

Abstract
Circular RNAs (circRNAs) are a class of covalently closed RNA molecules generated by backsplicing. circRNAs are expressed in a tissue-specific manner, accumulate with age in neural tissues, and are highly stable. In many cases, circRNAs are generated at the expense of a linear transcript as back-splicing competes with linear splicing. Some circRNAs regulate gene expression in cis, and some circRNAs can be translated into protein. The advent of deep sequencing and new bioinformatic tools has allowed detection of thousands of circRNAs in eukaryotes. Studying the functions of circRNAs is done using a combination of molecular and genetic methods. The unique genetic tools that can be used in studies of Drosophila melanogaster are ideal for deciphering the functions of circRNAs in vivo. These tools include the GAL4-UAS system, which can be used to manipulate the levels of circRNAs with exquisite temporal and spatial control, and genetic interaction screening, which could be used to identify pathways regulated by circRNAs. Research performed in Drosophila has revealed circRNAs production mechanisms, details of their translation, and their physiological functions. Due to their short lifecycle and the existence of excellent neurodegeneration models, Drosophila can also be used to study the role of circRNAs in aging and age-related disorders. Here, we review molecular and genetic tools and methods for detecting, manipulating, and studying circRNAs in Drosophila.
Introduction
Gene expression is highly regulated in most organisms, due to control at the transcriptional (pre-mRNA production), co-transcriptional (i.e. pre-mRNA processing) and post-transcriptional levels [1,2]. For example, RNA splicing, the process by which introns are removed and exons join together from the newly transcribed pre-mRNA, occurs co-transcriptionally in metazoans [3,4]. Interestingly, alternative splicing allows organisms to generate several mRNAs and proteins from a given gene [5–8]. Whereas linear splicing occurs with a 5’−3’ directionality, backsplicing allows “head-to-tail” joining in which a downstream 5’ splice donor is covalently linked with an upstream 3’ splice acceptor of the same or another upstream exon [9]. Backsplicing results in the generation of circular RNA molecules containing exons called exonic circular RNAs (circRNAs). There are also circular RNAs that are formed by canonical splicing and that contain introns, these molecules are called intronic circular RNAs [10–14]. This review will focus on exonic circRNAs. These circRNAs are evolutionarily conserved, highly abundant molecules that are expressed across the animal kingdom [15–17], in plants, fungi and protists [18–21].
Introns carry the information that directs exon circularization [17,22,23]. circRNA-forming exons are flanked by long and generally inefficiently spliced introns [17,22]. Moreover, many of these introns contain long or short regions of complementary sequence that are hypothesized to favor exon circularization by inhibiting linear splicing and by bringing the 5’ and 3’ backsplice sites into close proximity [17,22,24]. In eukaryotes, circRNAs are produced by the spliceosome [17,24,25] and their production levels are regulated by cis-regulatory elements as well as trans-acting factors [17,22,26,27]. Among the latter are RNA binding proteins like MUSCLEBLIND (MBL) and QUAKING. These proteins likely favor circRNA formation by inhibiting linear splicing and/or by facilitating the interaction between the introns flanking the circularizable exon (or exons) [17,26]. More general modifiers of RNA structure like protein factors ADAR and DHX9 and environmental factors like temperature can also modulate the levels of circRNAs [28,29].
circRNAs are generated in a cell- and tissue-specific manner [30] and are extremely stable as they are inherently resistant to degradation by exonucleases due to the absence of free 3’ or 5’ ends [16]. In particular circRNAs are highly expressed in neural tissues [28,31–33], upregulated during neurogenesis, and some of them are enriched in synaptoneurosomes, more so than their linear mRNA counterparts [28,32]. These features suggest that circRNAs carry out important functions in the brain. Importantly, circRNAs accumulate with age in the brains of flies, worms, and mice and in the human substantia nigra [34–38]. This led to the speculation that circRNAs might be involved in neurodegenerative diseases, and some studies have provided evidence for roles of circRNAs in these age-dependent disorders [37,39].
Probably the most striking example of circRNA regulatory conservation is the circRNA generated from the second exon of the gene muscleblind (mbl). The second exon of this gene expresses the most abundant circRNA in Drosophila; its levels are more than 10 times higher than that of the linear MBL mRNA, which encodes a splicing factor essential for fly development [40–42]. In mice and humans, circRNAs from the gene mbl are generated from the same exon of the homologous genes, while this is not the case for other genes. Interestingly, the levels of this circRNA produced from the second exon of muscleblind seem to be primarily regulated by MBL, the protein encoded by the locus. Cell-culture experiments with both fly and human cells showed that increases or decreases in levels of MBL result in higher or lower circRNA levels, respectively [17]. Moreover, MBL binds to circMbl in the cytoplasm, suggesting a highly conserved mutual regulatory mechanisms between the two products of the mbl locus [17]. Interestingly, circMbl can also be translated in vivo [43]. Generally and although a subset of circRNAs are translated both in flies and mammalian tissues, there is not much evidence of the functionality of the circRNA-encoded proteins [43–45].
Initially, circRNAs were thought to act as miRNA sponges. This was based on the finding that two circRNAs, ciRS7/CDR1as and circSry, have many sequences complementary to the seed regions of miR-7 and miR-138, respectively [16,46,47]. Although it is possible that circRNA-miRNA interactions regulate the localization or stability of miRNAs, there is evidence that these circRNAs do more than sequestering miRNAs [16,48,49]. Indeed, knockout of ciRS7/CDR1 results in lower levels of miR-7 and upregulation of miR-7 targets. This might be due to stabilization of miR-7 by ciRS7/CDR1as [50]. Importantly, there is no solid evidence for miRNA sponging functions for other circRNAs, suggesting that circSry and ciRS7/CDR1as are the exceptions rather than the rule even for the circRNA binding capabilities. Other putative functions of circRNAs include regulating intracellular transport of RNA binding proteins [9,17,49] or act as protein decoys [51]. There is evidence that some circRNAs play a role in cancer development by affecting cell cycle and growth [51,52], and there is growing evidence for circRNA involvement in response to viral infection [53–55].
One obstacle while working with circRNAs is their identification and validation. Standard RNA sequencing techniques depend on enrichment of poly(A)-containing RNAs. As circRNAs lack a poly(A) tail, they are not typically identified in datasets created using standard RNA sequencing library production procedures. However, RNA library preparation methods based on ribosomal RNA-depletion, RNase R treatment, and new computational pipelines have resulted in the detection of thousands of circRNAs in worms, Drosophila melanogaster [22,34], mouse, monkey, and humans [48,56–59]. D. melanogaster has been used as a model system for over a century and has proven to be a valuable organism for study of circRNAs, which are highly expressed in fly heads [17,34] and accumulate with age in the nervous system [34]. In this review, we discuss the tools available for accurate detection of the circRNA expression and characterization of their functions using Drosophila as a model organism.
Separating the wheat from the chaff: How to identify and validate circRNAs?
Genome-wide identification of circRNAs by RNA sequencing
Identifying and validating circRNAs is essential for determining potential functions. However, this is not an easy task, especially given the similitude of sequence to the mRNA of the hosting gene. Therefore, special consideration must be taken during detection and verification of circRNAs. The easiest and most common method for de novo detection of circRNAs in Drosophila is by RNA-seq. Indeed, the discovery of widespread circRNA production across the genome was made possible only with the advancement of high throughput sequencing together with the development of new computational pipelines. As circRNAs do not have poly(A) tails [15], the large majority are not detected by standard sequencing methods. However, some exons contained within circRNAs have short A-stretches, and those circRNAs can be detected in RNA-seq experiments based on poly(A) selection. Detection is highly unusual as it also requires that the A-stretch is near the backsplicing junction. Therefore, identification and quantification of circRNAs require sequencing of total RNA libraries (which are generally prepared either by ligation or random primer-based methods after depletion of rRNA) [16,60].
Enrichment for circRNAs: RNase R pre-treatment
The identification of backsplicing junctions by RNA-seq in Drosophila does not necessarily mean the existence of a circRNA. For example, some linear RNA species, like those generated by trans splicing can have junctions that resemble those of circRNAs. In additional, technical artifacts produced by reverse transcriptase due to the template switching activity can also suggest the presence of a circRNA when there is not one. Therefore, profiling of circRNAs by total RNA-seq in Drosophila should be followed by some type of verification. The most common way to determine whether a given junction is present involves pretreatment with RNase R.
RNase R is a 3’−5’ exoribonuclease, that degrades most linear RNAs [61,62] but leaves the circular transcripts intact due to lack of free 3’ end [9,47] (Fig1B). The resistance to RNase R of specific circRNA candidates in Drosophila can be assessed directly by comparing their levels in mock and RNase R treated RNA-seq libraries. This comparison allows high-accuracy identification of bona fide circRNAs [9]. However, some linear RNAs are not efficiently degraded due to their secondary structures so the method does result in some false positive identifications [63]. Further, samples treated with RNase R cannot be used for circRNA quantification. This is because of the high variability between replicates, likely due to variations on the efficiency of the enzyme. In theory, it should be possible to overcome this challenge by normalizing the degradation to endogenous or spiked-in RNAs. But this has not yet been carefully validated.
Figure 1. An illustration of techniques used for detecting circRNAs.
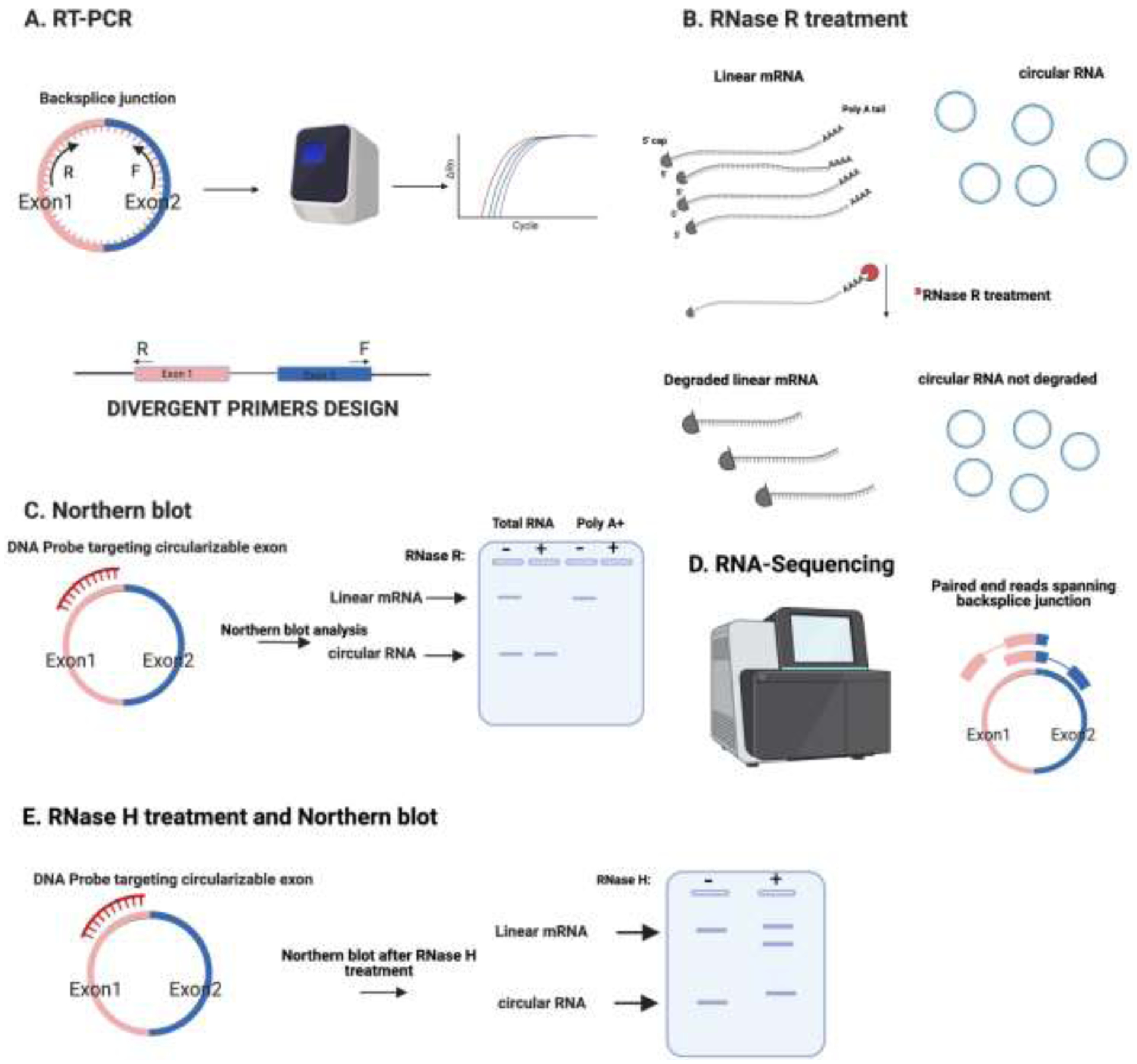
Some of the most utilized approaches include:
A) Reverse Transcription (RT)-PCR utilizing divergent primers for amplifying the backsplice junction of circRNAs. The levels of circRNAs are quantified using the cycle quantification (Cq) values.
B) RNase R treatment, this exoribonuclease degrades specifically the linear RNA, which results in the enrichment of circRNAs.
C) The design of a DNA probe targeting a circularizable exon, that allows the detection of the circRNA and its linear mRNA counterpart via northern blot. Samples from total and poly A+ RNA are either treated with RNase R or mock before running them on the agarose gel. The circRNA, which is resistant to RNase R treatment, prevails while its linear counterpart gets degraded.
D) Total RNA-sequencing with paired end reads that span the backsplice junction of circRNAs.
E) Design of DNA probes targeting a circularizable exon. RNase H treatment will degrade RNA in a DNA-RNA heterodimer which could be detected by northern blot: the linear mRNA has two bands and the migration of the circRNA changes.
One important consideration when analyzing circRNAs in flies or any other species after pretreatment with RNase R is that this treatment will degrade a substantial fraction of the RNA. A common mistake when performing circRNA validation is to use equivalent amounts of RNA in follow-up procedures (i.e., RT-qPCR or northern blot) without proper normalization. Many studies compare the amount of a given RNA (i.e., by qRT-PCR) from 1 ug of total RNA and compare it with 1ug of sample following RNase R treatment. However, the latter was generated from a much larger amount of RNA. Instead, similar amounts of RNA should be mock or RNase R treated and similar fraction of the resulting RNA should be used for the follow up applications. If performing RT-qPCR following the RNase R treatment, it is advisable to spike in RNAs from a different species in order to normalize for potential differences in reverse transcription efficiency due to differences in starting amount of RNA (the mock-treated samples generally has 5–10 higher RNA concentrations than the RNase R-treated samples) [28].
Bioinformatic tools to identify and quantify circRNAs
The advent of deep sequencing and advanced bioinformatic tools allowed profiling thousands of circRNAs from different tissues and cell samples. As the backsplice junction is the only sequence unique to the circRNAs, methods for identifying circRNAs in Drosophila from RNA-seq data rely on the detection of backsplice junction reads [60,64]. These reads do not align to the genome or transcriptome when stringent criteria are used, but the ends align head-to-tail to two exonic regions. There are a growing number of pipelines for circRNA identification including circRNA_finder [34], CIRCexplorer [23], CIRI [65], find_circ [16], MapSplice [66], DCC [67], KNIFE [33], and CIRCexplorer2 [68]. There is not yet a single gold-standard algorithm to detect bona fide circRNAs, and it is recommended that the pipelines be used in combination [64]. The main problem with this approach is that the metrics of the different pipelines are not always equivalent, making it difficult to compare the levels of circRNAs that are only detected by few pipelines. This is because the different pipelines utilize different alignment tools and parameters and have different thresholds for detection and filtering. In addition, not all algorithms are suited for both single-end [16] and paired-end [15,30,34] data. In addition to doubling the chances of finding a backsplicing junction, there is an additional advantage of using paired-end sequencing. The structure of potential circRNAs can be further validated if the paired-end mate read of a backsplicing junction read is within the candidate circRNA. Stringent expression thresholds for linear and circRNA read counts are also important [23,48,65,69]. Moreover, other additional criteria are generally used to filter out likely false positives. These include consideration of only circRNAs with characterized (annotated) splice junctions, resistance to RNase R, and size thresholds. The latter is sometimes complicated, as it is not clear how efficiently the introns within the circRNAs are spliced out.
A recent study has reported a sensitive computational tool called the Short Read CircRNA Pipeline (SRCP) that allows annotation and quantification of circRNAs from RNA-seq datasets from a combination of pipelines. The SRCP procedure consists of two steps. First, circRNAs are annotated by integrating the results of several pipelines in control and RNaseR-treated samples. This results in a list of likely circRNAs including their junction sequences. Second, the RNA-seq reads that do not align to the transcriptome are aligned to this list using a stringent but short-match strategy. This lead enables detection of circRNAs with very high sensitivity and quantification of all circRNAs, even those detected only by one or two pipelines. SRCP also quantifies the linear mRNA junctions, making it differential expression analysis possible [70].
Reverse-transcription PCR
A simple yet powerful method for identifying, validating, and quantifying circRNAs in Drosophila is RT-PCR. As circRNAs lack poly(A) tails, the cDNA must be synthesized using random primers (typically hexamers). Detection of circRNAs by RT-PCR relies on amplification of the unique backsplice junction using ‘divergent’ primers. The divergent primers align to the genome facing away from each other and cannot amplify linear transcripts [9] (Fig1A). The relative abundances of circRNAs can then be quantified by quantitative RT-PCR (qRT-PCR) [71].
Although RT-PCR is an efficient method for detecting circRNAs in flies there are chances of false positives due to template switching during the reverse transcription [72]. In addition, genomic duplication and trans-spliced transcripts [73] also produce PCR products that can be picked up by divergent primers [71]. Though the presence of backsplice junction can be confirmed by sequencing the PCR product, sequencing cannot distinguish real circRNA reads from false positives originating from trans-spliced or template-switched products. The circularity of the RNA can be confirmed using RNase R pretreatment [9]. It is advisable to sequence the amplified junction in mock- and RNase R-treated samples.
Northern blot
One of the most versatile and effective methods for detecting circRNAs is northern blot [74]. When using northern blots to visualize circRNAs in flies, it is possible to utilize probes complementary to the backsplice junction or to the exonic sequence that is circularized. The latter has the advantage of detecting both linear and circular transcripts. The circular and linear transcripts can sometimes be differentiated based on the distance migrated through the gel [75]. Usually, linear mRNAs are significantly longer than circRNAs, so they migrate more slowly during electrophoresis. Due to topological and resistance differences, however, if circular and linear molecules are the same size, the circRNA will migrate more slowly. It can, therefore, be difficult to identify the circular and linear molecules solely in their migration patterns, and linear- and circRNA-specific probes must be utilized [16,74,76]. The best way to identify circular and linear molecules by northern blot is to pretreat portions of the sample from Drosophila with RNase R or to selectively cleave the linear and circular transcripts with RNaseH. Pretreatment with RNase R will selectively degrade the linear RNAs, aiding in identification and serving as a validation step for the circRNA under study (Fig1C).
RNase H specifically cleaves the RNA in an RNA-DNA hybrid. Therefore, treatment of the sample with this enzyme in the presence of a short (20 nucleotides) DNA probe complementary to an exon contained within the circRNA will result in a change in mobility of the circRNA as well as in cleavage of the linear RNA isoform into two fragments (Fig1E). Therefore, when combined with northern blot, this pretreatment allows further verification of the circularity of a candidate circRNA in Drosophila, differentiation of linear and circular isoforms, and accurate determination of the size of the candidate circRNA.
Tools to Manipulate circRNA Levels
Challenges in manipulation of circRNA levels
The most common way to elucidate gene function is by manipulating the levels of the specific RNA. This is particularly challenging for circular RNAs for several reasons. First, typical mutagenesis approaches used for identifying genes cannot be generally used for circRNAs, as these molecules are generally produced from exons constitutively included in linear transcripts, so that deletions of the exons within circRNAs will result in de facto mutants of the linear mRNA encoded by the hosting gene. Second, almost all of a circRNA sequence is shared with the linear mRNAs, making silencing approaches problematic. Third, circRNAs are generated from genes with very long introns and by rules that are not fully understood, making it difficult to specifically inhibit (or upregulate) their expression. Despite these limitations, several approaches have been developed to manipulate circRNA levels even in whole animals.
Using small RNA to downregulate circRNAs
RNA interference (RNAi) is a post-transcriptional gene silencing mechanism [77] that regulates gene expression in eukaryotes [78]. Biologists have harnessed RNAi to enable silencing of specific genes [79]. The two types of RNAi effectors in mammalian systems are short interfering RNAs (siRNAs) and short-hairpin RNAs (shRNAs) [80]. The siRNAs are 21-base-pair, double stranded RNA molecules [81], and siRNAs have been used to downregulate circRNAs without altering linear transcript levels by targeting the backsplice junction [82]. Although siRNAs are specific, their efficiency depends on transfection efficiency, and these agents cause only to a transient knockdown of the target. In contrast, shRNAs can be stably expressed and have fewer off-target effects.
In Drosophila, in vivo RNAi screens have been performed using short and long dsRNAs [83]. In cell culture experiments dsRNA has proven to be a powerful tool for gene silencing due to efficient uptake and processing into many small interfering RNA (siRNA) effectors [84–86]. However, the production of different siRNA from single dsRNA can lead to an increased chance of off-target effects[83].
In Drosophila, long hairpins or shRNAs are used as silencing triggers. Long hairpins are less effective in silencing in the female germline than shRNAs and cannot be effectively restricted to the backsplice junction [87]. Use of the UAS-GAL4 system allows spatial and temporal expression of shRNAs in Drosophila. shRNA processing into microRNA-like double strands results in loading of the resulting effector into AGO1 and action through the major RNAi pathway in flies [87]. These developments set the stage for using endogenously expressed microRNA-like shRNAs to silence circRNAs in vivo [88].
One of the challenges of using silencing approaches, even when targeting the backsplicing junction with shRNAs, is that the siRNA or shRNA might target RNAs other than the desired circRNA, even RNAs with limited complementary to the so-called seed region of the siRNA. Off-targeting might result in partial degradation by the RNAi pathway or translational silencing by the miRNA pathway. The latter is unlikely in Drosophila as flies sort their small RNAs into relatively separate and independent pathways [89]. However, this possibility should be considered when performing shRNA experiments.
We recently generated the first flies in which circRNAs were specifically downregulated using shRNAs [88] (Fig2A(i)). In our case, we obtained very strong and specific knockdown by carefully selecting the shRNA sequences and by performing several controls. Specifically, during shRNA design, we filtered out guide or passenger strand sequences with more than 16 nucleotide matches with the fly genome [87]. This can be complicated if other regions of the genome have sequences with similarity to the circRNA as the region in the circRNA that can be targeted without considerable complementary to the linear mRNA is limited. Therefore, the linear mRNA expressed by the hosting gene is the main source of off-target effects. The presence of off targets were assessed as described previously [88]. Specifically, we (i) tested the silencing capacity of the shRNA, (ii) determined the effect of expression of the shRNA on mRNA and protein (when there are available antibodies) encoded by the linear mRNA, and (iii) determined by RNA-seq that no other mRNAs with partial complementarity or seed sequence complementarity were downregulated upon expression of the shRNA. Indeed, we found almost no off-targets for most of the shRNAs tested. In the few cases with off-target effects, we were able to redesign the shRNA by sliding it 1 or 2 bases in either the 5’ or 3’ direction. The generation of additional shRNAs also is important for validating phenotypes as it is unlikely that off-targets will prevail for different shRNAs. Although it could be argued that all the shRNAs that can be designed to target a given circRNA must be similar hence, an alternative approach is to rescue the knockdown. In conclusion, shRNAs can be used to efficiently downregulate circRNAs. However, it is good to confirm the authenticity of a phenotype caused by the knockdown of a circRNA by generating multiple shRNAs targeting the same backsplicing junction. In Drosophila, shRNA targeting of circRNAs (by miRNA-like shRNAs) seems to be very specific, likely due to the particular division of the small RNAs pathways in this insect.
Figure 2. An illustration of techniques used for modulating circRNAs.
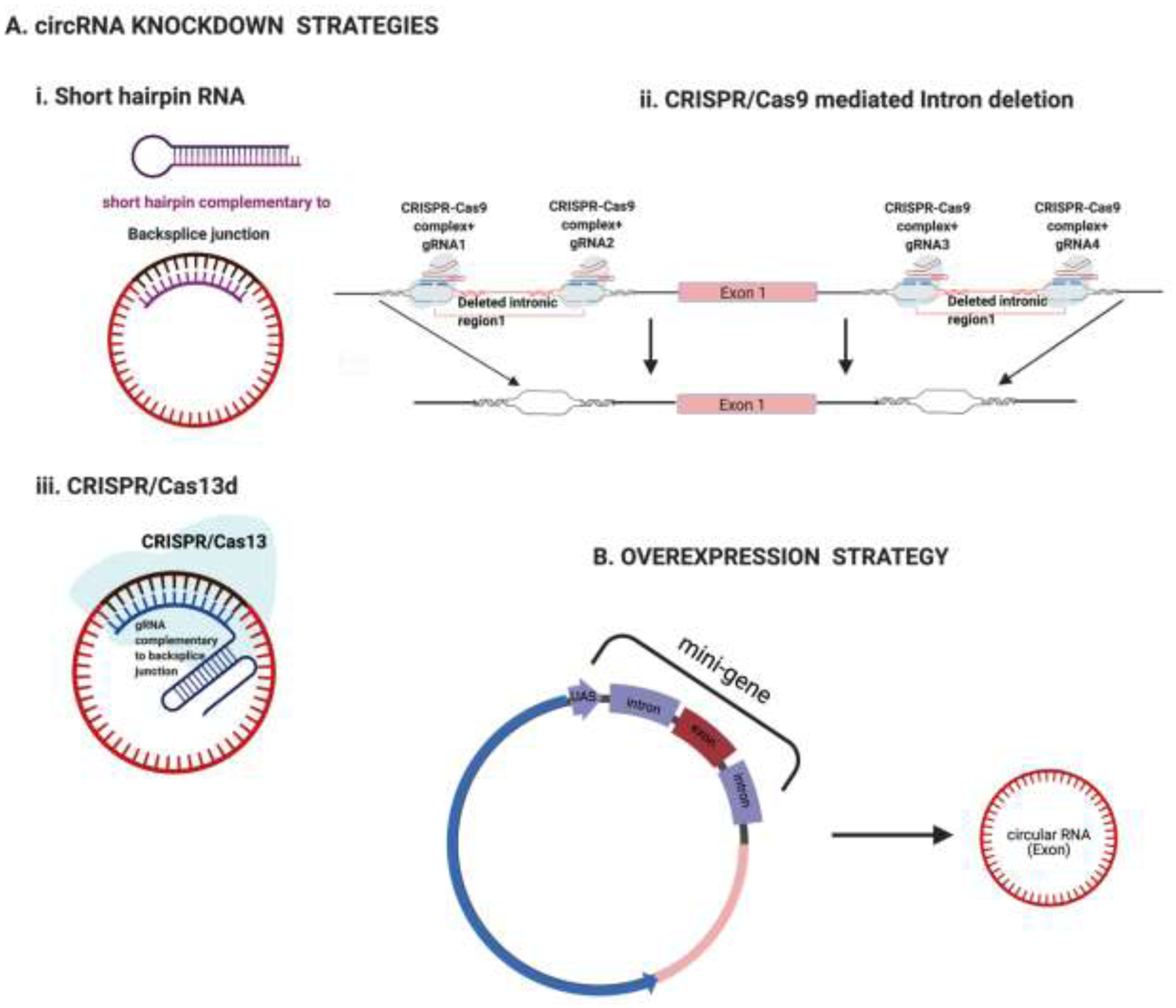
A. circRNA knockdown strategies: A schematic representation of
i) The design of short hairpin RNA targeting the backsplice junction of circRNAs, to knockdown the expression of circRNAs.
ii) CRISPR/Cas9 editing to modify or delete sequences in the flanking introns of a circularizable exon, to modulate the expression of circRNAs.
iii) CRISPR/Cas13 to downregulate circRNAs by designing a gRNA complementary to the backsplice junction.
B. circRNA overexpression strategy:
Schematic representation of the use of the common mini-gene system to artificially express circRNAs: entails the generation of a plasmid containing the circularizable exon, together with its flanking introns and canonical splice sites in a plasmid.
CRISPR-Cas13-mediated knockdown of circRNAs
CRISPR-Cas9 has revolutionized the field of genome editing. This system has been used by scientists to alter gene function in multiple ways from creating INDELs, to performing gene replacement or deletion, to activation of gene activity [90]. However, gene editing is not restricted to the DNA. Indeed, several Cas proteins with the ability to bind and cut RNA have been described, including CRISPR-Cas13 [90]. CRISPR-Cas13 is guided by specific RNAs that require longer complementarity than shRNAs (30–35 nucleotides instead of 22 nucleotides). This potentially means that CRISPR-Cas13 has less likelihood of off-target effects. Recent studies in Drosophila have demonstrated the efficiency of Cas13-mediated knockdown both ex vivo and in vivo [90,91].
Studies in mammalian cells established the possibility of using CRISPR effectors Cas13a, Cas13b, and Cas13d RNases [92,93] for the cleavage of single-stranded RNA target [94]. A recent study evaluated the efficiency and specificity of CRISPR-Cas13 as a tool to screen for circRNAs. CRISPR-RfxCas13d with single guide RNAs that spanned the backspliced junctions of various circRNAs, efficiently targeted the circRNAs without affecting the levels of the linear mRNAs [94] (Fig2A(iii)). Another report described use of the Cas13d enzyme CasRx to reduce levels of circRNAs both in the nucleus and cytosol [95]. Importantly, the authors showed that Cas13 was more specific for the targeted circRNAs than were shRNAs.
Genetic mutation of circRNA-promoting sequences
Complementary short repeats in flanking introns can facilitate circularization of an exon [48]. Deletion of an upstream intron using CRISPR/Cas9 in cell culture studies efficiently downregulated circRNA expression without altering the levels of the mRNA from the same gene locus [96]. Hence deleting circRNA-promoting sequences (or even whole introns) might be a straightforward way to inhibit circRNA biogenesis (Fig2A(ii)). This approach offers many advantages over silencing strategies. First is lack of off-target effects (if, of course, there are no major effects on the levels of the linear mRNA when an intron is deleted). Second, this strategy might uncover potential cis effects on the synthesis of the circRNA (if is highly produced and the biosynthesis is at the expense of the linear RNA). Notably, these two advantages might be intertwined as for those circRNAs with cis regulatory functions there might be strong effects on the levels of the linear RNA. This highlights the importance of using complementary approaches (i.e., CRISPR mutations and shRNAs).
It remains challenging to identify the sequences that influence circRNA production. Several groups have demonstrated that the sequences required for circularization of an exon are in flanking introns[17,22–26,28,48].Work performed in Drosophila S2 cells using minigenes has demonstrated that modifying or deleting flanking intronic sequences results in downregulation of circRNA expression. Deletion of up to 300 base pairs of flanking intronic sequence of the circularizing second exon of mbl in a minigene system resulted in decrease of circRNA expression. Interestingly, deletion of intronic regions with complementary sequences in the other flanking intron significantly modified the expression of the circRNA [17]. Further, mutating the binding sequences of the splicing factor MBL that promotes the circularization of its own exons by binding to flanking introns also modifies the expression of circRNA [17]. Also, another report in Drosophila S2 cells has shown that introns flanking the circularizable exon of Laccase2 was sufficient for circRNA production and deleting the repeats in the introns abolished circularization[27]. Similarly, endogenous circRNA expression can also modified by either deleting or modifying the flanking intronic sequences using CRISPR/Cas9-based genome editing tools [76]. However, deleting intronic sequences responsible for circularization does not allow for tissue-specific resolution unless is achieved in adults (e.g., by the use of specifically inserted FRT sites) and cannot be used for globally screening the functions of circRNAs [88].
Overexpression of circRNAs
Previous reports suggested that the circularization of an exon is favored by the presence of inverted repeat sequences, particularly Alu elements, in the flanking introns in mammals [48]. Further, exon circularization can be efficiently achieved when splice site sequences and short inverted repeats of about 40 nucleotides are present [24]. This property can be used to overexpress specific circRNAs in order to study their biological functions. An entire gene [71] or just a mini-gene system consisting on the circularizable exon flanked by sections of the flanking introns (Fig2B) or by long complementary repeats can be used to induce circRNA expression [17,22,24,25,27,47]. However, although highly effective in whole animals, overexpression strategies tend to generate linear concatemers when expressed in cultured cells [97]. This could be due to the generation of double-stranded RNA and trans splicing from different minigene transcripts or from artifacts generated by rolling-circle transcription. The latter occurs when RNA polymerase bypasses the termination signal and transcribes the entire plasmid generating a concatemer of RNA. When this leads to linear concatemers containing the circularized exon sequence or to RNA with scrambled junction sequences, spurious identification and overestimation of circRNA levels can occur [71]. In theory, overexpression of circRNA can also be achieved by introducing binding sites for circRNA-promoting proteins like MBL or QKI in the introns flanking the circularizable exon.
Spatial and temporal expression of circRNA in Drosophila melanogaster
Some of the key findings in the circRNA field have been made in Drosophila. These include the observations that circRNA biogenesis competes with linear splicing[17], that intronic sequences are key for exon circularization [17,22,24,34], that circRNAs are enriched in neural tissues, accumulate with age [34], and have potential functions in aging [38], as well as findings that some circRNAs are translated [43] and have functions in trans [88]. Drosophila melanogaster is an ideal organism for uncovering functions and mechanisms of action of new types of molecules because of the large number of genetic and molecular tools available for manipulation of gene expression and gene targeting. This includes the existence of inducible systems for spatial and temporal regulation of any gene of interest [98].
Tissue-specific transgene expression can be achieved using the bi-partite GAL4-UAS system [99]. Over the past few decades, biologists have generated collection of fly lines expressing GAL4 under different cell-type-specific promoters (GAL4 drivers) and also many different UAS transgenes for expression of proteins, RNAi libraries, and reporter genes [100,101]. These reagents allow performing mutant rescues, cell ablation, and interference experiments in a tissue-specific manner. In flies, the functions of circRNAs can be assessed in a cell-type specific manner, by using the UAS-GAL4 system to spatially silence by shRNAs [88] or to overexpress a specific circRNA [43].
Although, the UAS-GAL4 system allows for spatial control of gene expression, temporal control is more complicated as the promoters and enhancers used in the GAL4 are active at multiple stages of development [102]. To overcome this limitation, researchers have generated flies expressing the temperature sensitive GAL80 transgene (GAL80ts). GAL80 is an antagonist of GAL4 activity [103,104], so in presence of the GAL80ts protein, GAL4 is not active unless the temperature is raised at 29 °C. This allows for a system with exquisite temporal and spatial control (the latter resulting from the GAL4 driver). However, the temperature-sensitive GAL80 system is not ideal for studies with circRNAs, as circRNA levels dramatically increase with temperature in Drosophila [17]. Another way to conditionally express a transgene is by using a drug-inducible system. One example is a chimeric protein that contains the DNA binding domain from GAL4, the receptor ligand-binding domain from human progesterone, and the activation domain from human p65 [105,106]. In this system, the activation of transcription is achieved when the chimeric molecule binds to the UAS sequence in the presence of anti-progestin RU486. Importantly, there are well characterized GAL4-GS drivers available for expression in brain and muscle [106], the two tissues with the high expression levels of circRNA.
Phenotypic assays for study of circRNA functions in Drosophila
Advantages of fly behavior assays
Drosophila melanogaster is a very versatile model organism with a vast toolkit for genetic manipulations. In addition, flies perform a wide range of behaviors which are used to study functions of other classes of RNA or protein and can also be used to screen for potential functions of circRNAs. These include feeding, locomotion, and more complex social behaviors like courtship [107]. Importantly, behavioral screens in flies are easy, inexpensive, and can be done on a large scale [108]. Behavioral assays can be used to screen for phenotypes upon manipulation of circRNA levels in vivo. Some of the phenotypes that could be used to perform enhancer-suppressor genetic screenings for identifying modulators of circRNA functions are described below.
Viability assay
Viability assays not only help to gauge the fitness of a strain [109] but also allows one to know the lethal effects of downregulating or overexpressing circRNAs during different stages of development. To understand the effects of loss of function of circRNAs on viability, one can quantify progeny from embryonic to adult stages in both knockdown and control flies (Fig3A). A statistical test can be done to determine any significant changes in number at each developmental stage and evaluate for stage-specific lethality [88]. Additionally, counting the males and females separately can reveal sex-specific lethality caused by misexpression of circRNAs.
Figure 3. Schematic of Key behavioral assays to study the functions of circRNAs.
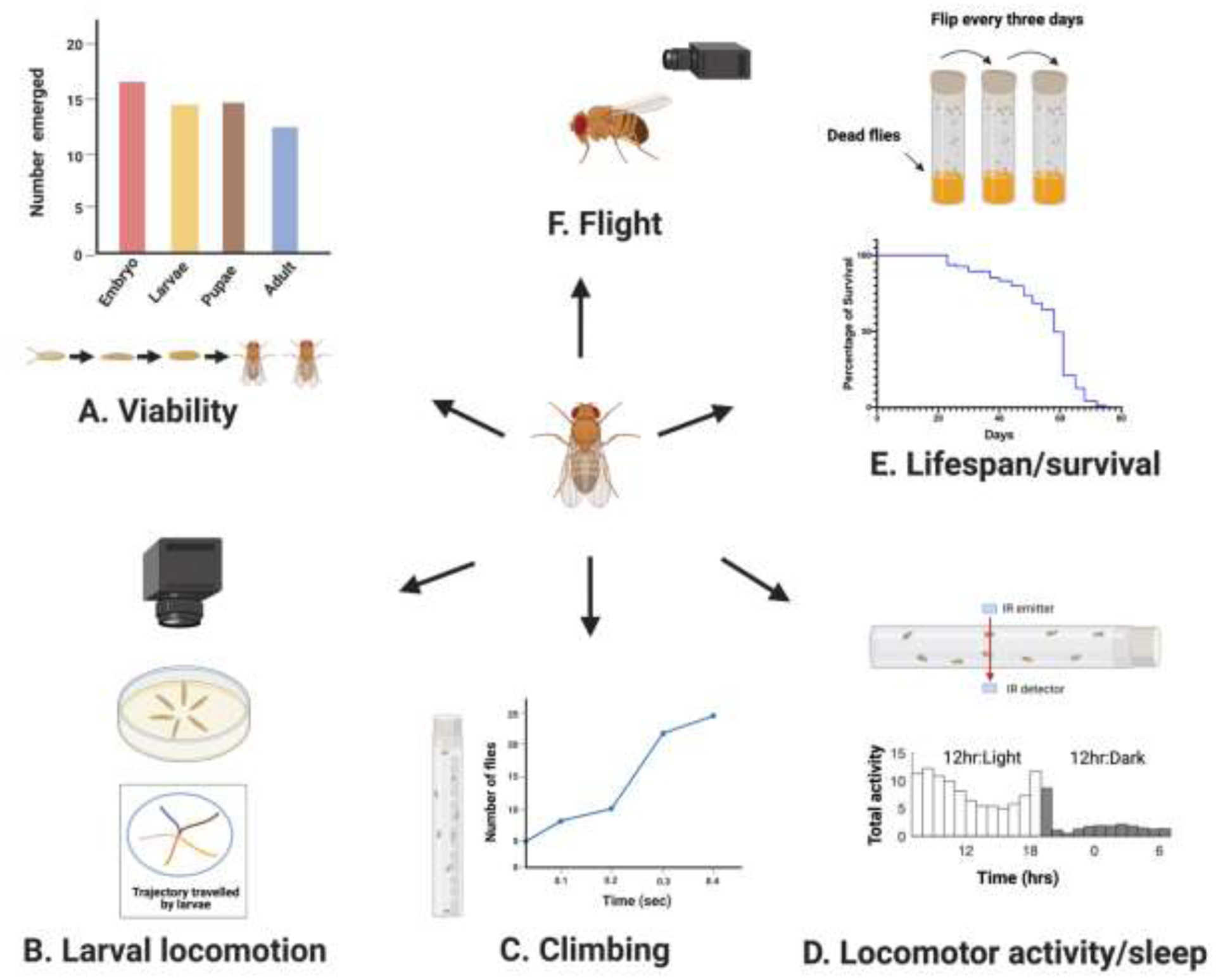
A) Viability assay: for tracking flies of interest from embryonic to adult stage in order to evaluate lethality levels for each developmental stage. This data can then be represented in a bar graph as shown in the example.
B) Larval locomotion assay: 3rd instar larvae placed in a 2% agar petri-dish are recorded for one minute. The larval trajectory can then be computationally drawn. In this example, the trajectories of five different larvae have been plotted.
C) Negative-geotaxis or climbing assay: An illustrative experimental setup for monitoring locomotor activity based on negative-geotaxis behavior of the fly. An example is represented in a line graph to illustrate the number of flies that reach a predetermined height in a given period of time.
D) Adult fly locomotor activity/ sleep: The Data Activity Monitor (DAM) system is used to record the activity of single flies in a tube by counting the number of times in which the fly interrupts the infra-red beam of the monitor. An example histogram of averaged total activity of flies in 12:12hr Light: Dark
E) Lifespan assay: flies are flipped every three days and the number of dead flies is recorded. This allows the generation of survival plots such as the example shown.
F) Flight assay: a high-speed video recording can be used to assess the wing beat frequency of the flies.
Morphological phenotypes
A severe effect caused by knockdown or overexpression of a gene or a circRNA can lead to visible adult phenotypes[88]. The measurable morphological defects range from deformity in body parts, to abnormality in wing posture or wing venation, to defects in eye size or appearance when compared to control flies. The types of morphological phenotypes depend on the GAL4 driver utilized to perturb the circRNA levels.
Behavioral assays to assess locomotor functions
The abilities to walk, climb, and fly have been monitored to shed light into muscular and neuronal processes involved in neuromuscular disorders in fly models [110,111]. These behaviors can be used to assess the functions of circRNAs in flies. Changes in crawling during the larval stage are reliable indicators of early developmental defects. The larval locomotion assay is especially useful when the genetic manipulation leads to lethality in the pupal stage or adult stage [108]. Defects in the contraction ability of 3rd instar larval muscle and speed of crawling are good indicators of muscle defects [112]. Larval crawling behavior assays can be very useful in understanding the role of circRNAs in muscular, neuronal, or neuromuscular tissues by restricting the expression of the circRNAs with the UAS-GAL4 system to the CNS and/or muscle cells. A simple, sensitive, and low-cost behavioral set up can be used to study effects of circRNA knockdown or overexpression versus controls (Fig3B).
Another behavioral assay called the climbing assay that is based on the principle that flies have an innate tendency to climb up against gravity [113,114] is a powerful tool to detect general motor defects in adult flies and also has been used in studies of fly models of Parkinson’s disease [115]. This assay can be performed in high-throughput screens to identify circRNAs with possible neuromuscular functions (Fig3C).
Lastly, Drosophila traverse long distances for foraging [116]. This crucial behavior requires fast and active control mechanisms [117]. The flight assay is an effective tool to study locomotor behavior in adult flies [118]. Flight performance like wing beat and flight during free fall can be used in screens for circRNAs that directly or indirectly alter the ability to fly (Fig3F).
Circadian and sleep behavior
Flies, like most animals, display strong daily rhythms in physiology, behavior, and gene expression. These circadian rhythms repeat every 24 hours and are self-sustaining and independent of external environmental cues [119,120]. Pioneering research in Drosophila uncovered the genetic bases of circadian rhythms [121,122]. In flies, the circadian clock controls physiologically important process like time of eclosion [122], sleep, and metabolism [123]. In insects, sleep is important in development [124], reproduction [125], memory, and learning [126]. Drosophila has been used as an model organism to dissect the genetic basis of sleep [127]. Locomotor rhythms and sleep can be assessed easily and simultaneously in the lab and can be used for screening purposes (Fig3D). To study the biological relevance of circRNAs, locomotor activity rhythms and sleep can be studied when the expression of circRNAs are modulated.
Lifespan assay
Aging leads to decline in crucial biological functions over a period of time. Aging results in reduction in fertility and finally death [128]. Drosophila has been used for almost 100 years as a model organism to study aging [129]. The normal average lifespan of flies maintained at 25 °C is 70 days, and they can live up to a maximum of 90 days [126]. Changes in lifespan can result from effects on metabolism, behavior, physical activity, stress resistance, fertility, and neuronal and immune functions [128]. A recent study demonstrated the role of an insulin-sensitive circRNA in regulating lifespan in Drosophila [38]. Lifespan is easy to measure, and this parameter can be used for effects of circRNA knockdown or overexpression and to identify genetic interactions (Fig3E).
Use of genetic-suppressor screening to identify genes in circRNA pathways
A repertoire of Drosophila genetic tools has been used for large-scale genetic enhancer-suppressor screens. This type of screening provides an unbiased approach for identifying genes involved in particular processes or pathways. As examples, embryonic lethal screens allowed identification of mutants in signaling pathways in embryo patterning like Wingless and Spitz [131–133]. Initial genetic screenings involved the use of homozygous mutations and hence have the limitation of genes with homozygous lethal phenotypes. However, overexpression or RNAi screenings are now standard and, when combined with spatial and temporal expression, can reveal the need for these genes in specific stages of development. Importantly, there are publicly available knockdown and overexpression transgenes for almost all genes in Drosophila. Therefore, following the identification of a specific phenotype associated with modulation of a circRNA, an enhancer suppressor screening can provide key insights into the mechanism of action of the circRNA.
Conclusions and future directions
Research on circRNAs has exploded in the last few years, but there is still much to learn about these molecules. Research has somewhat reached a plateau due to the complexity of specifically modulating circRNAs in vivo and performing perturbation experiments. However, new tools available in model organisms are likely to significantly increase the pace of advances in the field. In particular, Drosophila will likely prove to be a key model for circRNA study. Flies have been critical in a number of pioneering discoveries in the field and have the potential to help unravel molecular and behavioral functions of circRNAs. Moreover, fly models could also be utilized to test the usefulness of circRNAs in synthetic biology and as molecular memory tools. Last but not least, new research suggests that circRNAs may influence the development or progression of human neurodegenerative diseases like amyotrophic lateral sclerosis, Alzheimer’s disease, and Parkinson’s disease. As there are well-established models of those diseases in flies, this model organism could be used to study the roles of circRNAs in neurodegenerative diseases.
Some circRNAs are highly abundant in fly heads and of physiological relevance
Validating circRNAs requires consideration to computational & experimental artifacts
There are several methods for modulating levels of circRNAs in vivo in Drosophila
Drosophila provides a very versatile system to determine functions of circRNAs
ACKNOWLEDGMENTS:
We thank VV. Konakondla and J.Harris for the valuable discussions and comments on the manuscript. AM.Anduaga and N.Reddy for proof reading the manuscript. This work was supported by NIH R01 Grants: R01GM122406 and R01-AG057700. Figure illustrations were Created with BioRender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors declare that they have no conflict of interest
References
- [1].Halbeisen RE, Galgano A, Scherrer T, Gerber AP, Post-transcriptional gene regulation: From genome-wide studies to principles, Cell. Mol. Life Sci 65 (2008) 798–813. 10.1007/s00018-007-7447-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Neugebauer KM, On the importance of being co-transcriptional, J. Cell Sci 115 (2002) 3865–3871. 10.1242/jcs.00073. [DOI] [PubMed] [Google Scholar]
- [3].Khodor YL, Rodriguez J, Abruzzi KC, Tang CHA, Marr MT, Rosbash M, Nascent-seq indicates widespread cotranscriptional pre-mRNA splicing in Drosophila, Genes Dev 25 (2011) 2502–2512. 10.1101/gad.178962.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ameur A, Zaghlool A, Halvardson J, Wetterbom A, Gyllensten U, Cavelier L, Feuk L, Total RNA sequencing reveals nascent transcription and widespread co-transcriptional splicing in the human brain, Nat. Struct. Mol. Biol 18 (2011) 1435–1440. 10.1038/nsmb.2143. [DOI] [PubMed] [Google Scholar]
- [5].Black DL, Protein diversity from alternative splicing: A challenge for bioinformatics and post-genome biology, Cell 103 (2000) 367–370. 10.1016/S0092-8674(00)00128-8. [DOI] [PubMed] [Google Scholar]
- [6].Graveley BR, Alternative splicing: Increasing diversity in the proteomic world, Trends Genet 17 (2001) 100–107. 10.1016/S0168-9525(00)02176-4. [DOI] [PubMed] [Google Scholar]
- [7].Blencowe BJ, Alternative Splicing: New Insights from Global Analyses, Cell 126 (2006) 37–47. 10.1016/j.cell.2006.06.023. [DOI] [PubMed] [Google Scholar]
- [8].Naftelberg S, Schor IE, Ast G, Kornblihtt AR, Regulation of alternative splicing through coupling with transcription and chromatin structure, Annu. Rev. Biochem 84 (2015) 165–198. 10.1146/annurev-biochem-060614-034242. [DOI] [PubMed] [Google Scholar]
- [9].Jeck WR, Sharpless NE, Detecting and characterizing circular RNAs, Nat. Biotechnol 32 (2014) 453–461. 10.1038/nbt.2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhang Y, Zhang XO, Chen T, Xiang JF, Yin QF, Xing YH, Zhu S, Yang L, Chen LL, Circular Intronic Long Noncoding RNAs, Mol. Cell 51 (2013) 792–806. 10.1016/j.molcel.2013.08.017. [DOI] [PubMed] [Google Scholar]
- [11].Tay MLI, Pek JW, Maternally Inherited Stable Intronic Sequence RNA Triggers a Self-Reinforcing Feedback Loop during Development, Curr. Biol 27 (2017) 1062–1067. 10.1016/j.cub.2017.02.040. [DOI] [PubMed] [Google Scholar]
- [12].Lu Z, Filonov GS, Noto JJ, Schmidt CA, Hatkevich TL, Wen Y, Jaffrey SR, Gregory Matera A, Metazoan tRNA introns generate stable circular RNAs in vivo, RNA 21 (2015) 1554–1565. 10.1261/rna.052944.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Talhouarne GJS, Gall JG, Lariat intronic RNAs in the cytoplasm of Xenopus tropicalis oocytes, RNA 20 (2014) 1476–1487. 10.1261/rna.045781.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Talhouarne GJS, Gall JG, Lariat intronic RNAs in the cytoplasm of vertebrate cells, Proc. Natl. Acad. Sci. U. S. A 115 (2018) E7970–E7977. 10.1073/pnas.1808816115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO, Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types, PLoS One 7 (2012). 10.1371/journal.pone.0030733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, Loewer A, Ziebold U, Landthaler M, Kocks C, Le Noble F, Rajewsky N, Circular RNAs are a large class of animal RNAs with regulatory potency, Nature 495 (2013) 333–338. 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- [17].Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N, Kadener S, circRNA Biogenesis Competes with Pre-mRNA Splicing, Mol. Cell 56 (2014) 55–66. 10.1016/j.molcel.2014.08.019. [DOI] [PubMed] [Google Scholar]
- [18].Barrett SP, Wang PL, Salzman J, Circular RNA biogenesis can proceed through an exon-containing lariat precursor, Elife 4 (2015) 1–18. 10.7554/eLife.07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Broadbent KM, Broadbent JC, Ribacke U, Wirth D, Rinn JL, Sabeti PC, Strand-specific RNA sequencing in Plasmodium falciparum malaria identifies developmentally regulated long non-coding RNA and circular RNA, BMC Genomics 16 (2015) 1–22. 10.1186/s12864-015-1603-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lu T, Cui L, Zhou Y, Zhu C, Fan D, Gong H, Zhao Q, Zhou C, Zhao Y, Lu D, Luo J, Wang Y, Tian Q, Feng Q, Huang T, Han B, Transcriptome-wide investigation of circular RNAs in rice, Rna 21 (2015) 2076–2087. 10.1261/rna.052282.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang PL, Bao Y, Yee MC, Barrett SP, Hogan GJ, Olsen MN, Dinneny JR, Brown PO, Salzman J, Circular RNA is expressed across the eukaryotic tree of life, PLoS One 9 (2014). 10.1371/journal.pone.0090859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ivanov A, Memczak S, Wyler E, Torti F, Porath HT, Orejuela MR, Piechotta M, Levanon EY, Landthaler M, Dieterich C, Rajewsky N, Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals, Cell Rep 10 (2015) 170–177. 10.1016/j.celrep.2014.12.019. [DOI] [PubMed] [Google Scholar]
- [23].Zhang XO, Bin Wang H, Zhang Y, Lu X, Chen LL, Yang L, Complementary sequence-mediated exon circularization, Cell 159 (2014) 134–147. 10.1016/j.cell.2014.09.001. [DOI] [PubMed] [Google Scholar]
- [24].Liang D, Wilusz JE, Short intronic repeat sequences facilitate circular RNA production, Genes Dev 28 (2014) 2233–2247. 10.1101/gad.251926.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Starke S, Jost I, Rossbach O, Schneider T, Schreiner S, Hung LH, Bindereif A, Exon circularization requires canonical splice signals, Cell Rep 10 (2015) 103–111. 10.1016/j.celrep.2014.12.002. [DOI] [PubMed] [Google Scholar]
- [26].Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, Roslan S, Schreiber AW, Gregory PA, Goodall GJ, The RNA binding protein quaking regulates formation of circRNAs, Cell 160 (2015) 1125–1134. 10.1016/j.cell.2015.02.014. [DOI] [PubMed] [Google Scholar]
- [27].Kramer MC, Liang D, Tatomer DC, Gold B, March ZM, Cherry S, Wilusz JE, Combinatorial control of Drosophila circular RNA expression by intronic repeats, hnRNPs, and SR proteins., Genes Dev 29 (2015) 2168–82. 10.1101/gad.270421.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rybak-Wolf A, Stottmeister C, Glažar P, Jens M, Pino N, Giusti S, Hanan M, Behm M, Bartok O, Ashwal-Fluss R, Herzog M, Schreyer L, Papavasileiou P, Ivanov A, Öhman M, Refojo D, Kadener S, Rajewsky N, Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed., Mol. Cell 58 (2015) 870–85. 10.1016/j.molcel.2015.03.027. [DOI] [PubMed] [Google Scholar]
- [29].Aktaş T, Ilik IA, Maticzka D, Bhardwaj V, Pessoa Rodrigues C, Mittler G, Manke T, Backofen R, Akhtar A, DHX9 suppresses RNA processing defects originating from the Alu invasion of the human genome, Nature 544 (2017) 115–119. 10.1038/nature21715. [DOI] [PubMed] [Google Scholar]
- [30].Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO, Cell-type specific features of circular RNA expression., PLoS Genet 9 (2013) e1003777. 10.1371/journal.pgen.1003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hanan M, Soreq H, Kadener S, CircRNAs in the brain., RNA Biol 14 (2017) 1028–1034. 10.1080/15476286.2016.1255398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].You X, Vlatkovic I, Babic A, Will T, Epstein I, Tushev G, Akbalik G, Wang M, Glock C, Quedenau C, Wang X, Hou J, Liu H, Sun W, Sambandan S, Chen T, Schuman EM, Chen W, Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity, Nat. Neurosci 18 (2015) 603–610. 10.1038/nn.3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Szabo L, Morey R, Palpant NJ, Wang PL, Afari N, Jiang C, Parast MM, Murry CE, Laurent LC, Salzman J, Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development, Genome Biol 16 (2015) 1–26. 10.1186/s13059-015-0690-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Westholm JO, Miura P, Olson S, Shenker S, Joseph B, Sanfilippo P, Celniker SE, Graveley BR, Lai EC, Genome-wide Analysis of Drosophila Circular RNAs Reveals Their Structural and Sequence Properties and Age-Dependent Neural Accumulation, Cell Rep 9 (2014) 1966–1980. 10.1016/j.celrep.2014.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cortés-López M, Gruner MR, Cooper DA, Gruner HN, Voda AI, van der Linden AM, Miura P, Global accumulation of circRNAs during aging in Caenorhabditis elegans, BMC Genomics 19 (2018) 1–12. 10.1186/s12864-017-4386-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Knupp D, Miura P, CircRNA accumulation: A new hallmark of aging?, Mech. Ageing Dev 173 (2018) 71–79. 10.1016/j.mad.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hanan M, Simchovitz A, Yayon N, Vaknine S, Cohen-Fultheim R, Karmon M, Madrer N, Rohrlich TM, Maman M, Bennett ER, Greenberg DS, Meshorer E, Levanon EY, Soreq H, Kadener S, A Parkinson’s disease Circ RNA s Resource reveals a link between circ SLC 8A1 and oxidative stress, EMBO Mol. Med 12 (2020) 1–19. 10.15252/emmm.201911942. [DOI] [Google Scholar]
- [38].Weigelt CM, Sehgal R, Tain LS, Cheng J, Eßer J, Pahl A, Dieterich C, Grönke S, Partridge L, An Insulin-Sensitive Circular RNA that Regulates Lifespan in Drosophila, J. Clean. Prod (2020). 10.1016/j.molcel.2020.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Errichelli L, Dini Modigliani S, Laneve P, Colantoni A, Legnini I, Capauto D, Rosa A, De Santis R, Scarfò R, Peruzzi G, Lu L, Caffarelli E, Shneider NA, Morlando M, Bozzoni I, FUS affects circular RNA expression in murine embryonic stem cell-derived motor neurons, Nat. Commun 8 (2017) 1–11. 10.1038/ncomms14741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Begemann G, Paricio N, Artero R, Kiss I, Pérez-Alonso M, Mlodzik M, Muscleblind, a gene required for photoreceptor differentiation in Drosophila, encodes novel nuclear Cys3His-type zinc-finger-containing proteins, Development 124 (1997) 4321–4331. [DOI] [PubMed] [Google Scholar]
- [41].Artero R, Prokop A, Paricio N, Begemann G, Pueyo I, Mlodzik M, Perez-Alonso M, Baylies MK, The muscleblind gene participates in the organization of Z-bands and epidermal attachments of Drosophila muscles and is regulated by Dmef2, Dev. Biol 195 (1998) 131–143. 10.1006/dbio.1997.8833. [DOI] [PubMed] [Google Scholar]
- [42].Irion U, Drosophila muscleblind Codes for Proteins with One and Two Tandem Zinc Finger Motifs, PLoS One 7 (2012) e34248. 10.1371/journal.pone.0034248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Pamudurti NR, Bartok O, Jens M, Ashwal-Fluss R, Stottmeister C, Ruhe L, Hanan M, Wyler E, Perez-Hernandez D, Ramberger E, Shenzis S, Samson M, Dittmar G, Landthaler M, Chekulaeva M, Rajewsky N, Kadener S, Translation of CircRNAs, Mol. Cell 66 (2017) 9–21. e7. 10.1016/j.molcel.2017.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Legnini I, Di Timoteo G, Rossi F, Morlando M, Briganti F, Sthandier O, Fatica A, Santini T, Andronache A, Wade M, Laneve P, Rajewsky N, Bozzoni I, Circ-ZNF609 Is a Circular RNA that Can Be Translated and Functions in Myogenesis, Mol. Cell 66 (2017) 22–37. e9. 10.1016/j.molcel.2017.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, Jin Y, Yang Y, Chen LL, Wang Y, Wong CCL, Xiao X, Wang Z, Extensive translation of circular RNAs driven by N 6 -methyladenosine, Cell Res 27 (2017) 626–641. 10.1038/cr.2017.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Hansen TB, Wiklund ED, Bramsen JB, Villadsen SB, Statham AL, Clark SJ, Kjems J, MiRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA, EMBO J 30 (2011) 4414–4422. 10.1038/emboj.2011.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J, Natural RNA circles function as efficient microRNA sponges, Nature 495 (2013) 384–388. 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- [48].Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE, Erratum: Circular RNAs are abundant, conserved, and associated with ALU repeats (RNA (156)), Rna 19 (2013) 426. 10.1261/rna.035667.112.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hentze MW, Preiss T, Circular RNAs: splicing’s enigma variations., EMBO J 32 (2013) 923–5. 10.1038/emboj.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Piwecka M, Glažar P, Hernandez-Miranda LR, Memczak S, Wolf SA, Rybak-Wolf A, Filipchyk A, Klironomos F, Cerda Jara CA, Fenske P, Trimbuch T, Zywitza V, Plass M, Schreyer L, Ayoub S, Kocks C, Kühn R, Rosenmund C, Birchmeier C, Rajewsky N, Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function, Science (80-.). 357 (2017) eaam8526. 10.1126/science.aam8526. [DOI] [PubMed] [Google Scholar]
- [51].Du WW, Yang W, Liu E, Yang Z, Dhaliwal P, Yang BB, Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2, Nucleic Acids Res 44 (2016) 2846–2858. 10.1093/nar/gkw027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Patop IL, Kadener S, circRNAs in Cancer HHS Public Access, Curr Opin Genet Dev 48 (2018) 121–127. 10.1016/j.gde.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Liu CX, Li X, Nan F, Jiang S, Gao X, Guo SK, Xue W, Cui Y, Dong K, Ding H, Qu B, Zhou Z, Shen N, Yang L, Chen LL, Structure and Degradation of Circular RNAs Regulate PKR Activation in Innate Immunity, Cell 177 (2019) 865–880. e21. 10.1016/j.cell.2019.03.046. [DOI] [PubMed] [Google Scholar]
- [54].Chen YG, Chen R, Ahmad S, Verma R, Kasturi SP, Amaya L, Broughton JP, Kim J, Cadena C, Pulendran B, Hur S, Chang HY, N6-Methyladenosine Modification Controls Circular RNA Immunity, Mol. Cell 76 (2019) 96–109. e9. 10.1016/j.molcel.2019.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Chen YG, Kim MV, Chen X, Batista PJ, Aoyama S, Wilusz JE, Iwasaki A, Chang HY, Sensing Self and Foreign Circular RNAs by Intron Identity, Mol. Cell 67 (2017) 228–238. e5. 10.1016/j.molcel.2017.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Fan X, Zhang X, Wu X, Guo H, Hu Y, Tang F, Huang Y, Single-cell RNA-seq transcriptome analysis of linear and circular RNAs in mouse preimplantation embryos, Genome Biol 16 (2015) 1–17. 10.1186/s13059-015-0706-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Gao Y, Wang J, Zheng Y, Zhang J, Chen S, Zhao F, Comprehensive identification of internal structure and alternative splicing events in circular RNAs, Nat. Commun 7 (2016). 10.1038/ncomms12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Dong R, Ma XK, Chen LL, Yang L, Increased complexity of circRNA expression during species evolution, RNA Biol 14 (2017) 1064–1074. 10.1080/15476286.2016.1269999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Yang L, Duff MO, Graveley BR, Carmichael GG, Chen LL, Genomewide characterization of non-polyadenylated RNAs, Genome Biol 12 (2011) 1–14. 10.1186/gb-2011-12-2-r16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gao Y, Zhao F, Computational Strategies for Exploring Circular RNAs, Trends Genet 34 (2018) 389–400. 10.1016/j.tig.2017.12.016. [DOI] [PubMed] [Google Scholar]
- [61].Suzuki H, Zuo Y, Wang J, Zhang MQ, Malhotra A, Mayeda A, Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing, Nucleic Acids Res 34 (2006). 10.1093/nar/gkl151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Xiao MS, Wilusz JE, An improved method for circular RNA purification using RNase R that efficiently removes linear RNAs containing G-quadruplexes or structured 3’ ends, Nucleic Acids Res 47 (2019) 8755–8769. 10.1093/nar/gkz576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Panda AC, De S, Grammatikakis I, Munk R, Yang X, Piao Y, Dudekula DB, Abdelmohsen K, Gorospe M, High-purity circular RNA isolation method (RPAD) reveals vast collection of intronic circRNAs, Nucleic Acids Res 45 (2017) 1–13. 10.1093/nar/gkx297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Hansen TB, Improved circRNA Identification by Combining Prediction Algorithms, Front. Cell Dev. Biol 6 (2018) 20. 10.3389/fcell.2018.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Gao Y, Wang J, Zhao F, CIRI: An efficient and unbiased algorithm for de novo circular RNA identification, Genome Biol 16 (2015). 10.1186/s13059-014-0571-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou CM, MacLeod JN, Chiang DY, Prins JF, Liu J, MapSplice: Accurate mapping of RNA-seq reads for splice junction discovery, Nucleic Acids Res 38 (2010) e178–e178. 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Cheng J, Metge F, Dieterich C, Specific identification and quantification of circular RNAs from sequencing data, Bioinformatics 32 (2016) 1094–1096. 10.1093/bioinformatics/btv656. [DOI] [PubMed] [Google Scholar]
- [68].Zhang J, Chen S, Yang J, Zhao F, Accurate quantification of circular RNAs identifies extensive circular isoform switching events, Nat. Commun 11 (2020) 90. 10.1038/s41467-019-13840-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Guo JU, Agarwal V, Guo H, Bartel DP, Expanded identification and characterization of mammalian circular RNAs, Genome Biol 15 (2014) 1–14. 10.1186/s13059-014-0409-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Rabin A, Ashwal-Fluss R, Shenzis S, Apelblat D, Kadener S, A comprehensive pipeline for accurate annotation and quantification of circRNAs, BioRxiv (2019). 10.1101/2019.12.15.876755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Barrett SP, Salzman J, Circular RNAs: Analysis, expression and potential functions, Dev 143 (2016) 1838–1847. 10.1242/dev.128074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Kulpa D, Topping R, Telesnitsky A, Determination of the site of first strand transfer during Moloney murine leukemia virus reverse transcription and identification of strand transfer-associated reverse transcriptase errors, EMBO J 16 (1997) 856–865. 10.1093/emboj/16.4.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Agabian N, Trans splicing of nuclear pre-mRNAs, Cell 61 (1990) 1157–1160. 10.1016/0092-8674(90)90674-4. [DOI] [PubMed] [Google Scholar]
- [74].Capel B, Swain A, Nicolis S, Hacker A, Walter M, Koopman P, Goodfellow P, Lovell-Badge R, Circular transcripts of the testis-determining gene Sry in adult mouse testis, Cell 73 (1993) 1019–1030. 10.1016/0092-8674(93)90279-Y. [DOI] [PubMed] [Google Scholar]
- [75].Pfafenrot C, Preußer C, Establishing essential quality criteria for the validation of circular RNAs as biomarkers, Biomol. Detect. Quantif 17 (2019) 100085. 10.1016/j.bdq.2019.100085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Zhang Y, Xue W, Li X, Zhang J, Chen S, Zhang JL, Yang L, Chen LL, The Biogenesis of Nascent Circular RNAs, Cell Rep 15 (2016) 611–624. 10.1016/j.celrep.2016.03.058. [DOI] [PubMed] [Google Scholar]
- [77].Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC, 35888, Nature 391 (1998) 806–811. https://www.nature.com/articles/35888.pdf. [DOI] [PubMed] [Google Scholar]
- [78].Bartel DP, MicroRNAs: Genomics, Biogenesis, Mechanism, and Function, Cell 116 (2004) 281–297. 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- [79].Stojic L, Lun ATL, Mangei J, Mascalchi P, Quarantotti V, Barr AR, Bakal C, Marioni JC, Gergely F, Odom DT, Specificity of RNAi LNA and CRISPRi as loss-of-function methods in transcriptional analysis, Nucleic Acids Res 46 (2018) 5950–5966. 10.1093/nar/gky437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Huppi K, Martin SE, Caplen NJ, Defining and assaying RNAi in mammalian cells, Mol. Cell 17 (2005) 1–10. 10.1016/j.molcel.2004.12.017. [DOI] [PubMed] [Google Scholar]
- [81].Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T, Generation of target cells, 411 (2001) 1–5. www.nature.com. [DOI] [PubMed] [Google Scholar]
- [82].Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J, The biogenesis, biology and characterization of circular RNAs, Nat. Rev. Genet 20 (2019) 675–691. 10.1038/s41576-019-0158-7. [DOI] [PubMed] [Google Scholar]
- [83].Heigwer F, Port F, Boutros M, Rna interference (RNAi) screening in Drosophila, Genetics 208 (2018) 853–874. 10.1534/genetics.117.300077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Wakiyama M, Matsumoto T, Yokoyama S, Drosophila U6 promoter-driven short hairpin RNAs effectively induce RNA interference in Schneider 2 cells, Biochem. Biophys. Res. Commun 331 (2005) 1163–1170. 10.1016/j.bbrc.2005.03.240. [DOI] [PubMed] [Google Scholar]
- [85].Clemens JC, Worby CA, Simonson-Leff N, Muda M, Maehama T, Hemmings BA, Dixon JE, Use of double-stranded RNA interference in Drosophila cell lines to dissect signal transduction pathways, Proc. Natl. Acad. Sci 97 (2000) 6499–6503. 10.1073/pnas.110149597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Caplen NJ, Fleenor J, Fire A, Morgan RA, dsRNA-mediated gene silencing in cultured Drosophila cells: A tissue culture model for the analysis of RNA interference, Gene 252 (2000) 95–105. 10.1016/S0378-1119(00)00224-9. [DOI] [PubMed] [Google Scholar]
- [87].Ni J-Q, Zhou R, Czech B, Liu L-P, Holderbaum L, Yang-Zhou D, Shim H-S, Tao R, Handler D, Karpowicz P, Binari R, Booker M, Brennecke J, Perkins LA, Hannon GJ, Perrimon N, A genome-scale shRNA resource for transgenic RNAi in Drosophila, (2011). 10.1038/nmeth.1592. [DOI] [PMC free article] [PubMed]
- [88].Pamudurti NR, Patop IL, Krishnamoorthy A, Ashwal-Fluss R, Bartok O, Kadener S, An in vivo strategy for knockdown of circular RNAs, Cell Discov 6 (2020). 10.1038/s41421-020-0182-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Okamura K, Ishizuka A, Siomi H, Siomi MC, Distinct roles for Argonaute proteins in small RNA-directed RNA cleavage pathways, Genes Dev 18 (2004) 1655–1666. 10.1101/gad.1210204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Huynh N, Depner N, Larson R, King-Jones K, A versatile toolkit for CRISPR-Cas13-based RNA manipulation in Drosophila, Genome Biol 21 (2020) 1–29. 10.1186/s13059-020-02193-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Viswanatha R, CRISPR-Cas13 mediated Knock Down in Drosophila cultured cells, 21 (2020) 1–9. 10.1101/2020.11.01.364166. [DOI] [Google Scholar]
- [92].Abudayyeh OO, Gootenberg JS, Konermann S, Joung J, Slaymaker IM, Cox DBT, Shmakov S, Makarova KS, Semenova E, Minakhin L, Severinov K, Regev A, Lander ES, Koonin EV, Zhang F, C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector, Science (80-.). 353 (2016). 10.1126/science.aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Zhang Y, Nguyen TM, Zhang X-O, Wang L, Phan T, Clohessy JG, Pandolfi PP, Optimized RNA-targeting CRISPR/Cas13d technology outperforms shRNA in identifying functional circRNAs, Genome Biol 22 (2021) 41. 10.1186/s13059-021-02263-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Li S, Li X, Xue W, Zhang L, Yang LZ, Cao SM, Lei YN, Liu CX, Guo SK, Shan L, Wu M, Tao X, Zhang JL, Gao X, Zhang J, Wei J, Li J, Yang L, Chen LL, Screening for functional circular RNAs using the CRISPR–Cas13 system, Springer; US, 2020. 10.1038/s41592-020-01011-4. [DOI] [PubMed] [Google Scholar]
- [95].Konermann S, Lotfy P, Brideau NJ, Oki J, Shokhirev MN, Hsu PD, Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors, Cell 173 (2018) 665–676. e14. 10.1016/j.cell.2018.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Zheng Q, Bao C, Guo W, Li S, Chen J, Chen B, Luo Y, Lyu D, Li Y, Shi G, Liang L, Gu J, He X, Huang S, Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs, Nat. Commun 7 (2016). 10.1038/ncomms11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Wilusz JE, Circular RNAs: Unexpected outputs of many protein-coding genes, RNA Biol 14 (2017) 1007–1017. 10.1080/15476286.2016.1227905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Suster ML, Seugnet L, Bate M, Sokolowski MB, Refining GAL4-driven transgene expression inDrosophila with a GAL80 enhancer-trap, Genesis 39 (2004) 240–245. 10.1002/gene.20051. [DOI] [PubMed] [Google Scholar]
- [99].Brand AH, Perrimon N, Targeted gene expression as a means of altering cell fates and generating dominant phenotypes, Development 118 (1993) 401–415. [DOI] [PubMed] [Google Scholar]
- [100].Brand AH, Dormand EL, The GAL4 system as a tool for unravelling the mysteries of the Drosophila nervous system, Curr. Opin. Neurobiol 5 (1995) 572–578. 10.1016/0959-4388(95)80061-1. [DOI] [PubMed] [Google Scholar]
- [101].Phelps CB, Brand AH, Ectopic gene expression in Drosophila using GAL4 system, Methods A Companion to Methods Enzymol 14 (1998) 367–379. 10.1006/meth.1998.0592. [DOI] [PubMed] [Google Scholar]
- [102].Barwell T, DeVeale B, Poirier L, Zheng J, Seroude F, Seroude L, Regulating the UAS/GAL4 system in adult Drosophila with Tet-off GAL80 transgenes, PeerJ 2017 (2017). 10.7717/peerj.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Ma J, Ptashne M, The carboxy-terminal 30 amino acids of GAL4 are recognized by GAL80, Cell 50 (1987) 137–142. 10.1016/0092-8674(87)90670-2. [DOI] [PubMed] [Google Scholar]
- [104].Nogi Y, Fukasawa T, Functional domains of a negative regulatory protein, GAL80, of Saccharomyces cerevisiae., Mol. Cell. Biol 9 (1989) 3009–3017. 10.1128/mcb.9.7.3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Roman G, Endo K, Zong L, Davis RL, P{switch}, a system for spatial and temporal control of gene expression in drosophila melanogaster, Proc. Natl. Acad. Sci. U. S. A 98 (2001) 12602–12607. 10.1073/pnas.221303998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Osterwalder T, Yoon KS, White BH, Keshishian H, A conditional tissue-specific transgene expression system using inducible GAL4, Proc. Natl. Acad. Sci. U. S. A 98 (2001) 12596–12601. 10.1073/pnas.221303298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Lyu S, Tonoki A, Drosophila behavior genetics, Encycl. Anim. Behav (2019) 259–266. 10.1016/B978-0-12-809633-8.20689-4. [DOI] [Google Scholar]
- [108].Nichols CD, Becnel J, Pandey UB, Methods to Assay Drosophila Behavior, J. Vis. Exp (2012). 10.3791/3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Rockwell AL, Beaver I, Hongay CF, A direct and simple method to assess drosophila melanogaster’s viability from embryo to adult, J. Vis. Exp 2019 (2019) 1–9. 10.3791/59996. [DOI] [PubMed] [Google Scholar]
- [110].Takeyama KI, Ito S, Yamamoto A, Tanimoto H, Furutani T, Kanuka H, Miura M, Tabata T, Kato S, Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in Drosophila, Neuron 35 (2002) 855–864. 10.1016/S0896-6273(02)00875-9. [DOI] [PubMed] [Google Scholar]
- [111].de Haro M, Al-Ramahi I, De Gouyon B, Ukani L, Rosa A, Faustino NA, Ashizawa T, Cooper TA, Botas J, MBNL1 and CUGBP1 modify expanded CUG-induced toxicity in a Drosophila model of myotonic dystrophy type 1, Hum. Mol. Genet 15 (2006) 2138–2145. 10.1093/hmg/ddl137. [DOI] [PubMed] [Google Scholar]
- [112].Post Y, Paululat A, Muscle Function Assessment Using a Drosophila Larvae Crawling Assay, BIO-PROTOCOL 8 (2018). 10.21769/bioprotoc.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Benzer S, BEHAVIORAL MUTANTS OF Drosophila ISOLATED BY COUNTERCURRENT DISTRIBUTION, Proc. Natl. Acad. Sci 58 (1967) 1112–1119. 10.1073/pnas.58.3.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Madabattula ST, Strautman JC, Bysice AM, O’Sullivan JA, Androschuk A, Rosenfelt C, Doucet K, Rouleau G, Bolduc F, Quantitative analysis of climbing defects in a drosophila model of neurodegenerative disorders, J. Vis. Exp 2015 (2015) 52741. 10.3791/52741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Feany MB, Bender WW, A Drosophila model of Parkinson’s disease, Nature 404 (2000) 394–398. 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- [116].Leitch K, Ponce F, Van Breugel F, Dickinson MH, The long-distance flight behavior of Drosophila suggests a general model for wind-2 assisted dispersal in insects. 3 4 Authors and their affiliations 5, (n.d.). 10.1101/2020.06.10.145169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Whitehead SC, Beatus T, Canale L, Cohen I, Pitch perfect: how fruit flies control their body pitch angle, (2015). 10.1242/jeb.122622. [DOI] [PubMed]
- [118].Babcock DT, Ganetzky B, An Improved Method for Accurate and Rapid Measurement of Flight Performance in Drosophila, J. Vis. Exp 84 (2014) 51223. 10.3791/51223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Hardin PE, Molecular genetic analysis of circadian timekeeping in Drosophila, in: Adv. Genet, 2011: pp. 141–173. 10.1016/B978-0-12-387690-4.00005-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Hardin PE, Panda S, Circadian timekeeping and output mechanisms in animals, Curr. Opin. Neurobiol 23 (2013) 724–731. 10.1016/j.conb.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Pittendrigh CS, Circadian systems. I. The driving oscillation and its assay in Drosophila pseudoobscura., Proc. Natl. Acad. Sci. U. S. A 58 (1967) 1762–1767. 10.1073/pnas.58.4.1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Konopka RJ, Benzer S, Clock mutants of Drosophila melanogaster., Proc. Natl. Acad. Sci. U. S. A 68 (1971) 2112–2116. 10.1073/pnas.68.9.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Dubowy C, Sehgal A, Circadian Rhythms and Sleep in Drosophila melanogaster, (2017). 10.1534/genetics.115.185157. [DOI] [PMC free article] [PubMed]
- [124].Kayser MS, Biron D, Sleep and development in genetically tractable model organisms, Genetics 203 (2016) 21–33. 10.1534/genetics.116.189589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Potdar S, Daniel DK, Thomas FA, Lall S, Sheeba V, Sleep deprivation negatively impacts reproductive output in Drosophila melanogaster, J. Exp. Biol 221 (2018). 10.1242/jeb.174771. [DOI] [PubMed] [Google Scholar]
- [126].Beyaert L, Greggers U, Menzel R, Honeybees consolidate navigation memory during Sleep, J. Exp. Biol 215 (2012) 3981–3988. 10.1242/jeb.075499. [DOI] [PubMed] [Google Scholar]
- [127].Beckwith EJ, French AS, Sleep in Drosophila and Its Context, Front. Physiol 10 (2019) 1–19. 10.3389/fphys.2019.01167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Piper MDW, Partridge L, Drosophila as a model for ageing, Biochim. Biophys. Acta - Mol. Basis Dis 1864 (2018) 2707–2717. 10.1016/j.bbadis.2017.09.016. [DOI] [PubMed] [Google Scholar]
- [129].Hyde RR, Inbreeding, outbreeding, and selection with Drosophila melanogaster, J. Exp. Zool 40 (1924) 181–215. 10.1002/jez.1400400105. [DOI] [Google Scholar]
- [130].Ziehm M, Piper MD, Thornton JM, Analysing variation in Drosophila aging across independent experimental studies: A meta-analysis of survival data, Aging Cell 12 (2013) 917–922. 10.1111/acel.12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Müller HAJ, Samanta R, Wieschaus E, Wingless signaling in the Drosophila embryo: Zygotic requirements and the role of the frizzled genes, Development 126 (1999) 577–586. [DOI] [PubMed] [Google Scholar]
- [132].Brunner E, Peter O, Schweizer L, Basler K, pangolin encodes a Lef-1 homologue that acts downstream of Armadillo to transduce the Wingless signal in Drosophila, Nature 385 (1997) 829–833. 10.1038/385829a0. [DOI] [PubMed] [Google Scholar]
- [133].Van de Wetering M, Cavallo R, Dooijes D, Van Beest M, Van Es J, Loureiro J, Ypma A, Hursh D, Jones T, Bejsovec A, Peifer M, Mortin M, Clevers H, Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF, Cell 88 (1997) 789–799. 10.1016/S0092-8674(00)81925-X. [DOI] [PubMed] [Google Scholar]