

Abstract
Anti-tumor immunity through checkpoint inhibitors, specifically anti-programmed death 1 (PD-1)/programmed death ligand 1 (PD-L1) interaction, is a promising approach for cancer therapy. However, as early clinical trials indicate that colorectal cancers (CRCs) do not respond well to immune-checkpoint therapies, new effective immunotherapy approaches to CRC warrant further study. Simvastatin is an inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A (CoA) reductase (HMGCR), the rate-limiting enzyme of the mevalonate (MVA) pathway for the cholesterol biosynthesis. However, little is known about the functions of simvastatin in the regulation of immune checkpoints or long noncoding RNA (lncRNA)-mediated immunoregulation in cancer. Here, we found that simvastatin inhibited PD-L1 expression and promoted anti-tumor immunity via suppressing the expression of lncRNA SNHG29. Interestingly, SNHG29 interacted with YAP and inhibited phosphorylation and ubiquitination-mediated protein degradation of YAP, thereby facilitating downregulation of PD-L1 transcriptionally. Patient-derived tumor xenograft (PDX) models and the clinicopathological analysis in samples from CRC patients further supported the role of the lncRNA SNHG29-mediated PD-L1 signaling axis in tumor microenvironment reprogramming. Collectively, our study uncovers simvastatin as a potential therapeutic drug for immunotherapy in CRC, which suppresses lncRNA SNHG29-mediated YAP activation and promotes anti-tumor immunity by inhibiting PD-L1 expression.
Keywords: simvastatin, colorectal cancer, immune-checkpoint PD-L1, lncRNA SNHG29, YAP activation, immunotherapy
Graphical abstract
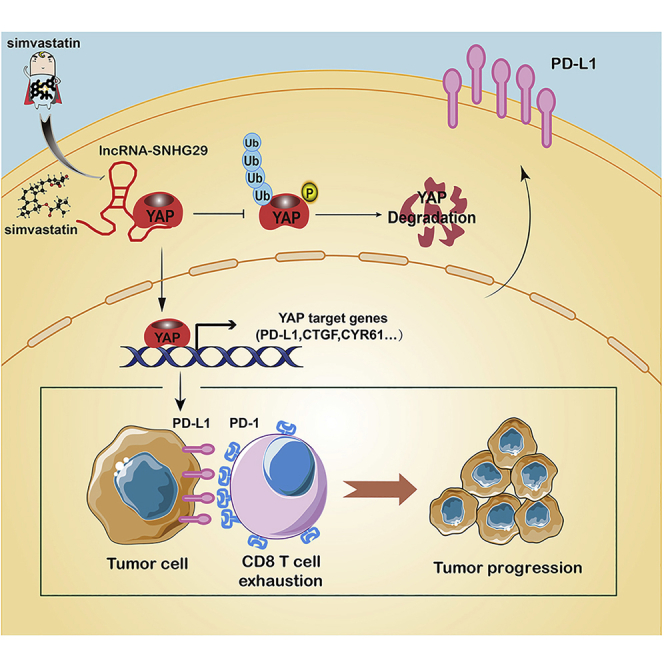
Ni et al. found that simvastatin suppresses lncRNA SNHG29-mediated YAP activation and promotes anti-tumor immunity by inhibiting PD-L1 expression, proposing simvastatin treatment as a potential therapy or an adjuvant therapy for immunotherapy in colorectal cancer.
Introduction
Colorectal cancer (CRC) is the third most commonly diagnosed cancer and the fourth leading cause of cancer-related deaths across the world.1 CRC was developed through a multistep of initiation and progression, including common mutations and transcriptional and post-transcriptional modification, which generate hallmarks of cancer.2, 3, 4 Notably, the reprogramming of energy metabolism and evading immune destruction have been accepted as core hallmarks of cancer cells.5 However, the underlying mechanisms of these emerging properties of cancer remain poorly understood.
The mevalonate (MVA) pathway is an essential metabolic pathway, which was also referred to as the cholesterol biosynthesis pathway critical to tumor progression.6,7 The 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR), a rate-limiting enzyme of the MVA pathway, is aberrantly high expressed in CRC tissues. Therefore, treatment of HMGCR inhibitors, such as simvastatin, significantly reduced cancer-specific mortality of CRC patients.8 Simvastatin may be involved in essential cellular functions, such as cell proliferation and apoptosis of tumor cells;9,10 however, little is known about the function of simvastatin in the dysregulation of immune checkpoints and immunotherapy.
Recently, immunotherapy has made great progress with promising results, which prolong the overall survival (OS) of patients.11, 12, 13 A prominent example of immune-checkpoint inhibitor (ICPi) is anti-programmed death 1 (PD-1), which could block PD-1 to enhance antitumor immunity, thereby causing tumor cell death.14,15 PD-1 is displayed on T, B, and natural killer (NK) cells, and its ligand programmed death ligand 1 (PD-L1) is displayed on cancer cell and antigen-presenting cells.16,17 However, the efficacy of immune-checkpoint therapy (ICT) is limited in CRC patients.18 Increasing results indicate that patients with mismatch repair deficient (dMMR) benefit from anti-PD-1 antibody (Ab) immunotherapy.19 Progression-free survival of CRC patients with high expression of PD-L1 and microsatellite instability-high (MSI-H) is significantly prolonged by anti-PD-1 antibody immunotherapy.20, 21, 22 However, as the MMR defect is only found in about 10% to 15% of CRC patients at stages II and III, the new effective approaches to cancer immunotherapy warrant further study.
In addition, alterations in noncoding RNAs (ncRNAs) such as long ncRNAs (lncRNAs) or microRNA (miRNA) could also contribute to multiple steps of the carcinogenesis process and can be used as predictive markers of patient outcome.23 lncRNAs are emerging as critical regulators of gene expression, including direct interactions with DNA, RNA, and proteins. Many studies including ours indicated that lncRNA GAS5,24 H19,25 and HOTAIR26 are dysregulated in CRC tissues and are associated with tumor progression and metastasis of CRC. lncRNAs might also have critical regulatory roles in various links of immune system development and function, especially in immune response.27 For example, lincR-Ccr2-5′AS is regulated by the T helper 2 (Th2) subset of Th cell transcription factor GATA-3, which upregulates numbers of key Th2 chemokines.28 linc-MAF-4 is reported specifically expressed in the Th1 cell and can negatively regulate transcription factor MAF to inhibit Th2 cell differentiation.29 Nevertheless, whether lncRNA mediates the expression of ICPis PD-1/PD-L1 remains unclear.
Here, we investigated the therapeutic implications of simvastatin in an anti-tumor immune response. We identified simvastatin as a new inhibitor of PD-L1 through repressing the expression of lncRNA SNHG29, suggesting the therapeutic implications of lncRNA SNHG29 for CRC patients.
Results
Simvastatin targets cholesterol biosynthesis rate-limiting enzyme HMGCR and reduces CRC-specific mortality
As cholesterol metabolism is vital for the progression of multi-cancers, we analyzed the primary rate-limiting enzyme in cholesterol biosynthesis via compiling The Human Protein Atlas database (http://www.proteinatlas.org). We found that HMGCR, the rate-limiting enzyme of the MVA pathway, which is referred to as the cholesterol biosynthesis pathway, was greatly upregulated in CRC (Figures 1A and 1B). Furthermore, we examined the expression of HMGCR by immunohistochemistry (IHC) staining on paraffin-embedded CRC specimens in cohort 1, which included 142 cases of CRC patients with clinical follow-up data collected at Sun Yat-sen Memorial Hospital. Results showed that the expression of HMGCR was significantly increased in the tumor tissues compared with the adjacent normal counterparts (Figures 1C and 1D; Figure S1). Furthermore, Kaplan-Meier analysis indicated that the higher HMGCR expression was correlated to poor OS of CRC patients (log rank = 4.158, p = 0.0414) (Figure 1E).
Figure 1.

Simvastatin targets HMGCR and reduces colorectal cancer (CRC)-specific mortality
(A and B) Exploring HMGCR mRNA and protein expression in most cancer categories by assembling The Human Protein Atlas (http://www.proteinatlas.org). (C) The expression of HMGCR by immunohistochemistry (IHC) staining on paraffin-embedded CRC specimens. (D) The expression levels of HMGCR in formalin-fixed paraffin-embedded (FFPE) colon cancers and normal tissues were showed in the indicated scattergram using ImageJ (n = 142). ∗∗∗p < 0.001. (E) Kaplan-Meier analysis of overall survival (OS) of CRC patients with HMGCR. (F) The mutually adjusted hazard ratios for the associations of characteristics with the risks of recurrence, CRC-specific mortality, and all-cause mortality from the DCCG database (2001–2011). (G) Kaplan-Meier analysis of OS in 415 CRC patients with and without statins therapy. (H) Cytotoxicity activity of simvastatin against CRC cell line. (I) The Gene Ontology analysis shows the enriched biological process of differentially expressed genes in the simvastatin-treated group (/fold change/ > 2; p value < 0.05). (J) Heatmap showing the difference transcripts between negative control and simvastatin-treated CRC cells (/fold change/ > 2; p value < 0.05).
Statin, the inhibitor of HMGCR, is a widely used drug in patients with hypercholesterolemia. The cohort from the Danish Colorectal Cancer Group (DCCG) database,30 a clinical registry of CRC patients with nearly complete overall mortality outcome and follow-up, demonstrated that the usage of statin reduced the hazard of death from CRC (Figure 1F). Furthermore, we analyzed the association between statin use and OS rates of CRC patients collected at Sun Yat-sen Memorial Hospital from January 2015 to December 2018 from cohort 2, which included 415 cases of CRC patients with clinical follow-up data. Kaplan-Meier analysis indicated that never-used statins the preceding year was related to poor OS in patients with CRC (log rank = 3.909, p = 0.048) (Figure 1G).
Simvastatin, a well-established HMGCR inhibitor, is recorded for safety and reduces cardiovascular risk at the maximal recommended dose of 80 mg/day. We added simvastatin at different concentrations into the culture medium of the CRC cell lines to study the inhibition efficiency of this drug against CRC tumor progression. The half-inhibitory concentration (IC50) was estimated, simultaneously employing all dose responses across different CRC cell lines. The dose-response curve was shown in Figure 1H, and the simvastatin drug dose was lower than the initiation-effective plasma concentration of patients in this study. We further performed mRNA sequencing to explore the function of simvastatin in CRC cell lines. The Gene Ontology analysis was used to identify the biological process of differentially expressed genes in the simvastatin-treated group (/fold change/ > 2; p value < 0.05), demonstrating that these genes were enriched for functions associated with the immune system process (Figure 1I; Table S1). Intriguingly, the expression of ICPi PD-L1 was significantly decreased in the simvastatin-treated group (Figure 1J). Collectively, these data suggested that the HMGCR inhibitor, simvastatin, reduces CRC-specific mortality and inhibits CRC cell expression of PD-L1.
Simvastatin represses tumor expression of PD-L1 and enhances cytotoxic T lymphocyte (CTL) infiltration in CRC patients
The mRNA-sequencing analysis showed that PD-L1 expression was decreased in simvastatin-treated CRC cells. Furthermore, qRT-PCR and western blotting assays showed that PD-L1 mRNA and protein level were significantly repressed in CRC cell lines or CRC tumor-derived xenograft tumor models treated with simvastatin compared with that from control groups (Figures 2A−2D). To determine whether simvastatin contributes to CTL exhaustion phenotype and function in vivo, a patient-derived tumor xenograft (PDX) model of CRC patients was obtained and analyzed (Figure 2E). Interestingly, simvastatin significantly decreased the tumor growth rate and the mean tumor volume compared to the negative control group (Figure 2F). Compared with that in negative group, the PD-L1 expression level was significantly decreased, and tumor-infiltrating CD8+ T cells had higher activation marker granzyme B expression in the simvastatin-treated PDX model of CRCs (Figures 2G and 2H). In addition, we examined PD-L1 expression and CD8+ T cells from the subcutaneous MC38 tumor model (Figure 2I). We found that the tumor volume and PD-L1 expression level were significantly repressed in the simvastatin-treated group compared to that in the negative group, and the combination treatment of simvastatin and PD-L1 antibody showed the enhanced inhibitory effects on tumor growth compared with simvastatin treatment alone (Figures 2J and 2K). When we sorted tumor-infiltrated CD45+ cells out and quantified tumor-infiltrating T cells and the immune-checkpoint expression level on CD8+ T cells, we found more CD8+ tumor-infiltrating T cells in the simvastatin-treated group than that in the negative control group. Analysis of the mouse peripheral blood mononuclear cell (PBMC) by flow cytometry also indicated that CD8+ T cells were significantly higher in the simvastatin-treated group than that in the control group (Figure 2L). Taken together, these results indicated that simvastatin inhibits tumor expression of PD-L1 and restores tumor-infiltrating CD8+ T cell function in the tumor microenvironment. Overall, these results implicate an unsuspected relevance of the MVA pathway inhibitor, simvastatin, in enhancing anti-tumor immunity, as well as a role for simvastatin as a new PD-L1 inhibitor.
Figure 2.

Simvastatin represses tumor expression of PD-L1 and enhances cytotoxic T lymphocyte (CTL) infiltration in CRC patients
(A) PD-L1 mRNA expression in HCT116 and RKO cells treated with simvastatin or negative control. (B) PD-L1 protein expression in HCT116 and RKO cells treated with simvastatin or negative control. (C) PD-L1 protein expression in xenograft tumors from HCT116 and RKO cells. (D) PD-L1 protein expression in xenograft tumors from RKO cells. (E) Graphic illustration of a human PDX model-based therapeutic study. PDX tumors were generated from CRC patients and treated with simvastatin or negative control. (F) In vivo analyses of tumor (left panel) and growth (right panel) in mice that were subcutaneously implanted with tumor tissues from CRC patients and treated with simvastatin or negative control (50 mg/kg) five times weekly for 3 weeks (n = 6). Results are presented as mean ± SD. ∗∗∗p < 0.001. (G) PD-L1 protein expression in the PDX model treated with simvastatin or negative control. (H) Representative immunohistochemical images were shown in randomly selected tumor. (I) Graphic illustration of C57BL/6 mice injected with 2 × 106 MC38 tumor cells. At day 7 after tumor inoculation, simvastatin or negative control was intragastric administrated into tumor-bearing mice (50 mg/kg, five times weekly for 3 weeks). Anti-PD-L1 antibody was intraperitoneally injected combined with simvastatin in addition (10 μg/g, two times weekly for 3 weeks). (J) In vivo analyses of tumors (upper panel) and their growth (bottom panel) in C57BL/6 mice that were subcutaneously injected with 2 × 106 MC38 tumor cells and treated with simvastatin or combined with anti-PD-L1 antibody (n = 6). Results are presented as mean ± SD. ∗∗∗p < 0.001. (K) PD-L1 protein expression in the C57BL/6 mice model treated with simvastatin or negative control. (L) CD8+ T cells in PBMC and TILs were analyzed by flow cytometry. Experiments were performed with at least three biological replicates and are representative of at least two independent experiments. The results are presented as mean ± SD. ∗∗p < 0.01, ∗∗∗p < 0.001.
Simvastatin inhibits tumor expression of lncRNA SNHG29, which promotes PD-L1 expression and tumorigenesis in vivo
Although we found that simvastatin plays a crucial role in inhibition of PD-L1, the underlying mechanism has yet to be elucidated. lncRNAs are involved in a variety of tumorigenic processes including immune response. We performed lncRNA sequencing to determine whether lncRNA mediates the function of simvastatin in CRC anti-tumor immunotherapy. We identified 39 lncRNAs that were significantly downregulated (≥2.0-fold) in CRC cells treated with simvastatin (Figure 3A; Table S2). To validate the data from RNA sequencing, the top ten promising lncRNA candidates (with annotated NR accession number) were detected using qRT-PCR in CRC cells with different concentrations of simvastatin (Figure 3B). Among these, lncRNA SNHG29, located in 17p11.2 with 7 exons and 3,206 bp in length, was highly expressed in CRC cells and significantly downregulated after simvastatin treatment, and the expression of PD-L1 was also impaired significantly (Figures 3C and 3D). We further transfected these small interfering RNAs (siRNAs) targeting lncRNA candidates into CRC cells and examined the expression of PD-L1. Among them, lncRNA SNHG29 knockdown significantly impaired PD-L1 expression, whereas overexpression of lncRNA SNHG29 enhanced PD-L1 expression (Figure 3E). Furthermore, we performed RNA in situ hybridization (ISH) and IHC in PDX models to study changes of lncRNA SNHG29 expression after simvastatin treatment. As expected, simvastatin inhibited lncRNA SNHG29 expression and Ki67 expression of tumor cells and enhanced CTL activity in vivo (Figure 3F). Collectively, these results indicated that simvastatin inhibits tumor expression of lncRNA SNHG29, which promotes PD-L1 expression and tumorigenesis in vivo.
Figure 3.

Simvastatin inhibits tumor expression of lncRNA SNHG29, which promotes PD-L1 expression
(A) lncRNA-sequencing analysis of simvastatin-treated and negative control cells is presented in a heatmap analysis. (B) Flow chart of screening-altered lncRNAs in indicated samples. (C) Schematic annotation of lncRNA SNHG29 genomic locus on chromosome 17 (p11.2) in humans. Green rectangles represent exons. (D) qRT-PCR analysis of differentially expressed lncRNA SNHG29 and PD-L1. (E) Western blotting tested PD-L1 level in lncRNA SNHG29-overexpressed or -knockdown CRC cells. (F) Representative ISH staining for SNHG29 and IHC staining for PD-L1, CD8, and Ki67 expression in the xenograft tumor tissues from CRC patients, treated with simvastatin or negative control (50 mg/kg) five times weekly for 3 weeks. The relative intensities of ISH and IHC staining were quantified by ImageJ software (n = 6). The density of immune cell infiltrates in the tumor was calculated as the number of positive cells per field of tissue. The results are presented as mean ± SD. ∗∗p < 0.01, ∗∗∗p < 0.001.
lncRNA SNHG29 knockdown enhances CTL killing targets in vitro and in vivo
lncRNA SNHG29 was downregulated in simvastatin-treated CRC cells and could upregulate PD-L1 expression, suggesting that lncRNA SNHG29 might serve as a molecular link between simvastatin and tumor-immune escape processes. Therefore, we investigated the effects of simvastatin treatment and lncRNA SNHG29 knockdown in CRC cells on the effector functions of tumor antigen-specific CD8+ T cells by cytotoxicity assays using the lactate dehydrogenase (LDH) release measurement. Either simvastatin or negative control was added to the cell culture medium of each CRC cell and incubated for 24 h, and then the tumor cells and mature monocyte-derived dendritic cell (Mo-DC; human leukocyte antigen [HLA]-DR+, CD86+, CD14−)-induced activated CTLs were co-cultured for 6 h to determine LDH activity (Figure 4A). The T cell subset (CD8+, CD69+, CD25+) was activated (Figure 4B), and Mo-DCs with typical DC phenotype were generated (Figure 4C) in vitro for the CTL assay. The CTL assay showed that simvastatin treatment increased killing effects of CTLs on tumor cells compared with control treatment (Figure 4D). Moreover, lncRNA SNHG29 knockdown also showed an increase cytotoxicity rate of CTLs toward tumor cells (Figure 4E).
Figure 4.

lncRNA-SNHG29 inhibition manifest enhances CTL killing targets in vitro and in vivo
(A) Graphic illustration of isolation, activation, and expansion of human cytotoxic T cells and Mo-DCs to perform cytotoxicity LDH assay. (B and C) Flow cytometry analysis of cell surface markers showed in vitro-generated and -activated CD8+ T cells (B) and mature Mo-DCs (C). (D and E) Cytotoxic activity of CD8+ T cells against CRC cells treated with simvastatin or negative control (D) and mock or lncRNA SNHG29-specific siRNAs (E). The graph shows percentage of specific killing of CRC cells with various effector-to-target (E:T) ratios measured by LDH assays. (F−H) Graphic illustration of a human PDX model-based therapeutic study. PDX tumors with negative control or SNHG29-specific shRNAs were generated from CRC patients (F). At day 7 after tumor inoculation, SNHG29-specific shRNAs or negative control were intratumor injected into tumor-bearing mice (109 TU, twice). Tumor volume was monitored every 7 days (G and H). (I) Representative ISH staining for SNHG29 and IHC staining for PD-L1, CD8, granzyme B, and Ki67 expression in the xenograft tumor tissues from CRC patients treated with SNHG29-specific shRNAs or negative control after 3 weeks. (J) The relative intensities of ISH and IHC staining were quantified by ImageJ software. The density of immune cell infiltrates in the tumor was calculated as the number of positive cells per field of tissue. All experiments were performed in triplicate, and results are presented as mean ± SD. ∗∗∗p < 0.001. (K) Tumor volume of PDX tumor tissues was monitored every 7 days with anti-PD-L1 antibody or immunoglobulin G (IgG) isotype intraperitoneal injection (10 μg/g, two times weekly for 3 weeks), and lncRNA SNHG29-specific shRNAs or negative control were intratumor injected (109 TU, twice) (n = 5). Results are presented as mean ± SD. ∗∗p < 0.05, ∗∗∗p < 0.001.
We next examined the role of lncRNA SNHG29 in tumorigenesis using PDX models of CRC, which retain the patient’s tumor microenvironment and heterogeneity (Figure 4F). In the PDX model, depletion of lncRNA SNHG29 by short hairpin (sh)RNA significantly reduced tumor growth (Figures 4G and 4H). In contrast to negative control, an sh-lncRNA SNHG29-treated tumor exhibited an elevated antigen-specific CD8+ T cell anti-tumor response (Figures 4I and 4J). Furthermore, the therapeutic efficacy of the PD-L1-checkpoint blockade was enhanced in the sh-lncRNA SNHG29 PDX mice model (Figure 4K), implicating lncRNA SNHG29 as a potential therapeutic target in anti-cancer immunotherapy. These results demonstrate that lncRNA SNHG29 induces immune evasion via PD-L1-dependent exhaustion of CD8+ T cells in vitro and in vivo.
lncRNA SNHG29 interacts with Yes-associated protein (YAP) to inhibit its phosphorylation and ubiquitination-mediated protein degradation
We next performed an RNA pulldown assay by mass spectrometry (MS) to identify lncRNA SNHG29-associated proteins that might be involved in regulation of PD-L1 expression. Among these proteins, the YAP protein was specifically enriched and confirmed by western blotting (Figure 5A; Table S3). Moreover, RNA immunoprecipitation (RIP) followed by qRT-PCR analysis of co-purified RNAs showed that lncRNA SNHG29 was specially enriched in YAP immunoprecipitates (Figure 5B). Further immunofluorescence staining showed the localization of YAP in different CRC cells (Figure S3). RNA fluorescent ISH (FISH) and an immunofluorescence co-staining assay showed that lncRNA SNHG29 was co-localized with the YAP protein in CRC cells (Figures S3 and S5). Further immunofluorescence staining and western blotting using cytoplasmic and nuclear protein fraction isolation showed that lncRNA SNHG29 promoted nuclear accumulation of YAP protein, whereas lncRNA SNHG29 knockdown arrested the YAP protein in the cytoplasm (Figures 5C and 5D). Western blotting showed that knockdown of lncRNA SNHG29 increased the level of YAP phosphorylation at serine 127 and reduced YAP target gene connective tissue growth factor (CTGF), whereas overexpression of lncRNA SNHG29 reduced YAP phosphorylation and increased CTGF expression (Figure 5E). It has been reported that phosphorylated YAP can be sequestered in the cytoplasm and subsequently degraded by a ubiquitin-proteasome system. The cycloheximide (CHX) chase assay was performed to detect whether lncRNA SNHG29 was related to YAP protein stability. Western blotting analysis showed that the half-life of the YAP protein was remarkably decreased in lncRNA SNHG29 knockdown CRC cells (Figure 5F). A ubiquitination assay showed a significant decrease in the polyubiquitinated YAP protein in lncRNA SNHG29-overexpressing cells and a significant increase in YAP ubiquitination in lncRNA SNHG29 knockdown cells (Figure 5G). Further mRNA-sequencing and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis demonstrated that the Hippo-YAP pathway was significantly enriched in simvastatin-treated CRC cells (Figure 5H; Table S4). Western blotting analysis showed that simvastatin led to YAP phosphorylation in Ser 127 and thus inhibited CTGF and PD-L1 expression, whereas serine/threonine-protein kinase LATS1, a pivotal protein in the Hippo-YAP pathway, was not changed (Figure 5I). Interestingly, an immunofluorescence staining assay indicated that simvastatin attenuated lncRNA SNHG29 and YAP protein nuclear accumulation, and lncRNA SNHG29 overexpression abolished decreasing accumulation of the nuclear YAP protein induced by simvastatin treatment (Figure 5J; Figure S4). Taken together, these results demonstrated that lncRNA SNHG29 inhibits YAP phosphorylation and ubiquitination-mediated protein degradation via facilitating YAP nuclear accumulation.
Figure 5.

lncRNA SNHG29 interacts with YAP to inhibit its phosphorylation and ubiquitination-mediated protein degradation
(A) lncRNA SNHG29-associated proteins identified by RNA pulldown and mass spectrometric analyses in HCT116 cells (upper panel). Western blot analysis of pulldown product (bottom panel). (B) RIP assays in HCT116 cells. qRT-PCR analysis of RIP (upper panel). Agarose electrophoresis of PCR products (bottom panel). Experiments were performed in triplicate, and data are presented as mean ± SD. ∗∗∗p < 0.001. (C and D) lncRNA SNHG29 facilities YAP nuclear accumulation as demonstrated by immunofluorescence staining (C) and western blot (D). (E) Silencing of SNHG29 caused downregulation of CTGF but increased phosphorylation of YAP in HCT116 cells. Overexpression of SNHG29 caused upregulation of CTGF but reduced YAP phosphorylation in DLD1 cells. (F) RKO cells were transfected with SNHG29-specific siRNAs or negative control and subjected to a cycloheximide (CHX) chase assay. Immunoblot detection of YAP (left panel); IB data were quantified using ImageJ software (right panel). After 24 h, CHX (10 μg/mL) was added to the cell culture medium, and incubation was continued for 0, 4, 8, or 12 h. Error bars indicate the mean ± SD. ∗∗∗p < 0.001. (G) Ubiquitination assays of CRC cells co-transfected YAP with SNHG29-specific siRNA (left) or SNHG29 plasmid (right). The bottom panels depict the input of the cell lysates. 24 h after transfection, 10 nM MG132 was added to the 1640 culture medium, and incubation was continued for 8 h. (H) KEGG enrichment analysis of pathways, which significantly enriched in simvastatin-treated CRC cells (adjusted p ≤ 0.05). (I) Western blot showed total and phosphorylated protein of YAP, LATS1, CTGF, and PD-L1 in RKO cells treated with simvastatin or negative control. (J) YAP localization as demonstrated by immunofluorescence staining. Cells were treated with simvastatin or negative control alone or co-cultured with SNHG29 plasmid for 24 h before fixation.
lncRNA SNHG29 promotes the expression of PD-L1 via facilitating YAP transcriptional activity
We used qRT-PCR and western blotting to analyze the expressions of YAP and PD-L1 in CRC cells. Notably, YAP expression was positively correlated with PD-L1 expression in CRC cells (Figures 6A and 6B). Furthermore, western blotting analysis showed that YAP knockdown remarkably decreased the expression of PD-L1, whereas YAP overexpression increased the PD-L1 expression significantly (Figure 6C). We used the PROMO database to predict the region from −2,000 to +100 bp upstream of the PD-L1 transcriptional start site containing the YAP binding site, and the chromatin immunoprecipitation (ChIP) assay in YAP-overexpressed CRC cells confirmed that YAP bound to the PD-L1 promoter region (Figure 6D). To determine the functionality of the predicted sites, we constructed the PD-L1 promoter luciferase (luc) reporter. The luciferase reporter assays indicated that the luciferase activity of the PD-L1 promoter was dramatically enhanced by YAP, whereas the luciferase activity was significantly inhibited by lncRNA SNHG29-specific siRNA co-transfected into CRC cells (Figure 6E). Further western blotting analysis indicated that SNHG29 overexpression promoted expression of CTGF and PD-L1, whereas YAP-specific siRNA abolished SNHG29-mediated upregulation of CTGF and PD-L1. Meanwhile, knockdown of SNHG29 inhibited expression of CTGF and PD-L1, whereas YAP overexpression abolished SNHG29 knockdown-mediated downregulation of CTGF and PD-L1 (Figure 6F). Overall, these findings demonstrated that lncRNA SNHG29 bound with YAP, sustained YAP nuclear localization, and promoted transcriptional activity of YAP. Together, our study showed that simvastatin impairs the binding of the transcription factor YAP to the PD-L1 promoter via inhibiting the expression of lncRNA SNHG29, thereby inhibiting PD-L1 transcription (Figure 6G).
Figure 6.

lncRNA SNHG29 promotes the expression of PD-L1 via facilitating YAP transcriptional activity
(A and B) qRT-PCR (A) and western blots analysis (B) showed a positive correlation between YAP and PD-L1 in various CRC cells. (C) IB detection using the indicated antibodies in CRC cells transfected with the indicated plasmid (left panel) or siRNAs (right panel). (D) Binding of YAP to the PD-L1 promoter was studied by the chromatin immunoprecipitation (ChIP) assay, and the coprecipitated DNA was subjected for analysis of PD-L1 by qRT-PCR (upper panel). The PCR procedures was shown by agarose gel electrophoresis (bottom panel). Experiments were performed in triplicate, and data are presented as mean ± SD. ∗∗∗p < 0.001. (E) Potential YAP binding sites in the human PD-L1 promoter between –2,000 and + 100 bp. YAP plasmid alone or with SNHG29-specific siRNA was transfected into HEK293 cells to detect the transcriptional activity of the PD-L1 promoter by a dual-luciferase reporter system. Experiments were performed in triplicate, and data are presented as mean ± SD. ∗∗p < 0.05, ∗∗∗p < 0.001. (F) IB detection using the indicated antibodies in CRC cells transfected with the indicated plasmid or siRNAs. Data shown represent three independent experiments. (G) The graphic illustration of simvastatin as a new inhibitor of PD-L1 to enhance anti-tumor immunity via inhibiting lncRNA-SNHG29-mediated YAP activation in CRC tumor progression. (H and I) The ISH staining of SNHG29 and IHC staining of YAP and PD-L1 in tumor tissues and adjacent normal tissues of CRC paraffin-embedded samples. (J) The expression levels of SNHG29, YAP, and PD-L1 in FFPE colon cancers and normal tissues were shown in an indicated scattergram using ImageJ (n = 163). ∗∗∗p < 0.001. (K) The correlation of expression of lncRNA SNHG29, YAP and PD-L1 in FFPE CRC tissues (n = 163 biologically independent samples); linear regression analysis.
lncRNA SNHG29 expression positively correlates with YAP and PD-L1 levels in tumors from a CRC patient
Because lncRNA SNHG29 functions closely with YAP activity to promote PD-L1 expression, we examined the expression of lncRNA SNHG29 by ISH staining in paraffin-embedded CRC specimens and subsequently categorized them as lncRNA SNHG29-low and lncRNA SNHG29-high groups by comparing their levels to the individual median. The ISH analysis and IHC staining in paraffin-embedded CRC specimens showed that the expression of lncRNA SNHG29, YAP, and PD-L1 was significantly increased in the tumor tissues compared with the adjacent tissues (p < 0.01) (Figures 6H and 6J). Among them, eighty-three tumor tissues (50.9%) exhibited a high expression, and the other 80 cases (49.1%) had low expression of lncRNA SNHG29. Eighty-seven of tumor tissues (53.4%) exhibited a high expression of YAP and one hundred (61.3%) exhibited a high expression of PD-L1. The relationship among these three markers and the clinicopathologic parameters of patients with CRC were analyzed (Tables S5−S7). Further, the expression of lncRNA SNHG29 was positively correlated with YAP and PD-L1 expressions in tumors from CRC patients (Figure 6I; Figure S1). In addition, correlations among the expressions of lncRNA SNHG29, YAP, and PD-L1 were analyzed with a Spearman’s rank correlation. The scatterplot showed a positive relationship between lncRNA SNHG29 and YAP (r2 = 0.853; p < 0.001) and YAP and PD-L1 (r2 = 0.585; p < 0.001) and a positive relationship between lncRNA SNHG29 and PD-L1 (r2 = 0.548; p < 0.001) in 163 CRC specimens (Figure 6K). Furthermore, we analyzed the association between lncRNA SNHG29 and OS rates of CRC patients in cohort 3, which included 94 cases of CRC patients with clinical follow-up data. Fifty-seven patients (60.6%) exhibited a higher expression of SNHG29 in tumor tissues, and the other 37 cases (39.4%) had lower expression of SNHG29 in tumor tissues. Kaplan-Meier analysis indicated that the higher SNHG29 expression was related to poor OS of CRC patients (log rank = 4.244, p = 0.0394). Kaplan-Meier and log-rank test analyses also suggested a positive correlation between the YAP expression and a significantly reduced OS rate of CRC patients (log rank = 6.467, p = 0.0110) (Figure S2).
Collectively, these results demonstrated that the expression of lncRNA SNHG29 is increased in CRC tissues compared with adjacent tissues, and higher expression of lncRNA SNHG29 is associated with elevated expressions of YAP and PD-L1 in tumor tissues from CRC patients. Elevated expression of lncRNA SNHG29 is a significant prognostic factor for poor OS in CRC patients, indicating that lncRNA SNHG29 may be a potential therapeutic target for cancer treatment.
Discussion
ICT has revolutionized the field of cancer therapy since ipilimumab, nivolumab, pembrolizumab, and atezolizumab were successively approved for clinical oncology. About 20% of patients who have not responded well to conventional chemoradiotherapy or target therapies have achieved satisfactory tumor remission and long-term survival after treatment with the ICPis such as PD-1/PD-L1 antibodies (nivolumab, pembrolizumab, and atezolizumab) or CTLA-4 antibodies (ipilimumab). However, the effect of ICT in the vast majority of treated individuals is still limited. Therefore, scientists are actively exploring and developing new therapeutic strategies to enhance the efficacy of tumor immunotherapy. Recently, the inhibition of PCSK9, a key protein in the regulation of cholesterol metabolism, was found to promote the response of tumors to ICT via increasing the expression of major histocompatibility complex (MHC) class I proteins on the tumor cell surface and promoting robust intratumoral infiltration of cytotoxic T cells,31 suggesting that targeting the lipid metabolism will be a promising approach for immunotherapy.
Cholesterol biosynthesis is an essential metabolic pathway for mammalian cell survival, and cholesterol-derived metabolites play vital roles in promoting tumorigenesis and suppressing immune responses.32, 33, 34, 35 Mammalian cells can synthesize cholesterol via the MVA pathway, which is upregulated in several cancers such as breast,36 hepatic,37 and CRC.38 Statins acting as inhibitors of the MVA pathway have been reported to reduce the cancer-specific mortality in CRC patients through their interactions with tumor cell proliferation or apoptosis.8 However, little is known about the functions of simvastatin in the regulation of immune checkpoints or lncRNA-mediated immunoregulation in cancer. A prospective observational study (UMIN: UMIN000021694) enrolled 73 patients who were treated with nivolumab for advanced non-small cell lung cancer (NSCLC) at the Tokyo Metropolitan Cancer and Infectious Diseases Center Komagome Hospital (Tokyo, Japan) between January 2016 and February 2017. The results demonstrated that a combination therapy of nivolumab and statins improves the survival rate of advanced NSCLC.39 However, the effects of statin treatment combined with PD-1/PD-L1 antibodies in CRC patients were still unclear. In this study, we identified simvastatin as a new inhibitor of PD-L1 through repressing the expression of lncRNA SNHG29. Mechanistically, we screened and verified that SNHG29 could interact with YAP to inhibit the ubiquitination-mediated protein degradation and promote the transcription of PD-L1. Our study demonstrated that combining simvastatin with anti-PD-L1 antibodies could enhance the ICT response and uncover a perspective therapy for future clinical trials in CRC patients.
Emerging studies demonstrate multiple and intricate regulation pathways of PD-L1 expression at the transcriptional, post-transcriptional, and protein level.40, 41, 42 For example, BRAFV600E that activates mitogen-activated protein kinase-extracellular signal-regulated kinase (MAPK-ERK) signaling could transcriptionally upregulate PD-L1 expression and enhance chemotherapy-induced apoptosis in CRC.43 In addition, a histone modifier, AT-rich interaction domain-containing protein 3B (ARID3B) promoted the stem-like features of CRC to escape the PD-1/PD-L1-checkpoint blockade.44 Interestingly, lncRNA SNHG14 was found to upregulate Zinc finger E-box binding homeobox 1 (ZEB1) and then transcriptionally activate SNHG14 and PD-L1 to promote the immune evasion of the diffuse large B cell lymphoma (DLBCL).45 Nevertheless, the underlying mechanism about lncRNAs and PD-L1 in CRC is still poorly characterized. Here, we identified a novel lncRNA SNHG29/YAP/PD-L1 axis, which was suppressed by statin treatment in CRC.
lncRNAs are involved in transcriptional, post-transcriptional, or epigenetic regulation through their interactions with DNA,46 RNA,47 and proteins.48,49 lncRNAs may shuttle between the nucleus and the cytoplasm to regulate signaling mediators that are critical for cancer development.50, 51, 52 In our study, we identified a new mechanism that lncRNA SNHG29 inhibits YAP phosphorylation and ubiquitination-mediated protein degradation via facilitating YAP nuclear accumulation. However, the mechanisms modulating the subcellular transport of the YAP protein in CRC progression still remain poorly understood. Intriguingly, our results showed that SNHG29 increased the total cholesterol production, whereas the expression of HMGCR was not altered with either overexpression or knockdown of SNHG29 (Figure S6). These findings suggest that the lncRNA SNHG29 may promote total cholesterol production through a mechanism that is independent of targeting the functions of HMGCR, which warrants further study in the future.
Herein, we found that that simvastatin inhibits PD-L1 expression and promotes anti-tumor immunity via suppressing the expression of lncRNA SNHG29. Mechanistically, SNHG29 interacts with YAP and inhibits phosphorylation and ubiquitination-mediated protein degradation of YAP, thereby facilitating downregulation of PD-L1 transcriptionally. Our study not only indicates lncRNA SNHG29 as a critical molecular link between simvastatin and PD-L1-dependent immune-escape processes but also suggests a potential therapeutic target for treating CRC.
Materials and methods
Human tissues management, ISH, IHC, and co-staining of RNA FISH and immune fluorescence
CRC tissues and the adjacent normal tissues were collected from Sun Yat-Sen Memorial Hospital from January 2015 to December 2018. The study was approved by the Ethics Committee of Sun Yat-Sen University, and informed consent was obtained from each patient. The locked nucleic acid (LNA)-modified oligonucleotide probe targeting lncRNA SNHG29 (5′-DiGN/TGATGCTGATTCTTCACGAAT/-3′DiG_N) was designed and synthesized at Exiqon (Vedbaek, Denmark). lncRNA SNHG29 expression was measured in paraffin-embedded CRC samples using the enhanced sensitive ISH Detection Kit (Boster, Wuhan, China) according to the manufacturer’s instructions.
For co-staining of RNA FISH and immune fluorescence, endogenous lncRNA SNHG29 expression in CRC cells was measured according to the manufacturer’s instructions of the enhanced sensitive ISH Detection Kit V (fluorescein isothiocyanate [FITC]) (Boster, Wuhan, China). In brief, the signal was detected by incubation with horseradish peroxidase (HRP)-conjugated anti-digoxigenin (DIG) antibodies. Then, the signals were amplified and stained using a strept avidin-biotin complex (SABC)-conjugated FITC kit, and then, cell samples were co-incubated overnight at 4°C with Alexa Fluor 647 anti-active YAP1 antibody. The images were acquired with a fluorescence microscope (LSM 800; Zeiss, Germany).
IHC was performed as we previously described.53,54 The ISH and IHC staining scores were evaluated by two individuals in a blinded fashion. The scoring system from 0 to 12 combined the intensity and percentage (signal: “0,” no staining; “1,” weak staining; “2,” intermediate staining; and “3,” strong staining; percentage: “0,” 0%; “1,” 1%–25%; “2,” 26%–50%; “3,” 51%–75%; “4,” >75%), which were used as we described previously. The median value of total staining scores was identified as the optimal cut-off value.
In vivo model
All animal studies were performed according to the NIH Guide for the Care and Use of Laboratory Animals, with the approval of the Institutional Animal Care and Use Committee of Sun Yat-Sen University.
For the subcutaneously injected tumor model, 2 × 106 human tumor cells or mice tumor cells were subcutaneously injected into the flanks of male BALB/c nude mice or C57BL/6 mice (age, 4–6 weeks; Guangdong Medical Laboratory Animal Center, China). For PDX models, fresh tumor tissues were obtained from CRC patients and cut into ∼0.5−1 mm. After subcutaneously injecting tumor tissues from CRC patients into the B-NDG mice (age, 4–6 weeks; Biocytogen, China), when the volume of xenograft reached 200 mm3, simvastatin (50 mg/kg, five times weekly) or sh-lncRNA SNHG29 (109 TU, twice) was administered. We monitored tumor growth for 4 weeks and performed ISH or IHC staining to measure indicated biomarkers.
Tissue dissociation, PBMC isolation, and flow cytometry
Tissue dissociation was automated using the gentleMACS Octo Dissociator according to the manufacturer’s instructions (Miltenyi Biotec, Bergisch Gladbach, Germany). Moreover, tumor-infiltrating leukocytes (TILs) were isolated by CD45 (TIL) MicroBeads. Briefly, the cell suspension was loaded onto a MACS column, and the magnetically retained CD45+ cells can be eluted as the positively selected cell fraction. Isolation of PBMCs used density centrifugation (Ficoll-Paque) (Solarbio Life Sciences, China) according to the manufacturer’s instructions.
We used flow cytometry to detect and quantify TILs and PBCM T cell subpopulations. For surface staining, cells were incubated with indicated antibody and analyzed within 30 min. For intracellular cytokine staining, stimulation by activation cocktail with GolgiPlug is needed before the fixation and permeabilization steps. Viability was assessed by staining with Fixable Viability Stain (FVS) Reagent FVS510. All flow cytometry was performed on a BD Fortessa flow cytometer. Analysis of flow cytometry data was performed using FlowJo (Tree Star).
The following antibodies were used in this study: CD45-phycoerythrin-cyanine 7 (PE-Cy7; Becton Dickinson [BD]; lot #552848), CD3-BUV395 (BD; lot #563565), CD4-Percp-cy5.5 (BioLegend; lot #100434), CD8-BB515 (BD; lot #564422), PD1-BV650 (BD; lot #744546), granzyme B-BV421 (BioLegend; lot #396414), CD14-FITC (Invitrogen; lot #11-0149-42), HLA-DR-BV605 (BD; lot #562845), CD86-PerCP-Cy5.5 (BioLegend; lot #305420), and CD11c-PE (Invitrogen; lot #12-0116-42).
Generation of Mo-DCs as stimulator cells
PBMCs were obtained from healthy donors and isolated using density centrifugation (Ficoll-Paque) (Solarbio Life Sciences, China) according to the manufacturer’s instructions. Monocytes were enriched with anti-CD14 MicroBeads according to the manufacturer’s protocol (Miltenyi Biotec, Bergisch Gladbach, Germany). Monocytes were differentiated in complete culture medium containing RPMI 1640 with 10% fetal bovine serum (FBS), 100 ng/mL granulocyte macrophage-colony-stimulating factor (GM-CSF; PeproTech, Cranbury, NJ, USA), and 50 ng/mL interleukin (IL)-4 (PeproTech, Cranbury, NJ, USA) for 6 days. DC maturation was induced by addition of 50 ng/mL tumor necrosis factor (TNF)-α (PeproTech, Cranbury, NJ, USA) for 1 day. Mature DCs (mDCs; HLA-DR+, CD86+, CD14−) were seeded into 6-well plates and incubated with tumor lysates for 1 day to stimulate mDC antigen presentation.
Isolation, activation, and expansion of CD8+ CTLs from PBMCs
CTLs were sorted from PBMCs by anti-CD8 MicroBeads according to the manufacturer’s protocol (Miltenyi Biotec, Bergisch Gladbach, Germany). In vitro stimulation of purified T cells used the T Cell Transact for 3 days according to the manufacturer’s protocol (Miltenyi Biotec, Bergisch Gladbach, Germany). Activated T cells were expanded in complete culture medium containing RPMI 1640 with 10% FBS and 50 IU/mL IL-2 (Miltenyi Biotec, Bergisch Gladbach, Germany) for 3 days. Incubated mDCs were used as antigen-presenting cells to stimulate CTLs for another 8 days. At day 14, they proceeded to downstream application.
In vitro LDH cytotoxicity assay
An LDH assay was performed to evaluate the specific cytotoxic effects of CTLs on CRC cells. CRC cells were used as target cells and seeded into 6-well culture plates at density of 1 × 104 cells/well for 24 h. We seeded activated CTLs into 6-wells in complete RPMI 1640. DC-induced activated CTLs were used as effector cells and added into target cells at variable effector to target (E:T) ratios (0:1, 10:1, 20:1, and 40:1) to conduct the LDH assay. After incubation for 6 h at 37°C in a 5% CO2 incubator, cell lysates were collected to test the amount of LDH according to the manufacturer’s instructions (Dojindo, Shanghai, China). The percentage of lysis was calculated using the following formula: 100% × ([experimental release − effector spontaneous release − target effector spontaneous release]/[target maximum release − target spontaneous release]).
Cell lines, cell culture, and transfection
CRC cell lines including HCT116, RKO, DLD1, LOVO, SW480, LS174T, SW620, and HT29 were obtained from the Cell Bank of Type Culture Collection (Guangzhou Cellcook Biotech, Guangzhou, China). Short tandem repeat (STR) identification was performed to show the cell line authentication with the ATCC STR database. The cell culture and transfection were performed as we previously described. Detailed descriptions of oligonucleotide sequences, primers, and antibodies can be found in Table S8.
RNA sequencing
RNA sequencing was performed to explore the function of simvastatin in the CRC cells. Total RNA was extracted using TRIzol reagent (Takara Bio, Dalian, China). The Agilent 2100 Bioanalyzer and NanoDrop 2000 were used to analyze the total RNA quality and quantity. Then rRNAs were removed to retain mRNAs and ncRNAs. The enriched mRNAs and ncRNAs were fragmented into short fragments by using fragmentation buffer and reverse transcripted into cDNA with random primers. The cDNA libraries were sequenced on the Illumina sequencing platform by Genedenovo Biotechnology (Guangzhou, China). Transcript abundances were quantified by software RSEM. The transcript expression level was normalized by using the FPKM (fragments per kilobase of transcript per million mapped reads) method. Therefore, the calculated transcript expression can be directly used for comparing the difference of transcript expression among samples.
RNA pulldown and RIP assay
For RNA pulldown, biotin-labeled lncRNA SNHG29 was transcribed in vitro with the Biotin RNA Labeling Mix (Roche) and T7 RNA polymerase (Roche) as we described previously. Biotinylated RNA was then mixed with streptavidin magnetic beads (Life Technologies, Gaithersburg, MD, USA) at 4°C for 16 h. Then cell lysates were added into the binding reaction, the RNA-protein binding mixture was incubated for 1 h, and the eluted proteins were detected by MS or western blotting.
For RIP assay, Magna RIP RNA Binding Protein Immunoprecipitation Kit (Millipore, Billerica, MA, USA) was used as we previously described. Briefly, magnetic beads were pre-incubated with indicated antibody, added into tumor cell lysates, and incubated at 4°C overnight. Then, purified RNA was eluted and quantified by qRT-PCR or RNA sequencing.
CHX chase and ubiquitination assay
For CHX chase, the YAP plasmids and lncRNA SNHG29-specific siRNAs were transiently transfected into CRC cells using jetPRIME according to the manufacturer’s instructions (Polyplus, Strasbourg, France). 24 h later, CHX (10 μg/mL) was added to the complete culture medium containing RPMI 1640 with 10% FBS, and incubation was continued for 0, 8, 12, or 24 h. Then, the cell lysates were submitted to western blotting, and analysis data were quantified by ImageJ software.
For ubiquitination assay, ubiquitin, YAP, and lncRNA SNHG29-specific siRNAs were transiently transfected into CRC cells using jetPRIME according to the manufacturer’s instructions (Polyplus, Strasbourg, France). 8 h later, 10 nM MG132 was added to the complete culture medium containing RPMI 1640 with 10% FBS, and incubation was continued for 8 h. Then, magnetic beads pre-incubated with indicated antibody were added into tumor cell lysates and incubated at 4°C overnight with rotation, and eluted proteins were detected by western blotting.
ChIP and dual-luciferase reporter assay
We performed the ChIP assay using the SimpleChIP Enzymatic Chromatin IP Kit according to the manufacturer’s instructions (Cell Signaling Technology, Beverly, MA, USA). As we previously described, tumor cells were fixed with formaldehyde and then harvested and chromatin fragmented by digestion. Finally, the fragmented chromatin is subjected to immunoprecipitation using YAP antibodies to detect the particular YAP-binding DNA sequences.
For dual-luciferase reporter assay, Dual-Glo Luciferase Assay System Kit (Promega, Madison, WI, USA) was used following the manufacturer’s instructions. A fragment from −2,000 to + 100 relative to the transcription start site of the PD-L1 genomic sequence of PD-L1 (−2,000/+100)-luciferase was fused to pGL3-basic vector. Briefly, we transfected YAP plasmids with constructed PD-L1 promoter-luciferase vector (0.4 μg per well) and pRL-TK vector (20 ng per well) for 24 h. The pGL3-basic vector was transfected as a negative control. 24 h later, prepared cell lysates were used to measure the luciferase activity on a Spark multimode microplate reader (Tecan, Mannedorf, Switzerland) as we previously described.
Statistical analysis
All statistical analyses in this study were carried out using SPSS 19.0 software. The data are presented as mean ± SD. The significance of mean values between two groups was analyzed by Student’s t test (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001). Pearson correlation analysis was performed to determine the correlation among lncRNA SNHG29, YAP, and PD-L1 expression. Pearson’s chi-square test was used to analyze the clinical variables. Kaplan-Meier survival analysis was utilized to compare CRC patient survival based on lncRNA SNHG29 expression by log-rank test. A p value < 0.05 was considered a significant difference. Each experiment was repeated independently with similar results at least three times.
Data availability
The RNA-sequencing data discussed in this paper have been deposited in NCBI Sequence Read Archive under accession and are accessible through SRA: SRP313172. The data will become public when this manuscript is published online.
Acknowledgments
This work was supported by National Key R&D Program of China (2017YFC1309000), National Natural Science Foundation of China (grant numbers U1801282, 81802382, and 81702322), Guangzhou Science and Technology Plan Projects (Health Medical Collaborative Innovation Program of Guangzhou; 201803040019), Key-Area Research and Development Program of Guangdong Province (2019B020229002), and Guangdong Basic and Applied Basic Research Foundation (2021A1515010224).
Author contributions
J.L. conducted and designed the research. W.N., H.M., Y. Liu, and Y.X. jointly performed most of the experiments. C.Q., Yuqing Li, and A.Z. contributed to the clinical sample collection and pathological analysis. Yuhui Li, R.Z., and L.C. contributed to immunohistochemistry analysis. S.Y., Y.Z., and J.H. performed the animal study. W.N. analyzed and interpreted the data and drafted the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.05.012.
Supplemental information
References
- 1.Dekker E., Tanis P.J., Vleugels J.L.A., Kasi P.M., Wallace M.B. Colorectal cancer. Lancet. 2019;394:1467–1480. doi: 10.1016/S0140-6736(19)32319-0. [DOI] [PubMed] [Google Scholar]
- 2.Punt C.J., Koopman M., Vermeulen L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat. Rev. Clin. Oncol. 2017;14:235–246. doi: 10.1038/nrclinonc.2016.171. [DOI] [PubMed] [Google Scholar]
- 3.Farooqi A.A., de la Roche M., Djamgoz M.B.A., Siddik Z.H. Overview of the oncogenic signaling pathways in colorectal cancer: Mechanistic insights. Semin. Cancer Biol. 2019;58:65–79. doi: 10.1016/j.semcancer.2019.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Crockett S.D., Nagtegaal I.D. Terminology, Molecular Features, Epidemiology, and Management of Serrated Colorectal Neoplasia. Gastroenterology. 2019;157:949–966.e4. doi: 10.1053/j.gastro.2019.06.041. [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Mullen P.J., Yu R., Longo J., Archer M.C., Penn L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer. 2016;16:718–731. doi: 10.1038/nrc.2016.76. [DOI] [PubMed] [Google Scholar]
- 7.Huang B., Song B.L., Xu C. Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat. Metab. 2020;2:132–141. doi: 10.1038/s42255-020-0174-0. [DOI] [PubMed] [Google Scholar]
- 8.Nielsen S.F., Nordestgaard B.G., Bojesen S.E. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 2012;367:1792–1802. doi: 10.1056/NEJMoa1201735. [DOI] [PubMed] [Google Scholar]
- 9.Mantha A.J., Hanson J.E.L., Goss G., Lagarde A.E., Lorimer I.A., Dimitroulakos J. Targeting the mevalonate pathway inhibits the function of the epidermal growth factor receptor. Clin. Cancer Res. 2005;11:2398–2407. doi: 10.1158/1078-0432.CCR-04-1951. [DOI] [PubMed] [Google Scholar]
- 10.Ingallina E., Sorrentino G., Bertolio R., Lisek K., Zannini A., Azzolin L., Severino L.U., Scaini D., Mano M., Mantovani F. Mechanical cues control mutant p53 stability through a mevalonate-RhoA axis. Nat. Cell Biol. 2018;20:28–35. doi: 10.1038/s41556-017-0009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Young K., Lawlor R.T., Ragulan C., Patil Y., Mafficini A., Bersani S., Antonello D., Mansfield D., Cingarlini S., Landoni L. Immune landscape, evolution, hypoxia-mediated viral mimicry pathways and therapeutic potential in molecular subtypes of pancreatic neuroendocrine tumours. Gut. 2020 doi: 10.1136/gutjnl-2020-321016. Published online September 3, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pollack S.M., Ingham M., Spraker M.B., Schwartz G.K. Emerging Targeted and Immune-Based Therapies in Sarcoma. J. Clin. Oncol. 2018;36:125–135. doi: 10.1200/JCO.2017.75.1610. [DOI] [PubMed] [Google Scholar]
- 13.Rowshanravan B., Halliday N., Sansom D.M. CTLA-4: a moving target in immunotherapy. Blood. 2018;131:58–67. doi: 10.1182/blood-2017-06-741033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu-Monette Z.Y., Zhou J., Young K.H. PD-1 expression and clinical PD-1 blockade in B-cell lymphomas. Blood. 2018;131:68–83. doi: 10.1182/blood-2017-07-740993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen L., Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J. Clin. Invest. 2015;125:3384–3391. doi: 10.1172/JCI80011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marabelle A., Tselikas L., de Baere T., Houot R. Intratumoral immunotherapy: using the tumor as the remedy. Ann. Oncol. 2017;28(Suppl 12):xii33–xii43. doi: 10.1093/annonc/mdx683. [DOI] [PubMed] [Google Scholar]
- 17.Liu B., Song Y., Liu D. Recent development in clinical applications of PD-1 and PD-L1 antibodies for cancer immunotherapy. J. Hematol. Oncol. 2017;10:174. doi: 10.1186/s13045-017-0541-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le D.T., Uram J.N., Wang H., Bartlett B.R., Kemberling H., Eyring A.D., Skora A.D., Luber B.S., Azad N.S., Laheru D. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chalabi M., Fanchi L.F., Dijkstra K.K., Van den Berg J.G., Aalbers A.G., Sikorska K., Lopez-Yurda M., Grootscholten C., Beets G.L., Snaebjornsson P. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat. Med. 2020;26:566–576. doi: 10.1038/s41591-020-0805-8. [DOI] [PubMed] [Google Scholar]
- 20.Kawazoe A., Kuboki Y., Shinozaki E., Hara H., Nishina T., Komatsu Y., Yuki S., Wakabayashi M., Nomura S., Sato A. Multicenter Phase I/II Trial of Napabucasin and Pembrolizumab in Patients with Metastatic Colorectal Cancer (EPOC1503/SCOOP Trial) Clin. Cancer Res. 2020;26:5887–5894. doi: 10.1158/1078-0432.CCR-20-1803. [DOI] [PubMed] [Google Scholar]
- 21.Chen E.X., Jonker D.J., Loree J.M., Kennecke H.F., Berry S.R., Couture F., Ahmad C.E., Goffin J.R., Kavan P., Harb M. Effect of Combined Immune Checkpoint Inhibition vs Best Supportive Care Alone in Patients With Advanced Colorectal Cancer: The Canadian Cancer Trials Group CO.26 Study. JAMA Oncol. 2020;6:831–838. doi: 10.1001/jamaoncol.2020.0910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luchini C., Bibeau F., Ligtenberg M.J.L., Singh N., Nottegar A., Bosse T., Miller R., Riaz N., Douillard J.Y., Andre F., Scarpa A. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD-1/PD-L1 expression and tumour mutational burden: a systematic review-based approach. Ann. Oncol. 2019;30:1232–1243. doi: 10.1093/annonc/mdz116. [DOI] [PubMed] [Google Scholar]
- 23.Jung G., Hernández-Illán E., Moreira L., Balaguer F., Goel A. Epigenetics of colorectal cancer: biomarker and therapeutic potential. Nat. Rev. Gastroenterol. Hepatol. 2020;17:111–130. doi: 10.1038/s41575-019-0230-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ni W., Yao S., Zhou Y., Liu Y., Huang P., Zhou A., Liu J., Che L., Li J. Long noncoding RNA GAS5 inhibits progression of colorectal cancer by interacting with and triggering YAP phosphorylation and degradation and is negatively regulated by the m6A reader YTHDF3. Mol. Cancer. 2019;18:143. doi: 10.1186/s12943-019-1079-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakagawa H., Chadwick R.B., Peltomaki P., Plass C., Nakamura Y., de La Chapelle A. Loss of imprinting of the insulin-like growth factor II gene occurs by biallelic methylation in a core region of H19-associated CTCF-binding sites in colorectal cancer. Proc. Natl. Acad. Sci. USA. 2001;98:591–596. doi: 10.1073/pnas.011528698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kogo R., Shimamura T., Mimori K., Kawahara K., Imoto S., Sudo T., Tanaka F., Shibata K., Suzuki A., Komune S. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011;71:6320–6326. doi: 10.1158/0008-5472.CAN-11-1021. [DOI] [PubMed] [Google Scholar]
- 27.Atianand M.K., Caffrey D.R., Fitzgerald K.A. Immunobiology of Long Noncoding RNAs. Annu. Rev. Immunol. 2017;35:177–198. doi: 10.1146/annurev-immunol-041015-055459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu G., Tang Q., Sharma S., Yu F., Escobar T.M., Muljo S.A., Zhu J., Zhao K. Expression and regulation of intergenic long noncoding RNAs during T cell development and differentiation. Nat. Immunol. 2013;14:1190–1198. doi: 10.1038/ni.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ranzani V., Rossetti G., Panzeri I., Arrigoni A., Bonnal R.J., Curti S., Gruarin P., Provasi E., Sugliano E., Marconi M. The long intergenic noncoding RNA landscape of human lymphocytes highlights the regulation of T cell differentiation by linc-MAF-4. Nat. Immunol. 2015;16:318–325. doi: 10.1038/ni.3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lash T.L., Riis A.H., Ostenfeld E.B., Erichsen R., Vyberg M., Ahern T.P., Thorlacius-Ussing O. Associations of Statin Use With Colorectal Cancer Recurrence and Mortality in a Danish Cohort. Am. J. Epidemiol. 2017;186:679–687. doi: 10.1093/aje/kww245. [DOI] [PubMed] [Google Scholar]
- 31.Liu X., Bao X., Hu M., Chang H., Jiao M., Cheng J., Xie L., Huang Q., Li F., Li C.Y. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature. 2020;588:693–698. doi: 10.1038/s41586-020-2911-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chimento A., Casaburi I., Avena P., Trotta F., De Luca A., Rago V., Pezzi V., Sirianni R. Cholesterol and Its Metabolites in Tumor Growth: Therapeutic Potential of Statins in Cancer Treatment. Front. Endocrinol. (Lausanne) 2019;9:807. doi: 10.3389/fendo.2018.00807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cruz P.M., Mo H., McConathy W.J., Sabnis N., Lacko A.G. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: a review of scientific findings, relevant to future cancer therapeutics. Front. Pharmacol. 2013;4:119. doi: 10.3389/fphar.2013.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wen Y.A., Xiong X., Zaytseva Y.Y., Napier D.L., Vallee E., Li A.T., Wang C., Weiss H.L., Evers B.M., Gao T. Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis. 2018;9:265. doi: 10.1038/s41419-018-0330-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma X., Bi E., Lu Y., Su P., Huang C., Liu L., Wang Q., Yang M., Kalady M.F., Qian J. Cholesterol Induces CD8+ T Cell Exhaustion in the Tumor Microenvironment. Cell Metab. 2019;30:143–156.e5. doi: 10.1016/j.cmet.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Borgquist S., Djerbi S., Pontén F., Anagnostaki L., Goldman M., Gaber A., Manjer J., Landberg G., Jirström K. HMG-CoA reductase expression in breast cancer is associated with a less aggressive phenotype and influenced by anthropometric factors. Int. J. Cancer. 2008;123:1146–1153. doi: 10.1002/ijc.23597. [DOI] [PubMed] [Google Scholar]
- 37.Sutter A.P., Maaser K., Höpfner M., Huether A., Schuppan D., Scherübl H. Cell cycle arrest and apoptosis induction in hepatocellular carcinoma cells by HMG-CoA reductase inhibitors. Synergistic antiproliferative action with ligands of the peripheral benzodiazepine receptor. J. Hepatol. 2005;43:808–816. doi: 10.1016/j.jhep.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 38.Caruso M.G., Notarnicola M., Santillo M., Cavallini A., Di Leo A. Enhanced 3-hydroxy-3-methyl-glutaryl coenzyme A reductase activity in human colorectal cancer not expressing low density lipoprotein receptor. Anticancer Res. 1999;19(1A):451–454. [PubMed] [Google Scholar]
- 39.Omori M., Okuma Y., Hakozaki T., Hosomi Y. Statins improve survival in patients previously treated with nivolumab for advanced non-small cell lung cancer: An observational study. Mol. Clin. Oncol. 2019;10:137–143. doi: 10.3892/mco.2018.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He X., Xu C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020;30:660–669. doi: 10.1038/s41422-020-0343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun C., Mezzadra R., Schumacher T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity. 2018;48:434–452. doi: 10.1016/j.immuni.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Payandeh Z., Khalili S., Somi M.H., Mard-Soltani M., Baghbanzadeh A., Hajiasgharzadeh K., Samadi N., Baradaran B. PD-1/PD-L1-dependent immune response in colorectal cancer. J. Cell. Physiol. 2020;235:5461–5475. doi: 10.1002/jcp.29494. [DOI] [PubMed] [Google Scholar]
- 43.Feng D., Qin B., Pal K., Sun L., Dutta S., Dong H., Liu X., Mukhopadhyay D., Huang S., Sinicrope F.A. BRAFV600E-induced, tumor intrinsic PD-L1 can regulate chemotherapy-induced apoptosis in human colon cancer cells and in tumor xenografts. Oncogene. 2019;38:6752–6766. doi: 10.1038/s41388-019-0919-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liao T.T., Lin C.C., Jiang J.K., Yang S.H., Teng H.W., Yang M.H. Harnessing stemness and PD-L1 expression by AT-rich interaction domain-containing protein 3B in colorectal cancer. Theranostics. 2020;10:6095–6112. doi: 10.7150/thno.44147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao L., Liu Y., Zhang J., Liu Y., Qi Q. LncRNA SNHG14/miR-5590-3p/ZEB1 positive feedback loop promoted diffuse large B cell lymphoma progression and immune evasion through regulating PD-1/PD-L1 checkpoint. Cell Death Dis. 2019;10:731. doi: 10.1038/s41419-019-1886-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Böhmdorfer G., Wierzbicki A.T. Control of Chromatin Structure by Long Noncoding RNA. Trends Cell Biol. 2015;25:623–632. doi: 10.1016/j.tcb.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomson D.W., Dinger M.E. Endogenous microRNA sponges: evidence and controversy. Nat. Rev. Genet. 2016;17:272–283. doi: 10.1038/nrg.2016.20. [DOI] [PubMed] [Google Scholar]
- 48.Lin C., Yang L. Long Noncoding RNA in Cancer: Wiring Signaling Circuitry. Trends Cell Biol. 2018;28:287–301. doi: 10.1016/j.tcb.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ni W., Zhang Y., Zhan Z., Ye F., Liang Y., Huang J., Chen K., Chen L., Ding Y. A novel lncRNA uc.134 represses hepatocellular carcinoma progression by inhibiting CUL4A-mediated ubiquitination of LATS1. J. Hematol. Oncol. 2017;10:91. doi: 10.1186/s13045-017-0449-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen L.L. Linking Long Noncoding RNA Localization and Function. Trends Biochem. Sci. 2016;41:761–772. doi: 10.1016/j.tibs.2016.07.003. [DOI] [PubMed] [Google Scholar]
- 51.Lin A., Hu Q., Li C., Xing Z., Ma G., Wang C., Li J., Ye Y., Yao J., Liang K. The LINK-A lncRNA interacts with PtdIns(3,4,5)P3 to hyperactivate AKT and confer resistance to AKT inhibitors. Nat. Cell Biol. 2017;19:238–251. doi: 10.1038/ncb3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin A., Li C., Xing Z., Hu Q., Liang K., Han L., Wang C., Hawke D.H., Wang S., Zhang Y. The LINK-A lncRNA activates normoxic HIF1α signalling in triple-negative breast cancer. Nat. Cell Biol. 2016;18:213–224. doi: 10.1038/ncb3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yao S., Zheng P., Wu H., Song L.M., Ying X.F., Xing C., Li Y., Xiao Z.Q., Zhou X.N., Shen T. Erbin interacts with c-Cbl and promotes tumourigenesis and tumour growth in colorectal cancer by preventing c-Cbl-mediated ubiquitination and down-regulation of EGFR. J. Pathol. 2015;236:65–77. doi: 10.1002/path.4502. [DOI] [PubMed] [Google Scholar]
- 54.Wu H., Yao S., Zhang S., Wang J.R., Guo P.D., Li X.M., Gan W.J., Mei L., Gao T.M., Li J.M. Elevated expression of Erbin destabilizes ERα protein and promotes tumorigenesis in hepatocellular carcinoma. J. Hepatol. 2017;66:1193–1204. doi: 10.1016/j.jhep.2017.01.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-sequencing data discussed in this paper have been deposited in NCBI Sequence Read Archive under accession and are accessible through SRA: SRP313172. The data will become public when this manuscript is published online.