

Abstract
Both mechanical and biochemical stimulation are required for maintaining the integrity of articular cartilage. However, chondrocytes respond differently to mechanical stimuli in osteoarthritic cartilage when biochemical signaling pathways, such as Insulin-like Growth Factor-1 (IGF-1), are altered. The Transient Receptor Potential Vanilloid 4 (TRPV4) channel is central to chondrocyte mechanotransduction and regulation of cartilage homeostasis. Here, we propose that changes in IGF-1 can modulate TRPV4 channel activity. We demonstrate that physiologic levels of IGF-1 suppress hypotonic-induced TRPV4 currents and intracellular calcium flux by increasing apparent cell stiffness that correlates with actin stress fiber formation. Disruption of F-actin following IGF-1 treatment results in the return of the intracellular calcium response to hypotonic swelling. Using point mutations of the TRPV4 channel at the microtubule-associated protein 7 (MAP-7) site shows that regulation of TRPV4 by actin is mediated via the interaction of actin with the MAP-7 domain of TRPV4. We further highlight that ATP release, a down-stream response to mechanical stimulation in chondrocytes, is mediated by TRPV4 during hypotonic challenge. This response is significantly abrogated with IGF-1 treatment. As chondrocyte mechanosensitivity is greatly altered during osteoarthritis progression, IGF-1 presents as a promising candidate for prevention and treatment of articular cartilage damage.
Keywords: Chondrocytes, Mechanosensitivity, IGF-1, TRPV4, Actin, Hypotonic Swelling
Graphical Abstract
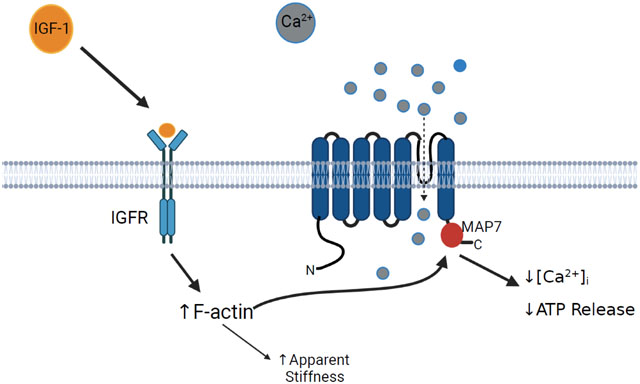
1. INTRODUCTION
Articular cartilage homeostasis requires a continuous balance between repair and degradation [3,4]. Loss of this remodeling balance can lead to degradation of the articular cartilage resulting in osteoarthritis (OA), the leading cause of disability among American adults [1,2]. Chondrocytes embedded within the extracellular matrix (ECM) of cartilage contribute to this remodeling process by responding to biomechanical and biochemical signals to synthesize or breakdown the ECM [5].
Mechanical forces experienced by chondrocytes can initiate both catabolic and anabolic activity [6,7] depending on the magnitude and type of stimuli [8,9]. These actions are mediated, in part, by signaling pathways that include growth factors and receptors [10,11], mitogen-activated protein (MAP) kinases [12], Rho guanosine-5’-triphosphate (GTP)ases [13], nitric oxide [9,14], integrins [15], and ion channels [16]. Among these biochemical signals, insulin-like growth factor-1 (IGF-1) is unusual in that it stimulates both mitogenic and anabolic functions while inhibiting catabolic activity in articular chondrocytes [17–20]. Mechanical forces and IGF-1signaling interact to regulate articular chondrocytes [21–23], however the mechanisms responsible for this interaction between the two remain unclear.
The mechanosensitive Transient Receptor Potential Vanilloid 4 (TRPV4) ion channel is integral in chondrocyte mechanotransduction through its regulation of Ca2+ influx. TRPV4-mediated Ca2+ influx regulates SOX9 expression [24], a key transcription factor involved in cartilage matrix synthesis. TRPV4 mediates the cellular response to changes in osmolarity [25–27] and mechanical stimulation [28–31], both of which are integral in cartilage integrity. The importance of the TRPV4 channel to cartilage homeostasis is illustrated by targeted deletion of TRPV4 in mice that results in severe joint damage resembling osteoarthritis and a loss of sensitivity to osmotic swelling [32]. However, the mechanism through which biochemical signaling and biomechanical stimuli influence TRPV4 gating remains unknown.
Chondrocyte stiffness has been shown to be significantly greater in chondrocytes isolated from OA cartilage than those isolated from normal articular cartilage [33]. However, it remains unclear what causes the changes in cell stiffness and how such changes influence cellular responses to mechanical stimuli. We have reported that osteoblasts subjected to fluid shear stress (FSS) increase the polymerization of F-actin through activation of RhoA GTPase that, in turn, increases the stiffness of the cell [34]. Previous studies suggest IGF-1 also increases cell stiffness and F-actin formation in chondrocytes [35] and this increase in F-actin by IGF-1 results from activation of a Rho GTPase [18]. We have also shown that actin organization regulates the activity of the mechanosensitive channels in osteoblasts [36]. Thus, IGF-1 may regulate TRPV4 activity in chondrocytes via reorganization of the actin cytoskeleton.
Suzuki et al. suggest that the TRPV4 channel is regulated through binding of F-actin to the Microtubule Associated Protein Binding 7 (MAP7: amino acids 798-809) domain of TRPV4 [37]. We previously reported that two point mutations within this domain (P799L and G800D) results in metatropic dysplasia, a severe skeletal dysplasia, in children [39]. These mutations cause a gain of function of the TRPV4 channel which alters chondrogenesis and chondrocyte intracellular calcium influx in human chondrocytes. This effect may be influenced by altering the regulation of the TRPV4 channel by the actin cytoskeleton. By leveraging these novel mutations, we highlight the role of the MAP7 binding in regulation of the TRPV4 channel during IGF-1 treatment.
2. Material and Methods
2.1. Clinical Information
Patients were recruited from the Skeletal Dysplasia Program at Alfred I. duPont Hospital for Children (AIDHC) in Wilmington, Delaware, under a Nemours IRB-approved protocol. Metatropic dysplasia was diagnosed based on clinical evaluation and radiographic information.
2.2. Culture of ATDC5 and HEK293 cells
ATDC5 mouse chondrocyte-like cells (Sigma-Aldrich, St. Louis, MO) were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Sigma), 10% FBS (Gibco, New York, NY), 100 U/ml penicillin G, and 100 μg/ml streptomycin (P/S, Mediatech, Manassas, VA). HEK 293 cells that were un-transfected (UT) or transfected with either wild-type TRPV4 (WT), a proline to leucine mutation at the 799 amino acid of TRPV4 (P799L), or a glycine to aspartate mutation at the 800 amino acid of TRPV4 (G800D) were grown in Minimal Essential Media (MEM, Corning) supplemented with 10% FBS and 1% P/S. Cells were maintained in a humidified incubator at 37°C with 5% CO2/95% air and passaged once cells reached 75% confluence. Before all experiments, cells were serum starved in reduced serum media (0.2% FBS, 1% P/S).
2.3. Electrophysiological recordings
ATDC5 cells were cultured in DMEM with 10% FBS and 100 U/ml penicillin G, and 100 μg/ml streptomycin on 15mm round coverglass (Ted Pella, Inc., Redding, CA). Pipettes were manufactured by pulling 100 μL capillary tubes (VWR, Arlington Heights, IL) to a tip diameter of ~0.5 micron (tip resistance = 1–5 MΩ) using a dual-stage pipette puller (Narishige, East Meadow, NY). Pipettes were flame polished and coated with wax to reduce capacitance artifacts. Cells were bathed in an extracellular-like solution (ECF) composed of (in mM): NaCl 150, CsCl 6, MgCl2 1, CaCl2 5, HEPES 10 and glucose 10, titrated to pH 7.4 with CsOH (total osmolarity 320mOsm). Pipettes were filled with an intracellular-like solution (ICF) containing (in mM): CsCl 20, CsAspartate 100, MgCl2 1, HEPES 10, Mg2ATP 4, CaCl2 0.08 and BAPTA 10, titrated to pH 7.4 with CsOH. Cesium was used in both the bath and pipette solutions to prevent activation of potassium currents [38]. Whole cell recordings were performed at room temperature with an Axopatch 200B amplifier (Axon Instruments, USA) equipped with a Digidata 1332A A/D converter. Recordings were made using a ramp protocol that clamps the cell at 0 mV, then ramps the membrane voltage from −80mV to +80 mV over 1000 ms. The cell then is returned to 0 mV for 12.5s prior to restarting the protocol. Data were collected on an IBM compatible PC using pClamp version 10.0 software (Axon Instruments).
To determine the effect of hypotonic swelling (HS) on TRPV4 whole cell currents, the ECF was diluted to approximately 110 mOsm (75% change in osmolarity), perfused into the patch chamber and changes in whole cell current recorded using the same ramp protocol as described above. To determine if the TRPV4 channel was responsible for the currents induced by hypotonic swelling, cells were pre-incubated with 10μM RN-1734 (Sigma), a specific inhibitor for TRPV4 channels, for 15 minutes prior to swelling. To determine the effects of IGF-1 on TRPV4 channel activity, the cell medium was replaced with reduced serum media supplemented with 300ng/mL IGF-1 (human recombinant, Sigma) for 3 hours. Electrophysiology measurements were then performed to determine if IGF-1 pretreatment altered TRPV4 currents induced by hypotonic swelling. All experiments were performed at room temperature to avoid the influence of TRPV4 activation at physiological temperatures.
2.4. Mechanical Stimuli and Pharmacological Treatment for Actin Immunofluorescence
ATDC5 cells were subjected to hypotonic swelling (HS) by adding equal volume of water. Cells were pre-treated with 300 ng/ml IGF-1 for 3 hours prior to HS or pre-treated for 10 min with the general TRPV channel inhibitor, ruthenium red (RR, 10 μM) or with the TRPV4-specific inhibitor, RN-1734. The concentrations of IGF-1, ruthenium red, and RN-1734 were maintained during HS. To determine the role of cytoskeleton organization on mechanosensitivity following IGF-1 treatment, cells were treated with 1 μM cytochalasin D for 30 minutes prior to HS in both IGF-1 treated and un-treated control cells. ATDC5 cells were washed with PBS for 5 minutes and then fixed in 4% paraformaldehyde (Electron Microscopy Sciences, PA, USA) in PBS containing 0.1% Triton X-100 (Sigma) for 30 min on ice. Cells were then washed with PBS and incubated with blocking buffer containing 3% BSA (Sigma) at room temperature for 1 hr, and then incubated with Alexa Fluor® 488 phalloidin (1:1000, Life Technologies, Grand Island, NY), mounted using the SlowFade® Antifade kit (Life Technologies) and imaged on a Zeiss LSM710 laser scanning confocal microscope with a 63X oil immersion lens at the midplane of the cells.
2.5. Atomic Force Microscopy
The apparent stiffness of individual cells was measured using an Atomic Force Microscope (AFM: BioScope II, Veeco Inc., Plainview, NY) mounted on an inverted optical microscope. Soft microlever probes (MLCT-AUNM, Veeco Inc.) with a conical tip having a spring constant of 0.01 N/m were first calibrated using a thermal fluctuation method in fluid. An area of 30 μm × 30 μm was first scanned at a speed of ~3 μm/sec to generate a topographic map of an individual cell. Seven to ten points were then selected over the cell body at the nuclear and peri-nuclear regions for indentation measurements. Cell membranes were indented at a speed of ~2.5 μm/sec until a force of 100 pN was reached, which typically resulted in an indentation depth of < 70 nm (10% of the cell height). The elastic modulus at each point was estimated from the recorded force-deflection curve using a Hertz based model as defined by the following:
| (Eq.1) |
where E is the apparent stiffness, F is the cantilever force measured by the AFM, υ is the Poisson ratio of the cytoplasm (υ = 0.4) [40], φ is the opening angle of the conical cantilever tip (φ = 35°). The indentation depth (δ) was calculated by subtracting the cantilever deflection from the piezo displacement of the probe. The apparent stiffness for a given cell was defined by the average elastic modulus across each point measured. A total of 5 to 6 cells were measured following a given treatment, and each experiment was then repeated 3 times. The average cell stiffness for each treatment was compared to static controls and reported as a fold increase. Static controls were not subjected to any form of treatment or mechanical stimuli.
2.6. Calcium Imaging
ATDC5 cells and HEK cells were washed with Hanks’ balanced saline solution (HBSS, Sigma) before being loaded with fluorescent Ca2+ indicators (3 μM of fura-2 AM or 5.69 μM of Fluo-4 AM, Life Technologies) in HBSS for 30 minutes at 37°C. The cells were rinsed with HBSS and incubated at room temperature for 15 minutes in HBSS. Using fura-2AM, changes in [Ca2+]i were recorded using a ratiometric video-image analysis apparatus (Intracellular Imaging, Cincinnati, OH, USA) on a Nikon inverted microscope with a Nikon 30× fluor objective. The ratio of emitted light at 340 and 380 nm excitation was determined from consecutive frames and used to determine the [Ca2+]i of selected cells based on previous calibration of known standards. When loading cells with Fluo-4 AM, fluorescence was measured every 500ms on a Zeiss 5 Live DUO Highspeed Confocal with a 10×/0.3A water immersion objective. For all experiments, a baseline of [Ca2+]i or relative fluorescence were first recorded for 1 minute, after which HS was applied. The influence of IGF-1 on the [Ca2+]i response was determined by treating either ATDC5 or HEK WT and TRPV4 mutant cells with 300 ng/ml of IGF-1 for 3 hours prior to loading with fura-2AM or Fluo-4AM. To determine how different doses of IGF-1 influence ATDC5s 0, 1, 100, and 300 ng/ml were added to the cells for 3 hr before challenging with HS. Time course studies of 300 ng/ml IGF-1 treatment on ATDC5 cells were completed with pretreatment times of 0, 30, 60, 120, and 180 minutes prior to loading with fura-2 AM. For a subset of experiments, the cytoskeleton was disrupted by treating ATDC5 cells with 5μM of cytochalasin D for 30 minutes prior to HS. Imaging was performed at room temperature to avoid the influence of physiological activation of TRPV4.
2.7. ATP Assay
Purinergic signaling in response to mechanical loading has a large influence on the metabolism of chondrocytes. The release of ATP was measured from media samples extracted 5 minutes after the onset of HS to chondrocytes. The ATP concentration of each sample was measured using the ATP Bioluminescence Assay kit HTS II (Roche). Emitted light was measured using a 96 well micro-injector plate reader (POLARstar OPTIMA, BMG LABTECH GmBH). To normalize the measured ATP release to cell protein, the ATDC5 cells were washed with PBS immediately after HS, and then incubated at −20°C with lysis buffer. The protein samples were stored at −80°C for further analysis. The protein concentration of each sample was determined using a BCA assay (BCA Protein Assay Kit, Pierce, Rockford, IL) and the 96-well micro-injector plate reader.
2.8. Generation and Validation of HEK293 Cells Expressing WT and Mutant TRPV4
2.8.1. Total RNA Preparation
Human fibroblast cells (from individuals known to express WT, P799L, and G800D variants) were grown to approximately 50% confluence and RNA was isolated using TRIzol Reagent (Invitrogen) following the manufacturer’s instructions.
2.8.2. RT-PCR
ProtoScript II Reverse Transcriptase and the First Strand cDNA Synthesis Kit (NEB) were used to make cDNA per the manufacturer’s instructions. Each reaction used 0.5 μg of total RNA, 500 μg oligo(dT) (Agilent) and 0.5 μl RNaseBlock (Agilent). The TRPV4 coding sequence was amplified by PCR using the following gene specific primers: forward primer 5’-CAAAGGAGGTACCCACCATGGCGGATTCCAGCGA-3’, reverse primer 5’-AGGAGGAGACAACTTCTAGATCAGAGCGGGGCGTCATCAGT-3’. The underlined sequences are found in the TRPV4 CDS. PCR was performed using Q5® High-Fidelity DNA Polymerase (NEB) with GC enhancer, per the manufacturer’s instructions. The reaction was amplified for 35 cycles using the following settings: denaturation −98°C for 10 s, annealing −65.7°C for 30 s, extension −72°C for 120 s. The PCR product was then purified using the SV gel and PCR Cleanup Kit (Promega).
2.8.3. Gibson cloning:
Gibson Assembly reactions were performed using the Gibson Assembly Cloning Kit (NEB). ~ 75 fMol of insert was combined with ~25 fMol of vector and incubated for 15 minutes at 50°C. Then 5-alpha Competent E. coli (High Efficiency) (NEB) were transformed, per the manufacturer’s instructions. After outgrowth in SOC, cells were plated onto LB agar plates containing Kanamycin. The plates were incubated overnight, and colonies were selected for further analysis.
2.8.4. Vector preparation:
One μg of the vector pSF-CMV-EMCV-NEO was digested in a 20 μl reaction with 1 μl each of XbaI and NcoI (FastDigest enzymes, Fermentas) in 1X FastDigest Green Buffer (Fermentas). The reactions were electrophoresed on a 0.8% agarose gel using TAE buffer. The bands that corresponded to the double digested vector were gel purified using the SV gel and PCR Cleanup Kit (Promega).
2.8.5. Plasmid Preparation
Individual E. coli colonies were selected and added to LB broth and shaken vigorously at 37°C overnight. Then 1000 μL of each sample was centrifuged for 1 minute at RT. The precipitate was resuspended in ice-cold GTE buffer (50 mM glucose, 25 mM Tris, 10 mM EDTA). RNAse A was added to each sample to a final concentration of 100 μg/mL. Next, SDS/NAOH lysis solution (1% SDS and 0.2 M NaOH) was added, and the samples were rapidly inverted. The suspension was chilled for 5 minutes. Each tube then received ice-cold Potassium Acetate/Acetic acid neutralizing solution. Tubes were centrifuged for 4 minutes, and an equal volume of chloroform was added to each solution to make a 1:1 final concentration. Next, isopropanol was added to each sample and the tubes were incubated at room temperature for 2 minutes. Tubes were centrifuged for five minutes (4°C at 10,000g), the precipitate was then washed with 95% ethanol and the samples were centrifuged again. Finally, each pellet was suspended in TE buffer (10 mM Tris and 1 mM EDTA).
2.8.6. Diagnostic Digestion
Identification of mutant and wild type TRVP4 expression was accomplished by digestion with restriction enzyme SmaI (NEB). Digestion with SmaI was accomplished according to the manufacturer’s instructions.
2.8.7. Transfection
HEK 293 cells were seeded at 8,000 –10,000 cells/cm2 on type 1 collagen (10 μg/cm2, Sigma-Aldrich) coated 35 mm polystyrene dishes (Falcon) and were cultured overnight in MEM (Corning) with 10% FBS and 1% P/S. Transfection was accomplished using unsupplemented MEM media and Lipofectin (Life Technologies), per the manufacturer’s instructions. Cells were incubated for 24 hr at 37°C in 5% CO2/95% air. Following this incubation, the transfection mixture was replaced with growth media and cells were grown for an additional 48 hr before being replaced with growth media supplemented with 200 μg/ml G418 sulfate (Gibco). Cells were cultured for an additional 48 hours before being plated as single cells in a 96-well plate (Corning). Colonies were cultured for 4 days before transferring to T25 flasks (Corning) and grown until 70% confluent.
2.8.8. DNA Sequencing
Samples and primers were prepared and sent to the University of Delaware Sequencing & Genotyping Center for BigDye sequencing (LifeTechnologies). Each of the samples were provided at a concentration of 50 μg/μL. Six samples were supplied for testing and three sets of primers were prepared. The first primer set, forward: 5’ - CCT ACC ATC CAC TCG ACA CA – 3’, reverse: 5’ - CGC TTT GCT TTT GGC ATA TC – 3’. The second primer set, forward: 5’ - CCC CAT CCT CAA AGT CTT CA – 3’, reverse: 5’ - GCA TGT TGA GGA GCA GCA C – 3’. The third primer set, forward: 5’ - GGG GGC TAC TTC TAC TTT GG – 3’, reverse: 5’ - CAG GTA GAG GGC TGC TGA GA – 3’.
2.9. Statistical Analysis
The change in cell stiffness and ATP release was reported as the average ± SD fold increase relative to static controls unless otherwise stated. Significance between groups was determined using one-way or two-way analysis of variance (ANOVA) with a Tukey posthoc test to determine significance when multiple comparisons in the study were made. Significance was defined by a p-value < 0.05. Data for a given outcome measurement was pooled together in the case that no significant difference was found for a given treatment between different passages of cells.
3. Results
3.1. TRPV4 channel activity is suppressed in the presence of IGF-1
To determine the effects of IGF-1 on TRPV4 channels in ATDC5 cells, changes in whole-cell currents were determined when the patch clamp voltage was ramped from −80 mV to +80 mV (Fig. 1A). Because experiments were conducted at room temperature, the temperature-sensitive TRPV4 channels exhibited very little current in response to the ramp protocol in control cells. However, when the cells were subjected to hypotonic swelling (HS), currents were increased. Pretreatment of the cells with 300ng/ml IGF-1 for 3 hours prior to the patch, completely inhibited the HS effects on whole cell currents. Conductance of the TRPV4 channel was calculated from the slope of the current/voltage (I/V) relationships under static conditions, HS or pretreatment with either IGF-1 or the TRPV4 inhibitor, RN1734. The average conductance changes of the channel in response to these treatments are shown in Fig. 1B. Hypotonic swelling increased the conductance of the TRPV4 channel almost 10-foldover that of static control cells (HS vs. static control: 3.65 nS ± 1.32 vs. 0.36 nS ± 0.06, ±SEM). Both IGF-1 and RN1734 completely blocked the increase in conductance in response to HS (0.36 nS ± 0.14 and 0.21 ± 0.17, respectively. These data indicate that the swelling activated currents are mediated by activation of the TRPV4 channel and that IGF-1 significantly reduced this response of the channel to mechanical stimulation.
Figure 1: TRPV4 channel activity is altered in the presence of IGF-1.
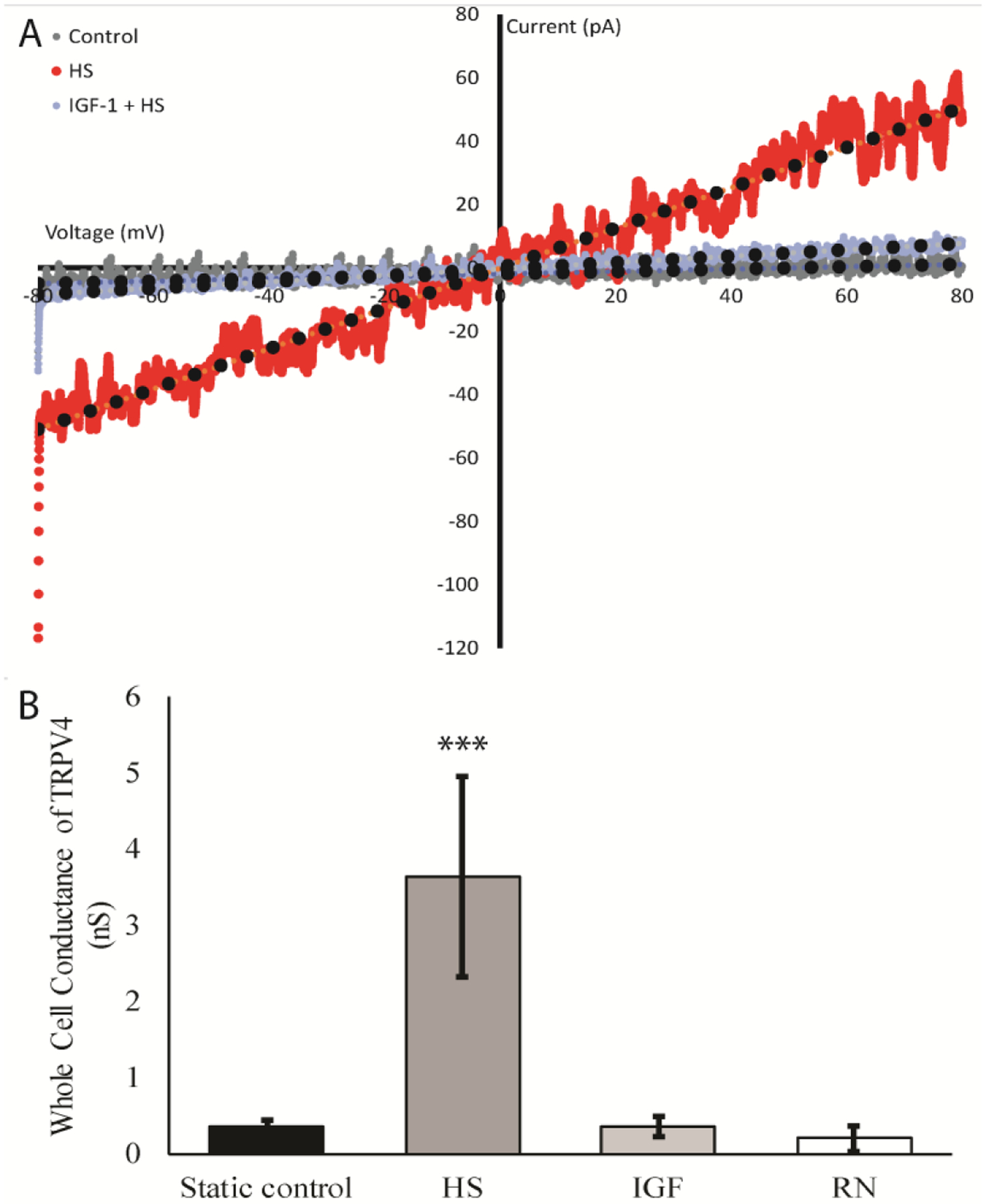
A) Representative IV curve ATDC5 cells during a voltage ramp conducted from −80mV to +80mV that were challenged with hypotonic swelling (HS, red) after treatment with the IGF-1 (300ng/mL) for 3 hours (blue), or static controls (grey). Current was greatly reduced when cells were pretreated with IGF-1. B) Mean whole cell conductance of TRPV4 in nS is shown during a voltage ramp from −80mV to +80mV. Cells were static controls (black), untreated cells (grey), or cells pretreated with IGF-1 (light grey) or the TRPV4 antagonist, RN1734 (10μM, white) for 15 minutes, before challenging with HS. IGF-1 treatment and inhibition of TRPV4 completely abolished the conductance in cells through TRPV4 when compared to cells challenged with HS alone. (Tukey-Kramer; *** indicates p-value is < 0.001 in comparison to all other conditions, error bars = ±SEM).
3.2. Actin Stress Fiber Formation and Cell Stiffness
The cytoskeleton is known to contribute to the mechanical behavior of chondrocytes, specifically cell stiffness [35], which can greatly influence mechanotransduction. IGF-1 increased actin stress fiber formation (ASFF) in ATDC5 cells as early as 2 hours after treatment (Fig. 2A). In addition, IGF-1 changed ATDC5 cell morphology to a flattened, elongated shape with stellate processes. ASFF appeared to peak after 3 hours of treatment with IGF-1 and was associated with ~ 4-fold (12 kPa) increase in cell stiffness compared to static untreated controls (3.5 kPa) (Fig. 2B). The increase in cell stiffness due to IGF-1 was dependent on F-actin based on the decrease in cell stiffness following disruption of the actin cytoskeleton with cytochalasin D treatment (Fig. 2B).
Figure 2: IGF-1 increases actin stress fiber formation and cell stiffness among ATDC5 cells.
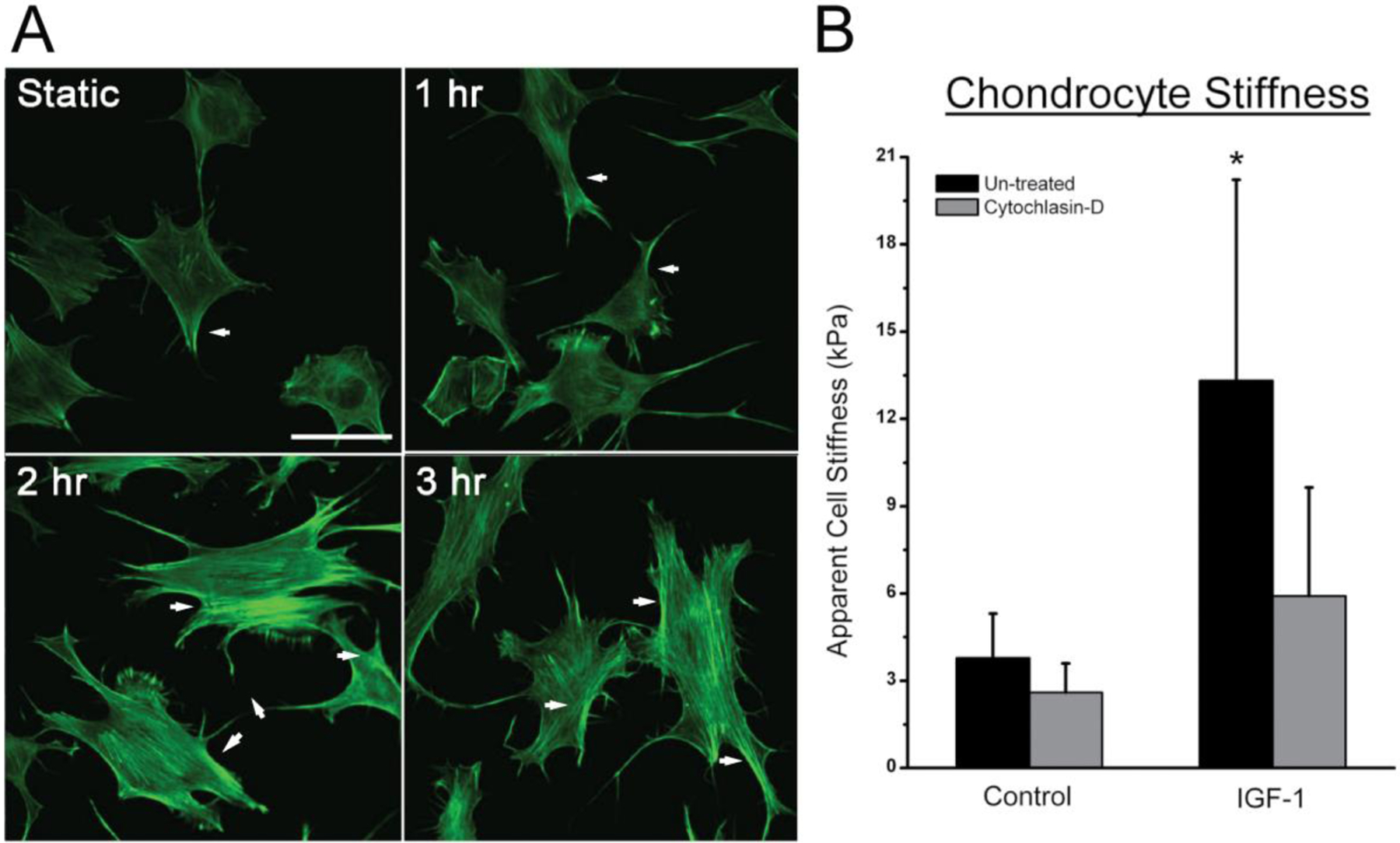
A) Time course of IGF-1 treatment indicates an increased stress fiber formation (white arrows) and cell spreading after 1 hour, with the peak amount occurring after 3 hours of treatment. Bar indicates 50 μm. B) Apparent cell stiffness of ATDC5 cells was significantly higher after 3 hours of treatment with IGF-1. The increased stiffness due to IGF-1 was dependent on F-actin organization based on the decrease in cell stiffness following cytochalasin D. (* indicates p-value is < 0.05 compared to controls)
3.3. TRPV4 Channel Mediated Calcium Response
One of the earliest responses to mechanical loading in chondrocytes is an influx of calcium into the cell that, in turn, initiates signaling cascades through a wide range of different intracellular pathways [65]. When subjecting ATDC5 cells to HS, we a found a significant increase in the number of cells exhibiting a calcium response, defined as a 50% increase in [Ca2+]i above baseline, as well as a significant increase in the magnitude of the [Ca2+]i response (Fig. 3). To determine if TRPV channels mediate the [Ca2+]i response to HS, ATDC5 cells were pre-treated with the general TRPV inhibitor, ruthenium red (RR). This non-specific inhibition of TRPV channels significantly decreased both the number of cells that responded to HS and the magnitude of the [Ca2+]i response to HS (Fig 3). Addition of RN1734 attenuated the number of cells responding and magnitude of [Ca2+]i response to HS to the same degree as ruthenium red.
Figure 3: The [Ca2+]i response to HS is mediated through TRPV4 channel.
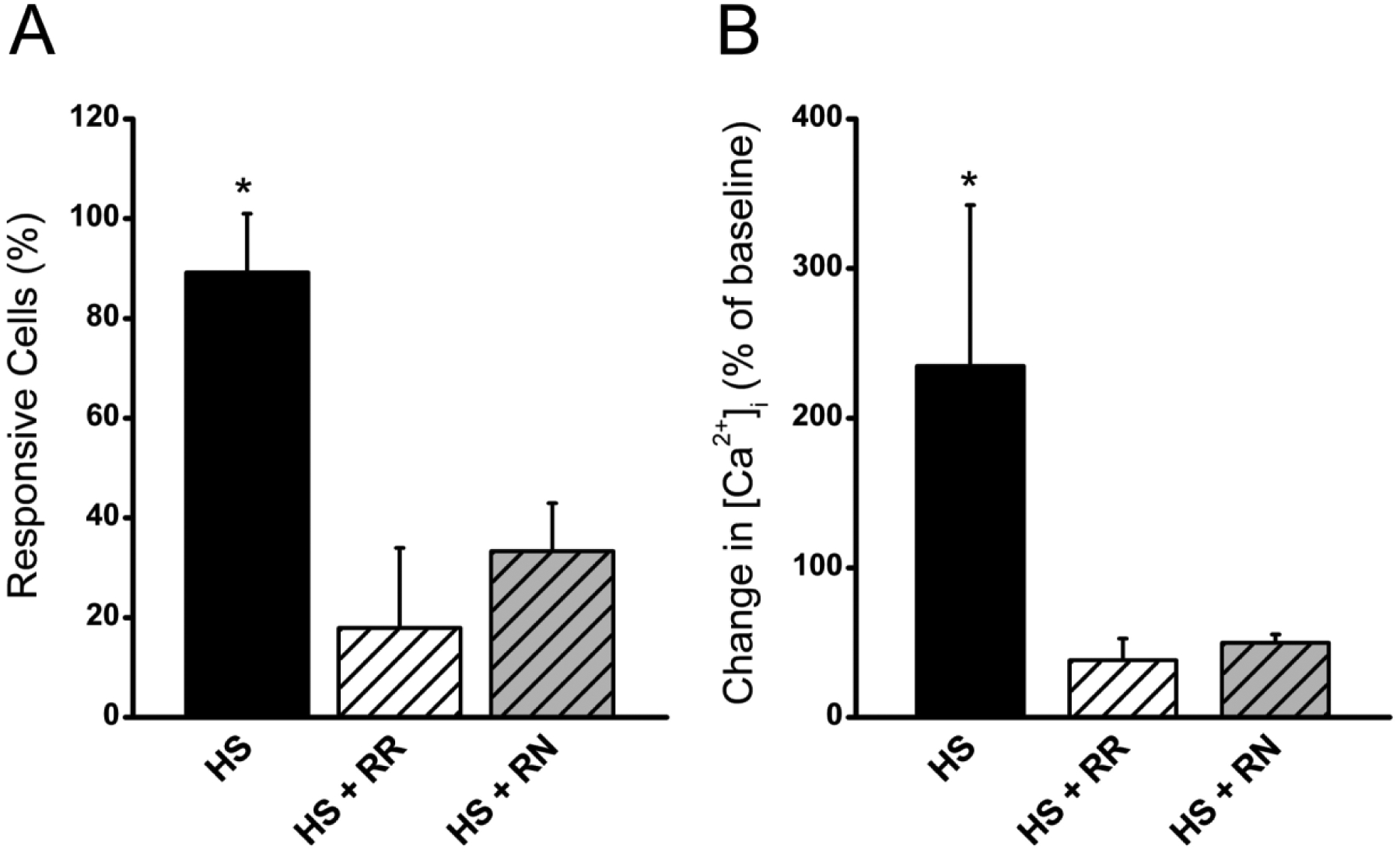
A) The number of cells responding and B) the change in [Ca2+]i as a percentage of baseline when ATDC5 cells were treated with 10 μM ruthenium red prior to and during hypotonic swelling (HS + RR) in order to inhibit all TRPV channels. Similar results were observed when only TRPV4 was inhibited by treating ATDC5 cells with 10 μM RN1734 prior to and during hypotonic swelling (HS + RN). (* indicates p-value < 0.05 compared to controls, error bars = ± SEM)
3.4. IGF-1 alters mechanosensitivity through actin cytoskeletal polymerization via actin’s interaction with TRPV4 at the MAP7 Binding Domain
Increases in cell stiffness induced by actin cytoskeletal polymerization exert a large influence on the sensitivity of cells to mechanical stimuli [34,41]. To determine if IGF-1 down-regulates channel activity during mechanical loading (Fig 1) by increasing ASFF (Fig 2), ATDC5 cells were pretreated with 300 ng/ml of IGF-1 for 3 hrs prior to application of mechanical stimulation with HS (Fig. 4A and B). IGF-1 pretreatment significantly reduced both the number of cells responding to HS and the magnitude of the [Ca2+]i response to this stimulation. Addition of 1 μM of cytochalasin D (Cyto-D) five minutes before HS stimulation, reversed the IGF-1-induced loss in mechanosensitivity, increasing both the number of responding cells and the magnitude of the [Ca2+]i response when compared to chondrocytes treated with IGF-1 alone. To determine the optimal dose of IGF-1 that suppresses TRPV4 activity, ATDC5 cells were treated with either 0, 1, 10, 100, or 300 ng/ml of IGF-1 for 3 hours prior to mechanical stimulation with HS. IGF-1 at a concentration of 100 ng/ml and above significantly reduced the magnitude of the [Ca2+]i response (Fig 4C). To determine the optimal time of IGF-1 treatment, a time course of 0, 30, 60, 120 and 180 min was used when cells were treated with 300 ng/ml of IGF-1. A significant attenuation in the response to HS was achieved within 1 hour of treatment and sustained over 3 hours of IGF-1 pretreatment (Fig. 4D).
Figure 4: The [Ca2+]i response to HS is altered by IGF-1 treatment and actin cytoskeleton organization.
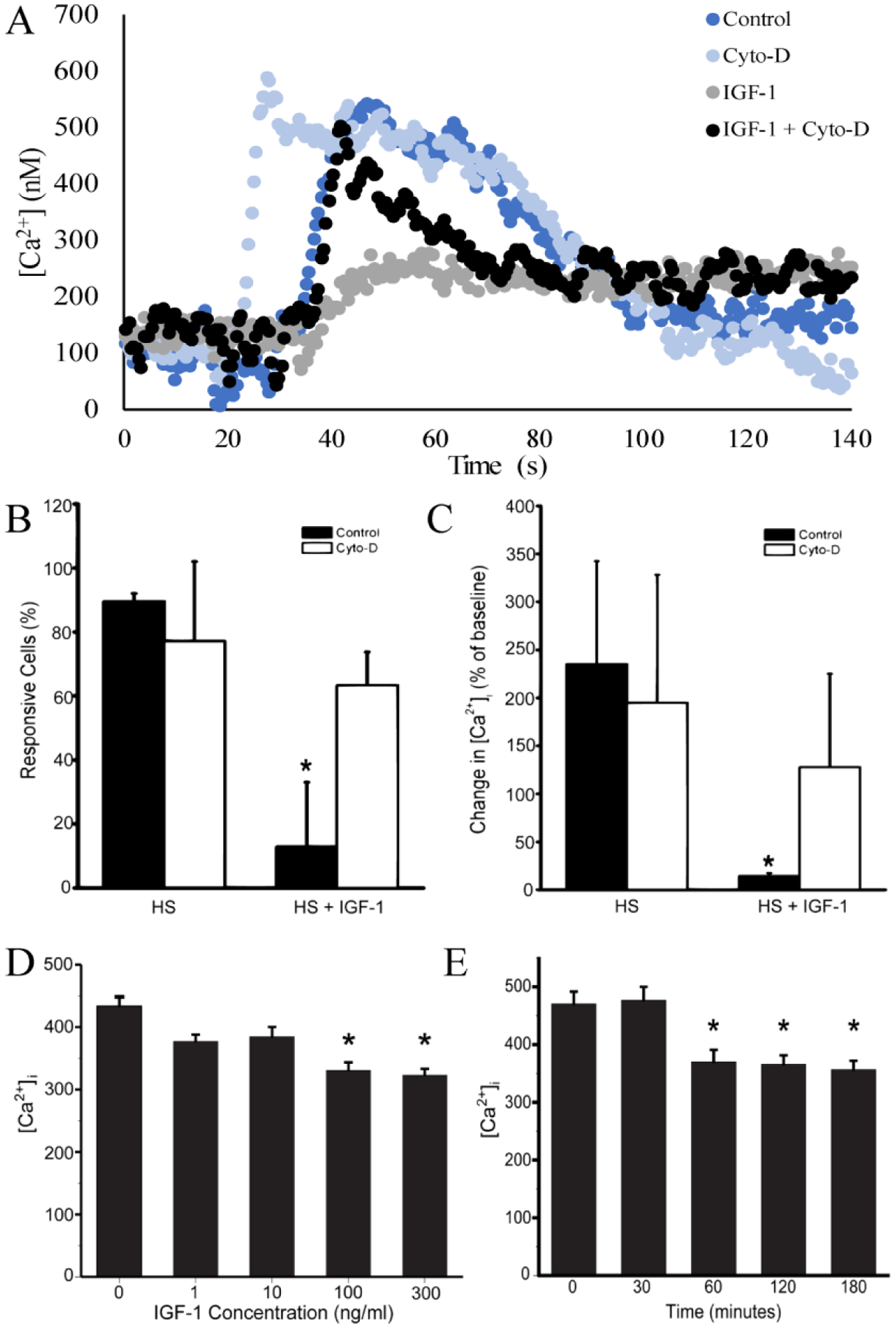
A. Representative [Ca2+]i traces for cells challenged with hypotonic swelling (blue) only or during 300ng/ml IGF-1 treatment (grey), IGF-1 + cytochalasin D (black), and cytochalasin D alone (light blue, Cyto-D). B) The number of cells responding to HS with a 50% or greater increase in [Ca2+]i compared to baseline was significantly attenuated following IGF-1 treatment. C) The change in [Ca2+]i compared to baseline in response to HS was significantly reduced following IGF-1 treatment. Disruption of the F-actin organization with 1 μM of cytochlasin D (Cyto-D) for 30 min after cells were treated with IGF-1 increased the number of cells exhibiting a [Ca2+]i response to HS and magnitude in response. D) [Ca2+]i of a dose response of ATDC5 to IGF-1 pre-treatment for 3 hr prior to challenge with 50% HS. Significant attenuation of [Ca2+]I during hypotonic challenge occurs at concentrations of 100 ng/ml and 300 ng/ml of IGF-1. E) Attenuation of [Ca2+]i when ATDC5 cells are challenged with HS starts within 60 minutes of IGF-1 treatment and is sustained over a 180 minute period. (* indicates p-value < 0.05 compared to controls)
We next sought to determine if the MAP7 binding domain of TRPV4 (aa. 798-809) is responsible for actin dependent regulation of TRPV4. We previously described how a novel mutation at aa. 800 and a previously known mutation at aa. 799, both within the MAP7 binding domain of TRPV4, alter TRPV4 mediated calcium kinetics, resulting in Metatropic Dysplasia [39]. By cloning the wild-type and mutant TRPV4 variants from these Metatropic Dysplasia patients, we created stably transfected HEK293 cells that express either wild-type TRPV4 (WT) or TRPV4 with mutations within the MAP7 binding domain (P799L or G800D). Stable TRPV4 expression in fibroblasts from human patients with mutations at either the 799 (P799L) or 800 (G800D) amino acids was confirmed using RT-PCR (Fig. 5A). We validated that WT and or mutant TRPV4 variants were cloned into the pSF-CMV-EMCV-daGFP plasmid, using SmaI digestion. The SmaI enzyme specifically cuts at specific sites with the given sequence: 5’-CCCGGG-3’. The WT TRPV4 coding sequence contains this specific sequence at the nucleotide positions 2397 – 2402, whereas the P799L and G800D mutants have the sequence 5’-CCTGGG-3’ and 5’- CCCGGA-3’ at this site, respectively. Thus, SmaI digests the WT variant transcript of TRPV4, resulting in the two fragments (Fig 5B). However, the P799L and G800D mutants lack this cut site, meaning that the enzyme does not fragment these variants. After transfecting the HEK293 cells with one of the three TRPV4 variants, the expression of functional TRPV4 channels was confirmed. Pharmacological activation of TRPV4 with 500 nM RN1747, a TRPV4 specific agonist, elicited an increase in [Ca2+]i in HEK cells expressing WT TRPV4. RN1747 failed to elicit a response in untransfected (UT) HEK cells, which do not endogenously express TRPV4 (Fig. 5C).
Figure 5: MAP7 Binding Domain mediates the response of HEK cells expressing WT, P799L, and G800D TRPV4 variants to IGF-1 treatment.
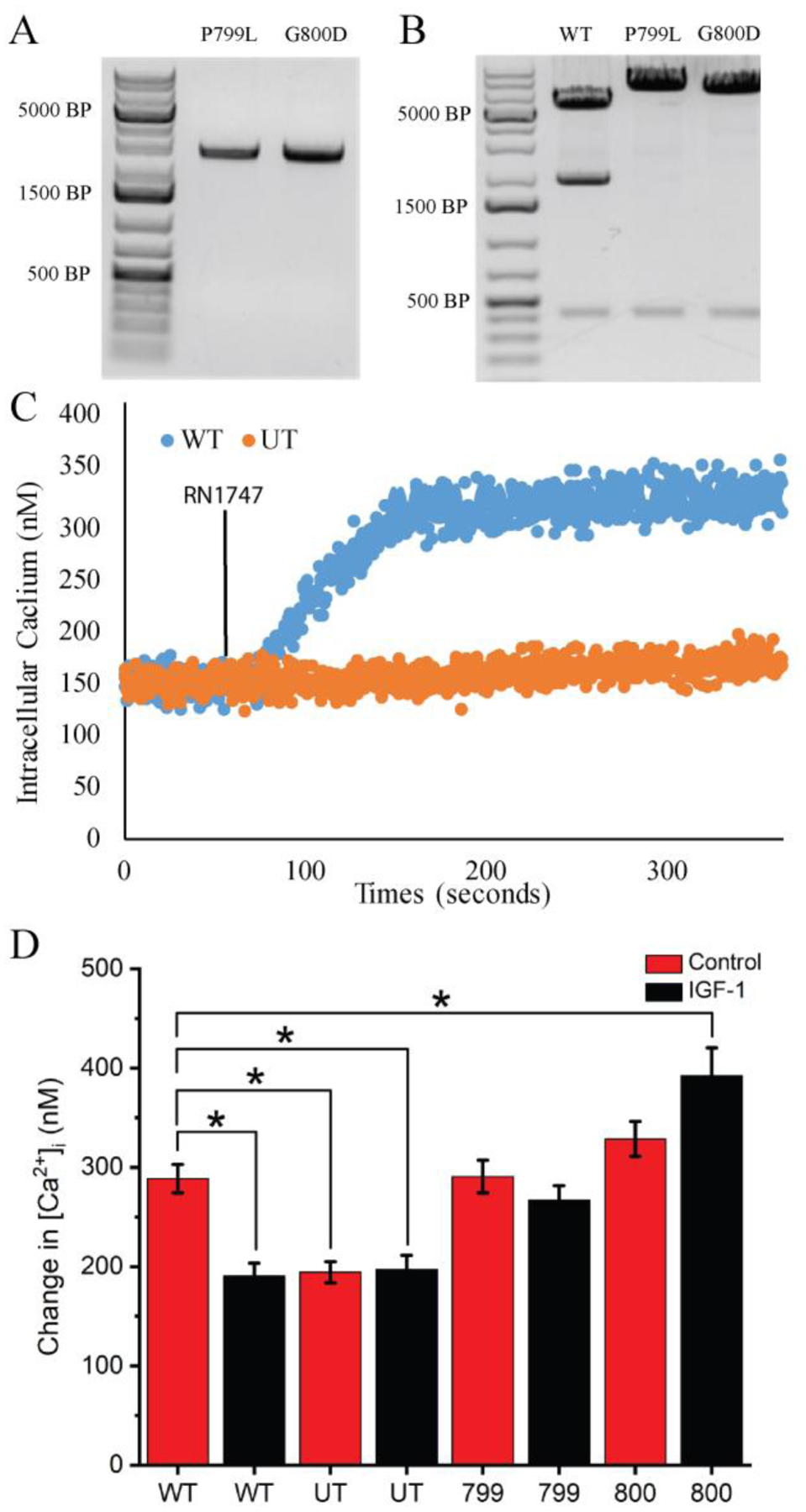
A) RT-PCR of mRNA from fibroblast expressing P799L and G800D TRPV4 mutations indicates that these cells stably express transcripts for mutant TRPV4. B) pSF-CMV-EMCV-daGFP plasmids containing WT, P799L, and G800D TRPV4 were successfully generated. SmaI digestion of WT, P799L, and G800D TRPV4 plasmids results only in cleavage of the WT TRPV4 clone (fragment ~2000bp). Mutations in the sequence of the P799L and G800D TRPV4 clones reside within the SmaI cleavage sequence, inhibiting enzymatic digestion of these clones by SmaI. D) Calcium imaging of HEK UT (orange) and HEK WT (blue) cells. When HEK UT and HEK WT cells are treated with the TRPV4 specific agonist RN1747 (500 nM) only HEK WT cells show elevated [Ca2+]i. This confirms the presence of functional TRPV4 protein in the HEK WT cells. Calcium traces represent the average of individual cells of HEK UT (n=77) or HEK WT (n=79). D) The change in [Ca2+]i compared to baseline in response to HS ± 300 ng/ml of IGF-1 in HEK 293 cells that were either UT or stably transfected with either WT TRPV4, a mutation of proline to lysine at aa. 799 (799) TRPV4, or a mutation of glycine to aspartate at aa. 800 (800) TRPV4. IGF-1 (black bars) treatment fails to suppress the response of cells to HS in the 799 and 800 cells. (* indicates p-value < 0.05 compared across all groups)
To test if IGF-1 regulates TRPV4 via the MAP7 binding domain of the channel, HEK cells expressing WT, P799L, or G800D TRPV4 variants and UT cells were pretreated with 300 ng/ml of IGF-1 for 3 hours, prior to challenge with HS. IGF-1 pretreatment significantly attenuated the magnitude of [Ca2+]i of the HEK WT cells (Fig. 5D). UT cells demonstrated a significantly lower [Ca2+]i in response to HS when compared to the WT cells. This indicates that TRPV4 predominantly mediates the response of the transfected HEK cells to hypo-osmotic pressures. Furthermore, UT cells exhibited no attenuation of the [Ca2+]i signal during IGF-1 pretreatment. IGF-1 pretreatment significantly reduced [Ca2+]i of WT transfected HEK cells compared to WT controls during HS and were not significantly different from UT cells. This suggests that IGF-1 blocked the response to HS in a similar manner to cells completely lacking TRPV4. The MAP7 binding domain of TRPV4 appears to be essential for regulation of the channel during IGF-1 treatment since IGF-1 pretreatment failed to attenuate [Ca2+]i in the cells expressing either of the TRPV4 mutants (Fig. 5D). P799L (799) cells responded similarly to HS, regardless of treatment with IGF-1. Interestingly, IGF-1 pretreatment appears to sensitize HEK G800D (800) cells to HS when compared to the WT control (Fig. 5D). These results highlight the importance of the MAP7 domain in regulating TRPV4 activity during IGF-1 treatment.
3.5. IGF-1 Alters Chondrocyte Purinergic Signaling
Purinergic signaling in response to mechanical loading is a vital component of the mechanotransduction pathway of chondrocytes [42,43]. In response to HS, ATDC5 cells exhibited a significant release of ATP 5 minutes after hypo-osmotic challenge (Fig 6A). This ATP release was significantly attenuated by pretreatment with IGF-1 for 3 hours, but was fully restored when the actin cytoskeleton was disrupted using cytochalasin D. In addition, the general TRPV4 inhibitor, ruthenium red, or the TRPV4 specific inhibitor, RN1734, attenuated this response (Fig. 6B) to similar levels as IGF-1 treatment. These data suggest that IGF-1 suppresses ATP release during osmotic challenge via the interaction of actin and TRPV4.
Figure 6: ATP release during HS is reduced following IGF-1 treatment and inhibition of TRPV channels.
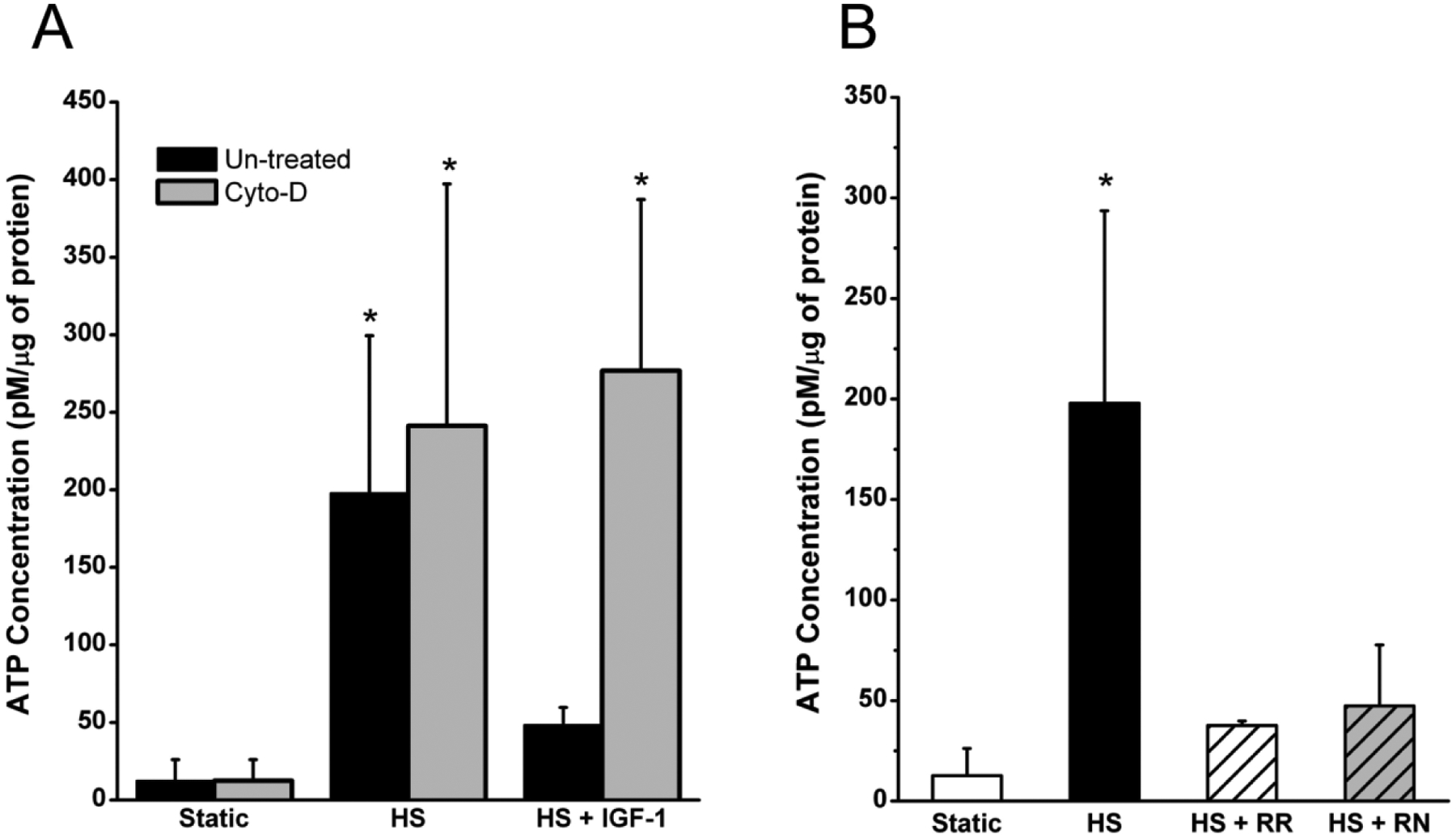
A) After 5 minutes of HS, ATP release was 12 times higher than static controls. IGF-1 treatment attenuated the release of ATP and was only 3 times greater than static controls. B) Inhibition of TRPV channels reduced ATP release during HS (HS + RR), and inhibition of TRPV4 (HS + RN) specifically reduced ATP by the same degree. (* indicates a p-value < 0.05 compared to controls)
4. Discussion
Both mechanical forces and biochemical factors regulate chondrocyte function and cartilage integrity. Mechanical loading of cartilage through exercise and physical therapy has been shown to enhance joint health in adults [66,67], relieving pain and improving joint mobility in OA patients [68–70]. This mechanical loading of the joint imposes several types of mechanical forces at the cellular level such as hydrostatic pressure, compressive and tensile strain, fluid shear and osmotic pressure suggesting mechanotransduction occurs at the membrane of these cells. Membrane structures such as ion channels, integrins and the primary cilium have all been associated with mechanotransduction in the chondrocyte [71]. However, transduction of mechanical signals into biochemical responses within the cell are complex and it is likely that different proteins can sense different types of mechanical signals.
Extracellular calcium influx is central to the response of cells to mechanical loading [72,73]. While several channels are expressed in chondrocytes that could affect calcium signaling, two types of mechanosensitive channels, TRPV4 and PIEZO 1 & 2, have been identified in chondrocytes and preferentially conduct calcium [74]. TRPV4 channels are expressed in a myriad of tissues and respond to low levels of mechanical stimulation such as osmotic pressure and membrane stretch, although it has been proposed that this channel may interact with other mechanosensors that could activate the channel [65]. In chondrocytes, TRPV4 channels have been associated with cartilage development [24], regulation of anabolic cell functions [58], suppression of pro-inflammatory responses [59] and participation in development of OA [60]. Piezo 1/2 channels were first characterized in 2010 [61] and are also expressed in many mechanosensitive tissues. Piezo 1 has been shown to respond to high levels of strain [75,62] however, Piezo 2 appears to be necessary for the full response to mechanical stimulation. Inhibition of these channels with GsMTx4 reduced chondrocyte apoptosis, suggesting that these channels may be important to the injury response in cartilage. While it is apparent that the roles of these channels in mechanotransduction in the chondrocyte are intertwined, we focused these studies on the TRPV4 channel due to the response of the channel to physiologic levels of load.
In these studies, we used osmotic challenge to stimulate the TRPV4 channel because degradation of proteoglycans during OA increases cartilage hydration through changes in the local osmotic pressure gradients, with swelling of cartilage representing one of the earliest macroscopic events during OA [49–51]. During OA, chondrocytes are unable to deform their membranes and experience impaired cell volume regulation [51,52]. Thus, elucidating how decreasing concentrations and/or insensitivity to IGF-1 in cartilage influences cartilage homeostasis may improve therapeutic targeting of OA
This study elucidates a novel mechanism by which IGF-1 interacts with TRPV4 channel to regulate the mechanical response of the chondrocyte. Specifically, our results indicate that IGF-1 increases cell stiffness through increased stress fiber formation of the actin cytoskeleton and thereby suppresses the activation of TRPV4 channels in response to osmotic stimuli. Several reports have suggested that actin and microtubules may be closely associated with this channel [37,44,45]. Perhaps the strongest indication that actin regulates this channel is that when actin is depolymerized, the osmotic response of the cell is lost [37,44]. Suzuki, et al. have shown the Microtubulin Associating Protein 7 (MAP7) domain at the amino acid sequence 798–809 of TRPV4 interacts with actin, but not microtubules, to regulate channel activity [37]. Our results confirm Suzuki’s finding that actin interacts with the MAP7 domain of TRPV4. Moreover, we show that IGF-1 suppresses TRPV4 activity via the MAP7 domain of the channel. When mutations in the MAP7 domain are expressed in HEK cells, we show that IGF-1 fails to suppress TRPV4 mediated [Ca2+]i and stimulates the response of G800D cells to HS. We hypothesize this attenuation in [Ca2+]i is caused by IGF-1 stimulating F-actin, which interacts with the MAP7 domain to modulate TRPV4 channel kinetics.
IGF-1 is normally located in the local matrix around chondrocytes, where it is available at relatively constant levels and signals to the cell to increase anabolic activity and for production of matrix molecules [46]. Importantly autologous implantation of chondrocytes overexpressing IGF-1 significantly improves collagen production, glycosaminoglycan, and mechanical properties of articular cartilage in equine PTOA models [47,48]. Although the mechanism by which IGF-1 alters actin polymerization is relatively unclear, Novakofski et al. have shown that IGF-1 suppresses RhoA GTPase activation in chondrocytes, while increasing cortical F-actin staining below the plasma membrane [18]. We show that IGF-1 stiffens ATDC5 cells via stimulation of F-actin. This increase in cell stiffness then reduces the deformation of the plasma membrane under hypotonic conditions and the subsequent activation of the TRPV4 channel.
IGF-1 levels fluctuate with age, reaching peak concentrations of over 300 ng/ml by the age of 15, and decreasing to less than a third of peak concentrations by the age of 65 [63]. Currently, few studies utilize IGF-1 concentrations above physiologic concentrations of 600 ng/ml in their experiments and some evidence suggests that chondrocytes differentially respond to IGF-1 administration. Phornphutkul et al. observed that 10 nM of IGF-1 treatment increased chondrocyte differentiation and proteoglycan production, while administration of 50 nM of IGF-1 promoted chondrocyte proliferation [64]. In our studies, we observed that IGF-1 attenuates the TRPV4 mediated response to osmotic stimuli starting at concentrations of 100 ng/ml and maximally suppresses chondrocyte osmosensation at 300 ng/ml. Contrary to our findings, Lee et al. show that IGF-1 sensitized TRPV4 activity through SGK1 phosphorylation of the serine 824 amino acid of TRPV4 in HEK293 cells [48]. However, Lee et al. did not challenge the HEK293 cells with HS and it is unclear if 100 μM or 1 μM of IGF-1 were utilized in these studies, due to differences reported in the concentrations of IGF-1 used between the results section and the figure legend of the paper. These results suggest that supraphysiologic concentrations of IGF-1 may sensitize TRPV4 but it remains unknown how these concentrations influence cells and, specifically, chondrocyte mechanotransduction. Thus, future studies may elucidate if there is a biphasic response in TRPV4 mediated mechanotransduction during IGF-1 treatment.
We have previously reported that ATP release from osteoblasts is dependent on calcium entry into the cell [54]. Thus, reducing the Ca2+ entry through the TRPV4 channel by IGF-1 could also impact ATP release in response to mechanical loading in chondrocytes. In this study, we highlight the link between IGF-1 regulation of TRPV4 and how it reduces extracellular ATP release from chondrocytes. This inhibition of extracellular ATP release appears to be mediated through the interaction of actin and TRPV4. While chondrocytes release ATP in response to mechanical stimuli to promote chondrogenesis, elevated levels of ATP have been found in the synovial fluid of patients with chondrocalcinosis and OA [55]. This indicates that differential responses to extracellular ATP in chondrocytes may be influenced by the pathological status of cartilage. With the loss of IGF-1 seen in OA patients, mechanical loading of chondrocytes may lead to excessive ATP release. Additionally, depletion of ATP in chondrocytes has previously been reported in spontaneous OA in Hartley Guinea Pigs [56] and exogenous adenosine administration reverses this disease state [57]. Ultimately, insensitivity to IGF-1 observed in OA patients may promote excessive ATP release during mechanical loading, leading to the depleted ATP status observed in OA.
Our study emphasizes IGF-1 as a promising candidate for the prevention and treatment of articular cartilage damage during aging and OA. Our data elucidate a potentially important mechanism by which IGF-1 regulates chondrocyte mechanosensitivity, a major contributor to articular chondrocyte function. This mechanism involves the coordinated control of actin organization, cell stiffness, TRPV4 mediated calcium flux and ATP release. However, because mechanical signals regulate both anabolic and catabolic chondrocyte functions, future studies are needed to define the relationship between this regulatory pathway and chondrocyte anabolic and catabolic activities.
Highlights.
IGF-1 suppresses hypotonic stimulation of the TRPV4 channel to reduce Ca2+influx in ATDC5 cells.
IGF-1 stimulates the f-actin stress fiber formation which binds to the TRPV4 MAP7 binding domain to elicit this suppression.
Mutation of the MAP7 site abrogates the response of cells to IGF-1.
TRPV4 mediated [Ca2+]i promotes ATP release from chondrocytes and is inhibited during IGF-1 treatment.
Acknowledgements
Voucher from NIH NIGMS P20 GM103446 INBRE IDeA grant and a grant from NIH U54 GM104941 DE-DTR ACCEL grant. Microscopy access was supported by grants from the NIH-NIGMS (P20 GM103446), the NSF (IIA-1301765) and the State of Delaware.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
The authors declare they have no conflicts of interest with the content of this manuscript.
References
- [1].Barbour KE, Helmick CG, Boring Michael, Brady TJ, Morbidity and Mortality Weekly Report Vital Signs: Prevalence of Doctor-Diagnosed Arthritis and Arthritis-Attributable Activity Limitation-United States, 2013–2015, n.d. s (accessed February 20, 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Helmick CG, Felson DT, Lawrence RC, Gabriel S, Hirsch R, Kwoh CK, Liang MH, Kremers HM, Mayes MD, Merkel PA, Pillemer SR, Reveille JD, Stone JH, Estimates of the prevalence of arthritis and other rheumatic conditions in the United States: Part I, Arthritis & Rheumatism. 58 (2008) 15–25. 10.1002/art.23177. [DOI] [PubMed] [Google Scholar]
- [3].Carter DR, Beaupre GS, Wong M, Smith RL, Andriacchi TP, Schurman DJ, The Mechanobiology of Articular Cartilage Development and Degeneration, Clinical Orthopaedics and Related Research. 427 (2004) S69–S77. 10.1097/01.blo.0000144970.05107.7e. [DOI] [PubMed] [Google Scholar]
- [4].Trippel SB, Growth factor actions on articular cartilage., The Journal of Rheumatology. Supplement. 43 (1995) 129–32. http://www.ncbi.nlm.nih.gov/pubmed/7752116 (accessed February 28, 2020). [PubMed] [Google Scholar]
- [5].Mariani E, Pulsatelli L, Facchini A, Signaling pathways in cartilage repair., International Journal of Molecular Sciences. 15 (2014) 8667–98. 10.3390/ijms15058667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].De Croos JNA, Dhaliwal SS, Grynpas MD, Pilliar RM, Kandel RA, Cyclic compressive mechanical stimulation induces sequential catabolic and anabolic gene changes in chondrocytes resulting in increased extracellular matrix accumulation, Matrix Biology. 25 (2006) 323–331. 10.1016/J.MATBIO.2006.03.005. [DOI] [PubMed] [Google Scholar]
- [7].Nebelung S, Gavenis K, Lüring C, Zhou B, Mueller-Rath R, Stoffel M, Tingart M, Rath B, Simultaneous anabolic and catabolic responses of human chondrocytes seeded in collagen hydrogels to long-term continuous dynamic compression, Annals of Anatomy - Anatomischer Anzeiger. 194 (2012) 351–358. 10.1016/J.AANAT.2011.12.008. [DOI] [PubMed] [Google Scholar]
- [8].Kim YJ, Sah RLY, Grodzinsky AJ, Plaas AHK, Sandy JD, Mechanical regulation of cartilage biosynthetic behavior: Physical stimuli, Archives of Biochemistry and Biophysics. 311 (1994) 1–12. 10.1006/abbi.1994.1201. [DOI] [PubMed] [Google Scholar]
- [9].Mawatari T, Lindsey DP, Harris AHS, Goodman SB, Maloney WJ, Smith RL, Effects of tensile strain and fluid flow on osteoarthritic human chondrocyte metabolism in vitro, Journal of Orthopaedic Research. 28 (2010) n/a-n/a. 10.1002/jor.21085. [DOI] [PubMed] [Google Scholar]
- [10].Neu CP, Khalafi A, Komvopoulos K, Schmid TM, Reddi AH, Mechanotransduction of bovine articular cartilage superficial zone protein by transforming growth factor β signaling, Arthritis & Rheumatism. 56 (2007) 3706–3714. 10.1002/art.23024. [DOI] [PubMed] [Google Scholar]
- [11].Vincent TL, McLean CJ, Full LE, Peston D, Saklatvala J, FGF-2 is bound to perlecan in the pericellular matrix of articular cartilage, where it acts as a chondrocyte mechanotransducer, Osteoarthritis and Cartilage. 15 (2007) 752–763. 10.1016/J.JOCA.2007.01.021. [DOI] [PubMed] [Google Scholar]
- [12].Fanning PJ, Emkey G, Smith RJ, Grodzinsky AJ, Szasz N, Trippel SB, Mechanical regulation of mitogen-activated protein kinase signaling in articular cartilage., The Journal of Biological Chemistry. 278 (2003) 50940–8. 10.1074/jbc.M305107200. [DOI] [PubMed] [Google Scholar]
- [13].Haudenschild DR, Nguyen B, Chen J, D’Lima DD, Lotz MK, Rho kinase-dependent CCL20 induced by dynamic compression of human chondrocytes., Arthritis and Rheumatism. 58 (2008) 2735–42. 10.1002/art.23797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Oh C-D, Chun J-S, Signaling mechanisms leading to the regulation of differentiation and apoptosis of articular chondrocytes by insulin-like growth factor-1., The Journal of Biological Chemistry. 278 (2003) 36563–71. 10.1074/jbc.M304857200. [DOI] [PubMed] [Google Scholar]
- [15].Lee H-S, Millward-Sadler SJ, Wright MO, Nuki G, Al-Jamal R, Salter DM, Activation of Integrin—RACK1/PKCα signalling in human articular chondrocyte mechanotransduction, Osteoarthritis and Cartilage. 10 (2002) 890–897. 10.1053/JOCA.2002.0842. [DOI] [PubMed] [Google Scholar]
- [16].Lee HS, Millward-Sadler SJ, Wright MO, Nuki G, Salter DM, Integrin and Mechanosensitive Ion Channel-Dependent Tyrosine Phosphorylation of Focal Adhesion Proteins and β-Catenin in Human Articular Chondrocytes After Mechanical Stimulation, Journal of Bone and Mineral Research. 15 (2000) 1501–1509. 10.1359/jbmr.2000.15.8.1501. [DOI] [PubMed] [Google Scholar]
- [17].Madry H, Zurakowski D, Trippel S, Overexpression of human insulin-like growth factor-I promotes new tissue formation in an ex vivo model of articular chondrocyte transplantation, Gene Therapy. 8 (2001) 1443–1449. 10.1038/sj.gt.3301535. [DOI] [PubMed] [Google Scholar]
- [18].Novakofski K, Boehm A, Fortier L, The small GTPase Rho mediates articular chondrocyte phenotype and morphology in response to interleukin-1α and insulin-like growth factor-I, Journal of Orthopaedic Research. 27 (2009) 58–64. 10.1002/jor.20717. [DOI] [PubMed] [Google Scholar]
- [19].Sah RL-Y, Doong J-YH, Grodzinsky AJ, Plaas AHK, Sandy JD, Effects of compression on the loss of newly synthesized proteoglycans and proteins from cartilage explants, Archives of Biochemistry and Biophysics. 286 (1991) 20–29. 10.1016/0003-9861(91)90004-3. [DOI] [PubMed] [Google Scholar]
- [20].Tyler JA, Insulin-like growth factor 1 can decrease degradation and promote synthesis of proteoglycan in cartilage exposed to cytokines., The Biochemical Journal. 260 (1989) 543–8. http://www.ncbi.nlm.nih.gov/pubmed/2788408 (accessed March 17, 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bonassar LJ, Grodzinsky AJ, Frank EH, Davila SG, Bhaktav NR, Trippel SB, The effect of dynamic compression on the response of articular cartilage to insulin-like growth factor-I, Journal of Orthopaedic Research. 19 (2001) 11–17. 10.1016/S0736-0266(00)00004-8. [DOI] [PubMed] [Google Scholar]
- [22].Bonassar LJ, Grodzinsky AJ, Srinivasan A, Davila SG, Trippel SB, Mechanical and Physicochemical Regulation of the Action of Insulin-Like Growth Factor-I on Articular Cartilage, Archives of Biochemistry and Biophysics. 379 (2000) 57–63. 10.1006/ABBI.2000.1820. [DOI] [PubMed] [Google Scholar]
- [23].Jin M, Emkey GR, Siparsky P, Trippel SB, Grodzinsky AJ, Combined effects of dynamic tissue shear deformation and insulin-like growth factor I on chondrocyte biosynthesis in cartilage explants., Archives of Biochemistry and Biophysics. 414 (2003) 223–31. 10.1016/s0003-9861(03)00195-4. [DOI] [PubMed] [Google Scholar]
- [24].Muramatsu S, Wakabayashi M, Ohno T, Amano K, Ooishi R, Sugahara T, Shiojiri S, Tashiro K, Suzuki Y, Nishimura R, Kuhara S, Sugano S, Yoneda T, Matsuda A, Functional gene screening system identified TRPV4 as a regulator of chondrogenic differentiation, Journal of Biological Chemistry. 282 (2007) 32158–32167. 10.1074/jbc.M706158200. [DOI] [PubMed] [Google Scholar]
- [25].Liedtke W, Transient receptor potential vanilloid channels functioning in transduction of osmotic stimuli, Journal of Endocrinology. 191 (2006) 515–523. 10.1677/joe.1.07000. [DOI] [PubMed] [Google Scholar]
- [26].Nilius B, Prenen J, Wissenbach U, Bödding M, Droogmans G, Differential activation of the volume-sensitive cation channel TRP12 (OTRPC4) and volume-regulated anion currents in HEK293 cells, Pflugers Archiv European Journal of Physiology. 443 (2001) 227–233. 10.1007/s004240100676. [DOI] [PubMed] [Google Scholar]
- [27].Nilius B, Watanabe H, Vriens J, The TRPV4 channel: structure-function relationship and promiscuous gating behaviour, Pflügers Archiv - European Journal of Physiology. 446 (2003) 298–303. 10.1007/s00424-003-1028-9. [DOI] [PubMed] [Google Scholar]
- [28].Flockerzi V, An Introduction on TRP Channels, in: Transient Receptor Potential (TRP) Channels, Springer Berlin Heidelberg, Berlin, Heidelberg, n.d.: pp. 1–19. 10.1007/978-3-540-34891-7_1. [DOI] [Google Scholar]
- [29].Gao X, Wu L, O’Neil RG, Temperature-modulated diversity of TRPV4 channel gating: activation by physical stresses and phorbol ester derivatives through protein kinase C-dependent and -independent pathways., The Journal of Biological Chemistry. 278 (2003) 27129–37. 10.1074/jbc.M302517200. [DOI] [PubMed] [Google Scholar]
- [30].Loukin S, Zhou X, Su Z, Saimi Y, Kung C, Wild-type and brachyolmia-causing mutant TRPV4 channels respond directly to stretch force., The Journal of Biological Chemistry. 285 (2010) 27176–81. 10.1074/jbc.M110.143370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mizoguchi F, Mizuno A, Hayata T, Nakashima K, Heller S, Ushida T, Sokabe M, Miyasaka N, Suzuki M, Ezura Y, Noda M, Transient receptor potential vanilloid 4 deficiency suppresses unloading-induced bone loss, Journal of Cellular Physiology. 216 (2008) 47–53. 10.1002/jcp.21374. [DOI] [PubMed] [Google Scholar]
- [32].Clark AL, Votta BJ, Kumar S, Liedtke W, Guilak F, Chondroprotective role of the osmotically sensitive ion channel transient receptor potential vanilloid 4: Age- and sex-dependent progression of osteoarthritis in Trpv4-deficient mice, Arthritis and Rheumatism. 62 (2010) 2973–2983. 10.1002/art.27624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Trickey WR, Vail TP, Guilak F, The role of the cytoskeleton in the viscoelastic properties of human articular chondrocytes, Journal of Orthopaedic Research. 22 (2004) 131–139. 10.1016/S0736-0266(03)0150-5. [DOI] [PubMed] [Google Scholar]
- [34].Gardinier J, Yang W, Madden GR, Kronbergs A, Gangadharan V, Adams E, Czymmek K, Duncan RL, P2Y 2 receptors regulate osteoblast mechanosensitivity during fluid flow, American Journal of Physiology-Cell Physiology. 306 (2014) C1058–C1067. 10.1152/ajpcell.00254.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Leipzig ND, Eleswarapu SV, Athanasiou KA, The effects of TGF-β1 and IGF-I on the biomechanics and cytoskeleton of single chondrocytes, Osteoarthritis and Cartilage. 14 (2006) 1227–1236. 10.1016/J.JOCA.2006.05.013. [DOI] [PubMed] [Google Scholar]
- [36].Zhang J, Ryder KD, Bethel JA, Ramirez R, Duncan RL, PTH-Induced Actin Depolymerization Increases Mechanosensitive Channel Activity to Enhance Mechanically Stimulated 2+ Signaling in Osteoblasts*, Journal of Bone and Mineral Research. 21 (2006) 1729–1737. 10.1359/jbmr.060722. [DOI] [PubMed] [Google Scholar]
- [37].Suzuki M, Hirao A, Mizuno A, Microtubule-associated [corrected] protein 7 increases the membrane expression of transient receptor potential vanilloid 4 (TRPV4)., The Journal of Biological Chemistry. 278 (2003) 51448–53. 10.1074/jbc.M308212200. [DOI] [PubMed] [Google Scholar]
- [38].Clay JR, Shlesinger MF, Analysis of the effects of cesium ions on potassium channel currents in biological membranes, Journal of Theoretical Biology. 107 (1984) 189–201. 10.1016/S0022-5193(84)80021-1. [DOI] [PubMed] [Google Scholar]
- [39].Hurd L, Kirwin SM, Boggs M, Mackenzie WG, Bober MB, Funanage VL, Duncan RL, A mutation in TRPV4 results in altered chondrocyte calcium signaling in severe metatropic dysplasia, American Journal of Medical Genetics, Part A. 167 (2015) 2286–2293. 10.1002/ajmg.a.37182. [DOI] [PubMed] [Google Scholar]
- [40].Mak AF, Lai WM, Mow VC, Biphasic indentation of articular cartilage—I. Theoretical analysis, Journal of Biomechanics. 20 (1987) 703–714. 10.1016/0021-9290(87)90036-4. [DOI] [PubMed] [Google Scholar]
- [41].Zhang J, Ryder KD, Bethel JA, Ramirez R, Duncan RL, PTH-Induced Actin Depolymerization Increases Mechanosensitive Channel Activity to Enhance Mechanically Stimulated 2+ Signaling in Osteoblasts*, Journal of Bone and Mineral Research. 21 (2006) 1729–1737. 10.1359/jbmr.060722. [DOI] [PubMed] [Google Scholar]
- [42].Millward-Sadler SJ, Wright MO, Flatman PW, Salter DM, ATP in the mechanotransduction pathway of normal human chondrocytes, IOS Press, 2004. http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.500.1421&rep=rep1&type=pdf (accessed October 25, 2019). [PubMed] [Google Scholar]
- [43].Pingguan-Murphy B, El-Azzeh M, Bader DL, Knight MM, Cyclic compression of chondrocytes modulates a purinergic calcium signalling pathway in a strain rate- and frequency-dependent manner, Journal of Cellular Physiology. 209 (2006) 389–397. 10.1002/jcp.20747. [DOI] [PubMed] [Google Scholar]
- [44].Becker D, Bereiter-Hahn J, Jendrach M, Functional interaction of the cation channel transient receptor potential vanilloid 4 (TRPV4) and actin in volume regulation, European Journal of Cell Biology. 88 (2009) 141–152. 10.1016/j.ejcb.2008.10.002. [DOI] [PubMed] [Google Scholar]
- [45].Goswami C, Kuhn J, Heppenstall PA, Hucho T, Importance of non-selective cation channel TRPV4 interaction with cytoskeleton and their reciprocal regulations in cultured cells, PLoS ONE. 5 (2010) 19–21. 10.1371/journal.pone.0011654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Martin JA, Buckwalter JA, The role of chondrocyte-matrix interactions in maintaining and repairing articular cartilage., Biorheology. 37 (2000) 129–40. http://www.ncbi.nlm.nih.gov/pubmed/10912185 (accessed February 28, 2020). [PubMed] [Google Scholar]
- [47].Ortved KF, Begum L, Mohammed HO, Nixon AJ, Implantation of rAAV5-IGF-I transduced autologous chondrocytes improves cartilage repair in full-thickness defects in the equine model, Molecular Therapy. 23 (2015) 363–373. 10.1038/mt.2014.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Griffin DJ, Ortved KF, Nixon AJ, Bonassar LJ, Mechanical properties and structure-function relationships in articular cartilage repaired using IGF-I gene-enhanced chondrocytes, Journal of Orthopaedic Research. 34 (2016) 149–153. 10.1002/jor.23038. [DOI] [PubMed] [Google Scholar]
- [49].Lee EJ, Shin SH, Chun J, Hyun S, Kim Y, Kang SS, The modulation of TRPV4 channel activity through its Ser 824 residue phosphorylation by SGK1, Animal Cells and Systems. 14 (2010) 99–114. 10.1080/19768354.2010.486939. [DOI] [Google Scholar]
- [50].Maroudas A, Venn M, Chemical composition and swelling of normal and osteoarthrotic femoral head cartilage. II. Swelling., Annals of the Rheumatic Diseases. 36 (1977) 399–406. 10.1136/ard.36.5.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Liess C, Lüsse S, Karger N, Heller M, Glüer C-C, Detection of changes in cartilage water content using MRI T2-mapping in vivo, Osteoarthritis and Cartilage. 10 (2002) 907–913. 10.1053/joca.2002.0847. [DOI] [PubMed] [Google Scholar]
- [52].Jones WR, Ping Ting-Beall H, Lee GM, Kelley SS, Hochmuth RM, Guilak F, Alterations in the Young’s modulus and volumetric properties of chondrocytes isolated from normal and osteoarthritic human cartilage, Journal of Biomechanics. 32 (1999) 119–127. 10.1016/S0021-9290(98)00166-3. [DOI] [PubMed] [Google Scholar]
- [53].Bush P, Hall A, The volume and morphology of chondrocytes within non-degenerate and degenerate human articular cartilage, Osteoarthritis and Cartilage. 11 (2003) 242–251. 10.1016/S1063-4584(02)00369-2. [DOI] [PubMed] [Google Scholar]
- [54].Genetos DC, Geist DJ, Liu D, Donahue HJ, Duncan RL, Fluid Shear-Induced ATP Secretion Mediates Prostaglandin Release in MC3T3-E1 Osteoblasts, Journal of Bone and Mineral Research. 20 (2004) 41–49. 10.1359/JBMR.041009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ryan LM, Kurup IV, Derfus BA, Kushnaryov VM, ATP-induced chondrocalcinosis, Arthritis & Rheumatism. 35 (1992) 1520–1525. 10.1002/art.1780351216. [DOI] [PubMed] [Google Scholar]
- [56].Johnson K, Svensson CI, Van Etten D, Ghosh SS, Murphy AN, Powell HC, Terkeltaub R, Mediation of spontaneous knee osteoarthritis by progressive chondrocyte ATP depletion in Hartley guinea pigs, Arthritis & Rheumatism. 50 (2004) 1216–1225. 10.1002/art.20149. [DOI] [PubMed] [Google Scholar]
- [57].Corciulo C, Lendhey M, Wilder T, Schoen H, Cornelissen AS, Chang G, Kennedy OD, Cronstein BN, Endogenous adenosine maintains cartilage homeostasis and exogenous adenosine inhibits osteoarthritis progression, Nature Communications. 8 (2017) 15019. 10.1038/ncomms15019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].O’Conor CJ, Leddy HA, Benefield HC, Liedtke WB, Guilak F, TRPV4-mediated mechanotransduction regulates the metabolic response of chondrocytes to dynamic loading, Proceedings of the National Academy of Sciences. 111 (2014) 1316–1321. 10.1073/pnas.1319569111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Fu S, Meng H, Inamdar S, Das B, Gupta H, Wang W, Thompson CL, Knight MM, Activation of TRPV4 by mechanical, osmotic or pharmaceutical stimulation is anti-inflammatory blocking IL-1β mediated articular cartilage matrix destruction, Osteoarthritis and Cartilage. 29 (2021) 89–99. 10.1016/j.joca.2020.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].O’Conor CJ, Ramalingam S, Zelenski NA, Benefield HC, Rigo I, Little D, Wu CL, Chen D, Liedtke W, McNulty AL, Guilak F, Cartilage-Specific Knockout of the Mechanosensory Ion Channel TRPV4 Decreases Age-Related Osteoarthritis, Scientific Reports. 6 (2016) 1–10. 10.1038/srep29053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, Dubin AE, Patapoutian A, Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels, Science. 330 (2010) 55–60. 10.1126/science.1193270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Servin-Vences MR, Moroni M, Lewin GR, Poole K, Direct measurement of TRPV4 and PIEZO1 activity reveals multiple mechanotransduction pathways in chondrocytes, ELife. 6 (2017) 1–24. 10.7554/eLife.21074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bidlingmaier M, Friedrich N, Emeny RT, Spranger J, Wolthers OD, Roswall J, Körner A, Obermayer-Pietsch B, Hübener C, Dahlgren J, Frystyk J, Pfeiffer AFH, Doering A, Bielohuby M, Wallaschofski H, Arafat AM, Reference Intervals for Insulin-like Growth Factor-1 (IGF-I) From Birth to Senescence: Results From a Multicenter Study Using a New Automated Chemiluminescence IGF-I Immunoassay Conforming to Recent International Recommendations, The Journal of Clinical Endocrinology & Metabolism. 99 (2014) 1712–1721. 10.1210/jc.2013-3059. [DOI] [PubMed] [Google Scholar]
- [64].Phornphutkul C, Wu KY, Yang X, Chen Q, Gruppuso PA, Insulin-like growth factor-I signaling is modified during chondrocyte differentiation, Journal of Endocrinology. 183 (2004) 477–486. 10.1677/joe.1.05873. [DOI] [PubMed] [Google Scholar]
- [65].Fodor J, Matta C, Oláh T, Juhász T, Takács R, Tóth A, Dienes B, Csernoch L, Zákány R, Store-operated calcium entry and calcium influx via voltage-operated calcium channels regulate intracellular calcium oscillations in chondrogenic cells, Cell Calcium. 54 (2013) 1–16. 10.1016/j.ceca.2013.03.003. [DOI] [PubMed] [Google Scholar]
- [66].Racunica TL, Teichtahl AJ, Wang Y, Wluka AE, English DR, Giles GG, O’Sullivan R, Cicuttini FM, Effect of physical activity on articular knee joint structures in community-based adults, Arthritis Care and Research. 57 (2007) 1261–1268. 10.1002/art.22990. [DOI] [PubMed] [Google Scholar]
- [67].Fitzgerald GK, Fritz JM, Childs JD, Brennan GP, Talisa V, Gil AB, Neilson BD, Abbott JH, Exercise, manual therapy, and use of booster sessions in physical therapy for knee osteoarthritis: a multi-center, factorial randomized clinical trial, Osteoarthritis and Cartilage. 24 (2016) 1340–1349. 10.1016/j.joca.2016.03.001. [DOI] [PubMed] [Google Scholar]
- [68].Fransen M, Mcconnell S, Ar H, Exercise for osteoarthritis of the knee (Review), (2015). 10.1002/14651858.CD004376.pub3. [DOI] [PubMed] [Google Scholar]
- [69].Bricca A, Wirth W, Juhl CB, Kemnitz J, Hunter DJ, Kwoh CK, Eckstein F, Culvenor AG, Moderate Physical Activity and Prevention of Cartilage Loss in People With Knee Osteoarthritis: Data From the Osteoarthritis Initiative, Arthritis Care and Research. 71 (2019) 218–226. 10.1002/acr.23791. [DOI] [PubMed] [Google Scholar]
- [70].Skou ST, Bricca A, Roos EM, The impact of physical activity level on the short- and long-term pain relief from supervised exercise therapy and education: a study of 12,796 Danish patients with knee osteoarthritis, Osteoarthritis and Cartilage. 26 (2018) 1474–1478. 10.1016/j.joca.2018.07.010. [DOI] [PubMed] [Google Scholar]
- [71].Zhao Z, Li Y, Wang M, Zhao S, Zhao Z, Fang J, Mechanotransduction pathways in the regulation of cartilage chondrocyte homoeostasis, Journal of Cellular and Molecular Medicine. 24 (2020) 5408–5419. 10.1111/jcmm.15204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Jones TJ, Nauli SM, Mechanosensory calcium signaling, Advances in Experimental Medicine and Biology. 740 (2012) 1001–1015. 10.1007/978-94-007-2888-2_46. [DOI] [PubMed] [Google Scholar]
- [73].Zhang K, Wang L, Liu Z, Geng B, Teng Y, Liu X, Yi Q, Yu D, Chen X, Zhao D, Xia Y, Mechanosensory and mechanotransductive processes mediated by ion channels in articular chondrocytes: Potential therapeutic targets for osteoarthritis, Channels. 15 (2021) 339–359. 10.1080/19336950.2021.1903184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Xu B-Y, Jin Y, Ma X-H, Wang C-Y, Guo Y, Zhou D, The potential role of mechanically sensitive ion channels in the physiology, injury, and repair of articular cartilage, (n.d.). 10.1177/2309499020950262. [DOI] [PubMed] [Google Scholar]
- [75].Lee W, Leddy HA, Chen Y, Lee SH, Zelenski NA, McNulty AL, Wu J, Beicker KN, Coles J, Zauscher S, Grandl J, Sachs F, Guilak F, Liedtke WB, Synergy between Piezo1 and Piezo2 channels confers high-strain mechanosensitivity to articular cartilage, Proceedings of the National Academy of Sciences of the United States of America. 111 (2014) E5114–E5122. 10.1073/pnas.1414298111. [DOI] [PMC free article] [PubMed] [Google Scholar]