

Abstract
Over the past decade, remarkable progress has been made towards elucidating the origin and genomic landscape of childhood high-grade brain tumours. It has become evident that paediatric high-grade gliomas differ from those in adults with respect to multiple defining aspects including: DNA copy number, gene expression profiles, tumour locations within the CNS and genetic alterations such as somatic histone mutations.
Despite these advances, clinical trials for children with gliomas have historically been based on ineffective adult regimens that fail to take into consideration the fundamental biological differences between the two. Additionally, although our knowledge of the intrinsic cellular mechanisms driving tumour progression has considerably expanded, little is known about the dynamic tumour immune microenvironment in paediatric high-grade gliomas.
In this review, we explore the genetic and epigenetic landscape of these gliomas and how this drives the creation of specific tumour subgroups with meaningful survival outcomes. Further, we provide a comprehensive analysis of the paediatric high-grade glioma tumour immune microenvironment and discuss emerging therapeutic efforts aimed at exploiting the immune functions of these tumours.
Keywords: paediatric glioma, diffuse intrinsic pontine glioma, tumour associated macrophage, inflammation, histone
Ross et al. provide a comprehensive review of the immune landscape of paediatric high-grade gliomas. They explore how different genetic alterations lead to the creation of specific tumour subgroups with distinct immune profiles, and discuss current and emerging immunotherapies.
Introduction
CNS neoplasms are the most common solid tumours in the paediatric population.1,2 Approximately 53% of all paediatric solid tumours are composed of low- (LGG) and high-grade (HGG) gliomas. Paediatric LGGs (pLGGs) account for approximately two-thirds, and paediatric HGGs (pHGGs) one-third, of all paediatric CNS tumours.3 High-grade gliomas include several disease entities such as anaplastic astrocytoma, glioblastoma, diffuse midline glioma (DMG) with the H3K27M mutation (WHO Grade IV) and diffuse intrinsic pontine glioma (DIPG). Less frequently encountered HGGs include anaplastic oligodendroglioma, oligoastrocytoma and gliosarcoma. Together, these gliomas occur in 1 per 100 000 children each year, yet despite their relative rarity, they are responsible for the most cancer-related deaths in children under the age of 19 years.1,2,4 To date, there have been very few predisposing conditions causally linked to the development of pHGG, but conditions such as Li-Fraumeni syndrome, constitutional mismatch repair deficiency, neurofibromatosis type 1 and Turcot syndrome have been implicated.5-7
Paediatric HGGs within the brain are found in two general locations, including the cortical hemispheres (∼50%) or the midline/brainstem (∼50%; collectively referred to as DMG) (Fig. 1A). Hemispheric pHGGs (supratentorial) are typically less aggressive than those found in the midline or brainstem (infratentorial). Furthermore, hemispheric pHGGs usually have a median age at diagnosis of 13 years and median survival of 18 months, compared with 7–9 years and 11 months, respectively, for DMGs.8,9 Much is known about the intrinsic cellular mechanisms driving tumour progression in pHGGs, yet the tumour immune microenvironment (TIME) has been less comprehensively characterized. In this review, we discuss the historical data describing the TIME of pHGGs and explore the implications that the TIME has on tumour dynamics and therapeutic efforts.
Figure 1.

Clinical features of pHGGs. (A) T2 FLAIR magnetic resonance image of a DIPG in the coronal plane and a post-contrast T1 MRI of H3/IDH wild-type hemispheric pHGG in the transverse plane. Corresponding haematoxylin and eosin stained (H&E) images displaying major histological characteristics, including microvascular proliferation (yellow arrow), pseudopalisading necrosis (red arrow) and brisk mitotic activity (black arrow). (B) Forty to sixty per cent of pHGGs have PDGFRA amplification. Half of the pHGGs have histone mutations, including H3.1/H3.3K27M mutations which arise in the brainstem and midline and H3.3G34R/V mutations, which are hemispheric tumours. The other half of pHGGs are histone wild-type, but commonly possess CDKN2A and TP53 mutations, and typically arise in the hemispheres of the brain.
Current standard of care
As with adult HGGs (aHGGs), the most effective treatment strategy for pHGGs is maximal safe tumour resection.10–13 However, the diffusely infiltrative nature of HGGs precludes complete tumour resection in nearly all cases, owing to inevitable tumour recurrence. In one study (CCG-945), patients who underwent gross total resection had a 5-year progression-free survival rate of 35%, while that of those who received subtotal resection was 17%.12 In cases where over 90% of the tumour is resected, the 5-year progression-free survival rate is significantly greater than in those with less successful resection.11 Because of the location of DMGs in delicate regions of the brain such as the brainstem and thalamus, these tumours are typically rendered inoperable, contributing to their dismal outlook. Treatment of DMGs thus relies heavily on radiotherapy with the use of corticosteroids for symptom management and cerebral oedema.14,15 A range of radiation doses are used to treat pHGGs, with most patients receiving between 50 Gy and 60 Gy of fractionated radiation spread over ∼6 weeks of 30 fractions.1,6,14 Unlike aHGGs, pHGGs typically do not respond to chemotherapies and the addition of chemotherapy to radiotherapy adds little benefit.16 Further, unlike aLGGs and aHGGs, which frequently harbour the prognostically favourable MGMT promoter methylation, the frequency of this genetic alteration is lower in pHGGs, and it is unclear whether this has prognostic value in the paediatric setting.16–20 The limited number of long-term pHGG survivors are often left with detrimental side effects, including endocrine morbidity, psychiatric and neurocognitive impairments, developmental disorders and a high incidence of secondary tumours.4–6 These deleterious outcomes further highlight the necessity for novel treatment modalities that extend the prognostic outcome while minimizing toxicities in children with pHGG.
Historically, therapeutic strategies for pHGG have been adapted from their adult counterparts and have therefore ignored the innate biological differences between the two. Immune checkpoint blockade strategies targeting adult systemic cancers have largely failed to produce benefits in pHGG, with the exception of patients harbouring constitutional mismatch repair deficiency.21,22 Furthermore, there have been few attempts to target the immunological properties of these tumours, most likely because the role of the TIME in pHGGs has overwhelmingly been ignored despite the abundance of non-neoplastic cell types found within them. Henceforth, we must put more emphasis on delineating the role of the TIME in pHGGs, including the cell types involved, their biological functions within the tumour and their potential for therapeutic targeting.
The genetic and epigenetic landscape of paediatric high-grade gliomas
The genetic differences between pHGGs and aHGGs are striking, highlighting the need to develop tailored therapeutic strategies specific to the former. Although both types of tumour share the same defining histological features, including diffusely infiltrating tumour margins, vigorous mitotic activity, microvascular hyperplasia, and pseudopalisading necrosis, the similarities diverge from there (Fig. 1A).23,24 Both pHGGs and aHGGs possess a high frequency of PDGFRA amplification and mutation; however, these alterations are more frequent in pHGGs (up to 40%) and constitute the most common genetic alteration in these tumours (Fig. 1B).9,25,26 Despite demonstrated activation of the PI3K pathway in pHGGs, alterations in the aHGG subtype-defining genes EGFR and PTEN are less frequent in pHGGs.26TERT promoter mutations which are frequent in aHGGs are present in only 2% of DMGs and 3% of hemispheric pHGGs.27 Moreover, chromosomal alterations are much more common in aHGGs.9,26
A prominently unique feature of pHGGs is the presence of mutations in histone encoding genes. Up to 50% of all cases possess mutations in H3F3A or HIST1H3B, including 60–80% of DIPGs, 60% of midline HGGs and 15% of hemispheric pHGGs.9,28 These mutations were first identified through whole-genome sequencing in 2012 by two separate groups, Schwartzentruber et al.29 and Wu et al.,30 who discovered that DIPGs exclusively possessed heterozygous somatic mutations encoding lysine to methionine alterations at lysine 27 (K27M) in the H3F3A and HIST1H3B genes.27 These mutations were not found in hemispheric pHGGs; however, somatic mutations encoding glycine to arginine and less frequently glycine to valine at glycine 34 (G34R/V) of the H3F3A gene are found in up to 15–20% of these tumours.29,31 K27M mutations are primarily found in brainstem/midline pHGGs, and G34R/V mutations are exclusively found in hemispheric pHGGs; yet, the exact mechanism for these location-based differences have yet to be revealed.
The presence of histone mutations appears to be the driving factor for the subtype-specific groupings of pHGGs. A recent meta-analysis of over 1000 pHGGs by Mackay et al.9 demonstrated unique survival statistics for each histone mutant subgroup. H3.1 and H3.3K27M mutations confer the worst survival and occur in younger patients, while H3.3G34R/V mutations confer slightly extended survival over K27M and histone wild-type tumours (Fig. 1B).9,29 Activating mutations in ACVR1 are frequently found in H3.1K27M tumours, and less frequently in H3.3K27M tumours.27,32 Almost all G34R/V tumours possess TP53 mutations and ∼50% also have ATRX-DAXX mutations.29,31 H3K27M tumours arising outside of the pons also possess ATRX loss, but at lower frequencies than G34R/V tumours.29,33,34 Despite possessing similar co-mutations with IDH1 mutant tumours, histone-mutant pHGGs do not harbour IDH1 mutations, and only ∼6% of all pHGGs do.31,34 Furthermore, histone-mutant pHGGs rarely have EGFR amplifications, BRAF V600E or NF1 mutations, and they typically do not possess receptor tyrosine kinase fusions (MET, FGFR2, NTRK2/NTRK3) that are otherwise found in hemispheric histone wild-type tumours.9,34,35 The heterozygous loss of chromosome arm 10q is also found in all subgroups of pHGG, including histone-mutant subgroups.9,34,35
Despite having a more indolent disease course, DMGs harbouring H3.1K27M and H3.3K27M mutations have similar overall survival and event-free survival rates compared with pHGGs and are therefore now classified as WHO Grade IV tumours even in the absence of high-grade histology.36,37 Tumours possessing K27M mutations are thought to arise from neural precursor cells during CNS development.38 These pHGGs usually possess TP53 overexpression or loss-of-function mutations as well as amplifications of PDGFRA.27–29 Histone H3.1 is produced during the S-phase of the cell cycle and is utilized for the packaging of newly synthesized DNA. H3.3 is synthesized during interphase and is deposited on actively transcribed genes by the ATRX/DAXX complex independent of the cell cycle.39–41 Although histone H3 mutant proteins make up only 3–17% of the total H3 pool in DIPGs, almost complete loss of H3K27me3 has been observed.42 Even though there is global loss of H3K27M methylation, it has been shown that patient-derived DMG cells harbouring the H3.3K27M mutation retain high methylation at a small number of domains such as the HOXD locus. This is in contrast to H3.1K27M-bearing cells, which do not retain methylation in these domains.41 The mutated histone proteins inhibit the enzymatic activity of the polycomb repressive complex 2 (PRC2) subunit EZH2, which leads to global loss of H3K27me3/me2 methylation and increased H3K27 acetylation (Fig. 2).42,43 PRC2 target genes are involved in developmental processes related to stem cell regulation, cellular proliferation and cellular differentiation.44,45 Through single-cell RNA sequencing of human and murine DIPG K27M mutant tumours, groups have shown that few genes are downregulated in comparison with other tumour types, and PRC2 target genes are upregulated as a result of hypomethylation.46,47 It has also been demonstrated that the majority of cells within DIPGs are most similar to self-renewing oligodendrocyte progenitor cells, indicating the H3K27M mutation likely prohibits differentiation into oligodendrocytes.46
Figure 2.

Histone mutations in pHGGs. (A) In histone wild-type pHGGs, there is maintenance of global H3K27me3 across the genome. (B) However, in H3.1 or H3.3K27M mutant pHGGs, the methyltransferase activity of the EZH2 enzymatic unit of the PRC2 complex is inhibited, resulting in global hypomethylation and increased acetylation. (C) In H3.3G34R/V mutant pHGGs, there is loss of H3K27me3 but only on affected histone proteins, due to the proximity of the G34 position to K36.
Much is known about histone wild-type and K27M mutant gliomas. Less is known about pHGGs that harbour H3G34R/V mutations, which arise predominately in the hemispheres of the brain. H3G34R/V mutations occur in up to 15% of hemispheric pHGGs, and in a similar manner to K27M tumours, they possess H3K36me3 hypomethylation; however, this is observed only on affected histone H3 mutant proteins.40,42 The loss in methylation is due to the close proximity of H3G34 to K36, leading to diminished SETD2 enzymatic activity and subsequent loss of H3K36me3.42,48 Interestingly, much like adult gioblastoma, H3G34R/V mutant tumours have been shown to possess a methylated MGMT promoter; however, this finding does not confer a survival advantage in patients treated with temozolomide.9,35,49 Unlike H3G34R/V mutant pHGGs, H3K27M pHGGs do not harbour MGMT promoter methylation.50 It has also been found that H3G34R/V tumours have a greater mutational burden than other pHGG subgroups, likely due to deficiencies in the DNA chaperone proteins ATRX and DAXX, which also aid in DNA repair.51 Although currently unexplored, these characteristics may be useful in the development of subtype-specific targeted immunotherapies.
In the absence of mutations in histone encoding genes, there are three main subgroups that arise from consensus clustering of 450k methylation data, and these groups have been termed WT-A, WT-B and WT-C.49 A large majority of WT-A samples were found to be hemispheric, resemble LGG subgroups and driven by BRAF V600E, NF1 and TP53 mutations, as well as receptor tyrosine kinase fusions such as MET, FGFR2 and NTRK2/NTRK3.49 Interestingly, these tumours showed upregulation of cytokine signalling and had the best overall survival of the three histone wild-type subgroups—reflective of historical findings, suggesting that pLGGs have increased cytokine and chemokine signalling compared with their pHGG counterparts.52 DNA methylation analysis by Grabovska et al.53 further demonstrated that WT-A tumours possessed greater monocyte infiltration and greater tumour infiltrating lymphocyte infiltration compared with WT-B and WT-C tumours. It was also found that lower B-cell and CD8 T-cell infiltration in this group correlated with worse survival. Other mutations commonly found in histone wild-type tumours that do not necessarily fall into specific subtypes include CDKN2A/CDKN2B deletions, RB1 mutations, receptor tyrosine kinase alterations (MET, IFG1R, NTRK2), PI3K pathway alterations (PTEN, PIK3CA, TSC2, PIK3R1) and MAPK pathway mutations (BRAF and NF1).9,49 Although not specifically discussed in this review, infant HGGs are also largely driven by receptor tyrosine kinase alterations (ALK, ROS, NTRK, MET) and possess their own subgroupings.54
Stratification between WT-B and WT-C tumours is less robust, yet differences do exist between them. WT-B tumours were found more evenly dispersed among the brain’s anatomical locations, had strong upregulation of MYC target genes and had the worst overall survival of the three histone wild-type subgroups.9 Furthermore, they demonstrated the lowest level of CD4 T-cell infiltration between the three subgroups.53 WT-C tumours were mostly hemispheric in nature yet their expression signature reflected that of midline tumours. These tumours harboured PDGFRA and MET amplifications and were associated with the proneural gene signature of adult gioblastoma, which is characterized by a relatively inert immunological landscape.9,55
It is well-established that pHGGs are highly heterogeneous in terms of region-specific differences in age at diagnosis, molecular alterations and survival. To date, in-depth studies investigating how the tumour location (midline versus cerebral hemispheres) or presence of onco-histones (H3K27M or H3G34R/V) impact the TIME have been lacking. We believe a focus on these areas will propel therapeutic discovery; therefore, we will now thoroughly discuss our current understanding of the pHGG TIME.
The immune landscape of paediatric high-grade gliomas
Studies specifically designed to enhance our understanding of the TIME of pHGGs are sparse yet informative. Several studies have broadly compared pLGGs to pHGGs; however, as the importance of the TIME in aHGG subtypes with respect to survival and response to therapy has dramatically increased, the pHGG TIME is becoming more heavily studied. A study carried out by Griesinger et al.56 demonstrated through flow cytometry that the number of infiltrating CD45+ and CD11b+ myeloid cells is significantly higher in paediatric pilocytic astrocytoma and ependymoma compared with gioblastoma and medulloblastoma samples. The latter two samples also displayed an immunosuppressed phenotype with a high proportion of CD163 and CD206 expression, as well as a smaller frequency of CD4+ and CD8+ T cells compared with pilocytic astrocytoma and ependymoma samples. It must be noted that the study did not provide sample sizes; however, it was one of the first to analyse the immune infiltrates of pHGGs.
Much of our knowledge about the pHGG TIME has been obtained through immunohistochemical analyses. Lieberman et al.52 used a set of pLGG (19 pilocytic astrocytoma, four ganglioglioma), pHGG (one anaplastic astrocytoma, 16 glioblastoma) and DIPG (n = 9, autopsy) samples for tissue microarray analysis and compared the findings with normal adjacent brain tissue (n = 12). There were minimal differences in the amount of CD68+CD163+ tumour-associated macrophages (TAMs) between tumour types; however, pLGG and pHGG samples had significantly more TAMs compared with the control tissue, while DIPG samples did not. The same trend held true for CD3+ and CD8+ T cells, highlighting the scarcity of lymphocytes in DIPGs. NanoString analysis of CCL2-4 demonstrated higher expression in pLGGs compared with pHGGs and DIPGs, while pHGGs and DIPGs had higher expression of CXCL8 than pLGG and non-tumour tissue. Interestingly, pHGG and DIPG samples demonstrated the highest expression of PDL1, again suggesting that the TIME of pHGGs is immunosuppressive in nature. Lastly, VEGFA expression was highest in DIPGs and pHGGs, likely indicative of the highly vascularized nature of these tumours.52 Robinson et al.57 performed multiplex immunohistochemical analysis of 18 pLGG (five pleomorphic xanthoastrocytoma, seven ganglioglioma and six pilocytic astrocytoma) and eight pHGG (four histone wild-type, three H3G34R and one H3K27M) tissue samples. This analysis revealed that pLGGs exhibit significantly higher amounts of CD3+ T-cell infiltration, although CD3 staining was highly variable among all samples. The same group also performed single-cell mass cytometry (CyTOF) on eight pLGGs and one pHGG (fresh tumours), and they found the predominant infiltrating T-cell subtype to be CD69+CD45RO+ tissue resident memory cells. Furthermore, TCF1+ T cells were observed to localize in perivascular areas, while CD103+ T cells were further away from CD31+ blood vessels.57 The number of high-grade samples in this study were low, yet they corroborated other findings of low T-cell infiltration in pHGGs.
A more integrated approach to comparing pLGGs and pHGGs was taken by Plant et al.,58 who performed immunohistochemistry on formalin fixed-paraffin embedded tumour samples and supplemented these data with flow cytometry on fresh tumour samples. The immunohistochemistry findings from 27 paediatric gioblastomas and 32 pLGGs demonstrated significantly greater infiltration of CD45+ myeloid cells, CD8+ T cells and PD1+ cells in pLGGs. The majority of samples were negative for PDL1 expression, and there were no correlations between immunohistochemical markers and overall survival in pHGGs. Additionally, OncoPanel™ results were available for a subset of patients which showed there to be seven or fewer mutations per tumour type, indicating that the mutational load of pLGGs and pHGGs was small. Flow cytometry on 10 pLGGs and three pHGGs further demonstrated a higher level of lymphocytic infiltration in pLGGs compared with pHGGs; however, statistical significance was not met. Interestingly, pHGG samples had significantly higher levels of CD45+CD19+ B cells and CD38+IgD+ activated B cells compared with pLGGs, yet the significance of these findings was not determined. In the same study, frozen blood samples from four pLGG and four pHGG patients were used for T-cell receptor sequencing. This analysis found little sharing of T-cell clones between tumour and corresponding blood samples, likely due to the limited T-cell infiltrate in these tumours.58 Overall, this study once again demonstrated the paucity of an immunogenic microenvironment in pHGGs, especially with respect to infiltrating T cells. These results suggest other immune cell types such as TAMs should take precedent when studying the TIME of pHGGs.
Studies carried out by our laboratory have further supported historical findings of scarce T-cell infiltration in pHGGs.59 Through the immunohistochemical analysis of human formalin fixed-paraffin embedded pHGG tissue samples, including 12 DIPG (7/12 K27M) and 11 paediatric gioblastoma (3/11 G34R) samples, we found few CD3+ and CD8+ T cells present and no differences between subtype or tumour location. In a larger cohort of 33 samples, including 17 DIPG (10/17 K27M) and 16 paediatric gioblastoma (4/16 G34R) samples, we did, however, find considerable infiltration of IBA1+ TAMs among all tumour types, with high sample-to-sample variability. Again, no differences were observed between tumour subtype, molecular subgroup or tumour location, but this does not preclude the existence of these differences. To make conclusive observations concerning subtype-specific differences, larger studies must be done. Regression analysis revealed significant correlations between CD3 and IBA1 positivity in matched samples, as well as for CD3 and CD31, CD3 and PDGFRB, IBA1 and CD31, PDGFB, and PDGFRB, suggesting the stromal compartment of the tumour may play a pivotal role in influencing immune cell infiltration.
To supplement our findings using immunohistochemistry, we also performed NanoString analysis on 22 human pHGG samples, including 10 DIPG (8/10 K27M) and 12 pHGG (3/12 G34R) samples. Interestingly, there were considerable differences in the inflammatory signalling pathways between the two HGG subtypes. Unsupervised hierarchical clustering of signalling pathway scores indicated that the majority of DIPG samples cluster together, while their hemispheric counterparts cluster together, with DIPGs possessing greater expression of genes involved in almost all inflammatory pathways. Moreover, cell type analysis indicated that DIPGs showed greater representation of genes associated with T cells, dendritic cells, Th1 cells, natural killer cells, neutrophils and macrophages. When examining differentially expressed genes, DIPGs showed significant expression of genes involved in the CXCR2 signalling pathway, including CXCL1, CXCL2, CXCL5, CXCL6 and IL8. These results were further supported by NanoString analysis on 11 murine pHGGs, including five H3.3K27M DIPGs and six H3.3WT hemispheric gioblastomas. This analysis again demonstrated that DIPG tumours found in the brainstem were more inflammatory than hemispheric pHGGs. Lastly, when we performed unsupervised clustering of human and murine pHGGs, we found that hemispheric pHGGs clustered together and DIPGs clustered together, regardless of species, indicating that the tumour location, molecular subtype or both influence the inflammatory microenvironment of pHGGs. Our results suggest that despite the lack of infiltrating T cells, pHGGs still possess an inflammatory TIME characterized by considerable TAM infiltration (Fig. 3), again highlighting the emerging role of TAMs in pHGG. We are currently investigating subtype-specific differences in pHGGs to determine how the molecular signature or tumour location dictates the TIME.
Figure 3.
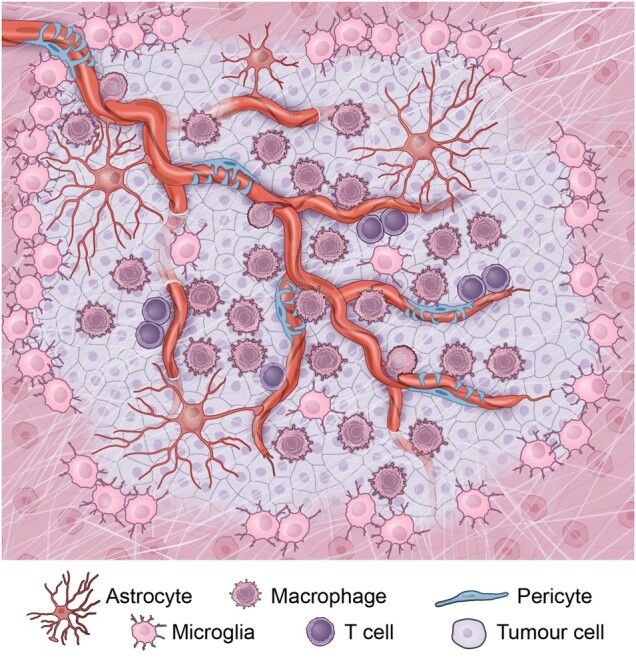
The tumour microenvironment of pHGGs. The pHGG tumour microenvironment is composed of a vascularized tumour bulk with invasive tumour margins. Infiltrating bone marrow-derived tumour associated macrophages are found in perivascular areas, while brain resident microglia are found at the periphery of the tumour. T cells are typically sparse, yet are found in perivascular areas.
A study by Mackay et al.49 on the HERBY phase II randomized trial in pHGGs further supported several key findings on our understanding of the immune response in pHGGs. The HERBY trial was designed to study the efficacy of adding an anti-angiogenic agent, bevacizumab, to the current standard of care, which included radiotherapy and/or temozolomide in 121 patients with newly diagnosed non-brainstem pHGG. As previously demonstrated in the field, they found histone mutant tumours (both K27M and G34R) were considerably immune cold, based on the absence of immunohistochemical staining of CD8+ T cells. PXA-like HGGs, however, were significantly enriched for CD8+ T cells, as were four hypermutator cases that were found to have mutations in POLE, POLD1 and MLH1. Patients with tumours possessing greater CD8+ T-cell infiltration had a significantly better overall survival and were more likely to respond to temozolomide/radiotherapy with the addition of bevacizumab. It is important to note, however, that 17/18 cases with a high presence of CD8+ T cells were hemispheric tumours, further supporting the notion of a relatively barren TIME of midline and brainstem pHGGs. Additionally, the impact of tumour resection on survival is difficult to control for, and ascribing positive results to high CD8+ T-cell infiltration must be done cautiously. These results contrast with findings from Bailey et al.,60 who demonstrated through CIBERSORT that the presence of CD8+ T cells in histone wild-type hemispheric tumours negatively correlated with survival. A case report by Bouffet et al.21 of two siblings aged 6 and 3.5 years, both harbouring PMS2 and POLE germline mutations, demonstrated that mismatch repair deficiency induces a significantly higher mutational load in paediatric gioblastoma. Because of their relatively high mutational burden (both over 20 000 mutations per exome), the children were selected for nivolumab (anti-PD1) treatment. After 9 and 5 months of therapy, both patients’ tumours were significantly reduced and a reduction in peritumoral oedema and nodular lesions was also observed. The patients were declared clinically stable after treatment, highlighting the importance of neoantigens and their influence on the response to immunotherapy. Although mismatch repair-deficient pHGGs are infrequent, evidence suggests a significant prognostic value exists in determining their presence in patients with pHGGs.
Another histological study by Jha et al.61 compared PDL1 expression in 126 adult and paediatric DMGs, and found PDL1 expression to be highest in H3K27M/IDH1 wild-type DMGs in both age groups. PDL1 expression significantly correlated with tumour-infiltrating lymphocyte infiltration in both age groups and patients whose tumours were PDL1-negative had a longer overall survival compared to PDL1 tumour-positive patients. Paediatric DMGs did not differ from adult DMGs in terms of CD3+ T cells; however, adult DMGs showed a significantly greater presence of CD8+ T cells, suggesting immunotherapeutic approaches must be tailored not only to specific tumour types, but also to specific age groups.
Other important findings from the Mackay et al.49 analysis of the HERBY trial showed that pHGGs with MAPK alterations had a greater T-cell signature and M2 macrophage response in RNA-seq data compared with those that did not have MAPK alterations. Furthermore, immunohistochemical staining for CD68 in pHGGs found heterogeneous infiltration of TAMs that were found to cluster around areas of necrosis and perivascular lymphocytes. Another study found in xenograft models of DIPG into NOD scid (severe combined immunodeficiency) gamma (NSG) mice that there were CD68+ and CD163+ cells resembling macrophages and microglia in the tumour parenchyma, with limited CD3+ T-cell infiltration.62 Becaue of the limited infiltration of T cells in pHGGs, analyses that focus on TAMs may prove to be more beneficial to our understanding of the TIME. Lin et al.63 performed immunofluorescence on DIPG tissue obtained from biopsy and found that DIPGs and adult gioblastomas showed high IBA1+ staining compared with normal cortical tissue. They also performed flow cytometry on early post-mortem DIPG tissue samples and found that DIPGs had a smaller CD45+CD11b− lymphoid population compared with adult gioblastomas. DIPGs however had significantly greater infiltration of CD45+CD11b+ TAMs compared with adult gioblastomas. TAMs from six DIPGs, four adult gioblastomas and three paediatric cortical control samples were flow sorted and RNA sequencing was performed. They did not find an enrichment of M1- or M2-associated genes in DIPG TAMs, indicating they may not necessarily fall discretely into classically- or alternatively-activated phenotypes. Compared with control samples, DIPG TAMs show upregulation of genes associated with interferon gamma (IFNγ) signalling, type 1 interferon signalling and antigen processing and presentation. Yet genes that were differentially expressed between adult gioblastomas and DIPGs were compared, inflammatory genes, including IL6, CCL4, IL1A, IL1B and CCL3, were upregulated in the adult gioblastoma samples. To determine if the DIPG tumour cells were producing inflammatory signals, they analysed the cytokine and chemokine secretome using an ELISA array on DIPG patient-derived cell cultures. Their findings again indicated that DIPG tumour cells produce significantly fewer cytokines and chemokines than adult gioblastomas.63 RNA sequencing data further supported these findings and in general point towards the conclusion that DIPGs are non-inflammatory when compared to adult gioblastomas; however, they did not directly compare DIPGs to non-brainstem pHGGs. These results highlight two key consistent findings; DIPGs possess sparse T-cell infiltration, yet are characterized by high TAM infiltration, and the inflammatory profile of DIPG-associated TAMs is quite different from that of adult gioblastoma TAMs.
Immunotherapeutic studies
Because of the limited efficacy of chemotherapy and a limited longitudinal response to radiotherapy, there has been increasing efforts to harness the immune system for antitumour treatment in pHGGs. Ideally, targeted immunotherapies would be designed to eliminate tumour cells, while sparing permanent neurological dysfunction. Targeting tumour-specific neoepitopes would achieve this and limit autoimmune reactions. Several therapeutic strategies are emerging as likely candidates for pHGG, including chimeric antigen receptor (CAR) T-cell therapy, tumour vaccines and viral therapies. All these approaches are designed to induce tumour-specific T-cell killing, while limiting pathological insults to adjacent normal brain tissue and vasculature, including blood–brain barrier integrity.
Checkpoint blockade
Checkpoint blockade in adult gioblastoma has historically shown no benefit to overall survival either alone or in combination with bevacizumab (anti-VEGF). The underlying mechanisms driving resistance to immunotherapy are yet to be determined; however, work with immunocompetent genetically engineered mouse models (GEMMs) of gioblastoma has shown that resistance is conserved across genetic subtypes and is not improved with adjuvant radiotherapy.64 Checkpoint blockade in the paediatric population has also shown little promise, with the exception of patients harbouring hypermutator phenotypes.21 The limited amount of tumour-infiltrating lymphocytes in pHGGs may contribute to the lack of efficacy of checkpoint blockade therapy; however, the pHGG TIME should be further examined to determine the exact underlying mechanisms responsible for resistance. There are several ongoing phase I and II clinical trials investigating the use of anti-PD1 and anti-CTLA4 therapies in children with newly diagnosed or refractory HGGs (Table 1).65–67 After the conclusion of these trials, it will be imperative to report both successful and unsuccessful cases. Combinatorial therapeutic strategies will likely be most efficacious and may be required for the treatment of pHGGs and need further investigation. Currently, other immune-modulatory strategies show promise and will therefore be further discussed in this review.
Table 1.
Actively recruiting immunotherapy trials for pHGGs as of March 2021
| NCT No. | Title of study | Therapeutic strategy | Phase | Objectives |
|---|---|---|---|---|
| NCT04196413 | GD2CART for the treatment of H3K27M mutated diffuse intrinsic pontine glioma or spinal diffuse midline glioma | GD2 CAR T cells | I | Determine feasibility of manufacturing autologous GD2 CAR T cells. Assess safety, determine maximum tolerated dose, and establish phase II dosing. Assess expansion, persistence and phenotype of GD2 CAR T cells. |
| NCT04185038 | Genetically engineered cells (B7-H3-specific CAR T cells) in treating pediatric patients with diffuse intrinsic pontine glioma, diffuse midline glioma, or recurrent or refractory central nervous system tumors | B7-H3 CAR T cells | I | Assess feasibility and safety of B7-H3 CAR T cells. Establish dose tolerability and define maximum tolerated dose. Assess B7-H3 CAR T-cell distribution within CSF. Assess disease response to B7-H3 CAR T cells. |
| NCT02442297 | HER2-Specific T cells in treating participants with recurrent or refractory HER2-positive glioblastoma | HER2 CAR T cells | I | Evaluate safety, persistence, expansion, and function of HER2 CAR T cells. |
| NCT03500991 | HER2-specific CAR T cell locoregional immunotherapy for HER2-positive recurrent/refractory pediatric CNS tumors | HER2 CAR T cells | I | Establish safety and feasibility of HER2-specific CAR T-cell infusions. Assess HER2 CAR T-cell distribution and function in CSF and peripheral blood. |
| NCT03638167 | EGFR806-specific CAR T cell locoregional immunotherapy for EGFR-positive recurrent or refractory pediatric CNS tumors | EGFR806-specific CAR T cells | I | Establish safety and feasibility of EGFR806-specific CAR T-cell infusions. Assess EGFR806-specific CAR T-cell distribution and function in CSF and peripheral blood. Assess expression of EGFR in relapsed treated tumours. |
| NCT02208362 | Genetically modified T-cells in treating patients with recurrent or refractory malignant glioma | IL13RA2 CAR T cells | I | Feasibility and safety of ex vivo expanded CAR T cells. Determine maximum tolerated dose schedule and phase II dosing. Assess post-immunotherapy responses. |
| NCT04099797 | Genetically engineered cells (C7R-GD2.CAR T cells) for the treatment of patients with GD2-expressing high grade glioma or diffuse intrinsic pontine glioma, The GAIL-B trial | GD2 CAR T cells with constitutively active IL7 receptor (C7R-GD2 CAR T cells) | I | Determine safety of escalating doses of CAR T cells. Estimate antitumour response. Evaluate fate and immunological effects of CAR T cells. |
| NCT03652545 | Multi-antigen T cell infusion against neuro-oncologic disease (REMIND) | TAA-T | I | Determine safety and feasibility of rapidly generated TAA-T. Determine optimal dosing schedule of TAA-T. |
| NCT03911388 | HSV G207 in children with recurrent or refractory cerebellar brain tumors | G207 HSV with or without radiotherapy | I | Determine safety and tolerability by frequency of grade 3 above adverse events. Assess immunological response via HSV-1 antibody titres. Assess progression free survival and overall survival. |
| NCT02960230 | H3.3K27M-specific Peptide Vaccine and Poly ICLC with or without Nivolumab in treating patients with newly diagnosed HLA-A2 positive, H3.3K27M positive diffuse intrinsic pontine glioma or other newly diagnosed gliomas | H3.3K27M-specific peptide vaccine and poly-ICLC with or without nivolumab (anti-PD1) | I | Assess safety of H3.3K27M-specific peptide vaccine with or without nivolumab in H3.3K27M+ DIPG or midline gliomas. Determine overall survival at 12 months. Assess H3.3K27M epitope-specific cytotoxic T lymphocyte response. |
| NCT03396575 | Brain stem gliomas treated with adoptive cellular therapy during focal radiotherapy recovery alone or with dose-intensified temozolomide (BRAVO) | TTRNA-DC with or without chemotherapy | I | Assess safety and feasibility of TTRNA-DC vaccines with or without chemotherapy. Determine the maximally achievable dose or maximum tolerated dose. Assess post-immunotherapy antitumour immune response in lymphocytes. |
| NCT01130077 | A pilot study of glioma associated antigen vaccines in conjunction with poly-ICLC in pediatric gliomas | HLA restricted glioma antigen peptides with poly-ICLC | I | Assess safety and tolerability. Determine glioma-associated antigen-specific T-cell responses via IFNγ activity. |
| NCT03389802 | CD40 agonistic monoclonal antibody APX005M in treating pediatric patients with recurrent or refractory brain tumors | CD40 agonistic monoclonal antibody | I | Evaluate safety of CD40 agonistic monoclonal antibody. Determine pharmacokinetics, maximum tolerated dose schedule, and phase II dosing. |
| NCT02359565 | Pembrolizumab in treating younger patients with recurrent, progressive, or refractory high-grade gliomas, diffuse intrinsic pontine gliomas, hypermutated brain tumors, ependymoma or medulloblastoma | Pembrolizumab (anti-PD1) | I | Establish safety and adverse effects with the administration of adult recommended dose. Estimate sustained objective response rate. Determine changes in immunophenotypic profile of CD8+ T cells. |
| NCT04323046 | Immunotherapy (Nivolumab and Ipilimumab) before and after surgery for the treatment of recurrent or progressive high grade glioma in children and young adults | Nivolumab (anti-PD1) and ipilimumab (anti-CTLA4) | I | Measure changes in the genetic signature of the tumour microenvironment after treatment. Safety and tolerability. |
| NCT03690869 | REGN2810 in pediatric patients with relapsed, refractory solid, or central nervous system (CNS) tumors and safety and efficacy of REGN2810 in combination with radiotherapy in pediatric patients with newly diagnosed or recurrent glioma | Cemiplimab (anti-PD1) with radiotherapy | I/II | Evaluate safety and anticipated phase II dose of cemiplimab. Determine pharmacokinetics of cemiplimab. Confirm safety and phase II dose of cemiplimab with radiotherapy. |
| NCT04049669 | Pediatric trial of indoximod with chemotherapy and radiation for relapsed brain tumors or newly diagnosed DIPG | Indoximod (indoleamine 2,3-dioxygenase inhibitor) with chemotherapy and/or radiation | II | Improve antitumour immune responses and measure outcomes according to iRANO criteria. |
DC = dendritic cell; iRANO = immune-related response assessment for neuro-oncology; poly-ICLC = polyinosinic-polycytidylic acid (poly[I:C]) stabilized by lysine and carboxymethylcellulose; TAA-T = tumour multi-antigen associated specific cytotoxic T lymphocytes; TTRNA-DC = total tumour RNA loaded dendritic cell vaccine.
CAR T-cell therapies and tumour peptides
The list of tumour-specific antigens for pHGGs is low, yet due to the distinct expression of mutated histone proteins in H3K27M and H3G34R pHGGs, there have been increasing efforts to develop CAR T-cell and peptide therapies specifically designed to target these proteins. Mutant onco-histone proteins are ideal targets, because normal brain tissue does not express these tumour-specific antigens; therefore, the risks of off-target toxicity are low. Moreover, due to the low mutational burden of these tumours, epitope spreading and antigen escape may be limited, allowing for a more durable response to immunotherapy. Because of the relatively impermeable blood–brain barrier, monoclonal antibodies do not easily infiltrate into the brain parenchyma, which may explain their lack of success in pHGG. However, activated immune cells, including T cells and dendritic cells, can cross the blood–brain barrier.68 CAR T cells are produced by collecting a patient’s lymphocytes via leukapheresis, engineering a single-chain variable fragment of a particular antibody with the T-cell receptor signalling domain CD3 and often including co-stimulatory domains in the engineered cells.69 The CAR T cells are then reintroduced into the patient, where they can recognize tumour-specific antigens without necessarily being stimulated by antigen-presenting cells, thus allowing for a robust and effective cytolytic immune response to take place (Fig. 4). To date, there have been few studies investigating the efficacy of CAR T-cell therapies in pHGG. There are currently multiple ongoing phase I clinical trials involving the use of IL13RA2, HER2 and EGFR modified CAR T cells for adult and paediatric HGG; however, to achieve maximum therapeutic responses, more tailored approaches might be necessary to account for the biological characteristics of pHGG.67,70
Figure 4.
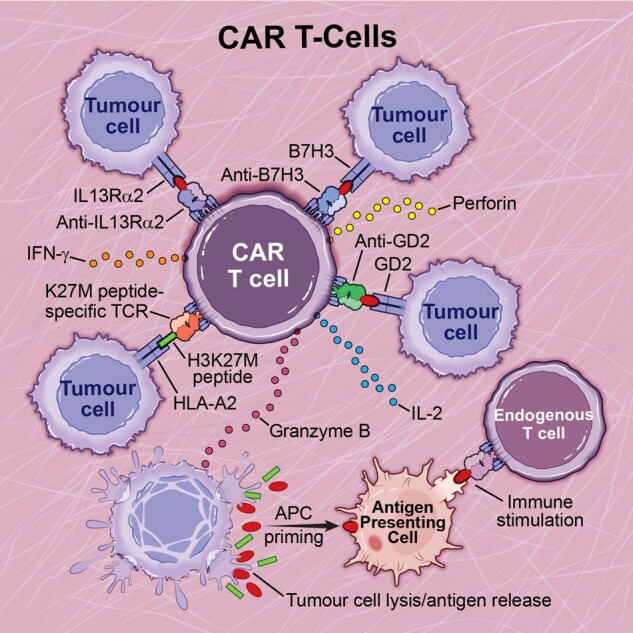
CAR T-cell therapy in pHGGs. CAR T-cell therapies that have been investigated for pre-clinical use in paediatric high-grade gliomas include anti-GD2, anti-IL13RA2, anti-B7H3 and K27M-peptide specific CARs. CAR T-cell therapy stimulates the adaptive immune response, allowing for direct tumour cell killing and priming of antigen presenting cells to further amplify the immune response.
Majzner et al.71 assessed the efficacy of B7-H3 CAR T cells for paediatric solid tumours. B7-H3 is believed to be a checkpoint molecule that is aberrantly expressed in a variety of cancers while being minimally expressed in normal tissue.72 The authors showed B7-H3 to be highly expressed in paediatric solid tumours, including HGGs and DIPG.71 Staining for B7-H3 demonstrated that it was ubiquitously expressed in all tumour cells, a desirable trait for CAR T-cell therapy. Interestingly, B7-H3 staining intensity was less in DIPG samples compared with HGGs, indicating that this therapy may be effective in both tumour types, although this was not tested. When co-cultured with patient-derived DIPG cultures, the B7-H3 CAR T cells produced high levels of IFNγ, TNFα and IL2, indicating that the CAR T cells possess high cytotoxic potential. The authors did not test the engineered B7-H3 CAR T cells in mouse models of pHGG or DIPG; however, they did observe significant reductions in tumour growth and a subsequent extension of survival in NSG medulloblastoma xenografts. A phase I clinical trial involving the use of B7-H3 CAR T cells is currently active for their use in DIPG or refractory pHGG (NCT04185038).
Another study by Mount et al.73 sought to identify potential targets for CAR T-cell therapy in DIPG. Using patient-derived cell cultures, they found that disialoganglioside GD2 was highly expressed on the surface of all H3K27M mutant DIPG cultures examined, while expression was much lower on H3-WT DIPG samples. Unlike other gangliosides, GD2 is minimally expressed in normal tissue and shows extensive expression in multiple tumour types.74 Mount et al.73 generated GD2-specific CAR T cells and demonstrated in vitro cytotoxic specificity for H3K27M DIPG tumour cells, but not H3WT DIPG tumour cells, through the production of IFNγ and IL2. They further demonstrated specificity by CRISPR-Cas9-mediated deletion of GD2 synthase in patient-derived cell cultures and found that the cytotoxic effects of GD2 CAR T cells were ablated. Using post-mortem DIPG patient tissue, they created mouse xenografts into NSG mice to test the therapeutic efficacy of their GD2 CAR T cells. Significant tumour clearance was observed in GD2 CAR intravenously-treated animals, resulting in prolonged survival; however, a small portion of H3K27M+ tumour cells were found to remain, which did not express GD2. It is possible that these GD2-negative tumour cells could survive through selective pressure and eventually repopulate the eradicated tumour. Combination therapies may reduce the likelihood of such events. Further histological analysis revealed extensive neuroinflammation within the brainstem and ventricles, including infiltration of CAR T cells and IBA1+ TAMs, especially around apoptosing cells. Unfortunately, ventriculomegaly and hydrocephalus were observed in a subset of animals with fourth ventricular compression. The authors hypothesized this treatment-induced cerebral oedema was due to on-target tumour-specific cytotoxic activity and not due to cytotoxicity on non-tumour cells.
Further analysis of GD2 CAR T cells and other CAR constructs in immunocompetent mouse models should be pursued to delineate the mechanisms underlying treatment induced oedema. This is especially important for tumours located in the brainstem, such as DIPGs; however, it would also be important for hemispheric pHGGs as well. Other investigational studies are currently being conducted on the use of HER2 and EGFR-specific CAR T cells in clinical trials; however, due to the limited frequency of EGFR amplifications in pHGG, the outcomes of these trials may not be as promising as pHGG-specific therapies.67,75 A phase I clinical trial for the use of GD2 CAR T cells modified with a constitutively signalling cytokine receptor, C7R, is currently active for children with DIPG or HGG (NCT04099797). The addition of the cytokine receptor allows for IL-7 signalling to occur within the engineered T cells without effecting bystander lymphocytes, thus causing the CAR T cells to survive longer and provide a more durable response.76
Approaches using tumour-specific peptides arising from oncohistone proteins have also been pursued to induce the cytotoxic T-cell response (Fig. 5). Ochs et al.77 recently described the identification of a H3K27M 27mer peptide (H3K27M p14-40) that binds to HLA-A*0201. Humanized mice expressing HLA-A*0201, HLA-DRA*0101 and HLA-DRB*0101, while lacking major histocompatibility complex (MHC) class I and II (A2.DR1 mice), were used to study vaccination with the identified peptide. Vaccination in a non-tumour setting induced mutation-specific CD4 and CD8+ T cell responses, including proliferation and the production of IFNγ. Utilizing a subcutaneous sarcoma model in A2.DR1 mice, H3K27M peptide vaccination resulted in reduced tumour size and increased infiltration of IFNγ producing CD4 and CD8+ T cells compared to vaccination with the wild-type H3 peptide. Ideally, vaccination into an MHC-humanized glioma model would be preferred; however, the authors stated that such a model does not exist. The authors also demonstrated the ability to detect H3K27M-specific cytotoxic T cells in the peripheral blood of three patients harbouring H3K27M mutant gliomas. Similarly, Chheda et al.78 discovered a 10mer peptide (H3K27M p26–35) that binds with high affinity to HLA-A*0201. Using human peripheral blood samples from HLA-A*0201+ DIPG patients and healthy donors, they found the peptide stimulated antigen-specific increases in IFNγ production. To demonstrate H3K27M cytotoxic specificity, they cloned a T-cell receptor specific for HLA-A*0201+H3K27M+ glioma cells and found that it significantly induced in vitro IL2 and IFNγ production in J76CD8+/− Jurkat T cells and primary human CD8+ T cells, respectively. They further demonstrated the cytotoxic capability of T-cell receptor-transduced T cells in response to HLA-A*0201+H3K27M+ glioma cells in vitro. Most importantly, in vivo activity of their T cell receptor-transduced T cells was tested in NSG mice bearing H3.3K27M mutant U87 glioblastoma cells. They found that mice receiving the T-cell receptor-transduced T cells showed significant reduction in tumour size compared with those receiving mock transduced T cells. The majority of the infiltrating CD4 and CD8+ T cells within the tumours were positive for the HLA-A*0201-H3K27M dextramer, and there were even dextramer-positive T cells found in the peripheral blood. Unfortunately, survival could not be assessed, as the animals receiving the mock-transduced and T-cell receptor-transduced T cells succumbed to graft-versus-host disease. The same group has recently implemented a pilot study to evaluate an H3.3K27M-26-35 peptide vaccine in children with H3.3K27M+ DIPG or HGG (NCT02960230) and their results are encouraging.79
Figure 5.
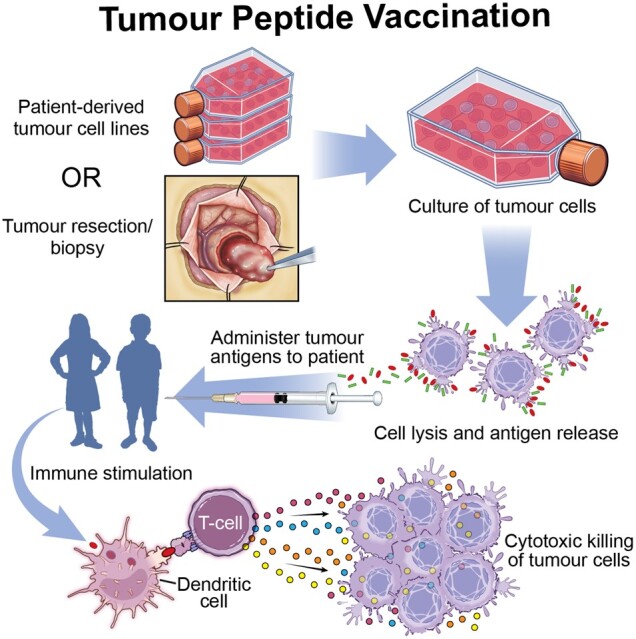
Tumour peptide vaccination in pHGGs. Patient-derived tumour cell lines from a tumour bank or tumour cells from surgical resection are cultured to produce tumour antigens. Tumour cells are lysed to release the tumour antigens, which are then administered to the patient to stimulate antigen-presenting cells and cytotoxic T cells to kill tumour cells.
There are multiple studies reporting the immune stimulatory effects of polyinosinic-polycytidylic acid (poly[I:C]) stabilized by lysine and carboxymethylcellulose (poly-ICLC) treatment alone against aHGG and pLGG, making it a potentially effective immune adjuvant for pHGG as well.80–84 A study by Pollack et al.85 investigated the combination of three glioma-associated antigen peptides combined into a vaccine administered to DIPG or non-brainstem HGG patients. They identified HLA-A2 restricted epitopes from IL13RA2, EPHA2 and survivin peptides and administered them in combination with poly-ICLC. No instances of high-grade reactions or autoimmunity occurred in the patient cohort. Vaccine-induced immune responses as denoted by IFNγ production were observed in 13 of 21 patients, with no discrimination between patients who received prior radiotherapy alone or in combination with chemotherapy. Interestingly the IL13RA2 epitope invoked the highest magnitude of response, and in one patient the immune response diminished after receiving high-dose dexamethasone and returned after dexamethasone tapering. This highlights the importance of determining optimal timing and therapeutic strategies in combining the use of immunotherapies with dexamethasone as a remedy for treatment-associated inflammation. The median survival of the overall patient cohort of this study was 13.3 months from diagnosis, 12.7 months for brainstem tumours and 25.1 months for non-brainstem HGGs. More importantly, two patients had disease-free status; these patients had undergone prior gross total resection, again emphasizing that the extent of tumour resection directly affects disease outcome in pHGGs. Although survival was negligibly extended, this study effectively showed the safety and immunogenic capability of combinatorial peptide vaccines for pHGG.
Dendritic cell therapy
Dendritic cells are the professional antigen-processing cells in the body and are responsible for priming cytotoxic lymphocytes during the adaptive immune response.86 Once dendritic cells have encountered an antigen, they migrate to draining lymph nodes via CCR5 and CCR7 upregulation, where they then activate the T-cell response.87 This process can be triggered or amplified through the isolation and ‘training’ of dendritic cells to specific antigens in a process known as dendritic cell vaccination (Fig. 6). There have been few studies investigating the use of dendritic cell vaccines in pHGG. Initial studies by Caruso et al.88 demonstrated the safety and feasibility of RNA-based dendritic cell vaccines in patients with recurrent high-grade brain tumours. While most patients in the study had eventual disease progression, one patient with anaplastic astrocytoma had stable disease after five vaccines and 21 months of follow-up. Unfortunately, the authors were unable to detect significant in vitro production of IFNγ or T-cell proliferation with treated patient samples. In a larger cohort of patients with relapsed HGG, De Vleeschouwer et al.89 demonstrated the efficacy of autologous dendritic cell adjuvant therapy after tumour resection in adults and children. No analysis of the immune response was performed; however, survival analysis indicated that patients under the age of 35 years had a better overall survival than older patients. Additionally, patients who had a greater extent of tumour resection had better progression-free survival and overall survival than those who had suboptimal resection. A similar study was performed by Ardon et al.90 in a cohort of 33 recurrent pHGGs using autologous dendritic cell vaccination. At the time of last follow-up, there were 7/33 patients still alive with multiple patients surviving 35–85 months. Five of the long-term survivors also received chemotherapy during or after immunotherapy, suggesting that combined therapy is more effective. Another study by Lasky et al.,91 in which 2/3 patients receiving autologous tumour lysate dendritic cell vaccination in combination with chemotherapy survived 40 and 51 months, respectively, further supported these findings. For a disease with such a dismal prognosis, these initial results were encouraging.
Figure 6.
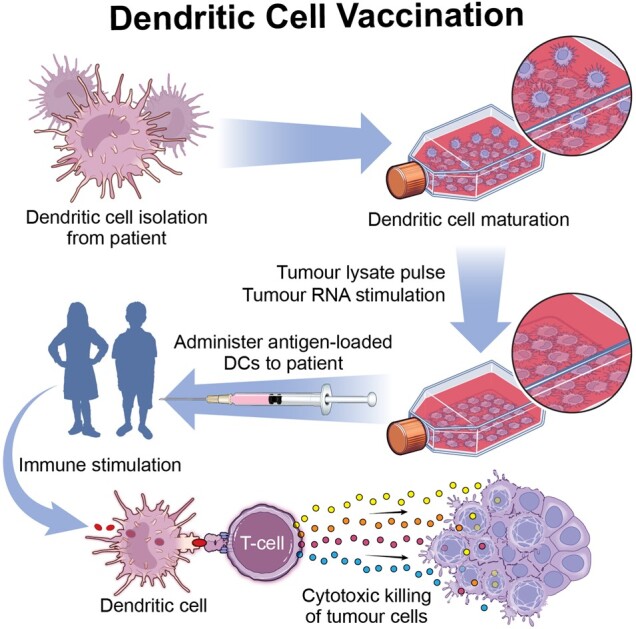
Dendritic cell vaccination in pHGGs. Dendritic cells are collected through leukapheresis, cultured in vitro and stimulated with tumour cell lysate or tumour RNA. Mature antigen-loaded dendritic cells are then readministered back into the patient to stimulate the adaptive immune response and cause cytotoxic killing of tumour cells.
A recent study by Benitez-Ribas et al.92 investigated the feasibility and safety of administering autologous dendritic cells pulsed with tumour cell line lysate to nine newly diagnosed children with DIPG. Patient dendritic cells were collected by leukapheresis after the discontinuation of dexamethasone and were loaded with lysate obtained from eight allogeneic DIPG tumour cell lines (ATCL) ex vivo. Mature, ATCL-pulsed dendritic cells were then administered intradermally 3–6 weeks after the completion of radiotherapy and then once every other week during the induction phase, with a subsequent booster 3 months later during the maintenance phase. Before administration, dendritic cells were confirmed to have increased expression of CD80 (co-stimulatory molecule), CD83 (activation), CCR7 (migration) and MHC-II (antigen presentation), indicating a mature and activated population of dendritic cells following ATCL loading. No adverse reactions were noted during the course of the study, demonstrating the safety of dendritic cell administration to patients. Analysis of peripheral blood and CSF indicated ATCL-specific proliferative responses and the production of IFNγ by patient T cells. Unfortunately, at the time of publication, there were no clinical outcome data available for this study; however, it was the first study to demonstrate the feasibility of ATCL pulsed dendritic cell therapy for DIPG. Although this method was adapted for the use of tumour cell line lysates, it could be applied to individual patient tumours as well. This would likely be costly and resource-intensive; however, it may offer a more effective and personalized approach compared with using a generalized cocktail of tumour cell lines. Since this approach would require a surgical biopsy or tumour sample after resection, further considerations would be required for tumours located in precarious regions such as the brainstem and midline regions (thalamus/hypothalamus), where surgery poses potential irreversible harm. The efficacy and feasibility of actively personalized vaccines has recently been demonstrated in adult gioblastoma and these findings will likely serve as templates for their use in pHGG.93,94 One challenge to consider for dendritic cell vaccination therapies is that there must be adequate time for vaccine production, which may allow for subsequent growth of remaining tumour to cause morbidity and mortality. There also must be enough time between the weaning of steroids and other chemotherapeutic agents before vaccination.
Viral therapies
To date, clinical trials investigating the use of oncolytic viruses for the treatment of pHGGs and DIPG are in the early stages and have not progressed to phase II studies. Most studies have been performed in vitro, with minimal supporting in vivo studies. Tejada et al.95,96 previously examined the safety and efficacy of DNX-2401 for the treatment of newly diagnosed DIPG. DNX-2401 (also known as Delta-24-RGD) is an oncolytic adenovirus that targets cells with a defective Rb pathway. The virus demonstrated therapeutic efficacy in recurrent human aHGG, murine models of pHGG and murine models of DIPG, and therefore was adapted for use in human pHGG.97–100 In an immunocompetent murine model of aHGG utilizing the GL261 glioma cell-line injected into C57BL/6 mice, Jiang et al.99 performed intratumoral injections of Delta-24-RGD. They observed an increase in natural killer cell, CD3+, CD4+ and CD8+ lymphocyte infiltration, as well as activated antigen-presenting cell populations after injections. Furthermore, they noted an increase in IFNγ production, as well as a significant survival advantage, in treated mice, demonstrating the effectiveness of the virus to elicit an antitumour immune response. Utilizing both immunodeficient and immunocompetent mouse models of pHGG and DIPG, Martina-Velez et al.98 further supported the immune stimulatory abilities of Delta-24-RGD. Intratumoral injection resulted in a significant increase in survival, accompanied by an increase in CD3+, CD4+ and CD8+ lymphocyte infiltration. They also analysed human tumour samples in silico and determined that the majority of pHGGs and DIPGs are susceptible to Delta-24-RGD infection, based on the aberrant expression of Rb pathway genes such as CCND1, CDKN2A and E2F1. These findings resulted in a study by the same authors in which they combined Delta-24-RGD with radiotherapy to study the synergistic effects in pHGG and DIPGs.98 They found combination therapy significantly extended survival in both pHGG- and DIPG-bearing mice compared with single treatments. Combination therapy resulted in the greatest infiltration of cytotoxic lymphocyte populations and increased expression of granzyme B and IFNG mRNA compared with viral monotherapy or radiotherapy alone. However, it is important to note that both monotherapies also induced an immune response. A case report on a patient with a H3.3K27M mutated DIPG treated with DNX-2401 validated the safety of administering the oncolytic virus to paediatric patients.96 The virus was injected immediately after tumour biopsy into the resection cavity using the biopsy tract and no virus-related toxicities were noted 4 weeks after injection. Unfortunately, the authors did not provide any immunological studies or survival statistics on the patient.
Multiple laboratories have studied the use of oncolytic herpes simplex virus (HSV) in vitro. Cockle et al.101 studied the efficacy of HSV1716 to inhibit cell migration and invasion of pHGG and DIPG cell lines in vitro. They found that HSV1716 significantly reduced both aspects of cell motility. In vitro time-lapse imaging demonstrated that HSV1716 treatment induced a loss of cell polarity and reduction in cell velocity compared with controls. Injection of an HSV1716-treated DIPG cell line into the fourth ventricle of NSG mice resulted in reduced infiltration of tumour cells into surrounding brain structures. However, no immune analyses were performed. Friedman et al.102 studied the oncolytic HSV G207 and HSV M002, due to their clinical significance for use in early phase I clinical trials (NCT02457845 and NCT02062827, respectively).102,103 They found that paediatric patient-derived tumour cells were more sensitive to both viruses than adult gioblastoma tumour cells in vitro. This sensitivity was accounted for by increased expression of the HSV entry molecule CD111 on the surface of the paediatric tumour cells compared with the adult tumour cells. Treatment of tumour-bearing mice resulted in an increase in survival with M002; however, these were nude mice lacking an intact immune system.102 These results suggest that CD111 expression may be a useful biomarker for predicting the efficacy of HSV therapy in the paediatric population.
The promising effects of a recombinant polio-rhinovirus chimera (PVSRIPO) vaccine in recurrent adult gioblastoma—in which eight patients survived to 24 months and five survived to 36 months—has prompted ongoing investigational use of this vaccine in recurrent malignant glioma in children (NCT03043391).104 Other viruses studied for oncolytic potential in pHGG include parvovirus and picornavirus. Because of the success of H1-parvovirus (H-1PV) in aHGG and rat glioma models, Josupeit et al. studied H-1PV in adult and paediatric HGG glioma stem cell neurosphere cultures to determine the ability of H-1PV to eradicate glioma stem cells in vitro and in vivo.105,106 Using four pHGG cell lines, including two DIPG and two paediatric gioblastoma lines, H-1PV was shown to induce cell death and reduce metabolic activity in a viral-dose dependent manner.107 Engraftment of virally-infected adult gioblastoma stem cells into NSG mice was reduced along with tumour volume compared with mock infected mice; however, again this analysis was performed in immunocompromised mice and no paediatric cell lines were investigated. Using seneca valley virus 001 (SSV-001), a member of the picornavirus family that has shown tumoricidal effects in medulloblastoma, Liu et al.108 demonstrated antitumour activity in pHGG.108,109 Freshly isolated human pHGG tumour cells from xenograft mouse models were treated with the virus in vitro (multiplicity of infection = 0.5), resulting in significant cell death in 4/6 models tested, with 2/6 resistant to SSV-001-induced cell death. Infection of glioma stem-like neurospheres was also demonstrated, indicating that SSV-001 can also target historically treatment-resistant populations. A single injection of SSV-001 into Rag2-SCID mice with orthotopically xenografted tumours significantly extended survival compared with placebo-treated mice. Analysis of sialic acids on permissive and resistant tumour cells indicated that SSV-001 infection was higher in cells with more α2,6- and α2,3-linked sialic acids, indicating SSV-001 viral therapy may have differential activity in humans and should therefore be investigated further to determine which patients will best respond to treatment. Future studies involving H-1PV and SSV-001 oncolytic tumour viruses need to be performed in immunocompetent mouse models to determine their oncolytic capacity when the innate and adaptive immune systems are intact.
An impressive study by Mendez et al.68 demonstrated the efficacy of adenoviral delivery of thymidine kinase and fms-like tyrosine kinase 3 ligand (Flt3L) gene therapy in an immunocompetent mouse model of ACVR1-mutant DIPG. This approach utilizes thymidine kinase to convert the prodrug ganciclovir into ganciclovir-triphosphate, which induces DNA damage and cell death (Fig. 7).110 Further administration of Flt3L recruits dendritic cells into the TME, allowing them to process antigens from dying tumour cells and inducing cytolytic T-cell activity.111 Using the sleeping beauty transposase system, which allows the integration of transposon DNA into the host genome through a cut-and-paste method, ACVR1 mutant or ACVR1 wild-type tumours were induced with the co-mutations short hairpin p53 and mutant NRAS in C57BL/6 mice. Primary neurospheres obtained from generated tumours were first treated in vitro with thymidine kinase and ganciclovir to demonstrate that thymidine kinase gene therapy induces the generation of damage associated molecular patterns. In response to thymidine kinase and ganciclovir treatment, the neurospheres released significant levels of ATP, calreticulin and HMGB1. To demonstrate the efficacy of thymidine kinase gene therapy in vivo, neurospheres were then injected into the pons of recipient C57BL/6 mice, and after tumour formation mice were intratumourally injected with thymidine kinase and Flt3L alone or in combination with radiotherapy. Thymidine kinase/Flt3L therapy significantly increased the median survival of mice from 18 days to 36 days compared with saline-treated animals. The addition of radiotherapy did not add an additional survival benefit to thymidine kinase/Flt3L therapy; however, it also did not reduce the efficacy of the viral therapy. Additionally, viral therapy induced a significant increase in intratumoral CD8 T-cell infiltration, while sparing deleterious neuroinflammation in normal brain tissue. The CD8 T cells had significantly greater IFNγ production, proliferation and cytolytic potential compared with CD8 T cells isolated from saline-treated animals. This study established a working immunocompetent mouse model for testing adenoviral gene therapy in DIPG. The same model could also be used for preclinical studies in other pHGG tumour types harbouring different genetic driver mutations. Moving forward, careful considerations must be made and diligent preclinical analysis done to ensure minimal neuroinflammation results after the administration of oncolytic viruses in pHGG and DIPG. Because of the unresectable nature of many DIPGs and pHGGs, therapeutic mechanisms stimulating the patient’s immune system may benefit current treatment strategies.
Figure 7.
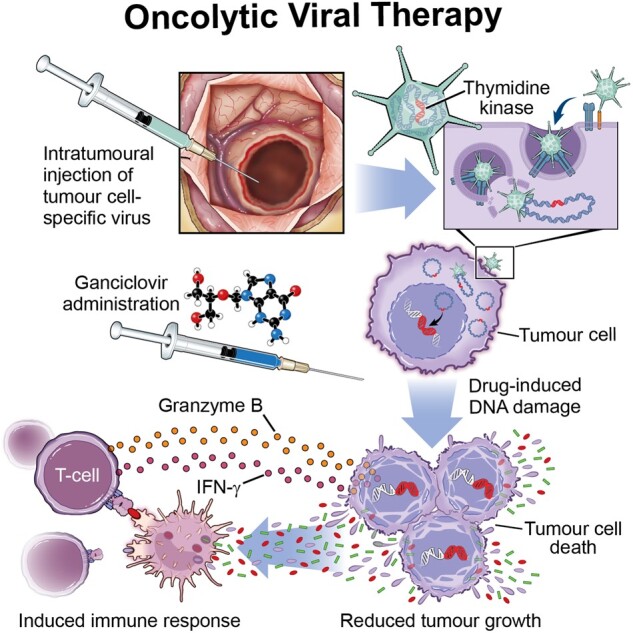
Viral therapy in pHGG. A tumour cell-specific virus is injected into the tumour bulk or resection cavity. A pro-drug such as ganciclovir is then administered and subsequently converted by the virus to a cytotoxic agent, which kills tumour cells and induces and amplifies an active immune response.
Concluding remarks
Remarkable progress in elucidating the origin and genomic landscape of childhood brain tumours has been made in the past decade. Much is known about the intrinsic cellular mechanisms driving tumour progression, yet the role of the TIME remains poorly understood. It is apparent that the TIME is a key driver of immunotherapy resistance through the orchestration of multiple complementary mechanisms blocking effector immune cell infiltration and activation. Therefore, the success of immunotherapy-based clinical trials and conventional cancer cell-targeted therapies hinges on a more mature understanding of the pHGG TIME. The utilization of pHGG GEMMs will help address these needs. Multiple groups have used these models to characterize the tumour microenvironment of pHGGs, demonstrating that they faithfully recapitulate the human disease with regard to histological hallmarks, tumour genetics and immune infiltrate.47,59,68,112,113 Because the majority of studies are carried out using patient-derived xenograft (PDX) models alone, which exhibit an immunocompromised status and species incompatibilities between immune cells and signalling molecules, there is a need for more studies investigating the use of immunotherapies in immunocompetent mouse models of pHGG.64
Despite the growing popularity of studying the TIME of pHGGs, we have yet to determine why they have fewer infiltrating immune cells than pLGGs and other tumour types. This review has demonstrated that pHGGs have low amounts of infiltrating T cells, yet they still recruit macrophages, including brain resident microglia and bone marrow-derived monocytes. Although efforts designed to efficiently target tumour cells have been studied, it is imperative to determine if microglia and infiltrating monocytes have differential functions in pHGG and how amenable they are for therapeutic use. These distinctions between microglia and bone marrow-derived macrophages are of paramount importance in light of recent advances illuminating substantial differences between the two populations in terms of their ontogeny, proliferative capacity, locations and biological functions.114–118 Determining whether the TAM lineage has functional consequences in pHGG is an important topic that needs to be addressed. Furthermore, although TAMs are genetically stable, their expression profiles are responsive to signals derived from tumour cells; therefore, cancer cell heterogeneity inevitably leads to heterogeneity within the TIME, impacting the effectiveness of therapeutic strategies. This leads to another important question; how do TAMs respond to chemotherapy, radiotherapy, and immunotherapy? Do they help accelerate cytotoxic clearing of tumour cells or do they contribute to therapeutic resistance? Answering these questions will enable us to design more effective therapeutic strategies that can easily be tested in immunocompetent mouse and PDX models of pHGG and eventually in human clinical trials.
Additional considerations should incorporate the possibility of cell-type specific, location-based differences within the brain, including microglia, myeloid cells and other cell types that are important for immune regulation. As previously discussed, tumour mutations can be specific to the location of the tumour within the brain, such as K27M mutations found in brainstem gliomas. It is now known that brain microenvironments are conducive to cultivating particular transcriptional and phenotypic profiles of the constituent cells present. For example, neuronal diversity has been documented through single cell RNA sequencing analysis, demonstrating that different subsets of neurons can be found in specific anatomic locations of the brain.119 Microglia have been demonstrated to be found in different densities, display distinct morphological features and possess unique transcriptomic profiles, depending on the brain region sampled.120–123 Myeloid cell diversity among brain compartments has also been described.124 This suggests a tumour found in the brainstem may possess a unique cellular milieu compared to a hemispheric tumour for more reasons than tumour genetics alone. We have demonstrated one example of this in our GEMMs of hemispheric pHGG and brainstem DIPG, where we found that blood–brain barrier permeability is influenced not by H3.3K27M status but rather by tumour location.125 Although these concepts are not highly novel, they have been largely ignored in brain tumour studies. Understanding how individual microenvironments are conducive for the phenotypic profiles of constituent cells will facilitate our understanding of the overall immune profile of pHGGs, therefore enhancing therapeutic discovery efforts and allowing us to better predict outcomes of immunotherapeutic strategies in clinical trials.
One example where our efforts utilizing GEMMs will lead to deeper understandings of therapeutic resistance in pHGG is by studying how and why patients with constitutional mismatch repair deficiency respond better to immunotherapy. Establishing a causal link between biallelic mismatch repair mutations and checkpoint inhibitor responses in pHGG will allow us to mechanistically dissect the underlying reasons for checkpoint inhibitor resistance in the broader pHGG population and will therefore enable us to design therapies that bypass these mechanisms. Moving forward, preclinical studies involving mouse models of pHGG will be critical to the advancement of immunotherapies as they will allow us to determine effective therapeutic combinations as well as delineate the mechanisms driving treatment resistance. Furthermore, because standard treatment protocols involve the use of immune-modulating steroids for the management of cerebral oedema, GEMMs will enable us to determine optimal timing and treatment strategies for pHGG.126,127 In conclusion, a deeper understanding of the pHGG TIME is essential to the development and progression of more efficacious therapeutic strategies for this dismal disease.
Acknowledgements
We thank Dr Adam Goldman-Yassen (Children’s Healthcare of Atlanta, Emory University) for his generous contributions of MRI images and radiological reports.
Funding
This study was funded by the National Cancer Institute F31CA232531 (J.L.R.), Cancer Research Instute (J.L.R.), and National Institutes of Health NS106554 (D.H.).
Competing interests
The authors report no competing interests.
Glossary
- CAR
chimeric antigen receptor
- DIPG
diffuse intrinsic pontine glioma
- DMG
diffuse midline glioma
- Flt3L
fms-like tyrosine kinase 3 ligand
- H/LGG
high/low-grade glioma
- NSG
nod scid gamma
- PRC2
polycomb repressive complex 2
- TAM
tumour-associated macrophage
- TIME
tumour immune microenvironment
References
- 1. Diwanji TP, Engelman A, Snider JW, Mohindra P.. Epidemiology, diagnosis, and optimal management of glioma in adolescents and young adults. Adolesc Health Med Ther. 2017;8:99–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ostrom QT, Cioffi G, Gittleman H, et al. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2012-2016. Neuro Oncol. 2019;21(Suppl 5):v1–v100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ostrom QT, de Blank PM, Kruchko C, et al. Alex's lemonade stand foundation infant and childhood primary brain and central nervous system tumors diagnosed in the United States in 2007-2011. Neuro Oncol. 2015;16(Suppl 10):x1–x36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Patil N, Kelly ME, Yeboa DN, et al. Epidemiology of brainstem high-grade gliomas in children and adolescents in the United States, 2000-2017. Neuro Oncol. 2021;23(6):990–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kyritsis AP, Bondy ML, Rao JS, Sioka C.. Inherited predisposition to glioma. Neuro Oncol. 2010;12(1):104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Braunstein S, Raleigh D, Bindra R, Mueller S, Haas-Kogan D.. Pediatric high-grade glioma: Current molecular landscape and therapeutic approaches. J Neurooncol. 2017;134(3):541–549. [DOI] [PubMed] [Google Scholar]
- 7. Guerrini-Rousseau L, Varlet P, Colas C, et al. Constitutional mismatch repair deficiency-associated brain tumors: Report from the European C4CMMRD consortium. Neurooncol Adv. 2019;1(1):vdz033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Juratli TA, Qin N, Cahill DP, Filbin MG.. Molecular pathogenesis and therapeutic implications in pediatric high-grade gliomas. Pharmacol Ther. 2018;182:70–79. [DOI] [PubMed] [Google Scholar]
- 9. Mackay A, Burford A, Carvalho D, et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell. 2017;32(4):520–537.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dorfer C, Czech T, Gojo J, et al. Infiltrative gliomas of the thalamus in children: The role of surgery in the era of H3 K27M mutant midline gliomas. Acta Neurochir (Wien). 2021;163(7):2025–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang T, Temkin N, Barber J, et al. Gross total resection correlates with long-term survival in pediatric patients with glioblastoma. World Neurosurg. 2013;79(3-4):537–544. [DOI] [PubMed] [Google Scholar]
- 12. Finlay JL, Wisoff JH.. The impact of extent of resection in the management of malignant gliomas of childhood. Childs Nerv Syst. 1999;15(11-12):786–788. [DOI] [PubMed] [Google Scholar]
- 13. Lukas RV, Wainwright DA, Ladomersky E, Sachdev S, Sonabend AM, Stupp R.. Newly diagnosed glioblastoma: A review on clinical management. Oncology (Williston Park). 2019;33(3):91–100. [PMC free article] [PubMed] [Google Scholar]
- 14. Cohen KJ, Jabado N, Grill J.. Diffuse intrinsic pontine gliomas-current management and new biologic insights. Is there a glimmer of hope? Neuro Oncol. 2017;19(8):1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aziz-Bose R, Monje M.. Diffuse intrinsic pontine glioma: Molecular landscape and emerging therapeutic targets. Curr Opin Oncol. 2019;31(6):522–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cohen KJ, Pollack IF, Zhou T, et al. Temozolomide in the treatment of high-grade gliomas in children: A report from the Children's Oncology Group. Neuro Oncol. 2011;13(3):317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. [DOI] [PubMed] [Google Scholar]
- 18. Pollack IF, Hamilton RL, Sobol RW, et al. O6-methylguanine-DNA methyltransferase expression strongly correlates with outcome in childhood malignant gliomas: Results from the CCG-945 Cohort. J Clin Oncol. 2006;24(21):3431–3437. [DOI] [PubMed] [Google Scholar]
- 19. Jakacki RI, Cohen KJ, Buxton A, et al. Phase 2 study of concurrent radiotherapy and temozolomide followed by temozolomide and lomustine in the treatment of children with high-grade glioma: A report of the Children's Oncology Group ACNS0423 study. Neuro Oncol. 2016;18(10):1442–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jones C, Karajannis MA, Jones DTW, et al. Pediatric high-grade glioma: Biologically and clinically in need of new thinking. Neuro Oncol. 2017;19(2):153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bouffet E, Larouche V, Campbell BB, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol. 2016;34(19):2206–2211. [DOI] [PubMed] [Google Scholar]
- 22. Johanns TM, Miller CA, Dorward IG, et al. Immunogenomics of hypermutated glioblastoma: A patient with germline POLE deficiency treated with checkpoint blockade immunotherapy. Cancer Discov. 2016;6(11):1230–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rong Y, Durden DL, Van Meir EG, Brat DJ.. ‘Pseudopalisading’ necrosis in glioblastoma: A familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol. 2006;65(6):529–539. [DOI] [PubMed] [Google Scholar]
- 24. Ross JL, Cooper LAD, Kong J, et al. 5-aminolevulinic acid guided sampling of glioblastoma microenvironments identifies pro-survival signaling at infiltrative margins. Sci Rep. 2017;7(1):15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koschmann C, Zamler D, MacKay A, et al. Characterizing and targeting PDGFRA alterations in pediatric high-grade glioma. Oncotarget. 2016;7(40):65696–65706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Paugh BS, Qu C, Jones C, et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol. 2010;28(18):3061–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu G, Diaz AK, Paugh BS, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014;46(5):444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Larson JD, Kasper LH, Paugh BS, et al. Histone H3.3 K27M accelerates spontaneous brainstem glioma and drives restricted changes in bivalent gene expression. Cancer Cell. 2019;35(1):140–155.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schwartzentruber J, Korshunov A, Liu XY, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482(7384):226–231. [DOI] [PubMed] [Google Scholar]
- 30. Wu G, Broniscer A, McEachron TA, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44(3):251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sturm D, Witt H, Hovestadt V, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22(4):425–437. [DOI] [PubMed] [Google Scholar]
- 32. Buczkowicz P, Hoeman C, Rakopoulos P, et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet. 2014;46(5):451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ebrahimi A, Skardelly M, Bonzheim I, et al. ATRX immunostaining predicts IDH and H3F3A status in gliomas. Acta Neuropathol Commun. 2016;4(1):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Solomon DA, Wood MD, Tihan T, et al. Diffuse midline gliomas with histone H3-K27M mutation: A series of 47 cases assessing the spectrum of morphologic variation and associated genetic alterations. Brain Pathol. 2016;26(5):569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Korshunov A, Ryzhova M, Hovestadt V, et al. Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol. 2015;129(5):669–678. [DOI] [PubMed] [Google Scholar]
- 36. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–820. [DOI] [PubMed] [Google Scholar]
- 37. Mosaab A, El-Ayadi M, Khorshed EN, et al. Histone H3K27M mutation overrides histological grading in pediatric gliomas. Sci Rep. 2020;10(1):8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pathania M, De Jay N, Maestro N, et al. H3.3(K27M) cooperates with Trp53 loss and PDGFRA gain in mouse embryonic neural progenitor cells to induce invasive high-grade gliomas. Cancer Cell. 2017;32(5):684–700.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Goldberg AD, Banaszynski LA, Noh KM, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140(5):678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jones C, Baker SJ.. Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma. Nat Rev Cancer. 2014;14(10):651–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sarthy JF, Meers MP, Janssens DH, et al. Histone deposition pathways determine the chromatin landscapes of H3.1 and H3.3 K27M oncohistones. Elife. 2020;9:e61090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lewis PW, Muller MM, Koletsky MS, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340(6134):857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bender S, Tang Y, Lindroth AM, et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell. 2013;24(5):660–672. [DOI] [PubMed] [Google Scholar]
- 44. Faria CM, Rutka JT, Smith C, Kongkham P.. Epigenetic mechanisms regulating neural development and pediatric brain tumor formation. J Neurosurg Pediatr. 2011;8(2):119–132. [DOI] [PubMed] [Google Scholar]
- 45. Margueron R, Reinberg D.. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469(7330):343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Filbin MG, Tirosh I, Hovestadt V, et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science. 2018;360(6386):331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cordero FJ, Huang Z, Grenier C, et al. Histone H3.3K27M represses p16 to accelerate gliomagenesis in a murine model of DIPG. Mol Cancer Res. 2017;15(9):1243–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu X, McEachron TA, Schwartzentruber J, Wu G.. Histone H3 mutations in pediatric brain tumors. Cold Spring Harb Perspect Biol. 2014;6(4):a018689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mackay A, Burford A, Molinari V, et al. Molecular, pathological, radiological, and immune profiling of non-brainstem pediatric high-grade glioma from the HERBY Phase II randomized trial. Cancer Cell. 2018;33(5):829–842.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang L, Li Z, Zhang M, et al. H3 K27M-mutant diffuse midline gliomas in different anatomical locations. Hum Pathol. 2018;78:89–96. [DOI] [PubMed] [Google Scholar]
- 51. Vinci M, Burford A, Molinari V, et al. Functional diversity and cooperativity between subclonal populations of pediatric glioblastoma and diffuse intrinsic pontine glioma cells. Nat Med. 2018;24(8):1204–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lieberman NAP, DeGolier K, Kovar HM, et al. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: Implications for development of immunotherapy. Neuro Oncol. 2019;21(1):83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Grabovska Y, Mackay A, O'Hare P, et al. Pediatric pan-central nervous system tumor analysis of immune-cell infiltration identifies correlates of antitumor immunity. Nat Commun. 2020;11(1):4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Guerreiro Stucklin AS, Ryall S, Fukuoka K, et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat Commun. 2019;10(1):4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kaffes I, Szulzewsky F, Chen Z, et al. Human Mesenchymal glioblastomas are characterized by an increased immune cell presence compared to Proneural and Classical tumors. Oncoimmunology. 2019;8(11):e1655360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Griesinger AM, Birks DK, Donson AM, et al. Characterization of distinct immunophenotypes across pediatric brain tumor types. J Immunol. 2013;191(9):4880–4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Robinson MH, Vasquez J, Kaushal A, et al. Subtype and grade-dependent spatial heterogeneity of T-cell infiltration in pediatric glioma. J Immunother Cancer. 2020;8(2):e001066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Plant AS, Koyama S, Sinai C, et al. Immunophenotyping of pediatric brain tumors: Correlating immune infiltrate with histology, mutational load, and survival and assessing clonal T cell response. J Neurooncol. 2018;137(2):269–278. [DOI] [PubMed] [Google Scholar]
- 59. Ross JL, Chen Z, Herting CJ, et al. Platelet-derived growth factor beta is a potent inflammatory driver in paediatric high-grade glioma. Brain. 2021;144(1):53–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bailey CP, Figueroa M, Gangadharan A, et al. Pharmacologic inhibition of lysine-specific demethylase 1 as a therapeutic and immune-sensitization strategy in pediatric high-grade glioma. Neuro Oncol. 2020;22(9):1302–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jha P, Manjunath N, Singh J, et al. Analysis of PD-L1 expression and T cell infiltration in different molecular subgroups of diffuse midline gliomas. Neuropathology. 2019;39(6):413–424. [DOI] [PubMed] [Google Scholar]
- 62. Caretti V, Sewing AC, Lagerweij T, et al. Human pontine glioma cells can induce murine tumors. Acta Neuropathol. 2014;127(6):897–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lin GL, Nagaraja S, Filbin MG, Suvà ML, Vogel H, Monje M.. Non-inflammatory tumor microenvironment of diffuse intrinsic pontine glioma. Acta Neuropathol Commun. 2018;6(1):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chen Z, Herting CJ, Ross JL, et al. Genetic driver mutations introduced in identical cell-of-origin in murine glioblastoma reveal distinct immune landscapes but similar response to checkpoint blockade. Glia. 2020;68(10):2148–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang SS, Bandopadhayay P, Jenkins MR.. Towards immunotherapy for pediatric brain tumors. Trends Immunol. 2019;40(8):748–761. [DOI] [PubMed] [Google Scholar]
- 66. Guerra-Garcia P, Marshall LV, Cockle JV, et al. Challenging the indiscriminate use of temozolomide in pediatric high-grade gliomas: A review of past, current, and emerging therapies. Pediatr Blood Cancer. 2020;67(1):e28011. [DOI] [PubMed] [Google Scholar]
- 67. Foster JB, Madsen PJ, Hegde M, et al. Immunotherapy for pediatric brain tumors: Past and present. Neuro Oncol. 2019;21(10):1226–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mendez F, Kadiyala P, Nunez FJ, et al. Therapeutic efficacy of immune stimulatory thymidine kinase and fms-like Tyrosine Kinase 3 Ligand (TK/Flt3L) gene therapy in a mouse model of high-grade brainstem glioma. Clin Cancer Res. 2020;26(15):4080–4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Patterson JD, Henson JC, Breese RO, Bielamowicz KJ, Rodriguez A.. CAR T cell therapy for pediatric brain tumors. Front Oncol. 2020;10:1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ahmed N, Brawley V, Hegde M, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: A phase 1 dose-escalation trial. JAMA Oncol. 2017;3(8):1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Majzner RG, Theruvath JL, Nellan A, et al. CAR T cells targeting B7-H3, a pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin Cancer Res. 2019;25(8):2560–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tang X, Zhao S, Zhang Y, et al. B7-H3 as a novel CAR-T therapeutic target for glioblastoma. Mol Ther Oncolytics. 2019;14:279–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mount CW, Majzner RG, Sundaresh S, et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M(+) diffuse midline gliomas. Nat Med. 2018;24(5):572–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Nazha B, Inal C, Owonikoko TK.. Disialoganglioside GD2 expression in solid tumors and role as a target for cancer therapy. Front Oncol. 2020;10:1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Paugh BS, Broniscer A, Qu C, et al. Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. J Clin Oncol. 2011;29(30):3999–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shum T, Omer B, Tashiro H, et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov. 2017;7(11):1238–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ochs K, Ott M, Bunse T, et al. K27M-mutant histone-3 as a novel target for glioma immunotherapy. Oncoimmunology. 2017;6(7):e1328340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Chheda ZS, Kohanbash G, Okada K, et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J Exp Med. 2018;215(1):141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mueller S, Taitt JM, Villanueva-Meyer JE, et al. Mass cytometry detects H3.3K27M-specific vaccine responses in diffuse midline glioma. J Clin Invest. 2020;130(12):6325–6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhu X, Fallert-Junecko BA, Fujita M, et al. Poly-ICLC promotes the infiltration of effector T cells into intracranial gliomas via induction of CXCL10 in IFN-alpha and IFN-gamma dependent manners. Cancer Immunol Immunother. 2010;59(9):1401–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Butowski N, Lamborn KR, Lee BL, et al. A North American brain tumor consortium phase II study of poly-ICLC for adult patients with recurrent anaplastic gliomas. J Neurooncol. 2009;91(2):183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Butowski N, Chang SM, Junck L, et al. A phase II clinical trial of poly-ICLC with radiation for adult patients with newly diagnosed supratentorial glioblastoma: A North American Brain Tumor Consortium (NABTC01-05). J Neurooncol. 2009;91(2):175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Salazar AM, Levy HB, Ondra S, et al. Long-term treatment of malignant gliomas with intramuscularly administered polyinosinic-polycytidylic acid stabilized with polylysine and carboxymethylcellulose: An open pilot study. Neurosurgery. 1996;38(6):1096–1103; discussion 1103–4. [PubMed] [Google Scholar]
- 84. Hartman LL, Crawford JR, Makale MT, et al. Pediatric phase II trials of poly-ICLC in the management of newly diagnosed and recurrent brain tumors. J Pediatr Hematol Oncol. 2014;36(6):451–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Pollack IF, Jakacki RI, Butterfield LH, et al. Antigen-specific immune responses and clinical outcome after vaccination with glioma-associated antigen peptides and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in children with newly diagnosed malignant brainstem and nonbrainstem gliomas. J Clin Oncol. 2014;32(19):2050–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Herz J, Filiano AJ, Smith A, Yogev N, Kipnis J.. Myeloid cells in the central nervous system. Immunity. 2017;46(6):943–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Thewissen K, Nuyts AH, Deckx N, et al. Circulating dendritic cells of multiple sclerosis patients are proinflammatory and their frequency is correlated with MS-associated genetic risk factors. Mult Scler. 2014;20(5):548–557. [DOI] [PubMed] [Google Scholar]
- 88. Caruso DA, Orme LM, Amor GM, et al. Results of a Phase I study utilizing monocyte-derived dendritic cells pulsed with tumor RNA in children with Stage 4 neuroblastoma. Cancer. 2005;103(6):1280–1291. [DOI] [PubMed] [Google Scholar]
- 89. De Vleeschouwer S, Fieuws S, Rutkowski S, et al. Postoperative adjuvant dendritic cell-based immunotherapy in patients with relapsed glioblastoma multiforme. Clin Cancer Res. 2008;14(10):3098–3104. [DOI] [PubMed] [Google Scholar]
- 90. Ardon H, De Vleeschouwer S, Van Calenbergh F, et al. Adjuvant dendritic cell-based tumour vaccination for children with malignant brain tumours. Pediatr Blood Cancer. 2010;54(4):519–525. [DOI] [PubMed] [Google Scholar]
- 91. Lasky JL 3rd, Panosyan EH, Plant A, et al. Autologous tumor lysate-pulsed dendritic cell immunotherapy for pediatric patients with newly diagnosed or recurrent high-grade gliomas. Anticancer Res. 2013;33(5):2047–2056. [PMC free article] [PubMed] [Google Scholar]
- 92. Benitez-Ribas D, Cabezon R, Florez-Grau G, et al. Immune response generated with the administration of autologous dendritic cells pulsed with an allogenic tumoral cell-lines lysate in patients with newly diagnosed diffuse intrinsic pontine glioma. Front Oncol. 2018;8:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Hilf N, Kuttruff-Coqui S, Frenzel K, et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. 2019;565(7738):240–245. [DOI] [PubMed] [Google Scholar]
- 94. Keskin DB, Anandappa AJ, Sun J, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565(7738):234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Tejada S, Alonso M, Patino A, Fueyo J, Gomez-Manzano C, Diez-Valle R.. Phase I trial of DNX-2401 for diffuse intrinsic pontine glioma newly diagnosed in pediatric patients. Neurosurgery 2018;83(5):1050–1056. [DOI] [PubMed] [Google Scholar]
- 96. Tejada S, Diez-Valle R, Dominguez PD, et al. DNX-2401, an oncolytic virus, for the treatment of newly diagnosed diffuse intrinsic pontine gliomas: A case report. Front Oncol. 2018;8:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lang FF, Conrad C, Gomez-Manzano C, et al. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: Replication and immunotherapeutic effects in recurrent malignant glioma. J Clin Oncol. 2018;36(14):1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Martinez-Velez N, Marigil M, Garcia-Moure M, et al. Delta-24-RGD combined with radiotherapy exerts a potent antitumor effect in diffuse intrinsic pontine glioma and pediatric high grade glioma models. Acta Neuropathol Commun. 2019;7(1):64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Jiang H, Clise-Dwyer K, Ruisaard KE, et al. Delta-24-RGD oncolytic adenovirus elicits anti-glioma immunity in an immunocompetent mouse model. PLoS One. 2014;9(5):e97407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Martinez-Velez N, Garcia-Moure M, Marigil M, et al. The oncolytic virus Delta-24-RGD elicits an antitumor effect in pediatric glioma and DIPG mouse models. Nat Commun. 2019;10(1):2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Cockle JV, Bruning-Richardson A, Scott KJ, et al. Oncolytic herpes simplex virus inhibits pediatric brain tumor migration and invasion. Mol Ther Oncolytics 2017;5:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Friedman GK, Bernstock JD, Chen D, et al. Enhanced sensitivity of patient-derived pediatric high-grade brain tumor xenografts to oncolytic HSV-1 virotherapy correlates with nectin-1 expression. Sci Rep. 2018;8(1):13930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Bernstock JD, Bag AK, Fiveash J, et al. Design and rationale for first-in-human phase 1 immunovirotherapy clinical trial of oncolytic HSV G207 to treat malignant pediatric cerebellar brain tumors. Hum Gene Ther 2020;31(19-20):1132–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Desjardins A, Gromeier M, Herndon JE 2nd, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med. 2018;379(2):150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Geletneky K, Kiprianova I, Ayache A, et al. Regression of advanced rat and human gliomas by local or systemic treatment with oncolytic parvovirus H-1 in rat models. Neuro Oncol. 2010;12(8):804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kiprianova I, Thomas N, Ayache A, et al. Regression of glioma in rat models by intranasal application of parvovirus h-1. Clin Cancer Res. 2011;17(16):5333–5342. [DOI] [PubMed] [Google Scholar]
- 107. Josupeit R, Bender S, Kern S, et al. Pediatric and adult high-grade glioma stem cell culture models are permissive to lytic infection with parvovirus H-1. Viruses. 2016;8(5):138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Liu Z, Zhao X, Mao H, et al. Intravenous injection of oncolytic picornavirus SVV-001 prolongs animal survival in a panel of primary tumor-based orthotopic xenograft mouse models of pediatric glioma. Neuro Oncol. 2013;15(9):1173–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Yu L, Baxter PA, Zhao X, et al. A single intravenous injection of oncolytic picornavirus SVV-001 eliminates medulloblastomas in primary tumor-based orthotopic xenograft mouse models. Neuro Oncol. 2011;13(1):14–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Fischer U, Steffens S, Frank S, Rainov NG, Schulze-Osthoff K, Kramm CM.. Mechanisms of thymidine kinase/ganciclovir and cytosine deaminase/5-fluorocytosine suicide gene therapy-induced cell death in glioma cells. Oncogene. 2005;24(7):1231–1243. [DOI] [PubMed] [Google Scholar]
- 111. Curtin JF, King GD, Barcia C, et al. Fms-like tyrosine kinase 3 ligand recruits plasmacytoid dendritic cells to the brain. J Immunol. 2006;176(6):3566–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Garcia-Fabiani MB, Comba A, Kadiyala P, et al. Isolation and characterization of immune cells from the tumor microenvironment of genetically engineered pediatric high-grade glioma models using the sleeping beauty transposon system. Methods Enzymol. 2020;632:369–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Hoeman CM, Cordero FJ, Hu G, et al. ACVR1 R206H cooperates with H3.1K27M in promoting diffuse intrinsic pontine glioma pathogenesis. Nat Commun. 2019;10(1):1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Chen Z, Feng X, Herting CJ, et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res. 2017;77(9):2266–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Herting CJ, Chen Z, Maximov V, et al. Tumour-associated macrophage-derived interleukin-1 mediates glioblastoma-associated cerebral oedema. Brain. 2019;142(12):3834–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Yona S, Kim KW, Wolf Y, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38(1):79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Shemer A, Jung S.. Differential roles of resident microglia and infiltrating monocytes in murine CNS autoimmunity. Semin Immunopathol. 2015;37(6):613–623. [DOI] [PubMed] [Google Scholar]
- 118. Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Tasic B, Menon V, Nguyen TN, et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat Neurosci. 2016;19(2):335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Silvin A, Ginhoux F.. Microglia heterogeneity along a spatio-temporal axis: More questions than answers. Glia. 2018;66(10):2045–2057. [DOI] [PubMed] [Google Scholar]
- 121. de Haas AH, Boddeke HW, Biber K.. Region-specific expression of immunoregulatory proteins on microglia in the healthy CNS. Glia. 2008;56(8):888–894. [DOI] [PubMed] [Google Scholar]
- 122. Doorn KJ, Breve JJ, Drukarch B, et al. Brain region-specific gene expression profiles in freshly isolated rat microglia. Front Cell Neurosci. 2015;9:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Grabert K, Michoel T, Karavolos MH, et al. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci. 2016;19(3):504–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Korin B, Ben-Shaanan TL, Schiller M, et al. High-dimensional, single-cell characterization of the brain's immune compartment. Nat Neurosci. 2017;20(9):1300–1309. [DOI] [PubMed] [Google Scholar]
- 125. Subashi E, Cordero FJ, Halvorson KG, et al. Tumor location, but not H3.3K27M, significantly influences the blood-brain-barrier permeability in a genetic mouse model of pediatric high-grade glioma. J Neurooncol. 2016;126(2):243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Vanan MI, Eisenstat DD.. Management of high-grade gliomas in the pediatric patient: Past, present, and future. Neurooncol Pract. 2014;1(4):145–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Brown NF, Carter TJ, Ottaviani D, Mulholland P.. Harnessing the immune system in glioblastoma. Br J Cancer. 2018;119(10):1171–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]