

Abstract
Mitochondria are dynamic organelles responsible for cellular energy production. Besides, regulating energy homeostasis, mitochondria are responsible for calcium homeostasis, signal transmission, and the fate of cellular survival in case of injury and pathologies. Accumulating reports have suggested multiple roles of mitochondria in neuropathologies, neurodegeneration, and immune activation under physiological and pathological conditions. Mitochondrial dysfunction, which occurs at the initial phase of brain injury, involves oxidative stress, inflammation, deficits in mitochondrial bioenergetics, biogenesis, transport, and autophagy. Thus, development of targeted therapeutics to protect mitochondria may improve functional outcomes following traumatic brain injury (TBI) and intracerebral hemorrhages (ICH). In this review, we summarize mitochondrial dysfunction related to TBI and ICH, including the mechanisms involved, and discuss therapeutic approaches with special emphasis on past and current clinical trials.
Keywords: Mitochondrial biogenesis, mitochondrial bioenergetics, immune activation, brain injury, mitophagy, therapeutic approach
1. Introduction
Mitochondria are dynamic double-membrane organelles that are involved in the regulation of cellular health. Mitochondria may vary in size, shape, and number in different cells, depending upon the cellular metabolism, stress, location, and function (Norat et al., 2020; Picard et al., 2011). The most significant role of mitochondria is energy production, as it is the key site for aerobic metabolism. Mitochondrial outer and inner membranes are structurally and functionally different. The inner membrane ports components of oxidative phosphorylation and respiratory chain, and therefore, regulates vital bioenergetics for cellular survival (Frey and Mannella, 2000; Norat et al., 2020). Apart from the adenosine 5′-triphosphate (ATP) production, mitochondria perform a central role in shaping cell signaling features through the production of reactive oxygen species (ROS), mediation of cell death, calcium homeostasis, immune signaling, neuroplasticity, neurotransmission, and monitoring of neural health (Cserep et al., 2020; Norat et al., 2020; Rose et al., 2017; West et al., 2011). Mitochondria across different cell types show differential properties such as density, shape, composition, distribution, and function and thus, contribute to cell specificity (Calvo and Mootha, 2010; McAvoy and Kawamata, 2019). Mitochondria isolated from different organs may vary in endogenous mitochondrial substrates, which in-turn affect the mitochondrial metabolism and byproducts. These substrates are present in negligible quantity in the brain as compared to heart, muscle or liver (Tretter and Ambrus, 2014). Mitochondria are adapted to perform diverse tasks in different cell types. For example, mitochondria control differentiation of immune and stem cells (Chen and Chan, 2017; MacIver et al., 2013), regulate neurite branching and regeneration (Misgeld and Schwarz, 2017), and synaptic strength, stability, and signaling (Devine and Kittler, 2018) in the nervous system. Some of these cell-type-specific functions of mitochondria are achieved by inflecting mitochondrial metabolic functions. However, few incredible functions of mitochondria such as immune signaling (Nunnari and Suomalainen, 2012; West et al., 2011) were discovered, while many remain unknown.
An average adult human brain weighs about 2% of total body weight. But it consumes about 20% of oxygen supply of the body to sustain its high rate of metabolic activity (Agranoff and Siegel, 1994; Raichle and Gusnard, 2002; Siegel and Albers, 1994; Steiner, 2019). Furthermore, younger and developing brain has shown striking demand of higher energy supply (Goyal et al., 2018; Goyal et al., 2015; Magistretti and Allaman, 2015; Steiner, 2019). Newborn human baby brain consumes approximately 60% of the body’s daily energy requirement, while 10 years old child’s brain utilizes around 50% of the total metabolic energy (Goyal et al., 2018; Goyal et al., 2015; Steiner, 2019). In the brain, neuronal cells consume around 75–80% of energy (Harris et al., 2012; Hyder et al., 2016; Magistretti and Allaman, 2015) as they perform neurotransmission that involves complex processes such as generation of action potential, ion movements, neurotransmitters release and reuptake, (Alle et al., 2009; Bordone et al., 2019; Harris et al., 2012; Magistretti and Allaman, 2015), and remodeling of synapses and neuronal network to support learning and memory (Bauernfeind et al., 2014; Harris et al., 2012; Magistretti, 2011; Sheng and Kim, 2011). Remaining 20–25% of basal metabolic energy are utilized by glia and other cells of the nervous system. While most of the basal metabolic energy is utilized by brain to maintain normal physiological activity, certain cognitive task may increase energy demand (Bauernfeind and Babbitt, 2014; Harris et al., 2012; Hyder et al., 2016; Magistretti and Allaman, 2015; Magistretti and Pellerin, 1999).
Beside cellular diversity, the brain also maintains spatial mitochondrial diversity. For example, the density of mitochondria differs among somatosensory cortical layers of juvenile rat. Santuy et al. reported the minimum volume fraction of cortical mitochondria (4.03%) in layer VI of somatosensory cortex of juvenile rat, while layer IV showed a maximum volume of cortical mitochondria (7.74%). Moreover, mitochondrial volume was found to be 44% in dendrites, 41% in non-synaptic elements, and 15% in axons (Santuy et al., 2018). Since, juvenile rat brain contains 38% of dendrites, 23% of non-synaptic element and 23% of axons of total volume of neuropil, dendrites seem to be richer in mitochondria. As a matter of fact, a positive correlation was found in the fraction of mitochondria in neuronal processes and the density of synapses (Santuy et al., 2018). Further, dendritic mitochondria were larger and densely packed than axonal mitochondria, which are small and sparsely distributed (Seager et al., 2020).
Apprehending the high demand for energy, CNS mitochondria have developed functional diversity across different brain cells (Bray, 2019). For example, Fecher et al. isolated mitochondria from three different cell types of cerebellum-granule cells (excitatory neurons), Purkinje cells (inhibitory neurons), and astrocytes, and found 200 of the mitochondrial proteins were differentially regulated among the groups, while 85% of proteins were conserved across cells (Fecher et al., 2019). Proteomic analyses revealed that mitochondria from granule cells were more efficient in buffering calcium through mitochondrial calcium uniporter (Mcu), while Purkinje cells showed more endoplasmic reticulum-mitochondria contacts via regulator of microtubule dynamics protein 3 (Rmdn3). Moreover, mitochondria from neuronal and glial cells perform specialized functions. Neuronal mitochondria are efficient to generate metabolite ‘ubiquinone’, while astrocytic mitochondria are specialized in β-oxidation to metabolize lipids faster than other cells (Bray, 2019; Fecher et al., 2019). Astrocytes, highly abundant glial cells, are best-studied cells in this connection, and co-exist with neuronal circuit and neurovascular unit in different parts of brain (Haim and Rowitch, 2017; Vasile et al., 2017). Astrocytes are rich in pyruvate decarboxylase, which regulate pyruvate metabolism, and therefore, are important to glutamine-glutamate cycling between neurons and astrocytes (McAvoy and Kawamata, 2019; Schousboe et al., 2019). Astrocytes account for approximately 15% of total oxygen utilized during brain network activation (Hyder et al., 2006). However, compared to neurons, which utilize oxidative phosphorylation (OXPHOS) to support neuronal transmission and activity (Rangaraju et al., 2019; Seager et al., 2020), astrocytes produce ATP predominantly through aerobic glycolysis rather than OXPHOS (Hyder et al., 2006; Magistretti, 2011; Magistretti and Allaman, 2015; McAvoy and Kawamata, 2019). A detailed discussion would be out of scope of this review. Therefore, we recommend readers to these reviews and articles to get an insight of these topics (Agrawal et al., 2018; Fecher et al., 2019; McAvoy and Kawamata, 2019; Misgeld and Schwarz, 2017; Picard and McEwen, 2014; Santuy et al., 2018; Seager et al., 2020; Yu et al., 2016)
CNS Mitochondria are susceptible to accumulating or acquired damage because of higher oxidative activity and complex biogenesis throughout the cellular lifespan. Mitochondrial damage results in dysfunctional cellular energy metabolism, loss of ATP production, increased oxidative stress, and reduced calcium buffering as seen in acute and chronic neuropathologies (Norat et al., 2020). For instance, a number of neuronal mitochondria around amyloid-β plaques were reduced more than those of glial origin in the cortex of APP/PS1 mice model of Alzheimer’s disease. Similarly, the spinal cord of pre-symptomatic SODG93A mice, predisposed to amyotropic lateral sclerosis (ALS) showed morphological alterations in neuronal mitochondria rather than glial mitochondria (Bray, 2019; Fecher et al., 2019). Similarly, early functional and structural alterations in mitochondria with cell death and poor cognitive functions have been reported in both clinical and experimental brain injury (Cheng et al., 2012; Fischer et al., 2016a; Gajavelli et al., 2015; Lifshitz et al., 2003; Lifshitz et al., 2004; Singh et al., 2006). Reduced cellular respiration and ATP production occur within 24 h post-injury and persist for up to 2 weeks in experimental TBI (Gilmer et al., 2009; Lifshitz et al., 2003; Singh et al., 2006; Xiong et al., 1997). In addition to cellular respiration, mitochondria dictate cellular fate by mediating apoptosis, necrosis, pyroptosis, and inflammasome activation (Breda et al., 2019). Mitochondrial injury initiates cell death pathways and apoptosis by releasing pro-apoptotic factors (Brustovetsky et al., 2002; Raghupathi et al., 2000).
Mitochondria continuously undergo fission and fusion to maintain a delicate balance to efficiently meet the energy demand of the cells (Bereiter-Hahn and Vöth, 1994; Chan, 2006; Van der Bliek et al., 2013; Westermann, 2012). While mitochondrial fusion enables maximum ATP production during high metabolic activity via increase in cristae (Westermann, 2012; Youle and Van Der Bliek, 2012), the mitochondrial fission, on the other hand increases the number of mitochondria to be transported to regions facing high energy demand and to replace worn out mitochondria (Otera et al., 2013; Youle and Van Der Bliek, 2012). Both processes are executed in perfect equilibrium in healthy cells. Any disruption in this balance between fission and fusion, cause catastrophic effect on energy homeostasis and has been reported in many neuropathologies and neurodegenerative diseases such as, TBI, stroke, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease (Archer, 2013; Burté et al., 2015; Fischer et al., 2016a; Knott and Bossy-Wetzel, 2008b; Knott et al., 2008). Since these mitochondrial dynamics (fission/fusion) are regulated by a complex system of genes and proteins, brain injury such as TBI and ICH cause alterations in these molecules. For example, genes and proteins related to fusion were upregulated in mild TBI while downregulated in severely injured traumatic brain. A reverse pattern was seen in these two models of TBI in relation to fission (Di Pietro et al., 2017; Fischer et al., 2016b). Similarly, increased mitochondrial fission was seen in brain post-ICH, and inhibition of mitochondrial division improved mitochondrial functional deficits and neuronal functions in rats (Xiao et al., 2021). A detailed discussion of mitochondrial fusion and fission in TBI and ICH is provided later in this review. As brain cells have high metabolic needs, uninterrupted energy supplies by functional and healthy mitochondria are essential for normal functioning of brain. Thus, improving mitochondrial function may offer a new therapeutic approach for the treatment in TBI and ICH, and in different neuropathologies e.g., Alzheimer’s disease, Parkinson’s disease, Huntington’s disease etc. In this review, we have summarized the mechanism behind mitochondrial damage following a brain injury keeping focus on TBI and ICH, and discussed the current therapeutic approach to improve mitochondrial function to rectify the injured brain.
2. Mitochondrial mechanism of brain damage in TBI and ICH
Mitochondrion, a powerhouse for the cell plays a critical role in maintaining cellular homeostasis and inevitably associated with the pathophysiology of brain injuries (Chen et al., 2020b). Humanin (HN), a small peptide transcribed from mitochondrial DNA has a cytoprotective role and profound downregulation of HN has been observed after cerebrovascular events such as ICH (Jung et al., 2020; Voigt and Jelinek, 2016). Following initial insult, secondary brain injuries are more aggravated, and are the main reasons for poor prognosis among the patients. Evidence of mitochondrial injury after brain injury such as ICH and TBI, have been reported in the past and various treatment have been proposed to alleviate mitochondrial damage by regulating oxidative stress, apoptosis, mitochondrial dynamics (fusion and fission) (Carinci et al., 2021; Chen et al., 2020b; Chen et al., 2021; Goodfellow et al., 2020; Salman et al., 2021; Wang et al., 2018; Xiao et al., 2021; Zhou et al., 2020). In the following sections, we will discuss the mitochondrial mechanisms that are exacerbated after brain injury (TBI and ICH) and pose a high risk of secondary brain injury such as oxidative stress, mitophagy, apoptosis, calcium homeostasis, mitochondrial fission, and fusion (Fig. 1).
Fig. 1. Mitochondrial injury-mediated pathways of cellular damage in TBI and ICH.

Mitochondria is a cellular powerhouse and is critical for cellular survival. However, because of their exposure to oxidant and active cellular respiration, mitochondria are vulnerable to injury (TBI and ICH)-induced stress. In this schematic diagram, we elucidated mitochondrial pathways of injury-induced cellular dysfunction. The main pathways leading to mitochondria-associated cellular dysfunction include: (1) Calcium overload causes mtPTP opening and increased mitochondrial membrane permeability. As a result of aggravated injury-mediated signaling, mitochondria go into overproduction of ROS and loss of ATP. (2) Injury-induced defects in mitochondrial biogenesis (fission and fusion) affects mitochondrial longevity. (3) Impaired intercellular transport leads to reduced number of mitochondria in affected cells. (4) When mitochondria become dysfunctional and pose a threat to cellular integrity, affected cell tries to remove mitochondria through mitophagy. 5) Finally, when survival attempts fade, the affected cell undergoes into cyt c-mediated apoptosis. Increased membrane permeability leads to cytochrome c release and activation of apoptosis. ATP, adenosine triphosphate; cyt c, cytochrome c; Drp1, dynamin-related protein 1; Mfn, mitofusin; mPTP, mitochondrial permeability transition pore; OPA, optic atrophy; ROS, reactive oxygen species; Ca2+, calcium ion.
2.1. Oxidative stress
Oxidative stress is a state of imbalance between the production of ROS and scavenging anti-oxidative agents (Higgins et al., 2010). The brain retains high oxidative metabolic activity, low antioxidant capacity, low repair mechanisms, and therefore, is highly vulnerable to oxidative damage. Under normal circumstances, about 4% oxygen is incompletely reduced to produce ROS via electron transport chain (ETC) reaction (Bhat et al., 2015). Basal ROS production is critical to drive normal cellular function such as response to anoxia, cellular signaling pathways, and induction of mitogenic response (Ristow and Schmeisser, 2014). In response to stress, mitochondrial ROS initiate transcriptional changes to enhance the expression of proteins involved in systemic defense (Ristow, 2014; Ristow and Schmeisser, 2014). Tolerable amount of mitochondrial stress leads to improve the health and viability of cells and tissue. This process is called as “mitohormesis” and depends on variety of signals including ROS, mitochondrial metabolites, released mitokines, and mitochondria-cytosol stress response (Bárcena et al., 2018; Ristow, 2014; Ristow and Schmeisser, 2014). While low level of ROS improve systemic defense and adaptive cellular response to injury, higher level of ROS act as a major contributing factor for cellular damage (Ristow and Schmeisser, 2014).
Oxidative phosphorylation is a major intracellular source of mitochondrial ROS generation (Koutsilieri et al., 2002). Superoxide (O2−) produced during oxidative phosphorylation is subsequently transferred to other ROS, such as hydrogen peroxide (H2O2), which in turn transformed into highly reactive hydroxyl radical (.OH) via Fenton reactions and targets nucleic acid, lipids, and proteins (Andersen, 2004). To counter the detrimental effect of ROS on irreversible injury including cellular debilitation, premature death, and functional impairment, mammalian cells have developed endogenous antioxidant defense mechanisms to limit oxidation of cellular biomolecules (Sayre et al., 2008). Glutathione (GSH) and its dependent enzymes (glutathione peroxidase, glutathione reductase) are important components of this endogenous antioxidant defense, and provides protection against oxidative stress in different brain pathologies (Hall et al., 2010; Khan et al., 2012; Koza and Linseman, 2019; Laird et al., 2008; Shrivastava et al., 2013; Sukumari-Ramesh et al., 2010; Vaibhav et al., 2013a; Vaibhav et al., 2013b). GSH in a reduced form acts as a scavenger of hydroxyl radicals, singlet oxygen, and various electrophiles. Glutathione peroxide converts hydrogen peroxide into the water with the help of GSH while, Oxidized glutathione (GSSG) gets reduced to GSH by glutathione reductase with the help of NADPH (Koza and Linseman, 2019). However, overwhelming oxidative stress along with molecular changes in cellular environment, mitochondrial damage, reduced ATP generation, neuroinflammation, and blood-brain barrier dysfunction is manifested as secondary injury post-TBI and results in the disruption of neural networks (Rizk et al., 2021).
Following brain injury, such as in the case of TBI, cellular metabolism becomes dysregulated and produces an excessive amount of ROS through injured mitochondria. This disturbs the cellular redox homeostasis, depletes the endogenous antioxidant, and consequently, builds up excessive ROS (Hall et al., 2010; Koza and Linseman, 2019). Reduction in brain GSH (Ansari et al., 2008) and its precursors such as S-adenosylmethionine (SAM) and methionine contents (Dash et al., 2016) post-TBI enhance the susceptibility of neurons to ROS, and correlates well with reduced GSH content in CSF of TBI patients (Bayir et al., 2002). Similarly, hemoglobin oxidation by-product hemin-induced peroxidative injury was associated with a rapid depletion of intracellular GSH in astrocytes, culminating in lipid peroxidation, inflammation, and cell death, effects that were reduced by co-treatment with exogenous GSH, N-acetyl-L-cysteine, or the glutathione peroxidase mimetic drug Ebselen in an in-vitro (hemin treatment) model of ICH (Laird et al., 2008). In TBI, the release of excitatory amino acid neurotransmitters (glutamate) causes Ca2+, Na+, and K+ influx leading to blood-brain barrier breakdown, excessive production of ROS, and exhaustion of endogenous antioxidants (Abdul-Muneer et al., 2015). Thus, TBI induces impaired mitochondrial respiration, cleavage of DNA, and lipid peroxidation (Omelchenko et al., 2019; Tewari et al., 2014). Similarly, hematoma and perihematomal region post-ICH are enriched with RBC lysis products particularly, hemin which is degraded into Fe2+. This drives Fenton reaction to generate hydroxyl radical, leading to oxidative stress (Bertero and Maack, 2018; Zille et al., 2017). Lack of nucleotide excision repair pathways and histone protection in mitochondrial DNA expose it to mutations causing a bioenergetic deficit thus reducing ATP production and increases the free radical production significantly (Szczesny et al., 2010). Alterations in activities of mitochondrial respiratory complexes of the ETC system, specially Complexes I and III may cause subsequent oxidative damage and apoptosis (Dröse et al., 2014; Fan et al., 2017a). Defected Complex I and III could release superoxide that could damage the mitochondrial matrix and inner mitochondrial membrane, which contributes to oxidative stress and apoptosis induction (Fan et al., 2017a). ICH leads to decreased production of ATP through suppression of Complex I and III activities in ETC. However, growth differentiation factor 11 (GDF11) protected the activity of ETC complex I and III via suppressing mitochondrial ROS productions and alterations in mitochondrial dynamics in in-vivo rat ICH model and in-vitro with freshly prepared erythrocyte homogenate treatment models as well (Xiao et al., 2021).
2.2. Altered bioenergetics and metabolic sensors
Mitochondria is the center of the cellular bioenergetics process. Reduced energy supply and disrupted bioenergetics have an important role in brain pathology including ischemia, stroke, and TBI (Khatri and Man, 2013). The ATP generated as a product of oxidative phosphorylation in mitochondria is utilized to meet demands for energy transfer as well as a second messenger in various neurological disease such as epilepsy, ischemia, TBI and pain. The damaged neurons release a large amount of ATP in the extracellular space and alter calcium signaling through modulation of purinergic receptors, and in turn aggravates inflammatory responses, excitotoxicity, and apoptosis (Roszek and Czarnecka, 2015; Yegutkin, 2008). At the molecular level, ATP and its precursor metabolites such as ADP and AMP serve as key intrinsic stimuli for the activation for multiple signaling pathways implicated in neuronal survival and regeneration such as Phosphatidylinositol-3-kinase (PI3K)/protein kinase B (PKB, also known as AKT), mechanistic target of rapamycin (mTOR) and AMP-activated protein kinase (AMPK) pathway (Gurusamy et al., 2010; Morita et al., 2017; Rodríguez-Vargas et al., 2012). In the brain, PI3K–Akt pathway regulates neuronal survival and synaptic plasticity via maintaining mitochondrial metabolism and cellular bioenergetics (Yin et al., 2013). However, oxidative stress activates c-Jun N-terminal kinase (JNK), an antagonizing pathway of PI3K-Akt and causes its translocation into mitochondria to impair energy metabolism (Yin et al., 2013).
AMPK is a master metabolic regulator that encourages the catalytic process to enhance energy production (Khatri and Man, 2013) and has shown a great impact on the immune cell polarization through metabolic regulation post-TBI and ICH (Baban et al., 2021; Cai et al., 2019; Vaibhav et al., 2018). AMPK is activated either by increased in the ratio of AMP to ATP originated from starvation and stressed or dysfunctional mitochondria (Frederich and Balschi, 2002; Kim et al., 2011; Qi and Young, 2015; Toyama et al., 2016) or by membrane depolarization and Ca2+-influx (Hawley et al., 2005). Further, AMPK regulates mitochondrial functions, such as fission, fusion, or the recycling of phospholipid membranes (Amadoro et al., 2014; Nardin et al., 2016; Toyama et al., 2016). Previously, mTOR protein, which promotes cellular growth, lipid, and protein synthesis was reported to be inhibited by activated AMPK (Gurusamy et al., 2010; Morita et al., 2017; Rodríguez-Vargas et al., 2012). In agreement, higher expression of phosphorylated AMPK was observed with low expression of phosphorylated-mTOR following the Hypoxic-ischemic injury (Cai et al., 2019). However, Morita et al. reported that inhibition of mTOR complex 1 (mTORC1) does not trigger mitochondrial dysfunction (Morita et al., 2017). AMPK activates DRP1 receptor mitochondrial fission factor (MFF) in conditions of excessive ATP loss through mitochondrial injury and inhibition of the electron transport chain (ETC) (Toyama et al., 2016). Further, AMPK up-regulates peroxisome proliferative activated receptor γ (PPARγ) coactivator 1α (PGC1-α), to promote mitochondrial biogenesis via heat shock protein 22 (Fan et al., 2021) and adiponectin receptor 1 (AdipoR1) (Yu et al., 2019).
Metabolic cofactors such as mitochondrial NAD+ have a critical role in cellular metabolism and energy homeostasis. Recently, it was reported that murine and human mitochondria can replenish NAD+/NAD(H) by importing either intact NAD directly or precursors nicotinamide (NAM)/nicotinamide mononucleotide (NMN) from cytosol through the unrecognized receptor (Davila et al., 2018). Reduced NAD+ (i.e., NADH) acts as the primary electron donor in the synthesis of ATP by oxidative phosphorylation in the mitochondrial respiratory chain. Various proteins e.g., Sirtuins, poly ADP-ribose polymerases (PARPs), and cyclic ADP-ribose synthases utilize NAD+. Further, NAD+ modulates metabolic, energetic and stress response with signaling and transcriptional events through activation of Sirtuins (Cantó et al., 2015; Srivastava, 2016). NAD+ can rewire metabolism, influence fuel selection, regulates circadian rhythms, maintains mitochondrial fitness, and helps in cell survival under stress conditions (Cantó et al., 2015; Davila et al., 2018). Intracellular NAD+ pool relies on the critical equilibrium between its metabolizing and biosynthetic pathways. NAD+/NADH ratio is a good indicator of cellular metabolism and regulates metabolic pathways such as glycolysis, Kreb’s cycle, and fatty acid oxidation. As mitochondrial damage leads to declined NAD+ levels, reduced NAD+/NADH ratio is associated with mitochondrial disorders, ischemia, cancer, ICH, and aging (Endres et al., 1997; He et al., 2010; Srivastava, 2016; Wei et al., 2017; Ying and Xiong, 2010; Zhao et al., 2010). While the detailed discussion of mitochondrial metabolism is beyond the scope of this review article, we refer interested readers to read the review articles published earlier (Jalloh et al., 2015; Liu et al., 2021; Thapa et al., 2021; Xu et al., 2020; Xu et al., 2021).
2.3. Autophagy/Mitophagy
Mitophagy, the autophagic breakdown of mitochondria, is a process of self-degradation of harmful or excess mitochondria. Double membrane autophagosomes sequester damaged/surplus organelles or portions of cytosol and fuse with lysosomes or vacuoles further broken down by hydrolytic enzymes (He and Klionsky, 2009). The process of mitophagy has long been speculated as a mechanism to remove dysfunctional mitochondria from the cell to conserve the mitochondrial DNA as evidenced in fluorescently labeled mitochondria in cultured mammalian cells (Twig et al., 2008). It was observed that mitochondrial division produced two uneven daughter organelles, among which the one with decreased membrane potential and reduced Optic Atrophy 1 (OPA1) levels were more likely to be removed by mitophagy. Inhibition of mitochondrial fission decreases mitophagy and results in oxidization of mitochondrial proteins, thereby decline respiratory capacity. By contrast, inhibition of autophagy leads to the accumulation of mitochondria with low membrane potential and low OPA1 (Twig et al., 2008). Generally, mitochondrial fission produces solitary mitochondria with intact membrane potential, which either re-fuse with the mitochondrial network or end up depolarized and depleted of OPA1. This prevents further fission and enables subsequent elimination by mitophagy (Westermann, 2010).
Mitophagy is essential for removing damaged mitochondria and its dysfunction in this process has deleterious effects in various brain diseases such as Alzheimer’s disease, Parkinson’s disease, ischemic stroke (Santos et al., 2010; Vives-Bauza and Przedborski, 2011; Zuo et al., 2014). Mitophagy alleviates injury and cellular dysfunction by maintaining mitochondrial dynamics in rodent TBI by removing dysfunctional or damaged mitochondria (Chen et al., 2016; Wu et al., 2018). In controlled cortical impact (CCI) model of TBI, the significant increase of autophagy markers have been observed (Niu et al., 2019; Zeng et al., 2020). It has evidently been stated that autophagy is upregulated after TBI since it is the chief pathway for the elimination of aberrant cell components, be it clinical or experimental model (Zhang et al., 2008).
Impaired mitophagy also accounts for damage after TBI as evidenced by the accumulation of damaged mitochondria inside the neuronal cells, resulting in high levels of ROS, causing damage to other mitochondria and ultimately inducing cell death (Fischer et al., 2016a). It has been observed that mitophagy reduces oxidative stress related neuronal death after subarachnoid hemorrhage in rats (Zhang et al., 2019; Zhou and Tan, 2020). Mechanically, PTEN induced putative kinase 1 (PINK1)/Parkin mediated fission is more characteristic related to mitophagy pathway along with mitochondrial damage and autophagy (Geisler et al., 2010). Although, under normal physiological conditions, mitophagy is cytoprotective but becomes lethal when over-activated in severe pathological stress, including ischemia (Huang et al., 2020). Mitophagy is regulated by BCL2 Interacting Protein 3 (BNIP3) and BNIP3 like (BNIP3L, also called NIX). NIX regulates basal mitophagy in normal conditions, while BNIP3 activates excessive mitophagy resulting in cell death after stroke (Shi et al., 2014). In the autologous blood model of mouse ICH, the FUN14 domain containing 1 (FUNDC1) was found to inhibit NLR family pyrin domain containing 3 (NLRP3) inflammasome-mediated inflammation through activating mitophagy. FUNDC1 is a kind of mitophagy receptor that mediates the elimination of dysfunctional mitochondria after hypoxia and mitochondrial stress. Inhibition of FUNDC1 suppresses mitophagy, potentiates inflammation, and exacerbates ICH injury (Zheng et al., 2021). Connexins (Cxs) are a crucial part of gap junctions, have also been shown to be regulated by autophagy (Bennett et al., 2003; Sun et al., 2015). However, TBI led to the accumulation of phosphorylated Cx43 in rat hippocampal astrocytes due to insufficient autophagy (Sun et al., 2015). Initially, TBI is a focal injury but progresses throughout the brain regions over a long period of time. In the CCI model of rats, the Cx 40 has been reported to decline 2 days after TBI and remained low until day 6. This decrease in CX40 mirrored neuronal degeneration and corresponded to the increased number of the autophagic vacuole (Chen et al., 2017).
2.4. Apoptosis
Damage due to injury of external nature such as TBI (force), ICH (hemorrhages), and stroke (restricted blood flow) can instigate necrosis, which causes inflammation that further aggravates the injury by expanding to various cells and organs. Conversely, apoptosis, in a more controlled manner is achieved as a result of biochemical changes in the cell (Kawane et al., 2014). Regulation of apoptosis involves interaction of pro apoptotic BCL-2 associated × protein (BAX) and Bcl-2 which inhibits apoptosis (Karbowski et al., 2006). Both Bax and Bcl-2 are located in the outer mitochondrial membrane, and its ratio reflects the tendency of cells to undergo apoptosis or survival. An increase in the ratio of Bcl-2/BAX increases survival while its decrease (e.g., in the case of ICH) leads to apoptosis (Luo et al., 2020). In response to various death stimuli, cells initiate mitochondria-dependent apoptotic pathway which involves mitochondrial outer membrane permeabilization, formation of apoptosome, and the activation of effector caspases (Luo et al., 2020). Mitochondrial damage is a major contributor to TBI-induced neuronal apoptosis and necrotic cell death (Harris et al., 2001). The role of mitochondria in the pre-apoptotic signaling pathway by releasing cytochrome c and apoptotic inducing factor (AIF) plays a central role in the survival or death of neurons (Sabirzhanov et al., 2016). Post TBI, cytochrome c binds with apoptotic protease activating factor (Apaf-1) and ATP in the outer membrane of mitochondria, which leads to apoptosome formation and neuronal apoptosis preceded by the release of small mitochondria-derived activator caspases (Hiebert et al., 2015; Kasahara and Scorrano, 2014). Failure in mitochondrial metabolism after changes in mitochondrial outer membrane permeability plays a critical role in the release of caspase activating molecules as well as caspase-dependent death effectors that lead to mitochondrial swelling and ultimately neuronal death (Chen et al., 2020a). Increased caspase 3 activation and cytochrome c translocation has been also observed in the perihematomal region after ICH (Chen et al., 2020a; Ding et al., 2017; Lu et al., 2015). Further, tumor necrosis factor (TNF) activated by macrophages can also induce apoptosis, which makes it the major extrinsic mediator of apoptosis (Ola et al., 2011). The excitotoxicity in TBI leads to cell damage and inflammation, resulting in necrosis and apoptosis (de Lores Arnaiz and Bersier, 2014). Similarly, early tissue injury after ICH is caused by apoptosis influenced by free-radical cascade reactions, inflammation, cytokines, thrombin, and erythrocyte lysis (Bobinger et al., 2018; Salihu et al., 2016).
2.5. Calcium homeostasis
Calcium homeostasis is important for the normal function of neurons and in neuronal processes such as growth and development, neurotransmission, differential gene expression in neurons, activity-dependent synaptic plasticity, learning, and memory. Since Ca2+ is a major signaling molecule in neurons, changes in its levels contribute to TBI and ICH pathology (Deshpande et al., 2008; Weber, 2012; Zheng et al., 2013). In brain injury such as in the case of TBI and subarachnoid hemorrhage (SAH), excessive release of excitatory amino acid neurotransmitters (e.g., glutamate) causes ionic disturbance by the high influx of Ca2+ into cytosol, and rise in intracellular Na+ (Abdul-Muneer et al., 2015; Omelchenko et al., 2019; Tewari et al., 2014). To maintain cytosolic Ca2+ levels, mitochondria take up excess cytosolic Ca2+ causing a reduction in mitochondrial potential, which can further lead to ATP depletion, mitochondrial membrane depolarization, and permeability, ROS generation, and finally apoptosis or necrosis (Ertel et al., 2000; Huang et al., 2015; Joshi et al., 2015). In a mild TBI model of rat induced by fluid percussion injury, significant cognitive impairment was observed in TBI rats at 7 days post-injury, and was positively correlated with elevated intracellular Ca2+ level (Deshpande et al., 2008). Excessive influx of Ca2+ post moderate or severe TBI is highly implicated in secondary injury post TBI through activation of Ca2+ dependent enzymes, and Ca2+ overload in mitochondria. The necrosis or apoptosis can be induced in cells depending on the passage of Ca2+ influx, and the Ca2+ load. An increase in intracellular Ca2+ after injury can activate caspases, phospholipases, nucleases, proteases, and increase the production of nitric oxide (NO) which can further exacerbate the Ca2+ load and neuronal damage (Weber, 2012). In a murine model of ICH, overactivation of transient receptor potential vanilloid 4 (TRPV4) altered Ca2+ homeostasis and caused neuronal apoptosis (Shen et al., 2019a).
Intracellular Ca2+ increase leads to activation of matrix metalloproteinase 9 (MMP9) which is physiologically important for remodeling of the extracellular matrix (ECM) components. Any alterations in MMP9 activities may distrupt ECM and neuronal synaptic activity (Averna et al., 2016; Christensen and Shastri, 2015; Mukhopadhyay et al., 2004; Reinhard et al., 2015). A paired microdialysis with CT scan study in TBI patients showed elevated MMP9 in the pericontusional brain with a maximal concentrations found within the first 72 h of injury as compared with the normal brain (Guilfoyle et al., 2015). Inhibition of MMP9 by selective inhibitor SB-3CT attenuated brain lesion and neuronal loss (Hadass et al., 2013). Upregulation of MMP9 has also been demonstrated after ICH, which can be triggered by ROS leading to BBB destruction (Katsu et al., 2010). Damage post-ICH is a multi-cascade effect evident by activation of inflammatory cells such as granulocytes, which also contribute to the pathogenesis of brain injury via production of ROS through nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and myeloperoxidase (Chen et al., 2020a). Immune activation through mitochondria is further discussed in a separate section in this review.
2.6. Mitochondrial fission and fusion
Being highly dynamic organelles, mitochondria undergo constant fusion and fission to maintain normal morphology and function. The whole process is categorized as fission, when a single mitochondrion separated into two or more daughter organelles. By contrast, fusion indicates when two or more organelles combine to form one mitochondrion. These processes are further accompanied by changes in morphology of the organelle (Fenton et al., 2021; Giacomello et al., 2020; Pekkurnaz et al., 2014). Both processes exist in equilibrium in healthy cells, and imbalance between these processes as seen in various neuropathologies such as TBI, stroke, Alzheimer’s disease etc. disrupts energy homeostasis (Archer, 2013; Burté et al., 2015; Fischer et al., 2016a; Knott and Bossy-Wetzel, 2008b; Knott et al., 2008). This disruption in fission/fusion equilibrium accounted for alterations in genes and proteins related to mitochondrial fusion and fission. For example, genes and proteins related to mitochondrial fusion were upregulated in mild TBI while downregulated in severely injured brain, while a reverse pattern of mitochondrial fission were seen in these two models of TBI (Di Pietro et al., 2017; Fischer et al., 2016a). Similarly, the rat brain showed increased mitochondrial fission post-ICH, while its inhibition improved mitochondrial deficits and neuronal functions in rats (Xiao et al., 2021).
Mitochondrial fission is regulated by phosphorylation of Drp1, a membrane protein recruited over the outer mitochondrial membrane. Phosphorylation at Ser616 (S616) accelerates the recruitment of Drp1 to the membrane while phosphorylation at S637 inhibits it (Fischer et al., 2016a). Recruitment of cytosolic Drp1 to outer mitochondrial membrane forms Drp1 oligomers at candidate fission site, marked as endoplasmic reticulum-mitochondria contact region where they constrict mitochondria by GTP hydrolysis, further dividing the mitochondria (Friedman et al., 2011; Westermann, 2010). Selective inhibition of Drp1 has reversed morphological changes in mitochondria and Drp1 translocation and as a result reduced ROS, BBB disruption, and downregulated MMP9 expression and apoptosis after subarachnoid hemorrhage (Fan et al., 2017b; Wu et al., 2020a; Wu et al., 2020b). Abnormal Drp1 activity accounts for excessive mitochondrial fission and neurodegeneration. TBI increased the expression of Drp1 in purified hippocampal mitochondria from rats as compared to sham controls. These mitochondria, as analyzed on a cryo-electron microscope had an initial increase in length at 24 hours post-TBI, followed by a significant decrease in length at 72 hours post-TBI. Interestingly, inhibition of fission by using a pharmacological inhibitor of dynamin-protein related protein 1 (Drp1), such as mitochondrial division inhibitor-1 (Mdivi-1; 3 mg/kg at 30 min, 24 hours, and 48 hours post-TBI)) limited mitochondrial length decrease, and improved novel object recognition (NOR) memory and context-specific fear memory in rats at 72 hours post-TBI (Fischer et al., 2016a). Previous studies also linked mitochondrial impairment to ICH (Xiao et al., 2021; Zheng et al., 2018). The initial hematoma after ICH induces glutamate release and then leads to mitochondrial dysfunction of neurons. Aberrant mitochondrial respiration was observed in the perihematomal region as early as 2 hours after ICH which declined progressively with time after ICH (Kim-Han et al., 2006). Extensive swelling and vacuolization of mitochondria were also observed in brain tissue of ICH diabetic rats (Zheng et al., 2018). In the in-vitro model of ICH, erythrocyte homogenate (EH) treatment resulted in increased mitochondrial fission in the neuronal SH-SY5Y human neuroblastoma cells with the fragmented, distorted, and bleb-like shape of mitochondria. Increased mitochondrial fragmentations were attributed to the up-regulation of DRP-1, p-DRP1 (at 616 residues) in EH treated cells (Xiao et al., 2021).
Conversely, mitochondrial fusion is initiated by mitofusin-1 and 2 (Mfn1 and Mfn2), which are also anchored on the outer membrane of mitochondria, and OPA1, a GTPase localized on the inner mitochondrial membrane. The long and short isoforms of OPA1 (L-OPA1 and S-OPA1) play a significant role in inner membrane mitochondrial fusion and mitochondrial fission, respectively (Westermann, 2010). Neuropathologies such as TBI and neurodegenerative diseases show a marked imbalance between mitochondrial fission and fusion. Excessive mitochondrial fusion induces mitochondrial respiratory dysfunction, increased ROS production, and release of apoptotic factors (Fischer et al., 2016a; Knott and Bossy-Wetzel, 2008a). A time-dependent shift from mitochondrial fission to fusion was observed in the hippocampus following TBI. An increase in expression of mitochondrial fusion related genes such as OPA1, MFN1, and MFN2 was observed in the injury region 120 days post-TBI, while mitochondrial fission related DRP1 reduced over time after TBI (Di Pietro et al., 2017).
2.7. Mitochondrial transfer
Horizontal transfer of mitochondria takes place between different cell types and can affect mitochondrial respiration defects in recipient cells besides regulating signaling, proliferation, or chemotherapy resistance (Spees et al., 2006; Torralba et al., 2016). Multiple mechanisms of mitochondrial transfer have been purposed and involve tunneling nanotubes (Onfelt et al., 2004; Rustom et al., 2004), melanosomes (Scott et al., 2002; Sugden et al., 2004), and exosomes (Denzer et al., 2000). The mitochondrial transfer is both unidirectional as well as bi-directional depending on cell types involved; however, the exact principle behind this is still not fully elucidated (Spees et al., 2006). Using a transient-focal cerebral ischemia model, Hayakawa and colleagues showed that the bi-directional transport of mitochondria between neurons and astrocytes. Astrocytes take up damaged mitochondria from neurons and recycle them (Davis et al., 2014), and release healthy mitochondria in the surrounding. These healthy mitochondria are further taken up by neuronal cells (Bambrick et al., 2004; Hayakawa et al., 2016). Besides astrocytes, other cell types such as endothelial-progenitor cells (EPCs) have been demonstrated to donate healthy mitochondria to injured neurons in an in-vitro oxygen-glucose deprivation (OGD) model (Hayakawa et al., 2018). It has been reported that Miro1, a mitochondrial Rho-GTPase regulates intercellular mitochondrial transfer from healthy stem cells to injured cells (Ahmad et al., 2014).
2.8. Mitochondria in immune activation
Mitochondrial dynamics and bioenergetics have emerged as important classifiers in the polarized states of immune cells in various pathologies (Boland et al., 2013; Devanney et al., 2020; Takeda et al., 2021; Wang et al., 2020; Yu et al., 2012). The crosstalk between mitochondrial dynamics in the form of fission/fusion can significantly influence immune cell metabolism and functions (Rambold and Pearce, 2018). Macrophages, particularly in their proinflammatory state predominantly utilize glycolysis, while in their anti-inflammatory state, as characterized during wound healing and tissue repair, switch to oxidative phosphorylation (O’Neill and Pearce, 2016; Rambold and Pearce, 2018). Further, mitochondria have displayed diverse functions in both infectious and sterile inflammation (West et al., 2011). In viral infections, mitochondrial antiviral signaling protein (MAVS) is involved in the recognition of viruses through RIG-I-like receptor (RLR) (Belgnaoui et al., 2011). Meanwhile, in sterile tissue damage, such as TBI, mitochondrial ROS trigger neuroinflammation through direct and indirect mediators of injury (Suliman and Piantadosi, 2016). Besides ROS, brain injury can also lead to release of mitochondrial damage-associated molecular patterns (mtDAMPs) such as mitochondrial DNA (mtDNA), adenosine triphosphate (ATP), succinate, cardiolipin, N-formylated peptides, and mitochondrial transcription factor (TFAM) to the immune system (Rodriguez-Nuevo and Zorzano, 2019). Further, the activation of inflammatory pathways through recognition of DAMPs requires the formation of cytosolic multi-protein inflammasome complexes. Inflammasome can be assembled through proteins such as nuclear oligomerization domain (NOD)-like receptor –pyrin-containing proteins (NLRP) and HIN domain-containing (PYHIN) protein families. Inflammasomes lead to activation of caspases, which promotes the maturation of cytokines such as interleukin IL-18, IL-1β and induces pyroptosis (Broz and Dixit, 2016).
Selective degradation of mitochondria known as mitophagy also plays a critical role in the maintenance of proper cellular functions (Ding and Yin, 2012). In experimentally induced TBI, the translocation of phospholipid cardiolipin, from the inner mitochondrial membrane to the outer membrane tags the mitochondria for mitophagy. Interestingly, this can also lead to activation of the inflammasome complex (Chu et al., 2013). Further, failure to induce mitophagy can lead to the release of mtDAMPs into extracellular spaces and causes inflammation (Grazioli and Pugin, 2018). Neural-mtDAMPs can diffuse through disrupted BBB into the circulation after TBI (Wang et al., 2014). A study in children with TBI founds mtDNA in cerebrospinal fluid (CSF) and its levels were correlated with neurological outcomes (Walko et al., 2014). Further, mitochondrial DNA and proteins can activate microglia, astrocyte, and exacerbate further tissue injury (Gyoneva and Ransohoff, 2015; Walko et al., 2014). Similarly, mtDAMPs can also act as activators of human neutrophils and lead to generation of ROS, secretion of pro-inflammatory cytokine such as IL-8 and activation of p38 and ERK1/2 MAPKs (Hazeldine et al., 2015).
The immune cells such as microglial cells and macrophages present in the brain and spinal cord act as damage sensors for the central nervous system. Microglial cells have been known to examine the brain microenvironments such as neuronal cell bodies and synapses (Li et al., 2012; Wake et al., 2009). In a recent study, the molecular mechanism involving mitochondria of neurons and the neuroprotective role of microglial cells was established. Interestingly, specialized communication sites were found between microglial cells and neuronal somatic membrane in both humans and mice. At the interface, the nanoarchitecture inside the neurons was found to be composed of juxtaposed mitochondria, reticular membrane, and vesicle-like membrane structures. As mitochondria are the immuno-metabolic hub of the cells, proximity between neuronal mitochondria and microglia mediate neuronal quality control through rapid sensing of neuronal health. In the event of brain injury, such an interaction promotes neural protection through the regulation of neural calcium load and functional connectivity (Cserep et al., 2020).
In recent times, the mitochondrial transfer has emerged as a therapeutic strategy (Han et al., 2020). In an interesting study, a transfer of mitochondria from engineered Mesenchymal stem cells (MSCs) have shown to affect inflammation through adaptive immune cells. Mitochondrial transfer from MSCs to T cells cause increased expression of several genes such as forkhead box P3 (FOXP3), interleukin 2RA (IL2RA), cytotoxic T-lymphocytes associated protein-4 (CTLA4), transforming growth factor β1 (TGFβ1), and led to activation and differentiation CD25+ FoxP3+ regulatory T cells (Court et al., 2020). In another recent study, mitochondrial transfer from neural stem cells (NSCs) through endoplasmic vesicles led to functional restoration in target cells. Additionally, mitochondrial transfer to inflammatory mononuclear phagocytes led to reduced expression of inflammatory genes and restoration of cellular metabolism (Peruzzotti-Jametti et al., 2021).
3. Mitochondria as a therapeutic target
In the brain, both acute and chronic injuries disrupt mitochondrial activities, and its biogenesis. Prior studies confirmed that alteration in mitochondrial number and functions are associated with secondary impairment characterized by progressive neuronal loss and astrogliosis and leads to cognitive impairment and mood disorders besides other post-injury complications. The rationale for targeting mitochondria during brain injury has been supported by several related preclinical studies and clinical trials on different human diseases and disorders such as ischemic stroke (Carinci et al., 2021), ICH (Chen et al., 2020b), TBI (Lifshitz et al., 2003; Lifshitz et al., 2004; Singh et al., 2006; Cheng et al., 2012; Walko et al., 2014; Gajavelli et al., 2015; Gyoneva and Ransohoff, 2015; Fischer et al., 2016a; Wang et al., 2018; Chen et al., 2021, Salman et al., 2021, Xiao et al., 2021; Zhao et al., 2021), acute and chronic neurodenegration (Goodfellow et al., 2020) and diabetic retinopathy (Zhou et al., 2020) and has been discussed in details in following sections (Fig. 2).
Fig. 2. Current treatment strategies for mitochondrial therapeutics in TBI and ICH.
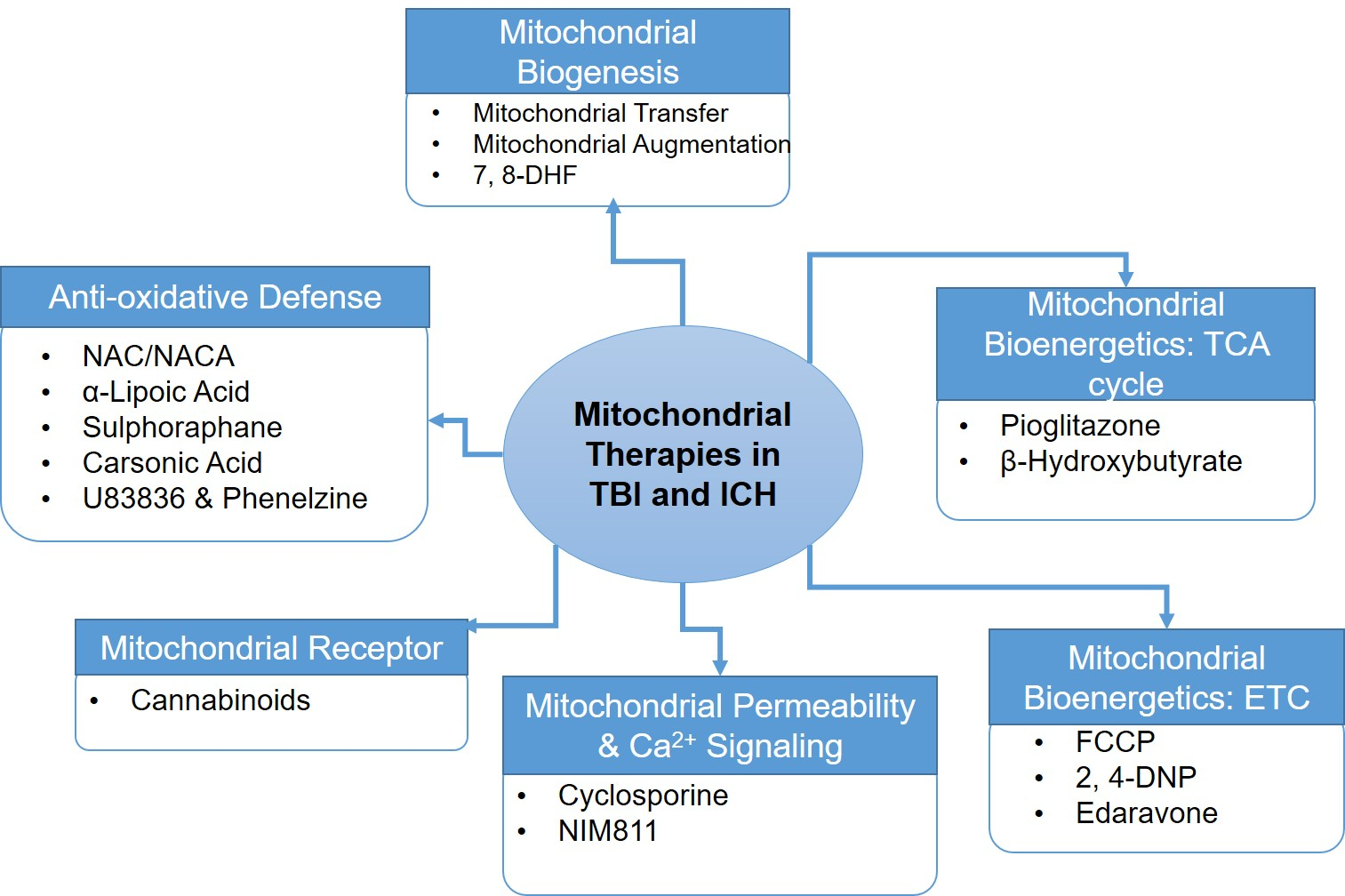
Mitochondrial rescue in case of brain injury can be achieved by using drugs that targets: (1) Mitochondrial homeostatic pathways such as mitochondrial biogenesis, and (2) Mitochondrial functional pathways such as mitochondrial bioenergetics including tri-carboxylic acid (TCA) cycle and electron transport chain (ETC), mitochondrial permeability & Ca2+ signaling, specific mitochondrial receptor and anti-oxidative defense mechanism. 7,8-DHF, 7,8-dihydroxyflavone; FCCP: Trifluoromethoxy carbonylcyanide phenylhydrazone; 2,4-DNP: 2, 4-dinitrophenol; NIM811, N-methyl-4-isoleucine-cyclosporine; NAC/NACA: N-acetylcysteine (NAC); N-acetylcysteine amide (NACA).
3.1. Therapeutics targeting mitochondrial biogenesis
As explained in earlier sections, the number of mitochondria in a cell is determined by the balance between mitochondrial fission and fusion. Mitochondrial biogenesis is the growth and division of pre-existing mitochondria, which can affect their number, size, and mass, and eventually its functions. Mitochondrial biogenesis is targeted in different neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and amyotrophic lateral sclerosis (Golpich et al., 2017). It includes targeting genes and proteins involved in mitochondrial complexes, and in the process such as mitochondrial fission and fusion.
3.1.1. Restoring mitochondrial number by mitochondrial transfer
A preclinical study suggests that mitochondrial transfer rescues neuronal apoptosis and effectively restores post-TBI cognitive impairment and mood disorder primarily by up-regulating brain-derived neurotropic factor (BDNF) signaling pathway (Zhao et al., 2021). Mitochondrial dysfunction in ischemia-reperfusion (I/R) condition is rescued by MSCs derived mitochondrial transfer under in-vitro conditions (Han et al., 2020; Liu et al., 2014). Miro1, a Rho-GTPase plays an important role in mitochondrial transfer. Overexpression of Miro1 in mesenchymal stem cells (MSCs) enhanced mitochondrial transfer from MSCs and improves lung epithelial injury, while its deletion leads to loss of beneficial effect (Ahmad et al., 2014). Using a transient-focal cerebral ischemia model, Hayakawa and colleagues showed bi-directional transport of mitochondria between neurons and astrocytes. Astrocytes take up damaged mitochondria from neurons and recycle them (Davis et al., 2014), and then released healthy mitochondria in the surrounding that is taken up by neuronal cells (Bambrick et al., 2004; Hayakawa et al., 2016). Besides astrocytes, other cell types such as endothelial-progenitor cells (EPCs) have been demonstrated to donate healthy mitochondria to injured brain endothelial cells in an in-vitro oxygen-glucose deprivation (OGD) model (Hayakawa et al., 2018).
3.1.2. Restoring mitochondrial number by mitochondrial augmentation
Mitochondrial augmentation (MAT) involves mitochondrial enrichment from the patient’s peripheral stem cells or from donor cells such as white blood cells and placental cells. Based on several preclinical studies on the rescuing effect of transfer of healthy mitochondria from mesenchymal and stromal cells (Ahmad et al., 2014; Paliwal et al., 2018), a phase 1/2 clinical trial to evaluate the safety and therapeutic effects of autologous CD34+ cells is currently recruiting patients (NCT03762512) (Almannai et al., 2020).
3.1.3. Drugs targeting mitochondrial biogenesis
3.1.3.1. 7, 8-dihydroxyflavone (7, 8-DHF):
The flavonoid 7, 8-dihydroxyflavone (7, 8-DHF) is a tropomyosin-receptor-kinase B (TrkB) agonist that mimics the effects of brain-derived neurotrophic factor (BDNF). 7, 8-DHF promotes neuronal survival and regeneration (Wu et al., 2014). It shows neuroprotective effect in a rodent model of ischemic-stroke (Jang et al., 2010), TBI (Wu et al., 2014), and in a variety of neuro-degenerative disorders such as Huntington’s (Jiang et al., 2013), and Alzheimer’s disease (Castello et al., 2014). Pretreatment with 7, 8-DHF protects immature neurons from excitotoxicity-mediated cell death in both in-vitro and in-vivo CCI injury models (Chen et al., 2015). Another study in CCI-TBI showed that TrkB activation reduces dendrite swellings in the cortex (Zhao et al., 2016). Uluc and co-authors have shown that 7, 8-DHF has more profound neuroprotective effects in female mice than male mice after hypoxia ischemia (HI) (Uluc et al., 2013). Through TrkB signaling, 7, 8-DHB also maintains mitochondrial homeostasis through PGC-1α-mediated transcription of nuclear respiratory factors (NRFs), cytochrome c oxidase II (COII) and mitochondrial transcription factor A (TFAM) (Agrawal et al., 2015). Though 7, 8-DHF can cross the BBB and is orally bioactive (Liu et al., 2013), yet no clinical trial has been reported so far to check its efficacy in humans.
3.2. Therapeutics targeting mitochondrial bioenergetics
Dysregulated mitochondrial bioenergetics mediates several of the pathophysiological features of brain injury and are, therefore, remain important therapeutic targets. These involve the drugs that target oxidative phosphorylation (also known as TCA cycle) and electron transport chain (ETC). Below, we have mentioned selected drugs that were actively investigated in the regulation of mitochondrial bioenergetics during brain injury.
3.2.1. Drugs targeting mitochondrial oxidative phosphorylation/TCA cycle
3.2.1.1. Pioglitazone:
Pioglitazone, FDA-approved anti-diabetic drug is a peroxisome proliferator-activated receptor-γ (PPARγ) agonist and known to bind a mitochondrial protein mitoNEET (CISD1) (Rabchevsky et al., 2017). MitoNEET is a mitochondrial outer surface zinc-finger protein, which contains 2Fe-2S iron-sulfur cluster. MitoNEET is a major role player in the mitochondrial function associated with metabolic disorders such as obesity and cancers (Geldenhuys et al., 2016). Loss of mitoNEET results in mitochondrial dysfunction, elevated ROS level, reduced ATP production, and loss of striatal dopamine and tyrosine hydroxylase (Geldenhuys et al., 2017). The binding of Pioglitazone as a ligand to mitoNEET prevented Ca2+-induced mitochondrial dysfunction and improved neurological outcomes after TBI (Yonutas et al., 2020) while, suppressed inflammation, rescued injured tissue, and improved locomotor activity after spinal cord injury (SCI) in animal models (Patel et al., 2017; Rabchevsky et al., 2017). However, some contradiction findings such as edema and fluid retention in patients treated with Pioglitazone may limit its use in treating brain injuries (Bełtowski et al., 2013).
3.2.2. Beta-hydroxybutyrate:
Adoptive metabolic response to brain injury includes utilization of non-glucose substrates such as lactate and ketones (e.g., Beta-hydroxybutyrate; BHB), to prevent further damage (Bernini et al., 2020; Prins, 2008; Veech, 2004). BHB can directly enter the TCA cycle and acts as an alternative source of energy. Experimental injury model, particularly after TBI, shows that exogenous BHB supplementation might attenuate brain damage (Prins et al., 2004). A recent completed clinical study on the levels of medium-chain triglycerides (MCTs) in patients with severe TBI suggests that nutritional ketosis was the main determinant of cerebral ketone bodies (KBs) metabolism (NCT02716532) (Bernini et al., 2020). Induction of ketosis in patients with TBI has not yet been examined in clinical trials, but an early phase 1 safety and feasibility study is currently recruiting patients (NCT03982602, NCT04079907) (Singhal et al., 2020).
3.2.3. Drugs targeting mitochondrial electron transport chain
3.2.3.1. FCCP:
Trifluoromethoxy carbonylcyanide phenylhydrazone (FCCP) is a mitochondrial uncoupler, which lowers ROS production and Ca2+ overload. A single dose of FCCP shows a neuroprotective effect in an animal model of moderate TBI (Pandya et al., 2009). However, its strong uncoupling effect narrows down its therapeutic index and its utility as an effective drug in brain injury (Lou et al., 2007).
3.2.3.2. 2, 4-DNP:
2, 4-dinitrophenol: Like FCCP, 2, 4-DNP is another protonophore-based mitochondrial uncoupler (Geisler, 2019). Studies show that 2, 4-DNP administration in different models of brain injuries improve mitochondrial homeostasis and attenuates tissue damage (Hubbard et al., 2018; Korde et al., 2005). Although the pharmacology of both FCCP and 2,4-DNP is quite similar, DNP is considered safe in comparison to FCCP (Lou et al., 2007). More recently, two prodrugs similar to 2,4-DNP have shown neuroprotective effects in the TBI animal model (Hubbard et al., 2018) and have undergone phase-1 clinical trial for multiple neurodegenerative disorders (Bando and Geisler, 2019; Geisler, 2019).
3.2.3.3. Edaravone:
Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one) is an antioxidative drug approved by the FDA for the treatment of Amyotrophic Lateral Sclerosis (ALS) (Cruz, 2018). Edaravone reduces brain edema, protect neuronal cells, and improves neurological impairment in rats with cerebral ischemia-reperfusion injury (CIRI) (Ren et al., 2019a). However, a meta-analysis of data from randomized controlled trials of Edaravone in acute stroke including ischemic stroke and intracerebral hemorrhage (ICH) does not show any promising results (Yang et al., 2015).
3.2.3. Nicotinamide adenine dinucleotide (NAD+):
Enhancing cellular NAD+ content by prompting either its biosynthesis from its precursor L-tryptophan (de novo synthesis) or dietary Niacin (salvage pathway), or activation of sirtuins, or inhibiting the activity of PARP and cADP-ribose synthases helps in cellular survival under stress (Cantó et al., 2015; Davila et al., 2018; Srivastava, 2016). Importantly, NAD+ administration significantly reduced brain damage in animal models of TBI (Ying et al., 2007a), cerebral ischemia (Ying et al., 2007a; Ying et al., 2007b), neonatal hypoxia-ischemia (He et al., 2010), neuroblastoma and glioma (Zhao et al., 2010). Similarly, prolonged treatment with nicotinamide mononucleotide (NMN), a key intermediate of NAD+ biosynthesis reduced oxidative stress and inflammation and improved neurological function post-ICH through activation of heme oxygenase 1 (HO-1) and nuclear factor-like 2 (Nrf2) (Wei et al., 2017). Further, NAD+ treatment reduced oxygen glucose deprivation (OGD)-induced neuronal death by enhancing DNA repair activity (Wang et al., 2008). PARP-1 is an abundant nuclear enzyme that is activated by DNA strand breaks or kinks (Amé et al., 2004). Activated PARP-1 repairs DNA nick by using NAD+. However, PARP-1 also promote cell death in case of extensive DNA damage, as seen during inflammation and ischemia (Alano and Swanson, 2006; Alano et al., 2004; Amé et al., 2004; Ying, 2007). NAD+ administration inhibited PARP-1-mediated cell death by preventing glycolytic inhibition, mitochondrial depolarization, mitochondrial permeability and apoptosis-inducing factor (AIF) translocation (Alano et al., 2004; Ying et al., 2003; Ying and Xiong, 2010).
3.3. Drugs targeting mitochondrial permeability
3.3.1. Cyclosporin:
Cyclosporin, originally described as an immunosuppressant of T-cell activation is also found neuroprotective in TBI animal model (Sullivan et al., 2000; Waldmeier et al., 2003). Cyclosporin acts as a ligand for mitochondrial cyclophilin D, an important component of mitochondrial permeability transition pore (mPTP). The binding of cyclosporine A that selectively targets synaptic mitochondria disrupts the formation of mPTP, thereby reversing the effect of Ca2+ overload in mitochondria post-TBI (Kulbe et al., 2017). A randomized, double-blind, dual-center, placebo-controlled trial in Italy has reported a good safety profile of cyclosporin at 5 mg/kg during the early phase in TBI patients (Mazzeo et al., 2009). NeuroVive lnc has conducted a phase II clinical trial on cyclosporin (NeuroSTAT®) in the treatment of TBI and reported promising results and is currently heading towards a phase III clinical trial (Nighoghossian et al., 2015). Another clinical study (Phase IIa) conducted by the Copenhagen Head Injury Cyclosporin (CHIC) has reported beneficial effects of cyclosporin in patients with severe TBI (Kelsen et al., 2019).
3.3.2. NIM811:
N-methyl-4-isoleucine-cyclosporin (NIM811), a non-immunosuppressive cyclophilin inhibitor has shown neuroprotective effect and improves behavioral outcome after TBI (Readnower et al., 2011) very much alike its analog cyclosporine (Mbye et al., 2009; Springer et al., 2018a). NIM811 improves open field locomotor performance, return of reflexive bladder control and significantly decrease the rostral-caudal extent of the lesion in the adult female rat after spinal cord contusion in a dose-dependent manner (Springer et al., 2018b). Another study in a severe unilateral controlled cortical impact model of TBI indicates dose-dependent effect of NIM811 and found an optimal dose of NIM811 (10 mg/kg) that improves cognition and mitochondrial functioning, and reduces oxidative damage following TBI (Readnower et al., 2011). NIM811 inhibits mPTP and ameliorates delayed neuronal death in the hippocampus in mice subjected to transient forebrain ischemia (Wilks et al., 2011). Despite favorable outcomes in brain injury in animal models, no clinical study evaluating the effect of NIM811 in patients with brain injury has been reported at the time of writing of this review article.
3.3.3. Methylene Blue:
Methylene Blue (MB), also known as methylthioninium chloride is an FDA-approved drug initially used in the treatment of malaria (Gensini et al., 2007), methemoglobinemia (Ashurst and Wasson, 2011) and as a prophylactic therapy against vasoplegic syndrome post-coronary artery bypass surgery and in septic shock (Stawicki et al., 2008; Tucker et al., 2018). It is validated for its neuroprotective properties for stroke (Watts et al., 2013) Parkinson’s disease, Alzheimer’s disease (Oz et al., 2009) and optic neuropathy (Rojas et al., 2012) Mechanistically, MB provides an alternative route for electron transfer between NADH (mitochondrial complex I) and cytochrome c (complex IV) (Wen et al., 2011), thereby reduces electron leakage and ROS production (Callaway et al., 2004). It promotes mitochondrial oxidative phosphorylation while decreases anaerobic glycolysis (Riha et al., 2005). Few preclinical studies examine the effect of MB in TBI. In a randomized double-blinded experiment in rats, treatment with MB significantly reduce MRI-lesion volume over multiple time points extending up to 14 days post-TBI. Scores on forelimb placement asymmetry and foot false test also improved following treatment (Talley Watts et al., 2016). A subsequent study from the same group further showed that MB treatment 24 hours post-TBI produced similar results (Talley Watts et al., 2014) In another study, MB treatment reverse neuronal mitochondrial dysfunction and improves BBB integrity in-vivo after TBI (Shen et al., 2019b). Despite promising results, no clinical trial on MB treatment in TBI or in ICH has been reported so far.
3.3.4: Kaempferol:
Kaempferol, also known as kaempferol-3 or kaempferide, is a phytoestrogen (flavonoid) that stimulates mitochondrial Ca2+ uniporter channel (mCU), enhances spontaneous/stimulus-evoked neural activity and neurovascular responses in the normal brain (Chitturi et al., 2019). It is present in common fruits such as apple, grape, and tomato (Kim and Choi, 2013) and in medicinal plants including Equisetum spp, Sophora japonica, Ginkgo biloba, Euphorbia pekinensis (Rupr.) (Calderón-Montaño et al., 2011). It has antioxidant and anti-inflammatory properties and is used in the treatment of various cancer diseases such as esophageal cancer, breast cancer, cervical cancer, hepatocellular carcinoma (HCC), ovarian cancer, etc. (Ren et al., 2019b). A pre-clinical study in a rat model of TBI, treatment with kaempferol (1 mg/kg, i.p., 3 doses 24 hours apart) significantly improved the long-term behavioral and brain function assessed two months post-TBI (Murugan et al., 2016). Using a similar experimental approach, Chitturi et al., performed a metabolic profile after treatment with kaempferol and found increase TCA cycle flux, sustained mitochondrial functional integrity, and improved neural viability and somatosensory behavioral outcome (Parent et al., 2020). Clinical trials with crude extracts containing kaempferol such as Ginkogo biloba, Bauhinia forficata have been completed for intrauterine growth restriction (IUGR) and Diabetes, however no clinical trial for TBI and ICH has been reported. For further reading on kaempferol, interested readers can refer to the review article published by Ren et al. (Ren et al., 2019b).
3.3.5: Ru360:
Ru360 is a high affinity selective mitochondrial calcium uptake inhibitor. In a pre-clinical study using a rat modal of fluid percussion injury (a model of TBI), intraperitoneal administration of Ru360 at a dose of 120 μg at 3 different time points (30 min, 24hours, and 48hours) improved neural viability and sensorimotor behavior. Metabolomics analysis further suggested that Ru360 upregulated glycolytic and pentose phosphate pathways, and reduced oxidative stress and NAD+ depletion (Chitturi et al., 2021).
3.4. Antioxidative defense
3.4.1. NAC/NACA:
N-acetylcysteine (NAC) and N-acetylcysteine amide (NACA), enhances the production of endogenous antioxidant, glutathione (GSH), which plays an important role in maintaining the cellular redox state during brain injury (Kawoos et al., 2019). A study on rat has shown beneficial effects of NAC on the integrity of the blood-brain barrier (BBB) following blast overpressure (BOP) induced TBI (Kawoos et al., 2019). NAC treatment increases the levels of anti-inflammatory M2 microglia in a controlled cortical impact (CCI) model (Haber et al., 2018). Several clinical trials investigating the effect of NAC on patients with head injury alone or in combination with other drugs are currently finished or undergoing (NCT03680911, Status: terminated, Data not reported; NCT03241732, Status: Recruiting; NCT04291066, Status: Active, Not recruiting, NCT00822263, Status: Completed, Results not posted;, NCT02791945, Status: Completed, Results: Successful; NCT01515839, Status: Completed, Results: Not posted,) (Deepmala et al., 2015). A randomized double-blind, placebo-controlled study on the effect of NAC on patients with mild TBI showed a beneficial effect (Bhatti et al., 2017; Hoffer et al., 2013). In contradiction, another phase I clinical trial of NAC in combination with probenecid on TBI patients showed no significant effect (NCT01322009) (Clark et al., 2017).
3.4.2. Alpha-lipoic acid:
Alpha Lipoic acid (ALA), a metal chelator, restores the BBB disruption and normalizes the astrocytic/microglial activation and glutathione (GSH) levels (Rocamonde et al., 2013; Sears, 2013). ALA exerts its antioxidant effect by downregulating the expression of caspase-3 and attenuating mitochondrial-mediated apoptosis (Wei et al., 2015). It also modulates the Nrf2 signaling pathway (Xia et al., 2019) and promotes angiogenesis after brain injury (Rocamonde et al., 2013). ALA restores limb I/R-induced structural damage of rat hippocampus (Hussein et al., 2018). In a rat model, ALA reduces TBI-induced neuronal cell death in the hippocampus, prefrontal, and parietal cortex (Ozbal et al., 2015). ALA treatment partially reverses the increase in brain water content and EB extravasation in weight-drop TBI model (Toklu et al., 2009). It is currently used in a clinical trial in combination with other drugs to support brain recovery after concussion (NCT01515839, Status: Completed, Results: Not posted).
3.4.3. Sulforaphane:
Sulforaphane is a naturally occurring compound and found in high concentrations in broccoli. Its antioxidant response is mediated by transcription factor nuclear factor E2-related factor (Nrf2) and antioxidant response element (ARE). Sulforaphane significantly reduced contusion volume and increased post-SCI coordination (Wang et al., 2012). It also protects neurons and astrocytes from oxygen and glucose deprivation (OGD) at a low dose (5 μM), while showing considerable toxicity at 50 μM in the astrocytes-neuronal co-culture (Ladak et al., 2021). Sulforaphane treatment improves cognitive functions following cortical impact injury in rat TBI model (Dash et al., 2009). Currently, a double-blind randomized controlled phase-II clinical trial on the effect of Sulforaphane on cognitive functions in patients with frontal brain damage is started in China (NCT04252261, Status: Not yet recruiting) (Liu et al., 2020).
3.4.4. Carnosic acid:
Carnosic acid (CA) is another Nrf2-ARE activating natural product synthesized by plants belonging to the Lamiaceae family (Satoh et al., 2008). The presence of di-phenolic catechol moiety in CA provides an additional electron-donating property, thereby, making it more potent than sulforaphane (Satoh et al., 2008). CA activates SIRT1/p66shc signaling pathway and showed a neuroprotective role in early brain injury secondary to SAH in rat (Teng et al., 2019). Administration of CA also improves motor and cognitive function and decrease Gfap and Iba1 immuno-reactivities within white matter track post-TBI in mice (Maynard et al., 2019).
3.4.5. U-83836E and Phenelzine:
U-83836E and Phenelzine can scavenge lipid peroxyl radicals such as 4-hydroxy-2-nonenal (4-HNE) that are produced during the acute phase of brain injury (Mustafa et al., 2018a; Mustafa et al., 2018b). 4-HNE binds to mitochondrial proteins and leads to mitochondrial dysfunction during brain injury (Sullivan et al., 2005). Phenelzine is a non-selective and irreversible inhibitor of the enzyme monoamine oxidase (MAO, both MAO-A and MAO-B), and potentiates monoaminergic neurotransmission. Phenelzine has been approved by FDA for the treatment of anxiety, atypical depression, depression that does not respond to other methods of treatment, bulimia, and social anxiety disorder (Kulbe et al., 2018).
3.5. Modulation of Calcium signaling:
Mitochondria controls and buffers excessive cytosolic calcium (Hatton et al., 2008). During brain injury, disruption in mitochondrial activity led to calcium imbalance and activation of cysteine proteases, calpain-1, and cathepsin-B, which cause both neuronal and vascular damage. A recent clinical trial (NCT02496975, status: Withdrawn) with calpain-inhibitors such as cyclosporin A in phase1/2 was withdrawn due to its sharp biphasic dose-response (Hatton et al., 2008).
3.6. Cannabinoids:
Cannabinoid receptors type-1 (CB-1) and type-2 (CB-2) were both implicated in the pathophysiology of brain injury particularly TBI (Braun et al., 2018; Reddy et al., 2020a). CB1 receptors were found on both neuronal and astrocytic mitochondria suggests a wider noncanonical action (Busquets-Garcia et al., 2018; Younts and Castillo, 2014), as CB1 receptors are known to regulate different cellular pathways, e.g., mTOR, the mitogen-activated protein kinases/extracellular signal-regulated kinase (MAPK/ERK), c-Jun N-terminal kinases (JNK), and PI3K/Akt pathways (Galve-Roperh et al., 2013). Neuronal mitochondrial type-CB1 (mtCB1) upregulates post-TBI and leads to mitochondrial dysfunction (Xu et al., 2016). The mtCB1 activity has been related to alterations in bioenergetics and mitochondrial respiration, which regulated hippocampal synaptic transmission, and subsequently, memory consolidation (Bénard et al., 2012; Busquets-Garcia et al., 2015; Hebert-Chatelain et al., 2016). The mtCB1 receptor decreased the phosphorylation of proteins involved in oxidative phosphorylation, and reduced ATP production (Hebert-Chatelain et al., 2016; Robledo-Menendez et al., 2021).
Endocannabinoids such as anandamide and 2-arachidonoyl glycerol, as well as some plant and synthetic cannabinoids, have shown neuroprotective effects following TBI (Braun et al., 2018; Stetson et al., 2001). Dexanabinol (HU-211 or ETS2101), a synthetic cannabinoid derivative had been tested in a phase-II clinical trial (NCT02054754, Status: Completed) for the treatment of closed head injury and showed positive outcome (Darlington, 2003; Knoller et al., 2002). Dexanabinol was found to be safe, but its multicentric phase III clinical trial (NCT00129857, Status: Completed, Results: Not posted) did not show any significant improvement over placebo-control (Maas et al., 2006). Conversely, β-caryophyllene, a selective CB2 agonist, effectively inhibited oxidative stress and mitochondrial dysfunction in brain disorders (Ullah et al., 2021). However, cannabinoid therapy targeting mitochondria in brain injury is still not evaluated completely. Until recently, modulation of different pathologies by phyto-cannabinoids or commercially available ligands have gained momentum and is also being actively evaluated by our group.
4. Limitation of the mitochondrial research and future perspectives
Most of the research on mitochondrial dysfunctions in the brain is done in the field of neurodegenerative disorders such as Alzheimer’s disease etc. Whilst the outcome from such studies is useful in determining the secondary pathological consequences of brain injuries such as, TBI and ICH in chronic conditions, the studies on acute mitochondrial response to TBI and ICH are still limited. Being an important organelle for cellular metabolism, mitochondria are particularly influenced by disease pathology and thus, contribute to cellular aging (Haas, 2019; Shin et al., 2021). Mitochondria play crucial role in the aging process and development of age-related diseases. Ageing is associated with a decline in mitochondrial activity and quality (Haas, 2019; Horan et al., 2012; Sun et al., 2016). The interaction of neuronal mitochondrial dysfunction and amyloid-β (Aβ) protein is determined as key mechanism as the concentration of N-acetyl-aspartate in the brain tissue, a marker of mitochondrial function has been found correlated with Aβ42 in CSF of AD patient (Jessen et al., 2011). Brain injury such as TBI has a great impact on mitochondrial integrity and function and leads to chronic pathology (Cheng et al., 2012; Hiebert et al., 2015). Exogenous or endogenously-induced brain injuries are associated with more profound adversarial symptoms in the elderly compared to younger individuals. Therefore, targeting mitochondria would be an effective therapeutic modality to prevent excessive tissue damages during the natural and brain injury-associated aging process.
Functional MRI (fMRI) differentiates abnormal brain mitochondrial functions as in Alzheimer’s disease from normal brain mitochondrial function (Bell et al., 2021). Along with fMRI, mitochondrial spare respiratory capacity (MSRC) can be used to establish biomarkers of TBI and ICH as deficits in MRSC in fibroblasts from sporadic Alzheimer’s disease patients have been shown correlated with Alzheimer’s disease early progression (Bell et al., 2021; Bell et al., 2020). ETC function is the core activity of mitochondrial respiration. Therefore, functions related to different complexes of ETC might be developed into surrogate markers of CNS mitochondrial dysfunction. For example, complex II activity is linked to MSRC (Pfleger et al., 2015). Therefore, estimation of the NAD+/NADH ratio may be a useful surrogate marker of mitochondrial function. A metabolomics approach to assess the function of ETC may also be a good choice. However, functional assessment of mitochondrial deficits requires several days of processing of samples and can be time-consuming and non-economical, which presents practical difficulties to find the biomarkers in quick time. TBI and ICH field is advancing at a faster rate, and peripheral tissues such as blood and CSF are actively being examined in search of pathological markers. Therefore, using peripheral samples to study metabolomics and mitochondrial markers such as NAD+/NADH ratio will certainly be beneficial over sampling tissue from CNS by invasive approach. Examining metabolic abnormalities that are common to all cell types in TBI, ICH, and other neuropathologies such as Alzheimer’s and Parkinson’s disease, may also be developed into to a tool for stratification of people at risk of progressive pathologies and may improve diagnostic accuracy.
Several validated drugs targeting mitochondria in brain injury were failed in human clinical trials. For example, two antioxidant agents namely polyethylene glycol-conjugated superoxide dismutase (PEG-SOD) and 21-aminosteroid LP inhibitor Tirilazad, which inhibits lipid peroxidation, (LP) were failed to improve 6-months neurological recovery in large phase III clinical trial in moderately- to severely injured TBI patients (Hall et al., 2019). This could be due to the short therapeutic window of antioxidant drug therapy after TBI and requires an understanding of the initial and chronic phase of cellular and molecular changes that lead to mitochondrial dysfunction following brain injury. Non-conventional treatments such as remote ischemic conditioning (RIC), cannabinoids and anti-diabetic drugs such as metformin (AMPK activator) have shown good results in brain injury by limiting inflammation, improving functional outcomes and cerebral blood flow (Baban et al., 2021; Braun et al., 2018; DiBona et al., 2021; Jarrahi et al., 2020; Reddy et al., 2020b; Vaibhav et al., 2018), and could be further evaluated in targeting mitochondria. While in this review article, we focused on mitochondrial therapies in TBI and ICH, a recent review article focused on recent clinical trials in mitochondrial therapeutics has been recently published (Almannai et al., 2020), and would be a good read for the audience.
In general, the functional state of mitochondria in a cell is regulated by multiple complex mechanisms. It is possible that metabolism of different cell types in the brain modulate differently during injury, thus there is a need to develop high-resolution techniques for cell-specific analysis of mitochondrial potential and functions (Doerrier et al., 2018). Further, understanding of cell-specific mitochondrial profile would help us to develop cell-targeted drug therapy to improve clinical outcomes.
5. Conclusion
Altered mitochondrial dynamics, morphological changes and defective mitochondrial bioenergetics play an important role in cellular death following brain injuries. Recent study showed that improving mitochondrial dynamics has a great effect in recovery from brain injury (Chimeh et al., 2018). Further, replacement of old and damaged mitochondria with new healthier ones have provided another promising avenue for treatment in various diseases including brain injuries, where mitochondria are abundant in distal axonal synapses and dendrites (He et al., 2020). However, a thorough understanding of mitochondrial selection and acceptance by donor cells, and ethical and technical complications associated with the artificial transfer of mitochondria still remain active areas of research. Failure of clinical trials targeting mitochondria encourage us to define the time window of mitochondrial administration to harness full benefits of transfer in preclinical and clinical settings. Further, a more in-depth study of mitochondrial injury during acute and chronic pathologies will help to lay down better treatment strategy for various diseases.
Table 1. Summary of drugs targeting mitochondria in TBI and ICH.
The given table summarizes the drugs and compounds used for therapeutically targeting mitochondria. We have included the mode of action of compounds with primary outcomes and suitable references.
| Agent | Mode of action | Primary outcome | Reference |
|---|---|---|---|
| 7, 8-dihydroxyflavone (7, 8-DHF) | Tropomyosin-receptor-kinase B (TrkB) agonist that mimics the effects of brain-derived neurotrophic factor (BDNF). Transcription of nuclear respiratory factors (NRFs), cytochrome c oxidase II (COII) and mitochondrial transcription factor A (TFAM). |
Neuronal survival and regeneration. Protection of immature neurons from excitotoxicity-mediated cell death. Reduction of dendrite swellings in the cortex. Maintenance of Mitochondrial homeostasis |
Wu et al, 2014
Chen et al. 2015 Zhao et al., 2016 Agrawal et al, 2015 |
| Pioglitazone | Peroxisome proliferator-activated receptor-γ (PPARγ) agonist and binds to mitochondrial protein mitoNEET (CISD1). | Prevention of Ca2+-induced mitochondrial dysfunction and improved neurological outcomes after TBI. Suppression of inflammation and tissue injury, and improve locomotor activity. Edema and fluid retention. |
Rabchevsky et al., 2017 Yonutas et al., 2020 Patel et al., 2017; Rabchevsky et al., 2017 Bełtowski et al., 2013 |
| Beta-hydroxybutyrate (BHB) | Non-glucose substrate for metabolic reprogramming. Upregulate cerebral ketone body metabolism. |
Attenuates brain damage. | Prins et al., 2004 |
| Trifluoromethoxy carbonylcyanide phenylhydrazone (FCCP) | Mitochondrial uncoupler | Lowers ROS production and Ca2+ overload. | Pandya et al. 2009 |
| 2, 4-dinitrophenol (2, 4- DNP) | Mitochondrial uncoupler | Improves mitochondrial homeostasis and attenuates tissue damage. |
Geisler, 2019 Hubbard et al., 2018; Korde et al., 2005 |
| Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one) | Antioxidative effect | Reduction in brain edema, protection of neuronal cells, and improvement in the neurological impairment. |
Cruz, 2018. Ren et al., 2019 |
| Nicotinamide adenine dinucleotide (NAD+) | Increase cellular NAD+ pool by multiple mechanisms. Activation of Sirtuins. Inhibition of PARP and cADP-ribose synthases Activation of heme oxygenase 1 (HO-1) and nuclear factor-like 2 (Nrf2). Prevents glycolytic inhibition, mitochondrial depolarization, mitochondrial permeability and apoptosis inducing factor (AIF) translocation. |
Promotes neuronal survival under stress. Reduces oxidative stress and inflammation. Enhance DNA-repair mechanism. |
Cantó et al., 2015; Davila et al., 2018; Srivastava, 2016 Wei et al., 2007a. Wang et al., 2008 Alano et al., 2004; Ying et al., 2003; Ying and Xiong, 2010 |
| Cyclosporin | Binds to cyclophilin D and disrupt the formation of mPTP. | Reversing the effect of Ca2+ overload in mitochondria. Suppress T-cell activation |
Sullivan et al., 2000; Waldmeier et al., 2003 |
| N-methyl-4-isoleucine-cyclosporin (NIM811) | Analog of cyclosporine, inhibits cyclophilin D and disrupt the formation of mPTP. | Improves open field locomotor performance, return of reflexive bladder control. Decrease the rostral-caudal extent of the lesion. Improves cognition and mitochondrial functioning, Reduces oxidative damage |
Mbye et al., 2009; Springer et al., 2018a Springer et al., 2018b Readnower et al., 2011 |
| Methylene Blue | Provides an alternative route for electron transfer between NADH (mitochondrial complex I) and cytochrome c (complex IV) | Reduce MRI-lesion volume Reverse neuronal mitochondrial dysfunction and improves BBB integrity |
Watts LT et al., 2016 Shen J et al, 2019 |
| Kaempferol | Stimulates mitochondrial Ca2+ uniporter channel (mCU). | Enhance spontaneous/stimulus-evoked neural activity and neurovascular response. Improved behavioral response and brain functions in Rat model. |
Murugan m et al, 2016 Chitturi et al., 2020 |
| Ru360 | High affinity selective mitochondrial calcium uptake inhibitor. | Increase neural viability and improve sensorimotor behavior score | Chitturi et al., 2021 |
| N-acetylcysteine (NAC) and N-acetylcysteine amide (NACA) | Enhances the production of endogenous antioxidant, glutathione (GSH). | Regulates cellular redox state during brain injury. Improves integrity of blood brain barrier. Increase levels of anti-inflammatory M2 microglia. |
Kawoos et al., 2019
Haber et al., 2018 |
| Alpha-lipoic acid (ALA) | Acts as metal chelator. Downregulation of caspase-3. Modulation of Nrf2 signaling. |
Restores the BBB disruption. Normalizes the astrocytic/microglial activation and glutathione (GSH) levels. Attenuates mitochondrial-mediated apoptosis. Promotes angiogenesis. |
Rocamonde et al., 2013; Sears, 2013 Wei et al., 2015 Rocamonde et al., 2013 |
| Sulforaphane | Regulates nuclear factor E2-related factor (Nrf2) and antioxidant response element (ARE). | Anti-oxidative response. Reduce contusion volume. Protects neurons and astrocytes from oxygen and glucose deprivation (OGD). Improves cognitive functions. |
Wang et al., 2012
Ladak et al., 2021 Dash et al., 2009 |
| Carnosic acid (CA) | Activates Nrf2-ARE signaling pathway. Activates SIRT1/p66shc signaling pathway. |
Improves motor and cognitive function. Decrease Gfap and Iba1 immunoreactivity. |
Satoh et al., 2008
Maynard et al., 2019 |
| U-83836E | Scavenge lipid peroxyl radicals such as 4-hydroxy-2-nonenal (4-HNE). | Improves mitochondrial functions. |
Mustafa et al., 2018a; Mustafa et al., 2018b |
| Phenelzine | Scavenge lipid peroxyl radicals such as 4-hydroxy-2-nonenal (4-HNE). Non-selective and irreversible inhibitor of the enzyme monoamine oxidase (MAO, both MAO-A and MAO-B) |
Reduce anxiety, atypical depression, depression | Kulbe et al., 2018 |
Highlights.
Mitochondria are powerhouse of the cell.
Mitochondria regulates bioenergetics, calcium homeostasis, signal transmission, and fate of cell in case of injury.
Mitochondrial dysfunction leads to oxidative stress, loss of ATP, inflammation, loss of mitochondrial dynamics and cell death.
Therapies targeting mitochondrial dynamics may improve acute and chronic outcomes in brain pathologies.
Mitochondrial transfer from stem cells to injured cells has shown promising results to restore cellular metabolism.
Funding
Authors’ research is supported by grants from the National Institutes of Neurological Diseases and Stroke (NS114560 to KV; NS065172, NS097825 to KMD; NS099455, NS113356, NS112511 to DCH; and NS110378 to BB/KMD), and AURI fund (MCGFD08343 to KV).
Footnotes
Conflicts of Interest
Authors declare no financial or competing conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Declarations of interest
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdul-Muneer PM, Chandra N, Haorah J, 2015. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol Neurobiol 51, 966–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agranoff BW, Siegel GJ, 1994. Basic neurochemistry: Molecular, cellular, and medical aspects. Raven Press. [Google Scholar]
- Agrawal A, Pekkurnaz G, Koslover EF, 2018. Spatial control of neuronal metabolism through glucose-mediated mitochondrial transport regulation. eLife 7, e40986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal R, Noble E, Tyagi E, Zhuang Y, Ying Z, Gomez-Pinilla F, 2015. Flavonoid derivative 7,8-DHF attenuates TBI pathology via TrkB activation. Biochim Biophys Acta 1852, 862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad T, Mukherjee S, Pattnaik B, Kumar M, Singh S, Kumar M, Rehman R, Tiwari BK, Jha KA, Barhanpurkar AP, Wani MR, Roy SS, Mabalirajan U, Ghosh B, Agrawal A, 2014. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J 33, 994–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alano CC, Swanson RA, 2006. Players in the PARP-1 cell-death pathway: JNK1 joins the cast. Trends Biochem Sci 31, 309–311. [DOI] [PubMed] [Google Scholar]
- Alano CC, Ying W, Swanson RA, 2004. Poly (ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. Journal of Biological Chemistry 279, 18895–18902. [DOI] [PubMed] [Google Scholar]
- Alle H, Roth A, Geiger JR, 2009. Energy-efficient action potentials in hippocampal mossy fibers. Science 325, 1405–1408. [DOI] [PubMed] [Google Scholar]
- Almannai M, El-Hattab AW, Ali M, Soler-Alfonso C, Scaglia F, 2020. Clinical trials in mitochondrial disorders, an update. Mol Genet Metab 131, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadoro G, Corsetti V, Florenzano F, Atlante A, Bobba A, Nicolin V, Nori SL, Calissano P, 2014. Morphological and bioenergetic demands underlying the mitophagy in post-mitotic neurons: the pink–parkin pathway. Frontiers in aging neuroscience 6, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amé JC, Spenlehauer C, de Murcia G, 2004. The PARP superfamily. Bioessays 26, 882–893. [DOI] [PubMed] [Google Scholar]
- Andersen JK, 2004. Oxidative stress in neurodegeneration: cause or consequence? Nat Med 10 Suppl, S18–25. [DOI] [PubMed] [Google Scholar]
- Ansari MA, Roberts KN, Scheff SW, 2008. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radical Biology and Medicine 45, 443–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer SL, 2013. Mitochondrial dynamics—mitochondrial fission and fusion in human diseases. New England Journal of Medicine 369, 2236–2251. [DOI] [PubMed] [Google Scholar]
- Ashurst J, Wasson M, 2011. Methemoglobinemia: a systematic review of the pathophysiology, detection, and treatment. Del Med J 83, 203–208. [PubMed] [Google Scholar]
- Averna M, Bavestrello M, Cresta F, Pedrazzi M, De Tullio R, Minicucci L, Sparatore B, Salamino F, Pontremoli S, Melloni E, 2016. Abnormal activation of calpain and protein kinase Cα promotes a constitutive release of matrix metalloproteinase 9 in peripheral blood mononuclear cells from cystic fibrosis patients. Arch Biochem Biophys 604, 103–112. [DOI] [PubMed] [Google Scholar]
- Baban B, Braun M, Khodadadi H, Ward A, Alverson K, Malik A, Nguyen K, Nazarian S, Hess DC, Forseen S, Post AF, Vale FL, Vender JR, Hoda MN, Akbari O, Vaibhav K, Dhandapani KM, 2021. AMPK induces regulatory innate lymphoid cells after traumatic brain injury. JCI Insight 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bambrick L, Kristian T, Fiskum G, 2004. Astrocyte mitochondrial mechanisms of ischemic brain injury and neuroprotection. Neurochem Res 29, 601–608. [DOI] [PubMed] [Google Scholar]
- Bando Y, Geisler JG, 2019. Disease modifying mitochondrial uncouplers, MP101, and a slow release ProDrug, MP201, in models of Multiple Sclerosis. Neurochem Int 131, 104561. [DOI] [PubMed] [Google Scholar]
- Bárcena C, Mayoral P, Quirós PM, 2018. Mitohormesis, an Antiaging Paradigm. Int Rev Cell Mol Biol 340, 35–77. [DOI] [PubMed] [Google Scholar]
- Bauernfeind AL, Babbitt CC, 2014. The appropriation of glucose through primate neurodevelopment. J Hum Evol 77, 132–140. [DOI] [PubMed] [Google Scholar]
- Bauernfeind AL, Barks SK, Duka T, Grossman LI, Hof PR, Sherwood CC, 2014. Aerobic glycolysis in the primate brain: reconsidering the implications for growth and maintenance. Brain Struct Funct 219, 1149–1167. [DOI] [PubMed] [Google Scholar]
- Bayir H, Kagan VE, Tyurina YY, Tyurin V, Ruppel RA, Adelson PD, Graham SH, Janesko K, Clark RS, Kochanek PM, 2002. Assessment of antioxidant reserves and oxidative stress in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatric research 51, 571–578. [DOI] [PubMed] [Google Scholar]
- Belgnaoui SM, Paz S, Hiscott J, 2011. Orchestrating the interferon antiviral response through the mitochondrial antiviral signaling (MAVS) adapter. Curr Opin Immunol 23, 564–572. [DOI] [PubMed] [Google Scholar]
- Bell SM, Barnes K, De Marco M, Shaw PJ, Ferraiuolo L, Blackburn DJ, Venneri A, Mortiboys H, 2021. Mitochondrial Dysfunction in Alzheimer’s Disease: A Biomarker of the Future? Biomedicines 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SM, De Marco M, Barnes K, Shaw PJ, Ferraiuolo L, Blackburn DJ, Mortiboys H, Venneri A, 2020. Deficits in mitochondrial spare respiratory capacity contribute to the neuropsychological changes of Alzheimer’s disease. Journal of personalized medicine 10, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bełtowski J, Rachańczyk J, Włodarczyk M, 2013. Thiazolidinedione-induced fluid retention: recent insights into the molecular mechanisms. PPAR Res 2013, 628628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bénard G, Massa F, Puente N, Lourenço J, Bellocchio L, Soria-Gómez E, Matias I, Delamarre A, Metna-Laurent M, Cannich A, 2012. Mitochondrial CB 1 receptors regulate neuronal energy metabolism. Nature neuroscience 15, 558–564. [DOI] [PubMed] [Google Scholar]
- Bennett MV, Contreras JE, Bukauskas FF, Sáez JC, 2003. New roles for astrocytes: gap junction hemichannels have something to communicate. Trends Neurosci 26, 610–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereiter-Hahn J, Vöth M, 1994. Dynamics of mitochondria in living cells: shape changes, dislocations, fusion, and fission of mitochondria. Microscopy research and technique 27, 198–219. [DOI] [PubMed] [Google Scholar]
- Bernini A, Masoodi M, Solari D, Miroz JP, Carteron L, Christinat N, Morelli P, Beaumont M, Abed-Maillard S, Hartweg M, Foltzer F, Eckert P, Cuenoud B, Oddo M, 2020. Modulation of cerebral ketone metabolism following traumatic brain injury in humans. J Cereb Blood Flow Metab 40, 177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertero E, Maack C, 2018. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ Res 122, 1460–1478. [DOI] [PubMed] [Google Scholar]
- Bhat AH, Dar KB, Anees S, Zargar MA, Masood A, Sofi MA, Ganie SA, 2015. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed Pharmacother 74, 101–110. [DOI] [PubMed] [Google Scholar]
- Bhatti J, Nascimento B, Akhtar U, Rhind SG, Tien H, Nathens A, da Luz LT, 2017. Systematic Review of Human and Animal Studies Examining the Efficacy and Safety of N-Acetylcysteine (NAC) and N-Acetylcysteine Amide (NACA) in Traumatic Brain Injury: Impact on Neurofunctional Outcome and Biomarkers of Oxidative Stress and Inflammation. Front Neurol 8, 744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobinger T, Burkardt P, H BH, Manaenko A, 2018. Programmed Cell Death after Intracerebral Hemorrhage. Curr Neuropharmacol 16, 1267–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland ML, Chourasia AH, Macleod KF, 2013. Mitochondrial dysfunction in cancer. Front Oncol 3, 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordone MP, Salman MM, Titus HE, Amini E, Andersen JV, Chakraborti B, Diuba AV, Dubouskaya TG, Ehrke E, Espindola de Freitas A, Braga de Freitas G, Gonçalves RA, Gupta D, Gupta R, Ha SR, Hemming IA, Jaggar M, Jakobsen E, Kumari P, Lakkappa N, Marsh APL, Mitlöhner J, Ogawa Y, Paidi RK, Ribeiro FC, Salamian A, Saleem S, Sharma S, Silva JM, Singh S, Sulakhiya K, Tefera TW, Vafadari B, Yadav A, Yamazaki R, Seidenbecher CI, 2019. The energetic brain - A review from students to students. J Neurochem 151, 139–165. [DOI] [PubMed] [Google Scholar]
- Braun M, Khan ZT, Khan MB, Kumar M, Ward A, Achyut BR, Arbab AS, Hess DC, Hoda MN, Baban B, Dhandapani KM, Vaibhav K, 2018. Selective activation of cannabinoid receptor-2 reduces neuroinflammation after traumatic brain injury via alternative macrophage polarization. Brain Behav Immun 68, 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray N, 2019. Many makes of mitochondria. Nature Reviews Neuroscience 20, 645–645. [DOI] [PubMed] [Google Scholar]
- Breda CNS, Davanzo GG, Basso PJ, Saraiva Camara NO, Moraes-Vieira PMM, 2019. Mitochondria as central hub of the immune system. Redox Biol 26, 101255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, Dixit VM, 2016. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16, 407–420. [DOI] [PubMed] [Google Scholar]
- Brustovetsky N, Brustovetsky T, Jemmerson R, Dubinsky JM, 2002. Calcium-induced Cytochrome c release from CNS mitochondria is associated with the permeability transition and rupture of the outer membrane. Journal of neurochemistry 80, 207–218. [DOI] [PubMed] [Google Scholar]
- Burté F, Carelli V, Chinnery PF, Yu-Wai-Man P, 2015. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nature reviews neurology 11, 11–24. [DOI] [PubMed] [Google Scholar]
- Busquets-Garcia A, Bains J, Marsicano G, 2018. CB 1 receptor signaling in the brain: extracting specificity from ubiquity. Neuropsychopharmacology 43, 4–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busquets-Garcia A, Desprez T, Metna-Laurent M, Bellocchio L, Marsicano G, Soria-Gomez E, 2015. Dissecting the cannabinergic control of behavior: The where matters. Bioessays 37, 1215–1225. [DOI] [PubMed] [Google Scholar]
- Cai C. c., Zhu J. h., Ye L. x., Dai Y. y., Fang M. c., Hu Y. y., Pan S. l., Chen S, Li P. j., Fu X. q., Lin Z. l., 2019. Glycine Protects against Hypoxic-Ischemic Brain Injury by Regulating Mitochondria-Mediated Autophagy via the AMPK Pathway. Oxidative Medicine and Cellular Longevity 2019, 4248529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderón-Montaño JM, Burgos-Morón E, Pérez-Guerrero C, López-Lázaro M, 2011. A review on the dietary flavonoid kaempferol. Mini Rev Med Chem 11, 298–344. [DOI] [PubMed] [Google Scholar]
- Callaway NL, Riha PD, Bruchey AK, Munshi Z, Gonzalez-Lima F, 2004. Methylene blue improves brain oxidative metabolism and memory retention in rats. Pharmacol Biochem Behav 77, 175–181. [DOI] [PubMed] [Google Scholar]
- Calvo SE, Mootha VK, 2010. The mitochondrial proteome and human disease. Annual review of genomics and human genetics 11, 25–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantó C, Menzies KJ, Auwerx J, 2015. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell metabolism 22, 31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carinci M, Vezzani B, Patergnani S, Ludewig P, Lessmann K, Magnus T, Casetta I, Pugliatti M, Pinton P, Giorgi C, 2021. Different Roles of Mitochondria in Cell Death and Inflammation: Focusing on Mitochondrial Quality Control in Ischemic Stroke and Reperfusion. Biomedicines 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello NA, Nguyen MH, Tran JD, Cheng D, Green KN, LaFerla FM, 2014. 7,8-Dihydroxyflavone, a small molecule TrkB agonist, improves spatial memory and increases thin spine density in a mouse model of Alzheimer disease-like neuronal loss. PloS one 9, e91453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC, 2006. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 22, 79–99. [DOI] [PubMed] [Google Scholar]
- Chen H, Chan DC, 2017. Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell metabolism 26, 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chan YL, Nguyen LT, Mao Y, de Rosa A, Beh IT, Chee C, Oliver B, Herok G, Saad S, Gorrie C, 2016. Moderate traumatic brain injury is linked to acute behaviour deficits and long term mitochondrial alterations. Clin Exp Pharmacol Physiol 43, 1107–1114. [DOI] [PubMed] [Google Scholar]
- Chen L, Gao X, Zhao S, Hu W, Chen J, 2015. The Small-Molecule TrkB Agonist 7, 8-Dihydroxyflavone Decreases Hippocampal Newborn Neuron Death After Traumatic Brain Injury. J Neuropathol Exp Neurol 74, 557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Guo C, Feng H, Chen Y, 2020a. Mitochondria: Novel Mechanisms and Therapeutic Targets for Secondary Brain Injury After Intracerebral Hemorrhage. Front Aging Neurosci 12, 615451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Guo C, Jia Z, Wang J, Xia M, Li C, Li M, Yin Y, Tang X, Chen T, Hu R, Chen Y, Liu X, Feng H, 2020b. Inhibition of Mitochondrial ROS by MitoQ Alleviates White Matter Injury and Improves Outcomes after Intracerebral Haemorrhage in Mice. Oxid Med Cell Longev 2020, 8285065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Guo Y, Yang W, Zheng P, Zeng J, Tong W, 2017. Involvement of autophagy in connexin 40 reduction in the late phase of traumatic brain injury in rats. Brain Res Bull 131, 100–106. [DOI] [PubMed] [Google Scholar]
- Chen Y, Gong K, Xu Q, Meng J, Long T, Chang C, Wang Z, Liu W, 2021. Phosphoglycerate Mutase 5 Knockdown Alleviates Neuronal Injury After Traumatic Brain Injury Through Drp1-Mediated Mitochondrial Dysfunction. Antioxid Redox Signal 34, 154–170. [DOI] [PubMed] [Google Scholar]
- Cheng G, Kong R.h., Zhang L.m., Zhang J.n., 2012. Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. British journal of pharmacology 167, 699–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimeh U, Zimmerman MA, Gilyazova N, Li PA, 2018. B355252, a novel small molecule, confers neuroprotection against cobalt chloride toxicity in mouse hippocampal cells through altering mitochondrial dynamics and limiting autophagy induction. International journal of medical sciences 15, 1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitturi J, Santhakumar V, Kannurpatti SS, 2019. Beneficial Effects of Kaempferol after Developmental Traumatic Brain Injury Is through Protection of Mitochondrial Function, Oxidative Metabolism, and Neural Viability. J Neurotrauma 36, 1264–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitturi J, Santhakumar V, Kannurpatti SS, 2021. Traumatic brain injury metabolome and mitochondrial impact after early stage Ru360 treatment. Mitochondrion 57, 192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen J, Shastri VP, 2015. Matrix-metalloproteinase-9 is cleaved and activated by Cathepsin K. BMC Research Notes 8, 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D, Wang KZQ, Zhu J, Klein-Seetharaman J, Balasubramanian K, Amoscato AA, Borisenko G, Huang Z, Gusdon AM, Cheikhi A, Steer EK, Wang R, Baty C, Watkins S, Bahar I, Bayir H, Kagan VE, 2013. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol 15, 1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RSB, Empey PE, Bayir H, Rosario BL, Poloyac SM, Kochanek PM, Nolin TD, Au AK, Horvat CM, Wisniewski SR, Bell MJ, 2017. Phase I randomized clinical trial of N-acetylcysteine in combination with an adjuvant probenecid for treatment of severe traumatic brain injury in children. PloS one 12, e0180280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court AC, Le-Gatt A, Luz-Crawford P, Parra E, Aliaga-Tobar V, Batiz LF, Contreras RA, Ortuzar MI, Kurte M, Elizondo-Vega R, Maracaja-Coutinho V, Pino-Lagos K, Figueroa FE, Khoury M, 2020. Mitochondrial transfer from MSCs to T cells induces Treg differentiation and restricts inflammatory response. EMBO Rep 21, e48052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz MP, 2018. Edaravone (Radicava): A Novel Neuroprotective Agent for the Treatment of Amyotrophic Lateral Sclerosis. P t 43, 25–28. [PMC free article] [PubMed] [Google Scholar]
- Cserep C, Posfai B, Lenart N, Fekete R, Laszlo ZI, Lele Z, Orsolits B, Molnar G, Heindl S, Schwarcz AD, Ujvari K, Kornyei Z, Toth K, Szabadits E, Sperlagh B, Baranyi M, Csiba L, Hortobagyi T, Magloczky Z, Martinecz B, Szabo G, Erdelyi F, Szipocs R, Tamkun MM, Gesierich B, Duering M, Katona I, Liesz A, Tamas G, Denes A, 2020. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 367, 528–537. [DOI] [PubMed] [Google Scholar]
- Darlington CL, 2003. Dexanabinol: a novel cannabinoid with neuroprotective properties. IDrugs 6, 976–979. [PubMed] [Google Scholar]
- Dash PK, Hergenroeder GW, Jeter CB, Choi HA, Kobori N, Moore AN, 2016. Traumatic brain injury alters methionine metabolism: implications for pathophysiology. Frontiers in Systems Neuroscience 10, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash PK, Zhao J, Orsi SA, Zhang M, Moore AN, 2009. Sulforaphane improves cognitive function administered following traumatic brain injury. Neurosci Lett 460, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila A, Liu L, Chellappa K, Redpath P, Nakamaru-Ogiso E, Paolella LM, Zhang Z, Migaud ME, Rabinowitz JD, Baur JA, 2018. Nicotinamide adenine dinucleotide is transported into mammalian mitochondria. eLife 7, e33246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CH, Kim KY, Bushong EA, Mills EA, Boassa D, Shih T, Kinebuchi M, Phan S, Zhou Y, Bihlmeyer NA, Nguyen JV, Jin Y, Ellisman MH, Marsh-Armstrong N, 2014. Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci U S A 111, 9633–9638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lores Arnaiz GR, Bersier MG, 2014. Relationship between Na+, K+-ATPase and NMDA receptor at central synapses. Curr Protein Pept Sci 15, 761–777. [DOI] [PubMed] [Google Scholar]
- Deepmala Slattery J., Kumar N, Delhey L, Berk M, Dean O, Spielholz C, Frye R, 2015. Clinical trials of N-acetylcysteine in psychiatry and neurology: A systematic review. Neurosci Biobehav Rev 55, 294–321. [DOI] [PubMed] [Google Scholar]
- Denzer K, Kleijmeer MJ, Heijnen HF, Stoorvogel W, Geuze HJ, 2000. Exosome: from internal vesicle of the multivesicular body to intercellular signaling device. J Cell Sci 113 Pt 19, 3365–3374. [DOI] [PubMed] [Google Scholar]
- Deshpande LS, Sun DA, Sombati S, Baranova A, Wilson MS, Attkisson E, Hamm RJ, DeLorenzo RJ, 2008. Alterations in neuronal calcium levels are associated with cognitive deficits after traumatic brain injury. Neurosci Lett 441, 115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devanney NA, Stewart AN, Gensel JC, 2020. Microglia and macrophage metabolism in CNS injury and disease: The role of immunometabolism in neurodegeneration and neurotrauma. Exp Neurol 329, 113310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine MJ, Kittler JT, 2018. Mitochondria at the neuronal presynapse in health and disease. Nature Reviews Neuroscience 19, 63–80. [DOI] [PubMed] [Google Scholar]
- Di Pietro V, Lazzarino G, Amorini AM, Signoretti S, Hill LJ, Porto E, Tavazzi B, Lazzarino G, Belli A, 2017. Fusion or Fission: The Destiny of Mitochondria In Traumatic Brain Injury of Different Severities. Sci Rep 7, 9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiBona VL, Shah MK, Krause KJ, Zhu W, Voglewede MM, Smith DM, Crockett DP, Zhang H, 2021. Metformin reduces neuroinflammation and improves cognitive functions after traumatic brain injury. Neurosci Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding W, Chen R, Wu C, Chen W, Zhang H, Fan X, Wang H, Ji Y, Xie L, Ning X, Shen L, 2017. Increased expression of HERPUD1 involves in neuronal apoptosis after intracerebral hemorrhage. Brain Res Bull 128, 40–47. [DOI] [PubMed] [Google Scholar]
- Ding WX, Yin XM, 2012. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem 393, 547–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerrier C, Garcia-Souza LF, Krumschnabel G, Wohlfarter Y, Mészáros AT, Gnaiger E, 2018. High-Resolution FluoRespirometry and OXPHOS Protocols for Human Cells, Permeabilized Fibers from Small Biopsies of Muscle, and Isolated Mitochondria. Methods Mol Biol 1782, 31–70. [DOI] [PubMed] [Google Scholar]
- Dröse S, Brandt U, Wittig I, 2014. Mitochondrial respiratory chain complexes as sources and targets of thiol-based redox-regulation. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics 1844, 1344–1354. [DOI] [PubMed] [Google Scholar]
- Endres M, Wang Z-Q, Namura S, Waeber C, Moskowitz MA, 1997. Ischemic brain injury is mediated by the activation of poly (ADP-ribose) polymerase. Journal of Cerebral Blood Flow & Metabolism 17, 1143–1151. [DOI] [PubMed] [Google Scholar]
- Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, Schwartz A, Snutch TP, Tanabe T, Birnbaumer L, Tsien RW, Catterall WA, 2000. Nomenclature of voltage-gated calcium channels. Neuron 25, 533–535. [DOI] [PubMed] [Google Scholar]
- Fan H, Ding R, Liu W, zhang X, Li R, Wei B, Su S, Jin F, Wei C, He X, Li X, Duan C, 2021. Heat shock protein 22 modulates NRF1/TFAM-dependent mitochondrial biogenesis and DRP1-sparked mitochondrial apoptosis through AMPK-PGC1α signaling pathway to alleviate the early brain injury of subarachnoid hemorrhage in rats. Redox Biology 40, 101856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L, Zhu C, Qiu R, Zan P, Zheng Z, Xu T, Li G, 2017a. MicroRNA-661 enhances TRAIL or STS induced osteosarcoma cell apoptosis by modulating the expression of cytochrome c1. Cellular Physiology and Biochemistry 41, 1935–1946. [DOI] [PubMed] [Google Scholar]
- Fan LF, He PY, Peng YC, Du QH, Ma YJ, Jin JX, Xu HZ, Li JR, Wang ZJ, Cao SL, Li T, Yan F, Gu C, Wang L, Chen G, 2017b. Mdivi-1 ameliorates early brain injury after subarachnoid hemorrhage via the suppression of inflammation-related blood-brain barrier disruption and endoplasmic reticulum stress-based apoptosis. Free Radic Biol Med 112, 336–349. [DOI] [PubMed] [Google Scholar]
- Fecher C, Trovò L, Müller SA, Snaidero N, Wettmarshausen J, Heink S, Ortiz O, Wagner I, Kühn R, Hartmann J, 2019. Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nature neuroscience 22, 1731–1742. [DOI] [PubMed] [Google Scholar]
- Fenton AR, Jongens TA, Holzbaur ELF, 2021. Mitochondrial dynamics: Shaping and remodeling an organelle network. Curr Opin Cell Biol 68, 28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer TD, Hylin MJ, Zhao J, Moore AN, Waxham MN, Dash PK, 2016a. Altered Mitochondrial Dynamics and TBI Pathophysiology. Front Syst Neurosci 10, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer TD, Hylin MJ, Zhao J, Moore AN, Waxham MN, Dash PK, 2016b. Altered Mitochondrial Dynamics and TBI Pathophysiology. Frontiers in Systems Neuroscience 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederich M, Balschi JA, 2002. The relationship between AMP-activated protein kinase activity and AMP concentration in the isolated perfused rat heart. Journal of Biological Chemistry 277, 1928–1932. [DOI] [PubMed] [Google Scholar]
- Frey TG, Mannella CA, 2000. The internal structure of mitochondria. Trends in biochemical sciences 25, 319–324. [DOI] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK, 2011. ER tubules mark sites of mitochondrial division. Science 334, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajavelli S, Sinha VK, Mazzeo AT, Spurlock MS, Lee SW, Ahmed AI, Yokobori S, Bullock RM, 2015. Evidence to support mitochondrial neuroprotection, in severe traumatic brain injury. Journal of bioenergetics and biomembranes 47, 133–148. [DOI] [PubMed] [Google Scholar]
- Galve-Roperh I, Chiurchiù V, Díaz-Alonso J, Bari M, Guzmán M, Maccarrone M, 2013. Cannabinoid receptor signaling in progenitor/stem cell proliferation and differentiation. Progress in lipid research 52, 633–650. [DOI] [PubMed] [Google Scholar]
- Geisler JG, 2019. 2,4 Dinitrophenol as Medicine. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W, 2010. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12, 119–131. [DOI] [PubMed] [Google Scholar]
- Geldenhuys WJ, Benkovic SA, Lin L, Yonutas HM, Crish SD, Sullivan PG, Darvesh AS, Brown CM, Richardson JR, 2017. MitoNEET (CISD1) Knockout Mice Show Signs of Striatal Mitochondrial Dysfunction and a Parkinson’s Disease Phenotype. ACS chemical neuroscience 8, 2759–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geldenhuys WJ, Yonutas HM, Morris DL, Sullivan PG, Darvesh AS, Leeper TC, 2016. Identification of small molecules that bind to the mitochondrial protein mitoNEET. Bioorganic & medicinal chemistry letters 26, 5350–5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gensini GF, Conti AA, Lippi D, 2007. The contributions of Paul Ehrlich to infectious disease. J Infect 54, 221–224. [DOI] [PubMed] [Google Scholar]
- Giacomello M, Pyakurel A, Glytsou C, Scorrano L, 2020. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol 21, 204–224. [DOI] [PubMed] [Google Scholar]
- Gilmer LK, Roberts KN, Joy K, Sullivan PG, Scheff SW, 2009. Early mitochondrial dysfunction after cortical contusion injury. Journal of neurotrauma 26, 1271–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golpich M, Amini E, Mohamed Z, Azman Ali R, Mohamed Ibrahim N, Ahmadiani A, 2017. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci Ther 23, 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodfellow MJ, Borcar A, Proctor JL, Greco T, Rosenthal RE, Fiskum G, 2020. Transcriptional activation of antioxidant gene expression by Nrf2 protects against mitochondrial dysfunction and neuronal death associated with acute and chronic neurodegeneration. Exp Neurol 328, 113247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal MS, Iannotti LL, Raichle ME, 2018. Brain Nutrition: A Life Span Approach. Annu Rev Nutr 38, 381–399. [DOI] [PubMed] [Google Scholar]
- Goyal MS, Venkatesh S, Milbrandt J, Gordon JI, Raichle ME, 2015. Feeding the brain and nurturing the mind: Linking nutrition and the gut microbiota to brain development. Proc Natl Acad Sci U S A 112, 14105–14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grazioli S, Pugin J, 2018. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front Immunol 9, 832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilfoyle MR, Carpenter KLH, Helmy A, Pickard JD, Menon DK, Hutchinson PJA, 2015. Matrix Metalloproteinase Expression in Contusional Traumatic Brain Injury: A Paired Microdialysis Study. Journal of neurotrauma 32, 1553–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurusamy N, Lekli I, Mukherjee S, Ray D, Ahsan MK, Gherghiceanu M, Popescu LM, Das DK, 2010. Cardioprotection by resveratrol: a novel mechanism via autophagy involving the mTORC2 pathway. Cardiovascular research 86, 103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyoneva S, Ransohoff RM, 2015. Inflammatory reaction after traumatic brain injury: therapeutic potential of targeting cell-cell communication by chemokines. Trends Pharmacol Sci 36, 471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas RH, 2019. Mitochondrial Dysfunction in Aging and Diseases of Aging. Biology (Basel) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber M, James J, Kim J, Sangobowale M, Irizarry R, Ho J, Nikulina E, Grin’kina NM, Ramadani A, Hartman I, Bergold PJ, 2018. Minocycline plus N-acteylcysteine induces remyelination, synergistically protects oligodendrocytes and modifies neuroinflammation in a rat model of mild traumatic brain injury. J Cereb Blood Flow Metab 38, 1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadass O, Tomlinson BN, Gooyit M, Chen S, Purdy JJ, Walker JM, Zhang C, Giritharan AB, Purnell W, Robinson CR II, Shin D, Schroeder VA, Suckow MA, Simonyi A, Y. Sun, G, Mobashery S, Cui J, Chang M, Gu Z, 2013. Selective Inhibition of Matrix Metalloproteinase-9 Attenuates Secondary Damage Resulting from Severe Traumatic Brain Injury. PloS one 8, e76904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haim LB, Rowitch DH, 2017. Functional diversity of astrocytes in neural circuit regulation. Nature Reviews Neuroscience 18, 31–41. [DOI] [PubMed] [Google Scholar]
- Hall ED, Vaishnav RA, Mustafa AG, 2010. Antioxidant therapies for traumatic brain injury. Neurotherapeutics 7, 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ED, Wang JA, Miller DM, Cebak JE, Hill RL, 2019. Newer pharmacological approaches for antioxidant neuroprotection in traumatic brain injury. Neuropharmacology 145, 247–258. [DOI] [PubMed] [Google Scholar]
- Han D, Zheng X, Wang X, Jin T, Cui L, Chen Z, 2020. Mesenchymal Stem/Stromal Cell-Mediated Mitochondrial Transfer and the Therapeutic Potential in Treatment of Neurological Diseases. Stem Cells Int 2020, 8838046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JJ, Jolivet R, Attwell D, 2012. Synaptic energy use and supply. Neuron 75, 762–777. [DOI] [PubMed] [Google Scholar]
- Harris LK, Black RT, Golden KM, Reeves TM, Povlishock JT, Phillips LL, 2001. Traumatic brain injury-induced changes in gene expression and functional activity of mitochondrial cytochrome C oxidase. J Neurotrauma 18, 993–1009. [DOI] [PubMed] [Google Scholar]
- Hatton J, Rosbolt B, Empey P, Kryscio R, Young B, 2008. Dosing and safety of cyclosporine in patients with severe brain injury. J Neurosurg 109, 699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG, 2005. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell metabolism 2, 9–19. [DOI] [PubMed] [Google Scholar]
- Hayakawa K, Chan SJ, Mandeville ET, Park JH, Bruzzese M, Montaner J, Arai K, Rosell A, Lo EH, 2018. Protective Effects of Endothelial Progenitor Cell-Derived Extracellular Mitochondria in Brain Endothelium. Stem Cells 36, 1404–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, Lo EH, 2016. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535, 551–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazeldine J, Hampson P, Opoku FA, Foster M, Lord JM, 2015. N-Formyl peptides drive mitochondrial damage associated molecular pattern induced neutrophil activation through ERK1/2 and P38 MAP kinase signalling pathways. Injury 46, 975–984. [DOI] [PubMed] [Google Scholar]
- He C, Klionsky DJ, 2009. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43, 67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Liu N, Xia W, Ying W, 2010. NAD+ administration can profoundly decrease the brain injury induced by hypoxia-ischemia. The FASEB Journal 24, 763.762–763.762. [Google Scholar]
- He Z, Ning N, Zhou Q, Khoshnam SE, Farzaneh M, 2020. Mitochondria as a therapeutic target for ischemic stroke. Free Radical Biology and Medicine 146, 45–58. [DOI] [PubMed] [Google Scholar]
- Hebert-Chatelain E, Desprez T, Serrat R, Bellocchio L, Soria-Gomez E, Busquets-Garcia A, Zottola ACP, Delamarre A, Cannich A, Vincent P, 2016. A cannabinoid link between mitochondria and memory. Nature 539, 555–559. [DOI] [PubMed] [Google Scholar]
- Hiebert JB, Shen Q, Thimmesch AR, Pierce JD, 2015. Traumatic brain injury and mitochondrial dysfunction. Am J Med Sci 350, 132–138. [DOI] [PubMed] [Google Scholar]
- Higgins GC, Beart PM, Shin YS, Chen MJ, Cheung NS, Nagley P, 2010. Oxidative stress: emerging mitochondrial and cellular themes and variations in neuronal injury. J Alzheimers Dis 20 Suppl 2, S453–473. [DOI] [PubMed] [Google Scholar]
- Hoffer ME, Balaban C, Slade MD, Tsao JW, Hoffer B, 2013. Amelioration of acute sequelae of blast induced mild traumatic brain injury by N-acetyl cysteine: a double-blind, placebo controlled study. PloS one 8, e54163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horan MP, Pichaud N, Ballard JW, 2012. Review: quantifying mitochondrial dysfunction in complex diseases of aging. J Gerontol A Biol Sci Med Sci 67, 1022–1035. [DOI] [PubMed] [Google Scholar]
- Huang CY, Wang LC, Wang HK, Pan CH, Cheng YY, Shan YS, Chio CC, Tsai KJ, 2015. Memantine alleviates brain injury and neurobehavioral deficits after experimental subarachnoid hemorrhage. Mol Neurobiol 51, 1038–1052. [DOI] [PubMed] [Google Scholar]
- Huang Y, Gao X, Zhou X, Xie B, Zhang Y, Zhu J, Zhu S, 2020. Mitophagy in the Hippocampus Is Excessive Activated After Cardiac Arrest and Cardiopulmonary Resuscitation. Neurochem Res 45, 322–330. [DOI] [PubMed] [Google Scholar]
- Hubbard WB, Harwood CL, Geisler JG, Vekaria HJ, Sullivan PG, 2018. Mitochondrial uncoupling prodrug improves tissue sparing, cognitive outcome, and mitochondrial bioenergetics after traumatic brain injury in male mice. J Neurosci Res 96, 1677–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein OA, Abdel-Hafez AMM, Abd El Kareim A, 2018. Rat hippocampal CA3 neuronal injury induced by limb ischemia/reperfusion: A possible restorative effect of alpha lipoic acid. Ultrastruct Pathol 42, 133–154. [DOI] [PubMed] [Google Scholar]
- Hyder F, Herman P, Bailey CJ, Møller A, Globinsky R, Fulbright RK, Rothman DL, Gjedde A, 2016. Uniform distributions of glucose oxidation and oxygen extraction in gray matter of normal human brain: No evidence of regional differences of aerobic glycolysis. J Cereb Blood Flow Metab 36, 903–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyder F, Patel AB, Gjedde A, Rothman DL, Behar KL, Shulman RG, 2006. Neuronal–glial glucose oxidation and glutamatergic–GABAergic function. Journal of Cerebral Blood Flow & Metabolism 26, 865–877. [DOI] [PubMed] [Google Scholar]
- Jalloh I, Carpenter KL, Helmy A, Carpenter TA, Menon DK, Hutchinson PJ, 2015. Glucose metabolism following human traumatic brain injury: methods of assessment and pathophysiological findings. Metab Brain Dis 30, 615–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang SW, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y, Wilson WD, Xiao G, Blanchi B, Sun YE, Ye K, 2010. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Natl Acad Sci U S A 107, 2687–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrahi A, Braun M, Ahluwalia M, Gupta RV, Wilson M, Munie S, Ahluwalia P, Vender JR, Vale FL, Dhandapani KM, Vaibhav K, 2020. Revisiting Traumatic Brain Injury: From Molecular Mechanisms to Therapeutic Interventions. Biomedicines 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen F, Lewczuk P, Gür O, Block W, Ende G, Frölich L, Hammen T, Arlt S, Kornhuber J, Kucinski T, 2011. Association of N-acetylaspartate and cerebrospinal fluid Aβ 42 in dementia. Journal of Alzheimer’s Disease 27, 393–399. [DOI] [PubMed] [Google Scholar]
- Jiang M, Peng Q, Liu X, Jin J, Hou Z, Zhang J, Mori S, Ross CA, Ye K, Duan W, 2013. Small-molecule TrkB receptor agonists improve motor function and extend survival in a mouse model of Huntington’s disease. Hum Mol Genet 22, 2462–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi DC, Tewari BP, Singh M, Joshi PG, Joshi NB, 2015. AMPA receptor activation causes preferential mitochondrial Ca(2)(+) load and oxidative stress in motor neurons. Brain Res 1616, 1–9. [DOI] [PubMed] [Google Scholar]
- Jung JE, Sun G, Bautista Garrido J, Obertas L, Mobley AS, Ting SM, Zhao X, Aronowski J, 2020. The Mitochondria-Derived Peptide Humanin Improves Recovery from Intracerebral Hemorrhage: Implication of Mitochondria Transfer and Microglia Phenotype Change. J Neurosci 40, 2154–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ, 2006. Role of Bax and Bak in mitochondrial morphogenesis. Nature 443, 658–662. [DOI] [PubMed] [Google Scholar]
- Kasahara A, Scorrano L, 2014. Mitochondria: from cell death executioners to regulators of cell differentiation. Trends Cell Biol 24, 761–770. [DOI] [PubMed] [Google Scholar]
- Katsu M, Niizuma K, Yoshioka H, Okami N, Sakata H, Chan PH, 2010. Hemoglobin-induced oxidative stress contributes to matrix metalloproteinase activation and blood-brain barrier dysfunction in vivo. J Cereb Blood Flow Metab 30, 1939–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawane K, Motani K, Nagata S, 2014. DNA degradation and its defects. Cold Spring Harb Perspect Biol 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawoos U, Abutarboush R, Zarriello S, Qadri A, Ahlers ST, McCarron RM, Chavko M, 2019. N-acetylcysteine Amide Ameliorates Blast-Induced Changes in Blood-Brain Barrier Integrity in Rats. Front Neurol 10, 650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsen J, Karlsson M, Hansson MJ, Yang Z, Fischer W, Hugerth M, Nordström CH, Åstrand R, Keep MF, Kilbaugh T, Wang KKW, Møller K, Juhler M, Elmér E, 2019. Copenhagen Head Injury Ciclosporin Study: A Phase IIa Safety, Pharmacokinetics, and Biomarker Study of Ciclosporin in Severe Traumatic Brain Injury Patients. J Neurotrauma 36, 3253–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Vaibhav K, Javed H, Khan MM, Tabassum R, Ahmed ME, Srivastava P, Khuwaja G, Islam F, Siddiqui MS, Safhi MM, Islam F, 2012. Attenuation of Aβ-induced neurotoxicity by thymoquinone via inhibition of mitochondrial dysfunction and oxidative stress. Mol Cell Biochem 369, 55–65. [DOI] [PubMed] [Google Scholar]
- Khatri N, Man H, 2013. Synaptic Activity and Bioenergy Homeostasis: Implications in Brain Trauma and Neurodegenerative Diseases. Frontiers in Neurology 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim-Han J, Kopp S, Dugan L, Diringer M, 2006. Perihematomal mitochondrial dysfunction after intracerebral hemorrhage (vol 37, pg 2457, 2006). Stroke 37, 3057–3057. [DOI] [PubMed] [Google Scholar]
- Kim AS, Miller EJ, Wright TM, Li J, Qi D, Atsina K, Zaha V, Sakamoto K, Young LH, 2011. A small molecule AMPK activator protects the heart against ischemia–reperfusion injury. Journal of molecular and cellular cardiology 51, 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Choi KC, 2013. Anti-cancer Effect and Underlying Mechanism(s) of Kaempferol, a Phytoestrogen, on the Regulation of Apoptosis in Diverse Cancer Cell Models. Toxicol Res 29, 229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoller N, Levi L, Shoshan I, Reichenthal E, Razon N, Rappaport ZH, Biegon A, 2002. Dexanabinol (HU-211) in the treatment of severe closed head injury: a randomized, placebo-controlled, phase II clinical trial. Crit Care Med 30, 548–554. [DOI] [PubMed] [Google Scholar]
- Knott AB, Bossy-Wetzel E, 2008a. Impairing the mitochondrial fission and fusion balance: a new mechanism of neurodegeneration. Ann N Y Acad Sci 1147, 283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott AB, Bossy-Wetzel E, 2008b. Impairing the mitochondrial fission and fusion balance: a new mechanism of neurodegeneration. Annals of the New York Academy of Sciences 1147, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E, 2008. Mitochondrial fragmentation in neurodegeneration. Nature Reviews Neuroscience 9, 505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korde AS, Pettigrew LC, Craddock SD, Maragos WF, 2005. The mitochondrial uncoupler 2,4-dinitrophenol attenuates tissue damage and improves mitochondrial homeostasis following transient focal cerebral ischemia. J Neurochem 94, 1676–1684. [DOI] [PubMed] [Google Scholar]
- Koutsilieri E, Scheller C, Tribl F, Riederer P, 2002. Degeneration of neuronal cells due to oxidative stress--microglial contribution. Parkinsonism Relat Disord 8, 401–406. [DOI] [PubMed] [Google Scholar]
- Koza L, Linseman DA, 2019. Glutathione precursors shield the brain from trauma. Neural regeneration research 14, 1701–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulbe JR, Hill RL, Singh IN, Wang JA, Hall ED, 2017. Synaptic Mitochondria Sustain More Damage than Non-Synaptic Mitochondria after Traumatic Brain Injury and Are Protected by Cyclosporine A. J Neurotrauma 34, 1291–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulbe JR, Singh IN, Wang JA, Cebak JE, Hall ED, 2018. Continuous Infusion of Phenelzine, Cyclosporine A, or Their Combination: Evaluation of Mitochondrial Bioenergetics, Oxidative Damage, and Cytoskeletal Degradation following Severe Controlled Cortical Impact Traumatic Brain Injury in Rats. J Neurotrauma 35, 1280–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladak Z, Garcia E, Yoon J, Landry T, Armstrong EA, Yager JY, Persad S, 2021. Sulforaphane (SFA) protects neuronal cells from oxygen & glucose deprivation (OGD). PloS one 16, e0248777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird MD, Wakade C, Alleyne CH Jr., Dhandapani KM, 2008. Hemin-induced necroptosis involves glutathione depletion in mouse astrocytes. Free Radic Biol Med 45, 1103–1114. [DOI] [PubMed] [Google Scholar]
- Li Y, Du XF, Liu CS, Wen ZL, Du JL, 2012. Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev Cell 23, 1189–1202. [DOI] [PubMed] [Google Scholar]
- Lifshitz J, Friberg H, Neumar RW, Raghupathi R, Welsh FA, Janmey P, Saatman KE, Wieloch T, Grady MS, McIntosh TK, 2003. Structural and functional damage sustained by mitochondria after traumatic brain injury in the rat: evidence for differentially sensitive populations in the cortex and hippocampus. Journal of Cerebral Blood Flow & Metabolism 23, 219–231. [DOI] [PubMed] [Google Scholar]
- Lifshitz J, Sullivan PG, Hovda DA, Wieloch T, McIntosh TK, 2004. Mitochondrial damage and dysfunction in traumatic brain injury. Mitochondrion 4, 705–713. [DOI] [PubMed] [Google Scholar]
- Liu F, Huang J, Hei G, Wu R, Liu Z, 2020. Effects of sulforaphane on cognitive function in patients with frontal brain damage: study protocol for a randomised controlled trial. BMJ Open 10, e037543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HY, Gale JR, Reynolds IJ, Weiss JH, Aizenman E, 2021. The Multifaceted Roles of Zinc in Neuronal Mitochondrial Dysfunction. Biomedicines 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Ji K, Guo L, Wu W, Lu H, Shan P, Yan C, 2014. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc Res 92, 10–18. [DOI] [PubMed] [Google Scholar]
- Liu X, Qi Q, Xiao G, Li J, Luo HR, Ye K, 2013. O-methylated metabolite of 7,8-dihydroxyflavone activates TrkB receptor and displays antidepressant activity. Pharmacology 91, 185–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou PH, Hansen BS, Olsen PH, Tullin S, Murphy MP, Brand MD, 2007. Mitochondrial uncouplers with an extraordinary dynamic range. Biochem J 407, 129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Jiang M, Lu L, Zheng G, Dong Q, 2015. Ultrastructural mitochondria changes in perihematomal brain and neuroprotective effects of Huperzine A after acute intracerebral hemorrhage. Neuropsychiatr Dis Treat 11, 2649–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, O’Neill KL, Huang K, 2020. The third model of Bax/Bak activation: a Bcl-2 family feud finally resolved? F1000Res 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas AI, Murray G, Henney H 3rd, Kassem N, Legrand V, Mangelus M, Muizelaar JP, Stocchetti N, Knoller N, 2006. Efficacy and safety of dexanabinol in severe traumatic brain injury: results of a phase III randomised, placebo-controlled, clinical trial. Lancet Neurol 5, 38–45. [DOI] [PubMed] [Google Scholar]
- MacIver NJ, Michalek RD, Rathmell JC, 2013. Metabolic regulation of T lymphocytes. Annual review of immunology 31, 259–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ, 2011. Neuron-glia metabolic coupling and plasticity. Exp Physiol 96, 407–410. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ, Allaman I, 2015. A cellular perspective on brain energy metabolism and functional imaging. Neuron 86, 883–901. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ, Pellerin L, 1999. Astrocytes Couple Synaptic Activity to Glucose Utilization in the Brain. News Physiol Sci 14, 177–182. [DOI] [PubMed] [Google Scholar]
- Maynard ME, Underwood EL, Redell JB, Zhao J, Kobori N, Hood KN, Moore AN, Dash PK, 2019. Carnosic Acid Improves Outcome after Repetitive Mild Traumatic Brain Injury. J Neurotrauma 36, 2147–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzeo AT, Brophy GM, Gilman CB, Alves OL, Robles JR, Hayes RL, Povlishock JT, Bullock MR, 2009. Safety and tolerability of cyclosporin a in severe traumatic brain injury patients: results from a prospective randomized trial. J Neurotrauma 26, 2195–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbye LH, Singh IN, Carrico KM, Saatman KE, Hall ED, 2009. Comparative neuroprotective effects of cyclosporin A and NIM811, a nonimmunosuppressive cyclosporin A analog, following traumatic brain injury. J Cereb Blood Flow Metab 29, 87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAvoy K, Kawamata H, 2019. Glial mitochondrial function and dysfunction in health and neurodegeneration. Molecular and Cellular Neuroscience 101, 103417. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Schwarz TL, 2017. Mitostasis in neurons: maintaining mitochondria in an extended cellular architecture. Neuron 96, 651–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Prudent J, Basu K, Goyon V, Katsumura S, Hulea L, Pearl D, Siddiqui N, Strack S, McGuirk S, St-Pierre J, Larsson O, Topisirovic I, Vali H, McBride HM, Bergeron JJ, Sonenberg N, 2017. mTOR Controls Mitochondrial Dynamics and Cell Survival via MTFP1. Mol Cell 67, 922–935.e925. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Munshi HG, Kambhampati S, Sassano A, Platanias LC, Stack MS, 2004. Calcium-induced matrix metalloproteinase 9 gene expression is differentially regulated by ERK1/2 and p38 MAPK in oral keratinocytes and oral squamous cell carcinoma. J Biol Chem 279, 33139–33146. [DOI] [PubMed] [Google Scholar]
- Murugan M, Santhakumar V, Kannurpatti SS, 2016. Facilitating Mitochondrial Calcium Uptake Improves Activation-Induced Cerebral Blood Flow and Behavior after mTBI. Front Syst Neurosci 10, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa AG, Al-Shboul O, Alfaqih MA, Al-Qudah MA, Al-Dwairi AN, 2018a. Phenelzine reduces the oxidative damage induced by peroxynitrite in plasma lipids and proteins. Arch Physiol Biochem 124, 418–423. [DOI] [PubMed] [Google Scholar]
- Mustafa AG, Alfaqih MA, Al-Shboul O, Al-Dwairi A, 2018b. Scavenging of lipid peroxyl radicals protects plasma lipids and proteins from peroxynitrite. Biomed Rep 9, 421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardin A, Schrepfer E, Ziviani E, 2016. Counteracting PINK/Parkin deficiency in the activation of mitophagy: a potential therapeutic intervention for Parkinson’s disease. Current neuropharmacology 14, 250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nighoghossian N, Berthezène Y, Mechtouff L, Derex L, Cho TH, Ritzenthaler T, Rheims S, Chauveau F, Béjot Y, Jacquin A, Giroud M, Ricolfi F, Philippeau F, Lamy C, Turc G, Bodiguel E, Domigo V, Guiraud V, Mas JL, Oppenheim C, Amarenco P, Cakmak S, Sevin-Allouet M, Guillon B, Desal H, Hosseini H, Sibon I, Mahagne MH, Ong E, Mewton N, Ovize M, 2015. Cyclosporine in acute ischemic stroke. Neurology 84, 2216–2223. [DOI] [PubMed] [Google Scholar]
- Niu F, Dong J, Xu X, Zhang B, Liu B, 2019. Mitochondrial Division Inhibitor 1 Prevents Early-Stage Induction of Mitophagy and Accelerated Cell Death in a Rat Model of Moderate Controlled Cortical Impact Brain Injury. World Neurosurg 122, e1090–e1101. [DOI] [PubMed] [Google Scholar]
- Norat P, Soldozy S, Sokolowski JD, Gorick CM, Kumar JS, Chae Y, Yağmurlu K, Prada F, Walker M, Levitt MR, Price RJ, Tvrdik P, Kalani MYS, 2020. Mitochondrial dysfunction in neurological disorders: Exploring mitochondrial transplantation. npj Regenerative Medicine 5, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A, 2012. Mitochondria: in sickness and in health. Cell 148, 1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LA, Pearce EJ, 2016. Immunometabolism governs dendritic cell and macrophage function. J Exp Med 213, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ola MS, Nawaz M, Ahsan H, 2011. Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol Cell Biochem 351, 41–58. [DOI] [PubMed] [Google Scholar]
- Omelchenko A, Shrirao AB, Bhattiprolu AK, Zahn JD, Schloss RS, Dickson S, Meaney DF, Boustany NN, Yarmush ML, Firestein BL, 2019. Dynamin and reverse-mode sodium calcium exchanger blockade confers neuroprotection from diffuse axonal injury. Cell Death Dis 10, 727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onfelt B, Nedvetzki S, Yanagi K, Davis DM, 2004. Cutting edge: Membrane nanotubes connect immune cells. J Immunol 173, 1511–1513. [DOI] [PubMed] [Google Scholar]
- Otera H, Ishihara N, Mihara K, 2013. New insights into the function and regulation of mitochondrial fission. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1833, 1256–1268. [DOI] [PubMed] [Google Scholar]
- Oz M, Lorke DE, Petroianu GA, 2009. Methylene blue and Alzheimer’s disease. Biochem Pharmacol 78, 927–932. [DOI] [PubMed] [Google Scholar]
- Ozbal S, Cankurt U, Tugyan K, Pekcetin C, Sisman AR, Gunduz K, Micili SC, 2015. The effects of α-lipoic acid on immature rats with traumatic brain injury. Biotech Histochem 90, 206–215. [DOI] [PubMed] [Google Scholar]
- Paliwal S, Chaudhuri R, Agrawal A, Mohanty S, 2018. Regenerative abilities of mesenchymal stem cells through mitochondrial transfer. J Biomed Sci 25, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya JD, Pauly JR, Sullivan PG, 2009. The optimal dosage and window of opportunity to maintain mitochondrial homeostasis following traumatic brain injury using the uncoupler FCCP. Exp Neurol 218, 381–389. [DOI] [PubMed] [Google Scholar]
- Parent M, Chitturi J, Santhakumar V, Hyder F, Sanganahalli BG, Kannurpatti SS, 2020. Kaempferol Treatment after Traumatic Brain Injury during Early Development Mitigates Brain Parenchymal Microstructure and Neural Functional Connectivity Deterioration at Adolescence. J Neurotrauma 37, 966–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, Cox DH, Gollihue JL, Bailey WM, Geldenhuys WJ, Gensel JC, Sullivan PG, Rabchevsky AG, 2017. Pioglitazone treatment following spinal cord injury maintains acute mitochondrial integrity and increases chronic tissue sparing and functional recovery. Exp Neurol 293, 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekkurnaz G, Trinidad JC, Wang X, Kong D, Schwarz TL, 2014. Glucose regulates mitochondrial motility via Milton modification by O-GlcNAc transferase. Cell 158, 54–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peruzzotti-Jametti L, Bernstock JD, Willis CM, Manferrari G, Rogall R, Fernandez-Vizarra E, Williamson JC, Braga A, van den Bosch A, Leonardi T, Krzak G, Kittel A, Beninca C, Vicario N, Tan S, Bastos C, Bicci I, Iraci N, Smith JA, Peacock B, Muller KH, Lehner PJ, Buzas EI, Faria N, Zeviani M, Frezza C, Brisson A, Matheson NJ, Viscomi C, Pluchino S, 2021. Neural stem cells traffic functional mitochondria via extracellular vesicles. PLoS Biol 19, e3001166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfleger J, He M, Abdellatif M, 2015. Mitochondrial complex II is a source of the reserve respiratory capacity that is regulated by metabolic sensors and promotes cell survival. Cell death & disease 6, e1835–e1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, McEwen BS, 2014. Mitochondria impact brain function and cognition. Proceedings of the National Academy of Sciences 111, 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, Taivassalo T, Gouspillou G, Hepple RT, 2011. Mitochondria: isolation, structure and function. The Journal of physiology 589, 4413–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins M, 2008. Diet, ketones, and neurotrauma. Epilepsia 49 Suppl 8, 111–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins ML, Lee SM, Fujima LS, Hovda DA, 2004. Increased cerebral uptake and oxidation of exogenous betaHB improves ATP following traumatic brain injury in adult rats. J Neurochem 90, 666–672. [DOI] [PubMed] [Google Scholar]
- Qi D, Young LH, 2015. AMPK: energy sensor and survival mechanism in the ischemic heart. Trends in Endocrinology & Metabolism 26, 422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabchevsky AG, Patel SP, Sullivan PG, 2017. Targeting mitoNEET with pioglitazone for therapeutic neuroprotection after spinal cord injury. Neural Regen Res 12, 1807–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghupathi R, Graham DI, McIntosh TK, 2000. Apoptosis after traumatic brain injury. Journal of neurotrauma 17, 927–938. [DOI] [PubMed] [Google Scholar]
- Raichle ME, Gusnard DA, 2002. Appraising the brain’s energy budget. Proceedings of the National Academy of Sciences 99, 10237–10239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Pearce EL, 2018. Mitochondrial Dynamics at the Interface of Immune Cell Metabolism and Function. Trends Immunol 39, 6–18. [DOI] [PubMed] [Google Scholar]
- Rangaraju V, Lauterbach M, Schuman EM, 2019. Spatially stable mitochondrial compartments fuel local translation during plasticity. Cell 176, 73–84. e15. [DOI] [PubMed] [Google Scholar]
- Readnower RD, Pandya JD, McEwen ML, Pauly JR, Springer JE, Sullivan PG, 2011. Post-injury administration of the mitochondrial permeability transition pore inhibitor, NIM811, is neuroprotective and improves cognition after traumatic brain injury in rats. J Neurotrauma 28, 1845–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy V, Grogan D, Ahluwalia M, Salles É L, Ahluwalia P, Khodadadi H, Alverson K, Nguyen A, Raju SP, Gaur P, Braun M, Vale FL, Costigliola V, Dhandapani K, Baban B, Vaibhav K, 2020a. Targeting the endocannabinoid system: a predictive, preventive, and personalized medicine-directed approach to the management of brain pathologies. Epma j 11, 217–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy V, Grogan D, Ahluwalia M, Salles ÉL, Ahluwalia P, Khodadadi H, Alverson K, Nguyen A, Raju SP, Gaur P, Braun M, Vale FL, Costigliola V, Dhandapani K, Baban B, Vaibhav K, 2020b. Targeting the endocannabinoid system: a predictive, preventive, and personalized medicine-directed approach to the management of brain pathologies. EPMA Journal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard SM, Razak K, Ethell IM, 2015. A delicate balance: role of MMP-9 in brain development and pathophysiology of neurodevelopmental disorders. Frontiers in Cellular Neuroscience 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H, Ma L, Gong X, Xu C, Zhang Y, Ma M, Watanabe K, Wen J, 2019a. Edaravone Exerts Brain Protective Function by Reducing the Expression of AQP4, APP and Aβ Proteins. Open Life Sci 14, 651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Lu Y, Qian Y, Chen B, Wu T, Ji G, 2019b. Recent progress regarding kaempferol for the treatment of various diseases. Exp Ther Med 18, 2759–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riha PD, Bruchey AK, Echevarria DJ, Gonzalez-Lima F, 2005. Memory facilitation by methylene blue: dose-dependent effect on behavior and brain oxygen consumption. Eur J Pharmacol 511, 151–158. [DOI] [PubMed] [Google Scholar]
- Ristow M, 2014. Unraveling the truth about antioxidants: mitohormesis explains ROS-induced health benefits. Nat Med 20, 709–711. [DOI] [PubMed] [Google Scholar]
- Ristow M, Schmeisser K, 2014. Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose Response 12, 288–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizk M, Vu J, Zhang Z, 2021. Impact of pediatric traumatic brain injury on hippocampal neurogenesis. Neural Regen Res 16, 926–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robledo-Menendez A, Vella M, Grandes P, Soria-Gomez E, 2021. Cannabinoid control of hippocampal functions: the where matters. The FEBS Journal n/a. [DOI] [PubMed] [Google Scholar]
- Rocamonde B, Paradells S, Barcia C, Garcia Esparza A, Soria JM, 2013. Lipoic acid treatment after brain injury: study of the glial reaction. Clin Dev Immunol 2013, 521939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Nuevo A, Zorzano A, 2019. The sensing of mitochondrial DAMPs by non-immune cells. Cell Stress 3, 195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Vargas JM, Ruiz-Magaña MJ, Ruiz-Ruiz C, Majuelos-Melguizo J, Peralta-Leal A, Rodríguez MI, Muñoz-Gámez JA, De Almodóvar MR, Siles E, Rivas AL, 2012. ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell research 22, 1181–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas JC, Bruchey AK, Gonzalez-Lima F, 2012. Neurometabolic mechanisms for memory enhancement and neuroprotection of methylene blue. Prog Neurobiol 96, 32–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose J, Brian C, Woods J, Pappa A, Panayiotidis MI, Powers R, Franco R, 2017. Mitochondrial dysfunction in glial cells: Implications for neuronal homeostasis and survival. Toxicology 391, 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roszek K, Czarnecka J, 2015. Is Ecto-nucleoside Triphosphate Diphosphohydrolase (NTPDase)-based Therapy of Central Nervous System Disorders Possible? Mini Rev Med Chem 15, 5–20. [DOI] [PubMed] [Google Scholar]
- Rustom A, Saffrich R, Markovic I, Walther P, Gerdes HH, 2004. Nanotubular highways for intercellular organelle transport. Science 303, 1007–1010. [DOI] [PubMed] [Google Scholar]
- Sabirzhanov B, Stoica BA, Zhao Z, Loane DJ, Wu J, Dorsey SG, Faden AI, 2016. miR-711 upregulation induces neuronal cell death after traumatic brain injury. Cell Death Differ 23, 654–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salihu AT, Muthuraju S, Idris Z, Izaini Ghani AR, Abdullah JM, 2016. Functional outcome after intracerebral haemorrhage - a review of the potential role of antiapoptotic agents. Rev Neurosci 27, 317–327. [DOI] [PubMed] [Google Scholar]
- Salman M, Kaushik P, Tabassum H, Parvez S, 2021. Melatonin Provides Neuroprotection Following Traumatic Brain Injury-Promoted Mitochondrial Perturbation in Wistar Rat. Cell Mol Neurobiol 41, 765–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos RX, Correia SC, Wang X, Perry G, Smith MA, Moreira PI, Zhu X, 2010. A synergistic dysfunction of mitochondrial fission/fusion dynamics and mitophagy in Alzheimer’s disease. J Alzheimers Dis 20 Suppl 2, S401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santuy A, Turégano-López M, Rodríguez JR, Alonso-Nanclares L, DeFelipe J, Merchán-Pérez A, 2018. A Quantitative Study on the Distribution of Mitochondria in the Neuropil of the Juvenile Rat Somatosensory Cortex. Cerebral Cortex 28, 3673–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Kosaka K, Itoh K, Kobayashi A, Yamamoto M, Shimojo Y, Kitajima C, Cui J, Kamins J, Okamoto S, Izumi M, Shirasawa T, Lipton SA, 2008. Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cysteines on Keap1. J Neurochem 104, 1116–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayre LM, Perry G, Smith MA, 2008. Oxidative stress and neurotoxicity. Chem Res Toxicol 21, 172–188. [DOI] [PubMed] [Google Scholar]
- Schousboe A, Waagepetersen HS, Sonnewald U, 2019. Astrocytic pyruvate carboxylation: status after 35 years. Journal of neuroscience research 97, 890–896. [DOI] [PubMed] [Google Scholar]
- Scott G, Leopardi S, Printup S, Madden BC, 2002. Filopodia are conduits for melanosome transfer to keratinocytes. J Cell Sci 115, 1441–1451. [DOI] [PubMed] [Google Scholar]
- Seager R, Lee L, Henley Jeremy M., Wilkinson Kevin A., 2020. Mechanisms and roles of mitochondrial localisation and dynamics in neuronal function. Neuronal Signaling 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears ME, 2013. Chelation: harnessing and enhancing heavy metal detoxification--a review. ScientificWorldJournal 2013, 219840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Tu L, Chen D, Tan T, Wang Y, Wang S, 2019a. TRPV4 channels stimulate Ca2+-induced Ca2+ release in mouse neurons and trigger endoplasmic reticulum stress after intracerebral hemorrhage. Brain Research Bulletin 146, 143–152. [DOI] [PubMed] [Google Scholar]
- Shen J, Xin W, Li Q, Gao Y, Yuan L, Zhang J, 2019b. Methylene Blue Reduces Neuronal Apoptosis and Improves Blood-Brain Barrier Integrity After Traumatic Brain Injury. Front Neurol 10, 1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Kim E, 2011. The postsynaptic organization of synapses. Cold Spring Harb Perspect Biol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi RY, Zhu SH, Li V, Gibson SB, Xu XS, Kong JM, 2014. BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci Ther 20, 1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin MK, Vázquez-Rosa E, Koh Y, Dhar M, Chaubey K, Cintrón-Pérez CJ, Barker S, Miller E, Franke K, Noterman MF, Seth D, Allen RS, Motz CT, Rao SR, Skelton LA, Pardue MT, Fliesler SJ, Wang C, Tracy TE, Gan L, Liebl DJ, Savarraj JPJ, Torres GL, Ahnstedt H, McCullough LD, Kitagawa RS, Choi HA, Zhang P, Hou Y, Chiang CW, Li L, Ortiz F, Kilgore JA, Williams NS, Whitehair VC, Gefen T, Flanagan ME, Stamler JS, Jain MK, Kraus A, Cheng F, Reynolds JD, Pieper AA, 2021. Reducing acetylated tau is neuroprotective in brain injury. Cell 184, 2715–2732.e2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava P, Vaibhav K, Tabassum R, Khan A, Ishrat T, Khan MM, Ahmad A, Islam F, Safhi MM, Islam F, 2013. Anti-apoptotic and anti-inflammatory effect of Piperine on 6-OHDA induced Parkinson’s rat model. J Nutr Biochem 24, 680–687. [DOI] [PubMed] [Google Scholar]
- Siegel GJ, Albers RW, 1994. Basic neurochemistry: molecular, cellular, and medical aspects. Raven Press. [Google Scholar]
- Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED, 2006. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. Journal of Cerebral Blood Flow & Metabolism 26, 1407–1418. [DOI] [PubMed] [Google Scholar]
- Singhal NS, Sun CH, Lee EM, Ma DK, 2020. Resilience to Injury: A New Approach to Neuroprotection? Neurotherapeutics 17, 457–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spees JL, Olson SD, Whitney MJ, Prockop DJ, 2006. Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci U S A 103, 1283–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer JE, Prajapati P, Sullivan PG, 2018a. Targeting the mitochondrial permeability transition pore in traumatic central nervous system injury. Neural Regen Res 13, 1338–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer JE, Visavadiya NP, Sullivan PG, Hall ED, 2018b. Post-Injury Treatment with NIM811 Promotes Recovery of Function in Adult Female Rats after Spinal Cord Contusion: A Dose-Response Study. J Neurotrauma 35, 492–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, 2016. Emerging therapeutic roles for NAD(+) metabolism in mitochondrial and age-related disorders. Clinical and translational medicine 5, 25–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stawicki SP, Sims C, Sarani B, Grossman MD, Gracias VH, 2008. Methylene blue and vasoplegia: who, when, and how? Mini Rev Med Chem 8, 472–490. [DOI] [PubMed] [Google Scholar]
- Steiner P, 2019. Brain Fuel Utilization in the Developing Brain. Annals of Nutrition and Metabolism 75(suppl 1), 8–18. [DOI] [PubMed] [Google Scholar]
- Stetson PD, McKnight LK, Bakken S, Curran C, Kubose TT, Cimino JJ, 2001. Development of an ontology to model medical errors, information needs, and the clinical communication space. Proc AMIA Symp, 672–676. [PMC free article] [PubMed] [Google Scholar]
- Sugden D, Davidson K, Hough KA, Teh MT, 2004. Melatonin, melatonin receptors and melanophores: a moving story. Pigment Cell Res 17, 454–460. [DOI] [PubMed] [Google Scholar]
- Sukumari-Ramesh S, Laird MD, Singh N, Vender JR, Alleyne CH Jr., Dhandapani KM, 2010. Astrocyte-derived glutathione attenuates hemin-induced apoptosis in cerebral microvascular cells. Glia 58, 1858–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suliman HB, Piantadosi CA, 2016. Mitochondrial Quality Control as a Therapeutic Target. Pharmacol Rev 68, 20–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Rabchevsky AG, Waldmeier PC, Springer JE, 2005. Mitochondrial permeability transition in CNS trauma: cause or effect of neuronal cell death? J Neurosci Res 79, 231–239. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Thompson M, Scheff SW, 2000. Continuous infusion of cyclosporin A postinjury significantly ameliorates cortical damage following traumatic brain injury. Exp Neurol 161, 631–637. [DOI] [PubMed] [Google Scholar]
- Sun LQ, Gao JL, Cui Y, Zhao MM, Jing XB, Li R, Tian YX, Cui JZ, Wu ZX, 2015. Neuronic autophagy contributes to p-connexin 43 degradation in hippocampal astrocytes following traumatic brain injury in rats. Mol Med Rep 11, 4419–4423. [DOI] [PubMed] [Google Scholar]
- Sun N, Youle RJ, Finkel T, 2016. The Mitochondrial Basis of Aging. Mol Cell 61, 654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesny B, Tann AW, Mitra S, 2010. Age- and tissue-specific changes in mitochondrial and nuclear DNA base excision repair activity in mice: Susceptibility of skeletal muscles to oxidative injury. Mech Ageing Dev 131, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda H, Yamaguchi T, Yano H, Tanaka J, 2021. Microglial metabolic disturbances and neuroinflammation in cerebral infarction. J Pharmacol Sci 145, 130–139. [DOI] [PubMed] [Google Scholar]
- Talley Watts L, Long JA, Boggs RC, Manga H, Huang S, Shen Q, Duong TQ, 2016. Delayed Methylene Blue Improves Lesion Volume, Multi-Parametric Quantitative Magnetic Resonance Imaging Measurements, and Behavioral Outcome after Traumatic Brain Injury. J Neurotrauma 33, 194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talley Watts L, Long JA, Chemello J, Van Koughnet S, Fernandez A, Huang S, Shen Q, Duong TQ, 2014. Methylene blue is neuroprotective against mild traumatic brain injury. J Neurotrauma 31, 1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng L, Fan L, Peng Y, He X, Chen H, Duan H, Yang F, Lin D, Lin Z, Li H, Shao B, 2019. Carnosic Acid Mitigates Early Brain Injury After Subarachnoid Hemorrhage: Possible Involvement of the SIRT1/p66shc Signaling Pathway. Front Neurosci 13, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari A, Mahendru V, Sinha A, Bilotta F, 2014. Antioxidants: The new frontier for translational research in cerebroprotection. J Anaesthesiol Clin Pharmacol 30, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapa K, Khan H, Singh TG, Kaur A, 2021. Traumatic Brain Injury: Mechanistic Insight on Pathophysiology and Potential Therapeutic Targets. Journal of Molecular Neuroscience. [DOI] [PubMed] [Google Scholar]
- Toklu HZ, Hakan T, Biber N, Solakoğlu S, Oğünç AV, Sener G, 2009. The protective effect of alpha lipoic acid against traumatic brain injury in rats. Free Radic Res 43, 658–667. [DOI] [PubMed] [Google Scholar]
- Torralba D, Baixauli F, Sánchez-Madrid F, 2016. Mitochondria Know No Boundaries: Mechanisms and Functions of Intercellular Mitochondrial Transfer. Front Cell Dev Biol 4, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama EQ, Herzig S, Courchet J, Lewis TL, Losón OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, 2016. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351, 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter L, Ambrus A, 2014. Chapter Twelve - Measurement of ROS Homeostasis in Isolated Mitochondria, In: Murphy AN, Chan DC (Eds.), Methods in Enzymology. Vol. 547,Academic Press, pp. 199–223. [DOI] [PubMed] [Google Scholar]
- Tucker D, Lu Y, Zhang Q, 2018. From Mitochondrial Function to Neuroprotection-an Emerging Role for Methylene Blue. Mol Neurobiol 55, 5137–5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS, 2008. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27, 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullah H, Di Minno A, Santarcangelo C, Khan H, Daglia M, 2021. Improvement of Oxidative Stress and Mitochondrial Dysfunction by β-Caryophyllene: A Focus on the Nervous System. Antioxidants (Basel) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uluc K, Kendigelen P, Fidan E, Zhang L, Chanana V, Kintner D, Akture E, Song C, Ye K, Sun D, Ferrazzano P, Cengiz P, 2013. TrkB receptor agonist 7, 8 dihydroxyflavone triggers profound gender- dependent neuroprotection in mice after perinatal hypoxia and ischemia. CNS Neurol Disord Drug Targets 12, 360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaibhav K, Braun M, Khan MB, Fatima S, Saad N, Shankar A, Khan ZT, Harris RBS, Yang Q, Huo Y, Arbab AS, Giri S, Alleyne CH Jr., Vender JR, Hess DC, Baban B, Hoda MN, Dhandapani KM, 2018. Remote ischemic post-conditioning promotes hematoma resolution via AMPK-dependent immune regulation. J Exp Med 215, 2636–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaibhav K, Shrivastava P, Khan A, Javed H, Tabassum R, Ahmed ME, Khan MB, Moshahid Khan M, Islam F, Ahmad S, Siddiqui MS, Safhi MM, Islam F, 2013a. Azadirachta indica mitigates behavioral impairments, oxidative damage, histological alterations and apoptosis in focal cerebral ischemia-reperfusion model of rats. Neurol Sci 34, 1321–1330. [DOI] [PubMed] [Google Scholar]
- Vaibhav K, Shrivastava P, Tabassum R, Khan A, Javed H, Ahmed ME, Islam F, Safhi MM, Islam F, 2013b. Delayed administration of zingerone mitigates the behavioral and histological alteration via repression of oxidative stress and intrinsic programmed cell death in focal transient ischemic rats. Pharmacol Biochem Behav 113, 53–62. [DOI] [PubMed] [Google Scholar]
- Van der Bliek AM, Shen Q, Kawajiri S, 2013. Mechanisms of mitochondrial fission and fusion. Cold Spring Harbor perspectives in biology 5, a011072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasile F, Dossi E, Rouach N, 2017. Human astrocytes: structure and functions in the healthy brain. Brain Structure and Function 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veech RL, 2004. The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot Essent Fatty Acids 70, 309–319. [DOI] [PubMed] [Google Scholar]
- Vives-Bauza C, Przedborski S, 2011. Mitophagy: the latest problem for Parkinson’s disease. Trends Mol Med 17, 158–165. [DOI] [PubMed] [Google Scholar]
- Voigt A, Jelinek HF, 2016. Humanin: a mitochondrial signaling peptide as a biomarker for impaired fasting glucose-related oxidative stress. Physiol Rep 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J, 2009. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci 29, 3974–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmeier PC, Zimmermann K, Qian T, Tintelnot-Blomley M, Lemasters JJ, 2003. Cyclophilin D as a drug target. Curr Med Chem 10, 1485–1506. [DOI] [PubMed] [Google Scholar]
- Walko TD 3rd, Bola RA, Hong JD, Au AK, Bell MJ, Kochanek PM, Clark RS, Aneja RK, 2014. Cerebrospinal fluid mitochondrial DNA: a novel DAMP in pediatric traumatic brain injury. Shock 41, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HC, Lin YJ, Tsai NW, Su BY, Kung CT, Chen WF, Kwan AL, Lu CH, 2014. Serial plasma deoxyribonucleic acid levels as predictors of outcome in acute traumatic brain injury. J Neurotrauma 31, 1039–1045. [DOI] [PubMed] [Google Scholar]
- Wang S, Xing Z, Vosler PS, Yin H, Li W, Zhang F, Signore AP, Stetler RA, Gao Y, Chen J, 2008. Cellular NAD replenishment confers marked neuroprotection against ischemic cell death: role of enhanced DNA repair. Stroke 39, 2587–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Zhao F, Ma X, Perry G, Zhu X, 2020. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol Neurodegener 15, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, de Rivero Vaccari JP, Wang H, Diaz P, German R, Marcillo AE, Keane RW, 2012. Activation of the nuclear factor E2-related factor 2/antioxidant response element pathway is neuroprotective after spinal cord injury. J Neurotrauma 29, 936–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhou F, Dou Y, Tian X, Liu C, Li H, Shen H, Chen G, 2018. Melatonin Alleviates Intracerebral Hemorrhage-Induced Secondary Brain Injury in Rats via Suppressing Apoptosis, Inflammation, Oxidative Stress, DNA Damage, and Mitochondria Injury. Transl Stroke Res 9, 74–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts LT, Lloyd R, Garling RJ, Duong T, 2013. Stroke neuroprotection: targeting mitochondria. Brain Sci 3, 540–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JT, 2012. Altered calcium signaling following traumatic brain injury. Front Pharmacol 3, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C-C, Kong Y-Y, Li G-Q, Guan Y-F, Wang P, Miao C-Y, 2017. Nicotinamide mononucleotide attenuates brain injury after intracerebral hemorrhage by activating Nrf2/HO-1 signaling pathway. Scientific Reports 7, 717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Wang H, Wu Y, Ding K, Li T, Cong Z, Xu J, Zhou M, Huang L, Ding H, Wu H, 2015. Alpha lipoic acid inhibits neural apoptosis via a mitochondrial pathway in rats following traumatic brain injury. Neurochem Int 87, 85–91. [DOI] [PubMed] [Google Scholar]
- Wen Y, Li W, Poteet EC, Xie L, Tan C, Yan LJ, Ju X, Liu R, Qian H, Marvin MA, Goldberg MS, She H, Mao Z, Simpkins JW, Yang SH, 2011. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J Biol Chem 286, 16504–16515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Shadel GS, Ghosh S, 2011. Mitochondria in innate immune responses. Nat Rev Immunol 11, 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B, 2010. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 11, 872–884. [DOI] [PubMed] [Google Scholar]
- Westermann B, 2012. Bioenergetic role of mitochondrial fusion and fission. Biochimica et Biophysica Acta (BBA)-Bioenergetics 1817, 1833–1838. [DOI] [PubMed] [Google Scholar]
- Wilks DC, Besson H, Lindroos AK, Ekelund U, 2011. Objectively measured physical activity and obesity prevention in children, adolescents and adults: a systematic review of prospective studies. Obes Rev 12, e119–129. [DOI] [PubMed] [Google Scholar]
- Wu CH, Hung TH, Chen CC, Ke CH, Lee CY, Wang PY, Chen SF, 2014. Post-injury treatment with 7,8-dihydroxyflavone, a TrkB receptor agonist, protects against experimental traumatic brain injury via PI3K/Akt signaling. PloS one 9, e113397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Gao C, Wang H, Zhang X, Li Q, Gu Z, Shi X, Cui Y, Wang T, Chen X, Wang X, Luo C, Tao L, 2018. Mdivi-1 alleviates blood-brain barrier disruption and cell death in experimental traumatic brain injury by mitigating autophagy dysfunction and mitophagy activation. Int J Biochem Cell Biol 94, 44–55. [DOI] [PubMed] [Google Scholar]
- Wu X, Cui W, Guo W, Liu H, Luo J, Zhao L, Guo H, Zheng L, Bai H, Feng D, Qu Y, 2020a. Acrolein Aggravates Secondary Brain Injury After Intracerebral Hemorrhage Through Drp1-Mediated Mitochondrial Oxidative Damage in Mice. Neurosci Bull 36, 1158–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Luo J, Liu H, Cui W, Guo K, Zhao L, Bai H, Guo W, Guo H, Feng D, Qu Y, 2020b. Recombinant Adiponectin Peptide Ameliorates Brain Injury Following Intracerebral Hemorrhage by Suppressing Astrocyte-Derived Inflammation via the Inhibition of Drp1-Mediated Mitochondrial Fission. Transl Stroke Res 11, 924–939. [DOI] [PubMed] [Google Scholar]
- Xia D, Zhai X, Wang H, Chen Z, Fu C, Zhu M, 2019. Alpha lipoic acid inhibits oxidative stress-induced apoptosis by modulating of Nrf2 signalling pathway after traumatic brain injury. J Cell Mol Med 23, 4088–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao A, Zhang Y, Ren Y, Chen R, Li T, You C, Gan X, 2021. GDF11 alleviates secondary brain injury after intracerebral hemorrhage via attenuating mitochondrial dynamic abnormality and dysfunction. Sci Rep 11, 3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Gu Q, Peterson P, Muizelaar JP, Lee C, 1997. Mitochondrial dysfunction and calcium perturbation induced by traumatic brain injury. Journal of neurotrauma 14, 23–34. [DOI] [PubMed] [Google Scholar]
- Xu F, Na L, Li Y, Chen L, 2020. Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci 10, 54. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Xu XJ, Yang MS, Zhang B, Niu F, Dong JQ, Liu BY, 2021. Glucose metabolism: A link between traumatic brain injury and Alzheimer’s disease. Chin J Traumatol 24, 5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Lv XA, Dai Q, Ge YQ, Xu J, 2016. Acute upregulation of neuronal mitochondrial type-1 cannabinoid receptor and it’s role in metabolic defects and neuronal apoptosis after TBI. Mol Brain 9, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Cui X, Li J, Zhang C, Zhang J, Liu M, 2015. Edaravone for acute stroke: Meta-analyses of data from randomized controlled trials. Dev Neurorehabil 18, 330–335. [DOI] [PubMed] [Google Scholar]
- Yegutkin GG, 2008. Nucleotide- and nucleoside-converting ectoenzymes: Important modulators of purinergic signalling cascade. Biochim Biophys Acta 1783, 673–694. [DOI] [PubMed] [Google Scholar]
- Yin F, Jiang T, Cadenas E, 2013. Metabolic triad in brain aging: mitochondria, insulin/IGF-1 signalling and JNK signalling. Biochem Soc Trans 41, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W, 2007. NAD+ and NADH in brain functions, brain diseases and brain aging. Front Biosci 12, 1863–1888. [DOI] [PubMed] [Google Scholar]
- Ying W, Garnier P, Swanson RA, 2003. NAD+ repletion prevents PARP-1-induced glycolytic blockade and cell death in cultured mouse astrocytes. Biochemical and biophysical research communications 308, 809–813. [DOI] [PubMed] [Google Scholar]
- Ying W, Wei G, Wang D, Shi J, Lu H, 2007a. Intranasal administration of NAD+ and the PARG inhibitor gallotannin can decrease both ischemic brain injury and traumatic brain injury, 37th American Society for Neuroscience Annual Meeting Abstracts. [Google Scholar]
- Ying W, Wei G, Wang D, Wang Q, Tang X, Shi J, Zhang P, Lu H, 2007b. Intranasal administration with NAD+ profoundly decreases brain injury in a rat model of transient focal ischemia. Front Biosci 12, 2728–2734. [DOI] [PubMed] [Google Scholar]
- Ying W, Xiong ZG, 2010. Oxidative stress and NAD+ in ischemic brain injury: current advances and future perspectives. Current medicinal chemistry 17, 2152–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonutas HM, Hubbard WB, Pandya JD, Vekaria HJ, Geldenhuys WJ, Sullivan PG, 2020. Bioenergetic restoration and neuroprotection after therapeutic targeting of mitoNEET: New mechanism of pioglitazone following traumatic brain injury. Exp Neurol 327, 113243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Van Der Bliek AM, 2012. Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younts TJ, Castillo PE, 2014. Endogenous cannabinoid signaling at inhibitory interneurons. Current opinion in neurobiology 26, 42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu E, Mercer J, Bennett M, 2012. Mitochondria in vascular disease. Cardiovasc Res 95, 173–182. [DOI] [PubMed] [Google Scholar]
- Yu J, Zheng J, Lu J, Sun Z, Wang Z, Zhang J, 2019. AdipoRon Protects Against Secondary Brain Injury After Intracerebral Hemorrhage via Alleviating Mitochondrial Dysfunction: Possible Involvement of AdipoR1–AMPK–PGC1α Pathway. Neurochemical research 44, 1678–1689. [DOI] [PubMed] [Google Scholar]
- Yu Y, Lee H-C, Chen K-C, Suhan J, Qiu M, Ba Q, Yang G, 2016. Inner membrane fusion mediates spatial distribution of axonal mitochondria. Scientific Reports 6, 18981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Z, Zhang Y, Jiang W, He L, Qu H, 2020. Modulation of autophagy in traumatic brain injury. J Cell Physiol 235, 1973–1985. [DOI] [PubMed] [Google Scholar]
- Zhang T, Wu P, Budbazar E, Zhu Q, Sun C, Mo J, Peng J, Gospodarev V, Tang J, Shi H, Zhang JH, 2019. Mitophagy Reduces Oxidative Stress Via Keap1 (Kelch-Like Epichlorohydrin-Associated Protein 1)/Nrf2 (Nuclear Factor-E2-Related Factor 2)/PHB2 (Prohibitin 2) Pathway After Subarachnoid Hemorrhage in Rats. Stroke 50, 978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YB, Li SX, Chen XP, Yang L, Zhang YG, Liu R, Tao LY, 2008. Autophagy is activated and might protect neurons from degeneration after traumatic brain injury. Neurosci Bull 24, 143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Chen H, Xia W, Li S, Ying W, 2010. NAD+ induces C6 glioma cell death by generating oxidative stress and increasing intracellular calcium concentrations. The FASEB Journal 24, lb470–lb470. [Google Scholar]
- Zhao J, Qu D, Xi Z, Huan Y, Zhang K, Yu C, Yang D, Kang J, Lin W, Wu S, Wang Y, 2021. Mitochondria transplantation protects traumatic brain injury via promoting neuronal survival and astrocytic BDNF. Transl Res. [DOI] [PubMed] [Google Scholar]
- Zhao S, Gao X, Dong W, Chen J, 2016. The Role of 7,8-Dihydroxyflavone in Preventing Dendrite Degeneration in Cortex After Moderate Traumatic Brain Injury. Mol Neurobiol 53, 1884–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Shi L, Liang F, Xu W, Li T, Gao L, Sun Z, Yu J, Zhang J, 2018. Sirt3 ameliorates oxidative stress and mitochondrial dysfunction after intracerebral hemorrhage in diabetic rats. Frontiers in neuroscience 12, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Gong Y, Wang X, Xie Q, Tang H, Wang D, Chen X, 2013. Alteration of intracellular calcium and its modulator SLC24A6 after experimental intracerebral hemorrhage. Acta Neurochir Suppl 118, 169–173. [DOI] [PubMed] [Google Scholar]
- Zheng S, Jian D, Gan H, Wang L, Zhao J, Zhai X, 2021. FUNDC1 inhibits NLRP3-mediated inflammation after intracerebral hemorrhage by promoting mitophagy in mice. Neuroscience Letters 756, 135967. [DOI] [PubMed] [Google Scholar]
- Zhou HR, Ma XF, Lin WJ, Hao M, Yu XY, Li HX, Xu CY, Kuang HY, 2020. Neuroprotective Role of GLP-1 Analog for Retinal Ganglion Cells via PINK1/Parkin-Mediated Mitophagy in Diabetic Retinopathy. Front Pharmacol 11, 589114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou ZD, Tan EK, 2020. Oxidized nicotinamide adenine dinucleotide-dependent mitochondrial deacetylase sirtuin-3 as a potential therapeutic target of Parkinson’s disease. Ageing Res Rev 62, 101107. [DOI] [PubMed] [Google Scholar]
- Zille M, Karuppagounder SS, Chen Y, Gough PJ, Bertin J, Finger J, Milner TA, Jonas EA, Ratan RR, 2017. Neuronal Death After Hemorrhagic Stroke In Vitro and In Vivo Shares Features of Ferroptosis and Necroptosis. Stroke 48, 1033–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo W, Zhang S, Xia CY, Guo XF, He WB, Chen NH, 2014. Mitochondria autophagy is induced after hypoxic/ischemic stress in a Drp1 dependent manner: the role of inhibition of Drp1 in ischemic brain damage. Neuropharmacology 86, 103–115. [DOI] [PubMed] [Google Scholar]