

Abstract
Protein aggregation is a feature of numerous neurodegenerative diseases. However, regulated, often reversible formation of protein aggregates, also known as condensates, helps control a wide range of cellular activities including stress response, gene expression, memory, cell development and differentiation. This review presents examples of aggregates found in biological systems, how they are used, and cellular strategies that control aggregation and disaggregation. We include features of the aggregating proteins themselves, environmental factors, co-aggregates, post-translational modifications and well-known aggregation-directed activities that influence their formation, material state, stability and dissolution. We highlight the emerging roles of biomolecular condensates in early animal development, and disaggregation processing proteins that have recently been shown to play key roles in gametogenesis and embryogenesis.
Keywords: biomolecular condensate, Amyloid, chaperone, ABCF gene family, RuvBL gene family
Graphical Abstract
Introduction: Order and Disorder as Natural and Dynamic Cellular States
Cellular aggregation has long been equated with current or impending health issues. However, accumulating evidence suggests that aggregation is important physiologically and is not exclusively pathological. Although native three-dimensional structure of proteins is largely encoded by the primary amino acid sequence, molecular chaperones help ensure co- and post-translational quality by promoting proper protein conformation as intracellular and extracellular environments change. If proteins fail to fold as predicted or assume an alternative fold, they may assemble into aggregates. Some of these aggregates, whether generated by exposure to stress, concentration-dependent precipitation, the failure of co-translational directed folding or modifications associated with the ribosome [1], are recognized as aberrant and either selected for degradation or refolding. In response to an elevated aggregate load, the cell may invoke degradation pathways, place the aggregates into storage compartments, or recover the aggregating proteins. For the latter process, chaperones known as disaggregases are employed. The role of disaggregase type chaperones is to establish a suitable balance between the aggregation process and protein solubilization and refolding.
Disaggregases are also important for processing another class of proteins, those that form ordered aggregates independent of stress. Ordered aggregation includes the formation of cross-ß structure that can stiffen condensates or form long unbranched fibers known as amyloids. Amyloids that form as the result of an underlying genomic change are frequently pathogenic and are characteristic of several neurodegenerative disorders including Huntington’s, Parkinson’s, Alzheimer’s and prion-related diseases.
However, the capacity to form amyloid or other ordered aggregate forms is an intrinsic property of many proteins [2, 3] and is not necessarily pathological. There are an increasing number of cases where aggregate formation is fundamental to essential biological activities. In recent years many of the biological “bodies”, “plasm”, ‘speckles’ and ‘non-membrane bound organelles’ identified by microscopic inspection by cell and developmental biologists have been shown to form through selective protein and nucleic acid aggregation. We and others have uncovered novel aggregates in the earliest stages of development, including the oocyte, embryo and gametogonia [4–13]. The functional significance of these previously unappreciated aggregate bodies is suggested by genetic perturbations that affect the aggregation phenotype while simultaneously impacting development [14].
The field of biological and pathological protein aggregation and the various mechanism that influence it is vast, and we regret not being able to do justice to every aspect of it. Some interesting and important topics related but not central to the main thrust of this review include the crucial role of yeast models in the study of disease-related amyloids [15], the full scope of functional amyloid examples in mammalian and plant systems [16–19], and growing evidence of the involvement of liquid-liquid phase separation in forming well-recognized as well as novel membraneless organelles that comprise a fundamental organizing principle in living cells ([20–25]), to name a few.
The goal of this review is to explore the topic of biological (as opposed to pathological) aggregation with special emphasis on the ways condensate formation, maintenance and dissolution can be influenced, highlighting examples from cell and developmental biology. The review begins with some fundamentals including a brief overview of various material states exhibited by biological aggregates, the distinction between ‘structural’ and ‘storage’ aggregates, the need for storage aggregate to be reversible, and a description of molecular chaperones whose enzymatic disaggregation activities are very well characterized. These enzymatic activities are paired with a discussion of the many non-chaperone activities that are central to regulation of biological aggregates including features of the aggregation-prone proteins themselves, as well as their regulators and modifiers. All of this sets the stage for a review of the subset of aggregates that are important in different aspects of animal development, and finally the search for aggregate processing activities specifically linked to development.
Diversity of Material States
Figure 1A provides a simplified framework representing a roster of states from liquid droplets, the most dynamic, through the formation of amyloid fibers, the most solid. The material states are depicted as part of an equilibrium that is influenced by protein (or RNA) concentration, co-aggregate complexity, modification, environment and varying levels of enzymatic activity. The phrase “biological condensate” was recently coined [26] as way of describing all non-stoichiometric assemblies of biomolecules that inhabit this broad spectrum of aggregate types without regard to stability or material state. Importantly, biomolecular condensate are membraneless. The constituent biological polymers in a condensate may undergo self-assembly via clustering that increases the local concentration of the assembling components. In living organisms, proteins, RNA and other polymers can form biomolecular condensates via liquid-liquid phase separation (LLPS) to generate colloidal emulsions or liquid crystals, in which material may flow like a liquid but have a crystalline molecular structure; or by liquid-solid phase separation to generate gels, sols or suspensions within cells or as extracellular secretions. In that the phrase ‘condensate’ makes no assumptions about either the physical mechanism through which assemblies are achieved, nor the material state of the resulting assembly, we have opted to use the term “condensate” throughout this review when information suggesting a more restrictive structure, like amyloid, may still be debated.
Figure 1. Material states and their biological correlates.
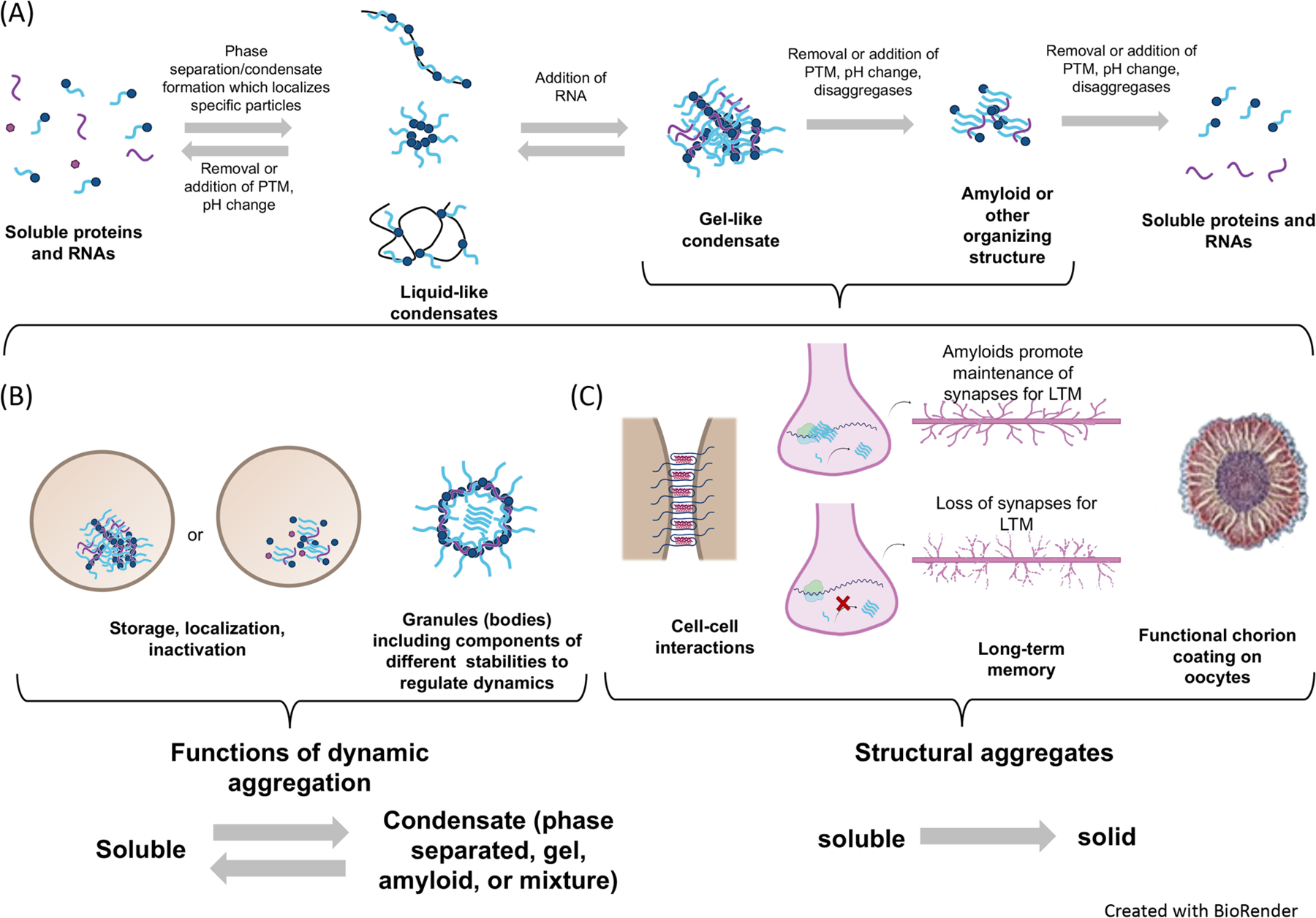
(A) Equilibria and reversibility between different condensate material states highlighting the range and diversity of their structural organization and the strategies used by the cell to transition from one state to the next. (B) Functional roles of reversible condensates. Left, yeast (Cdc19) has a stabilizing amyloid core. Right, granules such as the Balbiani body or the nucleolus consist of multiple material states, which may contribute to their stability and aggregation/disaggregation dynamics. (C) One-way changes to an amyloid or amyloid-like conformation are needed for some functions. Left, cell-cell interactions by adhesins in Candida albicans, Middle, aggregation of Orb2 in Drosophila melanogaster for long-term memory formation and right, structural amyloids such as the protective chorion layer of oocytes. Purple wavy lines represent RNA. Filled blue circles represent ordered domains and light blue lines represent intrinsically disordered domains of proteins. Black lines represent oligomers. Figure created with Biorender.com. Oocyte in panel C adapted from [72].
Examples of biomolecular condensates in the cytoplasm include physiological bodies such as stress granules [27], P-bodies [28], germline P granules [29], starch granules [30] as well as pathological or pre-pathological entities, such as cataracts [31], Lewy bodies [32], and amyloid fibrils among others. In the nucleus, examples include the nucleolus [33], heterochromatin, paraspeckles, transcriptional condensates, and replication compartments [34].
The challenges inherent in the definitive identification of the material state of condensates have been described in some recent reviews [35, 36]. However, as the list of biological condensates grows, it seems clear that material state may be mixed (liquid, gel, oligomer or fiber) and that transitions between states may be an important aspect of their function and regulation. For example, liquid droplets formed by LLPS are part of many membraneless compartments whose function may be to concentrate, localize, activate, inactivate, or filter its contents [37]. Furthermore, introduction of a co-aggregate like RNA during or after droplet formation, may shift the material state toward something more gel-like or change the concentration of protein needed to phase separate [38, 39]. The formation of a reinforcing structure, like the cross ß-sheets that characterize amyloids, may further solidify a condensate. Amyloid aggregates were historically characterized as being resistant to detergent solubilization, however, the cross ß-sheet structure can be found in much less resistant structures as well [40]. Amyloid assembly is self-perpetuating with soluble, native protein recruited into growing amyloid oligomers or fibers.
Functional (Physiological) Amyloids
The cross ß structure and fiber forming process have been highlighted in many pathological examples of protein condensates [41]. Nonetheless, the recent literature has abundant examples of regulated amyloids with physiological roles. For example, in response to starvation, the S. cerevisiae RNA binding protein Rim4 forms amyloid aggregates that repress translation of the cell cycle progression gene, CLB3 and other mRNAs. The active, amyloid form of Rim4 sequesters mRNAs that, though toxic if translated early in meiosis, are necessary for late meiotic events [42–44]. Temporally regulated phosphorylation of the intrinsically disordered C-terminal domain of Rim4 leads to amyloid disassembly causing the release of late meiotic mRNAs followed by the subsequent degradation of Rim4 [42–44]. Likewise, in metazoa, conversion of the human melanocyte protein, Pmel17, to the amyloid-forming conformation is pH-dependent, requiring the mildly acidic conditions found in the melanosome lumen [45]. Pmel17 fibrils disassociate at neutral pH to regenerate monomers. This aggregation/disaggregation cycle provides a safeguard against releasing Pmel17 fibrils into the cytosol thus circumventing their toxicity [45]. Another example of a regulated functional amyloid is found in memory storage attributed first to the neuronal isoform of Aplysia cytoplasmic polyadenylation element binding protein CPEB [46]. The self-sustaining amyloidogenic state of neuronal CPEB produces a persistent mark in the activated synapse required for long-term memory [47]. When Drosophila CPEB, encoded by the ORB2 gene, adopts the amyloid state it gains a new function, converting the translation-repressive monomeric Orb2 into a translation-activating amyloid [48].
Storage and Structure: Reversible and One-way Changes in State
The many roles carried out by biological condensates include basic cellular functions such as the modulation of cell growth and survival in response to the demands of the environment. The strategy of forming condensates rather than degrading molecules during exposure to a stress, means that protein availability during recovery does not require new synthesis. One example is the yeast Cdc19 pyruvate kinase, which is recruited and preserved as a condensate in stress granules that form in response to heat shock and other stressors [49]. Once stress conditions dissipate, disaggregation of the enzyme permits the rapid resumption of energy metabolism and protein synthesis. Metabolic enzymes from bacteria to animals form this type of reversible condensate, assembling into filaments in response to starvation [50, 51]. When conditions improve, the filaments disassemble and the enzymes are reused.
In contrast to reversible condensates like Cdc19, many of which are involved in storage, some amyloid forming proteins are structural. In the case of structural condensate, the transition to the amyloid conformation is a permanent (one-way) change in state. Examples of structural amyloids include the bacterial curli proteins involved in biofilm formation [52, 53], the adhesins of pathogenic yeast [54–56], which mediate cell-cell and cell-tissue interactions, and the eggshell chorion proteins of silkworm and related proteins in fish and mammals that protect the developing embryo from the environment [57]. Reversible and one-way changes in state are depicted in Fig. 1.
Chaperone-based Regulatory Activities for Condensate Processing
Proteins that have assumed a novel conformation with a free energy state lower than that of the normally folded protein may require the help of dedicated molecular chaperones to liberate and refold misfolded or denatured proteins. Molecular chaperones have key roles in folding newly synthesized proteins into their native states, in trafficking proteins to specific locations in cells and in the efficient assembly of molecular subunits into functional multimeric structures. Chaperone-mediated disaggregation and stimulation of proteolytic degradation are crucial aspects of proteostasis [58, 59]. Proteostasis seems to fail as organisms age, hence modulation of proteostasis pathways including prevention of amyloid formation, disaggregation of pre-existing condensates, and condensate sequestration [60–62] are promising targets for the treatment of degenerative diseases.
Disaggregation may take place enzymatically through the use of molecular chaperones with ATP-dependent disaggregase activity. Here we describe two major enzymatic activities, one encoded by potent Hsp100 proteins and the other encoded by a universal Hsp70 network that involves a collaboration between Hsp70, Hsp40 and Hsp110 proteins that has emerged as the likely Hsp100-equivalent in animals.
The Hsp100 family:
Hsp100 family genes (HSP104 in yeast, clpB in bacteria, HSP101 in plants) encode a potent disaggregase whose broad spectrum of substrates include pre-amyloid oligomers, phase-transitioned gels, as well as disordered aggregates and prions [63]. The Hsp100 family is frequently described as present in all taxa except animals. More precisely, animals lack cytoplasmic and nuclear Hsp100s [64] while maintaining a small repertoire of (diverged) Hsp100 relatives in the mitochondrial compartment (e.g., mitochondrial Skd3 [65]). The HSP100 genes encode spiral-shaped hexameric AAA+ chaperones that thread trapped polypeptides through a central pore using a ratchet-like mechanism that is powered by ATP hydrolysis. Fungal Hsp104 is not required under normal growth conditions but is essential for thermotolerance and is advantageous under stress conditions due to its ability to work in conjunction with Hsp70 and Hsp40 to mediate the recovery of properly folded proteins from disordered stress-denatured aggregates, disaggregate/fragment prion fibers into polymers which can be adaptive in stress [66–68], and participate in proteasomal degradation of select cytosolic proteins [69–72] among other activities. Hsp104 also confers longevity by promoting the retention of damaged protein aggregates in the mother cell [73–75].
Mechanistically (recent cryo-EM structure studies are nicely described in [63]), the Hsp104 nucleotide binding domains (NBD-1 and NBD-2) use energy from ATP hydrolysis to disaggregate proteins trapped in various higher order structures via translocation across its axial channel using tyrosine bearing pore loops that contact the substrate [76, 77]. The released polypeptides may refold spontaneously or may require assistance from molecular chaperones. Refolding and reactivation occur following release from Hsp104 [78, 79]. In cases such as the Sup35 prion, refolding isn’t necessary, as Hsp104 translocates the prion domain but stops short of the carboxy terminal GTPase domain, which remains correctly folded in the prion state [80].
In vitro work with purified Hsp104 suggest it has intrinsic ability to engage and disaggregate substrates [80, 81] however, it is clear that the in vivo disaggregase activity of Hsp104 (and the bacterial ortholog, ClpB) requires collaboration with the Hsp70 molecular chaperone system, which includes J-domain proteins (Hsp40s) and a nucleotide-exchange factor (Hsp110) in addition to the Hsp70 chaperone [78, 79, 82, 83]. The Hsp70 chaperone system acts at condensate surfaces to initiate the Hsp100 translocation process via partial solubilization and delivery of an Hsp70-bound unfolded polypeptide strand to the hexamer channel [84]. Hsp70s activate Hsp100s through direct interaction with the coiled-coil M domain located within NBD-1 of Hsp104/ClpB [84–88]. The association of Hsp70 releases Hsp104/ClpB from a repressed state, boosting its ATPase and protein processing activities [87]. Certain mutations in the M domain are hyperactive, displaying elevated activity, higher affinity for a protein substrate and disaggregation activity in the absence of Hsp70. These variant Hsp104s are highly toxic [89] presumably because they may not discriminate between improperly folded proteins and proteins that are properly folded proteins but have one or more intrinsically disordered domains [89, 90]. Analysis of single and double M domain mutants suggested that at least one role of Hsp70 is to shift substrate specificity toward unfolded proteins [84, 91, 92].
Hsp70 Chaperone Network:
Hsp100-type disaggregases are found in bacteria, and every compartment of fungi, plants, and protists. Although there are several Hsp100 type proteins that localize to the mitochondria in metazoans, there are no cytosolic or nuclear Hsp100 activities [64]. In metazoa, Hsp70 family members cooperate with a specific subset of J-proteins and nucleotide exchange factors to form a protein disaggregation machine capable of solubilizing a wide range of amorphous and amyloid-like aggregates.
The 70 kDa heat shock protein encoded by HSP70 genes participates in all aspects of protein life, including the folding of newly synthesized proteins, the translocation of polypeptides into mitochondria, chloroplasts and the endoplasmic reticulum (ER), the disassembly of protein complexes, and the regulation of protein activity. The stress-related activities of Hsp70 proteins include preventing protein aggregation, solubilizing aggregated proteins, promoting the refolding of misfolded or unfolded proteins and cooperating with cellular degradation machineries to clear aberrant proteins and protein aggregates [86].
Hsp70 ATPase activity is regulated by co-chaperones including J-proteins encoded by HSP40 genes, and nucleotide exchange factors (NEF) encoded by HSP110 genes among others [93–96]. As a result of J-protein mediated binding of substrate proteins to Hsp70·ATP, in conjunction with direct J-protein-Hsp70 interactions, ATP is hydrolyzed and Hsp70 undergoes a transition to the ADP-bound state, which has high affinity for the substrate. NEFs then induce ADP dissociation and rebinding of ATP, converting Hsp70 to the low- affinity state and causing substrate release [97–99].
Hsp70s recognize a degenerate motif consisting of 5 residues enriched in hydrophobic amino acids and flanked by positively charged amino acids, typically buried in the interior of natively folded proteins, but exposed when proteins become unfolded or misfolded. J-proteins confer selectivity. The A and B classes of J-proteins interact with Hsp70 separately with distinct condensate selection properties [100]. For example, the constitutively expressed Hsc70 (HSPA8) forms an ATP-dependent chaperone with J-protein DNAJB1 and Hsp110 class NEFs (HSPH1–3) to solubilize aggregates formed by protein denaturation. The expanded number of J-protein family members in animals (45) over yeast (22) or bacteria (7) [101] increases the activity profile within each organism. For example, Hsp110-Hsp70 complexed with DNAJA proteins cannot disaggregate fibrils, but can disaggregate disordered aggregates [101], while DNAJA2 allows disaggregation of smaller aggregates [102] and Hsp110-Hsp70 complexes that include DNAJB1 promote the release of monomers from amyloid fibers by end depolymerization as well as fibril fragmentation [103]. In eukaryotes but not prokaryotes, J-proteins also bind to each other to form “complexes” [100]. J-protein complexes have a wider substrate spectrum compared to the individual J-proteins or homo J-protein oligomers, because distinct substrate binding specificities are combined. How the different classes of J-proteins (single and in complex) recognize distinct clients remains unclear [104].
In addition to the Hsp40 co-chaperones which provide substrate selectivity, the Hsp70 chaperone network relies on the small heat shock proteins (sHSPs), a special class of molecular chaperones between 12 and 43 kDa that lack an ATPase domain. The sHSPs act early, holding proteins to facilitate their refolding or degradation by ATP-dependent chaperone complexes [105, 106].
The flexible hydrophobic surfaces of sHSPs interact with exposed hydrophobic surfaces of misfolded or denatured client proteins to sequester misfolded substrates into large inclusions to protect them from proteases [105]. sHsp chaperones are thus poised to handle early misfolding events prior to expression of other stress-inducible chaperones. sHsps also facilitate subsequent disaggregation and folding by Hsp70 and Hsp100 by displacing and releasing surface-bound sHsps from sHsp/substrate assemblies. sHSPs themselves are sequestered into large, dormant, multimeric structures called sHsp oligomers to prevent any deleterious effects due to their exposed hydrophobic surfaces. sHsp oligomers disperse into smaller oligomers as they become active [105], a transition that can be precipitated by stress and post-translational phosphorylation. The vast array of heterodimeric and heteromeric oligomer combinations generated by the ~10 sHSP genes in mammalian genomes lead to distinct client recognition patterns and other types of regulation.
A recent survey of chaperone expression across different tissues suggest that various disaggregation loads intrinsic to different tissues is supported by specific RNA expression profiles of chaperones and co-chaperones [107]. The many chaperone activities found in cells can be subdivided into those with core activities, found in all cells and those with variable activities, which have a more variable range of tissue expression. Interestingly, mutations identified in chaperones often lead to tissue-specific phenotypes, even when expression profiles of the chaperone may be the same in multiple tissues.
The in vivo relevance of the Hsp110-Hsp70-Hsp40 disaggregase activity was examined in C. elegans. Knock-down of Hsp110 in worms that were briefly heat shocked on day 1 of their lives caused persistent aggregates and reduced lifespan by 4.5 days, a phenotype that was exacerbated by simultaneous knockdown of the Hsp70 homolog, HSP-1 [108]. To investigate whether interfering with protein disaggregation had any organismal level impact, lifespan was examined in worms with reduced expression of HSP-110, HSP-1, or both. The knock-down of HSP-1 led to a developmental delay of about 1 day in 80% of animals. However, all animals reached adulthood and did not display any other obvious phenotypes. In related experiments, small Hsps were found to facilitate the disaggregation process and to be essential under some thermal stress conditions in C. elegans [108].
Non-chaperone Regulatory Activities for Condensate Control
Aggregation is an efficient and often self-propagating process [3] so it comes as no surprise that the formation of functional condensates must be highly regulated. In addition, condensates that function as storage compartments require reliable mechanisms for the release of resident molecules. In this section we enumerate and describe some of the many cellular mechanisms that are central to the effective cellular use of aggregation in normal physiological processes.
In addition to enzymatic disaggregation discussed above, there are numerous naturally occurring cellular or environmental events that control the flow of proteins and other biomolecules into and out of condensates. Hence, the concept of disaggregation must include disaggregation “activities” which are not enzymatic (disaggregases). The large repertoire of condensate regulatory strategies, some of which require substantial genomic (cellular) resources, suggests that condensate biology is an important (albeit late) addition to our understanding of cell and development processes. Some of the interesting condensate regulatory strategies are described below.
Gatekeeper residues:
The process of forming amyloid, both pathological and beneficial, is driven by the ability of individual protein segments to adopt cross β-strand conformations and assemble into fibrils, for example via tight, zipper-like interfaces. To ensure that condensates are formed or kept in check according to the demands of the system, many aggregation-prone stretches of amino acids in proteins are flanked by ‘‘gatekeeper’’ residues such as lysine, arginine and proline [109] that inhibit condensate formation. The positively charged residues are repulsive in close-packed condensates and their large flexible side chains are entropically unfavorable [109] and proline is incompatible with the β-sheet structure of amyloid type aggregates.
In a computational overview of all of the non-disease condensate-prone proteins in the human proteome, gatekeeper residues with low aggregation propensity like Pro, Arg, Lys, Asp and Glu were particularly highly represented at strategic (flanking) positions suggesting that molecular evolution has acted on protein sequences to finely modulate their aggregation propensities [2]. One example of gatekeeper regulation is found in the curli proteins encoded by the enterobacterial csgA and csgB genes. Curli protein is an extracellular amyloid fiber that mediates bacterial attachment to surfaces, cell-cell aggregation and biofilm formation. Aggregation is kept in check in part by five imperfect repeats in the major curli subunit protein, CsgA, thus rendering aggregation dependent on the CsgB protein [2, 110]. Repeats R1 and R5 promote responsiveness to CsgB nucleation and self-seeding by CsgA fibers [52, 53] but Repeats R2–R4 include aspartic and glycine residues that reduce aggregation propensity, and thus modulate polymerization efficiency and potential toxicity [111]. CsgA mutants lacking those gatekeeper residues polymerized in vitro significantly faster than the wild-type protein, and polymerized in vivo even in absence of its nucleator CsgB.
Interestingly, misregulation of spatial and temporal aspects of functional amyloid formation as a result of mutations in gatekeeper residues or regions seem to be a common disease mechanism in the case of hormones. Peptide hormones are concentrated in secretory granules as functional amyloids [112] (the environmental regulation of peptide hormone will be described in more detail below). While the details of proprotein sorting into granules is unclear, self-aggregation of regulated cargo at the trans-Golgi network (TGN) is known to contribute to granule formation. Dominant mutations in provasopressin (precursor to the water homeostasis hormone vasopressin) that cause cell degeneration and diabetes insipidus prevent native folding and produce fibrillar aggregates in the endoplasmic reticulum (ER) that might reflect mislocalized amyloid formation by sequences that are important for granule sorting [113].
Amyloid Nucleating Partners:
As seen in the bacterial CsgA-CsgB system for Curli fibers, the formation of certain amyloids requires a partner protein. One such set of partner proteins in animals is Rip1 and Rip3 which mediate necroptosis. Necrotopsis is a type of programmed cell death with necrotic cell morphology [114] that is the result of interaction between the serine/threonine kinase, Rip1 and its paralog, Rip3, under conditions of caspase-8 inhibition [17]. Rip1 and Rip3 interact via RHIM domains to form the necrosome consisting of heteromeric amyloid fibers [115]. The amyloid-like structures that form upon their interaction may act as a scaffold to activate multiple downstream pathways for necroptosis, lead to Rip3/Rip3 homo-oligomerization and Rip3 autophosphorylation. Phosphorylated Rip3 recruits and phosphorylates mixed lineage kinase domain like protein MLKL, which leads to membrane pore formation, loss of membrane integrity and, eventually, necrotic death [114].
Environmental control:
Some functional amyloid-prone proteins take advantage of specific environmental features of a subcellular compartment to sustain or break down condensates. One example is the pH regulation of Pmel17 [116, 117] discussed earlier. Another example is the highly concentrated membrane-enclosed secretory granules [118] that allow cells to stock hormone until a signal triggers its release, at which point hormone can be secreted much faster than it could be synthesized. The amyloid structure of the hormone enables a controlled release of monomeric, functional hormone [112]. Each hormone has its own dissociation rate which is controlled by extrinsic factors such as pH, ion concentration, and extracellular chaperones [119].
Spatial differences in ATP may also affect the formation of condensates. Initial studies by Patel et al. examining the solubilizing effect of ATP on a variety of condensates in vitro, specifically identified the changes within the 1 to 8 mM range of concentrations found within cells [120]. A role for ATP, beyond providing energy or serving as a substrate for chaperones or other enzymes, has been supported by subsequent studies [121, 122] that have examined the stability of cellular condensates in response to ATP concentration. It is interesting that earlier studies on proteasome activity [123] show a peak of activity at ATP levels that are less than a tenth of normal physiological levels. Their data suggest proteosome activity increases as ATP concentration drops.
RNA as a co-condensate:
The role of RNA as a co-condensate species in protein condensate formation was suggested by the identification of non-membrane bound nuclear particles where RNA processing, RNA transport and ribosome assembly occur, and the identification of C. elegans P-granules as centers of condensate formation [29]. A broader net was later cast using the chemical isoxazole which served as an RNA mimetic to capture an expanded roster of proteins whose aggregation status is influenced by RNA [124]. RNA may also serve as a substrate for protein activities collected together in condensates. For example, fibrillarin methylates rRNA while phase-separated within the dense fibrillar component of the nucleolus.
Recently, there has been more attention paid to the intrinsic property of RNA to aggregate [125]. The ability of RNA to form inter- and intra- strand secondary structure contributes to nucleic acid only as well as mixed protein-RNA complexes [38, 39]. Both the charge contribution from the phosphodiester containing backbone of the RNA and the sequence seem to play a role. Sequence-mediated stem-loop structures and chemical modifications of bases each contribute to specific protein binding. As discussed below, RNA is featured in multiple biological condensates.
RNA helicases and RNase activities:
RNA helicases provide a way to manage RNA structural changes that promote or discourage RNA aggregation as well as RNA:protein co-aggregation. Processive RNA helicases of the DEAH/RHA and Ski families as well as pro-processive RNA unwinding helicases of the DEAD box family are frequently associated with condensates [126]. These proteins may regulate RNA structure dynamically via hydrolysis of ATP, but in an ATP-independent manner may compete for RNA binding, thus influencing the association of RNA with other proteins. For example, the mRNA encoding the Wnt antagonist Draxin in RNA processing condensates found in neural crest cells is selectively degraded in a process that depends on Draxin microRNA and the RNA helicase, DDX6 [127]. The connection between RNA helicases and regulated formation and activity of condensates is an emerging theme in both nuclear and cytosolic particles [128–130].
Post-translational modification:
Entire reviews could be written on the role of post-translational modifications (PTM) such as phosphorylation in modulating aggregation. PTM of proteins can directly alter the shape and folding of a protein, affect its activity, and enhance or suppress protein quality control checkpoints. The reversibility of post-translational modification provides an opportunity to regulate protein aggregation behavior, and dysregulation of PTM activities is known to contribute to aberrant aggregation and disease. For instance, returning to the functional amyloids discussed earlier, Pmel17 glycosylation influences its sorting and fibril formation; Pmel17 fibrils fail to form in mutants lacking sialic acid and galactose modifications [131]. Yeast Rim4 is dephosphorylated and aggregated during starvation conditions and a threshold of phosphorylation events by kinase Ime2 leads to its dissolution and eventual degradation, thus allowing the progression through meiosis [44]. In the presence of mRNA, CPEB undergoes LLPS in vitro upon SUMOylation and, when in vivo SUMOylation is inhibited by ginkgolic acid, CPEB localization to the phase-separated P-body decreases [132]. It is likely that the numerous cellular and developmental pathways that employ reversible post-translational modifications mediated by enzymatic pairs including kinases and phosphatases, methyl-transferases, acetyl transferases, ubiquitylation and deubiquitylation or sumoylation and desumoylation as signaling molecules may be triggering protein condensate formation as one of their downstream responses. Finally, it is worth noting that the equilibrium between a protein in its soluble form and its condensate form can be influenced during synthesis via controlling accessibility of sites for post-translational modification or by direct regulation of the modifying enzymes.
Aggregation in Cell Division and Development
Temporal responsiveness and spatial organization are important during all cell divisions, but may take on added complexity during development. Taken broadly, cell division during development helps to mediate the acquisition of cell fates and the proliferation of a pluripotent and differentiating cell populations from gametogenesis through old age. Gametogenesis and the early stages of development have historically provided a rich source of observable cellular structures due to their accessibility and size compared to most somatic cells. Although some of the structures, like germ plasm, are not found in most cells, they highlight some of the general features that are found in many cellular condensates [133, 134]. In the following sections, we focus on the role aggregates play in distributing cellular contents to daughter cells and how cells in developing embryos use aggregates to filter cytosolic contents.
Condensate-based strategies for Symmetric and Asymmetric distribution of cellular contents:
Conceptually, condensates are especially useful in the context of the directed transport of cellular contents. When coupled with mother cell division, condensates can influence daughter cell inheritance and even cell specification when cytoplasmic determinants are involved.
Symmetry:
Although some condensates are destined to be retained by the mother yeast cell, others (known as prions) are distributed to daughter cells to confer advantages in fluctuating physical environments [135]. In many cases this requires the cleavage activity of ScHsp104 which reduces mature fibrils into fibril propagons that are transported through the bud neck into daughter cells [135]. Hence the chaperone network within S. cerevisiae ensures that daughter cells benefit from the distribution of endogenous prions. Interestingly, recent work has revealed a class of S. cerevisiae prions that are not amyloid in nature and that are Hsp104-independent [66, 136, 137]. These may be akin to the Het-S prion of P. anserina and the only known S. pombe prion, [CTR4+], neither of which can be cured by inactivating Hsp104 [138, 139]. It appears that some prions rely on other chaperones for their distribution.
Asymmetry:
Condensate asymmetry is important in aging and lifespan. The asymmetric distribution of condensates during mitosis promotes the birth of cells unburdened with potentially toxic condensates accumulated over a lifetime, and are instead afforded a clean slate and a normal lifespan. In budding yeast, mother cells actively retain condensates via cytoskeletal tethering to facilitate retrograde transport away from the bud [74, 140–142] as well as by compartmentalization of condensates in non-membrane bound entities that associate with specific organelles like the intranuclear INQ and vacuolar IPOD [140, 141].
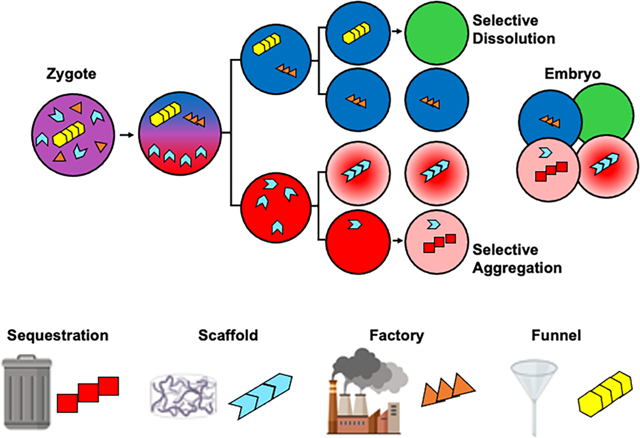
Strategies for creating asymmetry include extrinsic signaling cues and asymmetric partitioning of molecular determinants as well as differential RNA localization and biased microtubule-organizing center activity. For example, liquid-like condensates at the base of motile cilia [143] may facilitate the ciliary “sweeping” of developmental morphogens that are implicated in symmetry breaking events in the vertebrate node. Recent reports by the Mowery [144] and Huber labs [145] in Xenopus highlight the asymmetric packaging of mRNAs in mixed RNA-protein condensates. Interestingly, the tight sequestration of mRNAs in condensates such as the vegetally localized Vg1 mRNA, is reversed during early development, allowing relocalization from the cortex to the cytosol of cells that eventually form endoderm [146]. Cell divisions resulting in two distinct daughter cell fates may be especially attractive targets for condensate regulation. For instance, C. elegans embryonic development is characterized by sequential asymmetric cell divisions starting from the one-celled zygote [12, 147] and is accompanied by reiterated asymmetric P granule localization via polarized dissolution/condensation cycles. In A. gossypii Whi3 mediates RNA-specific condensates that help define domains within a syncytial, multinucleate space. Whi3 condensates located in fungal branches include BN1 and SPA2 mRNAs, while Whi3 condensates that form near nuclei have CLN3 mRNA [148].
Developmental condensates as molecular filters that generate spatial repositories:
Many of the large, well-characterized cytoplasmic condensates visible by low resolution microscopy seem to play a role in filtering cytoplasmic components, by which we mean enriching or depleting a subcellular locale of what would otherwise be freely diffusing molecules. Here we give some examples of subcellular bodies that are involved in this filtering mechanism, as this strategy is known to generate the asymmetry in embryonic cells important for germline development and clearing factors that could impede germ cell fate maintenance.
Vertebrate Balbiani bodies, associated with the grouping of mitochondria [145] endoplasmic reticulum, RNA and protein in the oocyte, is a transient non-membrane bound structure thought to preserve a low-activity state required for long-term oocyte storage [149]. In Xenopus, Balbiani body formation depends on the self-assembly of Xvelo protein into a solid amyloid aggregate. Once formed, Balbiani bodies filter the cytoplasm for specific proteins and RNA [150], which are then translocated to the vegetal cortex [151], where they play a role in germline specification. Current models suggest that the Balbiani body houses a liquid-like condensate within the cage-like Xvelo assembly that facilitates a diffusion-capture mechanism of germ plasm components to drive their asymmetric inheritance. Interestingly, the stably aggregated form of Xvelo is nonetheless reversible, as it dissolves in concert with phosphorylation events that may affect the ability of the Xvelo protein to form amyloid [4, 152] leading to Balbiani body dissolution early in oogenesis.
A second example of membraneless germline condensates with a filtering function required for germline specification is the Drosophila germ granules [153]. These germline condensates of protein and RNA have both liquid-like and gel-like attributes [7]. Granule formation occurs when posteriorly localized Oskar protein phase separates and recruits a germ granule core that includes the RNA helicase Vasa, the factors Pum and Dazl, and the argonaute family member Aubergine. This condensate structure then localizes hundreds of mRNA clients including nanos mRNA, which is required in the developing germ cells to repress somatic transcripts [126]. Balbiani bodies and germ granules also share some of the same clients that regulate germline specification despite the distinct core composition of each condensate.
A third developmental example of condensate-based filtration are the P granules of C. elegans, named for their localization to the germline (P) lineage [11]. P granule condensates consist of a gel-like core that includes the predicted RNA binding proteins PGL-1/3, the intrinsically disordered proteins MEG-3/4, and the helicase LAF-1[154]. Despite the non-dynamic core, P granules display liquid-like properties and dynamic dissolution/condensation cycles. Control of P granule formation is accomplished by preferential solubilization in the anterior of dividing cells while condensation occurs posteriorly [29]. Larval depletion of PGL-1/3 in combination with knockdown of two P granule helicases leads to sterile adults with oogenesis defects [155], suggesting a P granule role in germline maintenance rather than germline specification. Current models suggest that P granules function by localizing to the cytoplasmic side of the nuclear pore where they monitor mRNAs exiting the nucleus and prevent translation of transcripts that are not essential for germline function [155]. Therefore, while Drosophila germ granules and vertebrate Balbiani bodies appear to collect germ cell components and specify the germline, P granules may use a similar filtration mechanism to eliminate somatic factors that otherwise would cause spurious germ cell differentiation. Interestingly, introduction of the potent fungal disaggregase, Hsp104, into the C. elegans germline not only altered condensate distribution and abundance but also led to germline defects and embryonic lethality [14].
Emerging areas in developmental protein condensate formation.
The above examples are a few among the many that indicate that condensates in the adult germline, mature gametes and the developing embryo are important arbiters of early development. Recently, analyses of the material state of these developmentally significant condensates and visible nuclear and cytosolic particles indicate they contain more structure than previously appreciated. Specifically, we and others have applied various markers of amyloid oligomers and fibrils to the germlines, oocytes and early embryos of Xenopus and C. elegans and found that both these organisms begin life with an appreciable amount of amyloid material. However, it should be noted that the identity of the amyloidogenic proteins remains largely unknown.
The C. elegans germline and early embryo contain discrete amyloid positive domains:
Defects in C. elegans P granule aggregation following the introduction of fungal Hsp104, together with the ability of Hsp104 to solubilize higher order condensates, suggested the presence of possible endogenous amyloid-type substrates during normal C. elegans development. Furthermore, Hsp104 introduction caused cell division defects and eventual embryonic arrest and lethality, suggesting that this disaggregase was disassembling functional amyloid-like aggregates [14]. This is consistent with early work in which the use of Hsp104 was explored as a therapeutic to mitigate toxic protein aggregation in Drosophila models of Spinocerebellar Ataxia Type-3. In this study, Cushman-Nick et al. [156] showed that careful modulation of Hsp104 level was essential to avoid toxic effects of Hsp104 itself. Indeed, amyloid character was revealed in the worm germline, oocyte and early embryo by various amyloid markers including the antibody A11 (amyloid oligomers) and the antibody OC (amyloid fibrils) [14]. These markers revealed distinct subcellular locales including those previously associated with aggregation such as the nuclear membrane [157], the centrosome [13] and a subset of embryonic P granules [11]. The P granule localization pattern is especially intriguing as these are asymmetrically inherited structures known to influence germline-specific functions (see above). Further, analysis of late stage (3-fold) embryos showed that much of the early amyloid had been solubilized, disaggregated or degraded in the soma, but either maintained or re-aggregated de novo in the germline precursors Z2/Z3, suggesting the germline is already materially distinct from the soma at this early stage. These data indicate that amyloid aggregates are a normal aspect of C. elegans development and that their proper regulation is essential for normal embryogenesis.
Amyloid as part of condensates found in Xenopus:
The size of Xenopus oocytes and the cells of the early embryo give rise to large cellular structures which facilitate their detection and analysis. Examples of condensates with at least some amyloid structure in Xenopus cytosol include those formed by Xvelo in Balbiani bodies [4], particles that participate in RNA localization [145, 158], the base of cilia [143] and germ cell granules [159]. Developmentally, the Xenopus oocyte and early embryo use cytosolic condensation to help solve the problem of spatial inheritance as well as to create temporal control of specific activities.
The nucleus of Xenopus oocytes has provided a rich source for studies on non-membrane bound particles [33, 160–164]. Nucleoli, Cajal bodies involved in snRNP biogenesis, mRNA processing speckles, histone locus bodies and small RNA processing Pearls can all be detected with dyes and antibodies that recognize amyloid structure [6]. Among the differences between the nucleus and the cytosol is the steady state level of ATP, with the nucleus being 2 to 3 times higher, providing an environment where many aggregating proteins may tend to form more fluid condensates. This may be counterbalanced by the abundance of RNAs that may have roles as co-condensates. The nucleus may also be providing enhanced concentration of selected proteins, potentially pushing them toward the formation of condensates. The breakdown of the nucleus during oocyte maturation and later during embryonic divisions would drop the local concentration of proteins, allowing proteins in a more liquid condensation state to disperse and the non-membrane bound particles to come apart. As protein is reconcentrated in the nucleus after M phase, condensates would reform.
Next Steps and Discovery
What are the components of the condensates?
One area of opportunity is the identification of the materials that initiate, join, stabilize and persist as part of biological condensates. There is a gap between the observation of punctate structures in the cell and the identification of their composition and control of their material state. One strategy for identifying constituents is to take advantage of the growing proteome repositories as well as datasets identifying protein-protein interactions, often collected for other purposes, to develop specific hypotheses regarding condensate formation. Hundreds of proteins, typically those with regions of low complexity, have been implicated in vitro. Recent publications identify proteins that form condensates in vitro [124, 165] many of which appear in biological condensates. Many of these same proteins co-aggregate with RNA, therefore transcriptome data may also be useful. Furthermore, recent advances in proximity labeling in vivo provides ways to deconvolute close associations and potential condensate molecular partners under a variety of conditions [166]. There also remain challenges for direct observation of condensate structure. Techniques to identify material state range from amyloid detecting dyes and antibodies to detect structural epitopes to chemicals like 1, 6 hexanediol that probe liquid droplet state [37] have been used. Some condensates may not be stable during isolation or fixation, and may therefore require visualization and characterization in living tissues However, a simple failproof method of in vivo condensate identification awaits discovery.
What are the disaggregation activities that regulate early development?
The aggregation of proteins in animal germlines and embryos appears to be essential for development. Ectopic expression of the fungal Hsp104 disaggregase in C. elegans resulted in developmental defects as well as changes in aggregation phenotypes [14]. The challenge is to identify not only the developmentally important aggregation-prone proteins and co-aggregating RNAs, but also their regulators. Disaggregation activities may take the form of any of the regulatory pathways described above. Such activities should be expressed during animal development, localized to specific developmental bodies with condensate character, and cause developmental defects when lost.
Reassessment of the activity of familiar proteins using bioinformatic, phylogenetic and proteomic inquiry may identify candidate regulators. For example, the ABCF proteins (discussed below) were identified by their close phylogenetic relationship to the fungal-specific disaggregase, New1. A bioinformatic approach might be to identify the intersection of the set of proteins with requisite features such as RNA binding, ATPase, and helicase domains and regions of disorder with the set of proteins that are physically associated with developmental condensates. Beyond bioinformatic approaches, candidates can be mutated or knocked down in animal model systems and evaluated for their impact on development and on developmental condensates such as P-granules in C. elegans or nucleoli in X. laevis [14]. While studies using zebrafish, fruit flies, ascidians or filamentous fungi all provide their own special advantages, the transparency of worms and the large size of Xenopus oocytes provide distinct opportunities for the study of condensates with roles in development. However, candidates can also be evaluated in budding yeast, a system in which many well characterized reporters of disaggregation activity have been developed. Furthermore, many relevant proteins are conserved between fungi and metazoa, including the Hsp proteins as well as the proteins encoded by the ABCF gene family and the RuvBL proteins discussed below. Finally, complementary studies on disaggregation activity in vitro using purified aggregation-prone proteins or peptides will also be useful.
RuvBL proteins are aggregation regulators:
The RuvBL proteins have roles in chromatin remodeling [167] and DNA repair [168]. However, the RuvBL protein family was also identified in an siRNA screen in mammalian cell lines for proteins involved in the formation of aggresomes, a non-membrane bound condensate storage compartment that forms when the protein degradation system of the cell is overwhelmed [169]. The RuvBL proteins have DNA-dependent ATPase and DNA helicase activities and belong to the AAA+ (ATPases associated with diverse cellular activities) protein family. Like other AAA+ proteins, including the disaggregase Hsp104 [170], RuvBL1 and RuvBL2 proteins form a barrel-like structure. The RuvB proteins assemble into hetero-hexameric rings and together can form a mixed dodecamer made of two stacked hexameric rings [171]. Xenopus RuvBL was also identified in a proteomic screen for nuclear condensates [121] and is enriched in the proteome from neural ectoderm fated cells in an early embryo [172] consistent with earlier localization studies of RuvBL mRNAs [173]. The temporal and spatial analysis of RuvBL mRNAs suggests that the proteins may be important developmentally [173]. Without embryonic expression of RuvBL1 and RuvBL2 there are gastrulation and cell proliferation defects in Xenopus development [174] and the failure to express the C. elegans, RUVB-1, causes embryonic lethality [175].
Like Hsp104, RuvBL proteins can process amyloid fibers [169]. The ATPase activity of purified dodecamer RuvBL complex is stimulated by the fibrillized amyloidogenic peptide Aβ1–42 [169] and affects the formation of Aβ fibrils. Addition of small sub-stoichiometric amounts of purified dodecamer RuvBL complex in the absence of ATP significantly delayed Aβ fibril seeding and reduced the rate of fibril growth, while addition of ATP reversed this effect suggesting that RuvBL binding to oligomeric seeds is reduced or that seed propagation is promoted [169]. Unlike Hsp104, the RuvBL proteins do not refold denatured substrates and do not require Hsp40 and Hsp70 cofactors [169]. Additionally, overexpression of the yeast orthologs, RVB1 or RVB2 suppresses thermotolerance defects of hsp104 mutants [169]. The in vivo activities of RuvBL proteins include surveillance of condensate size. Very small condensates accumulating during normal growth conditions are kept in check by the RuvBL system which recognizes and clears condensates that are larger than a critical threshold [176]. In cells lacking this activity, aggregation kinetics are accelerated [176]. Because of its in vitro activity in disaggregating amyloid fibers, and the developmental phenotypes in knockdowns, RuvBL proteins are a potential member of the portfolio of developmental disaggregases in metazoa.
ABCF proteins play a role in protein disaggregation pathways:
The ABC family of ATPases are a large family of proteins with eight subclasses, A through H, of which all but two (E and F) have membrane-spanning domains and function in ATP-dependent transmembrane transport. The non-membrane associated ABCF1, 2 and 3 proteins were initially picked out as disaggregase candidates by their evolutionary relationship to the fungal New1 protein which has Hsp104-independent disaggregation activity in yeast [9, 177].
In cross-species rescue experiments carried out in S. cerevisiae, Human, Xenopus and C. elegans, ABCF proteins were able to correct defects in the processing of ordered or disordered condensates caused by the absence (Abcf3/Gcn20) or reduction in normal levels (Abcf2/Arb1) of the yeast orthologs [14]. Heat-denaturation of firefly luciferase (FFL-GFP) reporter provided a test of disordered condensate processing, and the expanded Q97 exon I of the Huntingtin protein (Htt-Q97-GFP) reporter provided a test of activity with respect to a classical amyloid structure. In yeast, Hsp104 acts on both disordered and ordered amyloid condensate types, and our analyses suggest that all three ABCF proteins engage in pathways directed at amyloid-like targets, whereas only ABCF1 and 2 targeted disordered FFL-GFP condensates [14]. Importantly, these assays support a role for ABCF proteins that go beyond the previously established translational and ribosome biogenesis and ribosome quality control roles for ARB1 and GCN20 [178–180].
Interestingly, ABCF proteins are implicated in spatial control of development. In Xenopus, maternal ABCF2 mRNA is predominantly inherited by cells that will migrate during gastrulation [14]. Without ABCF2, gastrulation fails [14]. In C. elegans, the loss of ABCF1 led to developmental delays, germline arrest, and enhanced the amyloid character of P-granules [14]. Of note is the recent finding that the RNA-protein condensates that localizes maternal mRNA to the vegetal hemisphere in Xenopus include ABCF1 [145]. Whether ABCF proteins have enzymatic activity toward condensates awaits definitive in vitro experiments. However, even if the activity is indirect, their inactivation nonetheless causes an increased cellular load of condensates, and the basis for this phenotype is a fertile field for future investigation.
Conclusions, Perspectives, Future Directions
Although robust methods for identification of the material state of physiological condensates are still being developed, it is clear that the reversible, regulated management of condensate state transitions and condensate dissolution may involve an expansive repertoire of disaggregation activities. These activities may promote or inhibit condensate formation, which may, in turn, initiate or delay cellular activity, maintain a specific cellular response, or allow for an adaptation to new cellular challenges. The cellular arsenal for aggregation management includes post-translational modification, local changes in the cellular environment, the incorporation of co-aggregates, as well as the activities of chaperones and disaggregases. The aggregation and disaggregation of protein complexes can be tailored to meet conditions that favor rapid exchange, substrate dependent assembly or structural stability. Condensates can be composed of dozens of components and can be transient or long-lasting.
Protein condensates are central to normal cellular function. In addition to acting as cytosolic filters (see above), protein condensates support many additional activities and could serve various physiological roles in developmental and cell biological contexts. For instance, they may serve as scaffolds for efficient signal transduction, localizing members of a developmental signaling pathway, and thereby optimizing the response time of the targeted cell. Similarly, biochemical pathways could be localized and concentrated into condensates to improve productivity, induce rapid polymerization or reduce toxic byproducts. During morphogenesis, condensates including cell adhesion components could stabilize cell-cell interactions facilitating tissue cohesion during the coordinated bending of epithelial sheets. Condensates could also sequester toxic cellular products, thus protecting developing embryos. The sequestration of proteins into condensates and subsequent inactivation of cell fate determinants could isolate somatic regulatory proteins until cell-type specific disaggregation mechanisms are employed, promoting a cell-specific soluble proteome that would function alongside traditional transcriptional regulatory mechanisms. Despite the long history that equates aggregation with the abnormal or pathologic consequences of misfolding, the observation that non-membrane bound condensates of protein and RNA are central to many cellular functions, coupled with in vitro observations that purified proteins can be induced to form similar condensates, has served to invigorate investigations that are attempting to bridge in vitro studies with what occurs in cells. We believe that the list of biologically important condensates will continue to grow and that the identification of their RNA and protein scaffolding, as well as the nature of their regulation, and their client lists, especially during development, will provide an opportunity to better understand determinative events during embryogenesis, as well as the adaptation of organisms to stress and environmental changes.
ACKNOWLEDGEMENTS
We gratefully acknowledge discussions and input by Dr. Paul Huber, University of Notre Dame and Dr. Daniel Summers, University of Iowa as well as by Dr. David Cooper and Tyler Atagozli of the University of Iowa. This work was supported by a University of Iowa Office of the Vice President for Research grant (DLW and JSF), a University of Iowa Center for Biocatalysis and Biotechnology grant (DLW, BTP, JSF), NIH award GM124063 (DLW), NIH award GM114007 (BTP) and NSF-IOS award 1917169 (JSF and BTP).
GLOSSARY
- Amyloid
A special type of protein aggregate formed by the recruitment and subsequent cross-polymerization of specific proteins into a special type of β-sheet secondary structure (known as cross-β), and the ability to be stained by dyes including Congo Red, and Thioflavin dyes.
- Amyloid Disease
Any disease in which the pathology is attributable to the conversion of a specific protein to its amyloid form. In humans, there are at least 37 amyloid prone proteins that cause disease including the β amyloid peptide, Aβ from the amyloid precursor protein, involved in Alzheimer’s disease, α-synuclein involved in Parkinson’s disease, PrP involved in transmissible spongiform encephalopathy, the microtubule associated protein Tau, involved in various tauopathies, and Huntingtin exon 1 involved in Huntington’s disease.
- Balbiani body
The Balbiani body is a non-membrane bound compartment found in animal oocytes. It forms during the early stages of oogenesis and disappears as the oocyte matures. It seems to assemble and contain Endoplasmic Reticuli, Golgi, RNA, mitochondria and proteins thus assisting spatial distribution and restriction of maternally derived molecules.
- Chaperones
Proteins that assist in the conformational folding or unfolding of proteins as well as the assembly or disassembly of other macromolecular structures. One major function is preventing newly synthesized polypeptide chains and assembled subunits from aggregating into nonfunctional structures. Many chaperones are induced by stress to mitigate the tendency of proteins to unfold and aggregate under such conditions.
- Condensate
Here used to describe an assemblage of proteins that have taken on a conformation that includes coalescence into a non-membrane bound cellular “compartment” that may have a number of material states from supersaturated “liquid-liquid” droplets to the most solid being “amyloid”.
- Disaggregase
Molecular chaperones with enzymatic activities leading to the resolubilization of protein aggregates with concomitant ATP hydrolysis.
- Functional (physiological, biological) Amyloid
Non-pathological amyloid with a well-defined physiological role and whose formation is regulated by one or more physiological signals [181].
- Phase separation
The creation of two distinct phases from a single homogeneous mixture as seen with oil and water. The effect of different types of compatible or incompatible/repulsive macromolecules within the cytoplasm or nucleus, magnified by the effects of macromolecular crowding leading to micro-compartmentalization [26, 37].
- P granule
P granules are liquid like RNA/protein membraneless condensates in the germline of C. elegans that are typically perinuclear [11].
- Prion
The word prion, coined in 1982 by Stanley Prusiner [182], is short for “proteinaceous infectious particle” and refers to misfolded proteins with the ability to self-replicate and confer their misfolded shape on normally folded molecules of the same protein and that is transmissible between individuals. The term “prionoid,” was introduced to describe amyloid aggregates that replicate within an organism and is transmissible between cells, but not between individuals [183, 184].
REFERENCES
- [1].Pechmann S, Willmund F, Frydman J. The ribosome as a hub for protein quality control. Mol Cell. 2013;49:411–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Monsellier E, Ramazzotti M, Taddei N, Chiti F. Aggregation propensity of the human proteome. PLoS Comput Biol. 2008;4:e1000199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med (Berl). 2003;81:678–99. [DOI] [PubMed] [Google Scholar]
- [4].Boke E, Ruer M, Wuhr M, Coughlin M, Lemaitre R, Gygi SP, et al. Amyloid-like Self-Assembly of a Cellular Compartment. Cell. 2016;166:637–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Guyonnet B, Egge N, Cornwall GA. Functional amyloids in the mouse sperm acrosome. Mol Cell Biol. 2014;34:2624–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hayes MH, Weeks DL. Amyloids assemble as part of recognizable structures during oogenesis in Xenopus. Biol Open. 2016;5:801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kistler KE, Trcek T, Hurd TR, Chen R, Liang FX, Sall J, et al. Phase transitioned nuclear Oskar promotes cell division of Drosophila primordial germ cells. eLife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pimentel RN, Navarro PA, Wang F, Robinson LG Jr., Cammer M, Liang F, et al. Amyloid-like substance in mice and human oocytes and embryos. J Assist Reprod Genet. 2019;36:1877–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Skuodas S, Clemons A, Hayes M, Goll A, Zora B, Weeks DL, et al. The ABCF gene family facilitates disaggregation during animal development. submitted. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Siniukova VA, Sopova JV, Galkina SA, Galkin AP. Search for functional amyloid structures in chicken and fruit fly female reproductive cells. Prion. 2020;14:278–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Seydoux G The P Granules of C. elegans: A Genetic Model for the Study of RNA-Protein Condensates. J Mol Biol. 2018;430:4702–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Strome S, Wood WB. Generation of asymmetry and segregation of germ-line granules in early C. elegans embryos. Cell. 1983;35:15–25. [DOI] [PubMed] [Google Scholar]
- [13].Zwicker D, Decker M, Jaensch S, Hyman AA, Julicher F. Centrosomes are autocatalytic droplets of pericentriolar material organized by centrioles. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E2636–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Skuodas S, Clemons A, Hayes M, Goll A, Zora B, Weeks DL, et al. The ABCF gene family facilitates disaggregation during animal development. Mol Biol Cell. 2020;31:1324–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rencus-Lazar S, DeRowe Y, Adsi H, Gazit E, Laor D. Yeast Models for the Study of Amyloid-Associated Disorders and Development of Future Therapy. Front Mol Biosci. 2019;6:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Loquet A, Saupe SJ, Romero D. Functional Amyloids in Health and Disease. J Mol Biol. 2018;430:3629–30. [DOI] [PubMed] [Google Scholar]
- [17].Rubel MS, Fedotov SA, Grizel AV, Sopova JV, Malikova OA, Chernoff YO, et al. Functional Mammalian Amyloids and Amyloid-Like Proteins. Life (Basel). 2020;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sergeeva AV, Galkin AP. Functional amyloids of eukaryotes: criteria, classification, and biological significance. Curr Genet. 2020;66:849–66. [DOI] [PubMed] [Google Scholar]
- [19].Santos J, Ventura S. Functional Amyloids Germinate in Plants. Trends Plant Sci. 2021;26:7–10. [DOI] [PubMed] [Google Scholar]
- [20].Nesterov SV, Ilyinsky NS, Uversky VN. Liquid-liquid phase separation as a common organizing principle of intracellular space and biomembranes providing dynamic adaptive responses. Biochim Biophys Acta Mol Cell Res. 2021:119102. [DOI] [PubMed] [Google Scholar]
- [21].Schneider N, Wieland FG, Kong D, Fischer AAM, Horner M, Timmer J, et al. Liquid-liquid phase separation of light-inducible transcription factors increases transcription activation in mammalian cells and mice. Sci Adv. 2021;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li W, Jiang H. Nuclear protein condensates and their properties in regulation of gene expression. J Mol Biol. 2021:167151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Xiao Q, McAtee CK, Su X. Phase separation in immune signalling. Nat Rev Immunol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wagh K, Garcia DA, Upadhyaya A. Phase separation in transcription factor dynamics and chromatin organization. Curr Opin Struct Biol. 2021;71:148–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Saito Y, Kimura W. Roles of Phase Separation for Cellular Redox Maintenance. Front Genet. 2021;12:691946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. 2017;18:285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Nover L, Scharf KD, Neumann D. Cytoplasmic heat shock granules are formed from precursor particles and are associated with a specific set of mRNAs. Mol Cell Biol. 1989;9:1298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sheth U, Parker R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science. 2003;300:805–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, et al. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science. 2009;324:1729–32. [DOI] [PubMed] [Google Scholar]
- [30].Closs CB, Conde-Petit B, Roberts ID, Tolstoguzov VB, Escher F. Phase separation and rheology of aqueous starch/galactomannan systems. Carbohydrate Polymers. 1999;39:67–77. [Google Scholar]
- [31].Ishimoto C, Goalwin PW, Sun ST, Nishio I, Tanaka T. Cytoplasmic phase separation in formation of galactosemic cataract in lenses of young rats. Proceedings of the National Academy of Sciences of the United States of America. 1979;76:4414–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Engelhardt E. Lafora and Tretiakoff: the naming of the inclusion bodies discovered by Lewy. Arq Neuropsiquiatr. 2017;75:751–3. [DOI] [PubMed] [Google Scholar]
- [33].Feric M, Vaidya N, Harmon TS, Mitrea DM, Zhu L, Richardson TM, et al. Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell. 2016;165:1686–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sabari BR, Dall’Agnese A, Young RA. Biomolecular Condensates in the Nucleus. Trends Biochem Sci. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kato M, McKnight SL. A Solid-State Conceptualization of Information Transfer from Gene to Message to Protein. Annu Rev Biochem. 2018;87:351–90. [DOI] [PubMed] [Google Scholar]
- [36].Peng A, Weber SC. Evidence for and against Liquid-Liquid Phase Separation in the Nucleus. Non-coding RNA. 2019; 5:50–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Alberti S, Gladfelter A, Mittag T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell. 2019;176:419–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Boeynaems S, Holehouse AS, Weinhardt V, Kovacs D, Van Lindt J, Larabell C, et al. Spontaneous driving forces give rise to protein-RNA condensates with coexisting phases and complex material properties. Proceedings of the National Academy of Sciences of the United States of America. 2019;116:7889–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Maharana S, Wang J, Papadopoulos DK, Richter D, Pozniakovsky A, Poser I, et al. RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science. 2018;360:918–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kato M, Lin Y, McKnight SL. Cross-beta polymerization and hydrogel formation by low-complexity sequence proteins. Methods. 2017;126:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Aguzzi A, Altmeyer M. Phase Separation: Linking Cellular Compartmentalization to Disease. Trends in cell biology. 2016;26:547–58. [DOI] [PubMed] [Google Scholar]
- [42].Berchowitz LE, Gajadhar AS, van Werven FJ, De Rosa AA, Samoylova ML, Brar GA, et al. A developmentally regulated translational control pathway establishes the meiotic chromosome segregation pattern. Genes Dev. 2013;27:2147–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Berchowitz LE, Kabachinski G, Walker MR, Carlile TM, Gilbert WV, Schwartz TU, et al. Regulated Formation of an Amyloid-like Translational Repressor Governs Gametogenesis. Cell. 2015;163:406–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Carpenter K, Bell RB, Yunus J, Amon A, Berchowitz LE. Phosphorylation-Mediated Clearance of Amyloid-like Assemblies in Meiosis. Dev Cell. 2018;45:392–405 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].McGlinchey RP, Lee JC. Why Study Functional Amyloids? Lessons from the Repeat Domain of Pmel17. J Mol Biol. 2018;430:3696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Alexandrov IM, Ivshina M, Jung DY, Friedline R, Ko HJ, Xu M, et al. Cytoplasmic polyadenylation element binding protein deficiency stimulates PTEN and Stat3 mRNA translation and induces hepatic insulin resistance. PLoS genetics. 2012;8:e1002457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Si K, Lindquist S, Kandel E. A possible epigenetic mechanism for the persistence of memory. Cold Spring Harbor symposia on quantitative biology. 2004;69:497–8. [DOI] [PubMed] [Google Scholar]
- [48].Khan MR, Li L, Perez-Sanchez C, Saraf A, Florens L, Slaughter BD, et al. Amyloidogenic Oligomerization Transforms Drosophila Orb2 from a Translation Repressor to an Activator. Cell. 2015;163:1468–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Saad S, Cereghetti G, Feng Y, Picotti P, Peter M, Dechant R. Reversible protein aggregation is a protective mechanism to ensure cell cycle restart after stress. Nat Cell Biol. 2017;19:1202–13. [DOI] [PubMed] [Google Scholar]
- [50].Petrovska I, Nüske E, Munder MC, Kulasegaran G, Malinovska L, Kroschwald S, et al. Filament formation by metabolic enzymes is a specific adaptation to an advanced state of cellular starvation. eLife. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Prouteau M, Loewith R. Regulation of Cellular Metabolism through Phase Separation of Enzymes. Biomolecules. 2018;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wang X, Chapman MR. Curli provide the template for understanding controlled amyloid propagation. Prion. 2008;2:57–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wang X, Hammer ND, Chapman MR. The molecular basis of functional bacterial amyloid polymerization and nucleation. J Biol Chem. 2008;283:21530–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lipke PN, Klotz SA, Dufrene YF, Jackson DN, Garcia-Sherman MC. Amyloid-Like beta-Aggregates as Force-Sensitive Switches in Fungal Biofilms and Infections. Microbiol Mol Biol Rev. 2018;82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Otoo HN, Lee KG, Qiu W, Lipke PN. Candida albicans Als adhesins have conserved amyloid-forming sequences. Eukaryot Cell. 2008;7:776–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ramsook CB, Tan C, Garcia MC, Fung R, Soybelman G, Henry R, et al. Yeast cell adhesion molecules have functional amyloid-forming sequences. Eukaryot Cell. 2010;9:393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Otzen D, Riek R. Functional Amyloids. Cold Spring Harb Perspect Biol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–32. [DOI] [PubMed] [Google Scholar]
- [59].Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16:574–81. [DOI] [PubMed] [Google Scholar]
- [60].Hipp MS, Kasturi P, Hartl FU. The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol. 2019;20:421–35. [DOI] [PubMed] [Google Scholar]
- [61].Ryno LM, Wiseman RL, Kelly JW. Targeting unfolded protein response signaling pathways to ameliorate protein misfolding diseases. Curr Opin Chem Biol. 2013;17:346–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Suzuki Y Emerging novel concept of chaperone therapies for protein misfolding diseases. Proc Jpn Acad Ser B Phys Biol Sci. 2014;90:145–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Shorter J, Southworth DR. Spiraling in Control: Structures and Mechanisms of the Hsp104 Disaggregase. Cold Spring Harb Perspect Biol. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Erives AJ, Fassler JS. Metabolic and Chaperone Gene Loss Marks the Origin of Animals: Evidence for Hsp104 and Hsp78 Chaperones Sharing Mitochondrial Enzymes as Clients. Plos One. 2015;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Cupo RR, Shorter J. Skd3 (human ClpB) is a potent mitochondrial protein disaggregase that is inactivated by 3-methylglutaconic aciduria-linked mutations. eLife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Chakravarty AK, Jarosz DF. More than Just a Phase: Prions at the Crossroads of Epigenetic Inheritance and Evolutionary Change. J Mol Biol. 2018;430:4607–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Halfmann R, Alberti S, Lindquist S. Prions, protein homeostasis, and phenotypic diversity. Trends in cell biology. 2010;20:125–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Shorter J, Lindquist S. Prions as adaptive conduits of memory and inheritance. Nat Rev Genet. 2005;6:435–50. [DOI] [PubMed] [Google Scholar]
- [69].Winkler J, Tyedmers J, Bukau B, Mogk A. Chaperone networks in protein disaggregation and prion propagation. Journal of structural biology. 2012;179:152–60. [DOI] [PubMed] [Google Scholar]
- [70].Chernova TA, Wilkinson KD, Chernoff YO. Prions, Chaperones, and Proteostasis in Yeast. Cold Spring Harbor Perspectives in Biology. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Lee DH, Goldberg AL. Hsp104 is essential for the selective degradation in yeast of polyglutamine expanded ataxin-1 but not most misfolded proteins generally. Biochemical and biophysical research communications. 2010;391:1056–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Knowles TP, Mezzenga R. Amyloid Fibrils as Building Blocks for Natural and Artificial Functional Materials. Adv Mater. 2016;28:6546–61. [DOI] [PubMed] [Google Scholar]
- [73].Andersson V, Hanzen S, Liu B, Molin M, Nystrom T. Enhancing protein disaggregation restores proteasome activity in aged cells. Aging (Albany NY). 2013;5:802–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Erjavec N, Larsson L, Grantham J, Nystrom T. Accelerated aging and failure to segregate damaged proteins in Sir2 mutants can be suppressed by overproducing the protein aggregation-remodeling factor Hsp104p. Genes Dev. 2007;21:2410–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Saarikangas J, Barral Y. Protein aggregates are associated with replicative aging without compromising protein quality control. eLife. 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Deville C, Carroni M, Franke KB, Topf M, Bukau B, Mogk A, et al. Structural pathway of regulated substrate transfer and threading through an Hsp100 disaggregase. Sci Adv. 2017;3:e1701726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Gates SN, Yokom AL, Lin J, Jackrel ME, Rizo AN, Kendsersky NM, et al. Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science. 2017;357:273–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. [DOI] [PubMed] [Google Scholar]
- [79].Goloubinoff P, Mogk A, Zvi AP, Tomoyasu T, Bukau B. Sequential mechanism of solubilization and refolding of stable protein aggregates by a bichaperone network. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:13732–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Sweeny EA, Jackrel ME, Go MS, Sochor MA, Razzo BM, DeSantis ME, et al. The Hsp104 N-terminal domain enables disaggregase plasticity and potentiation. Mol Cell. 2015;57:836–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Shorter J, Lindquist S. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science. 2004;304:1793–7. [DOI] [PubMed] [Google Scholar]
- [82].Kaimal JM, Kandasamy G, Gasser F, Andreasson C. Coordinated Hsp110 and Hsp104 Activities Power Protein Disaggregation in Saccharomyces cerevisiae. Mol Cell Biol. 2017;37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Shorter J The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS One. 2011;6:e26319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Mogk A, Kummer E, Bukau B. Cooperation of Hsp70 and Hsp100 chaperone machines in protein disaggregation. Front Mol Biosci. 2015;2:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Kummer E, Szlachcic A, Franke KB, Ungelenk S, Bukau B, Mogk A. Bacterial and Yeast AAA+ Disaggregases ClpB and Hsp104 Operate through Conserved Mechanism Involving Cooperation with Hsp70. J Mol Biol. 2016;428:4378–91. [DOI] [PubMed] [Google Scholar]
- [86].Rosenzweig R, Moradi S, Zarrine-Afsar A, Glover JR, Kay LE. Unraveling the mechanism of protein disaggregation through a ClpB-DnaK interaction. Science. 2013;339:1080–3. [DOI] [PubMed] [Google Scholar]
- [87].Seyffer F, Kummer E, Oguchi Y, Winkler J, Kumar M, Zahn R, et al. Hsp70 proteins bind Hsp100 regulatory M domains to activate AAA+ disaggregase at aggregate surfaces. Nat Struct Mol Biol. 2012;19:1347–55. [DOI] [PubMed] [Google Scholar]
- [88].Sielaff B, Tsai FT. The M-domain controls Hsp104 protein remodeling activity in an Hsp70/Hsp40-dependent manner. J Mol Biol. 2010;402:30–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Lipinska N, Zietkiewicz S, Sobczak A, Jurczyk A, Potocki W, Morawiec E, et al. Disruption of ionic interactions between the nucleotide binding domain 1 (NBD1) and middle (M) domain in Hsp100 disaggregase unleashes toxic hyperactivity and partial independence from Hsp70. J Biol Chem. 2013;288:2857–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Schirmer EC, Homann OR, Kowal AS, Lindquist S. Dominant gain-of-function mutations in Hsp104p reveal crucial roles for the middle region. Mol Biol Cell. 2004;15:2061–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Chamera T, Klosowska A, Janta A, Wyszkowski H, Obuchowski I, Gumowski K, et al. Selective Hsp70-Dependent Docking of Hsp104 to Protein Aggregates Protects the Cell from the Toxicity of the Disaggregase. J Mol Biol. 2019;431:2180–96. [DOI] [PubMed] [Google Scholar]
- [92].Zietkiewicz S, Lewandowska A, Stocki P, Liberek K. Hsp70 chaperone machine remodels protein aggregates at the initial step of Hsp70-Hsp100-dependent disaggregation. J Biol Chem. 2006;281:7022–9. [DOI] [PubMed] [Google Scholar]
- [93].Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–8. [DOI] [PubMed] [Google Scholar]
- [94].Karzai AW, McMacken R. A bipartite signaling mechanism involved in DnaJ-mediated activation of the Escherichia coli DnaK protein. J Biol Chem. 1996;271:11236–46. [DOI] [PubMed] [Google Scholar]
- [95].Liberek K, Marszalek J, Ang D, Georgopoulos C, Zylicz M. Escherichia coli DnaJ and GrpE heat shock proteins jointly stimulate ATPase activity of DnaK. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:2874–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Misselwitz B, Staeck O, Rapoport TA. J proteins catalytically activate Hsp70 molecules to trap a wide range of peptide sequences. Mol Cell. 1998;2:593–603. [DOI] [PubMed] [Google Scholar]
- [97].Liberek K, Skowyra D, Zylicz M, Johnson C, Georgopoulos C. The Escherichia coli DnaK chaperone, the 70-kDa heat shock protein eukaryotic equivalent, changes conformation upon ATP hydrolysis, thus triggering its dissociation from a bound target protein. J Biol Chem. 1991;266:14491–6. [PubMed] [Google Scholar]
- [98].Mayer MP. Hsp70 chaperone dynamics and molecular mechanism. Trends Biochem Sci. 2013;38:507–14. [DOI] [PubMed] [Google Scholar]
- [99].Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Nillegoda NB, Bukau B. Metazoan Hsp70-based protein disaggregases: emergence and mechanisms. Front Mol Biosci. 2015;2:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Nillegoda NB, Kirstein J, Szlachcic A, Berynskyy M, Stank A, Stengel F, et al. Crucial HSP70 co-chaperone complex unlocks metazoan protein disaggregation. Nature. 2015;524:247–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Kakkar V, Kuiper EF, Pandey A, Braakman I, Kampinga HH. Versatile members of the DNAJ family show Hsp70 dependent anti-aggregation activity on RING1 mutant parkin C289G. Sci Rep. 2016;6:34830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Nillegoda NB, Stank A, Malinverni D, Alberts N, Szlachcic A, Barducci A, et al. Evolution of an intricate J-protein network driving protein disaggregation in eukaryotes. eLife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Haslbeck M, Weinkauf S, Buchner J. Small heat shock proteins: Simplicity meets complexity. J Biol Chem. 2019;294:2121–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].McHaourab HS, Godar JA, Stewart PL. Structure and mechanism of protein stability sensors: chaperone activity of small heat shock proteins. Biochemistry. 2009;48:3828–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Shemesh N, Jubran J, Dror S, Simonovsky E, Basha O, Argov C, et al. The landscape of molecular chaperones across human tissues reveals a layered architecture of core and variable chaperones. Nature communications. 2021;12:2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Rampelt H, Kirstein-Miles J, Nillegoda NB, Chi K, Scholz SR, Morimoto RI, et al. Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J. 2012;31:4221–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Rousseau F, Serrano L, Schymkowitz JW. How evolutionary pressure against protein aggregation shaped chaperone specificity. J Mol Biol. 2006;355:1037–47. [DOI] [PubMed] [Google Scholar]
- [110].Yan Z, Yin M, Chen J, Li X. Assembly and substrate recognition of curli biogenesis system. Nature communications. 2020;11:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Wang X, Zhou Y, Ren JJ, Hammer ND, Chapman MR. Gatekeeper residues in the major curlin subunit modulate bacterial amyloid fiber biogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:163–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Maji SK, Perrin MH, Sawaya MR, Jessberger S, Vadodaria K, Rissman RA, et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science. 2009;325:328–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Beuret N, Hasler F, Prescianotto-Baschong C, Birk J, Rutishauser J, Spiess M. Amyloid-like aggregation of provasopressin in diabetes insipidus and secretory granule sorting. BMC Biol. 2017;15:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Liu Y, Liu T, Lei T, Zhang D, Du S, Girani L, et al. RIP1/RIP3-regulated necroptosis as a target for multifaceted disease therapy (Review). Int J Mol Med. 2019;44:771–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150:339–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Dean DN, Lee JC. pH-Dependent fibril maturation of a Pmel17 repeat domain isoform revealed by tryptophan fluorescence. Biochim Biophys Acta Proteins Proteom. 2019;1867:961–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].McGlinchey RP, Shewmaker F, McPhie P, Monterroso B, Thurber K, Wickner RB. The repeat domain of the melanosome fibril protein Pmel17 forms the amyloid core promoting melanin synthesis. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13731–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Badtke MP, Hammer ND, Chapman MR. Functional amyloids signal their arrival. Sci Signal. 2009;2:pe43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Greenwald J, Riek R. Biology of amyloid: structure, function, and regulation. Structure. 2010;18:1244–60. [DOI] [PubMed] [Google Scholar]
- [120].Patel A, Malinovska L, Saha S, Wang J, Alberti S, Krishnan Y, et al. ATP as a biological hydrotrope. Science. 2017;356:753–6. [DOI] [PubMed] [Google Scholar]
- [121].Hayes MH, Peuchen EH, Dovichi NJ, Weeks DL. Dual roles for ATP in the regulation of phase separated protein aggregates in Xenopus oocyte nucleoli. eLife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Sridharan S, Kurzawa N, Werner T, Gunthner I, Helm D, Huber W, et al. Proteome-wide solubility and thermal stability profiling reveals distinct regulatory roles for ATP. Nature communications. 2019;10:1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Huang H, Zhang X, Li S, Liu N, Lian W, McDowell E, et al. Physiological levels of ATP negatively regulate proteasome function. Cell Res. 2010;20:1372–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149:753–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Roden C, Gladfelter AS. RNA contributions to the form and function of biomolecular condensates. Nat Rev Mol Cell Biol. 2021;22:183–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Jarmoskaite I, Russell R. RNA helicase proteins as chaperones and remodelers. Annu Rev Biochem. 2014;83:697–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Hutchins EJ, Piacentino ML, Bronner ME. P-bodies are sites of rapid RNA decay during the neural crest epithelial-mesenchymal transition. BioRxiv. 2020. [Google Scholar]
- [128].Iserman C, Roden C, Boerneke M, Sealfon R, McLaughlin G, Jungreis I, et al. Specific viral RNA drives the SARS CoV-2 nucleocapsid to phase separate. bioRxiv. 2020. [Google Scholar]
- [129].Rhine K, Vidaurre V, Myong S. RNA Droplets. Annu Rev Biophys. 2020;49:247–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Tauber D, Tauber G, Parker R. Mechanisms and Regulation of RNA Condensation in RNP Granule Formation. Trends Biochem Sci. 2020;45:764–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Valencia JC, Rouzaud F, Julien S, Chen KG, Passeron T, Yamaguchi Y, et al. Sialylated core 1 O-glycans influence the sorting of Pmel17/gp100 and determine its capacity to form fibrils. J Biol Chem. 2007;282:11266–80. [DOI] [PubMed] [Google Scholar]
- [132].Ford L, Ling E, Kandel ER, Fioriti L. CPEB3 inhibits translation of mRNA targets by localizing them to P bodies. Proceedings of the National Academy of Sciences of the United States of America. 2019;116:18078–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Jamieson-Lucy A, Mullins MC. The vertebrate Balbiani body, germ plasm, and oocyte polarity. Curr Top Dev Biol. 2019;135:1–34. [DOI] [PubMed] [Google Scholar]
- [134].Weber SC, Brangwynne CP. Getting RNA and protein in phase. Cell. 2012;149:1188–91. [DOI] [PubMed] [Google Scholar]
- [135].Wickner RB, Shewmaker FP, Bateman DA, Edskes HK, Gorkovskiy A, Dayani Y, et al. Yeast prions: structure, biology, and prion-handling systems. Microbiol Mol Biol Rev. 2015;79:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Chakrabortee S, Byers JS, Jones S, Garcia DM, Bhullar B, Chang A, et al. Intrinsically Disordered Proteins Drive Emergence and Inheritance of Biological Traits. Cell. 2016;167:369–81 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Chakravarty AK, Smejkal T, Itakura AK, Garcia DM, Jarosz DF. A Non-amyloid Prion Particle that Activates a Heritable Gene Expression Program. Mol Cell. 2020;77:251–65 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Malato L, Dos Reis S, Benkemoun L, Sabate R, Saupe SJ. Role of Hsp104 in the propagation and inheritance of the [Het-s] prion. Mol Biol Cell. 2007;18:4803–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Sideri T, Yashiroda Y, Ellis DA, Rodriguez-Lopez M, Yoshida M, Tuite MF, et al. The copper transport-associated protein Ctr4 can form prion-like epigenetic determinants in Schizosaccharomyces pombe. Microb Cell. 2017;4:16–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Miller SB, Ho CT, Winkler J, Khokhrina M, Neuner A, Mohamed MY, et al. Compartment-specific aggregases direct distinct nuclear and cytoplasmic aggregate deposition. EMBO J. 2015;34:778–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Miller SB, Mogk A, Bukau B. Spatially organized aggregation of misfolded proteins as cellular stress defense strategy. J Mol Biol. 2015;427:1564–74. [DOI] [PubMed] [Google Scholar]
- [142].Tessarz P, Schwarz M, Mogk A, Bukau B. The yeast AAA+ chaperone Hsp104 is part of a network that links the actin cytoskeleton with the inheritance of damaged proteins. Mol Cell Biol. 2009;29:3738–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Huizar RL, Lee C, Boulgakov AA, Horani A, Tu F, Marcotte EM, et al. A liquid-like organelle at the root of motile ciliopathy. eLife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Cabral SE, Mowry KL. Organizing the oocyte: RNA localization meets phase separation. Curr Top Dev Biol. 2020;140:87–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Yang C, Wilson GM, Champion MM, Huber PW. Remnants of the Balbiani body are required for formation of RNA transport granules in Xenopus oocytes. bioRxiv. 2020;bioRxiv 2020.05.27.119487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Weeks DL, Melton DA. A maternal mRNA localized to the vegetal hemisphere in Xenopus eggs codes for a growth factor related to TGF-beta. Cell. 1987;51:861–7. [DOI] [PubMed] [Google Scholar]
- [147].Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol. 1983;100:64–119. [DOI] [PubMed] [Google Scholar]
- [148].Langdon EM, Qiu Y, Ghanbari Niaki A, McLaughlin GA, Weidmann CA, Gerbich TM, et al. mRNA structure determines specificity of a polyQ-driven phase separation. Science. 2018;360:922–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Woodruff JB, Hyman AA, Boke E. Organization and Function of Non-dynamic Biomolecular Condensates. Trends Biochem Sci. 2018;43:81–94. [DOI] [PubMed] [Google Scholar]
- [150].Chang CC, Lee WC, Cook CE, Lin GW, Chang T. Germ-plasm specification and germline development in the parthenogenetic pea aphid Acyrthosiphon pisum: Vasa and Nanos as markers. Int J Dev Biol. 2006;50:413–21. [DOI] [PubMed] [Google Scholar]
- [151].Houston DW, King ML. Germ plasm and molecular determinants of germ cell fate. Curr Top Dev Biol. 2000;50:155–81. [DOI] [PubMed] [Google Scholar]
- [152].Boke E, Mitchison TJ. The balbiani body and the concept of physiological amyloids. Cell cycle. 2017;16:153–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Trcek T, Lehmann R. Germ granules in Drosophila. Traffic (Copenhagen, Denmark). 2019;20:650–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Putnam A, Cassani M, Smith J, Seydoux G. A gel phase promotes condensation of liquid P granules in Caenorhabditis elegans embryos. Nat Struct Mol Biol. 2019;26:220–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Updike DL, Knutson AK, Egelhofer TA, Campbell AC, Strome S. Germ-granule components prevent somatic development in the C. elegans germline. Curr Biol. 2014;24:970–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Cushman-Nick M, Bonini NM, Shorter J. Hsp104 suppresses polyglutamine-induced degeneration post onset in a drosophila MJD/SCA3 model. PLoS genetics. 2013;9:e1003781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Ader C, Frey S, Maas W, Schmidt HB, Gorlich D, Baldus M. Amyloid-like interactions within nucleoporin FG hydrogels. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:6281–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Neil CR, Jeschonek SP, Cabral SE, O’Connell LC, Powrie EA, Wood TA, et al. L-bodies are novel RNA-protein condensates driving RNA transport in Xenopus oocytes. bioRxiv. 2020;bioRxiv 2020.05.08.084814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Aguero T, Zhou Y, Kloc M, Chang P, Houliston E, King ML. Hermes (Rbpms) is a Critical Component of RNP Complexes that Sequester Germline RNAs during Oogenesis. J Dev Biol. 2016;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Brangwynne CP, Mitchison TJ, Hyman AA. Active liquid-like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:4334–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Mais C, Scheer U. Molecular architecture of the amplified nucleoli of Xenopus oocytes. J Cell Sci. 2001;114:709–18. [DOI] [PubMed] [Google Scholar]
- [162].Franke WW, Kleinschmidt JA, Spring H, Krohne G, Grund C, Trendelenburg MF, et al. A nucleolar skeleton of protein filaments demonstrated in amplified nucleoli of Xenopus laevis. J Cell Biol. 1981;90:289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Gall JG, Wu Z, Murphy C, Gao H. Structure in the amphibian germinal vesicle. Exp Cell Res. 2004;296:28–34. [DOI] [PubMed] [Google Scholar]
- [164].Handwerger KE, Cordero JA, Gall JG. Cajal bodies, nucleoli, and speckles in the Xenopus oocyte nucleus have a low-density, sponge-like structure. Mol Biol Cell. 2005;16:202–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Han TW, Kato M, Xie S, Wu LC, Mirzaei H, Pei J, et al. Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell. 2012;149:768–79. [DOI] [PubMed] [Google Scholar]
- [166].Fazal FM, Han S, Parker KR, Kaewsapsak P, Xu J, Boettiger AN, et al. Atlas of Subcellular RNA Localization Revealed by APEX-Seq. Cell. 2019;178:473–90 e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Shen X, Mizuguchi G, Hamiche A, Wu C. A chromatin remodelling complex involved in transcription and DNA processing. Nature. 2000;406:541–4. [DOI] [PubMed] [Google Scholar]
- [168].Kanemaki M, Kurokawa Y, Matsu-ura T, Makino Y, Masani A, Okazaki K, et al. TIP49b, a new RuvB-like DNA helicase, is included in a complex together with another RuvB-like DNA helicase, TIP49a. J Biol Chem. 1999;274:22437–44. [DOI] [PubMed] [Google Scholar]
- [169].Zaarur N, Xu X, Lestienne P, Meriin AB, McComb M, Costello CE, et al. RuvbL1 and RuvbL2 enhance aggresome formation and disaggregate amyloid fibrils. EMBO J. 2015;34:2363–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Erzberger JP, Berger JM. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu Rev Biophys Biomol Struct. 2006;35:93–114. [DOI] [PubMed] [Google Scholar]
- [171].Gorynia S, Bandeiras TM, Pinho FG, McVey CE, Vonrhein C, Round A, et al. Structural and functional insights into a dodecameric molecular machine - the RuvBL1/RuvBL2 complex. Journal of structural biology. 2011;176:279–91. [DOI] [PubMed] [Google Scholar]
- [172].Baxi AB, Lombard-Banek C, Moody SA, Nemes P. Proteomic Characterization of the Neural Ectoderm Fated Cell Clones in the Xenopus laevis Embryo by High-Resolution Mass Spectrometry. ACS Chem Neurosci. 2018;9:2064–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Etard C, Wedlich D, Bauer A, Huber O, Kuhl M. Expression of Xenopus homologs of the beta-catenin binding protein pontin52. Mech Dev. 2000;94:219–22. [DOI] [PubMed] [Google Scholar]
- [174].Etard C, Gradl D, Kunz M, Eilers M, Wedlich D. Pontin and Reptin regulate cell proliferation in early Xenopus embryos in collaboration with c-Myc and Miz-1. Mech Dev. 2005;122:545–56. [DOI] [PubMed] [Google Scholar]
- [175].Simmer F, Moorman C, van der Linden AM, Kuijk E, van den Berghe PV, Kamath RS, et al. Genome-wide RNAi of C. elegans using the hypersensitive rrf-3 strain reveals novel gene functions. PLoS biology. 2003;1:E12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Narayanan A, Meriin A, Andrews JO, Spille JH, Sherman MY, Cisse, II. A first order phase transition mechanism underlies protein aggregation in mammalian cells. eLife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Inoue Y, Kawai-Noma S, Koike-Takeshita A, Taguchi H, Yoshida M. Yeast prion protein New1 can break Sup35 amyloid fibrils into fragments in an ATP-dependent manner. Genes to cells : devoted to molecular & cellular mechanisms. 2011;16:545–56. [DOI] [PubMed] [Google Scholar]
- [178].Dong J, Lai R, Jennings JL, Link AJ, Hinnebusch AG. The novel ATP-binding cassette protein ARB1 is a shuttling factor that stimulates 40S and 60S ribosome biogenesis. Mol Cell Biol. 2005;25:9859–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Su T, Izawa T, Thoms M, Yamashita Y, Cheng J, Berninghausen O, et al. Structure and function of Vms1 and Arb1 in RQC and mitochondrial proteome homeostasis. Nature. 2019;570:538–42. [DOI] [PubMed] [Google Scholar]
- [180].Ousalem F, Singh S, Chesneau O, Hunt JF, Boel G. ABC-F proteins in mRNA translation and antibiotic resistance. Res Microbiol. 2019;170:435–47. [DOI] [PubMed] [Google Scholar]
- [181].Si K, Kandel ER. The Role of Functional Prion-Like Proteins in the Persistence of Memory. Cold Spring Harb Perspect Biol. 2016;8:a021774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–44. [DOI] [PubMed] [Google Scholar]
- [183].Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron. 2009;64:783–90. [DOI] [PubMed] [Google Scholar]
- [184].Riek R The Three-Dimensional Structures of Amyloids. Cold Spring Harb Perspect Biol. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]