

Abstract
The Gab1 protein is tyrosine phosphorylated in response to various growth factors and serves as a docking protein that recruits a number of downstream signaling proteins, including phosphatidylinositol 3-kinase (PI-3 kinase). To determine the role of Gab1 in signaling via the epidermal growth factor (EGF) receptor (EGFR) we tested the ability of Gab1 to associate with and modulate signaling by this receptor. We show that Gab1 associates with the EGFR in vivo and in vitro via pTyr sites 1068 and 1086 in the carboxy-terminal tail of the receptor and that overexpression of Gab1 potentiates EGF-induced activation of the mitogen-activated protein kinase and Jun kinase signaling pathways. A mutant of Gab1 unable to bind the p85 subunit of PI-3 kinase is defective in potentiating EGFR signaling, confirming a role for PI-3 kinase as a downstream effector of Gab1. Inhibition of PI-3 kinase by a dominant-interfering mutant of p85 or by Wortmannin treatment similarly impairs Gab1-induced enhancement of signaling via the EGFR. The PH domain of Gab1 was shown to bind specifically to phosphatidylinositol 3,4,5-triphosphate [PtdIns(3,4,5)P3], a product of PI-3 kinase, and is required for activation of Gab1-mediated enhancement of EGFR signaling. Moreover, the PH domain mediates Gab1 translocation to the plasma membrane in response to EGF and is required for efficient tyrosine phosphorylation of Gab1 upon EGF stimulation. In addition, overexpression of Gab1 PH domain blocks Gab1 potentiation of EGFR signaling. Finally, expression of the gene for the lipid phosphatase PTEN, which dephosphorylates PtdIns(3,4,5)P3, inhibits EGF signaling and translocation of Gab1 to the plasma membrane. These results reveal a novel positive feedback loop, modulated by PTEN, in which PI-3 kinase functions as both an upstream regulator and a downstream effector of Gab1 in signaling via the EGFR.
The engagement of cell-surface receptors by ligand regulates a number of critical biological processes. In the case of receptor tyrosine kinases (RTKs), these processes include cell growth, survival, differentiation, and transformation, all of which depend on the activation of multiple signaling pathways (45). The mechanisms by which RTKs coordinate a wide variety of intracellular signaling events have been the subject of intense investigation. An emerging paradigm maintains that following dimerization and activation, signaling by RTKs is accomplished through the direct recruitment of signaling molecules containing appropriate recognition domains to specific sites on the receptor, where conformational changes, tyrosine phosphorylation, or translocation to the vicinity of effectors initiates signaling cascades (41, 46). However, a number of receptors also rely on a growing family of accessory, or docking, proteins that are recruited to the plasma membrane, become tyrosine phosphorylated, and act as centers for assembly of multiprotein complexes. This family includes the proteins Grb2-associated protein 1 (Gab1), Grb-2 associated protein 2 (Gab2), insulin receptor substrate 1 (IRS-1), IRS-2, IRS-3, Daughter of Sevenless (DOS), FGFR substrate 2 (FRS2), Downstream of tyrosine kinase (Dok), Linker for activation of T cells (LAT), and Dok-related (Dok-R) (7, 13, 17, 18, 24, 29, 44, 52, 55, 57). A number of genetic and biochemical experiments have provided evidence for the importance of docking proteins in cellular signaling. For example, DOS, a Drosophila docking protein, was first identified in a genetic screen for signaling components downstream of the protein tyrosine kinase Sevenless (17, 44). More recently, targeted disruption of the IRS-2 gene was shown to result in a generation of diabetic mice, demonstrating a role for this docking protein in signaling by the insulin receptor (54). Similarly, disruption of the gene for LAT, a docking protein, expressed in T cells, has demonstrated a critical role in T-cell activation and development (58). Other studies have shown that overexpression of docking proteins such as Gab1, Gab2, and FRS2 potentiates cellular signaling by receptors (13, 14, 19, 29, 50, 53), while overexpression of nonfunctional mutant docking proteins has been shown to block signaling (59).
Docking proteins share a number of common features, including some determinant that permits association with the plasma membrane. IRS-1, DOS, Dok, Dok-R, Gab1, and Gab2 all have PH domains at their amino termini that may direct interactions with specific phospholipids in the membrane (7, 13, 17, 24, 44, 55, 57). The LAT protein, on the other hand, has a transmembrane domain (59), whereas FRS2 contains an amino-terminal myristoylation site that is required for membrane localization (29, 40). Docking proteins also often have a specific receptor binding domain. IRS-1 contains an amino-terminal phosphotyrosine binding (PTB) domain that binds to the insulin receptor, insulin growth factor receptor 1, and other receptors (57), while the PTB domain of FRS2 interacts specifically with the fibroblast growth factor and nerve growth factor (NGF) receptors (29, 40). Similarly, the Dok-R PTB domain mediates association with the Tek-Tie2 receptor (24). It has been proposed that Gab1 contains a region that mediates binding to the activated Met receptor (53).
A key feature of all known docking proteins is the presence of multiple phosphorylated tyrosine residues in the carboxy termini of these proteins that serve as binding sites for SH2-containing proteins. IRS-1, FRS2, Dok, LAT, Dok-R, Gab1, and Gab2 all contain sites for such diverse SH2-containing proteins as Grb2, SHP2, phospholipase Cγ1, Nck, Crk, and Fyn (7, 13, 17, 18, 24, 29, 55, 57, 59). The ability to recruit diverse signaling molecules to a docking protein may help to expand the repertoire of signaling pathways that can be activated by a given receptor. In contrast, some docking proteins recruit a unique complement of downstream effectors, which may account for their specific signaling properties. For example, LAT binds SLP-76 and Vav, which are both expressed only in hematopoietic cells (59).
The Gab1 docking protein was first identified and cloned as a Grb2-associated protein and was shown to undergo tyrosine phosphorylation in response to epidermal growth factor (EGF) and insulin stimulation (18). Gab1 was subsequently isolated in a yeast two-hybrid screen for proteins capable of binding to the carboxy-terminal tail of the Met-hepatocyte growth factor (HGF) receptor (53). In this latter study as well as others, Gab1 was shown to be tyrosine phosphorylated in response to HGF (38, 53), and more recently, it was shown to be tyrosine phosphorylated in response to NGF (19). Gab1 binding to the Met receptor is mediated in part by a sequence termed the Met binding domain (MBD) (53). However, studies of Met receptor mutants have indicated that indirect association via Grb2 also contributes to receptor binding (3, 10, 38). In addition to the MBD domain, Gab1 is characterized by the presence of an amino-terminal PH domain and multiple tyrosine phosphorylation sites that include three p85–phosphatidylinositol 3-kinase (PI-3-kinase) binding sites (19). It has been shown that p85 association with Gab1 and activation of PI-3–kinase are required for the promotion of cell survival by NGF (19). Gab1 has also been implicated in cell transformation and epithelial cell scattering as well as in cell morphogenesis (18, 35, 53).
EGF has been shown to stimulate generation of PI-3 kinase products despite lacking a canonical PI-3 kinase binding site (5). However, activation of PI-3 kinase by EGF is relatively weak compared with activation induced by platelet-derived growth factor or insulin stimulation (20). A number of reports have suggested that another member of the EGF receptor (EGFR) family, ErbB3, which contains multiple YXXM motifs (canonical PI-3 kinase binding sites) that are transphosphorylated by the EGFR, acts as surrogate p85 adaptor upon heterodimerization with the EGFR (42, 47). The properties of Gab1 raise the possibility that this protein may facilitate signal-regulated recruitment of PI-3 kinase in cells that express or do not express ErbB3. In this report we demonstrate that Gab1 can bind directly to the EGFR and is capable of potentiating EGF-induced stimulation of mitogen-activated protein kinase (MAPK) and Jun kinase (JNK) in a PI-3 kinase-dependent fashion. Furthermore, we show that the PH domain of Gab1 binds primarily to phosphatidylinositol 3,4,5-triphosphate [PtdIns(3,4,5)P3], a major product of PI-3 kinase, and can target Gab1 to the plasma membrane in response to EGF stimulation. Both membrane recruitment and potentiation of the JNK and Erk signaling pathways are inhibited by Wortmannin treatment or by overexpression of a dominant-negative mutant of p85. Furthermore, overexpression of the isolated Gab1 PH domain could inhibit these effects. We therefore propose that the PH domain of Gab1 allows Gab1 recruitment to be controlled directly by the products of PI-3 kinase. In support of this, expression of PTEN, a lipid phosphatase that dephosphorylates PtdIns(3,4,5)P3, is able to inhibit EGFR signaling and translocation of Gab1. These findings suggest that Gab1 may participate in a positive feedback loop with respect to PI-3 kinase activation, to enhance signaling via the EGFR.
MATERIALS AND METHODS
Expression vectors.
cDNA fragments encoding the full-length Gab1 or the Gab1 MBD (amino acids 450 to 532) and PH domain (amino acids 14 to 116) were amplified by PCR with oligonucleotides containing a 5′ BamHI site and a 3′ EcoRI site. Following restriction digestion, these fragments were ligated in frame into a BamHI/EcoRI-digested pGEX2T or pGEX2TK bacterial expression vector to generate glutathione S-transferase (GST) fusion proteins or into BglII/EcoRI-digested pEGFP-C1 (Clonetech) to generate green fluorescent protein (GFP) fusion proteins.
To generate expression vectors for transient transfection of mammalian cells, the full-length Gab1 cDNA was subcloned from pBAT Flag (53) into pRK5 as a NotI fragment. Point mutations in Gab1 were generated by PCR mutagenesis using the QuickChange mutagenesis system (Stratagene). The ΔPH mutant was generated by PCR amplification of cDNA sequences encoding amino acids 122 to 698 of Gab1 by using oligonucleotides containing an EcoRI site and a 5′ ATG and then subcloned into pRK5.
Cells and DNA transfection.
293T cells, COS-1 cells, and HeLa cells were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum. Transfections were carried out by using Lipofectamine reagent (Gibco BRL) according to the manufacturer's instructions. HeLa cells were stimulated for 5 min with 100 ng of EGF (Intergen) per ml prior to harvesting 48 h posttransfection.
Antibodies.
Polyclonal anti-EGFR antibodies directed against an epitope in the carboxy terminus of the receptor have been described before (34). Antihemagglutinin (anti-HA) antibodies were obtained from Boehringer-Mannheim. Polyclonal anti-GFP antibodies were generated against a GST-GFP fusion protein. The polyclonal antiphosphotyrosine antibodies have been described previously (4). Monoclonal anti-Flag M2 antibodies were obtained from Sigma.
Immunoprecipitations and immunoblotting.
Cells were lysed with Triton lysis buffer (50 mM HEPES, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10% glycerol, and 1% Triton X-100). Immunocomplexes were collected on protein A- or protein G-Sepharose, washed with lysis buffer, resuspended in Laemmli sample buffer, boiled for 5 min, subjected to sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE), and transferred to nitrocellulose. The membranes were blocked in TBST (10 mM Tris [pH 7.5], 50 mM NaCl, and 0.1% Triton X-100) containing 5% bovine serum albumin for 1 h. Membranes were then incubated for 1.5 h with antibody. Membranes were washed extensively with TBST, and immunoreactive proteins were detected by incubation with horseradish peroxidase-conjugated protein A for detection of polyclonal antibodies or horseradish peroxidase-conjugated anti-mouse immunoglobulin G for detection of monoclonal antibodies. Proteins were visualized by enhanced chemiluminescence and autoradiography.
Expression and purification of the GST fusion proteins.
Following transformation of DH5 bacteria with various GST constructs, cells were grown to an optical density at 600 nm of about 0.3 to 0.4 and induced with isopropyl-1-thio-β-d-galactopyranoside (IPTG) for 3 h. Cells were centrifuged and lysed by sonication in lysis buffer (50 mM Tris [pH 8.0], 100 mM NaCl, 10% glycerol, 1 mM dithiothreitol) containing protease inhibitors. Triton X-100 was added to a concentration of 1%, and the cellular debris were removed by centrifugation. The cleared lysates were incubated with glutathione-agarose beads for 1 h at 4°C. The beads were then washed three times with phosphate-buffered saline and stored at 4°C.
Microscopy.
GFP fusion proteins were visualized following fixation of cells with 4% paraformaldehyde in phosphate-buffered saline for 10 min at room temperature by conventional fluorescence microscopy or by confocal microscopy using an upright SARASTRO 2000 CLSM (Molecular Dynamics).
In vivo labeling and analysis of phosphoinositides.
The labeling of cells with myo-[3H]inositol and the analysis of phosphoinositides extracted from these cells has been described elsewhere (8).
JNK and MAPK assays.
For JNK assays the GST-Jun protein, containing the first 79 amino acids of c-Jun fused to GST, [(GST-c-Jun(1-79)], was grown in bacteria and harvested following induction with IPTG. Approximately 2 μg of protein purified on glutathione-agarose beads was used as the substrate in kinase assays following immunoprecipitation of JNK 1 from lysates of transiently transfected cells with either anti-HA or anti-Flag antibodies. Kinase assays were carried out in 30 μl of kinase buffer (20 mM Hepes [pH 7.5], 20 mM B-glycerol phosphate, 10 mM PNPP, 10 mM MgCl2, 10 mM dithiothreitol, 50 μM sodium vanadate) containing 20 μM ATP and 0.5 ml of [γ-32P]ATP for 20 min at 30°C. Reactions were terminated by the addition of 2× sample buffer, and proteins were separated by SDS–10% PAGE.
Anti-HA antibodies were used to immunoprecipitate the transfected Erk2 kinase from transfected cells. In vitro kinase reactions were carried out in kinase buffer (20 mM Tris-HCl [pH 7.5], 10 mM MgCl2) with myelin basic protein as a substrate. Following incubation at 30°C for 15 min, reactions were terminated by the addition of sample buffer and proteins were resolved by SDS–12% PAGE.
RESULTS
Gab1 associates with the EGFR.
As EGF has been shown to stimulate tyrosine phosphorylation of Gab1, we were interested to know whether Gab1 becomes associated with the EGFR upon EGF stimulation. In lysates from A431 cells stimulated with EGF, we were able to identify a protein with an apparent molecular mass of 180 kDa that migrates in SDS gels by immunoblotting of Gab1 immunoprecipitates with antiphosphotyrosine antibodies (Fig. 1A). This phosphorylated protein corresponded to the EGFR, as revealed by immunoblotting of Gab1 immunoprecipitates with antibodies to the EGFR (Fig. 1A). Additionally, a 115-kDa phosphorylated protein that corresponds in size to Gab1 itself is detected in antiphosphotyrosine immunoprecipitates. Immunoblotting with anti-Gab1 antibodies confirmed the identity of this protein as Gab1 (Fig. 1A).
FIG. 1.

Association of Gab1 with the EGFR. (A and B) Anti-Gab1 antibodies (A) and a GST-Gab1 MBD fusion protein (B), respectively, were used to precipitate proteins from lysates prepared from untreated (−) or EGF-stimulated (+) (100 ng/ml for 5 min) NIH 3T3 cells expressing the EGFR (HER14 cells). Coprecipitating proteins were resolved by SDS-PAGE and immunoblotted with either anti-pTyr, anti-EGFR, or anti-Gab1 antibodies. (C) Lysates prepared from untreated or EGF-stimulated (100 ng/ml for 5 min) HER14 cells and NIH 3T3 cells expressing the CD63 mutant were incubated with anti-Gab1 antibodies. Immunocomplexes were resolved by SDS-PAGE and immunoblotted with anti-pTyr antibodies. (D) Lysates prepared from A431 cells were incubated with a GST-Gab1 MBD fusion protein in the presence of 10 mM concentrations of peptides corresponding to EGFR sequences containing a particular tyrosine phosphorylation site. Complexes were resolved by SDS-PAGE and immunoblotted with anti-pTyr antibodies (top) or anti-EGFR antibodies (bottom).
Gab1 has been previously shown to associate with the Met-HGF receptor in the yeast two-hybrid system via an 83-amino-acid stretch termed the MBD (53). To test whether the MBD can mediate association of Gab1 with the EGFR, a GST fusion protein of the MBD was used to determine whether it could precipitate the EGFR. The results presented in Fig. 1B show that the GST-Gab1 MBD fusion protein associates with the EGFR in lysates from EGF-stimulated cells. These results demonstrate a phosphorylation-dependent association between the EGFR and Gab1 that is mediated by the MBD. To determine which phosphotyrosines in the activated EGFR are responsible for this interaction we next used a cell line expressing a deletion mutant of the EGFR that is lacking the last 63 amino acids, which include two known tyrosine phosphorylation sites, Y1148 and Y1173. Figure 1C shows that both the wild type and the CD63 mutant can be detected in a pTyr immunoblot of GST-Gab1 MBD precipitates from lysates of cell lines expressing wild-type EGFR or the CD63 mutant. This experiment suggests that binding of the MBD of Gab1 to the EGFR involves one or more of the three phosphorylated tyrosine residues (at positions 992, 1068, and 1086) that are not deleted in CD63.
To determine which of these remaining tyrosine autophosphorylation sites are important for Gab1 binding we used a series of phosphopeptides encompassing each of the five known tyrosine autophosphorylation sites of EGFR to compete for binding of the MBD to the EGFR. Consistent with the CD63 immunoprecipitation result, phosphopeptides containing Y1148 or Y1173 were unable to compete for Gab1 MBD binding (Fig. 1D). However, phosphopeptides containing phosphorylated tyrosine Y1068 or Y1086 were able to effectively compete for Gab1 MBD binding, suggesting that either one or both of these pTyr residues are required for association of the Gab1 MBD to the EGFR. It is noteworthy that pY1068 and pY1086 function as primary and secondary binding sites for the Grb2 SH2 domain (4).
EGF-induced MAPK and JNK activation are enhanced by overexpression of Gab1.
Previous reports have implicated Gab1 in activation of MAPK in response to cytokines and by simple overexpression in transfected cells (50, 53). In addition to activating the well-known MAPK pathway, the EGFR has been shown to activate the JNKs (32, 36). Thus, we wanted to determine whether overexpression of Gab1 could modulate the activity of JNK as well as MAPK.
Transient overexpression of the EGFR in 293T cells causes autophosphorylation without the need for EGF stimulation (data not shown) and leads to activation of Erk2 and JNK when HA-tagged versions of these kinases are coexpressed (Fig. 2). The activities of Erk2 and JNK were measured following immunoprecipitation with anti-HA antibodies by using an in vitro kinase reaction with myelin basic protein and c-Jun as the respective substrates (Fig. 2). When increasing amounts of Gab1 were transfected with fixed amounts of EGFR and HA-tagged versions of Erk2 or JNK 1, a proportional increase in the enzymatic activities of these kinases was observed (Fig. 2; see also Fig. 5). Control immunoblots to measure expression of Gab1 and EGFR show appropriate protein levels (Fig. 2). These results demonstrate that overexpression of Gab1 enhances the capacity of EGFR to stimulate MAPK and JNK and suggests that Gab1 plays a role in signaling through the EGFR.
FIG. 2.

Potentiation of EGFR-mediated activation of MAPK and JNK by overexpression of Gab1. 293T cells were transiently transfected with increasing amounts (0.5, 2, and 4 μg) of Gab1 expression vector and a constant amount (100 ng) of EGFR (four rightmost lanes) and HA-tagged Erk2 (left) or HA-tagged JNK1 (right) (1.5 μg each). At 48 h posttransfection, cells were lysed and kinase assays were performed as described in Materials and Methods (top blots). Expression of the transfected proteins was determined by immunoblotting total cell lysates with the antibodies indicated beside the lower rows of blots.
FIG. 5.

Lipid binding specificity of the Gab1 PH domain. (A) GST-agarose beads containing the Gab1 PH domain were incubated with a deacylated lipid extract prepared from [3H]inositol-labeled IMR33 fibroblasts. The beads were washed, and the bound glycerophosphoinositols were eluted with 0.1 N HCl separated by HPLC and quantitated by liquid scintillation counting. The amount of radioactivity of each compound was calculated as a percentage of the total radioactivity present in the extract and then represented as a percentage relative to the amount of gPI(3,4,5)P3 bound. The error bars represent the standard deviations of three independent experiments. (B) A pool of equal amounts of soluble lipids were mixed with GST-agarose beads containing the Gab1 PH domain, and the bound lipids were analyzed by HPLC. The amount of radioactivity of each compound was corrected for minor differences of the starting material present in the pool and then represented as a percentage relative to the amount of Ins(1,3,4,5)P4 bound. Results are the means of three independent experiments. (C) Displacement of binding of the Gab1 PH binding to PtdIns(3,4,5)P3 by the soluble head group Ins(1,3,4,5)P4. [32P]gPI(3,4,5)P3 was generated in vitro by using PI-3 kinase and PtdIns(4,5)P2 as a substrate and bound to the Gab1 PH domain in the presence of increasing concentrations of Ins(1,3,4,5)P4. gPI(3,4,5)P3, glycerophosphoinositol(3,4,5)phosphate.
PI-3 kinase activation is required for signaling through Gab1.
Experiments using membrane-targeted versions of the p110 catalytic subunit of PI-3 kinase have demonstrated that PI-3 kinase is an upstream activator of the JNK signaling pathway (27, 32) and that EGF-induced JNK activation is in part mediated by PI-3 kinase (32). As Gab1 was previously shown to bind PI-3 kinase and mediate PI-3 kinase activation in response to EGF and NGF treatment (18, 19), we were interested to know whether PI-3 kinase activity is required for the ability of Gab1 to potentiate JNK activation by the EGFR.
A dominant-interfering mutant of PI-3 kinase was utilized to block PI-3 kinase activation. This mutant consists of sequences from the inter-SH2 domain of p85, a region necessary for p85 association with the catalytic 110 subunit, and has been previously shown to be effective in blocking both PI-3 kinase in vivo (21) and activation of JNK by the EGFR (32). We also generated a mutant of Gab1 in which the three tyrosines that function as p85 binding sites were mutated to phenyalanines (3F mutant). This Gab1 3F mutant has been previously shown not to associate with activated PI-3 kinase in response to NGF stimulation (19). As PI-3 kinase was implicated in JNK activation, mutations affecting PI-3 kinase activity would be expected to inhibit EGF-induced JNK stimulation. As shown in Fig. 3, the 3F mutant was defective in its ability to potentiate JNK activation induced by EGFR. Moreover, overexpression of the p85INT blocked enhancement of JNK signaling mediated by wild-type Gab1 (Fig. 3). This result confirms that Gab1 potentiation of JNK activation by EGFR requires PI-3 kinase activity.
FIG. 3.

Enhancement of EGFR-mediated activation of JNK by Gab1 requires PI-3 kinase. As shown on the left, an expression vector encoding an HA-tagged dominant-negative mutant of PI-3 kinase (p85INT) (2 μg in the third lane from the left and 4 μg in the fourth lane from the left) was cotransfected with the EGFR, Gab1, and Flag-tagged JNK 1. Anti-Flag immunoprecipitates were divided and one half was used in a JNK assay (top) and the other half was blotted with anti-Flag antibodies to show equal amounts of JNK 1 (bottom). For the two middle blots, total cell lysates were blotted with anti-Gab1 and anti-HA antibodies. As shown on the right, wild-type Gab1 (1.5 μg 3 μg in the second end third lanes from the left, respectively) and a mutant of Gab1 (3 μg) (last lane) containing substitutions of the three putative p85 binding sites (Y448, Y473, and Y590) were introduced with the EGFR and HA-JNK 1, and their relative ability to enhance EGFR activation of this kinase was measured in vitro as described in Materials and Methods.
Gab1 is translocated to the plasma membrane upon EGFR stimulation in a PI-3 kinase-dependent fashion.
Numerous studies have demonstrated a role for PH domains in the membrane recruitment of signaling proteins (30, 31). The presence of a PH domain in Gab1 prompted us to examine the cellular localization of Gab1 upon EGFR activation. To that end, we generated a traceable Gab1 fusion protein using GFP. The GFP-Gab1 construct was transfected into COS-1 cells, and membrane recruitment was assessed by fluorescence microscopy following EGF treatment. Figure 4A shows that in unstimulated cells the GFP-Gab1 fusion was distributed diffusely in the cytoplasm. However, upon stimulation with EGF, a distinct pattern of fluorescence was observed at the plasma membrane (Fig. 4B). Additionally, many cells displayed intense staining at sites of membrane ruffling (data not shown).
FIG. 4.

Gab1 is translocated to the plasma membrane in response to EGF stimulation. (A and B) COS1 cells fixed and analyzed by fluorescence microscopy after a cDNA encoding the GFP-Gab1 fusion protein was transfected into them. At 48 h posttransfection, the cells were left untreated (A) or stimulated with 100 ng of EGF per ml (B). (C to E) COS-1 cells fixed and analyzed by confocal fluorescence microscopy after a cDNA encoding the GFP-Gab1 PH domain fusion protein was transfected into them. At 48 h posttransfection, the cells were left untreated (C), stimulated with 100 ng of EGF per ml (D), or pretreated with 100 nM Wortmannin for 10 min prior to stimulation with EGF (E). Arrows indicate fluorescence staining of the plasma membrane. Magnification, ×660.
We also assessed the ability of the Gab1 PH alone fused to GFP to translocate in response to EGF and observed a similar membrane pattern of staining, suggesting that the PH domain mediates membrane localization of Gab1. However, we did note more pronounced staining of membrane ruffles with the isolated Gab1 PH domain than with the full-length Gab1 (Fig. 4C). We also detected nuclear staining in both unstimulated and EGF-treated cells with the Gab1 PH domain (Fig. 4C and D). This nuclear staining has been observed for other isolated PH domains, including the PH domains of PLCγ1, Grp1, and ARNO, and most likely represents diffusion of the fusion protein through the nuclear pore due to its relatively smaller size (8, 51).
It has been previously reported that a membrane-targeted version of the p110 catalytic subunit of PI-3 kinase can promote membrane localization of a GFP-Gab1 PH domain (22). To test whether PI-3 kinase is required for membrane recruitment of Gab1 PH domain in response to EGF, COS-7 cells were pretreated with 100 nM Wortmannin prior to stimulation with EGF. Figure 4E shows that the GFP-Gab1 PH domain was localized only in the cytoplasm of cells that had been pretreated with Wortmannin. These results implicate PI-3 kinases in membrane recruitment of Gab1.
Gab1 PH domain binds PtdIns(3,4,5)P3.
The results with the GFP-Gab1 PH fusion protein suggested that the Gab1 PH domain is involved in membrane recruitment of Gab1. This result prompted examination of the lipid binding properties of the Gab1 PH domain. Comparison of the sequence of the Gab1 PH domain with other PH domains revealed a strong similarity with PH domains of proteins known to bind products of PI-3 kinase. Specifically, a strong sequence identity was noted in the β1 and β2 strands of a region previously shown to be involved in lipid binding (9, 16). To examine the lipid binding properties of the Gab1 PH domain, a GST fusion protein of the Gab1 PH domain was generated, bound to glutathione beads, and mixed with deacylated lipid extracts from platelet-derived growth factor-stimulated [3H]inositol-labeled IMR33 fibroblasts. The associated glycerophosphoinositides were eluted and analyzed by high-performance liquid chromatography (HPLC). The results, shown in Fig. 5A, demonstrate that the Gab1 PH domain binds most strongly to PtdIns(3,4,5)P3. Smaller amounts of binding to phosphatidylinositol 3-phosphate, phosphatidylinositol 3,4-biphosphate [PtdIns(3,4)P2], and phosphatidylinositol 4,5-biphosphate [PtdIns(4,5)P2] were also detected. To correct for differences in the amount of each lipid species in the starting extract, the binding experiments were repeated using approximately equal amounts of the corresponding soluble inositol phosphate headgroup for each lipid species. Again, the results shown in Fig. 5C confirm a preference of the Gab1 PH domain for PtdIns(3,4,5)P3 {inositol(1,3,4,5)tetraphosphate [Ins(1,3,4,5)P4]}, followed by PtdIns(3,4)P2 {inositol(1,3,4)triphosphate[Ins(1,3,4)P3]}, PtdIns(4,5)P2 {inositol(1,4,5)triphosphate [Ins(1,4,5)P3]}, and PtdIns(3)P {inositol(1,3)biphosphate [Ins(1,3)P2]}.
Displacement experiments were conducted to determine the 50% inhibitory concentration (IC50) of binding of PtdIns(3,4,5)P3 to the Gab1 PH domain by its soluble head group, Ins(1,3,4,5)P4. The water-soluble PtdIns(3,4,5)P3 head group, Ins(1,3,4,5)P4, was used to compete the binding of [32P]gPI(3,4,5)P3 generated in vitro using purified PI-3 kinase and gPI(4,5)P2 as a substrate to the Gab1 PH domain (Fig. 5C). The IC50 was determined to be approximately 10 μM, in the range previously observed for the binding of PtdIns(3,4,5)P3 to other PH domains (8, 25).
Gab1 PH domain is required for Gab1-mediated activation of JNK.
As the PH domain of Gab1 is capable of binding membrane lipids and can potentially mediate translocation of Gab1 to the plasma membrane, we were interested to know whether this domain is required for Gab1 potentiation of signaling via EGFR. To test this we generated a mutant of Gab1 with a deletion of the PH domain and compared its ability to potentiate JNK activation with the wild-type Gab1. HeLa cells were transfected with wild type or the deletion mutant and then stimulated with EGF. Figure 6A shows that, whereas the wild-type Gab1 potentiates activation of JNK by EGF, expression of the PH deletion mutant leads to no further activation of JNK beyond what is observed upon EGF stimulation. This result demonstrates that the PH domain is critical for EGFR signaling mediated by Gab1.
FIG. 6.

The PH domain of Gab1 is required for signaling. (A) HeLa cells were transfected with increasing amounts (0.5 and 2 μg) of Gab1 or Gab1ΔPH expression vector and HA-tagged JNK 1 (1.5 μg). At 48 h posttransfection cells were stimulated with EGF (100 ng/ml) for 5 min and lysed, and kinase assays were performed as described in Materials and Methods (top blot). Expression levels of the transfected components was determined by immunoblotting of total cell lysates with the indicated antibodies (lower blots). (B) HeLa cells were transfected with increasing amounts (0.5, 1.5, and 3 μg) of Gab1 or Gab1ΔPH expression vector. At 48 h posttransfection cells were stimulated with EGF (100 ng/ml) for 5 min and lysed, and immunoblots of anti-Gab1 immunoprecipitates were conducted using antiphosphotyrosine (top blot) or anti-Gab1 (bottom blot) antibodies.
To determine the effect of the PH domain deletion on the ability of Gab1 to be tyrosine phosphorylated in response to EGF, immunoprecipitates of the wild-type Gab1 and the PH domain deletion mutant were blotted with antiphosphotyrosine antibodies. Figure 6B shows that the deletion mutant can still be tyrosine phosphorylated but somewhat less efficiently than the wild-type Gab1, as demonstrated by a reduction in the intensity of phosphorylated Gab1 deletion mutant bands.
PI-3 kinase is an upstream activator of Gab1.
The results above demonstrate a requirement for PI-3 kinase in Gab1 signaling. In addition, the ability of the Gab1 PH domain to bind the products of PI-3 kinase as well as translocate to the plasma membrane in response to EGF suggest that PI-3 kinase not only is required for signaling through Gab1 but also may function as an upstream activator of Gab1. We therefore tested whether Wortmannin, an inhibitor of PI-3 kinase, could repress the potentiation of EGFR signaling by Gab1. By contrast with activation of JNK, MAPK activation is independent of PI-3 kinase and would not be expected to be directly affected by an inhibitor of PI-3 kinase function unless PI-3 kinase is acting upstream of Gab1. As shown in Fig. 7A, pretreatment of transfected cells with 100 nM Wortmannin resulted in significant inhibition of the Gab1-mediated increase in EGFR-induced JNK activation. Significantly, Gab1-mediated potentiation of MAPK was also potently inhibited by Wortmannin (Fig. 7A).
FIG. 7.

Wortmannin and the Gab1 PH domain inhibit Gab1 potentiation of EGFR signaling. (A) An expression vector for EGFR was transfected into 293T cells together with vectors encoding HA-Erk2 (right) or HA-JNK 1 (left) with (second and third lanes from left) or without (leftmost lane) Gab1. At 48 h posttransfection untreated cells or cells pretreated with 100 nM Wortmannin were lysed and kinase assays were performed following immunoprecipitation with anti-HA antibodies (top blot). Immunoblots were performed with the indicated antibodies on total cell lysates (lower blots). (B) HeLa cells were transfected with vectors encoding Gab1. At 48 h posttransfection untreated cells or cells pretreated with 100 nM Wortmannin were lysed and immunoblots were performed using antiphosphotyrosine antibodies (top blot) or anti-Gab1 antibodies (bottom blot) following immunoprecipitation with anti-Gab1 antibodies. (C) HeLa cells were transfected with vectors encoding HA-JNK 1 and increasing concentrations of the Gab1 PH domain. Cells not stimulated (leftmost lane) or stimulated with EGF (100 ng/ml) for 5 min (three rightmost lanes) and then lysed 48 h posttransfection. A JNK assay was performed as described in Materials and Methods following immunoprecipitation with anti-HA antibodies (top blot). Immunoblots of total cell lysates with the indicated antibodies are shown in the lower blots. (D) Results from panel C were quantitated by densitometric scanning of the kinase assay and plotted. The vertical scale is the measurement of the density of the bands in arbitrary units.
We next tested the effect of Wortmannin on the tyrosine phosphorylation of Gab1. Figure 7B shows that treatment of HeLa cells with Wortmannin prior to stimulation with EGF resulted in a significant decrease in the level of tyrosine phosphorylation of Gab1. These results are consistent with the hypothesis that PI-3 kinase regulation of Gab1 function is mediated by binding of specific phosphoinositides to the Gab1 PH domain facilitating plasma membrane recruitment of Gab1.
To further test this hypothesis we investigated the possibility that the PH domain could act as a dominant-negative mutant of Gab1. We predicted that expression of the isolated PH domain of Gab1 might inhibit PI-3 kinase activation by endogenous EGFRs. HeLa cells were transiently cotransfected with Gab1 PH domain and with HA-JNK 1 expression vectors and stimulated with EGF. The results presented in Fig. 7C reveal inhibition of the EGF-stimulated increase in JNK activity by overexpression of Gab1 PH domain. This result not only confirms the importance of the PH domain but also suggests that Gab1 may be the primary mediator via which EGFR activates PI-3 kinase. Together, these results demonstrate that activation of PI-3 kinase by EGFR is mediated by Gab1. Further, they argue not only that PI-3 kinase is required as a downstream effector of Gab1 but also that it acts as an upstream activator of Gab1. This is likely to involve recruitment of Gab1 to the plasma membrane by the binding of Gab1 PH domain to PtdIns(3,4,5)P3, which in turn will stimulate PI-3 kinase activation by p85 recruitment to phosphorylated tyrosines on Gab1.
PTEN regulates EGFR signaling and Gab1 phosphorylation.
The PTEN gene is a tumor suppressor gene whose product has been recently shown to have lipid phosphatase activity (33). As the Gab1 PH domain can mediate translocation of Gab1 through its binding of PtdIns(3,4,5)P3, we wanted to know whether PTEN, which converts PtdIns(3,4,5)P3 to PtdIns(4,5)P2 could modulate EGFR signaling and Gab1 translocation. HeLa cells were transiently cotransfected with expression vectors containing either a wild-type or mutant PTEN defective in its lipid phosphatase activity with an HA-Erk2 or HA-JNK1 expression vector and stimulated with EGF. The results presented in Fig. 8A show an approximately twofold inhibition of EGF-stimulated MAPK and JNK activity by expression of the wild-type PTEN but not the mutant (despite relatively higher (1.5×) levels of expression of the mutant compared to wild-type PTEN. This result demonstrates that PTEN can modulate EGFR signaling.
FIG. 8.

PTEN regulates Gab translocation and phosphorylation. (A) HeLa cells were transfected with a Gab1 expression vector and HA-tagged ERK2 (left) or HA-tagged JNK 1 (right) with either the wild-type or a phosphatase-negative PTEN (PTEN C/S) expression vector. At 48 h posttransfection, cells were untreated (rightmost lanes) or stimulated with EGF (100 ng/ml) for 5 min (three rightmost lanes) and lysed, and kinase assays were performed as described in Materials and Methods. Expression levels of PTEN were determined by immunoblotting of total cell lysates (lower blots). The kinase assay was quantitated by densitometric scanning, and the percentages, relative to the amount of EGF-stimulated activity, are indicated below each lane. (B) GFP-Gab1 expression vector and a fivefold excess of either wild-type or mutant PTEN were transfected into COS-1 cells. At 48 h posttransfection the cells were stimulated with 100 ng of EGF per ml, fixed, and analyzed by fluorescence microscopy.
To determine whether this modulation might be due to effects on Gab1 we tested whether translocation of Gab1 to the plasma membrane might be inhibited by PTEN. The GFP-Gab1 construct was cotransfected into COS-1 cells with either wild-type or mutant PTEN, the cells were stimulated with EGF, and membrane recruitment was assessed by fluorescence microscopy. Figure 8B shows that in cells cotransfected with wild-type PTEN, the GFP-Gab1 fusion protein was distributed diffusely in the cytoplasm. However, fluorescence was observed at the plasma membrane, especially at sites of membrane ruffling, when GFP-Gab1 was introduced with mutant PTEN (Fig. 8B). These results provide evidence that Gab1 translocation and EGFR signaling can both be affected by PTEN expression and suggest that PTEN may regulate EGFR signaling by modulating Gab1 localization.
DISCUSSION
A growing family of docking proteins characterized by the presence of membrane targeting and receptor binding domains have been recently identified. Among these is Gab1, which is tyrosine phosphorylated in response to activation of a number receptor tyrosine kinases, including EGFR and the hepatocyte growth factor, fibroblast growth factor, and NGF receptors (18, 19, 53). Gab1 serves as the docking site for a number of downstream signaling molecules, including PI-3 kinase (18, 19). The importance of Gab1 as a docking protein is illustrated by the fact that Gab1 mediates NGF-induced cell survival and HGF-induced epithelial cell scattering and morphogenesis (19, 35, 53). In this paper we examine the role of Gab1 in EGFR signaling.
Previously, it was proposed that EGF activation of PI-3 kinase is regulated at least in part by heterodimerization with ErbB3, a membrane receptor containing canonical binding sites for PI-3 kinase (42, 47). Here, we show that Gab1 can also couple the EGFR directly to activation of the PI-3 kinase and JNK signaling pathway. Gab1 can bind to the activated EGFR, thereby recruiting p85 to the receptor. Association of Gab1 with the EGFR is mediated in part by the MBD, a region previously shown to mediate binding to the carboxy-terminal tail of the HGF-Met receptor (53). We have shown that pTyr 1068 and 1086 of the activated EGFR can function as sites for interaction with the Gab1 MBD. We have not identified any obvious similarities between these two sites and the tyrosines on the HGF-Met receptor that are responsible for direct association with Gab1. Studies with Met receptor mutants have attributed much of the observed Gab1 binding to indirect association via Grb2, with little direct binding that is mediated by other sites (3, 10, 38). As position 1068 of the EGFR serves as the primary binding site for Grb2 (Grb2 binds more weakly to pTyr 1086 [4]), we cannot rule out the possibility that part of the binding is indirect and mediated by Grb2. It is possible that, as suggested for the HGF-Met receptor, Gab1 binding to the EGFR requires a primary Grb2 site (Y1068) plus a secondary direct binding site (Y1086) that is recognized by the MBD.
PI-3 kinase appears to act as not only a downstream effector of Gab1 but also an upstream regulator. A mutant of Gab1 that fails to associate with PI-3 kinase is defective in potentiating JNK signaling (Fig. 3). Similarly, a dominant-negative mutant of p85 inhibits Gab1 potentiation of JNK stimulation by Gab1 (Fig. 7). Moreover, Wortmannin can block PH domain-mediated translocation to the plasma membrane and tyrosine phosphorylation of Gab1 as well as MAPK and JNK signaling in response to EGF stimulation (Fig. 7). Since activation of MAPK is thought to lie on a pathway independent of PI-3 kinase we conclude that PI-3 kinase must be acting as an upstream activator of Gab1. Consistent with this hypothesis the PH domain of Gab1 is capable of binding to PtdIns(3,4,5)P3, a major product of PI-3 kinase (Fig. 3). Significantly, the β1-β2 loop region of the Gab1 PH domain shows strong sequence identity with PH domains of other proteins that are known to bind lipids generated through the action of PI-3 kinase (9, 16). Furthermore, expression of the isolated Gab1 PH domain acts as a dominant-interfering mutant that blocks Gab1 potentiation of EGF-induced JNK activation (Fig. 7). Although, we cannot rule out the potential for disruption of other signaling pathways leading to JNK activation, this result together with our binding data and experiments with Wortmannin present strong evidence for the importance in PI-3 kinase regulation of Gab1 through its PH domain. These results are also consistent with a recent report by Maroun et al. (35), who similarly demonstrated a critical role for the PH domain and a requirement for PI-3 kinase in the translocation of Gab1 to the plasma membrane.
Thus, on the basis of these experiments, we propose a model by which PI-3 kinase is both an upstream activator and downstream effector of Gab1 (Fig. 9). According to our model, Gab1 recruitment is initiated upon binding of the MBD to phosphorylated tyrosine residues on the EGFR and/or indirect recruitment via Grb2. Subsequent phosphorylation of Gab1 by the EGFR facilitates its association with downstream effectors, including the p85 subunit of PI-3 kinase. Activation of PI-3 kinase then leads to the production of PtdIns(3,4,5)P3, which promotes further membrane recruitment of Gab1 and additional enhancement of PI-3 kinase signaling. Such a positive feedback loop may act to create a sustained PI-3 kinase signal necessary to achieve a specific biological effect. Since binding of the Gab1 MBD to the EGFR is relatively weak compared with binding of the PTB domain of Shc or the SH2 domain of Grb2 to the EGFR (data not shown), the concerted action of both the PH domain and the MBD may be required to achieve stable membrane recruitment of Gab1 of sufficient duration to enhance its tyrosine phosphorylation and association with downstream signaling molecules. Consistent with this idea, deletion of the Gab1 PH domain reduces but does not eliminate tyrosine phosphorylation of Gab1 in response to EGF (Fig. 6). A similar scenario may also apply for tyrosine phosphorylation of IRS-1 by insulin receptors, as it has been shown that the presence of both the PTB domain and the PH domain is required for efficient tyrosine phosphorylation of IRS-1 in response to insulin stimulation (38, 52, 56). Also, recent evidence suggests that stable association of Sos with the EGFR requires, in addition to the carboxy-terminal Grb2 binding site, the N-terminal PH domain (43).
FIG. 9.
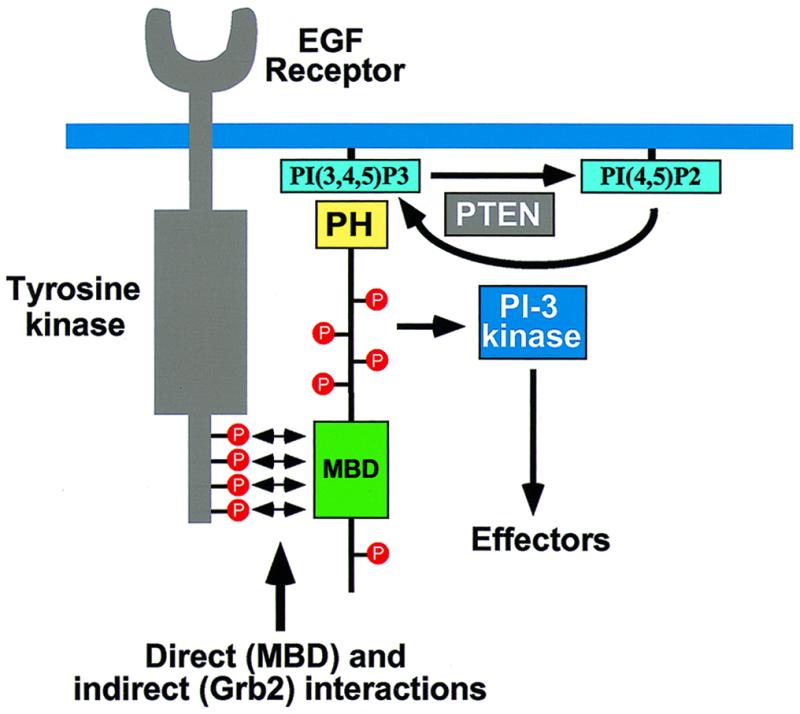
Model outlining the role of Gab1 in receptor tyrosine kinase signaling. Gab1 is recruited to the plasma membrane in response to growth factor stimulation through the concerted action of binding of its MBD to the EGFR directly and through Grb2 and binding of its PH domain to the products of PI-3 kinase. Tyrosine phosphorylation of Gab1 by EGFR leads to recruitment and activation of signaling molecules, including PI-3 kinase. Activation of PI-3 kinase in turn leads to the generation of phosphoinositides that enhance downstream signaling as well as initiate a positive feedback by recruiting more Gab1 molecules to the EGFR. Conversion of PtdIns(3,4,5)P3 to PtdIns(4,5)P2 by PTEN allows for dissociation of Gab1 from the plasma membrane and termination of signaling through Gab1.
It is not yet clear how the positive feedback loop mediated by Gab1 and PI-3 kinase is terminated. Since the Gab1 PH domain binds preferentially to PtdIns(3,4,5)P3, the most reasonable mechanism would be that membrane association of Gab1 is down-modulated by the action of 5′ phosphatidylinositol phosphatases such as PTEN, which generates PtdIns(4,5)P2 from PtdIns(3,4,5)P3. Since the Gab1 PH domain binds significantly more strongly to PtdIns(3,4,5)P3 than to PtdIns(3,4)P2, this would lead to dissociation of Gab1 from the cell membrane and hence signal termination. Indeed, we observe that PTEN not only can block translocation of Gab1 but also can block EGF activation of JNK (Fig. 8). This mode of regulation is similar to what has been observed for AKT/protein kinase B (PKB), where high levels of PtdIns(3,4,5)P3 and constitutive activation of AKT/PKB were seen in fibroblasts derived from PTEN-deficient mice (reviewed in reference 6).
The importance of PH domains in regulating membrane association and biological function has been documented for a number of proteins (31). Our experiments demonstrating the importance of PI-3 kinase in Gab1 signaling and the role of the PH domain in binding specific products of PI-3 kinase is in agreement with other recent evidence showing that PI-3 kinase can regulate the activity of many proteins through their PH domains. For example PI-3 kinase can regulate PLCγ1 activity through its PH domain (8) and modulate the Rac exchange activity of Sos through the PH domain (39). Similarly, Vav and Grp1 PH domain-containing guanine nucleotide exchange factors are regulated by PI-3 kinase (15, 25, 26). PI-3 kinase activates AKT/PKB via a more elaborate process involving binding of (3,4,5)P3 to its PH domain (11, 12, 23, 28) as well as the PH domain of another kinase, 3-phosphoinositide-dependent protein kinase 1 (PDK1) (1, 2, 48, 49). Thus, specific PI-3 kinase-derived phosphoinositides are emerging as an important class of second messengers that act as mediators of plasma membrane association of signaling molecules and perhaps also as allosteric regulators of enzyme activity. The experiments presented in this report underscore the central role that PI-3 kinases provide in generation of intricate intracellular circuitry that controls cell growth, differentiation, and metabolism as well as other important intracellular processes.
ACKNOWLEDGMENTS
We thank Walter Birchmeier for supplying the Gab1 cDNA and Mark Lemmon for critical reading of the manuscript.
The financial support of Telethon Italy (grant no. 328/bi) is gratefully acknowledged by M.F.
REFERENCES
- 1.Alessi D R, Deak M, Casamayor A, Caudwell F B, Morrice N, Norman D G, Gaffney P, Reese C B, MacDougall C N, Harbison D, Ashworth A, Bownes M. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr Biol. 1997;7:776–789. doi: 10.1016/s0960-9822(06)00336-8. [DOI] [PubMed] [Google Scholar]
- 2.Alessi D R, James S R, Downes C P, Holmes A B, Gaffney P R, Reese C B, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase B. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 3.Bardelli A, Longati P, Gramaglia D, Stella M C, Comoglio P M. Gab1 coupling to the HGF/Met receptor multifunctional docking site requires binding of Grb2 and correlates with the transforming potential. Oncogene. 1997;15:3103–3111. doi: 10.1038/sj.onc.1201561. [DOI] [PubMed] [Google Scholar]
- 4.Batzer A G, Rotin D, Ureña J M, Skolnik E Y, Schlessinger J. Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol Cell Biol. 1994;14:5192–5201. doi: 10.1128/mcb.14.8.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bjorge J D, Chan T O, Antczak M, Kung H J, Fujita D J. Activated type I phosphatidylinositol kinase is associated with the epidermal growth factor (EGF) receptor following EGF stimulation. Proc Natl Acad Sci USA. 1990;87:3816–3820. doi: 10.1073/pnas.87.10.3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cantley L C, Neel B G. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA. 1999;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carpino N, Wisniewski D, Strife A, Marshak D, Kobayashi R, Stillman B, Clarkson B. p62dok: a constitutively tyrosine-phosphorylated, GAP-associated protein in chronic myelogenous leukemia progenitor cells. Cell. 1997;88:197–204. doi: 10.1016/s0092-8674(00)81840-1. [DOI] [PubMed] [Google Scholar]
- 8.Falasca M, Logan S K, Lehto V P, Baccante G, Lemmon M A, Schlessinger J. Activation of phospholipase Cγ by PI 3-kinase-induced PH domain-mediated membrane targeting. EMBO J. 1998;17:414–422. doi: 10.1093/emboj/17.2.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferguson K M, Lemmon M A, Schlessinger J, Sigler P B. Structure of the high affinity complex of inositol trisphosphate with a phospholipase C pleckstrin homology domain. Cell. 1995;83:1037–1046. doi: 10.1016/0092-8674(95)90219-8. [DOI] [PubMed] [Google Scholar]
- 10.Fixman E D, Holgado-Madruga M, Nguyen L, Kamikura D M, Fournier T M, Wong A J, Park M. Efficient cellular transformation by the Met oncoprotein requires a functional Grb2 binding site and correlates with phosphorylation of the Grb2-associated proteins, Cbl and Gab1. J Biol Chem. 1997;272:20167–20172. doi: 10.1074/jbc.272.32.20167. [DOI] [PubMed] [Google Scholar]
- 11.Franke T F, Kaplan D R, Cantley L C, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997;275:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- 12.Frech M, Andjelkovic M, Ingley E, Reddy K K, Falck J R, Hemmings B A. High affinity binding of inositol phosphates and phosphoinositides to the pleckstrin homology domain of RAC/protein kinase B and their influence on kinase activity. J Biol Chem. 1997;272:8474–8481. doi: 10.1074/jbc.272.13.8474. [DOI] [PubMed] [Google Scholar]
- 13.Gu H, Pratt J C, Burakoff S J, Neel B G. Cloning of p97/Gab2, the major SHP2-binding protein in hematopoietic cells, reveals a novel pathway for cytokine-induced gene activation. Mol Cell. 1998;2:729–740. doi: 10.1016/s1097-2765(00)80288-9. [DOI] [PubMed] [Google Scholar]
- 14.Hadari Y R, Kouhara H, Lax I, Schlessinger J. Binding of Shp2 tyrosine phosphatase to FRS2 is essential for fibroblast growth factor-induced PC12 cell differentiation. Mol Cell Biol. 1998;18:3966–3973. doi: 10.1128/mcb.18.7.3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han J, Luby-Phelps K, Das B, Shu X, Xia Y, Mosteller R D, Krishna U M, Falck J R, White M A, Broek D. Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science. 1998;279:558–560. doi: 10.1126/science.279.5350.558. [DOI] [PubMed] [Google Scholar]
- 16.Harlan J E, Hajduk P J, Yoon H S, Fesik S W. Pleckstrin homology domains bind to phosphatidylinositol-4,5-bisphosphate. Nature. 1994;371:168–170. doi: 10.1038/371168a0. [DOI] [PubMed] [Google Scholar]
- 17.Herbst R, Carroll P M, Allard J D, Schilling J, Raabe T, Simon M A. Daughter of sevenless is a substrate of the phosphotyrosine phosphatase corkscrew and functions during sevenless signaling. Cell. 1996;85:899–909. doi: 10.1016/s0092-8674(00)81273-8. [DOI] [PubMed] [Google Scholar]
- 18.Holgado-Madruga M, Emlet D R, Moscatello D K, Godwin A K, Wong A J. A Grb2-associated docking protein in EGF- and insulin-receptor signalling. Nature. 1996;379:560–564. doi: 10.1038/379560a0. [DOI] [PubMed] [Google Scholar]
- 19.Holgado-Madruga M, Moscatello D K, Emlet D R, Dieterich R, Wong A J. Grb2-associated binder-1 mediates phosphatidylinositol 3-kinase activation and the promotion of cell survival by nerve growth factor. Proc Natl Acad Sci USA. 1997;94:12419–12424. doi: 10.1073/pnas.94.23.12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu P, Margolis B, Skolnik E Y, Lammers R, Ullrich A, Schlessinger J. Interaction of phosphatidylinositol 3-kinase-associated p85 with epidermal growth factor and platelet-derived growth factor receptors. Mol Cell Biol. 1992;12:981–990. doi: 10.1128/mcb.12.3.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu P, Schlessinger J. Direct association of p110β phosphatidylinositol 3-kinase with p85 is mediated by an N-terminal fragment of p110β. Mol Cell Biol. 1994;14:2577–2583. doi: 10.1128/mcb.14.4.2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Isakoff S J, Cardozo T, Andreev J, Li Z, Ferguson K M, Abagyan R, Lemmon M A, Aronheim A, Skolnik E Y. Identification and analysis of PH domain-containing targets of phosphatidylinositol 3-kinase using a novel in vivo assay in yeast. EMBO J. 1998;17:5374–5387. doi: 10.1093/emboj/17.18.5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.James S R, Downes C P, Gigg R, Grove S J, Holmes A B, Alessi D R. Specific binding of the Akt-1 protein kinase to phosphatidylinositol 3,4,5-trisphosphate without subsequent activation. Biochem J. 1996;315:709–713. doi: 10.1042/bj3150709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones N, Dumont D J. The Tek/Tie2 receptor signals through a novel Dok-related docking protein, Dok-R. Oncogene. 1998;17:1097–1108. doi: 10.1038/sj.onc.1202115. [DOI] [PubMed] [Google Scholar]
- 25.Klarlund J K, Guilherme A, Holik J J, Virbasius J V, Chawla A, Czech M P. Signaling by phosphoinositide-3,4,5-trisphosphate through proteins containing pleckstrin and Sec7 homology domains. Science. 1997;275:1927–1930. doi: 10.1126/science.275.5308.1927. [DOI] [PubMed] [Google Scholar]
- 26.Klarlund J K, Rameh L E, Cantley L C, Buxton J M, Holik J J, Sakelis C, Patki V, Corvera S, Czech M P. Regulation of GRP1-catalyzed ADP ribosylation factor guanine nucleotide exchange by phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:1859–1862. doi: 10.1074/jbc.273.4.1859. [DOI] [PubMed] [Google Scholar]
- 27.Klippel A, Reinhard C, Kavanaugh W M, Apell G, Escobedo M A, Williams L T. Membrane localization of phosphatidylinositol 3-kinase is sufficient to activate multiple signal-transducing kinase pathways. Mol Cell Biol. 1996;16:4117–4127. doi: 10.1128/mcb.16.8.4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klippel A, Kavanaugh W M, Pot D, Williams L T. A specific product of phosphatidylinositol 3-kinase directly activates the protein kinase Akt through its pleckstrin homology domain. Mol Cell Biol. 1997;17:338–344. doi: 10.1128/mcb.17.1.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kouhara H, Hadari Y R, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, Schlessinger J. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell. 1997;89:693–702. doi: 10.1016/s0092-8674(00)80252-4. [DOI] [PubMed] [Google Scholar]
- 30.Lemmon M A, Ferguson K M, Schlessinger J. PH domains: diverse sequences with a common fold recruit signaling molecules to the cell surface. Cell. 1996;85:621–624. doi: 10.1016/s0092-8674(00)81022-3. [DOI] [PubMed] [Google Scholar]
- 31.Lemmon M A, Falasca M, Ferguson K M, Schlessinger J. Regulatory recruitment of signalling molecules to the cell membrane by pleckstrin-homology domains. Trends Cell Biol. 1997;7:237–242. doi: 10.1016/S0962-8924(97)01065-9. [DOI] [PubMed] [Google Scholar]
- 32.Logan S K, Falasca M, Hu P, Schlessinger J. Phosphatidylinositol 3-kinase mediates epidermal growth factor-induced activation of the c-Jun N-terminal kinase signaling pathway. Mol Cell Biol. 1997;17:5784–5790. doi: 10.1128/mcb.17.10.5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maehama T, Dixon J E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 34.Margolis B, Li N, Koch A, Mohammadi M, Hurwitz D R, Zilberstein A, Ullrich A, Pawson T, Schlessinger J. The tyrosine phosphorylated carboxy terminus of the EGF receptor is a binding site for GAP and PLC-γ. EMBO J. 1990;9:4375–4380. doi: 10.1002/j.1460-2075.1990.tb07887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maroun C R, Holgado-Madruga M, Royal I, Naujokas M A, Fournier T M, Wong A J, Park M. The Gab1 PH domain is required for localization of Gab1 at sites of cell-cell contact and epithelial morphogenesis downstream from the Met receptor tyrosine kinase. Mol Cell Biol. 1999;19:1784–1799. doi: 10.1128/mcb.19.3.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Minden A, Lin A, Claret F X, Abo A, Karin M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- 37.Myers M G J, Grammer T C, Brooks J, Glasheen E M, Wang L M, Sun X J, Blenis J, Pierce J H, White M F. The pleckstrin homology domain in insulin receptor substrate-1 sensitizes insulin signaling. J Biol Chem. 1995;270:11715–11718. doi: 10.1074/jbc.270.20.11715. [DOI] [PubMed] [Google Scholar]
- 38.Nguyen L, Holgado-Madruga M, Maroun C, Fixman E D, Kamikura D, Fournier T, Charest A, Tremblay M L, Wong A J, Park M. Association of the multisubstrate docking protein Gab1 with the hepatocyte growth factor receptor requires a functional Grb2 binding site involving tyrosine 1356. J Biol Chem. 1997;272:20811–20819. doi: 10.1074/jbc.272.33.20811. [DOI] [PubMed] [Google Scholar]
- 39.Nimnual A S, Yatsula B A, Bar-Sagi D. Coupling of Ras and Rac guanosine triphosphatases through the Ras exchanger Sos. Science. 1998;279:560–563. doi: 10.1126/science.279.5350.560. [DOI] [PubMed] [Google Scholar]
- 40.Ong S H, Guy G R, Hadari Y, Laks S, Gotoh N, Schlessinger J, Lax I. FRS2 proteins recruit intracellular signaling pathways by binding to diverse targets on fibroblast growth factor and nerve growth factor receptors. 1999. Mol. Cell. Biol., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pawson T. Protein modules and signalling networks. Nature. 1995;373:573–580. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- 42.Prigent S A, Gullick W J. Identification of c-erbB-3 binding sites for phosphatidylinositol 3′- kinase and SHC using an EGF receptor/c-erbB-3 chimera. EMBO J. 1994;13:2831–2841. doi: 10.1002/j.1460-2075.1994.tb06577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qian X L, Vass W C, Papageorge A G, Anborgh P H, Lowy D R. N terminus of Sos1 Ras exchange factor: critical roles for the Dbl and pleckstrin homology domains. Mol Cell Biol. 1998;18:771–778. doi: 10.1128/mcb.18.2.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raabe T, Riesgo-Escovar J, Liu X D, Bausenwein B S, Deak P, Maröy P, Hafen E. DOS, a novel pleckstrin homology domain-containing protein required for signal transduction between sevenless and RAS1 in Drosophila. Cell. 1996;85:911–920. doi: 10.1016/s0092-8674(00)81274-x. [DOI] [PubMed] [Google Scholar]
- 45.Schlessinger J, Ullrich A. Growth factor signaling by receptor tyrosine kinases. Neuron. 1992;9:383–391. doi: 10.1016/0896-6273(92)90177-f. [DOI] [PubMed] [Google Scholar]
- 46.Schlessinger J. SH2/SH3 signaling proteins. Curr Opin Genet Dev. 1994;4:25–30. doi: 10.1016/0959-437x(94)90087-6. [DOI] [PubMed] [Google Scholar]
- 47.Soltoff S P, Carraway K L R, Prigent S A, Gullick W G, Cantley L C. ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol Cell Biol. 1994;14:3550–3558. doi: 10.1128/mcb.14.6.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter G F, Holmes A B, Gaffney P R J, Reese C B, McCormick F, Tempst P, Coadwell J, Hawkins P T. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science. 1998;279:710–714. doi: 10.1126/science.279.5351.710. [DOI] [PubMed] [Google Scholar]
- 49.Stokoe D, Stephens L R, Copeland T, Gaffney P R J, Reese C B, Painter G F, Holmes A B, McCormick F, Hawkins P T. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science. 1997;277:567–570. doi: 10.1126/science.277.5325.567. [DOI] [PubMed] [Google Scholar]
- 50.Takahashi-Tezuka M, Yoshida Y, Fukada T, Ohtani T, Yamanaka Y, Nishida K, Nakajima K, Hibi M, Hirano T. Gab1 acts as an adapter molecule linking the cytokine receptor gp130 to ERK mitogen-activated protein kinase. Mol Cell Biol. 1998;18:4109–4117. doi: 10.1128/mcb.18.7.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Venkateswarlu K, Oatey P B, Tavare J M, Cullen P J. Insulin-dependent translocation of ARNO to the plasma membrane of adipocytes requires phosphatidylinositol 3-kinase. Curr Biol. 1998;8:463–466. doi: 10.1016/s0960-9822(98)70181-2. [DOI] [PubMed] [Google Scholar]
- 52.Voliovitch H, Schindler D G, Hadari Y R, Taylor S I, Accili D, Zick Y. Tyrosine phosphorylation of insulin receptor substrate-1 in vivo depends upon the presence of its pleckstrin homology region. J Biol Chem. 1995;270:18083–18087. doi: 10.1074/jbc.270.30.18083. [DOI] [PubMed] [Google Scholar]
- 53.Weidner K M, Di Cesare S, Sachs M, Brinkmann V, Behrens J, Birchmeier W. Interaction between Gab1 and the c-Met receptor tyrosine kinase is responsible for epithelial morphogenesis. Nature. 1996;384:173–176. doi: 10.1038/384173a0. [DOI] [PubMed] [Google Scholar]
- 54.Withers D J, Gutierrez J S, Towery H, Burks D J, Ren J M, Previs S, Zhang Y T, Bernal D, Pons S, Shulman G I, Bonner-Weir S, White M F. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391:900–904. doi: 10.1038/36116. [DOI] [PubMed] [Google Scholar]
- 55.Yamanashi Y, Baltimore D. Identification of the Abl- and rasGAP-associated 62 kDa protein as a docking protein, Dok. Cell. 1997;88:205–211. doi: 10.1016/s0092-8674(00)81841-3. [DOI] [PubMed] [Google Scholar]
- 56.Yenush L, Makati K J, Smith-Hall J, Ishibashi O, Myers M G J, White M F. The pleckstrin homology domain is the principal link between the insulin receptor and IRS-1. J Biol Chem. 1996;271:24300–24306. doi: 10.1074/jbc.271.39.24300. [DOI] [PubMed] [Google Scholar]
- 57.Yenush L, White M F. The IRS-signalling system during insulin and cytokine action. Bioessays. 1997;19:491–500. doi: 10.1002/bies.950190608. [DOI] [PubMed] [Google Scholar]
- 58.Zhang W, Sommers C L, Burshtyn D N, Stebbins C C, DeJarnette J B, Trible R P, Grinberg A, Tsay H C, Jacobs H M, Kessler C M, Long E O, Love P E, Samelson L E. Essential role of LAT in T cell development. Immunity. 1999;10:323–332. doi: 10.1016/s1074-7613(00)80032-1. [DOI] [PubMed] [Google Scholar]
- 59.Zhang W G, Sloan-Lancaster J, Kitchen J, Trible R P, Samelson L E. LAT: the ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell. 1998;92:83–92. doi: 10.1016/s0092-8674(00)80901-0. [DOI] [PubMed] [Google Scholar]