

ABSTRACT
The fungus Paracoccidioides is a prevalent human pathogen endemic to South America. The genus is composed of five species. In this report, we use 37 whole-genome sequences to study the allocation of genetic variation in Paracoccidioides. We tested three genome-wide predictions of advanced speciation, namely, that all species should be reciprocally monophyletic, that species pairs should be highly differentiated along the whole genome, and that there should be low rates of interspecific gene exchange. We find support for these three hypotheses. Species pairs with older divergences show no evidence of gene exchange, while more recently diverged species pairs show evidence of modest rates of introgression. Our results indicate that as divergence progresses, species boundaries become less porous among Paracoccidioides species. Our results suggest that species in Paracoccidioides are at different stages along the divergence continuum.
KEYWORDS: speciation, gene exchange, hidden Markov model (HMM), introgression
INTRODUCTION
Paracoccidioides, a genus of temperature-dimorphic fungi, causes paracoccidioidomycosis (PCM), which is a systemic endemic mycosis that occurs across in most countries of Latin America from mideastern Mexico to Argentina (1, 2). Multiple genetic surveys have revealed extensive genetic variability within Paracoccidioides (3–7). This variation, coupled with the extensive geographic range of the fungus—and that of the disease it causes—led to the hypothesis of population structure and cryptic speciation within the group. Initial studies reveal the existence of at least three species (8), but more recent analyses have suggested the existence of five different species of Paracoccidioides (9–11). Clearly, the use of genetic markers holds the potential to reveal key aspects of the evolutionary biology of the pathogen. Yet, the genetic characterization of most isolates has been modest.
Species from the genus Paracoccidioides show a range of divergence that make the group promising to understand how diversification occurs in pathogenic fungi. One of the species of Paracoccidioides, Paracoccidioides lutzii, seems to have diverged from the rest of the species at least 30 million years ago (12). The species show extensive differences in terms of morphology and physiology. Five more species, all within the brasiliensis complex, form a monophyletic group. Paracoccidioides restrepiensis and Paracoccidioides venezuelensis are the most closely related dyad with recent divergence (less than 0.2 million years ago [MYA]) (10). Paracoccidioides brasiliensis sensu stricto is sister to the P. restrepiensis/P. venezuelensis dyad, while Paracoccidioides americana is sister to the ingroup. Paracoccidioides brasiliensis sensu stricto has also been proposed to be formed by two cryptic species, S1a and S1b (13), but no formal test of this divergence has been performed. A combination of yeast and conidial morphology differentiates between all Paracoccidioides species pairs (10, 11, 14).
Whole-genome sequences can be used to identify species boundaries in fungi. Three tests jointly indicate the existence of species boundaries (15). First, genome variation must reflect genetic differentiation in cases where speciation has taken place. In cases of advanced divergence, genomes of putative species should show reciprocal monophyly; this can be measured as the proportion of loci that show a phylogenetic history concordant with the hypothesized species history. Second, genetic variation should be partitioned across putative species; the extent of genetic differentiation between individuals from different putative species should be larger than the differentiation between individuals within each of the putative species. Finally, the genomes of putative species should show low or moderate levels of gene exchange. So far, all studies on species boundaries in the genus Paracoccidioides have focused on detecting genealogical congruence among a modest number of gene genealogies (8, 10–12). Incorporating genomic information is sorely needed to understand the magnitude of differentiation among Paracoccidioides pathogens. In this report, we use phylogenetics and population genetics to bridge that gap. We find that the species of Paracoccidioides are extensively differentiated, which suggests an advanced stage along the divergence continuum.
RESULTS
Paracoccidioides is haploid and shows little evidence for aneuploidy.
We tested two aspects regarding the ploidy of Paracoccidioides. First, we assessed whether the coverage in the genome was homogenous. Significant deviations from homogeneity indicate aneuploidy. Second, we tested the global and local ploidy by scoring the mean coverage and exploring for the presence of polymorphic sites. Haploid genomes, unlike higher ploidies, show no variation in each locus and should show no minor—or major—alleles. Figure 1 shows the results of this analyses. First, the global distribution of coverage is distributed evenly around 1, with a few sites getting no coverage with respect to the reference genome (Fig. 1A). Permutation tests show that this distribution is not significantly different from 1 (i.e., the expectation of homogenous ploidy along the genome; two-sample Fisher-Pitman permutation test, P = 0.08). When we ran similar analyses to study the mean coverage and minor allele frequency locally (i.e., in 5-kb windows) along the Paracoccidioides genome, we found that the expectations of haploidy were fulfilled as well (i.e., there no regions with abnormally high coverage or that were systematically polymorphic; Fig. 1B to E). Figures S2 to S5 in the supplemental material show similar analyses for all species of Paracoccidioides. Overall, these results confirm previous observations that Paracoccidioides is haploid (16); for all analyses from now on, we treat Paracoccidioides as such.
FIG 1.
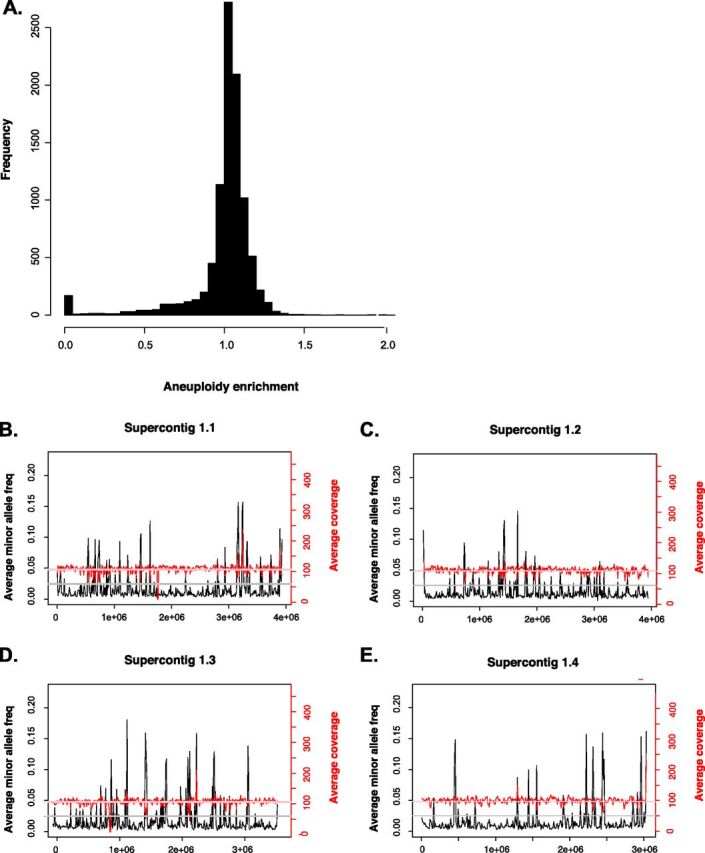
Paracoccidioides is haploid and shows no detectable levels of aneuploidy. (A) Mean coverage per site shows no large and consistent deviations from the expected sequencing coverage, suggesting little or no aneuploidy in Paracoccidioides genomes. (B) Per-site estimations of coverage (in red) show close adherence to the genome-wide mean coverage for the whole genome of the P. lutzii isolate EE. Per-site estimations of the extent of polymorphism across the genome suggest that no single site shows evidence of polymorphism (cutoff: minor allele frequency > 20%), suggesting that besides having no local aneuploidy, Paracoccidioides is haploid. The four panels on the right (B to E) show the coverage and minor allele frequency for the four largest supercontigs. Figure S2 to S5 in the supplemental material show similar plots for other Paracoccidioides species.
All proposed species show reciprocal monophyly and strong genealogical concordance.
We evaluated the phylogenetic relationships between all species of the Paracoccidioides genus using genome-wide variation and established how much of the genome supports these relationships. First, we built a maximum likelihood (ML) tree in which we used concatenated loci from the whole genome as the unit for phylogenetic analysis (this approach has important limitations [17]; see below). Figure 2A shows the resulting topology. Individual analyses of the 6 largest supercontigs yields similar topologies (Fig. 2B to G and Fig. S6 in the supplemental material), with some minor exceptions (see Fig. S6 and S7 in the supplemental material). Two results stand out from these analyses. As predicted by smaller efforts (10), all five named species of Paracoccidioides are reciprocally monophyletic. The tree shows that, on average, all scaffolds show an evolutionary history consistent with the clusters previously described as species, including S1a and S1b. Four out of the six supercontigs show an identical branching pattern to that of the whole-genome genealogy (Robinson-Foulds [RF] distance = 0.00). The topologies from supercontigs 2.2, 2.3, and 2.5 are slightly different from the genome-wide topology (RF distances = 24, 23, and 3.00, respectively; Fig. S7). The maximum RF distance in these trees is 70, following Bryant and Steel (18). Similarly, genome-wide phylogenetic reconstruction shows similar results to previous approaches and suggest that P. restrepiensis and P. venezuelensis are the most closely related species of the group. Paracoccidioides brasiliensis sensu stricto is sister to the dyad P. venezuelensis/P. restrepiensis, and P. americana is an outgroup to the other three species of the brasiliensis species complex.
FIG 2.
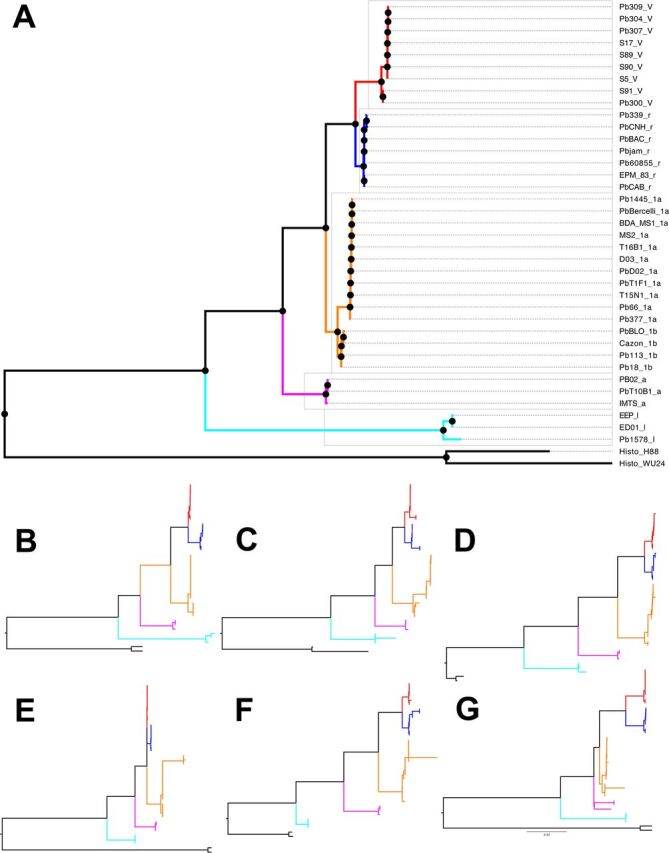
Reciprocal monophyly between Paracoccidioides species. (A) Maximum likelihood rooted phylogram using concatenated genome-wide loci. Paracoccidioides venezuelensis isolates are marked in red and have a “V” after the strain name. Paracoccidioides restrepiensis isolates are marked in blue and have a “r” after the strain name. Paracoccidioides brasiliensis isolates are marked in orange and have a “1a” or a “1b” after the strain name. Paracoccidioides americana isolates are marked in pink and have an “a” after the strain name. Paracoccidioides lutzii are isolates marked in cyan and have a “l” after the strain name. (B to G) Phylograms for the six largest supercontigs in the Pb18 genome show the same topology. We follow the same color scheme as that in in panel A. (B) Supercontig 1.1. (C) Supercontig 1.2. (D) Supercontig 1.3. (E) Supercontig 1.4. (F) Supercontig 1.5. (G) Supercontig 1.6. The results are consistent to those shown in Fig. 1 of Muñoz et al. (13) and Teixeira et al. (25).
Additionally, we calculated the concordance factors for the each of the five Paracoccidioides species and the putative cryptic species S1a and S1b. If speciation has proceeded to extensive genetic differentiation, then most of the genome should show the signature of reciprocal monophyly in each of the proposed species. Figure 3 shows the results of a gene genealogy concordance analysis using BUCKy. The obtained topology is identical to that produced using maximum likelihood. The concordance factors for the five proposed species is in all cases greater than 90%, which suggests that the vast majority of the genome shows concordance in the existence of the five species of Paracoccidioides. The only exceptions to this high level of concordance are the nodes that separate S1a and S1b, which had concordance factor (CF) values of 0.53 and 0.61, respectively. There is no metric on how high a CF value must be to elevate a group to species level (19, 20), but the genome-wide concordance of this groups is much lower than that of the other Paracoccidioides species. For all analyses that follow, we treat P. brasiliensis sensu stricto as a single species with strong population structure.
FIG 3.

The species tree of the genus Paracoccidioides suggests that the majority of the genome of Paracoccidioides shows similar phylogenetic signal. The tree shown (built with BUCKy and a prior α of 5) is based on individual benchmarking universal single-copy ortholog (BUSCO) genes. Values above each branch show the point estimate of the concordance factor (CF) for each node; values below branches show the 95% Bayesian credibility intervals for this estimate. The majority of the genome signals reciprocal monophyly for the previously proposed species of Paracoccidioides.
Next, we calculated the mean genetic distance between individuals of the same species and between pairs of individuals from different species. The expectation is that pairwise comparisons between individuals from different species should show much higher differentiation than individuals from the same species (15, 21). We found that genetic variability is partitioned among species and that in all cases the magnitude of interspecific distances is at least 2× higher than that of intraspecific distances for all species pairs in genome-wide estimations (Fig. 4 and Fig. S8 in the supplemental material). All pairwise comparisons between intraspecific and interspecific differentiation were significant (two-sample Fisher-Pitman permutation test, P < 1 × 10−10). As expected from the concordance analysis, the differentiation—measured as DXY— occurs along the whole genome (leftmost panels of Fig. 5 and Fig. S9 in the supplemental material).
FIG 4.

Genetic variation is partitioned across species in Paracoccidioides. (A and C) Distribution of pairwise divergence within four species of Paracoccidioides. Blue and red bars show the two mean values of π (one for each species). (B and D) Distribution of average pairwise DXY values between two species pairs, P. venezuelensis/P. restrepiensis and P. brasiliensis/P. lutzii. Black lines show the mean value of pairwise DXY. In all cases, the distributions of intraspecific and interspecific pairwise differences are nonoverlapping (two-sample Fisher-Pitman permutation test; P < 1 × 10−10).
FIG 5.

Differentiation between species of Paracoccidioides occurs genome wide. DXY along the whole genome between P. venezuelensis and P. restrepiensis (left) is larger than π in either of the two species (center and left). Red solid lines show the median values; dashed orange lines show the 90th and 10th percentiles.
The joint results from the phylogenetic analyses and the genetic distance calculations indicate that the genetic diversity within Paracoccidioides is partitioned across species. The five proposed species of Paracoccidioides fulfill the expectations of being advanced in the speciation continuum in terms of genomic divergence (22, 23). Next, we tested whether such divergence is accompanied by a reduction in the amount of gene flow between species.
Low rates of detectable gene exchange between species.
If speciation has proceeded to advanced stages, as seems to be the case for the species in Paracoccidioides, the magnitude of gene exchange between species should be limited. To detect potential alleles that have crosses species boundaries, we used two different methods, D-statistics and Int-HMM, a method that detects haplotypes likely to have crossed species boundaries. We describe the results for each species pair as follows.
(i) Paracoccidioides lutzii and the species from the brasiliensis complex. Using Int-HMM, we found no evidence of introgression in any of these species pairs or in any reciprocal direction. If hybridization and admixture has occurred between P. lutzii and the other Paracoccidioides species, it has left no trace in the genomes of any of the involved species.
(ii) Paracoccidioides americana and the rest of the species from the brasiliensis complex.
D and fD, two metrics to detect introgression from phylogenetic trees (see Materials and Methods for details), suggest that P. americana has donated more genetic material to P. brasiliensis and P. venezuelensis than it has donated to P. restrepiensis. In all cases the proportion of introgression is small (i.e., fD is lower than 0.04; Table 1). We followed up with Ancestry-HMM and found no evidence of large haplotypes (over 500 bp) between P. americana and the other species from the brasiliensis complex. This result suggest that introgressions are small and are probably old or strongly selected against. Regardless of the explanation for this low proportion of admixture, the joint results indicate that the magnitude of gene exchange between P. americana and the other species of the brasiliensis species complex is low.
TABLE 1.
D and fD values, two metrics to detect introgression for species tetrads involving P. americana as a donora
| Species tetrad (P1-P2-P3)b | D | fD |
|---|---|---|
| P. restrepiensis-P. venezuelensis-P. americana | 0.415 | 0.040 |
| P. restrepiensis-P. brasiliensis-P. americana | 0.304 | 0.019 |
| P. brasiliensis-P. venezuelensis-P. americana | 0.011 | 4.954 × 10−3 |
A positive D value means more introgression between P3 (P. americana) and P2 (the second species listed) than between P3 and P1 (the first species in the list); a negative D value means introgression between P3 and P1.
In all cases, P. lutzii is the outgroup.
(iii) Paracoccidioides brasiliensis/P. restrepiensis and P. brasiliensis/P. venezuelensis. Unlike pairwise comparisons involving the more divergent species pairs, we found evidence of limited gene exchange using both methods in these two species pairs. The only tetrad that fulfilled the requirements for the calculation of D was [(((venezuelensis, restrepiensis), brasiliensis), lutzii)]. The evidence of gene flow in this case was strong and showed that introgression between P. brasiliensis and P. venezuelensis is more common than between P. brasiliensis and P. restrepiensis (D = 0.331, P < 0.001; degrees of freedom [df] = 0.078). This amount of introgression is higher than that observed between P. americana and other species in the brasiliensis species group.
Next, we used Int-HMM to study the characteristics of the introgressed haplotypes. Table 2 shows the percentage of genome that has crossed species boundaries in each sequenced genome. We found no overlap of the introgression regions between reciprocal directions or species pairs. In P. brasiliensis/P. restrepiensis, introgression mean length did not differ between reciprocal directions (Welch two-sample t test data; t = −0.857, df = 4.135, P = 0.438) and in both cases was ∼15 kb, suggesting similar times of admixture or similar selection against introgression (Fig. 6A). We found a similar pattern in P. brasiliensis/P. venezuelensis. The haplotype length did not differ between reciprocal directions (Welch two-sample t tests; P. brasiliensis-restrepiensis: t = 0.449, df = 3.456, P = 0.680; P. brasiliensis-P. venezuelensis: t = 0.536, df = 50.795, P = 0.594) and in both cases was ∼15 kb (Fig. 6B). In all cases, introgressions occur at low frequency (i.e., present in a single isolate per species) and are mostly located in intergenic regions (Table 3). Figure 7A to D shows the location of introgressed haplotypes in the two directions of the cross. Introgressions were distributed along most supercontigs, suggesting that most supercontigs are equally permissive—or refractory—to introgression (Fig. 7).
TABLE 2.
Percentage of the genome that shows introgression in each sequenced P. restrepiensis, P. venezuelensis, and P. brasiliensis isolate
| Speciesa | Isolate | % of genome showing introgression fromb: |
||
|---|---|---|---|---|
| P. restrepiensis | P. brasiliensis | P. venezuelensis | ||
| P. restrepiensis | EPM_83 | NA | 0 | 1.20 |
| P. restrepiensis | Pb60855 | NA | 0.18 | 1.14 |
| P. restrepiensis | PbBAC | NA | 0 | 0 |
| P. restrepiensis | PbCAB | NA | 0 | 0 |
| P. restrepiensis | PbCNH | NA | 8.10 × 10−3 | 0 |
| P. restrepiensis | PbJAM | NA | 0.07 | 0.14 |
| P. brasiliensis sensu stricto | MS1 | 0.61 | NA | 1.51 |
| P. brasiliensis sensu stricto | DO3 | 0.13 | NA | 2.33 |
| P. brasiliensis sensu stricto | Pb1445 | 0.87 | NA | 1.14 |
| P. brasiliensis sensu stricto | Pb377 | 0.0 | NA | 0 |
| P. brasiliensis sensu stricto | PbBercelli | 0.66 | NA | 0 |
| P. brasiliensis sensu stricto | PbD02 | 0.10 | NA | 0.95 |
| P. brasiliensis sensu stricto | PbT1F1 | 0.14 | NA | 1.21 |
| P. brasiliensis sensu stricto | T15N1 | 0.05 | NA | 0.78 |
| P. brasiliensis sensu stricto | T16B1 | 0.26 | NA | 0.54 |
| P. brasiliensis sensu stricto | MS2 | 0.67 | NA | 0.06 |
| P. brasiliensis sensu stricto | Pb_66 | 0.29 | NA | 0.02 |
| P. venezuelensis | Pb309 | 1.20 | 0.87 | NA |
| P. venezuelensis | Pb304 | 0.45 | 1.10 | NA |
| P. venezuelensis | Pb307 | 0 | 0 | NA |
| P. venezuelensis | PbS89305 | 1.41 | 0.95 | NA |
| P. venezuelensis | PbS90384 | 0.55 | 0.05 | NA |
| P. venezuelensis | PbS5387 | 0.14 | 0.13 | NA |
| P. venezuelensis | PbS91444 | 0.13 | 0.04 | NA |
| P. venezuelensis | Pb300 | 0 | 0.11 | NA |
Isolates from P. lutzii and P. americana show no evidence of large (over 500 bp) introgressed haplotypes and are not listed.
NA, not applicable.
FIG 6.
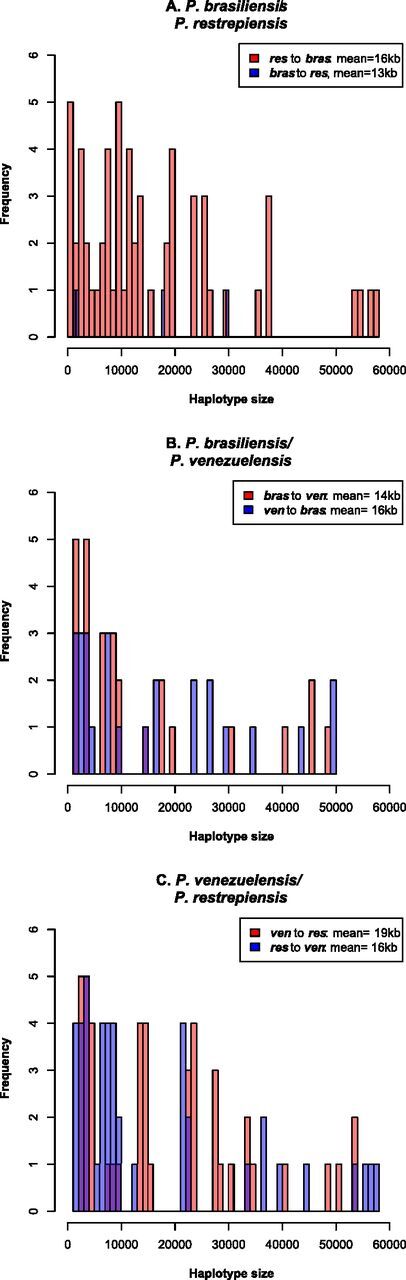
Distributions of the size of introgressed haplotypes in three species pairs of Paracoccidioides identified using Int‐HMM. The Int‐HMM algorithm was run on each individual separately, and the distribution of haplotype sizes was computed for each of the two directions of introgression. (A) Size distribution of introgressed haplotypes between P. brasiliensis sensu stricto and P. restrepiensis. (B) Size distribution of introgressed haplotypes between P. brasiliensis sensu stricto and P. venezuelensis. (C) Size distribution of introgressed haplotypes between P. venezuelensis and P. restrepiensis. Even though there are large introgressions (over 50 kb) in most crosses, the majority of the introgressions are small.
TABLE 3.
Introgressions between P. restrepiensis and P. brasiliensis sensu stricto are mostly found in intergenic regions
| Direction | Sequence typea | Length (kb) | Introgressed %b | Genomic %c | Enrichmentd |
|---|---|---|---|---|---|
| P. restrepiensis into P. brasiliensis | 10-kb inter | 9.78 | 42.8565 | 23.990 | 1.786 |
| P. restrepiensis into P. brasiliensis | 2-kb upstream inter | 6.28 | 27.4918 | 29.759 | 0.9245 |
| P. restrepiensis into P. brasiliensis | 3′ UTR | 0.03 | 0.1227 | 1.817 | 0.068 |
| P. restrepiensis into P. brasiliensis | 5′ UTR | 0 | 0 | 1.019 | 0 |
| P. restrepiensis into P. brasiliensis | CDS | 5.92 | 25.9365 | 44.598 | 0.582 |
| P. restrepiensis into P. brasiliensis | Intergenic | 0 | 0 | 1.904 | 0 |
| P. restrepiensis into P. brasiliensis | Intron | 0.82 | 3.5926 | 12.225 | 0.294 |
| P. brasiliensis into P. restrepiensis | 10-kb inter | 980.29 | 26.1924 | 23.990 | 1.092 |
| P. brasiliensis into P. restrepiensis | 2-kb upstream inter | 1,436.81 | 38.39 | 29.759 | 1.290 |
| P. brasiliensis into P. restrepiensis | 3′ UTR | 24.31 | 0.6495 | 1.817 | 0.358 |
| P. brasiliensis into P. restrepiensis | 5′ UTR | 10.12 | 0.2704 | 1.019 | 0.266 |
| P. brasiliensis into P. restrepiensis | CDS | 1,071.78 | 28.637 | 44.598 | 0.642 |
| P. brasiliensis into P. restrepiensis | Intergenic | 113.07 | 3.0212 | 1.904 | 1.587 |
| P. brasiliensis into P. restrepiensis | Intron | 106.27 | 2.8395 | 12.225 | 0.232 |
To study whether any particular type of sequence was over- or underrepresented, we partitioned the genome by sequence type with each region being assigned to one of the following eight sequence types: coding sequence (CDS), exon, 5′ untranslated region (UTR), 3′ UTR, intron, 2-kb upstream inter (intergenic sequence 2 kb upstream of a gene), 10-kb inter (intergenic sequence within 10 kb of a gene), and intergenic (intergenic sequence more than 10 kb from a gene).
The introgressed percentage is the percentage of introgressions overlapping a given sequence type for that direction.
The genomic percentage is the percentage of the genome represented by a given sequence type.
Enrichment = (introgressed percentage)/(genomic percentage).
FIG 7.
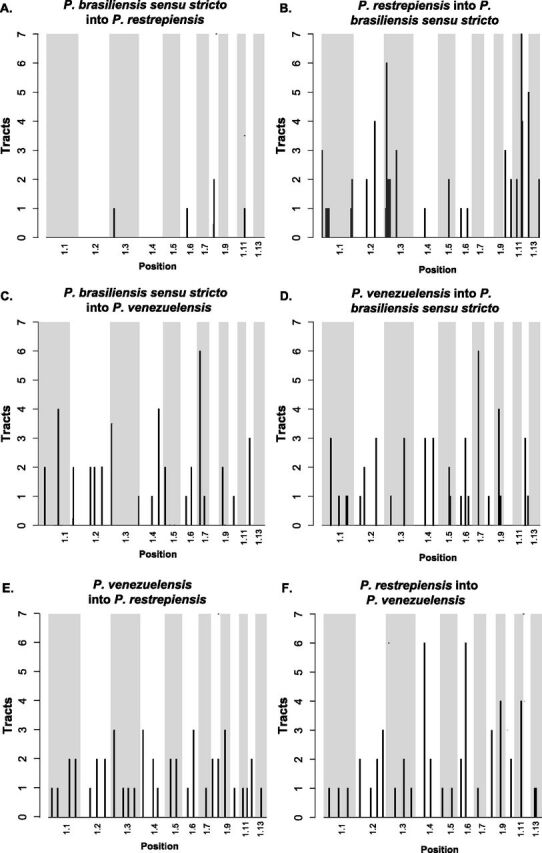
Location of the introgression tracts between species of Paracoccidioides. Each pair of panels shows the two reciprocal directions of introgression for a species pair. (A and B) P. restrepiensis and P. brasiliensis sensu stricto. (C and D) P. venezuelensis and P. brasiliensis sensu stricto. (E and F) P. restrepiensis and venezuelensis. The two directions show differences in the amount of introgression, hence the differences in the scale of the y axis. We plotted the largest 13 supercontigs of the genome. Histograms show the number of introgression tracts found in 500-kb windows.
(iv) P. venezuelensis/P. restrepiensis. Finally, we studied the most recently diverged species pair in Paracoccidioides using Int-HMM. We found no overlap in the location of haplotypes between the two reciprocal directions or with any of the other dyads of Paracoccidioides. There was no difference in the genome proportion introgressed per individual between reciprocal directions (Welch two-sample t test; t = 0.11, df = 9.95, P = 0.91). The mean haplotype length did not differ between reciprocal directions (Welch two-sample t test; t = 0.741, df = 85.12, P = 0.461) and in both cases was ∼16 kb (Fig. 6C). As in the case for the other Paracoccidioides dyads, introgressions were at a low frequency and were largely in intergenic regions (Table 3). Notably, we found similar amounts of introgression in this pair as we found in the more divergent pairs (Welch two-sample t test; P. venezuelensis/P. restrepiensis versus P. venezuelensis/P. brasiliensis sensu stricto, t = 1.205, df = 31.962, P = 0.237; P. venezuelensis/P. restrepiensis versus P. restrepiensis/P. brasiliensis sensu stricto, t = −1.372, df = 21.731, P = 0.184). Figure 6C shows the haplotype size frequency distribution of introgressions in the two reciprocal directions. Introgressions were distributed along the whole genome and did not follow a particular clustering pattern (Fig. 7E and F).
DISCUSSION
Our study uses genomic data to confirm previous observations that five species of Paracoccidioides (i) are all haploid, and (ii) are genetically differentiated. We also present results that suggest that the genomes of these species show strong levels of genealogical concordance genome wide and rarely exchange genes. The species of Paracoccidioides show considerable divergence and reciprocal monophyly which in turn suggest these five species are at an advanced stage on the speciation continuum (22–24).
Our analyses of the magnitude of gene flow between species confirm that despite extensive geographic overlap, the Paracoccidioides species rarely exchange genes. In the more divergent pairs, those of the brasiliensis complex and P. lutzii, we found no evidence of introgression. This is consistent with the high levels of divergence between P. lutzii and the species from the brasiliensis complex, which have been hypothesized to be over 30 million years apart (10, 12). We observed a similar—but not identical— pattern between P. americana and the other species of the brasiliensis complex (P. brasiliensis, P. restrepiensis, and P. venezuelensis). Potential introgressions between these species are rare and of very small size, making them potentially indistinguishable from incomplete lineage sorting, as there was no evidence of gene exchange in any of the pairs. This paucity of gene exchange is not caused by lack of contact. Paracoccidioides brasiliensis, P. lutzii, and P. americana coexist in Brazil and have even been found in the same host (8, 25). Paracoccidioides venezuelensis and P. americana share their geographic range in Venezuela as well. This extensive geographic overlap suggests that there is ample opportunity for gene exchange, but it does not occur.
We do find evidence of moderate gene exchange in the triad P. brasiliensis-P. restrepiensis-P. venezuelensis. These low levels of gene exchange are consistent with advanced divergence among Paracoccidioides species. Our scans for gene exchange pose two additional questions. First, the rate of gene exchange is symmetrical in two Paracoccidioides species pairs, P. brasiliensis/P. venezuelensis and P. venezuelensis/P restrepiensis. The third species pair, P. brasiliensis sensu stricto/P. restrepiensis, shows strongly asymmetric introgression that is mostly found in intergenic regions. The reasons behind this pattern remain unknown. We formulate two possibilities. First, the direction of migration might be asymmetric between these two species. If P. brasiliensis migrants come into the range of P. restrepiensis more often than the reciprocal type of migration, then P. brasiliensis alleles should be found more frequently in the P. restrepiensis background than the reciprocal. A second possibility is that the P. restrepiensis background is less permissive of introgression because the introgressed alleles might have more deleterious effects. Since P. restrepiensis has a much smaller effective population size (8), variants that might ameliorate the potentially deleterious effect of introgressed alleles should be rarer. On the other hand, small populations might harbor fewer deleterious mutations (26). The rates of migration, hybridization—or even intraspecific recombination— and of potential hybrid incompatibilities are unknown in Paracoccidioides, and we cannot disentangle these possibilities.
A second intriguing pattern is that P. venezuelensis and P. restrepiensis show a similar level of introgression to those of the other species pairs. As divergence increases, so should the number of incompatibilities (27–29), which in turn should reduce the proportion of genome that can flow from one species to the other (30, 31). Since the P. venezuelensis/P. restrepiensis dyad is more closely related than other species pairs within the brasiliensis complex, we expected a higher level of gene exchange. Our results do not support this hypothesis. Even though the precise reasons for this pattern remain unexplored, the role of geography might be of particular importance. The ranges of P. venezuelensis and P. restrepiensis are contiguous but have not been reported to overlap. This differs from all other species pairs in Paracoccidioides, which show some degree of geographic overlap (25). A precise assessment of the range and opportunity for hybridization will be crucial to establish the genetic, environmental, and demographic factors that govern the patterns of introgression in Paracoccidioides.
The identification of species boundaries and introgression in fungal pathogens has human health-related implications. Paracoccidioides lutzii and P. brasiliensis sensu stricto show differences in the immunological response they elicit (14, 32), the strength of the disease they cause (14), and in traits involved in diagnostic tools (33–35). Introgression, then, can be a vehicle to transfer virulence factors and antifungal resistance in Paracoccidioides. Gene exchange can also be a source of variation in other fungal pathogens (36–38). A systematic survey to characterize the virulence and resistance of differentiated species across their whole geographic range could reveal the extent to which diversification of the ethological agents of PCM has also led to divergence in virulence strategies. The combination of phenotypic studies and population genetics can also reveal whether gene exchange plays a role on the transfer of virulence factors and antifungal resistance strategies.
Our results are in line with those of other studies that show that species boundaries in fungi are semipermeable (15, 39, 40) and that introgression might not be rare. On the other hand, they also reveal that introgression is not an unavoidable outcome of secondary contact. Geographic overlap is not synonymous with hybridization, and in cases of diverged species (such as P. lutzii and the species of the brasiliensis species complex), hybrids might not occur even when species share a close geographic range. Hybrids might also be sterile or inviable (41). Genome factors such as the amount of divergence between hybridizing species (30) and the landscape of recombination (42, 43) affect whether an introgression persists after hybridization. The different levels of divergence and the ample opportunity for hybridization among Paracoccidioides species provide for a system to test the relative importance of genomic factors in determining the amount of introgression occurs in nature.
MATERIALS AND METHODS
Public data.
All of the data used here have been previously published. The SRA numbers are listed in Table S1 in the supplemental material. To root our trees (see below), we obtained sequencing reads from two species of Histoplasma: Histoplasma capsulatum sensu stricto and Histoplasma mississippiense (SRA BioProject accession number PRJNA416769) (44). These species are among some of the closest relatives of Paracoccidioides (45).
SRA numbers of the genome sequences used in this study. Download Table S1, DOCX file, 0.02 MB (22.2KB, docx) .
Copyright © 2020 Mavengere et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Read mapping and variant calling.
Reads were mapped to the Paracoccidioides brasiliensis strain Pb18 genome (BioProject accession number PRJNA28733 and BioSample accession number SAMN02953720), currently assembled into 57 supercontigs, using Burrows-Wheeler Aligner (BWA) version 0.7.12 (46). BAM files were then merged using SAMtools version 0.1.19. Indels were identified and reads locally remapped in the merged BAM files using the GATK version 3.2-2 RealignerTargetCreator and IndelRealigner functions (47, 48). Subsequently, single-nucleotide polymorphisms (SNPs) were called using the GATK UnifiedGenotyper function with the parameter “het” set to 0.01 and all others left as default. The following filters were applied to the resulting VCF file: QD = 2.0, FS_filter = 60.0, MQ_filter = 30.0, MQ_Rank_Sum_filter = −12.5, and Read_Pos_Rank_Sum_filter = −8.0. Sites were excluded if the coverage was less than 5 or greater than the 99th quantile of the genomic coverage distribution for the given line or if the SNP failed to pass one of the GATK filters.
Ploidy estimation.
To detect admixture between species of Paracoccidioides, we used Int-HMM (49), an algorithm to detect introgression that requires information on the ploidy of an individual (i.e., it can be run to detect introgression in diploid or haploid organisms; see below). We used genome-wide data to determine the most likely ploidy of the Paracoccidioides isolates. We used Illumina short reads from the five species of Paracoccidioides (described above) to do two ploidy tests. First, we plotted the per-site sequencing coverage. In cases in which there is partial aneuploidy in the form of chromosomal duplications, there will be a bimodal distribution. In cases where the genome does not harbor aneuploid regions, there will be a single mode in the distribution. To compare the observed distribution of per-site coverage with the null hypothesis of uniform sequencing coverage, we used a two-sample Fisher-Pitman permutation test (function oneway_test, library coin [50]). We used the “hist” function (library graphics [51]) in R to plot the distribution of the per site coverage and of allele frequencies per site across the whole genome for each strain.
Next, we studied the ploidy of Paracoccidioides at a local level. We used the same two metrics described above. Sites with the ploidy of the rest of the genome should show a mean per-window coverage. Once-duplicated segments (either as copy number variation or as changes in ploidy) should have twice the coverage of the genome average. We thus calculated the coverage and mean minor allele frequency for each 5-kb window in the genome to assess whether there were segments of the genome with evidence for changes in ploidy.
Phylogenetic reconstructions.
Our goal was to determine whether the species from Paracoccidioides were reciprocally monophyletic and thus satisfy the requirements to be considered phylogenetic species. We followed a phylogenetic species concept (15, 52, 53) to recognize species, defining species as genetic clusters that are reciprocally monophyletic and for which there was genealogical concordance across genome-wide unlinked loci. We used two parallel approaches: (i) maximum likelihood trees at the genome and at the supercontig level, and (ii) Bayesian concordance analysis of the genealogies of orthologous genes. We describe each of these two approaches as follows.
(i) Maximum likelihood phylogenetics. To determine whether the proposed Paracoccidioides species were monophyletic, we first used maximum likelihood phylogenetics. Reciprocal monophyly is a trademark of speciation (15, 22); as divergence accrues, the likelihood of reciprocal monophyly across the whole genome increases for two reasons. First, as divergence increases, the likelihood of incomplete lineage sorting decreases (54, 55). Likewise, in diverged lineages, the magnitude of retained introgression is lower even in cases where hybridization might occur frequently (30, 31, 49). Since recombination in Paracoccidioides occurs but seems to be rare, and there is a high level of linkage disequilibrium across the genome (see Results), we studied each supercontig as an unlinked locus. Since mitochondrial DNA (mtDNA) shows evidence of interspecific gene transfer (10, 56), we focused only on nuclear genomes. We obtained whole supercontig sequences for each individual from the VCF file using the FastaAlternateReferenceMaker tool in GATK, realigned them using Mafft version 7 (57), and used them to build maximum likelihood (ML) trees using RAxML version 8.2.9 (58). We inferred individual trees for each of the largest six supercontigs, which encompass 62% of the genome. We also generated a genome-wide tree of a concatenated alignment of all supercontigs. Analyses were run under the GTR + Γ model, with 1,000 bootstrap pseudoreplicates to assess support for each node. The genome-wide analysis was partitioned by supercontig, with each partition having its own set of GTR + Γ model parameters. All trees were rooted using Histoplasma mississippiense and H. capsulatum sensu stricto (44). To determine the extent of congruence among supercontig trees and the whole-genome tree, we used the Robinson-Foulds (RF) (or symmetric difference [59]) distance (function treedist, library phangorn [60]), which counts how many partitions are in one tree but not the other. Even though this is a simplistic approach that does not take into account branch length information (18, 61, 62), it reveals whether there are large-scale levels of incongruency in the topology. We also plotted the concatenated tree topology and superimposed the trees from each supercontig using the R function compare.chronograms (library phytools [63]) and the nodes that are present in the whole-genome sequence tree but not in the supercontig genealogies using the function comparePhylo (library ape [64]).
(ii) Bayesian concordance analyses. In cases where speciation has occurred and genetic divergence has accrued, the phylogenetic signal across the whole genome should be congruent across loci. We measured the genome-wide genealogical concordance at a finer scale using a Bayesian concordance analysis (BCA). First, we identified orthologous genes using the BUSCO annotation pipeline (65, 66), which encompasses 1,316 benchmarking universal single-copy orthologs. These groups of genes have been curated in 25 different species of ascomycetous fungi (65, 66). Next, we used MrBayes version 3.2.6 (67, 68) to generate posterior tree distributions for each single gene. We summarized the gene trees using the command mbsum in the BUCKy program (69–71) with a burn-in of 1,000 trees. We then fed individual gene genealogies to BUCKy version 1.4.2, with four independent runs and four Markov chain Monte Carlo (MCMC) chains, each with 10 million generations with a burn-in period of 100,000. Five values of the α parameter (0.1, 0.5, 1, 5, and 10) were tested, which correspond to the prior probability distribution for the number of distinct gene trees (70). The level of support for each node is expressed as a concordance factor (CF), which ranges between 0 (no concordance between genealogies) and 1 (complete concordance). This approach allowed us to infer the phylogenetic relationships between putative species, while estimating the genome-wide genealogical support for their monophyly.
Genetic distance.
To further assess the extent of genetic differentiation between phylogenetic species, we used the metric , the average number of nucleotide differences between one sequence randomly chosen from a population and another sequence randomly chosen from a second population or species. DXY, or , followed the form:
Mean was the mean of all pairwise comparisons between individuals from two species. We calculated 20 mean values. We also calculated , the average pairwise genetic distance between individuals of the same species. followed the same form as , but instead of calculating the average number of differences between species, it calculates the average number of differences between two randomly selected individuals of the same species. Mean was the mean within-species value for each of the five species. We used Python for all calculations.
In cases were speciation is complete, is expected to be much larger than . For each species pair, can take two values (i.e., from each of the two species), so for the pairwise comparisons is the pooled set of the two intraspecific distances. To compare the values of and for each species pair (10 pairwise comparisons), we used two-sample Fisher-Pitman permutation tests as implemented by the function oneway_test in the coin library in R (9,999 Monte Carlo resamplings) (50). We also calculated for each of the 10 pairwise comparisons and for the five Paracoccidioides species for each of the largest 6 supercontigs.
Gene exchange between species of Paracoccidioides.
Previous work based on coding and microsatellite data suggested the possibility of gene exchange between Paracoccidioides species (10). However, although microsatellite makers have the potential to reveal genealogical relationships between very closely related individuals, they are also prone to homoplasy, as they mutate quickly and their identity might not be caused by descent (72–74). To address the possibility of gene exchange with better resolution, we used whole-genome data and two complementary methods, D-statistics and Int-HMM.
(i) D-statistics. First, we calculated the excess of variants shared between potentially admixed species using D-statistics (75–78). D is a metric to detect introgression from phylogenetic trees. The metric requires a four-taxon topology [(((P1, P2), P3), O)]. The allele in the outgroup (O) is labeled A, while the derived allele in the ingroup is labeled B. D compares the occurrence of two discordant site patterns, ABBA and BABA, representing sites in which an allele is derived in P3 relative to O and is derived in one but not both of the sister lineages P1 and P2. These discordant patterns are most likely to arise if introgression occurs between P3 and either P2 or P1, in which case one site pattern will occur more frequently than the other. A positive D value means introgression between P3 and P2; a negative D value means introgression between P3 and P1. Due to the need for a sorted topology, we focused on four species tetrads where [(((P1, P2), P3), O)] were as follows: [(((venezuelensis, restrepiensis), americana), lutzii)], [(((venezuelensis, restrepiensis), brasiliensis), lutzii)], [(((venezuelensis, brasiliensis), americana), lutzii)], [(((restrepiensis, brasiliensis), americana), lutzii)].
For each species pair, we measured the standard deviation of D from 1,000 bootstrap replicates. The observed genome-wide D was converted to a Z‐score measuring the number of standard deviations it deviates from 0, and significance was assessed from a P value using an α of 0.01 as a cutoff after Holm-Bonferroni correction for multiple testing. We also calculated a variation of D, fD, which estimates the proportion of admixture by dividing the observed difference between the ABBA and BABA counts to the expected difference when the entire genome is introgressed. Besides the genome-wide average of D and fD, we calculated both metrics for 5-kb windows along the genome. We used DSuite for all calculations (79), and used the allele frequencies within each species, as recommended in reference 77.
(ii) Identification of introgressed haplotypes with Ancestry-HMM. We used a hidden Markov model (HMM) able to detect introgression in diploids and haploids (i.e., Int-HMM [36, 37, 49]) to identify introgressed regions between all pairs of Paracoccidioides species. The HMM identifies introgressions between a pair of diverged populations or species, a donor and a recipient (i.e., the admixed individual), by inferring the ancestry of every SNP in the genome. It then identifies a consecutive group of SNPs from the donor in the recipient background. Donor SNPs were selected such that they were monomorphic in the donor species and the allele frequency differences between the two species was greater than or equal to 30%. We also required that every individual in the donor species and at least one individual in the recipient species had a called genotype. Transition and emission probabilities of the HMM have been described elsewhere (36, 37).
(iii) Identifying introgression tracts. Int-HMM determined the most probable genotype for each marker in each individual. We defined tracts as contiguous markers with the same genotype (species 1 or species 2). Introgressed SNPs are defined as those within a tract where the HMM probability for an introgression state (d) (i.e., originating from the donor) was ≥50%. In cases where we identified a region of d with at least 10 introgressed SNP markers flanked on one side by a small tract (under 10 SNPs) from the recipient that in turn was flanked by a single larger tract that was completely d, the two introgressed regions were merged and consolidated into a single tract. We did four consecutive rounds of filtering to allow identification of larger introgressed tracts that were broken up by small sections of the recipient species. These broken regions might be caused by gene conversion, double recombination events, or sequencing error (Fig. S1 in the supplemental material shows an example).
Example of an introgression from P. restrepiensis to P. brasiliensis defined by Int-HMM. Blue bars represent P. brasiliensis ancestry; red bars represent P. restrepiensis ancestry. Light red bars represent potential P. restrepiensis introgressions with low support (i.e., few markers). Download FIG S1, PDF file, 0.1 MB (109.5KB, pdf) .
Copyright © 2020 Mavengere et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Paracoccidioides americana is haploid and shows no detectable levels of aneuploidy. Coverage and minor allele frequency plots for the largest 16 supercontigs. Per-site estimations of coverage (in red) show close adherence to the genome-wide mean coverage for the whole genome. Per-site estimations of the extent of polymorphism across the genome suggest that no single site shows evidence of polymorphism (cutoff: minor allele frequency > 20%), suggesting that besides having no local aneuploidy, Paracoccidioides is haploid. The plot shows results for isolate T10B1; plots for other lines can be found in the Dryad package (https://doi.org/10.5061/dryad.w3r2280pj). Download FIG S2, PDF file, 0.1 MB (79KB, pdf) .
Copyright © 2020 Mavengere et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Paracoccidioides brasiliensis is haploid and shows no detectable levels of aneuploidy. Coverage and minor allele frequency plots for the largest 16 supercontigs. Per-site estimations of coverage (in red) show close adherence to the genome-wide mean coverage for the whole genome. Per-site estimations of the extent of polymorphism across the genome suggest that no single site shows evidence of polymorphism (cutoff: minor allele frequency > 20%), suggesting that besides having no local aneuploidy, Paracoccidioides is haploid. The plot shows results for isolate T1F1; plots for other lines can be found in the Dryad package (https://doi.org/10.5061/dryad.w3r2280pj). Download FIG S3, PDF file, 0.1 MB (78.6KB, pdf) .
Copyright © 2020 Mavengere et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Paracoccidioides restrepiensis is haploid and shows no detectable levels of aneuploidy. Coverage and minor allele frequency plots for the largest 16 supercontigs. Per-site estimations of coverage (in red) show close adherence to the genome-wide mean coverage for the whole genome. Per-site estimations of the extent of polymorphism across the genome suggest that no single site shows evidence of polymorphism (cutoff: minor allele frequency > 20%), suggesting that besides having no local aneuploidy, Paracoccidioides is haploid. The plot shows results for isolate EPM83; plots for other lines can be found in the Dryad package (https://doi.org/10.5061/dryad.w3r2280pj). Download FIG S4, PDF file, 0.1 MB (78.3KB, pdf) .
Copyright © 2020 Mavengere et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Paracoccidioides venezuelensis is haploid and shows no detectable levels of aneuploidy. Coverage and minor allele frequency plots for the largest 16 supercontigs. Per-site estimations of coverage (in red) show close adherence to the genome-wide mean coverage for the whole genome. Per-site estimations of the extent of polymorphism across the genome suggest that no single site shows evidence of polymorphism (cutoff: minor allele frequency > 20%), suggesting that besides having no local aneuploidy, Paracoccidioides is haploid. The plot shows results for isolate Pb300; plots for other lines can be found in the Dryad package (https://doi.org/10.5061/dryad.w3r2280pj). Download FIG S5, PDF file, 0.1 MB (83.4KB, pdf) .
Copyright © 2020 Mavengere et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Graphic representation of tree concordance between the whole-genome topology and the genealogy produced from each supercontig. Arrows show differences in branch length between trees built with the whole genome (light red) and data from each supercontig (light blue). The plots were made using compare.chronograms (library phytools [63]). Download FIG S6, PDF file, 0.2 MB (167.4KB, pdf) .
Copyright © 2020 Mavengere et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Differences in topology between the trees from the whole genome and those from each supercontig. We only show the results for the tree supercontigs that showed differences with the whole-genome tree, as follows: (A) supercontig 2.2, (B) supercontig 2.3, and (C) supercontig 2.5. Blue circles show clades absent in the supercontig tree but present in the whole-genome tree. Download FIG S7, PDF file, 0.1 MB (152KB, pdf) .
Copyright © 2020 Mavengere et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
π and DXY calculations for pairwise comparisons that included P. lutzii. Color scheme and arrangement are similar to those in Fig. 4. Note that panels A and B are identical to panels C and D in Fig. 4. Download FIG S8, PDF file, 0.3 MB (268.8KB, pdf) .
Copyright © 2020 Mavengere et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Differentiation between species of Paracoccidioides occurs genome wide. DXY along the whole genome between P. brasiliensis and P. restrepiensis (left) is larger than π in either of the two species (center and left). Red solid lines show the median values; dashed orange lines show the 90th and 10th percentiles. Download FIG S9, PDF file, 0.8 MB (880.2KB, pdf) .
Copyright © 2020 Mavengere et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Enrichment by sequence type.
In cases where introgression is deleterious, selection will operate most efficiently against regions encoding functional elements (e.g., coding sequences and promoters [49, 80, 81]). To test if a particular type of sequence was more or less prone to appearing in introgressed regions, we partitioned the genome by sequence type into one of the following seven categories using the P. brasiliensis Pb18 genome annotations (BioProject accession number PRJNA28733): CDS (coding sequence), 5′ untranslated region [UTR], 3′ UTR, intron, 2-kb upstream inter (intergenic sequence 2 kb upstream of a gene), 10-kb inter (intergenic sequence within 10 kb of a gene excluding 2 kb upstream of a gene), and intergenic (intergenic sequence more than 10 kb from a gene [82]). Introgressions present in more than one individual but with different endpoints among isolates were broken into blocks, and these blocks were treated separately in the permutation test (described immediately below).
We calculated a summary statistic for each of the seven categories using the following definitions: “introgressed percentage” is the percentage of introgressions overlapping a given sequence type that occurred in any of the four possible introgression directions (two different species pairs and two reciprocal directions), “genomic percentage” is the proportion of the genome of any given type of sequence, and “Enrichment” is the ratio of the percentage of a given sequence type that has crossed species boundaries and the percentage of the genome encompassed by the same sequence type. We used a permutation test in which each introgression block was randomly assigned to a new position in the genome to calculate P values. For each permutation assay, we calculated the percentage of the randomly reassigned blocks overlapped with type of sequence. We repeated this procedure 10,000 times and generated a null-distribution for enrichment. If introgressions are more likely to occur within a certain type of sequence than in the rest of the genome, enrichment will be greater than 1. Conversely, if introgressions are less likely to occur in a given type of sequence, enrichment will be less than 1. We determined whether introgressions were significantly enriched for any sequence type (i.e., a significant departure from 1), by comparing the observed enrichment and the distribution of resampled enrichments.
ACKNOWLEDGMENTS
We thank the members of the Matute lab for helpful scientific discussions and comments. The work was extensively improved by the comments of two anonymous reviewers.
M.M.T. was supported by Conselho Nacional de Ciência e Tecnologia (CNPq) under contract 43460/2018-2. This work was supported by NIH award R01GM121750.
We have no conflicts of interest.
Footnotes
Citation Mavengere H, Mattox K, Teixeira MM, Sepúlveda VE, Gomez OM, Hernandez O, McEwen J, Matute DR. 2020. Paracoccidioides genomes reflect high levels of species divergence and little interspecific gene flow. mBio 11:e01999-20. https://doi.org/10.1128/mBio.01999-20.
REFERENCES
- 1.Brummer E, Castaneda E, Restrepo A. 1993. Paracoccidioidomycosis: an update. Clin Microbiol Rev 6:89–117. doi: 10.1128/cmr.6.2.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Restrepo A, Tobón AM. 2009. Paracoccidioides brasiliensis. In Mandell, Douglas, and Bennett’s principles and practice of infectious diseases, 7th ed. Churchill Livingstone Elsevier, Philadelphia, PA. [Google Scholar]
- 3.Roberto TN, Rodrigues AM, Hahn RC, De Camargo ZP. 2016. Identifying Paracoccidioides phylogenetic species by PCR-RFLP of the alpha-tubulin gene. Med Mycol 54:240–247. doi: 10.1093/mmy/myv083. [DOI] [PubMed] [Google Scholar]
- 4.Niño-Vega GA, Calcagno AM, San-Blas G, San-Blas F, Gooday GW, Gow NAR. 2000. RFLP analysis reveals marked geographical isolation between strains of Paracoccidioides brasiliensis. Med Mycol 38:437–441. doi: 10.1080/714030970. [DOI] [PubMed] [Google Scholar]
- 5.Kurokawa CS, Lopes CR, Sugizaki MF, Kuramae EE, Franco MF, Peraçoli MTS. 2005. Virulence profile of ten Paracoccidioides brasiliensis isolates: association with morphologic and genetic patterns. Rev Inst Med Trop Sao Paulo 47:257–262. doi: 10.1590/s0036-46652005000500004. [DOI] [PubMed] [Google Scholar]
- 6.Feitosa LDS, Cisalpino PS, Machado Dos Santos MR, Mortara RA, Barros TF, Morais F V, Puccia R, Da Silveira JF, De Camargo ZP. 2003. Chromosomal polymorphism, syntenic relationships, and ploidy in the pathogenic fungus Paracoccidioides brasiliensis. Fungal Genet Biol 39:60–69. doi: 10.1016/s1087-1845(03)00003-3. [DOI] [PubMed] [Google Scholar]
- 7.Morais FV, Barros TF, Fukada MK, Cisalpino PS, Puccia R. 2000. Polymorphism in the gene coding for the immunodominant antigen gp43 from the pathogenic fungus Paracoccidioides brasiliensis. J Clin Microbiol 38:3960–3966. doi: 10.1128/JCM.38.11.3960-3966.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matute DR, McEwen JG, Puccia R, Montes BA, San-Blas G, Bagagli E, Rauscher JT, Restrepo A, Morais F, Niño-Vega G, Taylor JW. 2006. Cryptic speciation and recombination in the fungus Paracoccidioides brasiliensis as revealed by gene genealogies. Mol Biol Evol 23:65–73. doi: 10.1093/molbev/msj008. [DOI] [PubMed] [Google Scholar]
- 9.Teixeira MM, Theodoro RC, Nino-Vega G, Bagagli E, Felipe MSS. 2014. Paracoccidioides species complex: ecology, phylogeny, sexual reproduction, and virulence. PLoS Pathog 10:e1004397. doi: 10.1371/journal.ppat.1004397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turissini DA, Gomez OM, Teixeira MM, McEwen JG, Matute DR. 2017. Species boundaries in the human pathogen Paracoccidioides. Fungal Genet Biol 106:9–25. doi: 10.1016/j.fgb.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Melo Teixeira M, Theodoro RC, De Oliveira FFM, Machado GC, Hahn RC, Bagagli E, San-Blas G, Felipe MSS. 2015. Paracoccidioides lutzii sp. nov.: biological and clinical implications. Med Mycol 52:19–28. [DOI] [PubMed] [Google Scholar]
- 12.Teixeira MM, Theodoro RC, de Carvalho MJA, Fernandes L, Paes HC, Hahn RC, Mendoza L, Bagagli E, San-Blas G, Felipe MSS. 2009. Phylogenetic analysis reveals a high level of speciation in the Paracoccidioides genus. Mol Phylogenet Evol 52:273–283. doi: 10.1016/j.ympev.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 13.Muñoz JF, Farrer RA, Desjardins CA, Gallo JE, Sykes S, Sakthikumar S, Misas E, Whiston EA, Bagagli E, Soares CM, Teixeira MM, Taylor JW, Clay OK, McEwen JG, Cuomo CA. 2016. Genome diversity, recombination and virulence across the major lineages of Paracoccidioides. mSphere 1:e00213-16. doi: 10.1128/mSphere.00213-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siqueira IM, Fraga CLF, Amaral AC, Souza ACO, Jerônimo MS, Correa JR, Magalhães KG, Inácio CA, Ribeiro AM, Burguel PH, Felipe MS, Tavares AH, Bocca AL. 2016. Distinct patterns of yeast cell morphology and host responses induced by representative strains of Paracoccidioides brasiliensis (Pb18) and Paracoccidioides lutzii (Pb01). Med Myco 54:177–188. doi: 10.1093/mmy/myv072. [DOI] [PubMed] [Google Scholar]
- 15.Matute DR, Sepúlveda VE. 2019. Fungal species boundaries in the genomics era. Fungal Genet Biol 131:103249. doi: 10.1016/j.fgb.2019.103249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Almeida AJ, Matute DR, Carmona JA, Martins M, Torres I, McEwen JG, Restrepo A, Leão C, Ludovico P, Rodrigues F. 2007. Genome size and ploidy of Paracoccidioides brasiliensis reveals a haploid DNA content: flow cytometry and GP43 sequence analysis. Fungal Genet Biol 44:25–31. doi: 10.1016/j.fgb.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 17.Mendes FK, Hahn MW. 2018. Why concatenation fails near the anomaly zone. Syst Biol 67:158–169. doi: 10.1093/sysbio/syx063. [DOI] [PubMed] [Google Scholar]
- 18.Bryant D, Steel M. 2009. Computing the distribution of a tree metric. IEEE/ACM Trans Comput Biol Bioinform 6:420–426. doi: 10.1109/TCBB.2009.32. [DOI] [PubMed] [Google Scholar]
- 19.Baum DA. 2007. Concordance trees, concordance factors, and the exploration of reticulate genealogy. Taxon 56:417–426. doi: 10.1002/tax.562013. [DOI] [Google Scholar]
- 20.Ané C, Larget B, Baum DA, Smith SD, Rokas A. 2007. Bayesian estimation of concordance among gene trees. Mol Biol Evol 24:412–426. doi: 10.1093/molbev/msl170. [DOI] [PubMed] [Google Scholar]
- 21.Hughes KW, Petersen RH, Lickey EB. 2009. Using heterozygosity to estimate a percentage DNA sequence similarity for environmental species’ delimitation across basidiomycete fungi. New Phytol 182:795–798. doi: 10.1111/j.1469-8137.2009.02802.x. [DOI] [PubMed] [Google Scholar]
- 22.Hendry AP, Bolnick DI, Berner D, Peichel CL. 2009. Along the speciation continuum in sticklebacks. J Fish Biol 75:2000–2036. doi: 10.1111/j.1095-8649.2009.02419.x. [DOI] [PubMed] [Google Scholar]
- 23.Roux C, Fraïsse C, Romiguier J, Anciaux Y, Galtier N, Bierne N. 2016. Shedding light on the grey zone of speciation along a continuum of genomic divergence. PLoS Biol 14:e2000234. doi: 10.1371/journal.pbio.2000234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hudson RR, Coyne JA. 2002. Mathematical consequences of the genealogical species concept. Evolution 56:1557–1565. doi: 10.1111/j.0014-3820.2002.tb01467.x. [DOI] [PubMed] [Google Scholar]
- 25.Teixeira M, de M, Cattana ME, Matute DR, Muñoz JF, Arechavala A, Isbell K, Schipper R, Santiso G, Tracogna F, de los Sosa MÁ, Cech N, Alvarado P, Barreto L, Chacón Y, Ortellado J, de Lima CM, Chang MR, Niño-Vega G, Yasuda MAS, Felipe MSS, Negroni R, Cuomo CA, Barker B, Giusiano G. 2020. Genomic diversity of the human pathogen Paracoccidioides across the South American continent. Fungal Genet Biol 140:103395. doi: 10.1016/j.fgb.2020.103395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kyriazis CC, Wayne RK, Lohmueller KE. 2020. Strongly deleterious mutations are a primary determinant of extinction risk due to inbreeding depression. biorXiv doi: 10.1101/678524. [DOI] [PMC free article] [PubMed]
- 27.Wang RJ, White MA, Payseur BA. 2015. The pace of hybrid incompatibility evolution in house mice. Genetics 201:229–242. doi: 10.1534/genetics.115.179499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matute DR, Butler IA, Turissini DA, Coyne JA. 2010. A test of the snowball theory for the rate of evolution of hybrid incompatibilities. Science 329:1518–1521. doi: 10.1126/science.1193440. [DOI] [PubMed] [Google Scholar]
- 29.Moyle LC, Nakazato T. 2010. Hybrid incompatibility “snowballs“ between Solanum species. Science 329:1521–1523. doi: 10.1126/science.1193063. [DOI] [PubMed] [Google Scholar]
- 30.Hamlin JAP, Hibbins MS, Moyle LC. 2020. Assessing biological factors affecting postspeciation introgression. Evol Lett 4:137–154. doi: 10.1002/evl3.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muirhead CA, Presgraves DC. 2016. Hybrid incompatibilities, local adaptation, and the genomic distribution of natural introgression between species. Am Nat 187:249–261. doi: 10.1086/684583. [DOI] [PubMed] [Google Scholar]
- 32.Lenhard-Vidal A, Assolini JP, Ono MA, Bredt CSO, Sano A, Itano EN. 2013. Paracoccidioides brasiliensis and P lutzii antigens elicit different serum IgG responses in chronic paracoccidioidomycosis. Mycopathologia 176:345–352. doi: 10.1007/s11046-013-9698-0. [DOI] [PubMed] [Google Scholar]
- 33.Comparato Filho OO, Morais FV, Bhattacharjee T, Castilho ML, Raniero L. 2019. Rapid identification of Paracoccidioides lutzii and P brasiliensis using Fourier transform infrared spectroscopy. J Mol Struct 1177:152–159. doi: 10.1016/j.molstruc.2018.09.016. [DOI] [Google Scholar]
- 34.Nobrega de Almeida J, Del Negro GMB, Grenfell RC, Vidal MSM, Thomaz DY, de Figueiredo DSY, Bagagli E, Juliano L, Benard G. 2015. Matrix-assisted laser desorption ionization-time of flight mass spectrometry for differentiation of the dimorphic fungal species Paracoccidioides brasiliensis and Paracoccidioides lutzii. J Clin Microbiol 53:1383–1386. doi: 10.1128/JCM.02847-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arantes TD, Theodoro RC, Teixeira M de M, Bagagli E. 2017. Use of fluorescent oligonucleotide probes for differentiation between Paracoccidioides brasiliensis and Paracoccidioides lutzii in yeast and mycelial phase. Mem Inst Oswaldo Cruz 112:140–145. doi: 10.1590/0074-02760160374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maxwell CS, Sepulveda VE, Turissini DA, Goldman WE, Matute DR. 2018. Recent admixture between species of the fungal pathogen Histoplasma. Evol Lett 2:210–220. doi: 10.1002/evl3.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maxwell CS, Mattox K, Turissini DA, Teixeira MM, Barker BM, Matute DR. 2019. Gene exchange between two divergent species of the fungal human pathogen, Coccidioides. Evolution 73:42–58. doi: 10.1111/evo.13643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Desjardins CA, Giamberardino C, Sykes SM, Yu CH, Tenor JL, Chen Y, Yang T, Jones AM, Sun S, Haverkamp MR, Heitman J, Litvintseva AP, Perfect JR, Cuomo CA. 2017. Population genomics and the evolution of virulence in the fungal pathogen Cryptococcus neoformans. Genome Res 27:1207–1219. doi: 10.1101/gr.218727.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schardl CL, Craven KD. 2003. Interspecific hybridization in plant-associated fungi and oomycetes: a review. Mol Ecol 12:2861–2873. doi: 10.1046/j.1365-294x.2003.01965.x. [DOI] [PubMed] [Google Scholar]
- 40.Steenkamp ET, Wingfield MJ, McTaggart AR, Wingfield BD. 2018. Fungal species and their boundaries matter—definitions, mechanisms and practical implications. Fungal Biol Rev 32:104–116. doi: 10.1016/j.fbr.2017.11.002. [DOI] [Google Scholar]
- 41.Giraud T, Gourbière S. 2012. The tempo and modes of evolution of reproductive isolation in fungi. Heredity (Edinb) 109:204–214. doi: 10.1038/hdy.2012.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schumer M, Xu C, Powell DL, Durvasula A, Skov L, Holland C, Blazier JC, Sankararaman S, Andolfatto P, Rosenthal GG, Przeworski M. 2018. Natural selection interacts with recombination to shape the evolution of hybrid genomes. Science 360:656–660. doi: 10.1126/science.aar3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martin SH, Davey JW, Salazar C, Jiggins CD. 2019. Recombination rate variation shapes barriers to introgression across butterfly genomes. PLoS Biol 17:e2006288. doi: 10.1371/journal.pbio.2006288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sepúlveda VE, Márquez R, Turissini DA, Goldman WE, Matute DR. 2017. Genome sequences reveal cryptic speciation in the human pathogen Histoplasma capsulatum. mBio 8:e01339-17. doi: 10.1128/mBio.01339-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dukik K, Muñoz JF, Jiang Y, Feng P, Sigler L, Stielow JB, Freeke J, Jamalian A, Gerrits van den Ende B, McEwen JG, Clay OK, Schwartz IS, Govender NP, Maphanga TG, Cuomo CA, Moreno LF, Kenyon C, Borman AM, de Hoog S. 2017. Novel taxa of thermally dimorphic systemic pathogens in the Ajellomycetaceae (Onygenales). Mycoses 60:296–309. doi: 10.1111/myc.12601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 1303.3997 [q-bio.GN] https://arxiv.org/abs/1303.3997.
- 47.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Turissini DA, Matute DR. 2017. Fine scale mapping of genomic introgressions within the Drosophila yakuba clade. PLoS Genet 13:e1006971. doi: 10.1371/journal.pgen.1006971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hothorn T, Hornik K, Van De Wiel MA, Zeileis A. 2008. Implementing a class of permutation pests: the coin package. J Stat Softw 28:1–23.27774042 [Google Scholar]
- 51.R Core Team. 2016. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/. [Google Scholar]
- 52.Taylor JW, Jacobson DJ, Kroken S, Kasuga T, Geiser DM, Hibbett DS, Fisher MC. 2000. Phylogenetic species recognition and species concepts in fungi. Fungal Genet Biol 31:21–32. doi: 10.1006/fgbi.2000.1228. [DOI] [PubMed] [Google Scholar]
- 53.De Queiroz K, De Queiroz K. 2007. Species concepts and species delimitation. Syst Biol 56:879–886. doi: 10.1080/10635150701701083. [DOI] [PubMed] [Google Scholar]
- 54.Gao Z, Przeworski M, Sella G. 2015. Footprints of ancient-balanced polymorphisms in genetic variation data from closely related species. Evolution 69:431–446. doi: 10.1111/evo.12567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Joly S, McLenachan PA, Lockhart PJ. 2009. A statistical approach for distinguishing hybridization and incomplete lineage sorting. Am Nat 174:E54–E70. doi: 10.1086/600082. [DOI] [PubMed] [Google Scholar]
- 56.Salgado-Salazar C, Jones LR, Restrepo Á, McEwen JG. 2010. The human fungal pathogen Paracoccidioides brasiliensis (Onygenales: Ajellomycetaceae) is a complex of two species: phylogenetic evidence from five mitochondrial markers. Cladistics 26:613–624. doi: 10.1111/j.1096-0031.2010.00307.x. [DOI] [PubMed] [Google Scholar]
- 57.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 59.Robinson DF, Foulds LR. 1981. Comparison of phylogenetic trees. Math Biosci 53:131–147. doi: 10.1016/0025-5564(81)90043-2. [DOI] [Google Scholar]
- 60.Schliep KP. 2011. phangorn: phylogenetic analysis in R. Bioinformatics 27:592–593. doi: 10.1093/bioinformatics/btq706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bryant D, Steel M. 2001. Constructing optimal trees from quartets. J Algorithms 38:237–259. doi: 10.1006/jagm.2000.1133. [DOI] [Google Scholar]
- 62.Steel MA, Penny D. 1993. Distributions of tree comparison metrics: some new results. Syst Biol 42:126–141. doi: 10.2307/2992536. [DOI] [Google Scholar]
- 63.Revell LJ. 2012. phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol Evol 3:217–223. doi: 10.1111/j.2041-210X.2011.00169.x. [DOI] [Google Scholar]
- 64.Paradis E, Claude J, Strimmer K. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 65.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 66.Waterhouse RM, Seppey M, Simao FA, Manni M, Ioannidis P, Klioutchnikov G, Kriventseva EV, Zdobnov EM. 2018. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol 35:543–548. doi: 10.1093/molbev/msx319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 68.Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Larget BR, Kotha SK, Dewey CN, Ané C. 2010. BUCKy: gene tree/species tree reconciliation with Bayesian concordance analysis. Bioinformatics 26:2910–2911. doi: 10.1093/bioinformatics/btq539. [DOI] [PubMed] [Google Scholar]
- 70.Ané C. 2010. Gene tree reconciliation: new developments in Bayesian concordance analysis with BUCKy. Nat Preced (2010). doi: 10.1038/npre.2010.4625.1. [DOI] [PubMed] [Google Scholar]
- 71.Chung Y, Ané C. 2011. Comparing two Bayesian methods for gene tree/species tree reconstruction: simulations with incomplete lineage sorting and horizontal gene transfer. Syst Biol 60:261–275. doi: 10.1093/sysbio/syr003. [DOI] [PubMed] [Google Scholar]
- 72.Liepelt S, Kuhlenkamp V, Anzidei M, Vendramin GG, Ziegenhagen B. 2001. Pitfalls in determining size homoplasy of microsatellite loci. Mol Ecol Notes 1:332–335. doi: 10.1046/j.1471-8278.2001.00085.x. [DOI] [Google Scholar]
- 73.Jarne P, Lagoda PJL. 1996. Microsatellites, from molecules to populations and back. Trends Ecol Evol 11:424–429. doi: 10.1016/0169-5347(96)10049-5. [DOI] [PubMed] [Google Scholar]
- 74.Ellegren H. 2004. Microsatellites: simple sequences with complex evolution. Nat Rev Genet 5:435–445. doi: 10.1038/nrg1348. [DOI] [PubMed] [Google Scholar]
- 75.Durand EY, Patterson N, Reich D, Slatkin M. 2011. Testing for ancient admixture between closely related populations. Mol Biol Evol 28:2239–2252. doi: 10.1093/molbev/msr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH-Y, Hansen NF, Durand EY, Malaspinas A-S, Jensen JD, Marques-Bonet T, Alkan C, Prüfer K, Meyer M, Burbano HA, Good JM, Schultz R, Aximu-Petri A, Butthof A, Höber B, Höffner B, Siegemund M, Weihmann A, Nusbaum C, Lander ES, Russ C, Novod N, Affourtit J, Egholm M, Verna C, Rudan P, Brajkovic D, Kucan Ž, Gušic I, Doronichev VB, Golovanova LV, Lalueza-Fox C, de la Rasilla M, Fortea J, Rosas A, Schmitz RW, Johnson PLF, Eichler EE, Falush D, Birney E, Mullikin JC, Slatkin M, et al. 2010. A draft sequence of the Neandertal genome. Science 328:710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martin SH, Davey JW, Jiggins CD. 2015. Evaluating the use of ABBA/BABA statistics to locate introgressed loci. Mol Biol Evol 32:244–257. doi: 10.1093/molbev/msu269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Martin SH, Dasmahapatra KK, Nadeau NJ, Salazar C, Walters JR, Simpson F, Blaxter M, Manica A, Mallet J, Jiggins CD. 2013. Genome-wide evidence for speciation with gene flow in Heliconius butterflies. Genome Res 23:1817–1828. doi: 10.1101/gr.159426.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Malinsky M. 2019. Dsuite—fast D-statistics and related admixture evidence from VCF files. bioRxiv doi: 10.1101/634477. [DOI] [PMC free article] [PubMed]
- 80.Sankararaman S, Mallick S, Dannemann M, Prüfer K, Kelso J, Pääbo S, Patterson N, Reich D. 2014. The genomic landscape of Neanderthal ancestry in present-day humans. Nature 507:354–357. doi: 10.1038/nature12961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Juric I, Aeschbacher S, Coop G. 2016. The strength of selection against Neanderthal introgression. PLoS Genet 12:e1006340. doi: 10.1371/journal.pgen.1006340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Desjardins CA, Champion MD, Holder JW, Muszewska A, Goldberg J, Bailão AM, Brigido MM, da Silva Ferreira ME, Garcia AM, Grynberg M, Gujja S, Heiman DI, Henn MR, Kodira CD, León-Narváez H, Longo LVG, Ma LJ, Malavazi I, Matsuo AL, Morais FV, Pereira M, Rodríguez-Brito S, Sakthikumar S, Salem-Izacc SM, Sykes SM, Teixeira MM, Vallejo MC, Walter MEMT, Yandava C, Young S, Zeng Q, Zucker J, Felipe MS, Goldman GH, Haas BJ, McEwen JG, Nino-Vega G, Puccia R, San-Blas G, de Soares CMA, Birren BW, Cuomo CA. 2011. Comparative genomic analysis of human fungal pathogens causing paracoccidioidomycosis. PLoS Genet 7:e1002345. doi: 10.1371/journal.pgen.1002345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SRA numbers of the genome sequences used in this study. Download Table S1, DOCX file, 0.02 MB (22.2KB, docx) .
Copyright © 2020 Mavengere et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Example of an introgression from P. restrepiensis to P. brasiliensis defined by Int-HMM. Blue bars represent P. brasiliensis ancestry; red bars represent P. restrepiensis ancestry. Light red bars represent potential P. restrepiensis introgressions with low support (i.e., few markers). Download FIG S1, PDF file, 0.1 MB (109.5KB, pdf) .
Copyright © 2020 Mavengere et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Paracoccidioides americana is haploid and shows no detectable levels of aneuploidy. Coverage and minor allele frequency plots for the largest 16 supercontigs. Per-site estimations of coverage (in red) show close adherence to the genome-wide mean coverage for the whole genome. Per-site estimations of the extent of polymorphism across the genome suggest that no single site shows evidence of polymorphism (cutoff: minor allele frequency > 20%), suggesting that besides having no local aneuploidy, Paracoccidioides is haploid. The plot shows results for isolate T10B1; plots for other lines can be found in the Dryad package (https://doi.org/10.5061/dryad.w3r2280pj). Download FIG S2, PDF file, 0.1 MB (79KB, pdf) .
Copyright © 2020 Mavengere et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Paracoccidioides brasiliensis is haploid and shows no detectable levels of aneuploidy. Coverage and minor allele frequency plots for the largest 16 supercontigs. Per-site estimations of coverage (in red) show close adherence to the genome-wide mean coverage for the whole genome. Per-site estimations of the extent of polymorphism across the genome suggest that no single site shows evidence of polymorphism (cutoff: minor allele frequency > 20%), suggesting that besides having no local aneuploidy, Paracoccidioides is haploid. The plot shows results for isolate T1F1; plots for other lines can be found in the Dryad package (https://doi.org/10.5061/dryad.w3r2280pj). Download FIG S3, PDF file, 0.1 MB (78.6KB, pdf) .
Copyright © 2020 Mavengere et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Paracoccidioides restrepiensis is haploid and shows no detectable levels of aneuploidy. Coverage and minor allele frequency plots for the largest 16 supercontigs. Per-site estimations of coverage (in red) show close adherence to the genome-wide mean coverage for the whole genome. Per-site estimations of the extent of polymorphism across the genome suggest that no single site shows evidence of polymorphism (cutoff: minor allele frequency > 20%), suggesting that besides having no local aneuploidy, Paracoccidioides is haploid. The plot shows results for isolate EPM83; plots for other lines can be found in the Dryad package (https://doi.org/10.5061/dryad.w3r2280pj). Download FIG S4, PDF file, 0.1 MB (78.3KB, pdf) .
Copyright © 2020 Mavengere et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Paracoccidioides venezuelensis is haploid and shows no detectable levels of aneuploidy. Coverage and minor allele frequency plots for the largest 16 supercontigs. Per-site estimations of coverage (in red) show close adherence to the genome-wide mean coverage for the whole genome. Per-site estimations of the extent of polymorphism across the genome suggest that no single site shows evidence of polymorphism (cutoff: minor allele frequency > 20%), suggesting that besides having no local aneuploidy, Paracoccidioides is haploid. The plot shows results for isolate Pb300; plots for other lines can be found in the Dryad package (https://doi.org/10.5061/dryad.w3r2280pj). Download FIG S5, PDF file, 0.1 MB (83.4KB, pdf) .
Copyright © 2020 Mavengere et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Graphic representation of tree concordance between the whole-genome topology and the genealogy produced from each supercontig. Arrows show differences in branch length between trees built with the whole genome (light red) and data from each supercontig (light blue). The plots were made using compare.chronograms (library phytools [63]). Download FIG S6, PDF file, 0.2 MB (167.4KB, pdf) .
Copyright © 2020 Mavengere et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Differences in topology between the trees from the whole genome and those from each supercontig. We only show the results for the tree supercontigs that showed differences with the whole-genome tree, as follows: (A) supercontig 2.2, (B) supercontig 2.3, and (C) supercontig 2.5. Blue circles show clades absent in the supercontig tree but present in the whole-genome tree. Download FIG S7, PDF file, 0.1 MB (152KB, pdf) .
Copyright © 2020 Mavengere et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
π and DXY calculations for pairwise comparisons that included P. lutzii. Color scheme and arrangement are similar to those in Fig. 4. Note that panels A and B are identical to panels C and D in Fig. 4. Download FIG S8, PDF file, 0.3 MB (268.8KB, pdf) .
Copyright © 2020 Mavengere et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Differentiation between species of Paracoccidioides occurs genome wide. DXY along the whole genome between P. brasiliensis and P. restrepiensis (left) is larger than π in either of the two species (center and left). Red solid lines show the median values; dashed orange lines show the 90th and 10th percentiles. Download FIG S9, PDF file, 0.8 MB (880.2KB, pdf) .
Copyright © 2020 Mavengere et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.