitor phenylbutyrate lessened TB disease severity, in particular among patients with low vitamin D levels and moderate-to-severe TB disease(125). Could supplemental Vitamin D be targeted to the TB endotypes most in need? Would exogenous IFN-γ benefit IFN-γ deficient patients, but be detrimental to those with excessive IFN-γ? Similarly, TB endotypes likely determine the duration of antimicrobial therapy needed by individual patients. Recent studies using antibiotic regimens that shortened time to mycobacterial clearance were associated with increased risk of relapse(1). These studies considered TB a uniform disease and new studies are being developed to evaluate if certain TB clinical phenotypes could successfully be treated with shorter antimicrobial regimens(1). Incorporating evaluation of TB endotypes, in particular persistent epigenetic-mediated immune exhaustion, is likely to help identify individuals at risk of relapse versus those who could successfully be treated with shorter regimens. For 48 years, TB treatment has not changed; applying these powerful bioinformatic tools promises to identify endotype-specific therapies and treatment durations.
Acknowledgements
ARD is supported by NIAID K23 AI141681-02; CC by the Cancer Prevention Institute of Texas (CPRIT) RP170005, NIH P30 shared resource grant CA125123, and NIEHS grants 1P30ES030285 and 1P42ES0327725; AMM is supported by NIH/NIAID R01AI137527-01A1 and NIH/DOD W81XWH1910026; CL is funded by the German Center for Infection Research (DZIF). JDC is funded in part from funds provided by the Texas A&M University System and National Institutes of Health grant AI104960; MGN is supported by an ERC Advanced Grant (#833247) and a Spinoza grant of the Netherlands Organization for Scientific Research. Figures were created with BioRender.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Imperial MZ, Nahid P, Phillips PPJ, Davies GR, Fielding K, Hanna D, et al. A patient-level pooled analysis of treatment-shortening regimens for drug-susceptible pulmonary tuberculosis. Nature medicine. 2018;24(11):1708–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tobin DM, Roca FJ, Oh SF, McFarland R, Vickery TW, Ray JP, et al. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell. 2012;148(3):434–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaufmann SHE, Dorhoi A, Hotchkiss RS, Bartenschlager R. Host-directed therapies for bacterial and viral infections. Nat Rev Drug Discov. 2018;17(1):35–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meintjes G, Stek C, Blumenthal L, Thienemann F, Schutz C, Buyze J, et al. Prednisone for the Prevention of Paradoxical Tuberculosis-Associated IRIS. The New England journal of medicine. 2018;379(20):1915–25. [DOI] [PubMed] [Google Scholar]
- 5.Lotvall J, Akdis CA, Bacharier LB, Bjermer L, Casale TB, Custovic A, et al. Asthma endotypes: a new approach to classification of disease entities within the asthma syndrome. The Journal of allergy and clinical immunology. 2011;127(2):355–60. [DOI] [PubMed] [Google Scholar]
- 6.Kuruvilla ME, Lee FE, Lee GB. Understanding Asthma Phenotypes, Endotypes, and Mechanisms of Disease. Clin Rev Allergy Immunol. 2019;56(2):219–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braido F, Tiotiu A, Kowal K, Mihaicuta S, Novakova P, Oguzulgen IK. Phenotypes/endotypes-driven treatment in asthma. Curr Opin Allergy Clin Immunol. 2018;18(3):184–9. [DOI] [PubMed] [Google Scholar]
- 8.Kaufmann SH. How can immunology contribute to the control of tuberculosis? Nature reviews Immunology. 2001;1(1):20–30. [DOI] [PubMed] [Google Scholar]
- 9.Kornfeld H, Singhal A. Enlisting the Host to Fight TB. Chest. 2018;153(6):1292–3. [DOI] [PubMed] [Google Scholar]
- 10.Nunes-Alves C, Booty MG, Carpenter SM, Jayaraman P, Rothchild AC, Behar SM. In search of a new paradigm for protective immunity to TB. Nature reviews Microbiology. 2014;12(4):289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Etna MP, Giacomini E, Severa M, Coccia EM. Pro- and anti-inflammatory cytokines in tuberculosis: a two-edged sword in TB pathogenesis. Semin Immunol. 2014;26(6):543–51. [DOI] [PubMed] [Google Scholar]
- 12.Roy Chowdhury R, Vallania F, Yang Q, Lopez Angel CJ, Darboe F, Penn-Nicholson A, et al. A multi-cohort study of the immune factors associated with M. tuberculosis infection outcomes. Nature. 2018;560(7720):644–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rozot V, Vigano S, Mazza-Stalder J, Idrizi E, Day CL, Perreau M, et al. Mycobacterium tuberculosis-specific CD8+ T cells are functionally and phenotypically different between latent infection and active disease. European journal of immunology. 2013;43(6):1568–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lalvani A, Brookes R, Wilkinson RJ, Malin AS, Pathan AA, Andersen P, et al. Human cytolytic and interferon gamma-secreting CD8+ T lymphocytes specific for Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(1):270–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawahara JY, Irvine EB, Alter G. A Case for Antibodies as Mechanistic Correlates of Immunity in Tuberculosis. Frontiers in immunology. 2019;10:996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bustamante J, Boisson-Dupuis S, Abel L, Casanova JL. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-gamma immunity. Semin Immunol. 2014;26(6):454–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dorman SE, Picard C, Lammas D, Heyne K, van Dissel JT, Baretto R, et al. Clinical features of dominant and recessive interferon gamma receptor 1 deficiencies. Lancet. 2004;364(9451):2113–21. [DOI] [PubMed] [Google Scholar]
- 18.Hoeve MA, de Boer T, Langenberg DM, Sanal O, Verreck FA, Ottenhoff TH. IL-12 receptor deficiency revisited: IL-23-mediated signaling is also impaired in human genetic IL-12 receptor beta1 deficiency. European journal of immunology. 2003;33(12):3393–7. [DOI] [PubMed] [Google Scholar]
- 19.Waddell SJ, Popper SJ, Rubins KH, Griffiths MJ, Brown PO, Levin M, et al. Dissecting interferon-induced transcriptional programs in human peripheral blood cells. PloS one. 2010;5(3):e9753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Jong R, Altare F, Haagen IA, Elferink DG, Boer T, van Breda Vriesman PJ, et al. Severe mycobacterial and Salmonella infections in interleukin-12 receptor-deficient patients. Science. 1998;280(5368):1435–8. [DOI] [PubMed] [Google Scholar]
- 21.Cleary AM, Tu W, Enright A, Giffon T, Dewaal-Malefyt R, Gutierrez K, et al. Impaired accumulation and function of memory CD4 T cells in human IL-12 receptor beta 1 deficiency. Journal of immunology. 2003;170(1):597–603. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Q, Cao X. Epigenetic regulation of the innate immune response to infection. Nature reviews Immunology. 2019;19(7):417–32. [DOI] [PubMed] [Google Scholar]
- 23.Browne SK, Burbelo PD, Chetchotisakd P, Suputtamongkol Y, Kiertiburanakul S, Shaw PA, et al. Adult-onset immunodeficiency in Thailand and Taiwan. The New England journal of medicine. 2012;367(8):725–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joosten SA, van Meijgaarden KE, Arend SM, Prins C, Oftung F, Korsvold GE, et al. Mycobacterial growth inhibition is associated with trained innate immunity. The Journal of clinical investigation. 2018;128(5):1837–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harari A, Rozot V, Enders FB, Perreau M, Stalder JM, Nicod LP, et al. Dominant TNF-alpha+ Mycobacterium tuberculosis-specific CD4+ T cell responses discriminate between latent infection and active disease. Nature medicine. 2011;17(3):372–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adekambi T, Ibegbu CC, Cagle S, Kalokhe AS, Wang YF, Hu Y, et al. Biomarkers on patient T cells diagnose active tuberculosis and monitor treatment response. The Journal of clinical investigation. 2015;125(5):1827–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ehlers S Role of tumour necrosis factor (TNF) in host defence against tuberculosis: implications for immunotherapies targeting TNF. Ann Rheum Dis. 2003;62 Suppl 2:ii37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gardam MA, Keystone EC, Menzies R, Manners S, Skamene E, Long R, et al. Anti-tumour necrosis factor agents and tuberculosis risk: mechanisms of action and clinical management. The Lancet Infectious diseases. 2003;3(3):148–55. [DOI] [PubMed] [Google Scholar]
- 29.Busch M, Herzmann C, Kallert S, Zimmermann A, Hofer C, Mayer D, et al. Lipoarabinomannan-Responsive Polycytotoxic T Cells Are Associated with Protection in Human Tuberculosis. American journal of respiratory and critical care medicine. 2016;194(3):345–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Esparza I, Mannel D, Ruppel A, Falk W, Krammer PH. Interferon gamma and lymphotoxin or tumor necrosis factor act synergistically to induce macrophage killing of tumor cells and schistosomula of Schistosoma mansoni. The Journal of experimental medicine. 1987;166(2):589–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garcia I, Miyazaki Y, Marchal G, Lesslauer W, Vassalli P. High sensitivity of transgenic mice expressing soluble TNFR1 fusion protein to mycobacterial infections: synergistic action of TNF and IFN-gamma in the differentiation of protective granulomas. European journal of immunology. 1997;27(12):3182–90. [DOI] [PubMed] [Google Scholar]
- 32.DiNardo AR, Guy E. Reactivation tuberculosis: role of surveillance. Expert Rev Anti Infect Ther. 2016;14(5):501–9. [DOI] [PubMed] [Google Scholar]
- 33.Botha T, Ryffel B. Reactivation of latent tuberculosis infection in TNF-deficient mice. Journal of immunology. 2003;171(6):3110–8. [DOI] [PubMed] [Google Scholar]
- 34.Olsen A, Chen Y, Ji Q, Zhu G, De Silva AD, Vilcheze C, et al. Targeting Mycobacterium tuberculosis Tumor Necrosis Factor Alpha-Downregulating Genes for the Development of Antituberculous Vaccines. MBio. 2016;7(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harris J, Hope JC, Keane J. Tumor necrosis factor blockers influence macrophage responses to Mycobacterium tuberculosis. The Journal of infectious diseases. 2008;198(12):1842–50. [DOI] [PubMed] [Google Scholar]
- 36.Barber DL, Mayer-Barber KD, Feng CG, Sharpe AH, Sher A. CD4 T cells promote rather than control tuberculosis in the absence of PD-1-mediated inhibition. Journal of immunology. 2011;186(3):1598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sakai S, Kauffman KD, Sallin MA, Sharpe AH, Young HA, Ganusov VV, et al. CD4 T Cell-Derived IFN-gamma Plays a Minimal Role in Control of Pulmonary Mycobacterium tuberculosis Infection and Must Be Actively Repressed by PD-1 to Prevent Lethal Disease. PLoS pathogens. 2016;12(5):e1005667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nair S, Huynh JP, Lampropoulou V, Loginicheva E, Esaulova E, Gounder AP, et al. Irg1 expression in myeloid cells prevents immunopathology during M. tuberculosis infection. The Journal of experimental medicine. 2018;215(4):1035–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, et al. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nature immunology. 2013;14(11):1173–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mizunoe K, Hiraki M, Nagano Y, Maehara N. Suppression of intracellular multiplication of Mycobacterium tuberculosis by virus-inhibiting factor or interferon. Jpn J Microbiol. 1975;19(3):235–6. [DOI] [PubMed] [Google Scholar]
- 41.DiNardo AR NT, Mace EM, Rajapakshe K, Mtetwa G, Kay A, Maphalala GP, Secor WE, Mejia RA, Coarfa CC, Bhalla KN, Graviss EA, Orange JS, Makedonas G, Mandalakas AM. Schistosomiasis Induces Persistent DNA Methylation and Tuberculosis-specific Immune Changes. Journal of immunology. 2018;In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Crevel R, Karyadi E, Preyers F, Leenders M, Kullberg BJ, Nelwan RH, et al. Increased production of interleukin 4 by CD4+ and CD8+ T cells from patients with tuberculosis is related to the presence of pulmonary cavities. The Journal of infectious diseases. 2000;181(3):1194–7. [DOI] [PubMed] [Google Scholar]
- 43.Braverman J, Sogi KM, Benjamin D, Nomura DK, Stanley SA. HIF-1alpha Is an Essential Mediator of IFN-gamma-Dependent Immunity to Mycobacterium tuberculosis. Journal of immunology. 2016;197(4):1287–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Winje BA, White R, Syre H, Skutlaberg DH, Oftung F, Mengshoel AT, et al. Stratification by interferon-gamma release assay level predicts risk of incident TB. Thorax. 2018. [DOI] [PubMed] [Google Scholar]
- 45.Andrews JR, Nemes E, Tameris M, Landry BS, Mahomed H, McClain JB, et al. Serial QuantiFERON testing and tuberculosis disease risk among young children: an observational cohort study. The Lancet Respiratory medicine. 2017;5(4):282–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell. 2013;153(3):521–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen M, Divangahi M, Gan H, Shin DS, Hong S, Lee DM, et al. Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. The Journal of experimental medicine. 2008;205(12):2791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tezera LB, Bielecka MK, Ogongo P, Walker NF, Ellis M, Garay-Baquero DJ, et al. Anti-PD-1 immunotherapy leads to tuberculosis reactivation via dysregulation of TNF-alpha. Elife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Apt A, Kramnik I. Man and mouse TB: contradictions and solutions. Tuberculosis. 2009;89(3):195–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reece ST, Loddenkemper C, Askew DJ, Zedler U, Schommer-Leitner S, Stein M, et al. Serine protease activity contributes to control of Mycobacterium tuberculosis in hypoxic lung granulomas in mice. The Journal of clinical investigation. 2010;120(9):3365–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Padhi S, Ravichandran K, Sahoo J, Varghese RG, Basheer A. Hemophagocytic lymphohistiocytosis: An unusual complication in disseminated Mycobacterium tuberculosis. Lung India. 2015;32(6):593–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shakoory B, Carcillo JA, Chatham WW, Amdur RL, Zhao H, Dinarello CA, et al. Interleukin-1 Receptor Blockade Is Associated With Reduced Mortality in Sepsis Patients With Features of Macrophage Activation Syndrome: Reanalysis of a Prior Phase III Trial. Crit Care Med. 2016;44(2):275–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Winchell CG, Mishra BB, Phuah JY, Saqib M, Nelson SJ, Maiello P, et al. Evaluation of IL-1 Blockade as an Adjunct to Linezolid Therapy for Tuberculosis in Mice and Macaques. Frontiers in immunology. 2020;11:891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gupta S, Weitzman S. Primary and secondary hemophagocytic lymphohistiocytosis: clinical features, pathogenesis and therapy. Expert Rev Clin Immunol. 2010;6(1):137–54. [DOI] [PubMed] [Google Scholar]
- 55.Ahmed R, Salmi A, Butler LD, Chiller JM, Oldstone MB. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. The Journal of experimental medicine. 1984;160(2):521–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity. 2016;45(2):358–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trebak M, Kinet JP. Calcium signalling in T cells. Nature reviews Immunology. 2019;19(3):154–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, et al. Transcription Factor IRF4 Promotes CD8(+) T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity. 2017;47(6):1129–41 e5. [DOI] [PubMed] [Google Scholar]
- 59.Mak TW, Grusdat M, Duncan GS, Dostert C, Nonnenmacher Y, Cox M, et al. Glutathione Primes T Cell Metabolism for Inflammation. Immunity. 2017;46(6):1089–90. [DOI] [PubMed] [Google Scholar]
- 60.Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science. 2019;366(6468):1013–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Velica P, et al. S-2-hydroxyglutarate regulates CD8(+) T-lymphocyte fate. Nature. 2016;540(7632):236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic Instruction of Immunity. Cell. 2017;169(4):570–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rambold AS, Pearce EL. Mitochondrial Dynamics at the Interface of Immune Cell Metabolism and Function. Trends Immunol. 2018;39(1):6–18. [DOI] [PubMed] [Google Scholar]
- 64.Valdor R, Macian F. Induction and stability of the anergic phenotype in T cells. Semin Immunol. 2013;25(4):313–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature. 2019;576(7786):293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen J, Lopez-Moyado IF, Seo H, Lio CJ, Hempleman LJ, Sekiya T, et al. NR4A transcription factors limit CAR T cell function in solid tumours. Nature. 2019;567(7749):530–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu X, Wang Y, Lu H, Li J, Yan X, Xiao M, et al. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature. 2019;567(7749):525–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weber EW, Lynn RC, Parker KR, Anbunathan H, Lattin J, Sotillo E, et al. Transient “rest” induces functional reinvigoration and epigenetic remodeling in exhausted CAR-T cells. bioRxiv. 2020:2020.01.26.920496. [Google Scholar]
- 69.Dan L, Liu L, Sun Y, Song J, Yin Q, Zhang G, et al. The phosphatase PAC1 acts as a T cell suppressor and attenuates host antitumor immunity. Nature immunology. 2020;21(3):287–97. [DOI] [PubMed] [Google Scholar]
- 70.Youngblood B, Reich NO. The early expressed HIV-1 genes regulate DNMT1 expression. Epigenetics. 2008;3(3):149–56. [DOI] [PubMed] [Google Scholar]
- 71.DiNardo AR, Nishiguchi T, Mace EM, Rajapakshe K, Mtetwa G, Kay A, et al. Schistosomiasis Induces Persistent DNA Methylation and Tuberculosis-Specific Immune Changes. Journal of immunology. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.DiNardo A, Rajapakshe K, Nishiguchi T, Mtetwa G, Grimm SL, Dlamini Q, et al. DNA hyper-methylation during Tuberculosis dampens host immune responsiveness. The Journal of clinical investigation. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity. 2016;45(2):374–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bello S, Afolabi RF, Ajayi DT, Sharma T, Owoeye DO, Oduyoye O, et al. Empirical evidence of delays in diagnosis and treatment of pulmonary tuberculosis: systematic review and meta-regression analysis. BMC Public Health. 2019;19(1):820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell. 2017;170(1):142–57 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mognol GP, Spreafico R, Wong V, Scott-Browne JP, Togher S, Hoffmann A, et al. Exhaustion-associated regulatory regions in CD8(+) tumor-infiltrating T cells. Proceedings of the National Academy of Sciences of the United States of America. 2017;114(13):E2776–E85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Seo H, Chen J, Gonzalez-Avalos E, Samaniego-Castruita D, Das A, Wang YH, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proceedings of the National Academy of Sciences of the United States of America. 2019;116(25):12410–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Coussens AK, Wilkinson RJ, Martineau AR. Phenylbutyrate Is Bacteriostatic againstMycobacterium tuberculosis and Regulates the Macrophage Response to Infection, Synergistically with 25-Hydroxy-Vitamin D3. PLoS pathogens. 2015;11(7):e1005007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Khor CC, Chapman SJ, Vannberg FO, Dunne A, Murphy C, Ling EY, et al. A Mal functional variant is associated with protection against invasive pneumococcal disease, bacteremia, malaria and tuberculosis. Nat Genet. 2007;39(4):523–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ni Cheallaigh C, Sheedy FJ, Harris J, Munoz-Wolf N, Lee J, West K, et al. A Common Variant in the Adaptor Mal Regulates Interferon Gamma Signaling. Immunity. 2016;44(2):368–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rosen LB, Freeman AF, Yang LM, Jutivorakool K, Olivier KN, Angkasekwinai N, et al. Anti-GM-CSF autoantibodies in patients with cryptococcal meningitis. Journal of immunology. 2013;190(8):3959–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Desvignes L, Weidinger C, Shaw P, Vaeth M, Ribierre T, Liu M, et al. STIM1 controls T cell-mediated immune regulation and inflammation in chronic infection. The Journal of clinical investigation. 2015;125(6):2347–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bell LCK, Noursadeghi M. Pathogenesis of HIV-1 and Mycobacterium tuberculosis co-infection. Nature reviews Microbiology. 2018;16(2):80–90. [DOI] [PubMed] [Google Scholar]
- 84.Babu S, Nutman TB. Helminth-Tuberculosis Co-infection: An Immunologic Perspective. Trends Immunol. 2016;37(9):597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chatterjee S, Nutman TB. Helminth-induced immune regulation: implications for immune responses to tuberculosis. PLoS pathogens. 2015;11(1):e1004582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Critchley JA, Restrepo BI, Ronacher K, Kapur A, Bremer AA, Schlesinger LS, et al. Defining a Research Agenda to Address the Converging Epidemics of Tuberculosis and Diabetes: Part 1: Epidemiology and Clinical Management. Chest. 2017;152(1):165–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ronacher K, van Crevel R, Critchley JA, Bremer AA, Schlesinger LS, Kapur A, et al. Defining a Research Agenda to Address the Converging Epidemics of Tuberculosis and Diabetes: Part 2: Underlying Biologic Mechanisms. Chest. 2017;152(1):174–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Segura-Cerda CA, Lopez-Romero W, Flores-Valdez MA. Changes in Host Response to Mycobacterium tuberculosis Infection Associated With Type 2 Diabetes: Beyond Hyperglycemia. Frontiers in cellular and infection microbiology. 2019;9:342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Prada-Medina CA, Fukutani KF, Pavan Kumar N, Gil-Santana L, Babu S, Lichtenstein F, et al.Systems Immunology of Diabetes-Tuberculosis Comorbidity Reveals Signatures of Disease Complications. Sci Rep. 2017;7(1):1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Restrepo BI, Twahirwa M, Rahbar MH, Schlesinger LS. Phagocytosis via complement or Fc-gamma receptors is compromised in monocytes from type 2 diabetes patients with chronic hyperglycemia. PloS one. 2014;9(3):e92977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Martinez N, Vallerskog T, West K, Nunes-Alves C, Lee J, Martens GW, et al. Chromatin decondensation and T cell hyperresponsiveness in diabetes-associated hyperglycemia. Journal of immunology. 2014;193(9):4457–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lachmandas E, Thiem K, van den Heuvel C, Hijmans A, de Galan BE, Tack CJ, et al. Patients with type 1 diabetes mellitus have impaired IL-1beta production in response to Mycobacterium tuberculosis. Eur J Clin Microbiol Infect Dis. 2018;37(2):371–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Restrepo BI, Fisher-Hoch SP, Pino PA, Salinas A, Rahbar MH, Mora F, et al. Tuberculosis in poorly controlled type 2 diabetes: altered cytokine expression in peripheral white blood cells. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2008;47(5):634–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ferlita S, Yegiazaryan A, Noori N, Lal G, Nguyen T, To K, et al. Type 2 Diabetes Mellitus and Altered Immune System Leading to Susceptibility to Pathogens, Especially Mycobacterium tuberculosis. J Clin Med. 2019;8(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lachmandas E, van den Heuvel CN, Damen MS, Cleophas MC, Netea MG, van Crevel R. Diabetes Mellitus and Increased Tuberculosis Susceptibility: The Role of Short-Chain Fatty Acids. J Diabetes Res. 2016;2016:6014631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vrieling F, Wilson L, Rensen PCN, Walzl G, Ottenhoff THM, Joosten SA. Oxidized low-density lipoprotein (oxLDL) supports Mycobacterium tuberculosis survival in macrophages by inducing lysosomal dysfunction. PLoS pathogens. 2019;15(4):e1007724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Singhal A, Jie L, Kumar P, Hong GS, Leow MK, Paleja B, et al. Metformin as adjunct antituberculosis therapy. Sci Transl Med. 2014;6(263):263ra159. [DOI] [PubMed] [Google Scholar]
- 98.Lachmandas E, Eckold C, Bohme J, Koeken V, Marzuki MB, Blok B, et al. Metformin Alters Human Host Responses to Mycobacterium tuberculosis in Healthy Subjects. The Journal of infectious diseases. 2019;220(1):139–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Degner NR, Wang JY, Golub JE, Karakousis PC. Metformin Use Reverses the Increased Mortality Associated With Diabetes Mellitus During Tuberculosis Treatment. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2018;66(2):198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jee SH, Golub JE, Jo J, Park IS, Ohrr H, Samet JM. Smoking and risk of tuberculosis incidence, mortality, and recurrence in South Korean men and women. Am J Epidemiol. 2009;170(12):1478–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Marais BJ, Lonnroth K, Lawn SD, Migliori GB, Mwaba P, Glaziou P, et al. Tuberculosis comorbidity with communicable and non-communicable diseases: integrating health services and control efforts. The Lancet Infectious diseases. 2013;13(5):436–48. [DOI] [PubMed] [Google Scholar]
- 102.Torrelles JB, Schlesinger LS. Integrating Lung Physiology, Immunology, and Tuberculosis. Trends Microbiol. 2017;25(8):688–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Reimann M, Schaub D, Kalsdorf B, Runge C, Carballo PS, Terhalle E, et al. Cigarette smoking and culture conversion in patients with susceptible and M/XDR-TB. The international journal of tuberculosis and lung disease : the official journal of the International Union against Tuberculosis and Lung Disease. 2019;23(1):93–8. [DOI] [PubMed] [Google Scholar]
- 104.Yen YF, Yen MY, Lin YS, Lin YP, Shih HC, Li LH, et al. Smoking increases risk of recurrence after successful anti-tuberculosis treatment: a population-based study. The international journal of tuberculosis and lung disease : the official journal of the International Union against Tuberculosis and Lung Disease. 2014;18(4):492–8. [DOI] [PubMed] [Google Scholar]
- 105.Stampfli MR, Anderson GP. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nature reviews Immunology. 2009;9(5):377–84. [DOI] [PubMed] [Google Scholar]
- 106.O’Leary SM, Coleman MM, Chew WM, Morrow C, McLaughlin AM, Gleeson LE, et al. Cigarette smoking impairs human pulmonary immunity to Mycobacterium tuberculosis. American journal of respiratory and critical care medicine. 2014;190(12):1430–6. [DOI] [PubMed] [Google Scholar]
- 107.Forrellad MA, Klepp LI, Gioffre A, Sabio y Garcia J, Morbidoni HR, de la Paz Santangelo M, et al. Virulence factors of the Mycobacterium tuberculosis complex. Virulence. 2013;4(1):3–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tram TTB, Nhung HN, Vijay S, Hai HT, Thu DDA, Ha VTN, et al. Virulence of Mycobacterium tuberculosis Clinical Isolates Is Associated With Sputum Pre-treatment Bacterial Load, Lineage, Survival in Macrophages, and Cytokine Response. Frontiers in cellular and infection microbiology. 2018;8:417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ngabonziza JCS, Loiseau C, Marceau M, Jouet A, Menardo F, Tzfadia O, et al. A sister lineage of the Mycobacterium tuberculosis complex discovered in the African Great Lakes region. Nat Commun. 2020;11(1):2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Portevin D, Gagneux S, Comas I, Young D. Human macrophage responses to clinical isolates from the Mycobacterium tuberculosis complex discriminate between ancient and modern lineages. PLoS pathogens. 2011;7(3):e1001307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Subbian S, Bandyopadhyay N, Tsenova L, O’Brien P, Khetani V, Kushner NL, et al. Early innate immunity determines outcome of Mycobacterium tuberculosis pulmonary infection in rabbits. Cell Commun Signal. 2013;11:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Manca C, Reed MB, Freeman S, Mathema B, Kreiswirth B, Barry CE 3rd, et al. Differential monocyte activation underlies strain-specific Mycobacterium tuberculosis pathogenesis. Infection and immunity. 2004;72(9):5511–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Howard NC, Marin ND, Ahmed M, Rosa BA, Martin J, Bambouskova M, et al. Mycobacterium tuberculosis carrying a rifampicin drug resistance mutation reprograms macrophage metabolism through cell wall lipid changes. Nat Microbiol. 2018;3(10):1099–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Holt KE, McAdam P, Thai PVK, Thuong NTT, Ha DTM, Lan NN, et al. Frequent transmission of the Mycobacterium tuberculosis Beijing lineage and positive selection for the EsxW Beijing variant in Vietnam. Nat Genet. 2018;50(6):849–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sweeney TE, Braviak L, Tato CM, Khatri P. Genome-wide expression for diagnosis of pulmonary tuberculosis: a multicohort analysis. The Lancet Respiratory medicine. 2016;4(3):213–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gupta RK, Turner CT, Venturini C, Esmail H, Rangaka MX, Copas A, et al. Concise whole blood transcriptional signatures for incipient tuberculosis: a systematic review and patient-level pooled meta-analysis. The Lancet Respiratory medicine. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.DiNardo AR, Rajapakshe K, Gandhi T, Grimm S, Nishiguchi T, Heyckendorf J, et al. Discerning divergent tuberculosis endotypes: A meta-analysis and systematic review of individual patient data. medRxiv. 2020:2020.05.13.20100776. [Google Scholar]
- 118.Sahiratmadja E, Alisjahbana B, de Boer T, Adnan I, Maya A, Danusantoso H, et al. Dynamic changes in pro- and anti-inflammatory cytokine profiles and gamma interferon receptor signaling integrity correlate with tuberculosis disease activity and response to curative treatment. Infection and immunity. 2007;75(2):820–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Singh A, Mohan A, Dey AB, Mitra DK. Inhibiting the programmed death 1 pathway rescues Mycobacterium tuberculosis-specific interferon gamma-producing T cells from apoptosis in patients with pulmonary tuberculosis. The Journal of infectious diseases. 2013;208(4):603–15. [DOI] [PubMed] [Google Scholar]
- 120.Mounir M, Lucchetta M, Silva TC, Olsen C, Bontempi G, Chen X, et al. New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Comput Biol. 2019;15(3):e1006701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wu J, Cui Y, Sun X, Cao G, Li B, Ikeda DM, et al. Unsupervised Clustering of Quantitative Image Phenotypes Reveals Breast Cancer Subtypes with Distinct Prognoses and Molecular Pathways. Clin Cancer Res. 2017;23(13):3334–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wu HX, Chen YX, Wang ZX, Zhao Q, He MM, Wang YN, et al. Alteration in TET1 as potential biomarker for immune checkpoint blockade in multiple cancers. J Immunother Cancer. 2019;7(1):264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Consortium CR, the GP, Allix-Beguec C, Arandjelovic I, Bi L, Beckert P, et al. Prediction of Susceptibility to First-Line Tuberculosis Drugs by DNA Sequencing. The New England journal of medicine. 2018;379(15):1403–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dheda K, Schwander SK, Zhu B, van Zyl-Smit RN, Zhang Y. The immunology of tuberculosis: from bench to bedside. Respirology. 2010;15(3):433–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bekele A, Gebreselassie N, Ashenafi S, Kassa E, Aseffa G, Amogne W, et al. Daily adjunctive therapy with vitamin D3 and phenylbutyrate supports clinical recovery from pulmonary tuberculosis: a randomized controlled trial in Ethiopia. J Intern Med. 2018;284(3):292–306. [DOI] [PMC free article] [PubMed] [Google Scholar]