

Abstract
A major challenge of analyzing the compositional structure of microbiome data is identifying its potential origins. Here, we introduce fast expectation-maximization microbial source tracking (FEAST), a ready-to-use scalable framework that can simultaneously estimate the contribution of thousands of potential source environments in a timely manner, thereby helping unravel the origins of complex microbial communities (https://github.com/cozygene/FEAST). The information gained from FEAST may provide insight into quantifying contamination, tracking the formation of developing microbial communities, as well as distinguishing and characterizing bacteria-related health conditions.
Knowledge of the diverse functions and distributions of microbial life and their effect on human health has rapidly increased due to the unprecedented expansion of microbiome data repositories such as the ‘Earth Microbiome Project’1–4. Such rich datasets provide the opportunity to study the relationships between the abundance profiles of taxa in different habitats. Nonetheless, one critical challenge in analyzing microbiome communities is due to their composition; each of them is typically comprised of several source environments, including different contaminants as well as other microbial communities that interacted with the sampled habitat. To account for this structure, methods for ‘microbial source tracking’ have been proposed5–11. These methods quantify the fraction, or proportion, of different microbial samples (sources) in a target microbial community (sink).
While traditionally framed in the context of quantifying contamination10, microbial source tracking has been used in a variety of other contexts (for example, characterizing patients in intensive care units (ICUs), gauging partial restoration of the microbiota of cesarean-born infants via vaginal microbial transfer and quantifying the contribution of certain sources to disease outbreaks)12–14. Microbial source tracking may also serve to quantify source contributions to ecological patches. In this use case, microbial source tracking could help unveil compositional patterns of microbial communities in habitats ranging from the human gut to soil. These examples demonstrate that learning the origins of microbial communities may not only significantly improve our current understanding of how microbial communities are formed, but could also inform disease prevention, agricultural practices and care-taking for newborns.
Current methods for microbial source tracking, however, are not without limitations. Some earlier methods5–7 typically limited their context to contamination, focusing on detecting only specific, predetermined contaminating species. More recent methods that leverage the entire community structure often lack a proper probabilistic framework or depend on the identification of indicator species, whose abundance reflects a specific environmental condition8,9. One notable exception is SourceTracker10, the most widely used method for microbial source tracking thus far. Unlike previous methods, SourceTracker uses a Bayesian approach to estimate proportions of contaminants in a given community by leveraging its structure and measuring the respective similarities between a sink community and potential source environments. By directly modeling the sink as a mixture of potential source environments, SourceTracker made a seminal contribution to the field. Nevertheless, this method is based on Markov chain Monte Carlo (MCMC), a computationally expensive procedure, and is therefore only applicable to small- to medium-size datasets with a small number of sources.
To address these limitations, we developed fast expectation-maximization microbial source tracking (FEAST). FEAST partitions microbial samples into their source components 30–300-fold faster than state-of-the-art methods, where, in some cases, it reduces running time from days or weeks to hours. The computational efficiency of FEAST allows it to simultaneously estimate thousands of potential source environments in a timely manner, and thus help unravel the origins of complex microbial communities. Moreover, we found that FEAST is more accurate than previous methods, particularly when the target microbial community contains taxa from an unknown, uncharacterized source.
Results
A brief description of FEAST.
FEAST is a highly efficient expectation-maximization-based method that takes as input a microbial community, the sink, as well as a separate group of potential source environments and estimates the fraction of the sink community that was contributed by each of the source environments. By virtue of these mixing proportions often summing to less than the entire sink, FEAST also reports the potential fraction of the sink attributed to other origins, collectively referred to as the unknown source. The statistical model used by FEAST assumes each sink is a convex combination of known and unknown sources. FEAST is agnostic to the sequencing data type (that is, 16S ribosomal RNA or shotgun sequencing) and can efficiently estimate up to thousands of source contributions to a sample.
Model evaluation using data-driven synthetic mixtures.
We compared the accuracy of FEAST to both SourceTracker10, and the random forest classifier used in previous source-tracking work9. We simulated source communities based on distributions in real source environments from the Earth Microbiome Project1, while varying the level of divergence between sources (see Methods). In each of our simulations, FEAST exhibited higher accuracy than SourceTracker and the random forest classifier across all levels of divergence (Fig. 1a and Supplementary Fig. 1). Since both SourceTracker and FEAST substantially improve accuracy over the random forest approach, we focused on these two methods for all subsequent benchmarks shown. Next, we examined the robustness of FEAST and SourceTracker through varying levels of sequencing depth, when disambiguation between sources is trivial (high divergence). As expected, the accuracy of both algorithms increased as sequencing depth increased. Nonetheless, we observed that FEAST still compared favorably across all levels of sequencing depth (Supplementary Fig. 2). Finally, as it may be nearly impossible to obtain sequencing data for all potential sources in a study, we sought to evaluate FEAST’s ability to estimate the contribution of the unknown source. To this end, we used real source environments from Lax et al.15, while varying the unknown source contribution from absent to exclusive. Across these experiments, FEAST was significantly more accurate in estimating the unknown source proportion (two-sided t-test P < 10−14). Notably, by properly adjusting its estimates for the unknown source, FEAST also produces more accurate mixing proportions for the observed sources as well as low variance (Fig. 1b and Supplementary Figs. 3 and 4).
Fig. 1 |. Methods comparison.
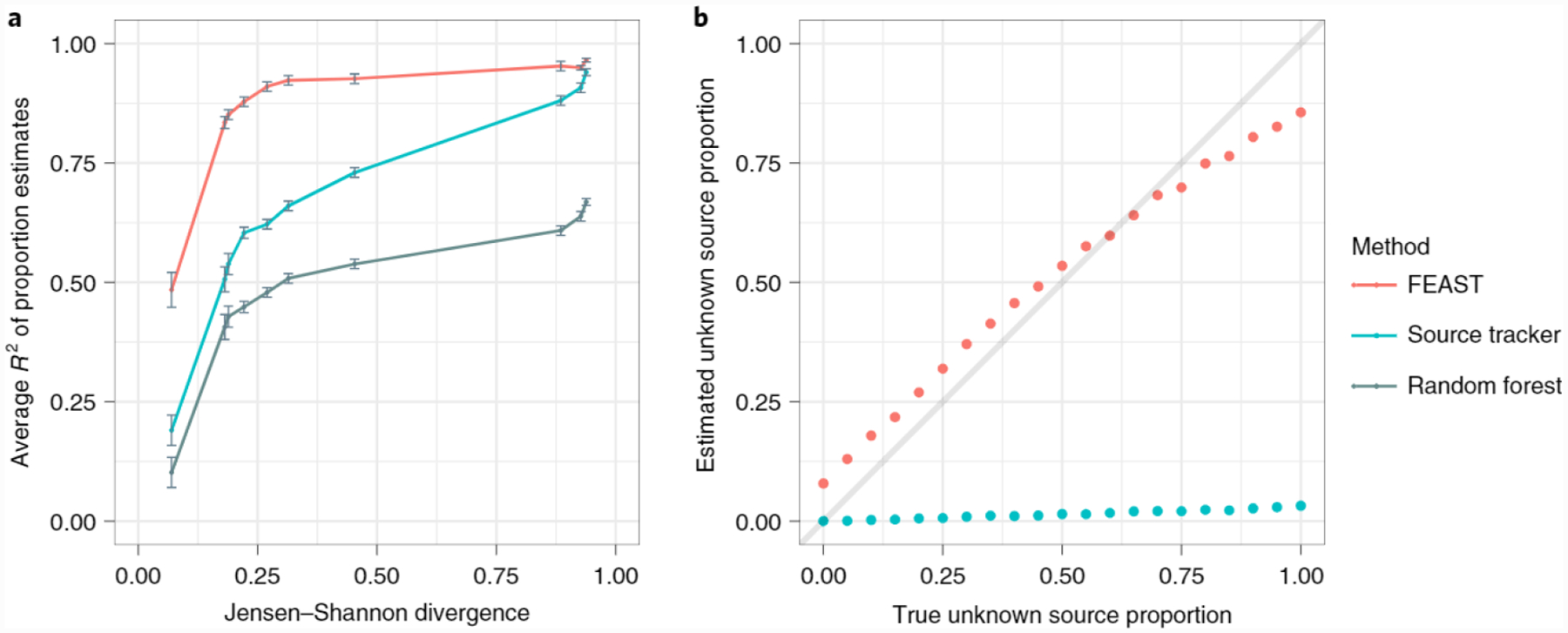
a, The accuracy of FEAST, the random forest classifier and SourceTracker on simulated data. Each simulation was performed using 20 real source environments and simulated sinks. The x axis is average Jensen-Shannon divergence value across known sources (that is, the degree of overlap between the sources from completely identical to completely non-overlapping). The y axis represents correlation across all source environments between true and estimated mixing proportions; error bars show the standard error of the mean (n = 30). b, Evaluation of FEAST and SourceTracker through varying levels of unknown source proportions.
Running time.
One of FEAST’s distinct advantages over other methods is its speed (Fig. 2 and Supplementary Table 1). Specifically, across all experiments, FEAST reduced running time by a factor of 30–300 compared to SourceTracker, while maintaining and even improving the accuracy. Consequently, FEAST can simultaneously estimate thousands of potential source environments on the order of minutes to hours, where SourceTracker may take anything upward of days (Supplementary Table 1). We note that SourceTracker’s accuracy may potentially be improved by increasing the number of burn-in iterations or otherwise increasing the number of iterations of the Markov chain, however, this comes at the expense of additional running time (see Methods for a comprehensive discussion of the tradeoff between time and accuracy in MCMC).
Fig. 2 |. Running time comparison to current state-of-the-art.
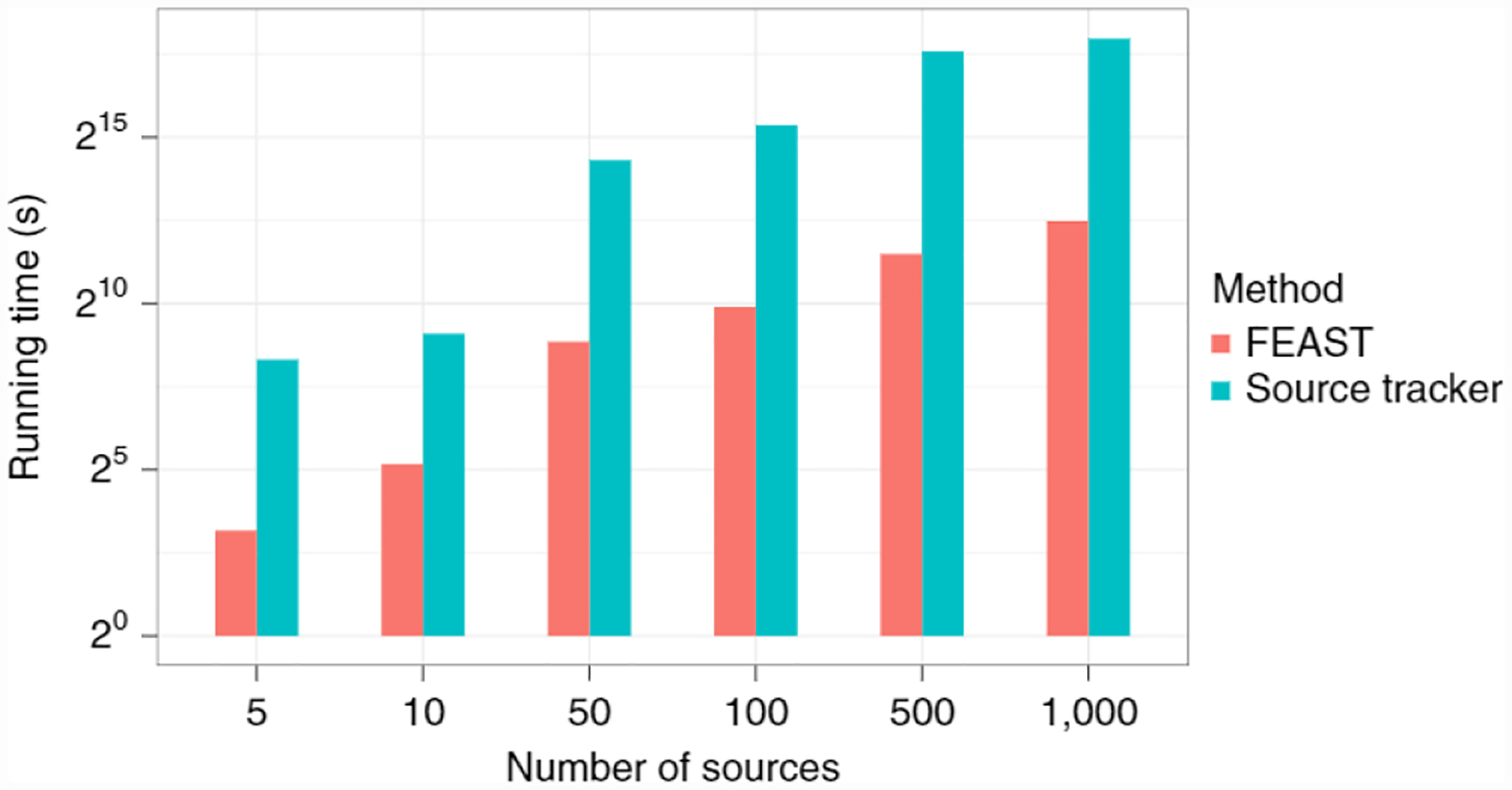
Running time (log scale, seconds) comparison across all simulation studies, using a sequencing depth of 10,000 reads per source.
Real data applications.
We applied FEAST to five real datasets to demonstrate the utility of microbial source tracking methods across different contexts. We first use FEAST as it was originally intended—to quantify the contribution of sources to specific sink environments.
Succession and initial colonization in infants.
Using FEAST for time-series analysis offers a quantitative way to characterize developmental microbial populations, such as the infant gut. In this context, we can leverage previous time points and external sources to understand the origins of a specific, temporal community state. For instance, we can estimate if taxa in the infant gut originate from the birth canal, or if they are derived from some other external source at a later time point. To demonstrate this capability, we used longitudinal data from Backhed et al.16, which contains gut microbiome samples from infants as well as from their corresponding mothers. In this analysis, we treated samples taken from the infants at age 12 months as sinks, considering respective earlier time points and maternal samples as sources. In these settings, FEAST revealed a significantly larger maternal contribution (two-sided t-test, P = 0.03161) in vaginally delivered infants over cesarean-delivered infants (Fig. 3), where other methods did not (Supplementary Fig. 5). These results are consistent with the results of Backhed et al.16. We further explored whether biological mothers were more likely to be identified as sources of their infant’s microbiome than other potential source communities. We considered all maternal and early infant samples as potential sources, and found that for over 83% of the sink samples, the top contributing sources were from the same family (Supplementary Material).
Fig. 3 |. FEAST estimations of source contribution to the sink; that is, gut microbiome of focal infant at 12-months of age.
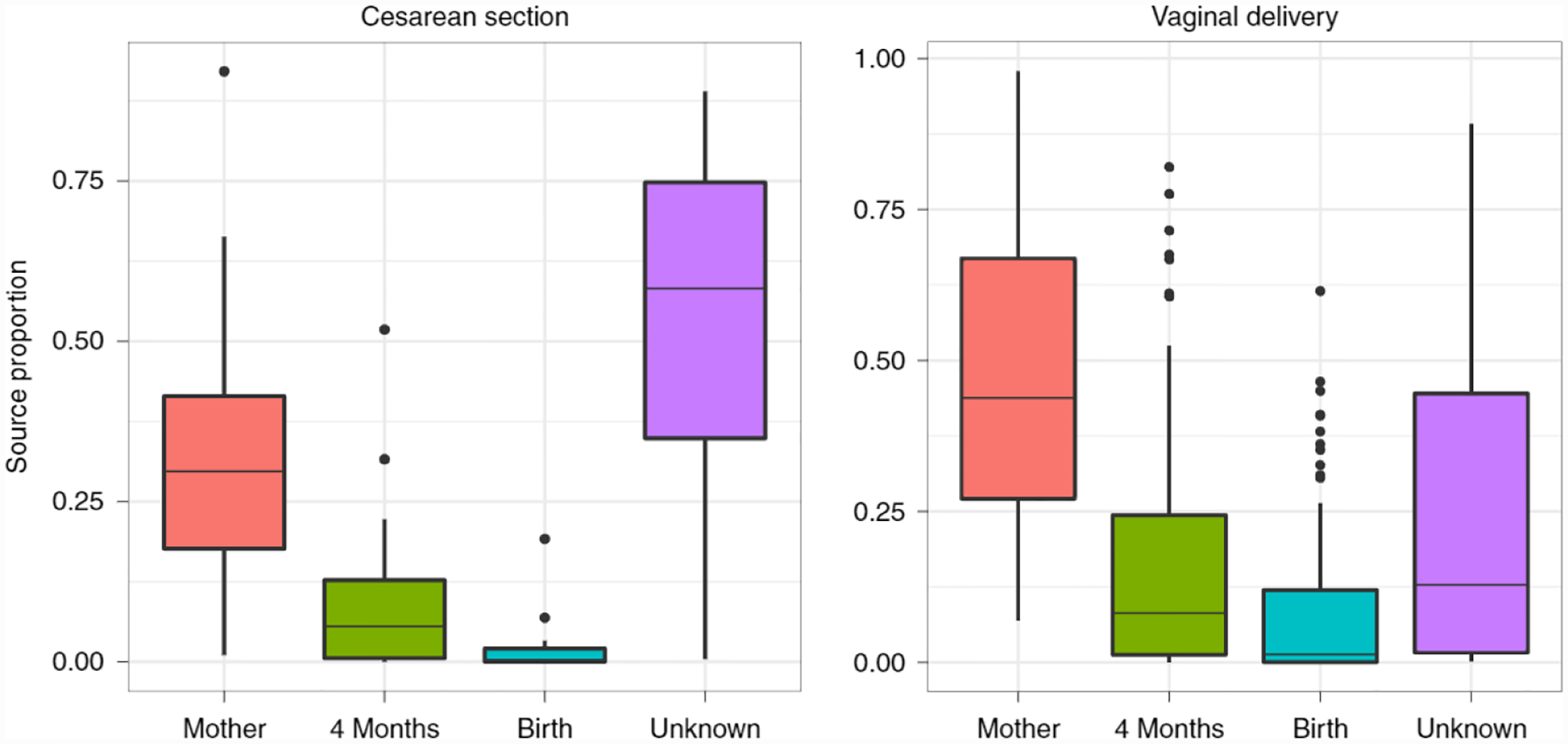
Box plots indicate the median (central lines), IQR (hinges) and the 5th and 95th percentiles (whiskers). Sources: gut microbiome of mother, focal infant at 4 months and focal infant at birth. (n = 98 sinks).
Detecting contamination.
To validate FEAST’s utility in detecting contamination, we first replicated the analysis of Knights et al.10 who investigated contamination in settings such as office buildings, hospitals and research laboratories. In these settings, where disambiguation between sources was relatively easy, FEAST estimated source contributions consistent with those reported by Knights et al.10, despite minor discrepancies (Supplementary Fig. 6). Next, we analyzed longitudinal data collected by Lax et al.15. In this analysis, we investigated one household, where the inhabitants were genetically related. We used skin samples of inhabitants from several body parts as sources and indoor house surfaces as sinks. Our analysis using FEAST shows that surfaces in home-settings are more diverse than their human sources and might not be entirely composed of bacteria originated from humans (Fig. 4). Our results stand in qualitative contrast to those of Lax et al.15, where they found that an overwhelming majority of microbial communities on these surfaces originated from humans. We believe that the difference stems from an underestimation of the unknown source by SourceTracker, which was used in the original analysis of Lax et al. Such underestimation is exacerbated in cases like this, when disambiguation of sources is challenging, that is, due to all individuals living in the same house. We further investigated whether we could explain the composition of these unknown sources, at the first time point, by including additional source environments from the Earth Microbiome Project. In addition to the contribution of the four inhabitants, we find potential evidence for contributions from avian egg product (8%), freshwater fish (8%) and soil (1%). As a consequence, the unknown source contribution was reduced to 5.8% (from approximately 25%, see Fig. 4).
Fig. 4 |. The proportion of the unknown sources in kitchen counter samples using FEAST and SourceTracker.
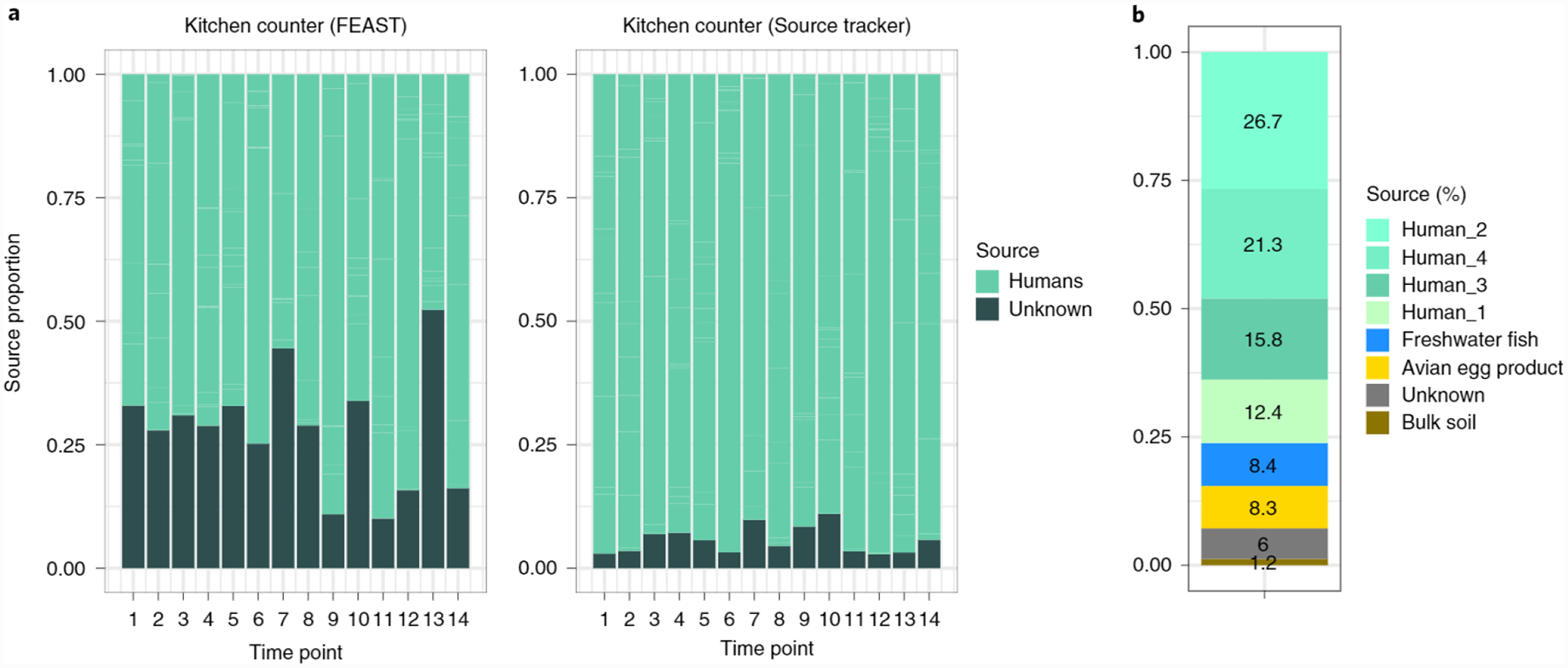
a, Source estimates considering 12 known human sources (hand, foot and nose across four inhabitants) using data from Lax et al.15 b, FEAST estimations of source contribution in one house kitchen counter, at the first time point, using additional sources from the Earth Microbiome Project.
Microbial source tracking as a metric of similarity.
In the following experiments we used FEAST in a different context—as a metric of similarity. To the best of our knowledge this is a novel application of microbial source tracking. In these experiments, we focused on the human gut microbiome, but rather than seeking among sources the contributors to a sink sample, we seek to represent each sink as a mixture of ‘characteristic environments’—source environments that are similar in composition to the sink and therefore capture its characteristics. We then quantify the similarities between the sink and its characteristic environments using mixing proportions reported by FEAST.
FEAST distinguishes patients in ICU from healthy adults.
To demonstrate FEAST’s utility in distinguishing and characterizing bacteria-related health conditions, we first replicated the analysis of McDonald et al.12 (Supplementary Fig. 7) in which they characterized a cohort of patients from an ICU. We found that our results using FEAST were consistent with the analysis of McDonald et al.12; that is, gut samples from patients in ICU are markedly different from those of healthy individuals. Next, we performed an additional analysis that was not included in the original study of McDonald et al.12: we used a bidirectional approach, randomly assigning gut samples from the American Gut Project (healthy controls) as either sources or sinks, in addition to assigning the gut microbiome of ICU patients as sinks (see Methods for a complete description). In doing so, we aimed to quantify the similarity between the gut microbiome of patients in ICU and healthy controls by comparing their source composition. Using FEAST, we found significant differences in the source composition between the two sink types (two-sided t-test, P = 0.02551; Supplementary Fig. 8). To verify our findings, we used UniFrac distance17, Jensen-Shannon divergence and the Bray-Curtis dissimilarity (Fig. 5 and Supplementary Fig. 8), which also captured the differences between the patients in ICU and healthy controls (that is, healthy sources are more similar to healthy sinks). However, we note that there is a large variance in the microbiome similarities among healthy controls, whether they are sources or sinks. We hypothesize that this variance stems from differences between individuals’ microbiomes unrelated to their health (for example, diet). We also note that these results should be interpreted with caution, since the healthy controls and patients in ICU are not matched and therefore batch effects or other confounders may affect the results. Nevertheless, if indeed the prediction accuracy is driven by confounders, these results demonstrate that FEAST can capture such confounder information better than existing methods.
Fig. 5 |. The receiver operating characteristic curve using FEAST, weighted UniFrac and Jensen-Shannon divergence to classify healthy individuals and patients in ICU with dysbiosis.
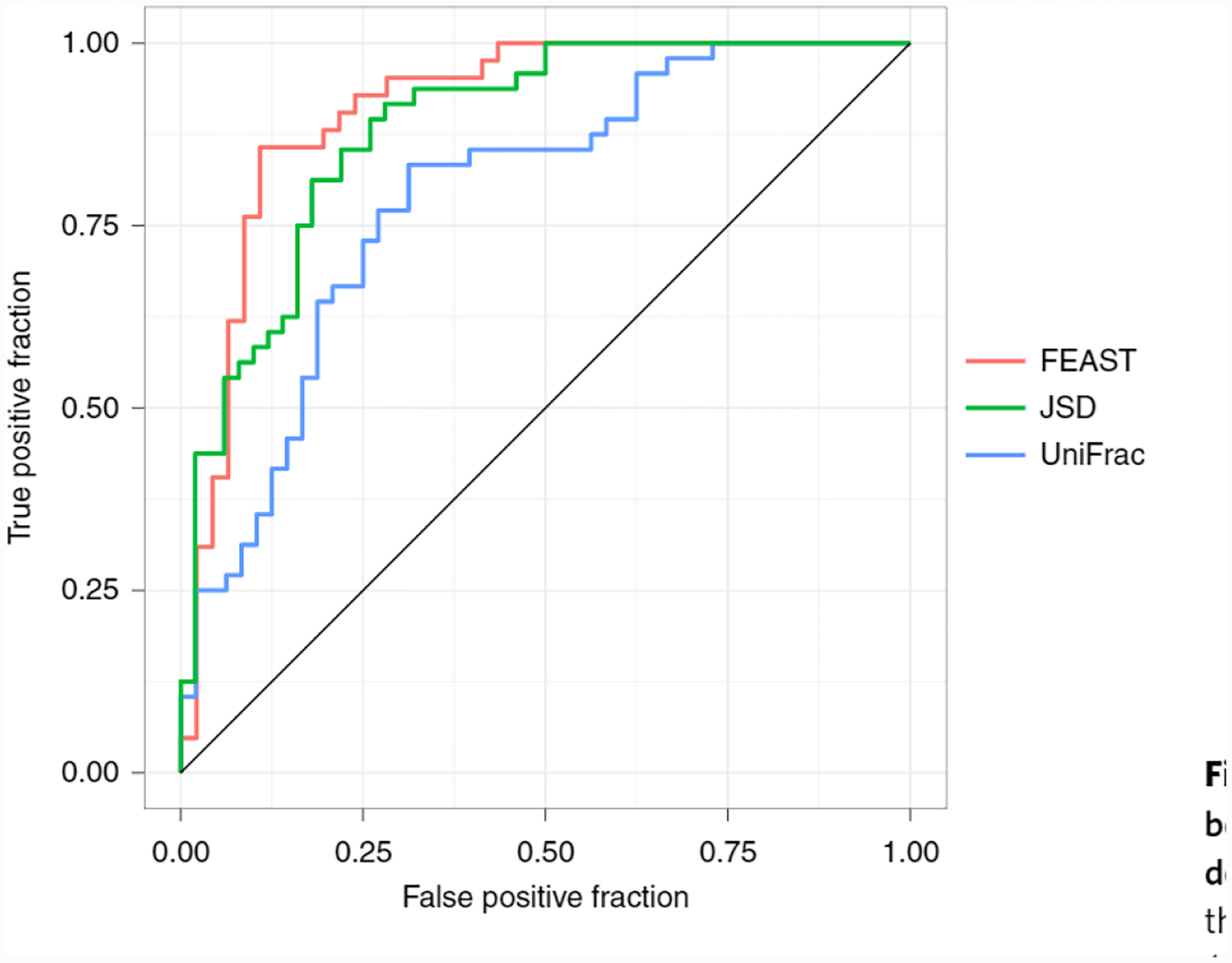
FEAST area under curve (AUC), 0.91; weighted UniFrac AUC, 0.78 and Jensen-Shannon divergence (JSD) AUC, 0.87.
FEAST implicates time-related compositional shifts in a cancer longitudinal study.
Considering the utility of FEAST as a method for classifying phenotypes, we sought to also characterize a cohort of patients with cancer undergoing allogeneic hematopoietic stem cell transplantation (allo-HSCT). In a study by Taur et al.18, it was suggested that assessing the gut microbiome of patients undergoing allo-HSCT may identify those at high risk for bloodstream infection (that is, bacteremia). Many of the patients were found to have intestinal domination, a condition in which at least 30% of the microbiome consists of a single bacterial taxon. As the exact nature of the association between compositional shifts in the microbiome and bacteremia is unclear, it is crucial to explain the dynamics of microbial community composition in patients undergoing allo-HSCT This led us to examine whether FEAST can be used as a tool for such an assessment. To this end, we labeled the two consecutive samples from before and during the first event of intestinal domination as sinks, and all corresponding samples from earlier time points as sources (per patient). FEAST revealed a significantly larger proportion of the unknown source in the sink samples with intestinal domination in comparison to the sink samples before intestinal domination (two-sided t-test P < 0.001; Fig. 6 and Supplementary Fig. 9). This is expected, as bacterial domination is defined in terms of abundance fractions, so by definition would be reflected in mixture proportions. Nonetheless, this result was not significant using other methods (two-sided t-test P = 0.09). We therefore demonstrated FEAST’s ability to capture shifts in microbial community composition that may underlie differences between pathogenic and neutral phenotypes.
Fig. 6 |. Significant differences in the distribution of the unknown source between sink samples before and during the first event of intestinal domination across 94 patients undergoing allo-HSCT.
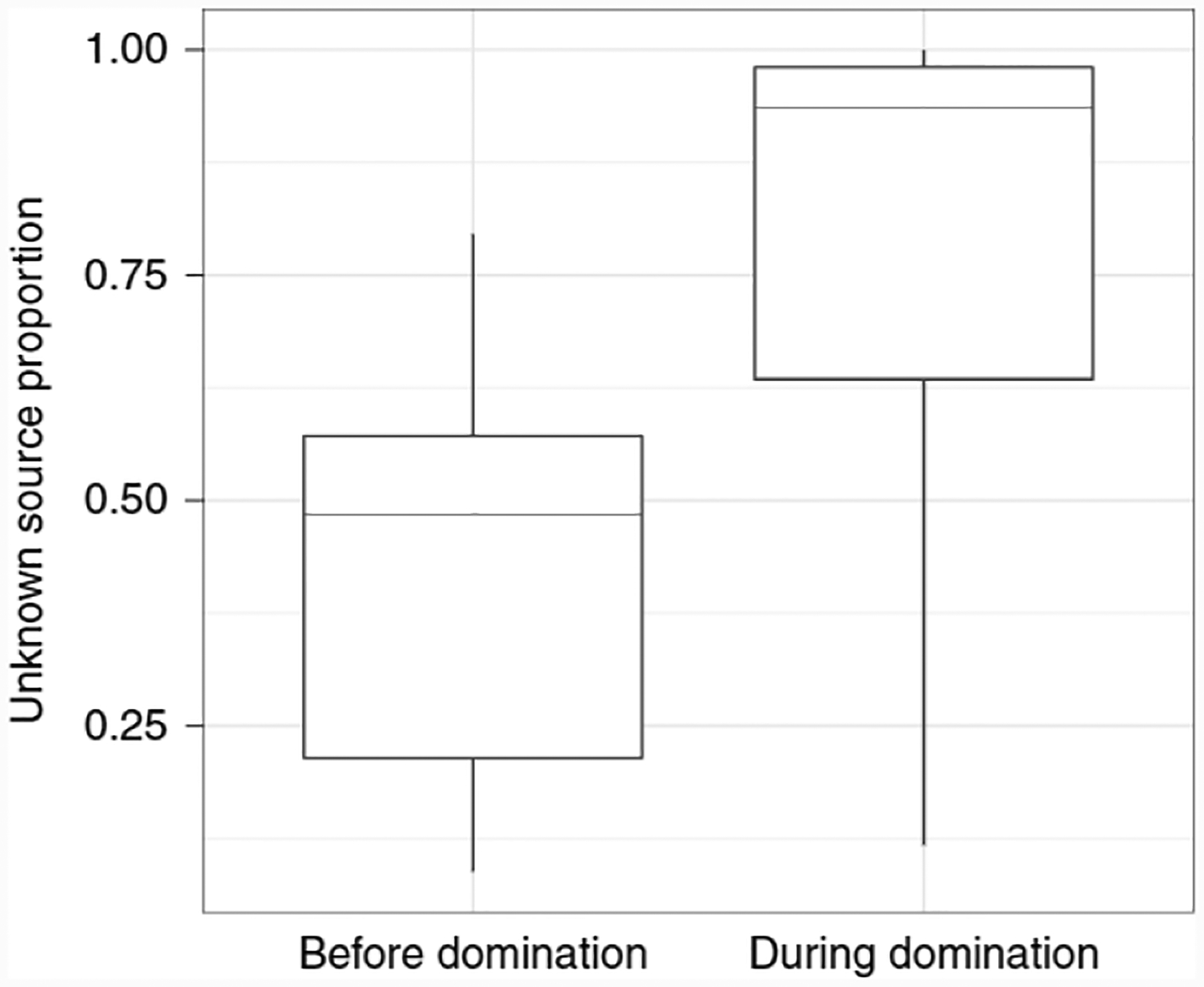
Box plots indicate the median (central lines), IQR (hinges) and the 5th and 95th percentiles (whiskers).
Discussion
FEAST was designed to address an important need in the rapidly evolving field of microbiome research—namely, to quantify the fraction of each source environment in a target microbial community (sink), through a natural, scalable statistical model. As a result, it provides a computationally efficient tool that can simultaneously evaluate hundreds to thousands of potential source environments, as well as the contribution of an unknown, uncharacterized source, outperforming state-of-the-art methods in terms of both speed and accuracy.
The utility of FEAST is established in two different contexts. First, we used FEAST as it was originally intended—to quantify the contribution of different source environments to a target microbial community. In this context, we were able to address questions surrounding succession and initial colonization of microbial species. Specifically, using FEAST we quantitatively reaffirmed the findings of Backhed et al.16, who demonstrated that gut microbiota of infants delivered by cesarean section showed significantly less resemblance to their mothers’ compared to vaginally delivered infants. Second, we used FEAST as a metric of similarity. In this context, FEAST can help researchers better understand the compositional characteristics of the human microbiome—an important task given that it has been linked to many aspects of human physiology and health including obesity, inflammatory diseases, cancer, metabolic diseases and aging2–4,19–29.
We showed the ability of FEAST to differentiate between the gut microbiome of ICU patients experiencing dysbiosis and that of healthy controls. The results from FEAST show that patients with dysbiosis and controls without dysbiosis have differences between their microbial source composition, namely that the gut microbiome of healthy adults demonstrates a greater resemblance to other healthy gut communities than to those of patients experiencing dysbiosis. Additionally, we investigated the characterization of patients with intestinal domination. Source contribution estimates produced by FEAST show increased contribution and reduced variability of the unknown source in patients experiencing intestinal domination compared to patients who are not. These results suggest that FEAST may be useful in distinguishing and characterizing phenotypes or conditions related to microbial injury. Furthermore, by highlighting novel differences among source composition, FEAST may contribute insight to downstream analyses aiming to implicate differences between healthy and diseased phenotypes at the taxa level.
We note that in some contexts, for example, patients with cancer undergoing allo-HSCT, the underlying assumption of FEAST is violated. In these situations, the sink is not a convex combination of its (known and unknown) sources due to significant differences between some of the source environments. The gut microbiome of patients with cancer, for example, can considerably change over-time due to antibiotics and immune system shutdown or restart. Additionally, we note that the ability to differentiate between the gut microbiome of patients in ICU experiencing dysbiosis and healthy controls may be attributed to technical confounders separating these two distinct datasets (healthy control from the American Gut Project30 and patients in ICU12), which, if true, are better detected using FEAST.
Methods
The FEAST probabilistic model.
Consider a single sink sample represented by a vector x, where xj corresponds to the abundance of taxa, j, 1 ≤ j ≤ N. Let K be the number of known sources. Each known source is represented by a vector yi, where yij is the observed abundance of taxa, j, in source i (1 ≤ i ≤ K). Additionally, we assume there is an unobserved source (K + 1). Let and be the total taxa counts of the known sources and sink, respectively. With this notation, the generative model is as follows: we assume that there are mixture proportions α—a vector of length K + 1—where αi corresponds to the fraction of source i in the sink, hence . We also assume that there is an unknown relative abundance for each of the sources. For each source, 1 ≤ i ≤ K + 1, we have a vector γ, where . Each γij represents the true relative abundance of taxa j in source i.
α and γ are not observed and are parameters of the model.
Fast inference via expectation-maximization.
FEAST uses an expectation-maximization approach31 to infer the model parameters. The likelihood is given by
E step: The log likelihood is given by
The expected complete log likelihood (Q) is given by
where
A more detailed derivation can be found in the Supplementary Material.
M step: Since the γij are required to sum to 1, we use Lagrange multipliers δi to constrain the γij values. The Lagrangian is given by
Taking partial derivatives of L and solving gives the optimal update
The update for the mixing proportions is given by
FEAST has two hyperparameters: the convergence threshold and the maximum number of iterations. In all our experiments we set these to default values of 10−6 and 1,000, respectively. We used the multinomial distribution to model the data generating process since it is particularly relevant when analyzing microbiome datasets. Specifically, it addresses count uncertainty rather than directly transforming counts to relative abundances, and also models the competition to be counted (between taxa) instead of treating the counts of each taxon as independent32.
Simulation studies.
Parameters and settings.
To construct realistic simulation scenarios, we used real microbiome data as sources and simulated sinks as convex combinations thereof. Therefore, our simulations are representative of the abundance, over-dispersion of zeros and technical noise mostly observed in real microbiome data. We designed our simulation parameters to reflect the wide range of Jensen-Shannon divergences and potential sources observed across the real datasets we investigated. For a detailed description of the parameters and settings in each simulation study, see Supplementary Material.
Main simulation study.
To examine the accuracy of FEAST, we used multiple source environments with varying degrees of overlap in their distribution by randomly sampling from the Earth Microbiome Project. Each source environment was sub-sampled to contain 10,000 reads. In each iteration of the simulation we sampled K + 1 known environments and used them to build a synthetic sink with different mixing proportions. To simulate an unknown source, only K source environments are designated as known sources. We used 30 mixing proportions (corresponding to 30 simulated sinks) and K = 20 known sources in each iteration. For a detailed description of the simulation, see Supplementary Material.
Sequencing depth simulations.
To examine the robustness of FEAST to varying levels of sequencing depth, we used multiple source environments from the Earth Microbiome Project while varying their sequencing depth. In each iteration of our simulation we sampled environments (with median Jensen-Shannon divergence of 0.95) and used them to build a synthetic sink, with different mixing proportions and a set sequencing depth ranging from 100 through 10,000. Notably, by choosing a median Jensen-Shannon divergence of 0.95 we wanted to emphasize that even under the scenario in which the sources are non-overlapping and thus trivial to disambiguate, the sequencing depth will have an effect. Additionally, in these simulations, we only varied the sequencing depth of the sources. However, since the sink samples are a linear combination of the sources, these samples are also, indirectly, affected. To simulate an unknown source, only K source environments are designated as known sources. We used 30 mixing proportions (corresponding to 30 simulated sinks) and K = 20 known sources in each iteration. For a detailed description of the simulation, see Supplementary Material.
Unknown source simulations.
To evaluate FEAST’s ability to estimate the contribution of the unknown source, we used real source environments from Lax et al.15 and created synthetic sink communities. Given that any source not sampled should, theoretically, be accounted for in the unknown source, realistic values of the unknown source can therefore span the range of percentages occupied by the observed sources. Specifically, there are scenarios in which the known sources comprise the entirety of the sink (unknown source contribution, 0), or on the other hand, scenarios in which the known sources did not contribute any taxa to the sink (unknown source contribution, 1). Therefore, the unknown source contribution values in our simulation ranges from 0 to 1. As a measure of accuracy, we used the squared Pearson correlation between the estimated mixing proportions and the true mixing proportions for the unknown source across repeated simulation runs. We used 30 mixing proportions (corresponding to 30 simulated sinks) and five sources (four known sources) in each iteration. For a detailed description of the simulation, see Supplementary Material.
Noisy samples among sources.
As source assignment is discretionary (that is, multiple samples can be pooled to a single source or considered as individual sources), we sought to examine the robustness of FEAST in the case where we have noisy realizations of the sources and their effect on prediction accuracy. We used K + 1 distinct source environments by randomly sampling from the Earth Microbiome Project (that is, soil, fresh water, feces, sebum and so on), where each source was represented by ten different samples (for example, soil1, soil2 and so on.). We then amalgamated these ten samples (per source environment) and used the amalgamation of each source to build simulated sinks, with 30 different mixing proportions (corresponding to 30 simulated sinks). In each iteration of our simulation, we aggregated s ∊ {1, …, 10} samples from the representative samples of each source environment to estimate the different mixing proportions.
Prediction accuracy.
To measure accuracy, we used the squared Pearson correlation coefficient between the estimated and true mixing proportions for each individual source across repeated simulation runs (that is, different mixing proportions) for the same Jensen-Shannon divergence value. In each iteration, we varied the degree of similarity of the source environments.
Running time measurements.
In each iteration, we used K randomly selected source environments from the Earth Microbiome Project, where K ∊ {5,10,50,100,500,100}. Each source environment was down-sampled to contain 10,000 reads. We recorded the running-time of each method, for each number of source environments, each iteration. The running time of hundreds of samples using the random forest classifier is relatively short. However, given that both SourceTracker and FEAST substantially improve accuracy over the random forest approach, we focused on these two methods for all subsequent benchmarks shown.
Comparing model performance.
We evaluated the performance of our model against common approaches widely used for microbial source tracking—namely, SourceTracker10 and the random forest classifier9. Both methods use community structure to measure the similarity between sink samples and potential source environments. The statistical model used by FEAST shares many similarities with the model proposed by SourceTracker10, namely that both models assume each sink is a convex combination of the known and unknown sources. Additionally, in both methods, source assignment is discretionary (that is, multiple samples can be pooled to a single source or considered as individual sources). Thus, the main difference between the methods lies in their optimization procedure. FEAST uses an expectation-maximization algorithm to evaluate the proportions of source contribution, whereas SourceTracker uses a Gibbs Sampler (MCMC). In other fields in genomics it has been demonstrated that such optimization can be critical in terms of the reduction of running time. For example, in statistical genetics, the original method for the inference of population structure, STRUCTURE33, uses MCMC for the parameter estimation, while other methods such as FRAPPE34 and ADMIXTURE35use expectation-maximization and quasi-Newton optimization techniques respectively to reach similar accuracy, but considerably more efficiently. This improvement in running time eventually may translate to improvement in accuracy. Particularly, the accuracy achieved by SourceTracker may be improved by increasing the number of burn-in iterations; however, this comes at the expense of additional running time.
Distinguishing patients in ICU from healthy adults.
The objective of this set of experiments is to classify each sink (patient in ICU or a healthy adult) using its overall dissimilarity to all sources (healthy adults). The dependent variable (y) is a binary vector of cases (patients in ICU) and controls (healthy adults) yi ∊ {0,1}, i = {1, …, N} where N is the number of sink samples. When classifying using FEAST or SourceTracker, we designate the proportion of the unknown source as a predictor for each sink’s class label. When classifying using Jensen-Shannon and UniFrac, we designate the average of the dissimilarity measurements between the sink and all the other sources as the predictor.
FEAST.
We applied FEAST to every sink sample (ICU or healthy), where the known sources are 100 distinct healthy individuals from the American Gut Project. We next used the estimated proportions of the unknown source as the input to the classifier.
SourceTracker.
We applied SourceTracker to every sink sample (ICU or healthy), where the known sources are 100 distinct healthy individuals from the American Gut Project. We next used the estimated proportions of the unknown source as the input to the classifier.
Jensen-Shannon divergence.
We calculated the Jensen-Shannon divergence value between each sink sample (ICU or healthy) and the known source samples used in FEAST and SourceTracker (for example, 100 distinct healthy individuals from the American Gut Project). We next used the average Jensen-Shannon divergence value (across known sources) as the input to the classifier.
UniFrac.
We calculated the Weighted UniFrac distance between each sink sample (ICU or healthy) and the known source samples used in FEAST and SourceTracker (for example, 100 distinct healthy individuals from the American Gut Project). We next used the average Weighted UniFrac distance (across known sources) as the input to the classifier.
Data distribution.
Throughout the paper, the box-plot elements are: center line, median; box limits, upper and lower quartiles; whiskers, 1.5X interquartile range (IQR); points and outliers.
Datasets.
We evaluated the performance of FEAST using five datasets collected using both 16S rRNA gene and whole metagenome shotgun sequencing.
The first dataset was collected and studied by Backhed et al.16 (accession number ERP005989), which characterizes the temporal gut microbiome of 98 Swedish infants, each sampled at birth, 4 months after birth and 12 months after birth. This dataset also contains gut microbiome samples collected from the infants’ corresponding mothers during the first few days after delivery. Eighty-three infants were delivered vaginally and the remaining 15 by cesarean section. In this dataset, shotgun sequencing reads were assembled into contigs using SOAPdenovo2 (ref.36). The contigs were binned according to their abundance variations across samples and GC-depth pattern for further assembly into draft genomes. The draft genomes were then clustered into MetaOTUs based on MUMi37 and the Spearman distance38 and their taxa were determined in relation to the NCBI genomes.
The second dataset was collected and studied by Lax et al.15 (accession number ERP005806). This study used the V4 region of the 16S rRNA gene to evaluate the microbial contamination from seven groups of individuals in their respective residences over the course of 6 weeks. In our analysis, we investigated one house, where the inhabitants were genetically related. We used skin samples of inhabitants from several body parts (hand, foot and nose) as sources and indoor house surfaces (for example, kitchen floor, kitchen counter) as sinks.
The third dataset was collected and studied by Knights et al.10 (data from this study are stored in https://github.com/danknights/sourcetracker). This study used datasets of bacterial 16S rRNA39,40 (V2 region of the 16S rRNA gene) to investigate contamination in settings such as office buildings, hospitals and research laboratories. As potential contaminants, human skin, oral cavities, feces and temperate soils were considered.
The fourth dataset was collected and studied by McDonald et al.12 (accession number ERP012810), the American Gut Project30 (EBI project number PRJEB11419). Using the V4 region of the 16S rRNA gene, McDonald et al. characterized a cohort of patients from an ICU. The study collected samples from the skin, mouth and feces (gut) of 115 US and Canadian patients in ICU at time of admission (within 48 h) to the ICU as well as at time of discharge from the ICU.
The fifth dataset was collected and studied by Taur et al.18 (data from this study are stored in http://www.ncbi.nlm.nih.gov/sra). In this study by Taur et al.18, fecal specimens were collected longitudinally from 94 patients undergoing allo-HSCT from before treatment up to 35 d after treatment. This study used the VI–V3 region of bacterial 16S rRNA genes.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All of the datasets analyzed in this paper are public and can be referenced at the following accession numbers: The first dataset was collected and studied by Backhed et al.16 (accession number ERP005989). The second dataset was collected and studied by Lax et al.15 (accession number ERP005806). The third dataset was collected and studied by Knights et al.10 (data from this study are stored in https://github.com/danknights/sourcetracker). The fourth dataset was collected and studied by McDonald et al.12 (accession number ERP012810) and the American Gut Project30 (EBI project number PRJEB11419). The fifth dataset was collected and studied by Taur et al.18 (data from this study are stored in http://www.ncbi.nlm.nih.gov/sra). In our simulations we used the Earth microbiome project (ftp://ftp.microbio.me/emp/release1/otu_tables/closed_ref_greengenes/).
Code availability
Code is available at https://github.com/cozygene/FEAST
Supplementary Material
Acknowledgements
We thank S. Mukherjee for insightful comments on the manuscript. This research was partially supported by European Research Council under the European Union’s Horizon 2020 research and innovation program, project number 640384. This work was partially supported by the National Science Foundation (grant number 1705197). T.A.J. was supported by National Science Foundation (grant no. DGE-1644869).
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, statements of code and data availability and associated accession codes are available at https://doi.org/10.1038/s41592-019-0431-x.
Competing interests
The authors declare no competing interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41592-019-0431-x.
References
- 1.Thompson LR et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 551, 457–463 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kau AL, Ahern PP, Griffin NW, Goodman AL & Gordon JI Human nutrition, the gut microbiome and the immune system. Nature 474, 327–336 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turnbaugh PJ & Gordon JI The core gut microbiome, energy balance and obesity. J. Physiol 587, 4153–4158 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ridaura VK et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341, 1241214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simpson JM, Santo Domingo JW & Reasoner DJ Microbial source tracking: state of the science. Environ. Sci. Technol 36, 5279–5288 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Wu CH et al. Characterization of coastal urban watershed bacterial communities leads to alternative community-based indicators. PLoS ONE 5, e11285 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greenberg J, Price B & Ware A Alternative estimate of source distribution in microbial source tracking using posterior probabilities. Water Res. 44, 2629–2637 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Dufrêne M & Legendre P Species assemblages and indicator species: the need for a flexible asymmetrical approach. Ecol. Monogr 67, 345–366 (1997). [Google Scholar]
- 9.Smith A, Sterba-Boatwright B & Mott J Novel application of a statistical technique, Random Forests, in a bacterial source tracking study. Water Res. 44, 4067–4076 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Knights D et al. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 8, 761–763 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Devane ML, Weaver L, Singh SK & Gilpin BJ Fecal source tracking methods to elucidate critical sources of pathogens and contaminant microbial transport through New Zealand agricultural watersheds—a review. J. Environ. Manag 222, 293–303 (2018). [DOI] [PubMed] [Google Scholar]
- 12.McDonald D et al. Extreme dysbiosis of the microbiome in critical illness. mSphere 1, pii: e00199–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dominguez-Bello MG et al. Partial restoration of the microbiota of cesarean-born infants via vaginal microbial transfer. Nat. Med 22, 250–253 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teaf CM, Flores D, Garber M & Harwood VJ Toward forensic uses of microbial source tracking. Microbiol. Spectr 6, 10.1128/microbiolspec.EMF-0014-2017 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Lax S et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science 345, 1048–1052 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Backhed F et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 17, 690–703 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Lozupone C & Knight R UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol 71, 8228–8235 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taur Y et al. Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin. Infect. Dis 55, 905–914 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turnbaugh PJ et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Ley RE Obesity and the human microbiome. Curr. Opin. Gastroenterol 26, 5–11 (2010). [DOI] [PubMed] [Google Scholar]
- 21.Turnbaugh PJ, Bäckhed F, Fulton L & Gordon JI Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 3, 213–223 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ley RE et al. Obesity alters gut microbial ecology. Proc. Natl Acad. Sci. USA 102, 11070–11075 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koren O et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc. Natl Acad. Sci. USA 108, 4592–4598 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clemente JC, Ursell LK, Parfrey LW & Knight R The impact of the gut microbiota on human health: an integrative view. Cell 148, 1258–1270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le Chatelier E et al. Richness of human gut microbiome correlates with metabolic markers. Nature 500, 541–546 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Clarke SF et al. The gut microbiota and its relationship to diet and obesity: new insights. Gut Microbes 3, 186–202 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeffery IB, Quigley EMM, Öhman L, Simrén M & O’Toole PW The microbiota link to irritable bowel syndrome: an emerging story. Gut Microbes 3, 572–576 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marchesi JR et al. Towards the human colorectal cancer microbiome. PLoS ONE 6, e20447 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qin J et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60 (2012). [DOI] [PubMed] [Google Scholar]
- 30.McDonald D et al. American Gut: an open platform for citizen science microbiome research. mSystems 3, e00031–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moon TK The expectation-maximization algorithm. IEEE Signal Process. Mag 13, 47–60 (1996). [Google Scholar]
- 32.Silverman JD, Shenhav L, Halperin EA, Mukherjee SA & David LA Statistical considerations in the design and analysis of longitudinal microbiome studies. Preprint at bioRxiv: 10.1101/448332 (2018). [DOI] [Google Scholar]
- 33.Pritchard JK, Stephens M & Donnelly P Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang H, Peng J, Wang P & Risch NJ Estimation of individual admixture: analytical and study design considerations. Genet. Epidemiol 28, 289–301 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Alexander DH, Novembre J & Lange K Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo R et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deloger M, El Karoui M & Petit M-A A genomic distance based on MUM indicates discontinuity between most bacterial species and genera. J. Bacteriol 191, 91–99 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leung HCM et al. A robust and accurate binning algorithm for metagenomic sequences with arbitrary species abundance ratio. Bioinformatics 27, 1489–1495 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Costello EK et al. Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lauber CL, Hamady M, Knight R & Fierer N Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol 75, 5111–5120 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All of the datasets analyzed in this paper are public and can be referenced at the following accession numbers: The first dataset was collected and studied by Backhed et al.16 (accession number ERP005989). The second dataset was collected and studied by Lax et al.15 (accession number ERP005806). The third dataset was collected and studied by Knights et al.10 (data from this study are stored in https://github.com/danknights/sourcetracker). The fourth dataset was collected and studied by McDonald et al.12 (accession number ERP012810) and the American Gut Project30 (EBI project number PRJEB11419). The fifth dataset was collected and studied by Taur et al.18 (data from this study are stored in http://www.ncbi.nlm.nih.gov/sra). In our simulations we used the Earth microbiome project (ftp://ftp.microbio.me/emp/release1/otu_tables/closed_ref_greengenes/).