

Eukaryotic translation elongation factor 1 alpha (eEF1A), a subunit of the essential eukaryotic elongation factor 1 (eEF1) complex, coordinates the recruitment of aminoacyl-transfer-RNA (aa-tRNA) to ribosomes during protein synthesis1. Human eEF1A has two distinct isoforms, eEF1A1 and eEF1A22. While eEF1A1 is ubiquitously expressed, eEF1A2 expression is limited to cardiac and skeletal muscle, as well as neurons3, suggesting tissue-specific functionality. Patients carrying a homozygous missense mutation p.P333L in eEF1A2 exhibited global developmental delay and dilated cardiomyopathy (DCM), ultimately leading to death in early childhood4. However, the physiological role of eEF1A2 in heart and the effect of the P333L mutation on eEF1A2 protein function are unknown.
To investigate the role of eEF1A2 in heart, we used Cre-LoxP technology to delete eEF1A2 in cardiomyocytes. The data, analytical methods, and study materials that support the findings of this study will be available to other researchers from the corresponding author on reasonable request. All mouse protocols were approved by the Institutional Animal Care and Use Committee. EEF1A2flox/flox mice, in which exon 5 of eEF1A2 is flanked by loxP sites, were generated using CRISPR-Cas9 technology. EEF1A2flox/flox mice were crossed with Xenopus laevis myosin light-chain 2 (Xmlc2)-Cre transgenic mice to ultimately produce cardiomyocyte-conditional knockout eEF1A2flox/flox; Xmlc2-Cre+ (hereafter cKO) mice. Among the tissues examined, eEF1A2 protein was specifically reduced in cKO hearts while remaining comparable to eEF1A2flox/flox (hereafter control) mice in other tissues (Figure [A]). Notably, loss of eEF1A2 markedly increased eEF1A1 protein expression in heart (Figure [A]).
Figure. Loss or P333L mutation of eEF1A2 in murine myocardium results in dilated cardiomyopathy.
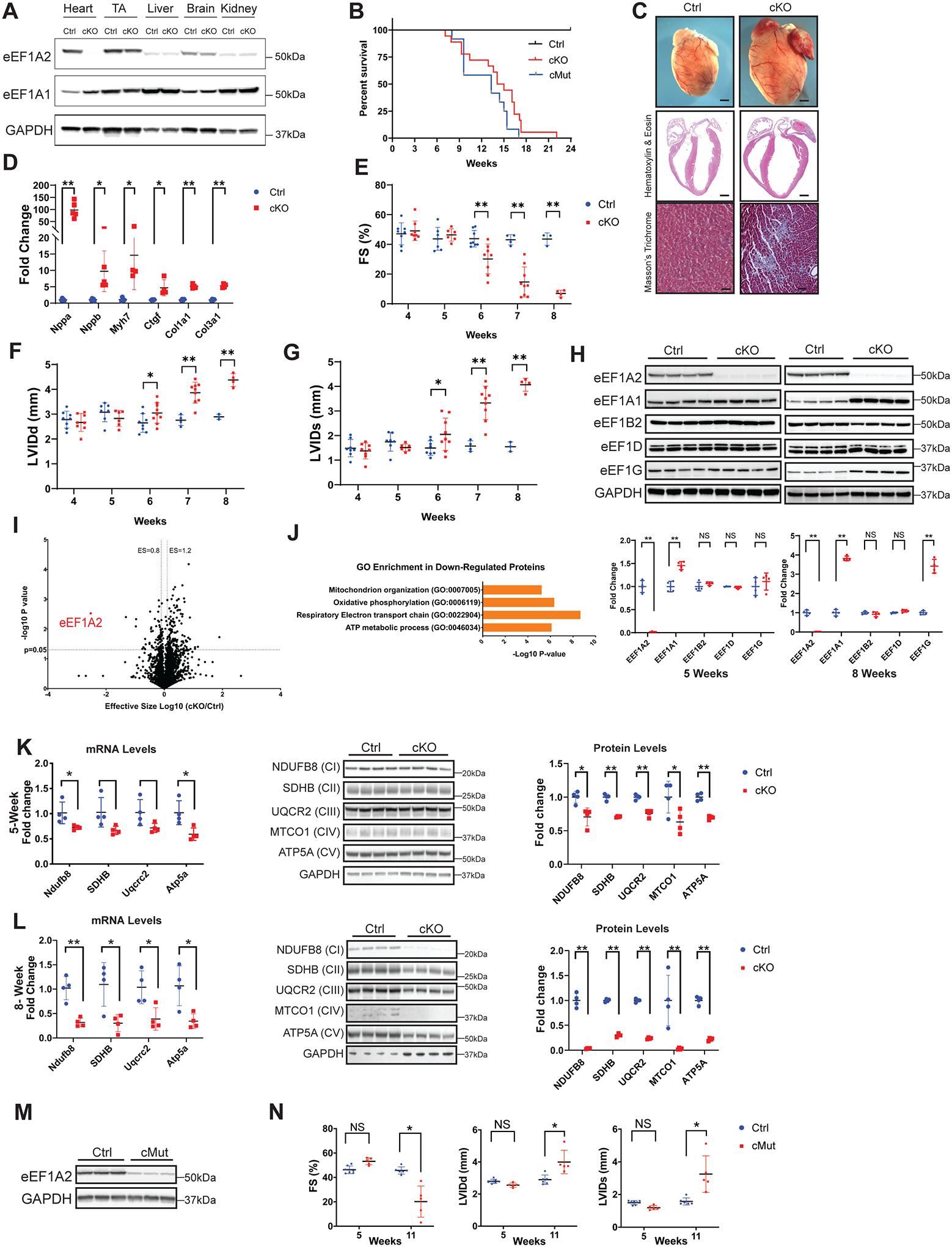
A, Western blot analysis of eEF1A2 and eEF1A1 in indicated tissues isolated from Cre-negative eEF1A2flox/flox (Ctrl) and eEF1A2flox/flox; Xmlc2-Cre+ (cKO) mice at 8 weeks of age. TA, tibialis anterior muscle. B, Kaplan-Meier survival curves of Ctrl (n=10), eEF1A2 cKO (n=18), and eEF1A2 P333L cMut (n=12) mice. C, Representative microscopic images of whole mouse hearts (top, scale bars: 1 mm), and cross-sectional views of H&E-stained (middle, scale bars: 1 mm) and Masson’s trichrome-stained (bottom, scale bars: 100 μm) hearts isolated from Ctrl and cKO mice at 8 weeks of age. D, qRT-PCR analysis of cardiac remodeling and fibrosis gene markers in Ctrl (blue, n=4–5) and cKO (red, n=4–5) mouse hearts at 8 weeks of age. E-G, Echocardiography measurements of (E) left ventricular (LV) systolic function (% of fractional shortening, FS) and LV internal diameter at (F) end-diastole (LVIDd) and (G) end-systole (LVIDs) in Ctrl (blue, n=4–6) and cKO (red, n=4–9) mice at 4, 5, 6, 7, and 8 weeks of age. H, Western blot (top) and corresponding quantification (bottom) analysis of eEF1B complex components in Ctrl (blue) and cKO (red) mouse hearts at 5 (left) and 8 (right) weeks of age. I, Volcano plot of protein expression changes between Ctrl and cKO 5-week-old ventricles (n=3). The region above the horizontal dashed line is the adjusted Bonferroni P value <0.05. Proteins with adjusted P<0.05 and effective size (ES) >1.2 or <0.8 are considered significantly upregulated or downregulated, respectively. J, Gene Ontology (GO) enrichment analysis of significantly downregulated proteins in cKO hearts by mass spectrometry (I) with ES <0.8 and P<0.05. GO terms for significantly downregulated biological processes are listed. K, Analysis of complex components of the mitochondrial respiratory electron transport chain (ETC) by qRT-PCR (left), western blot (middle), and corresponding quantification (right) analysis of ETC complex (C) proteins in Ctrl (blue, n=4) and cKO (red, n=4) hearts at 5 weeks of age. L, Analysis of ETC complex components by qRT-PCR (left), western blot (middle), and corresponding quantification (right) analysis of ETC complex (C) proteins in Ctrl (blue, n=4) and cKO (red, n=4) hearts at 8 weeks of age. M, Western blot analysis of eEF1A2 in eEF1A2flox/+; Xmlc2-Cre+ (Ctrl) and eEF1A2mut/flox; Xmlc2-Cre+ (cMut) mouse hearts at 8 weeks of age. N, Echocardiography measurements of LV systolic function (% FS), LVIDd, and LVIDs in Ctrl (blue, n=5) and cMut (red, n=4) mice at 5 and 11 weeks of age. Statistical analysis: Survival data were calculated using Kaplan-Meier survival analysis with a log-rank statistical method. For all other data, values are presented as mean ± standard deviation. For qRT-PCR and western blot analyses, data were normalized to corresponding Gapdh and GAPDH levels, respectively, and cKO or cMut was expressed as fold change versus Ctrl. A representative example of 3–5 independent experiments is shown. Statistical analysis was performed with a Student’s t-test or analysis of variance followed by Tuckey’s post-hoc test for multiple comparisons. P values of less than 0.05 were considered statistically significant (NS=not significant; *P<0.05; **P<0.01).
CKO mice were born at expected Mendelian ratios and appeared normal at birth. Over time, cKO mice began to die from 7 weeks after birth, with no surviving cKOs observed past 22 weeks (Figure [B]). CKO hearts exhibited marked cardiac enlargement with severe left ventricular (LV) dilation and fibrosis at 8 weeks (Figure [C]), with increased expression of cardiac remodeling and fibrosis gene markers (Figure [D]). Echocardiography revealed an age-dependent decrease in LV systolic function in cKO mice (Figure [E]) with a significant increase in LVIDd and LVIDs (Figure [F]–[G]), indicating that cKO mice started developing DCM 5 weeks after birth.
Since eEF1A2 functions in canonical mRNA translation elongation, we investigated the effect of eEF1A2 deletion on components of the eEF1B complex, a multiprotein guanine nucleotide exchange factor required for eEF1A to participate in elongation1. Protein levels of eEF1B subunits, eEF1Bα (eEF1B2), eEF1Bδ (eEF1D), and eEF1Bγ (eEF1G) remained unchanged in cKO hearts at 5 weeks, with an increase in eEF1Bγ observed only at 8 weeks (Figure [H]). We then utilized a puromycin incorporation assay to measure the effect of eEF1A2 deletion on global protein synthesis in the heart. Puromycin incorporation, and thus nascent peptide synthesis, was comparable in control and cKO hearts at 5 and 8 weeks (not shown). These data suggest that neither eEF1B complex expression, nor protein synthesis, were decreased by cardiac-specific loss of eEF1A2.
To gain a better understanding of how cKO mice develop the observed phenotypes, we performed mass spectrometry analysis of control and cKO hearts at 5 weeks of age and identified 78 proteins with significantly reduced expression (Figure [I]). Functional classification by Gene Ontology analysis revealed that the most significantly downregulated proteins were primarily involved in processes regulating mitochondrial energy production (Figure [J]). We therefore performed qRT-PCR and western blot analyses of the major oxidative phosphorylation (OXPHOS) complex (CI-V) components of the mitochondrial respiratory electron transport chain (ETC). Both mRNA and protein levels of NDUFB8 (CI), SDHB (CII), UQCR2 (CIII), MTCO1 (CIV), and ATP5A (CV) were slightly downregulated in cKO hearts at 5 weeks compared to controls (Figure [K]). At 8 weeks, all complex components were largely diminished in cKO hearts, at both the mRNA and protein level (Figure [L]), suggesting that impairment of ETC function is a secondary, rather than causative, effect of the observed DCM phenotype in cKO mice.
To investigate the effect of the P333L mutation on eEF1A2 protein function, we generated eEF1A2 P333L knock-in mice using CRISPR-Cas9 technology, in which proline 333 was mutated to leucine. Homozygous global eEF1A2 P333L mutant mice appeared normal until weaning, then quickly exhibited tremors, ataxia, weight loss, and lethality at 4 weeks of age, reminiscent of wasted mutant mice, in which eEF1A2 expression is abolished by a spontaneous genetic deletion5. To investigate cardiac-specific consequences of eEF1A2 P333L mutation, we generated cardiac-specific eEF1A2 P333L mutant (eEF1A2mut/flox; Xmlc2-Cre+, hereafter cMut) mice. eEF1A2 protein expression was markedly reduced in cMut hearts (Figure [M]), yet mRNA level was comparable to controls (not shown), suggesting that P333L mutation impairs eEF1A2 protein stability. Similar to observations with cKO mice, cMut mice began to die from 8 weeks after birth, with no surviving mutants observed past 17 weeks (Figure [B]). cMut mice also had diminished systolic function and marked LV chamber dilation (Figure [N]). Similar to cKO hearts, expression of the major mitochondrial ETC complex components was dramatically decreased at both mRNA and protein levels in cMut hearts at 8 weeks (not shown). Taken together, our results demonstrate that eEF1A2 is indispensable for cardiac development and function, and eEF1A2 P333L mutation constitutes a loss-of-function mutation by impairing eEF1A2 protein stability.
Sources of Funding:
JC is funded by grants from the NHLBI and holds an American Heart Association Endowed Chair in Cardiovascular Research.
Conflict of interest:
JC consulted for and received research funding from MyoKardia Inc.
Non-standard Abbreviations and Acronyms:
- eEF1A
Eukaryotic translation elongation factor 1 alpha
- CRISPR-Cas9
Clustered Regularly Interspaced Short Palindromic Repeats-Cas9
- aa-tRNA
aminoacyl-transfer-RNA
- DCM
dilated cardiomyopathy
- Xmlc2
Xenopus laevis myosin light-chain 2
- OXPHOS
oxidative phosphorylation
- ETC
electron transport chain
References
- 1.Sasikumar AN, Perez WB, Kinzy TG. The many roles of the eukaryotic elongation factor 1 complex. Wiley Interdiscip Rev RNA. 2012; 3:543–55. doi: 10.1002/wrna.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lund A, Knudsen SM, Vissing H, Clark B, Tommerup N. Assignment of human elongation factor 1alpha genes: EEF1A maps to chromosome 6q14 and EEF1A2 to 20q13.3. Genomics. 1996; 36:359–61. doi: 10.1006/geno.1996.0475. [DOI] [PubMed] [Google Scholar]
- 3.Khalyfa A, Bourbeau D, Chen E, Petroulakis E, Pan J, Xu S, Wang E. Characterization of elongation factor-1A (eEF1A-1) and eEF1A-2/S1 protein expression in normal and wasted mice. J Biol Chem. 2001; 276:22915–22. doi: 10.1074/jbc.M101011200. [DOI] [PubMed] [Google Scholar]
- 4.Cao S, Smith LL, Padilla-Lopez SR, Guida BS, Blume E, Shi J, Morton SU, Brownstein CA, Beggs AH, Kruer MC, Agrawal PB. Homozygous EEF1A2 mutation causes dilated cardiomyopathy, failure to thrive, global developmental delay, epilepsy and early death. Hum Mol Genet. 2017; 26:3545–3552. doi: 10.1093/hmg/ddx239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chambers DM, Peters J, Abbott CM. The lethal mutation of the mouse wasted (wst) is a deletion that abolishes expression of a tissue-specific isoform of translation elongation factor 1alpha, encoded by the Eef1a2 gene. Proc Natl Acad Sci USA. 1998; 95:4463–8. doi: 10.1073/pnas.95.8.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]