

Abstract
Pathogenic variants in GJB2, the gene encoding connexin 26, are the most common cause of autosomal-recessive hereditary deafness. Despite this high prevalence, pathogenic mechanisms leading to GJB2-related deafness are not well understood, and cures are absent. Humans with GJB2-related deafness retain at least some auditory hair cells and neurons, and their deafness is usually stable. In contrast, mice with conditional loss of Gjb2 in supporting cells exhibit extensive loss of hair cells and neurons and rapidly progress to profound deafness, precluding the application of therapies that require intact cochlear cells. In an attempt to design a less severe Gjb2 animal model, we generated mice with inducible Sox10iCreERT2-mediated loss of Gjb2. Tamoxifen injection led to reduced connexin 26 expression and impaired function, but cochlear hair cells and neurons survived for 2 months, allowing phenotypic rescue attempts within this time. AAV-mediated gene transfer of GJB2 in mature mutant ears did not demonstrate threshold improvement and in some animals exacerbated hearing loss and resulted in hair cell loss. We conclude that Sox10iCreERT2;Gjb2flox/flox mice are valuable for studying the biology of connexin 26 in the cochlea. In particular, these mice may be useful for evaluating gene therapy vectors and development of therapies for GJB2-related deafness.
Keywords: transgenic mouse, connexin 26, gap junction, cochlear GJB-2, DFNB1 deafness, hearing loss, AAV-Anc80L65 gene therapy, viral vector, inner hair cell
Graphical abstract
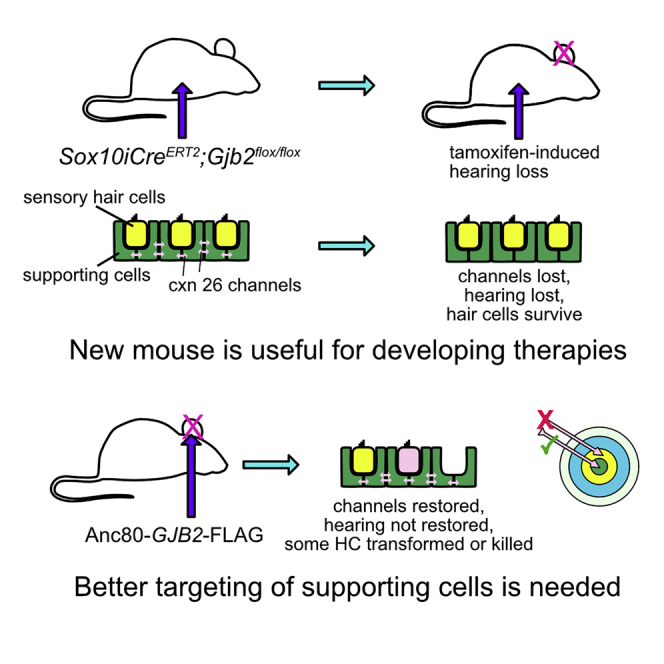
We generated mice with inducible Sox10iCreERT2-mediated loss of Gjb2. Tamoxifen led to reduced connexin 26 expression and impaired function, but hair cells and neurons survived for 2 months. AAV-mediated GJB2 gene transfer in mature mutant ears restored connexin 26 expression, induced hair cell loss, and failed to improve hearing.
Introduction
Hereditary deafness is diagnosed in approximately half of individuals who report hearing loss symptoms and receive medical attention and/or rehabilitation.1, 2, 3 The most common form of autosomal-recessive hereditary deafness (DFNB1) is due to pathogenic variants in GJB2, the gene encoding connexin 26 protein.4 Globally, GJB2 is the most common gene involved in hereditary deafness.5, 6, 7
Temporal bones of individuals with GJB2-related deafness may appear normal, and individuals with subtle defects in the temporal bone show no correlation between hearing ability and morphological changes.8 The cochlear sensory epithelium likely develops normally because persons homozygous for the 35delG are sometimes born with normal or near-normal hearing and then develop a profound loss,9 which is reflected by degeneration of the sensory epithelium.10 In addition, robust neuronal connectivity would not be likely to form and preserve unless there was initial sensory epithelium development. Because of the relatively good survival of auditory neurons (spiral ganglion neurons [SGNs]), individuals with GJB2-related deafness usually perform very well with cochlear implants that bypass the biological transduction of sound into action potentials. Still, molecular therapies aimed at phenotypic rescue and restoration of cochlear function are desirable, and mouse models have been generated to recapitulate the disease and provide a platform for testing molecular therapies.
Several mouse models have been generated to explore GJB2-related hearing loss and to aid in the design of future therapies. Homozygous germline deletion of connexin 26 is embryonic lethal;11 thus, loss-of-function mouse models for GJB2-related deafness have instead relied upon conditional deletion by Cre recombinase expression in specific cellular populations. The design of conditional deletion approaches is informed by spatial and temporal localization of connexin 26 in the cochlea. Connexin 26 is a main component in gap junctions, which are common in the inner ear and several other organs.12 The most prominent expression of Gjb2 occurs in non-sensory supporting cells (SCs), whereas hair cells (HCs) are devoid of connexin 26 and do not normally form gap junctions.13 Non-sensory cells that express Gjb2 are located outside the auditory epithelium, with the most prominent expression in the inner and outer sulcus regions that flank the organ of Corti medially and laterally, respectively. SCs in the organ of Corti also have gap junctions, but most of their junctions contain connexin 30 and not connexin 26.13 Connexin 26 is also present in other areas of the cochlea, most notably in fibrocytes and spiral ligament regions associated with the stria vascularis.13
To date, germline and somatic Gjb2 Cre-mediated conditional deletion mice have exhibited severe hearing loss and cochlear epithelial degeneration.14, 15, 16, 17 In contrast, human patients with GJB2-related deafness have variable hearing loss and perform well with cochlear implants, suggesting intact SGNs. Loss of Gjb2 throughout the ear, using Pax2Cre or Foxg1Cre mice16 or R26RCreERT2 mice,18 results in severe hearing loss and degeneration of cochlear HCs, SCs, and SGNs, indicating critical roles for connexin 26 during development. Restriction of Gjb2 deletion to cochlear non-sensory cells using Sox10Cre mice also results in hearing loss and severe cochlear pathology,14,17 suggesting that non-sensory cells are uniquely sensitive to Gjb2 disruption during development. Taken together, these observations show that deletion of Gjb2 in non-sensory cells during or after development results in hearing loss with cellular degeneration. The severe cochlear phenotypes observed in these mouse models has precluded their use for studies of pharmacological treatment or gene replacement therapy, which depend upon an intact epithelium and neural tissues.
To address the need for a less severe animal model in which critical cells in the cochlea survive, we sought to generate mice with Sox10iCreERT2-mediated Gjb2 loss only in a subset of SCs. We predicted this would allow comparison of the effects of Gjb2 deletion in developing versus mature cochleae and provide a window of opportunity for treatment. Sox10iCreERT2 mice19 express Cre in non-sensory cells in the cochlea after induction by tamoxifen.
We injected tamoxifen to delete Gjb2 at postnatal day (P) 1 (early stage of differentiation for cochlear epithelium) and P14 (after the onset of hearing) and determined that following the deletion at both time points, mice displayed elevated hearing thresholds, reduced distortion product otoacoustic emissions (DPOAEs), reduced endocochlear potential (EP), and intact HC and SGNs up to 2 months postinduction. The functional deficits were more severe in the P1 injected animals. HCs and SGNs began degenerating soon after P60 and showed marked degeneration starting 4 months postinduction. Delivery of an ancestral adeno-associated viral vector (AAV), Anc80, with the human GJB2 (hGJB2) cDNA into P28 cochleae resulted in robust transgene expression in SCs. The transgene was also expressed in HCs, and inner HC (IHC) degeneration was apparent. These studies present a window for phenotypic rescue intervention, as HCs and SGNs survive for approximately 2 months after deletion of connexin 26, showing the potential for AAV gene therapy and confirming that connexin 26 has important roles both during development and in the mature cochlea.
Results
Sox10iCre is expressed in the auditory epithelium
To assess Sox10iCreERT2 activity in the inner ear, we crossed Sox10iCreERT2 male and Rosa26R(R26R)-LacZ reporter female mice and injected pregnant R26R dams intraperitoneally with tamoxifen at embryonic day (E) 11.5. Cryo-sections of inner ears from embryos dissected at E17.5 were stained for β-galactosidase activity, which was detected in the cochlear epithelium (Figures 1A and 1B) and in the cochleovestibular ganglion, primarily in neurons but also in some Schwann cells (Figure 1C). We also crossed Sox10iCreERT2 male and Rosa26R(R26R)-EYFP reporter female mice and analyzed their progeny. Whole mounts of the cochlear epithelium were obtained from P16 Sox10iCreERT2;EYFP mice that were injected with tamoxifen at P14. Ears were stained with phalloidin to visualize F-actin in adherens junctions, which helps depict organ of Corti organization and identify IHCs and outer HCs (OHCs) (Figure 1D). EYFP+ reporter staining was visible in several types of non-sensory cells, including the pillars and Deiters cells, consistent with Sox10 expression restricted to SCs, as previously shown.20 These studies confirm that Sox10iCreERT2 mice express Cre recombinase in cochlear non-sensory cells in response to tamoxifen treatment and demonstrate that the level of expression is variable among these cells.
Figure 1.

Sox10iCreERT2 is expressed in developing inner ear SCs and cochlear ganglion.
(A–C) Sox10iCreERT2 mice crossed with Rosa26R(R26R)-LacZ reporter mice were injected with tamoxifen at E11.5 and dissected at E17.5. Shown are sagittal cryo-sections through the ear of a Sox10iCreERT2;Rosa26-LacZ mouse stained with X-gal and counter-stained with eosin. (B) and (C) are enlargements of the regions shown in (A). Blue indicates cells expressing the inducible Sox10iCreERT2 allele, including the epithelium of the cochlear duct (in B) and the ganglion (g; in C) where both neurons and Schwann cells are positive. (D) Sox10iCreERT2 mice crossed with Rosa26-EYFP reporter mice were injected with tamoxifen at postnatal day 14 (P14) and dissected at P16. Whole mount of the cochlear epithelium stained for phalloidin (red) and EYFP (green) shows Cre reporter label in inner sulcus cell (isc), apical region of inner hair cell (ap), inner hair cell (ihc), inner phalangeal cell (iph), inner pillar cell (ipc), outer pillar cell (opc), Hensen cell (H), Deiters cell (D), and rows of outer hair cells (1, 2, and 3). Scale bars, 100 μm (A) and 25 μm (B–D).
Gjb2 contributes to coat color
Sox10 plays an important role in the development and maintenance of the melanocyte lineage,21 and some individuals with heterozygous missense variants in GJB2 exhibit skin abnormalities, including keratitis and ichthyosis.22 In mice, Gjb2 is expressed in the stratum spinosum and granulosum of the skin at E19 but is downregulated in the adult.23 Gjb2 is also expressed in P3 mouse hair follicles, on the basis of next-generation sequencing.24 We observed that Sox10CreERT2;Gjb2flox/flox (inducible conditional knockout [iCKO]) mice treated with tamoxifen at P1 exhibit a silver coat color that is visible as early as P28 and becomes more pronounced with age (Figure S1). Interestingly, iCKO mice not treated with tamoxifen (thus retaining connexin 26) have black coat color, as do iCKO mice injected with tamoxifen at P14. Together, these observations suggest that Sox10-expressing melanocytes are sensitive to loss of Gjb2 at P1 but not at P14. These observations also suggest a previously unreported role for connexin 26 in melanocyte development in the skin.
Early or late postnatal deletion of connexin 26 has minimal effects on HC survival
We next tested the effects of Gjb2 deletion on the function and structure of the auditory periphery using tamoxifen injections in the developing cochlea, at P1, or in the structurally near-mature cochlea, at P14, and then analyzed the effects 21 days after injection (Figure 2). The same group of mice was used for histology, auditory brainstem response (ABR) audiometry, DPOAE, and EP tests. Whole mounts of the auditory epithelium from tamoxifen-injected control mice (defined in Materials and methods) stained for phalloidin and connexin 26 revealed the three rows of OHCs and the actin-rich pillar cells, along with the typical distribution of connexin 26 plaques in SC-SC junctions (Figure 2A). The connexin 26-rich junctions could be seen both medial and lateral to the sensory epithelium. The expression of connexin 26 was seen in all non-sensory cells, but due to focal plane differences, the intensity in a given image appears stronger in some cells than others. In comparison, iCKO mice injected with tamoxifen at P1 and analyzed at P21 had almost complete lack of connexin 26 in the sensory epithelium but normal-appearing cochlear HCs and organ of Corti cytoarchitecture (Figure 2B). Tamoxifen injection of iCKO mice at P14 induced a partial loss of connexin 26, such that some cells retained punctate staining and others were completely devoid of connexin 26 (Figure 2C). In addition, HC loss was minimal in iCKO mice injected with tamoxifen at P14, with absence of a few first-row OHCs. Together, these data suggest that inner ear HCs can sustain severe early (P1) or mild later (P14) loss of Gjb2 without major effects on their survival or integrity at least until 21 days postinduction.
Figure 2.

Tamoxifen injection to iCKO mice leads to reduction in connexin 26, elevation of ABR, and DPOAE thresholds and reduction in EP.
(A–C) Whole mounts of the organ of Corti and adjacent SC areas stained for connexin 26 (green, arrows) and F-actin (phalloidin, red) to depict HCs and their borders. (A) Abundant connexin 26 is seen in the inner sulcus area (medial aspect of the organ of Corti, top of image), and the outer sulcus (bottom of image) in control cochlea. In iCKO cochleae, early tamoxifen injection (B) leads to nearly complete loss of connexin 26 (arrow), whereas late injection results in partial loss (C). Scale bar in A (for A–C), 100 μm. (D–I) Deterioration of functional measures seen after both early and late tamoxifen injections in iCKO mice (CKO) compared with uninjected controls (wild-type [WT]) is summarized here; see text for detailed statistical analyses. (D) and (E) Significant elevation of ABR thresholds was seen after both early (D; P1) and late (E; P14) tamoxifen injections (CKO versus WT) at both tested frequencies (p < 0.0001). Loss of hearing was slightly greater in mice injected at the earlier age (P1 CKO [D] > P14 CKO [E], p < 0.05). (F) and (G) Significant elevation of DPOAE thresholds was seen after both early (F; P1) and late (G; P14) tamoxifen injections (CKO versus WT) at both tested frequencies (p < 0.0001). The differences between ages of injection (P1 CKO [F] versus P14 CKO [G]) were not meaningful. (H and I) Significant depression of endocochlear potential was seen after both early (H; P1, p < 0.02) and late (I; P14, p < 0.01) tamoxifen injections (CKO versus WT). Because of large variances and small sample sizes, the difference in loss of function between ages of injection (P1 [H] versus P14 [I]) was not significant (p > 0.05). ∗p < 0.02, ∗∗p < 0.01, and ∗∗∗∗p < 0.001.
Loss of Gjb2 in developing or near-mature ears results in hearing loss
We next assayed whether loss of Gjb2 at P1 or P14 disrupts hearing thresholds. ABR audiometry was used to determine the amount of sound exposure necessary to elicit electrical responses in the auditory brainstem (Figures 2D and 2E). Deletion of Gjb2 at P1 in iCKO mice resulted in threshold shifts at both 12 and 24 kHz, consistent with profound hearing loss (F = 460.2, df = 2 and 14, p < 0.0001); the increase was significant for each tested frequency (12 kHz: F = 811.9, df = 1 and 15, p < 0.0001; 24 kHz: F = 407.6, df = 1 and 15, p < 0.0001). Deletion of Gjb2 at P14 in iCKO mice resulted in a slightly less severe threshold shift (overall: F = 51.0, df = 2 and 19, p < 0.0001; 12 kHz: F = 101.3, df = 1 and 20, p < 0.0001; 24 kHz: F = 94.7, df = 1 and 20, p < 0.0001). Control mice injected at P14 had higher hearing thresholds than control mice injected at P1, whereas iCKO mice injected at P14 had lower thresholds than iCKO mice injected at P1, indicating a significantly smaller effect of the later injection (F = 4.58, df = 2 and 34, p = 0.0173). These data suggest that Gjb2 is required both in developing and near-mature ears for proper hearing function in adults.
Mild loss of OHCs in iCKO mice, along with increased ABR hearing thresholds, led us to ask whether other functions of the inner ear may be sensitive to Gjb2 loss. Connexin 26 protein forms gap junctions that facilitate potassium transfer between cells in the cochlear epithelium. We theorized that deficient potassium transfer leading to reduction in EP would correlate with impaired active cochlea. We therefore measured DPOAEs (Figures 2F and 2G) at two frequencies, 12 and 24 kHz. Deletion of Gjb2 in iCKO mice resulted in high thresholds for eliciting DPOAEs at 12 and 24 kHz following both P1 and P14 tamoxifen injections. For mice injected at P1, thresholds were significantly elevated in iCKO mice relative to control (F = 375.2, df = 2 and 10, p < 0.0001), and the increase was significant for each frequency tested (12 kHz: F = 72.4, df = 1 and 11, p < 0.0001; 24 kHz: F = 730.1, df = 1 and 11, p < 0.0001). Results were similar for mice injected at P14 (overall: F = 43.0, df = 2 and 18, p < 0.0001; 12 kHz: F = 50.0, df = 1 and 19, p < 0.0001; 24 kHz: F = 80.6, df = 1 and 19, p < 0.0001). Direct comparisons between the two ages of deletion were not meaningful because thresholds for both groups of iCKO mice were near or above the upper limit of the stimulus intensity. Both groups clearly exhibited severe hearing loss, but it was not possible to determine which condition was worse.
We measured EP in controls and iCKO mice that had been injected with tamoxifen at P1 or P14. EP was measured at P21 for the mice injected at P1 and at P35 for the mice injected at P14 (Figures 2H and 2I). For both time points, iCKO mice had significantly lower EP compared with WT controls, with the earlier time point injection resulting in more severe EP reduction. For mice injected at P1, EP was significantly reduced in iCKO mice relative to controls (F = 55.6, df = 1 and 2, p = 0.0175). The results were similar for mice injected at P14 (F = 13.1, df = 1 and 7, p = 0.0085). Analysis of the combined dataset revealed that the difference in EP between genotypes was not significantly affected by the age of injection (F = 0.501, df = 1 and 9, p = 0.497).
Taken together, these results suggest that deletion of Gjb2 early in development or close to maturation of the inner ear leads to reduction in EP, implicating potassium recycling and dysfunction of the stria vascularis in the loss of hearing. The severity of elevation in thresholds, especially in the earlier deletion of Gjb2, suggests that other pathologies also arise because of Gjb2 loss, possibly affecting the active cochlear amplification, as previously suggested.18
Balance
Previous studies using Sox10Cre;Gjb2 knockout mice showed that loss of Gjb2 in SCs results in severe hearing loss and degeneration of epithelial and neuronal cochlear components but does not disrupt vestibular function.25 The lack of vestibular phenotypes in Sox10Cre;Gjb2 knockout mice cannot be explained by compensation by other connexins13 or reconciled with the prolonged time course of development of vestibular structures.26 Nevertheless, we found it important to determine the vestibular function of the novel mutant described here. Specifically, we asked whether loss of Gjb2 disrupts vestibular function in ears depleted of endogenous connexin 26 in the mature state. To test this, we considered that the vestibular periphery matures several days after the auditory part of the inner ear26 and therefore injected tamoxifen at P21 and performed rotarod tests at P49 and P71 (Table 1). At P49 (trials 1a–1c), there was not a statistically significant difference in rotarod performance between iCKO and control mice (F = 4.08, df = 1 and 8, p = 0.078). At P71 (trials 2a–2c), iCKO mice had significantly worse performance on the rotarod compared with control mice (F = 23.6, df = 1 and 8, p = 0.0013). Some iCKO mutant mice performed similar to controls in early trials but exhibited worse performance (shortened duration) on the rotarod in later trials. Thus, Gjb2 deletion at P21 led to a moderate decrease in vestibular function, with variation among mice in both the extent of functional impairment and timing, with some mice showing defects at the late, but not earlier, time points.
Table 1.
The time span (in seconds) mice stayed on the rotating rod in each of the trials
| Trial 1 (P49) |
Trial 2 (P71) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| ID | Genotype | 1a | 1b | 1c | Mean | 2a | 2b | 2c | Mean |
| 3336 | iCKO | 56 | 76 | 65 | 66 | 71 | 70 | 65 | 69 |
| 3337 | iCKO | 57 | 67 | 122 | 82 | 52 | 62 | 73 | 62 |
| 3334 | iCKO | 125 | 54 | 91 | 90 | 78 | 94 | 60 | 77 |
| 3343 | iCKO | 38 | 68 | 77 | 61 | 48 | 94 | 60 | 67 |
| 3345 | iCKO | 76 | 111 | 107 | 98 | 49 | 100 | 111 | 87 |
| Mean | 70 | 75 | 92 | 79 | 60 | 84 | 74 | 72 | |
| 3338 | control | 112 | 80 | 134 | 109 | 112 | 76 | 108 | 99 |
| 3339 | control | 76 | 79 | 106 | 87 | 83 | 73 | 106 | 87 |
| 3340 | control | 92 | 118 | 50 | 87 | 61 | 120 | 111 | 97 |
| 3341 | control | 86 | 82 | 118 | 95 | 89 | 111 | 128 | 109 |
| 3342 | control | 112 | 89 | 96 | 99 | 106 | 130 | 99 | 112 |
| Mean | 96 | 90 | 101 | 95 | 90 | 102 | 110 | 101 | |
At P49, the difference between genotypes was not statistically significant (F = 4.08, df = 1 and 8, p = 0.078). At P71, the difference between genotypes was statistically significant (F = 23.6, df = 1 and 8, p = 0.0013). Connexin 26 depletion led to a slight decrease in vestibular function.
Long-term effects of Gjb2 deletion
We next asked whether prolonged loss of Gjb2 (beyond 21 days) in near-mature ears would be deleterious to inner ear structure. To test this, we injected iCKO mice with tamoxifen at P14 and assessed cochlear morphology 2 or 4 months later. Specimens were processed either as whole mounts or plastic sections to facilitate evaluation of both sensory epithelium and SGNs. Time-matched control mice received tamoxifen at P14. Cochlear structures in tamoxifen-injected iCKO mice were normal appearing at 2 months, whereas at 4 months postinjection, degenerative changes were observed in the inner sulcus cells and the organ of Corti in a base-to-apex gradient (Figure 3). In the base of the cochlea, the sensory epithelium had degenerated into a flat epithelium, and many SGNs were absent. Inner ears of control mice appeared normal at 2 and 4 months of age. Taken together, these data suggest that the mild inner ear phenotype caused by Gjb2 deletion at P14 involves no degeneration of HCs and SGNs at 2 months, but progressive degeneration starts later in the organ of Corti and the auditory nerve. This also suggests that there is a 6 week window of opportunity that may be targeted for phenotypic rescue.
Figure 3.

iCKO ears retain normal morphology for 2 months, but pathology develops later.
Whole mounts of the organ of Corti area stained with phalloidin (A, D, and G), plastic sections of the organ of Corti area (B, E, and H), and plastic sections of Rosenthal’s canal (C, F, and I) from iCKO mice that received tamoxifen at P14 and were analyzed at 2 months of age (A, B, and C), or 4 months (D–I). HCs and SGNs appear intact at the 2 month time point (A–C). In (A), bracket shows OHCs and arrow shows pillar cells. In (B), arrows show presence of OHCs (on left) and IHC. In (C), arrow points to a SGN. In the apical area of a 4 months post-tamoxifen ear, some HC loss is seen (arrows in D and E), but Rosenthal’s canal is nearly normal (F). The basal part of this ear has a flat epithelium replacing the organ of Corti (G and H), and the SGNs in Rosenthal’s canal are almost entirely absent (I). Scale bars, 25 μm.
Delivery of Anc80-eGFP to the cochlea results in SC and HC transduction
Having determined that iCKO mice treated with tamoxifen exhibit hearing loss and delayed-onset HC degeneration (by 2 months), we hypothesized that restored expression of Gjb2 during this 2 month time period might prevent or slow the progression of cochlear pathology. To test this, we first used Anc80-eGFP viral vectors, which have been shown to express GFP in SCs after surgical injection into perilymph in neonates.27 Control animals (1–4 months of age) received Anc80-eGFP, and cochlear whole mounts were prepared 2 weeks later. Preparations were evaluated by immunofluorescence for GFP expression and staining of myosin VIIa (HC marker). In HCs, intense GFP expression was detected throughout the cytoplasm and nucleus (Figure 4). GFP was detected in all IHCs and many OHCs throughout the cochlea. Robust GFP staining was also observed in SCs and in the cytoplasm and nucleus (Figure 4). Among the types of SCs that were positive for GFP were inner sulcus cells, inner phalangeal cells, inner pillar cells, outer pillar cells, Deiters cells, Hensen cells, and Claudius cells.
Figure 4.

Anc80-eGFP expression is seen in HCs and SCs in wild-type mice.
Two weeks after virus injection into mature mouse ears, in mid-cochlea (A), all IHCs (I in A) and some OHCs (O), and many SCs (S), express GFP. In the apex (B), all IHCs and most OHCs are GFP positive. Green, GFP; blue, myosin VIIa. Scale bar, 20 μm.
Anc80-GJB2-FLAG localizes to membranous subcellular structures in cochlear SCs and IHCs
We then tested the activity of an AAV-hGJB2 vector. We used an Anc80-GJB2-FLAG vector in control mice (7–16 weeks old), and stained with antibodies either to connexin 26 or the FLAG. On the basis of the design of the vector, detection of the FLAG should localize vector-mediated connexin 26, but not the endogenous connexin 26. We then assessed expression of Anc80-GJB2-FLAG injected control cochleae 1 week, 2 weeks, and 1 month after surgery, staining for myosin VIIa, FLAG or connexin 26, and F-actin (phalloidin). In contrast with Anc80-eGFP, Anc80-GJB2-FLAG localized mainly to the membrane of SCs and IHCs (Figures 5A and 5B, with connexin 26 and FLAG antibody, respectively). FLAG expression was detected in SCs both medial and lateral to the sensory epithelia. Furthermore, anti-FLAG staining in SCs of mice injected with Anc80-GJB2-FLAG appeared as a dotted line (Figure 5C, FLAG antibody), similar to the expression pattern of connexin 26 in control mice. HCs do not normally express GJB2, but after Anc80-GJB2-FLAG injection, some IHCs were absent, and the remaining IHCs displayed anti-FLAG at their lateral membrane, appearing as a continuous line (Figures 5D and 5E, connexin 26 antibody).
Figure 5.

Distribution and effects of Anc80-GJB2-FLAG in control cochleae.
AAV was injected into mature ears and analysis done after 1 week or 1 month. Staining is with connexin 26 antibody except for (B) and (C), which are stained for FLAG. Most OHCs (O) and IHCs (I) are present at 1 week after Anc80-GJB2-FLAG injection in both apex (A) and mid-cochlea (B), but some IHC loss is evident, especially in the base (arrows point to sites of missing IHCs). FLAG-tagged connexin 26 (green) is expressed in many SCs, in association with the plasma membranes of IHCs, and in the cytoplasm of OHCs. Red is myosin VIIa. (C) Higher magnification image of inner sulcus area 1 month after injection of Anc80-GJB2-FLAG. Green, FLAG; blue, phalloidin. (D) Confocal image with cross-sections (D′, D′′) showing membranal connexin 26 in IHCs (green). (E) High magnification showing connexin 26 (green) at a junction between two IHC; absence of intercalating SC is indicated by the lack of a visible gap in the red channel (myosin VIIa; E′). One month after AAV injection, in both apex (F) and mid-cochlea (G), nearly all IHCs are lost (arrows point to their normal sites), but most OHCs survive. Many OHCs continue to exhibit cytoplasmic connexin 26 (yellow arrowheads). Scale bars, 20 μm (A, B, D, F, and G), 10 μm (C), and 5 μm (E).
The fluorescence was especially intense in areas between two adjacent IHCs (Figure 5D′′ and 5E, connexin 26 antibody). The expression pattern of Anc80-GJB2-FLAG in OHCs was mostly cytoplasmic, similar to that of Anc80-eGFP. These data also demonstrate the similar pattern and localization of the viral-mediated transgene when stained for the FLAG sequence or for connexin 26 (compare Figures 5B and 5C stained for the FLAG with Figures 5A and 5D–5G stained for connexin 26).
The degeneration of IHCs following Anc80-GJB2-FLAG administration into inner ears of control mice (7–16 weeks of age) was quantified, with Anc80-eGFP serving as control (Figure 6). One week after the administration (Figure 6A), the population of OHCs appeared normal, but a few IHCs were already missing (compare with a control in Figure 2A). The average numbers of surviving IHCs at 1 week were 10.5, 9.9, and 9.4 in apex, middle, and basal turn, respectively (Figure 6D′, light gray). The numbers of IHCs continued to decline 2 weeks and 1 month after the surgery (Figures 6B and 6C, respectively). The decline in IHC number over time is plotted in Figure 6 D′. We compared the data at 2 weeks post-vector injection between the group that received Anc80-GJB2-FLAG and the control group receiving Anc80-eGFP (Figure 6D). The number of IHCs after Anc80-GJB2-FLAG administration was significantly lower than the GFP group (4.9 versus 21.8 in apical turn, 4.2 versus 18.9 in middle turn, 3.6 versus 14.5 in basal turn). In contrast, to the loss of IHCs, OHCs appeared intact (Figures 6A–6C).
Figure 6.

HC survival after administration of Anc80-GJB2-FLAG or Anc80-eGFP into control mice.
AAV was injected into mature ears and analysis done after 1 or 2 weeks or 1 month. (A) Most IHCs (I) and OHCs (O and bracket) present at 1 week. Red, myosin VIIa. (B) Most IHCs are absent at 2 weeks, but OHCs appear intact. (C) OHCs are still present at 1 month, but IHCs are absent. Scale bar in A (for A–C), 100 μm. (D) Bar charts showing differences between vectors (Anc80-GJB2-FLAG, blue; Anc80-eGFP, green) in the number of IHCs present per field of view (mean ± SE; fields are as shown in A–C) at the 2 week time point in apex, middle, and base of the cochlea. D′ shows the change in IHC survival over time after injection of Anc80-GJB2-FLAG. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p = 0.0001. There is significantly greater loss of IHCs at 2 weeks after Anc80-GJB2-FLAG injection than after Anc80-eGFP injection, both overall (F = 9.43, df = 3 and 9, p = 0.0039) and in each region that was sampled (apex, F = 33.60, df = 1 and 11, p = 0.0001; middle, F = 24.52, df = 1 and 11, p = 0.0004; base, F = 14.52, df = 1 and 11, p = 0.0029). Although the number of surviving IHCs tended to decrease over time (D′), variances were large, and the overall pattern was not significant (F = 2.21, df = 2 and 15, p = 0.0717). The only significant difference detected between subgroups was a large loss of IHCs in the base between 1 and 2 weeks (F = 6.64, df = 1 and 10, p = 0.0276).
Anc80-GJB2-FLAG in iCKO ears restores expression of connexin 26 in SCs
It was next necessary to test the effects of Anc80-GJB2-FLAG expression in GJB2-iCKO mice. For that purpose, we injected tamoxifen at one of two time points, P1 or P14. Mice in the P1 induction group, assessed 1 month after administration of Anc80-GJB2-FLAG into the left ear, exhibited a connexin 26 staining pattern (Figure 7A, connexin 26 staining) similar to control mice (Figure 2A). In contrast, the contralateral ears showed a nearly complete depletion of connexin 26 (Figure 7B). This suggests that injecting the Anc80-GJB2-FLAG vector restores connexin 26 in SCs (Figure 7A). Expression in the membrane of SCs was robust in the inner sulcus and outer sulcus areas, presenting as a dotted line-pattern similar to connexin 26 expression in control mice. In addition, there was weak connexin 26 staining in the area of pillar cells, Deiters cells, and Hensen cells. Myosin VIIa staining showed substantial loss of IHCs (Figure 7A), similar to that seen in control ears 1 month after Anc80-GJB2-FLAG injection. In addition, mild OHC loss was observed in some segments along the entire cochlear duct. Because there was no OHC loss in control mice injected with Anc80-GJB2-FLAG, we conclude that the mild loss of OHCs in iCKO mice injected with Anc80-GJB2-FLAG reflects the combined impact of GJB2 ablation in SCs and cytoplasmic expression of GJB2 in OHCs.
Figure 7.

Anc80-GJB2-FLAG delivery results in increased connexin 26 expression and IHC loss.
Ear of iCKO mouse given tamoxifen at P1, Anc80-GJB2-FLAG in the left ear at P28, and euthanized 1 month later (A) showing increased connexin 26 expression (green, arrows) and reduced IHC survival (rectangles show areas of missing IHCs) compared with the respective contralateral ear (B). HCs are labeled with myosin VIIa antibody (red). Similarly, left ear of iCKO mouse given tamoxifen at P14, Anc80-GJB2-FLAG in the left ear at P28 and euthanized 2 months later (C) shows more connexin 26 in SCs (arrow) and pronounced IHC loss (rectangle) compared with the contralateral ear (D). Connexin 26 detection is by antibody. Scale bar, 20 μm (A–D).
Partial restoration of connexin 26 was also seen in SCs of ears of the P14 induction group cochleae (Figure 7C) along with loss of IHCs. Because ears in Figure 7 are stained with an antibody to connexin 26, which does not distinguish between the endogenous ligand and the vector-mediated connexin 26, the contralateral ear control is essential. However, the contralateral ears may underestimate the degree of depletion of the endogenous protein, because AAV contralateral spread28 may be responsible for some of the connexin 26 staining in these ears. In the contralateral control ears, depletion of connexin 26 was near complete and HCs remained intact (Figure 7D). Thus, administration of Anc80-GJB2-FLAG at P28 enhanced the connexin 26 expression in the SCs in both P1 and P14 tamoxifen groups, and in parallel, resulted in the loss of IHCs.
Delivery of Anc80-GJB2-FLAG results in connexin 26 expression in IHC, IHC loss, and no physiological recovery in iCKO mice
Having determined that Anc80-GJB2-FLAG administration can result in expression of GJB2 transgene in SCs, where it was deleted after tamoxifen, and that in addition to SCs, IHCs also express connexin 26, it became important to assess the functional outcome of this gene transfer procedure. For that purpose, ABR thresholds were measured in three groups of animals: injected with normal saline, Anc80-eGFP, and Anc80-GJB2-FLAG. Control animals that received a saline injection in the left ear had significantly higher hearing thresholds in that ear at 1 week after injection than did uninjected control animals (Table 2; Figure 8A). Thresholds of the injected ears remained high at 2 weeks and were not significantly different from the 1 week thresholds. Injection of the Anc80-eGFP vector resulted in elevated thresholds at 1 week that were not significantly different from those induced by saline injection. However, hearing loss after Anc80-eGFP injection increased further between 1 and 2 weeks, but not between 2 weeks and 1 month, indicating that minimal post-surgical trauma may exist and manifest in a progressive way. Injection of the Anc80-GJB2-FLAG vector produced a significantly larger hearing loss at 1 week than the Anc80-eGFP treatment did, but the Anc80-GJB2-FLAG vector did not induce significant changes at later time points, indicating that the detrimental effects may start before apparent tissue damage.
Table 2.
Results of ANOVA tests (F statistic, degrees of freedom [df], and p values) assessing effects of Anc-80 vectors on control and iCKO mice
| Test | N1, N2 | F | df | p | |
|---|---|---|---|---|---|
| 1 | saline versus normal, 1 weeka | 4, 4 | 13.52 | 3,7 | 0.003 |
| 2 | saline, 1 week versus 2 weeks | 4, 4b | 0.25 | 3,1 | 0.860 |
| 3 | GFP versus saline, 1 week | 5, 4 | 0.10 | 3,5 | 0.954 |
| 4 | GFP, 1 week versus 2 weeksa | 5, 5b | 25.41 | 3,2 | 0.038 |
| 5 | GFP, 2 weeks versus 1 month | 5, 5b | 4.76 | 3,2 | 0.179 |
| 6 | GJB2 versus GFP, 1 weeka | 5, 5 | 40.04 | 3,6 | <0.001 |
| 7 | GJB2, 1 week versus 2 weeks | 5, 6 | 1.04 | 3,7 | 0.431 |
| 8 | GJB2, 2 weeks versus 1 month | 6, 6 | 1.62 | 3,8 | 0.261 |
| 9 | P1, GJB2 versus contralaterala | 7, 7b | 6.82 | 3,4 | 0.047 |
| 10 | P14, GJB2 versus contralateral | 4, 4 | 0.66 | 3,1 | 0.692 |
| 11 | GJB2, P1 versus P14 | 7, 4 | 2.20 | 3,7 | 0.175 |
N1 and N2 are sample sizes for the respective groups.
Significant differences were found only for these comparisons.
Tests in which the same subjects were evaluated in both ears or at both time points.
Figure 8.

Anc80-GJB2-FLAG does not improve hearing in iCKO mouse ears.
Thresholds detected by ABR audiometry in three frequencies in control animals (A) and iCKO with tamoxifen given at P1 or P14 (B). The green line in (A) depicts the thresholds for un-injected iCKO mice. All treatments induced significant hearing loss compared with normal ears (A, green line), as demonstrated by saline-treated ears at 1 week (cyan, solid line; Table 2, row 1, p = 0.003). Saline injection elevated thresholds at 16 and 32 kHz, but GFP treatment at 2 weeks or later (pink dashed and dotted lines) and GJB2 treatment at all time points (black lines) had elevated thresholds at all three tested frequencies. Thresholds in GFP-treated ears were not significantly higher than in saline-treated ears at 1 week after injection (Table 2, row 3, p = 0.954) but were significantly higher by 2 weeks after injection (Table 2, row 4, p = 0.038). Hearing loss at 1 week was greater after GJB2 treatment (solid black line) than GFP (solid pink line), which was very significant (Table 2, row 6, p < 0.001). Hearing loss at later time points after GJB2 treatment (dashed and dotted black lines) was not significantly greater (Table 2, rows 7 and 8, p > 0.2). Ears that received GJB2 treatment after loss of connexin 26 was induced at P1 (part B, red) had greater hearing loss after GJB2 treatment (Table 2, row 9, p = 0.047) because of elevated thresholds at 16 and 32 kHz. The effect of GJB2 treatment was not significant in ears with connexin 26 loss induced at P14 (purple lines; Table 2, row 10, p = 0.692), primarily because of marginally greater loss in contralateral ears. Thresholds for GJB2 treated ears (B, solid lines) did not differ as function of the age at which connexin loss was induced (Table 2, row 11, p = 0.175).
In iCKO mice that received tamoxifen at P1 or P14, contralateral ears received no further treatment and had elevated thresholds as reported above. There was no significant difference in hearing thresholds between contralateral ears of P1 and P14 tamoxifen-injected iCKO ears (Figure 8B). In P1 group animals, the ears treated with the Anc80-GJB2-FLAG vector had significantly higher thresholds than the contralateral ears at 32 kHz. Although the average thresholds of Anc80-GJB2-FLAG vector-treated ears were slightly higher than in the contralateral ears of P14 iCKOs, the difference was not significant. The lack of threshold improvement after delivery of Anc80-GJB2-FLAG vector may be correlated with the loss of IHCs in these experimental ears, and in the high frequencies, also to surgical trauma. Prevention of IHC expression and reduction of surgical related damage may be required for accomplishing a practical level of phenotypic rescue.
Discussion
In this paper, we present novel findings on the design and characterization of a new mouse model for inducible deletion of Gjb2 in SCs of the inner ear sensory epithelia. We determined that tamoxifen induction of Gjb2 deletion in a nearly mature ear is less disruptive than deletion at an earlier developmental stage, but the pathology is still significant. These results confirm a role for connexin 26 in the function and maintenance in the mature ear. Gjb2 deletion in the nearly mature ear did not disrupt survival of key cells in the cochlea such as HCs and neurons for at least 2 months, providing a window of opportunity for experimenting with molecular therapies. AAV-mediated gene transfer partially restored connexin 26 in SCs but also resulted in expression in IHCs, which was detrimental for their survival. Taken together, our results indicate that gene replacement protocols for phenotypic rescue of Gjb2-related hearing loss will need to specifically target SCs and induce minimal or no expression in HCs.
The auditory epithelium contains an abundant network of gap junctions formed mostly by connexin 26 and connexin 30.13 These connexins are also present elsewhere in the cochlea, with prominent staining in the lateral wall. HCs do not have gap junctions or connexins. The presumed function of SC-SC gap junctions is ionic coupling between the cells; this process has been shown to be disrupted in Gjb2-null mice and restored by gene transfer during cochlear development.16 The physiological role of gap junctions for hearing is thought to be related to recycling potassium, which is needed for generation of the EP and HC function. Consistent with this, Gjb2 knockout mice exhibit hearing loss and reduced EP, as reported here and elsewhere.29,30 Additional roles for connexins in the cochlea have been proposed.13 For instance, gap junctions in SCs within the organ of Corti have been implicated in the generation of DPOAEs as part of active cochlear amplification, and DPOAEs are reduced in Gjb2 mutants.18 To therapeutically correct the loss of connexin 26 in the mammalian cochlea, it is therefore necessary to replace it in several populations of non-sensory cells. Such approaches for therapy can be developed by using genetically induced mouse models that mimic GJB2 deafness in humans.
Deletion using Cre lines for Pax2 or Foxg1 leads to severe disruption of the cochlear epithelial cells with closed tunnel of Nuel and major HC loss.31 As Sox10 is expressed in a relatively restricted area of the otocyst, we and others used a Sox10Cre line to delete Gjb2, hoping to generate a less severe inner ear and hearing phenotype that better mimics humans with GJB2 loss. However, Sox10Cre;Gjb2 conditional knockout mice exhibited early degeneration of HCs and auditory neurons, rendering them unhelpful for testing the impacts of Gjb2 gene replacement therapies.14,17 We therefore shifted our efforts to the design of an inducible conditional deletion mouse model as reported here. Inducible deletion approaches have been used previously to delete Gjb2, using ROSA26CreEsr1 (R26RCreERT2) and Gjb2 flox alleles.18 However, tamoxifen injections at P10 in those mice (ROSA26CreEsr1;Gjb2flox/flox) resulted in threshold shifts in ABRs and DPOAEs that were larger than could be explained by the mild change in EP. These studies demonstrated the utility of using an inducible deletion and indicated that a specific role for connexin 26 may be directly related to active cochlear amplification.
In contrast, Sox10iCreERT2;Gjb2flox/flox mice induced by tamoxifen retain HCs and SGNs for at least 2 months yet show a time-sensitive variance in phenotype severity after deletion of Gjb2 at P1 (more severe) versus P14 (less severe). The difference in outcomes between the two injection times confirms the well-established fact that connexin 26 is needed for proper development of the cochlea and reinforces the notion that gap junctions containing connexin 26 are needed beyond development for normal function and for maintaining HCs and neurons.
The time between induction of gene deletion and onset of functional degeneration is important because it defines the time window for testing molecular therapies to restore gap junctions and hearing. More important is the question of why HCs and SGNs degenerate when connexin 26 and gap junctions are not present in those cells. This distribution implies that the loss of connexin 26 in SCs must have an indirect effect on HCs and SGNs. The reduced potassium recycling may lead to reduced activity in the organ of Corti, which ends up leading to the demise of HCs. Alternatively, it is possible that long-term presence of potassium in the vicinity of the IHC synapse, because of lack of clearance by SCs, ends up poisoning the HCs, similar to the fate of cells incubated in potassium rich solutions or injected into endolymph.32 The loss of SGNs may be secondary to the loss of HCs, as shown in other models of cochlear pathology.33,34 These possibilities need to be tested in future studies.
The normal performance on rotarod testing in Sox10iCreERT2;Gjb2flox/flox mice is interesting, especially considering the abundance of connexin 26 in the vestibular epithelium.13,35 The reason for preservation of vestibular function with loss of connexin 26 or connexin 30 in mice is unclear but is also observed in human patients with GJB2 pathogenic variants.36, 37, 38
The gray coat color observed with loss of Gjb2 is interesting. We found no reports of other mouse models of Gjb2 that exhibit coat color changes. We also observed no coat color changes in the constitutive (not tamoxifen-inducible) Sox10Cre;Gjb2flox/flox mutants that were previously studied in our lab.17 The unique experimental paradigm used in the current study allows for temporal loss of Gjb2 in Sox10+ melanocytes and appears to have revealed a critical window for connexin 26 function in early postnatal mouse pups. In humans, pathogenic variants in GJB2 are associated with unique phenotypes, some which cause only deafness and others of which are associated with skin disorders. More than 150 pathogenic variants in GJB2 have been identified and grouped into four main categories: non-syndromic hearing loss, syndromic hearing loss (including keratitis-ichthyosis/keratoderma), Vohwinkel syndrome with palmoplantar keratoderma, and normal hearing with hidrotic ectodermal dysplasia.39, 40, 41 The underlying mechanism by which these variants contribute to deafness and pigmentation defects is an area for future exploration and may include loss or gain of function.
We show here that loss of connexin 26 can be reversed in SCs by AAV.Anc80-mediated gene transfer, demonstrating the utility of this vector for phenotypic rescue by restoring connexin 26 in SCs. We also present novel and important findings indicating ectopic GJB2 expression in IHCs and degeneration detected in these cells. AAVs in the cochlea tend to be most effective for gene transfer into HCs, with SCs being a secondary target. It is therefore encouraging that robust connexin 26 expression was detected in vector-treated ears in multiple types of SCs alongside that in HCs. The ectopic GJB2 expression in HCs is interesting in several aspects. First, OHCs exhibit cytoplasmic localization of connexin 26, whereas IHCs exhibit the normal junctional localization of connexin 26. Second, in addition to placing connexin 26 at their periphery, IHCs also appear to form contacts with each other and establish junctions between them. This light microscope level observation has to be confirmed and further characterized with transmission electron microscopy, which may also shed light on the differences in connexin 26 localization between IHCs and OHCs.
Our data are different from results obtained by Yu et al.,16 who injected AAVs to neonatal ears and showed that ectopic connexin 26 expression did not lead to formation of ectopic junctions and had no impact on hearing. It is possible that the immature ear is better able to regulate expression and localization of ectopic proteins than the mature ear. Additionally, as expression in the OHCs was not as robust as in the IHCs, it is possible that lower expression in IHCs may prevent junctional localization of connexin 26 and subsequent IHC loss. We have evaluated only one dose, and it is possible that lower vector doses may result in transduction and expression of connexin 26 in SCs without IHC effects. Further studies are necessary to shed light on the underlying mechanisms that give rise to HC loss with exogenous connexin 26 expression. Although our data show that gene transfer approach of GJB2 to SCs of the cochlea is feasible, changes in AAV surface morphology or composition may be helpful for further enhancing tropism to SCs. Nevertheless, as multiple capsid variants present high affinity to IHCs, de-targeting transgene expression in HCs using selective regulatory sequences should be considered as well.
The elevation of thresholds seen with the Anc80-eGFP (control) vector points to a limitation in using reporter genes. Although these genes are valuable for research purposes, they are not included in vectors used for human therapy. Still, it is important to elucidate the toxic mechanism in detail in future studies. The negative effects of GFP have been detected before in other systems42, 43, 44 and may have been related to the high titer of the vector we used in the present study.
Conclusions
We have designed and tested a mouse model for loss of Gjb2 inducible by tamoxifen injection at a selected age. Tamoxifen was injected at P1 or P14 and assayed 1 or 3 weeks later, as well as at 2, 4, and 8 months of age. After tamoxifen induction, we detected reduced connexin 26 staining in non-sensory cochlear cells, accompanied by decreased EP and elevated ABR and DPAOE thresholds, but the cytoarchitecture of the organ of Corti was preserved. However, starting around 2 months after tamoxifen, cochlear structure and function displayed progressive and severe degeneration, thereby limiting the window for experimental treatment to several weeks. The effects of Gjb2 loss on balance were less severe than on hearing. AAVAnc80-CAG.hGJB2.FLAG delivery resulted in robust expression of connexin 26 in multiple SC types. Additional expression in HCs was also observed. Although IHC loss was detected, OHCs survived. Gene replacement using AAV vector did not result in threshold improvement. Sox10iCreERT2;Gjb2flox/flox mice can therefore be used for phenotypic rescue experimentation within the time window or 2 months after tamoxifen injection. To enhance the suitability of AAV gene transfer for treating Gjb2 mutations in mature ears, more selective expression in SCs may be needed.
Methods and materials
Mice and breeding
All animal studies were performed at the University of Michigan under the guidance of protocols approved by the Institutional Animal Care and Use Committee (IACUC). Animals were cared for by laboratory personnel and the Unit for Laboratory Animal Medicine. To generate mice for this study, we bred Sox10iCreERT2 mice from Leda Dimou19 with Gjb2flox/flox mice provided by Prof. Klaus Willecke to generate Sox10iCreERT2;Gjb2flox/flox (iCKO) and Sox10iCreERT2;Gjb2flox/+ (conditional heterozygous) mice. Gjb2flox/+ and Gjb2flox/flox mice were used for some experiments as control mice.
Cre reporter assays and tamoxifen injections
Cre expression in Sox10iCreERT2 mice during development was examined using R26R-LacZ JAX 003474 reporter mice and after birth using R26REYFP JAX 006148 reporter mice. Timed pregnancies were established by crossing Sox10iCreERT2 males and reporter females. Females were checked for plugs, and the morning of plug detection was marked as P0.5. Tamoxifen (Sigma-Aldrich) was prepared as a 10 mg/mL stock by dissolving the powder in corn oil (Sigma-Aldrich) using water-bath sonication for 30 min. The solution was then aliquoted and stored at −20°C. Pregnant dams were injected intraperitoneally with 0.1 mg/g body weight (b.w.) tamoxifen at E11.5. Dams were euthanized and embryos dissected at E17.5 then processed for cryo-sectioning, X-gal staining, and counter-staining with eosin. For postnatal Cre reporter assays, crosses were established between Sox10iCreERT2 male and EYFP+ female mice. Sox10iCreERT2;EYFP pups were injected intraperitoneally with tamoxifen at P14 and analyzed using whole-mount epifluorescence at P16.
ABRs
Mice were anesthetized by intraperitoneal injection of ketamine 65 mg/kg, xylazine 7 mg/kg, and acepromazine 2 mg/kg. Mouse core body temperature was maintained using a water-circulating heating pad. ABRs were recorded in an electrically and acoustically shielded chamber. Sub-dermal needle electrodes were placed at vertex (active) and ventral to the pinnae of the test ear (reference) and contralateral ear (ground). Tucker Davis Technologies (TDT) System III hardware and SigGen/BioSig software (TDT, Alachua, FL) were used to present the stimulus and record responses. Tones were delivered through an EC1 driver (TDT, aluminum enclosure made in-house), with the speculum placed just inside the tragus. Stimulus presentation was 15 ms tone bursts, with 1 ms rise/fall times, 10 bursts per second. Up to 1,024 responses were averaged for each stimulus level. Responses were collected for stimulus levels in 10 dB steps at higher stimulus levels, with additional 5 dB steps near threshold. Thresholds were interpolated between the lowest stimulus level at which a response was observed, and 5 dB lower, at which no response was observed. Frequencies measured were 12 and 24 kHz for the initial baseline studies and 8, 16, and 32 kHz for subsequent study after AAV-mediated gene transfer. In all studies, animals showing no responses at 105 dB SPL (the maximal output of the system) were designated as “no response.”
DPOAEs
DPOAE testing was performed on the same animals immediately after ABR recording. DPOAE tests were measured in left ears at 12 and 24 kHz as previously described.45,46 Briefly, primary tones, f1 and f2, were set at a ratio of f2/f1 = 1.2. Intensity of f1 was varied in 5–10 dB SPL steps, while f2 intensity was held 10 dB SPL quieter than f1. Tones were presented via two EC1 drivers (Beyerdynamic, Farmingdale, NY; aluminum-shielded enclosure made in-house) connected through an Etymotic microphone (ER 10B+; Etymotic Research, Elk Grove Village, IL). TDT System III hardware and SigGen/BioSig software were used to present the stimuli and record responses. DPOAE thresholds were recorded as the lowest stimulus intensity to elicit a response amplitude above the noise floor. Transient responses above the noise floor (response to a single stimulus intensity with no growth of response amplitude at the next highest stimulus intensity) were discarded. In the majority of iCKO mice, DPOAE thresholds indicated that there were responses at only the one or two highest stimulus intensities; therefore, it was not possible to estimate the average slope representing the growth of the response amplitude with increasing stimulus intensity for comparison with controls.
EP
Mice were deeply anesthetized by intraperitoneal injection of ketamine 120 mg/kg and xylazine 7 mg/kg and placed on a water-circulating heating pad to maintain body temperature. The external pinna was removed and soft tissue dissected away from the bulla. The ossicles and tympanic membrane were removed, and some of the bulla wall was removed to allow clear visualization of the cochlea. A small opening was made in the otic capsule over the stria vascularis. A glass micropipette filled with 150 mM KCl was connected to a capacity-compensated direct-current preamplifier, and a ground electrode was inserted into the shoulder muscle. The micropipette was attached to a micro-manipulator and slowly advanced through the lateral wall into scala media. Amplified electrical signals were recorded using TDT hardware and chart-recorder custom software. EP was calculated as the maximum change in voltage as the micropipette tip passed from lateral wall tissue into endolymph. Animals were euthanized at the end of the recording.
Histology
Mice were deeply anesthetized with ketamine (120 mg/kg, intraperitoneal [i.p.]) and xylazine (7 mg/kg, i.p.) and euthanized, and inner ear tissues were dissected and fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 2 h. After rinses in PBS, cochleae and utricles were collected, permeabilized in 0.3% Triton X-100 in PBS for 15 min, and then blocked with 5% normal goat serum for 30 min. Samples were then reacted with primary rabbit or mouse antibodies overnight at 4°C. Primary antibodies were rabbit anti-myosin VIIa (1:300; Proteus Biosciences, Ramona, CA), mouse anti-FLAG (1:400; Sigma-Aldrich, St. Louis, MO), and mouse anti-connexin 26 (1:200; Invitrogen, Waltham, MA). After rinsing the primary antibody in PBS, tissues were incubated with the appropriate secondary antibodies (Invitrogen; 1:300 in PBS) for 1 h. Samples were counter-stained with Alexa Fluor 647 phalloidin (1:300 in PBS; Invitrogen) to label F-actin for depicting cellular borders in the epithelium. Tissues were rinsed three times with PBS and mounted on glass slides with ProLong Gold Antifade Mountant (Invitrogen).
Whole-mount specimens were initially examined using a Leica DMRB epifluorescence microscope (Leica, Eaton, PA) equipped with a RT39M5 digital camera (Diagnostic Instruments, Sterling Heights, MI). Images were processed with Adobe Photoshop software (Adobe Systems, San Jose, CA). Selected areas were then analyzed on a confocal microscope (Leica SP8; Leica, Eaton, PA). LAS X imaging software was used to obtain z stack images.
Quantitative analyses
EP values and ABR and DPOAE thresholds were analyzed using similar statistical designs. For each age of tamoxifen injection, overall differences between genotypes were evaluated using ANOVA. For ABR and DPOAE thresholds, the ANOVA included data for both tested frequencies and was followed by post hoc comparisons of each tested frequency. Data for the two injection ages were combined to test whether there was a significant interaction, which would indicate that the effect of the deletion depended on the age at which it was induced by tamoxifen injection. For all multivariate tests, Pillai’s trace was used to approximate the value of the F ratio. All analyses were performed in R version 4.0.2.
AAV vector and injections
iCKO mice (n = 17) and their littermate controls (n = 31) were used in this study. Animals were anesthetized with intraperitoneal injection of xylazine (7 mg/kg) and ketamine (120 mg/kg). Ketoprofen (10 mg/kg; Zoetis, Parsippany, NJ) was given subcutaneously prior to the surgery. A solution consisting of 3 μL of viral vector mixed with 0.3 μL of 0.25% fast green dye was loaded into a glass pipette, which was pulled to a final outer diameter of ∼20 μm before surgery. A 1.5 cm incision was made in the left post-auricular region after shaving and disinfecting the area. Once the posterior semicircular canal (PSC) was exposed, a small opening was made using a 26-gauge needle until a slow leakage of perilymph was observed. The round window (RW) was then surgically exposed as described.47,48 In preparation for the injection, a small piece of muscle was inserted into the RW niche before the RW membrane was punctured, and the RW niche was loaded with 1 μL Vetbond tissue adhesive (3M, St. Paul, MN) to prevent potential leakage. A Nanoject system (Nanoject III Programmable Pump; Drummond Scientific Company, Broomall, PA) was used to control the injecting rate at 5 nL/s, 100 nL per cycle. Ten cycles with an interval of 10 s between cycles were performed, resulting in a total amount of 1 μL for each animal. Successful injection was indicated by efflux of blue-tinged fluid from the PSC vent. The pipette was removed immediately after the injection, then the RW niche and the PSC vent were sealed with tissue adhesive. The right ear of each animal remained intact and served as a control. Sham surgical controls were injected with normal saline containing fast green dye at the same concentration as above. Animals were euthanized at several time points between 1 and 8 weeks after surgery, as detailed in the Results.
The viral vectors were AAVAnc80-CAG.eGFP.bGHpA (Anc80-eGFP, 1E13 vg/mL), and AAVAnc80-CAG.hGJB2.3Flag.bGHpA (Anc80-hGJB2-FLAG, 1.2E13 vg/mL). The CAG promoter is composed of the cytomegalovirus (CMV) early enhancer, the chicken β-actin promoter, and the rabbit β-globin splice acceptor site. The bovine growth hormone (bGH) polyadenylation (pA) signal was also included. On the basis of this vector design, detection of the FLAG should localize viral-mediated connexin 26 transgene, but not the endogenous connexin 26, thereby indicating which cells are not only transduced with the vector but also expressing the transgene.
The human GJB2 coding sequence was used because the sequence identity with the mouse coding sequence is 87%, and it is more translationally relevant for future studies. Use of human genes in experimental animals is common practice when translational goals are involved, as seen in other organs and mutations.49 The vectors were produced at SABTech (Philadelphia, PA) in HEK293 cells following triple transfection and using standard purification methods. The vector titers were determined using Droplet Digital PCR (ddPCR).
Rotarod test
We used the rotarod test to evaluate overall vestibular function at different ages. Mice (iCKO or control, n = 5 per group) were placed on a stationary rod suspended 15 inches over the surface for 30 s to get accustomed to the environment. Mice were then prompted to remain on the rod during a constant low speed spinning (5 rpm) for 60 s for preliminary screening. At the next step, 1 min after the first trial, mice were presented with a more complex balance task using an accelerating spinning rod increasing from 5 to 45 rpm over 100 s, which was then maintained at 45 rpm for another 200 s. The total time a mouse spent on the rod before falling or reaching 200 s was recorded. Tests were performed three consecutive times and the differences between groups were evaluated by repeated-measures ANOVA in R, which is similar to the ANOVAs described above but also accounts for variations among repetitions by the same mouse.
Acknowledgments
This research was supported by National Institutes of Health (NIH)-National Institute on Deafness and Other Communication Disorders (NIDCD) grants R01-DC014832, R01-DC010412, R01-DC009410, R01-DC014456, and P30-DC005188; Akouos; the Ravitz Foundation Professorship in Pediatrics and Communicable Diseases (D.M.M.); and the R. Jamison and Betty Williams Professorship (Y.R.). J.G. is supported by the National Natural Science Foundation of China (81900929) and Beijing Natural Science Foundation (7194256). We thank Leda Dimou for the Sox10iCreERT2 mice, Klaus Willecke for the Gjb2flox/flox mice and Lisa Kabara for technical support.
Author contributions
Y.R. and D.M.M. conceived the design of the mouse and planned the experiments, then supervised data collection and interpretation and generation of the manuscript. J.G., X.M., and J.M.S. performed the experiments. L.A.B. and J.C. helped with mouse colony management. D.M.P. supervised the surgical work. D.L.S. designed the statistical aspect of the work and performed the statistical analyses as well as image preparation and assembling the figures. D.F.D. supervised the physiological tests. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interest.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.09.009.
Supplemental information
References
- 1.Ahituv N., Avraham K.B. Mouse models for human deafness: current tools for new fashions. Trends Mol. Med. 2002;8:447–451. doi: 10.1016/s1471-4914(02)02388-2. [DOI] [PubMed] [Google Scholar]
- 2.Holme R.H., Steel K.P. Genes involved in deafness. Curr. Opin. Genet. Dev. 1999;9:309–314. doi: 10.1016/s0959-437x(99)80046-x. [DOI] [PubMed] [Google Scholar]
- 3.Shearer A.E., Hildebrand M.S., Sloan C.M., Smith R.J. Deafness in the genomics era. Hear. Res. 2011;282:1–9. doi: 10.1016/j.heares.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelsell D.P., Dunlop J., Stevens H.P., Lench N.J., Liang J.N., Parry G., Mueller R.F., Leigh I.M. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature. 1997;387:80–83. doi: 10.1038/387080a0. [DOI] [PubMed] [Google Scholar]
- 5.Lalwani A.K. Evaluation of childhood sensorineural hearing loss in the post-genome world. Arch. Otolaryngol. Head Neck Surg. 2002;128:88–89. doi: 10.1001/archotol.128.1.88. [DOI] [PubMed] [Google Scholar]
- 6.Carvalho G.J., Lalwani A.K. The effect of cochleostomy and intracochlear infusion on auditory brain stem response threshold in the guinea pig. Am. J. Otol. 1999;20:87–90. [PubMed] [Google Scholar]
- 7.Tsukada K., Nishio S.Y., Hattori M., Usami S. Ethnic-specific spectrum of GJB2 and SLC26A4 mutations: their origin and a literature review. Ann. Otol. Rhinol. Laryngol. 2015;124(Suppl 1):61S–76S. doi: 10.1177/0003489415575060. [DOI] [PubMed] [Google Scholar]
- 8.Kenna M.A., Rehm H.L., Frangulov A., Feldman H.A., Robson C.D. Temporal bone abnormalities in children with GJB2 mutations. Laryngoscope. 2011;121:630–635. doi: 10.1002/lary.21414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prera N., Löhle E., Birkenhäger R. [Progressive hearing impairment with deletion in GJB2 gene despite normal newborn hearing screening] Laryngorhinootologie. 2014;93:244–248. doi: 10.1055/s-0033-1355343. [DOI] [PubMed] [Google Scholar]
- 10.Jun A.I., McGuirt W.T., Hinojosa R., Green G.E., Fischel-Ghodsian N., Smith R.J. Temporal bone histopathology in connexin 26-related hearing loss. Laryngoscope. 2000;110:269–275. doi: 10.1097/00005537-200002010-00016. [DOI] [PubMed] [Google Scholar]
- 11.Gabriel H.D., Jung D., Bützler C., Temme A., Traub O., Winterhager E., Willecke K. Transplacental uptake of glucose is decreased in embryonic lethal connexin26-deficient mice. J. Cell Biol. 1998;140:1453–1461. doi: 10.1083/jcb.140.6.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia-Vega L., O’Shaughnessy E.M., Albuloushi A., Martin P.E. Connexins and the epithelial tissue barrier: a focus on connexin 26. Biology (Basel) 2021;10:59. doi: 10.3390/biology10010059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jagger D.J., Forge A. Connexins and gap junctions in the inner ear—it’s not just about K+ recycling. Cell Tissue Res. 2015;360:633–644. doi: 10.1007/s00441-014-2029-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crispino G., Di Pasquale G., Scimemi P., Rodriguez L., Galindo Ramirez F., De Siati R.D., Santarelli R.M., Arslan E., Bortolozzi M., Chiorini J.A., Mammano F. BAAV mediated GJB2 gene transfer restores gap junction coupling in cochlear organotypic cultures from deaf Cx26Sox10Cre mice. PLoS ONE. 2011;6:e23279. doi: 10.1371/journal.pone.0023279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iizuka T., Kamiya K., Gotoh S., Sugitani Y., Suzuki M., Noda T., Minowa O., Ikeda K. Perinatal Gjb2 gene transfer rescues hearing in a mouse model of hereditary deafness. Hum. Mol. Genet. 2015;24:3651–3661. doi: 10.1093/hmg/ddv109. [DOI] [PubMed] [Google Scholar]
- 16.Yu Q., Wang Y., Chang Q., Wang J., Gong S., Li H., Lin X. Virally expressed connexin26 restores gap junction function in the cochlea of conditional Gjb2 knockout mice. Gene Ther. 2014;21:71–80. doi: 10.1038/gt.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takada Y., Beyer L.A., Swiderski D.L., O’Neal A.L., Prieskorn D.M., Shivatzki S., Avraham K.B., Raphael Y. Connexin 26 null mice exhibit spiral ganglion degeneration that can be blocked by BDNF gene therapy. Hear. Res. 2014;309:124–135. doi: 10.1016/j.heares.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu Y., Chen J., Liang C., Zong L., Chen J., Jones R.O., Zhao H.B. Connexin26 (GJB2) deficiency reduces active cochlear amplification leading to late-onset hearing loss. Neuroscience. 2015;284:719–729. doi: 10.1016/j.neuroscience.2014.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simon C., Lickert H., Götz M., Dimou L. Sox10-iCreERT2 : a mouse line to inducibly trace the neural crest and oligodendrocyte lineage. Genesis. 2012;50:506–515. doi: 10.1002/dvg.22003. [DOI] [PubMed] [Google Scholar]
- 20.Watanabe K., Takeda K., Katori Y., Ikeda K., Oshima T., Yasumoto K., Saito H., Takasaka T., Shibahara S. Expression of the Sox10 gene during mouse inner ear development. Brain Res. Mol. Brain Res. 2000;84:141–145. doi: 10.1016/s0169-328x(00)00236-9. [DOI] [PubMed] [Google Scholar]
- 21.Harris M.L., Buac K., Shakhova O., Hakami R.M., Wegner M., Sommer L., Pavan W.J. A dual role for SOX10 in the maintenance of the postnatal melanocyte lineage and the differentiation of melanocyte stem cell progenitors. PLoS Genet. 2013;9:e1003644. doi: 10.1371/journal.pgen.1003644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richard G., Rouan F., Willoughby C.E., Brown N., Chung P., Ryynänen M., Jabs E.W., Bale S.J., DiGiovanna J.J., Uitto J., Russell L. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. Am. J. Hum. Genet. 2002;70:1341–1348. doi: 10.1086/339986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Butterweck A., Elfgang C., Willecke K., Traub O. Differential expression of the gap junction proteins connexin45, -43, -40, -31, and -26 in mouse skin. Eur. J. Cell Biol. 1994;65:152–163. [PubMed] [Google Scholar]
- 24.Turkoz M., Townsend R.R., Kopan R. The Notch intracellular domain has an RBPj-independent role during mouse hair follicular development. J. Invest. Dermatol. 2016;136:1106–1115. doi: 10.1016/j.jid.2016.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee M.Y., Takada T., Takada Y., Kappy M.D., Beyer L.A., Swiderski D.L., Godin A.L., Brewer S., King W.M., Raphael Y. Mice with conditional deletion of Cx26 exhibit no vestibular phenotype despite secondary loss of Cx30 in the vestibular end organs. Hear. Res. 2015;328:102–112. doi: 10.1016/j.heares.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burns J.C., On D., Baker W., Collado M.S., Corwin J.T. Over half the hair cells in the mouse utricle first appear after birth, with significant numbers originating from early postnatal mitotic production in peripheral and striolar growth zones. J. Assoc. Res. Otolaryngol. 2012;13:609–627. doi: 10.1007/s10162-012-0337-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Landegger L.D., Pan B., Askew C., Wassmer S.J., Gluck S.D., Galvin A., Taylor R., Forge A., Stankovic K.M., Holt J.R., Vandenberghe L.H. A synthetic AAV vector enables safe and efficient gene transfer to the mammalian inner ear. Nat. Biotechnol. 2017;35:280–284. doi: 10.1038/nbt.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kho S.T., Pettis R.M., Mhatre A.N., Lalwani A.K. Safety of adeno-associated virus as cochlear gene transfer vector: analysis of distant spread beyond injected cochleae. Mol. Ther. 2000;2:368–373. doi: 10.1006/mthe.2000.0129. [DOI] [PubMed] [Google Scholar]
- 29.Wu X., Zhang W., Li Y., Lin X. Structure and function of cochlear gap junctions and implications for the translation of cochlear gene therapies. Front. Cell. Neurosci. 2019;13:529. doi: 10.3389/fncel.2019.00529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mei L., Chen J., Zong L., Zhu Y., Liang C., Jones R.O., Zhao H.B. A deafness mechanism of digenic Cx26 (GJB2) and Cx30 (GJB6) mutations: reduction of endocochlear potential by impairment of heterogeneous gap junctional function in the cochlear lateral wall. Neurobiol. Dis. 2017;108:195–203. doi: 10.1016/j.nbd.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y., Chang Q., Tang W., Sun Y., Zhou B., Li H., Lin X. Targeted connexin26 ablation arrests postnatal development of the organ of Corti. Biochem. Biophys. Res. Commun. 2009;385:33–37. doi: 10.1016/j.bbrc.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park Y.H., Wilson K.F., Ueda Y., Tung Wong H., Beyer L.A., Swiderski D.L., Dolan D.F., Raphael Y. Conditioning the cochlea to facilitate survival and integration of exogenous cells into the auditory epithelium. Mol. Ther. 2014;22:873–880. doi: 10.1038/mt.2013.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leake P.A., Hradek G.T. Cochlear pathology of long term neomycin induced deafness in cats. Hear. Res. 1988;33:11–33. doi: 10.1016/0378-5955(88)90018-4. [DOI] [PubMed] [Google Scholar]
- 34.Jyung R.W., Miller J.M., Cannon S.C. Evaluation of eighth nerve integrity by the electrically evoked middle latency response. Otolaryngol. Head Neck Surg. 1989;101:670–682. doi: 10.1177/019459988910100610. [DOI] [PubMed] [Google Scholar]
- 35.Klimpel K.E., Lee M.Y., King W.M., Raphael Y., Schacht J., Neitzel R.L. Vestibular dysfunction in the adult CBA/CaJ mouse after lead and cadmium treatment. Environ. Toxicol. 2017;32:869–876. doi: 10.1002/tox.22286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dodson K.M., Blanton S.H., Welch K.O., Norris V.W., Nuzzo R.L., Wegelin J.A., Marin R.S., Nance W.E., Pandya A., Arnos K.S. Vestibular dysfunction in DFNB1 deafness. Am. J. Med. Genet. A. 2011;155A:993–1000. doi: 10.1002/ajmg.a.33828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kasai M., Hayashi C., Iizuka T., Inoshita A., Kamiya K., Okada H., Nakajima Y., Kaga K., Ikeda K. Vestibular function of patients with profound deafness related to GJB2 mutation. Acta Otolaryngol. 2010;130:990–995. doi: 10.3109/00016481003596508. [DOI] [PubMed] [Google Scholar]
- 38.Todt I., Hennies H.C., Basta D., Ernst A. Vestibular dysfunction of patients with mutations of Connexin 26. Neuroreport. 2005;16:1179–1181. doi: 10.1097/00001756-200508010-00009. [DOI] [PubMed] [Google Scholar]
- 39.Lee J.R., White T.W. Connexin-26 mutations in deafness and skin disease. Expert Rev. Mol. Med. 2009;11:e35. doi: 10.1017/S1462399409001276. [DOI] [PubMed] [Google Scholar]
- 40.Giugliano R.P., Ruff C.T., Braunwald E., Murphy S.A., Wiviott S.D., Halperin J.L., Waldo A.L., Ezekowitz M.D., Weitz J.I., Špinar J., ENGAGE AF-TIMI 48 Investigators Edoxaban versus warfarin in patients with atrial fibrillation. N. Engl. J. Med. 2013;369:2093–2104. doi: 10.1056/NEJMoa1310907. [DOI] [PubMed] [Google Scholar]
- 41.Sanchez H.A., Verselis V.K. Aberrant Cx26 hemichannels and keratitis-ichthyosis-deafness syndrome: insights into syndromic hearing loss. Front. Cell. Neurosci. 2014;8:354. doi: 10.3389/fncel.2014.00354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Detrait E.R., Bowers W.J., Halterman M.W., Giuliano R.E., Bennice L., Federoff H.J., Richfield E.K. Reporter gene transfer induces apoptosis in primary cortical neurons. Mol. Ther. 2002;5:723–730. doi: 10.1006/mthe.2002.0609. [DOI] [PubMed] [Google Scholar]
- 43.Howard D.B., Powers K., Wang Y., Harvey B.K. Tropism and toxicity of adeno-associated viral vector serotypes 1, 2, 5, 6, 7, 8, and 9 in rat neurons and glia in vitro. Virology. 2008;372:24–34. doi: 10.1016/j.virol.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klein R.L., Dayton R.D., Leidenheimer N.J., Jansen K., Golde T.E., Zweig R.M. Efficient neuronal gene transfer with AAV8 leads to neurotoxic levels of tau or green fluorescent proteins. Mol. Ther. 2006;13:517–527. doi: 10.1016/j.ymthe.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karolyi I.J., Dootz G.A., Halsey K., Beyer L., Probst F.J., Johnson K.R., Parlow A.F., Raphael Y., Dolan D.F., Camper S.A. Dietary thyroid hormone replacement ameliorates hearing deficits in hypothyroid mice. Mamm. Genome. 2007;18:596–608. doi: 10.1007/s00335-007-9038-0. [DOI] [PubMed] [Google Scholar]
- 46.Hurd E.A., Adams M.E., Layman W.S., Swiderski D.L., Beyer L.A., Halsey K.E., Benson J.M., Gong T.W., Dolan D.F., Raphael Y., Martin D.M. Mature middle and inner ears express Chd7 and exhibit distinctive pathologies in a mouse model of CHARGE syndrome. Hear. Res. 2011;282:184–195. doi: 10.1016/j.heares.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshimura H., Shibata S.B., Ranum P.T., Smith R.J.H. Enhanced viral-mediated cochlear gene delivery in adult mice by combining canal fenestration with round window membrane inoculation. Sci. Rep. 2018;8:2980. doi: 10.1038/s41598-018-21233-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chien W.W., McDougald D.S., Roy S., Fitzgerald T.S., Cunningham L.L. Cochlear gene transfer mediated by adeno-associated virus: Comparison of two surgical approaches. Laryngoscope. 2015;125:2557–2564. doi: 10.1002/lary.25317. [DOI] [PubMed] [Google Scholar]
- 49.Tolmachova T., Tolmachov O.E., Barnard A.R., de Silva S.R., Lipinski D.M., Walker N.J., Maclaren R.E., Seabra M.C. Functional expression of Rab escort protein 1 following AAV2-mediated gene delivery in the retina of choroideremia mice and human cells ex vivo. J. Mol. Med. (Berl.) 2013;91:825–837. doi: 10.1007/s00109-013-1006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.