

Abstract
Structure-based virtual screening approaches have the ability to dramatically reduce the time and costs associated to the discovery of new drug candidates. Studies have shown that the true hit rate of virtual screenings improves with the scale of the screened ligand libraries. Therefore, we have recently developed an open source drug discovery platform (VirtualFlow), which is able to routinely carry out ultra-large virtual screenings. One of the primary challenges of molecular docking is the circumstance when the protein is highly dynamic or when the structure of the protein cannot be captured by a static pose. To accommodate protein dynamics, we report the extension of VirtualFlow to allow the docking of ligands using a grey wolf optimization algorithm using the docking program GWOVina, which substantially improves the quality and efficiency of flexible receptor docking compared to AutoDock Vina. We demonstrate the linear scaling behavior of VirtualFlow utilizing GWOVina up to 128 000 CPUs. The newly supported docking method will be valuable for drug discovery projects in which protein dynamics and flexibility play a significant role.
Keywords: ultra-large virtual screening, molecular docking, drug discovery, COVID-19, structure-based drug design, CADD, computer aided drug design, AutoDock, grey wolf optimization, cloud computing
Introduction
In structure based drug design one common goal is to design and optimize a small molecule (compound), such that it fits optimally, with favorable energetics into the binding pocket of a target protein. The conventional approach is to test the compounds individually in an experimental wet lab setting via high throughput screens. Besides these experiments, computer-aided drug design (CADD) has been established, which allows to compute the binding affinity between the small molecule and the target protein. In the realm of structure-based virtual screenings, for a given binding pocket on the surface of the protein, ligands from a large databases of prospective candidate molecules are screened, i.e. the binding strength of individual ligands from the large database to a given target are computationally predicted by molecular docking. Docking programs such as AutoDock Vina [22] fit these candidate molecules into the binding pocket and score the resulting binding geometry with regard to the binding affinity. The scoring function associated with the docking program assigns a docking score, that is a marker of the calculated binding affinity, to each tested interaction geometry (Fig. 1a). The docking score is a measure of the predicted binding affinity of the small molecule to the protein (Fig. 1b). The interaction geometry (conformation) with the best docking score is the final score for the compound. Docking methods should estimate the binding affinity as precisely as possible on the one hand, and as quickly as possible on the other hand. Even with fast docking programs, ultra-large virtual screenings involving hundreds of millions or billions of molecules require a massive amount of computation time with millions of CPU hours. Thus resources such as supercomputer or cloud-based computational platforms are required. With increased scale of the ligand library screened, the true hit rate (the fraction of hits which bind to the target protein in experimental binding assays) of the screening improves, as was shown by theoretical and experimental studies [8, 9, 13]. Since the chemical space of small molecules suitable for drug discovery is estimated to contain more than 1060 molecules [1], even billion of compounds represent only a minuscule fraction of the possible chemical space to be explored.
Figure 1.

Principle of virtual screening and scoring of small molecules
We describe here an extension of a platform allowing to screen billions of compounds (VirtualFlow), focusing on the new GWOVina docking program which is based on the novel global optimization algorithm.
1. Materials and Methods
VirtualFlow is an open source parallel workflow platform that we recently developed for executing virtual screenings [9, 23], which for the first time allowed to routinely screen billions of compounds. VirtualFlow can be employed on any type of Linux-based computer clusters managed by a batchsystem, as well as on cloud computing platforms such as the Google Cloud. VirtualFlow consists of two different modules, one for the ligand preparation (VirtualFlow for Ligand Preparation – VFLP) and one for the virtual screenings (VirtualFlow for Virtual Screening – VFVS). Although both methods are used for different tasks, they share the same core technology.
1.1. VirtualFlow for Ligand Preparation
Before the ligands can be docked to the target protein, they must be prepared into a ready-to-dock format. VFLP is dedicated for this task, and is able to prepare ultra-large ligand libraries which can be readily used by VFVS. In addition to the correct file format and the three dimensional spatial structure of the ligand, it can compute the tautomerization and protonation states of the molecules via tools like ChemAxon’s JChem package [2] and Open Babel [17]. Once a library is prepared, it can be used again and again with VFVS. However, since VFLP can prepare the molecules in almost any output format, the prepared ligand libraries can also be used for any other purpose and other docking platforms. We have previously used VFLP to prepare the REAL library from Enamine (2018 version) containing 1.4 billion compounds, as well as the ZINC15 library (2018 version) containing 1.2 billion compounds into a ready-to-dock format (VirtualFlow versions of these libraries) [9, 21]. The VirtualFlow versions of these libraries are freely available on our website [23].
1.2. VirtualFlow for Virtual Screening
A primary goal of virtual screenings is to create a hit list which contains the top scoring compounds ranked by their docking score, essentially sorting the molecules in the library based on their propensity to bind to the target site on the protein. The docking score correlates with the predicted binding affinity to the target protein. The scoring (and thus the ranking) is based on the calculation of the free energy of the small molecules to the receptor. While for a single compound the docking can be done relatively quickly, for a large ligand library containing millions or billions of compounds, this requires a substantial amount of computing capacity and time. The computational costs associated with the docking routines dramatically increase further if docking procedures with high accuracy (e.g. including receptor flexibility, exhaustive search of the docking space) are used. To solve the first challenge caused by the large number of ligands, VirtualFlow uses a massive parallelization approach to speed up the virtual screening (vide infra). To deal with the second challenge involving the increased computational costs in the high accuracy implementation, VirtualFlow can be deployed in a multi-staged manner. In a multi-staged virtual screen, the entire collection of ligands that are planned to be screened is at first docked with a fast method at reduced accuracy. In the next stage, the top X% of compounds of the first stage are transferred to the next stage, and screened with higher accuracy. In principle, any number of stages with subsequent increase in computational time, which in turn increases accuracy, can be employed.
One of the key characteristics of VirtualFlow is that it is able to scale very efficiently up to hundreds of thousands of CPUs, which it achieves by employing an embarrassingly/perfectly parallelization strategy. In this context, VirtualFlow uses an advanced task list approach, which completely eliminates the need for any communication between individual workers. The tasks are distributed at the beginning in advance to the individual workers by a workload balancer, which removes the need to access the task list during the runtime of a job. To reduce the number of tasks, the ligands which need to be processed are grouped into collections (of e.g. 1000 ligands), and each collection represents a task in the central task list. The input and output databases of the workflow consist of a multi-level file structure, which involves folders, and (compressed and uncompressed) tar archives.
VFVS supports different docking programs such as AutoDock Vina [22], Smina [11], Vina-Carb [16], and VinaXB [10]. All of them are based on AutoDock Vina and improve different aspects. Vina-Carb for instance improves the accuracy of carbohydrate docking. Here, we have added support for GWOVina [24], which is able to handle protein side chain flexibility more efficiently than AutoDock Vina. It is able to efficiently handle (in terms of computational speed) considerably more number of flexible side chains compared to AutoDock Vina. In addition, in terms of the quality of the results, GWOVina samples the conformational space of the side chains much more effectively due to the utilization of a new swarm-optimization-based optimization algorithm, the grey wolf optimizer (GWO) [24].
1.3. Grey Wolf Optimization-Based Docking Algorithm
We have added support of GWOVina [24] to VFVS. GWOVina uses the recently developed grey wolf optimization algorithm, which has turned out to be highly efficient for flexible ligand docking and other types of tasks [12, 14, 24]. Finding the optimal or best orientation of the ligand in the target site of the protein is equivalent to finding the absolute minimum of an energy landscape in the high-dimensional conformational space, similar to the protein folding problem [18]. The conformational space is defined by the degrees of freedom of both the ligand and the flexible side chains of the individual amino acid that constitute the docking site on the receptor, and all the degrees of freedom are treated equally by the algorithm. The degrees of freedom of the receptor side chains which are selected to be flexible consists of the torsion angles around the rotatable bonds. The degrees of freedom of the ligands includes the translation and rotation in three dimensions in addition to the torsion angles around the rotatable bonds.
Inspired by the hunting behavior of the grey wolf pack, the grey wolf algorithm is an optimization algorithm using swarm intelligence, i.e. it takes advantage of collective behavior of a self-organized system (wolf pack). Members of a grey wolf pack are either α, β, δ, or ω wolves, and each of them has a different function during the hunting of a prey. Within the hierarchy, the α wolf is the dominant wolf, making the decisions when hunting. The β wolf supports the α wolf in its decisions and also in the enforcement of the orders of the α wolf. The δ wolves can be considered as a collection of specialist such as scouts (monitoring the territory), hunters (helping α/β wolves in hunting) and elders (former experienced α and β wolves) are among with them. The δ wolves are subordinates of the α and β wolves. The ω wolves are the lowest wolves which have to submit to the α, β and δ wolves. In the GWO algorithm [14], which models the hunting strategy of m grey wolves, each wolf represents a search agent, and the prey is the optimum (in our case the energy minimum, i.e. the “best” orientation of the ligand and the flexible site chains in the target site). More precisely the α, β and δ wolves guide the hunting and the ω wolves follow. We outline the core algorithm below.
Let be the position of a wolf and the position of the prey at time step t. The distance vector of the wolf from the prey p is then set by
| (1) |
Here, the product c ☉ xp as well as the absolute value is understood element-wise (component-by-component), implying the former is the Hadamard product. The vector c = 2(rand[0, 1])n, where (rand[0, 1])n is a vector with n random numbers between 0 and 1 as its entries. Thus d is a vector with positive entries. With this, the position x(t + 1) of the wolf in the next time step is given by
| (2) |
where a = 2b ☉ (rand[0, 1])n − b and b is a vector with entries decreasing linearly from 2 to 0 in each iteration step. Note, that the absolute value of a determines the moving direction of the wolf with respect to the prey. In case of ∣a∣ > 1 the wolf veers away from the prey and in case ∣a∣ < 1 it moves towards the prey. However in our context, the position of the optimum (prey) is not known. In the first step the energy (fitness) for each wolf position is evaluated and arranged in increasing order of fitness. The algorithm then assigns the first three α, β and δ to the positions with the lowest energy since it is supposed that they are the closest to the prey (optimum). In a next step the three best updates are computed via
| (3) |
where c1, c2, and c3 are defined analogue to c. With this the estimated prey positions are obtained by
| (4) |
where a1, a2, a3 are defined analogue to a. The next position of the wolf is then given as
| (5) |
The original GWO algorithm was extended in GWOVina by an additional random walk mechanism which is sometimes employed for the movement of a wolf to improve the docking program.
For running the algorithm, the number m of search agents (wolves), the objective function (energy function), the dimension n as well as the search space (conformational space) has to be provided. The GWO algorithm replaces the global Monte Carlo-based optimizer used by the original AutoDock Vina, while the original scoring function of AutoDock Vina is used as the objective function. The computation time needed by GWOVina is proportional to the number of wolves used according to the authors, and the quality of the docking results increases with the number of wolves due to the more elaborate exploration of the conformation space [24]. To fully harness the capabilities of GWO, at least four wolves need to be employed when using this docking algorithm, as the grey wolf algorithm is based on a four level hierarchy of wolves (α, β, δ, and ω wolves). GWOVina was previously compared with AutoDock Vina via several benchmarks [24], and GWOVina has shown a substantial advantage over AutoDock Vina in terms of finding better docking poses in less time.
2. Results
Due to the large surface area that constitutes the RNA binding interface of nsp12 (Fig. 2a), there are a large number of protein side chains to account for at the interaction surface that are critical in engaging the RNA. Most docking programs cannot handle the flexibility of such large numbers of side chains efficiently, but for GWOVina this does not pose a challenge. We have allowed a total of 12 residue side chains at the RNA binding interface to be flexible during the benchmarks (Lys593, Phe594, Tyr595, Leu854, Glu857, Arg858, Val860, Ser861, Leu862, Ile864, Asp865, Tyr915), though GWOVina could handle significantly more [24]. An example compound docked to the target site can be seen in Fig. 2b). The computations were run on a Slurm cluster in the Google Cloud [7], and the compute nodes which were used employed second generation AMD EPYC Rome CPUs. The cluster file system which was used is an Elastifile Cloud File System [3], which is a Network File System (NFS) server. For this benchmark we have created a test library, which consists of compounds from the REAL library from Enamine which we had previously prepared (see above). The entire test library had a size of 1 billion compounds, which is large enough for not being depleted during the benchmarks. The test library consists of 10 metatranches, each containing 1000 tranches, of which each tranche contains 10 000 collections, and each collection contains 10 different compounds. The 10 compounds are the same in each collection, meaning each collection in the test library is identical but nonetheless treated independently by VirtualFlow, which makes the benchmark more reproducible. The 10 distinct test molecules are relatively flexible, each containing between 9 and 10 rotatable bonds, and have a molecular weight between 400 and 425 daltons.
Figure 2.

Biomolecular test system consisting of nsp12 used for the benchmarks
We have tested the scaling behavior and virtual screening speed of VirtualFlow using GWOVina with up to 128 000 CPUs, and the speedup was roughly linear up to the maximum number of CPUs tested (Fig. 3a). The test system which was used is the RNA-dependent RNA polymerase (RdRP) of the SARS-CoV-2 virus, and the targeted site on this protein is the RNA binding interface (Fig. 2). RdRPs have been effectively targeted to develop antiviral therapeutics in several viral infections in the past such as HCV, Zika virus (ZIKV), and HCoVs this an attractive target for therapeutic intervention of COVID-19 [4-6]. The receptor structure which was used is the cryo-EM structure with PDB code 7BV1 [25]. The RNA was removed, and the structure prepared with Maestro from Schrödinger by adding hydrogen atoms at physiological pH value [19]. The command line tool prepare_flexreceptor4.py of AutoDockTools was used to merge nonpolar hydrogen atoms, to split the receptor into rigid and flexible parts, and to convert the two parts into the PDBQT format [15]. The number of wolves used in the first benchmark is one, because this setting puts the most stress on the computational infrastructure, such as the cluster file system, due to the minimal docking time per ligand and thus maximum amount of file transfers and related activities.
Figure 3.
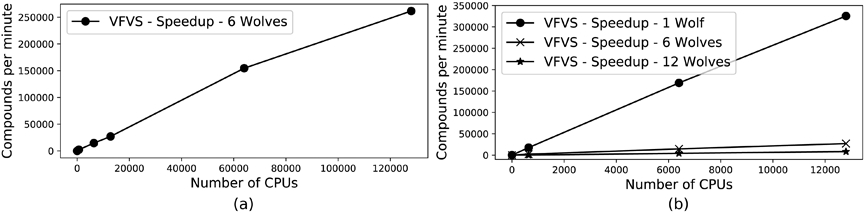
Scaling behavior of VFVS using GWOVina as the docking program
We have also tested the virtual screening speed for different numbers of wolves which are utilized by the grey wolf optimization algorithm of GWOVina. With only one wolf the docking speed is by far the fastest, while the computational costs of using six wolves is roughly double as fast compared to that of using 12 wolves (Fig. 3b).
Conclusion
Virtual screenings have an enormous potential in making drug discovery faster and more affordable in the future, and in allowing to find cures for diseases which so far were incurable. With ultra-large virtual screenings now in the accessible computational range, their power has substantially increased, and they could soon become a standard approach for finding new initial hit and lead compounds. Furthermore, due to the vast chemical spaces which can be screened in comparison to traditional approaches, tight binders to even highly challenging targets can be identified. And with that, the generally more challenging class of protein-protein interactions can be targeted via virtual screening approaches. Protein-protein interactions play a role in almost any disease, and are expected to become the most important class of targets in the future. For any docking efforts the choice of the protein structure and the target site on the protein is critical to the success of the screen. Normally the starting points for the docking efforts are structures derived from X-ray crystallography, cryo-EM or NMR. These structures normally represent a single snapshot of the protein and in most cases the lowest energy conformation. However in solution, inside the cell, where the hit molecule from the screen should function, the structure could be dynamic and accommodating this information about the protein dynamics will substantially improve the true hit rate. Capturing protein dynamics is not a trivial task. NMR studies can provide information about protein dynamics over a wide timescale (nanoseconds to hours), but it is not simple to relate it back to precise structural information. Molecular dynamics simulations can provide structural information but are limited in the timescale they can sample, typically up to a few microseconds. The interesting conformational changes and allosteric changes happen in the microsecond to millisecond time regime. There have been a few simulation that have been extended to milliseconds by the group of DE Shaw using the custom designed Anton computer [20]. In the absence of detailed structural information to capture dynamics, which is the case in most efforts, incorporating side chain dynamics is a good alternative to account for dynamics. The combination of GWOVina and VirtualFlow unifies the best of both worlds, to account for dynamics and identify genuine hits, facilitated by the power of supercomputing platforms. VFVS, including the new feature, is available on GitHub (https://github.com/VirtualFlow/VFVS).
Acknowledgments
We thank ChemAxon for a free academic license for the JChem package, and Google for computing time on the Google Cloud. H.A. acknowledges funding from the Claudia Adams Barr Program for Innovative Cancer Research. G.W. acknowledges support from NIH grants CA200913 and AI037581. K.F. would like to thank the Math+. We would like to thank Arthur Jaffe for his support. This research was supported in part by grant TRT 0159 from the Templeton Religion Trust and by ARO Grant W911NF1910302 to Arthur Jaffe.
Footnotes
Conflicts of interest: G.W. and C.G. are cofounders of the company Virtual Discovery, Inc., which provides virtual screening services. G.W. serves as the director of this company.
References
- 1.Bohacek RS, McMartin C, Guida WC: The art and practice of structure-based drug design: A molecular modeling perspective. Medicinal Research Reviews 16(1), 3–50 (1996), DOI: [DOI] [PubMed] [Google Scholar]
- 2.ChemAxon: JChem Suite 18.20.0. https://chemaxon.com/products/jchem-engines, accessed: 2020-09-06 [Google Scholar]
- 3.Elastifile Cloud File System. https://console.cloud.google.com/marketplace/details/elastifile/elastifile-cloud-file-system, accessed: 2020-09-06
- 4.Elfiky AA: Zika viral polymerase inhibition using anti-HCV drugs both in market and under clinical trials. Journal of Medical Virology 88(12), 2044–2051 (2016), DOI: 10.1002/jmv.24678 [DOI] [PubMed] [Google Scholar]
- 5.Elfiky AA, Elshemey WM: IDX-184 is a superior HCV direct-acting antiviral drug: a QSAR study. Medicinal Chemistry Research 25(5), 1005–1008 (2016), DOI: 10.1007/s00044-016-1533-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ganesan A, Barakat K: Applications of computer-aided approaches in the development of hepatitis C antiviral agents. Expert Opinion on Drug Discovery 12(4), 407–425 (2017), DOI: 10.1080/17460441.2017.1291628 [DOI] [PubMed] [Google Scholar]
- 7.Google Cloud. https://cloud.google.com, accessed: 2020-09-06
- 8.Gorgulla C: Free Energy Methods Involving Quantum Physics, Path Integrals, and Virtual Screenings. Ph.D. thesis, Freie Universität Berlin; (2018), DOI: 10.17169/refubium-11597 [DOI] [Google Scholar]
- 9.Gorgulla C, Boeszoermenyi A, Wang ZF, et al. : An open-source drug discovery platform enables ultra-large virtual screens. Nature 580(7805), 663–668 (2020), DOI: 10.1038/s41586-020-2117-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koebel MR, Schmadeke G, Posner RG, et al. : AutoDock VinaXB: implementation of XBSF, new empirical halogen bond scoring function, into AutoDock Vina. Journal of Cheminformatics 8(1), 27 (2016), DOI: 10.1186/s13321-016-0139-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koes DR, Baumgartner MP, Camacho CJ: Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. Journal of Chemical Information and Modeling 53(8), 1893–1904 (2013), DOI: 10.1021/ci300604z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lal DK, Barisal A, Tripathy M: Grey Wolf Optimizer Algorithm Based Fuzzy PID Controller for AGC of Multi-area Power System with TCPS. Procedia Computer Science 92, 99–105 (2016), DOI: 10.1016/j.procs.2016.07.329 [DOI] [Google Scholar]
- 13.Lyu J, Wang S, Balius TE, et al. : Ultra-large library docking for discovering new chemotypes. Nature 566, 224–229 (2019), DOI: 10.1038/s41586-019-0917-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mirjalili S, Mirjalili S, Lewis A: Grey wolf optimizer. Advances in Engineering Software 69, 46–61 (2014), DOI: 10.1016/j.advengsoft.2013.12.007 [DOI] [Google Scholar]
- 15.Morris GM, Huey R, Lindstrom W, et al. : AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry 30(16), 2785–2791 (2009), DOI: 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nivedha AK, Thieker DF, Makeneni S, et al. : Vina-Carb: Improving Glycosidic Angles during Carbohydrate Docking. Journal of Chemical Theory and Computation 12(2), 892–901 (2016), DOI: 10.1021/acs.jctc.5b00834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Boyle NM, Banck M, James CA, et al. : Open Babel: An open chemical toolbox. Journal of Cheminformatics 3(1), 1–14 (2011), DOI: 10.1186/1758-2946-3-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Papoian GA, Wolynes PG: The physics and bioinformatics of binding and folding – an energy landscape perspective. Biopolymers 68(3), 333–349 (2003), DOI: 10.1002/bip.10286 [DOI] [PubMed] [Google Scholar]
- 19.Schrödinger LLC, New York: Maestro Release 2020-3. https://www.schrodinger.com/maestro, accessed: 2020-09-06 [Google Scholar]
- 20.Shaw DE, Deneroff MM, Dror RO, et al. : Anton, a special-purpose machine for molecular dynamics simulation. Commun. ACM 51(7), 91–97 (2008), DOI: 10.1145/1364782.1364802 [DOI] [Google Scholar]
- 21.Sterling T, Irwin JJ: ZINC 15 – ligand discovery for everyone. Journal of Chemical Information and Modeling 55(11), 2324–2337 (2015), DOI: 10.1021/acs.jcim.5b00559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trott O, Olson AJ: AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of computational chemistry 31(2), 455–461 (2010), DOI: 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.VirtualFlow. https://virtual-flow.org/ (2020), accessed: 2020-09-06
- 24.Wong KM, Tai HK, Siu SWI: GWOVina: A grey wolf optimization approach to rigid and flexible receptor docking. Chemical Biology & Drug Design (2020), DOI: 10.1111/cbdd.13764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yin W, Mao C, Luan X, et al. : Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 368(6498), 1499–1504 (2020), DOI: 10.1126/science.abc1560 [DOI] [PMC free article] [PubMed] [Google Scholar]