

Abstract
Drug transporters are integral membrane proteins that play a critical role in drug disposition by affecting absorption, distribution, and excretion. They translocate drugs, as well as endogenous molecules and toxins, across membranes using ATP hydrolysis, or ion/concentration gradients. In general, drug transporters are expressed ubiquitously, but they function in drug disposition by being concentrated in tissues such as the intestine, the kidneys, the liver, and the brain. Based on their primary sequence and their mechanism, transporters can be divided into the ATP-Binding Cassette (ABC), Solute-Linked Carrier (SLC), and the SoLute Carrier Organic anion (SLCO) superfamilies. Many X-ray crystallography and cryo-electron microscopy (cryo-EM) structures have been solved in the ABC and SLC transporter superfamilies or of their bacterial homologs. The structures have provided valuable insight into the structural basis of transport. This chapter will provide particular focus on the promiscuous drug transporters because of their effect on drug disposition and the challenges associated with them.
Keywords: Cryo-electron microscopy, X-ray crystallography, Drug Transporters, ATP-Binding Cassette, Solute-Linked Carrier
1. Introduction
Drug transporters are protein pumps that are involved in drug disposition and drug distribution (1–3). They are integral membrane proteins that can translocate drugs and other molecules across membranes through active transport using ATP hydrolysis or through ion or concentration gradients (4). Drug transporters can be divided into three superfamilies: ATP-Binding Cassette (ABC), Solute-Linked Carrier (SLC), and the solute carrier organic anion (SLCO) superfamilies (2, 5, 6). Over the last couple of decades, the number of high-resolution drug transporter structures solved by X-ray crystallography and cryo-electron microscopy (cryo-EM) has exploded (e.g., 7–9). The high-resolution transporter structures have provided insight into their transport mechanisms (10–12). High-resolution structures have been solved in the ABC and SLC transporter superfamilies or their bacterial homologs (8, 10, 13). As far as we are aware, no high-resolution structures or their protein domains exist for members of the SLCO superfamily.
1.1. Transporter Types
Fig. 1 shows the types of protein-mediated transport processes versus passive diffusion. In passive diffusion (Fig. 1A), a molecule moves across the membrane based on the concentration gradient without the assistance of a protein transporter (4). Drug transporters can undergo two types of transport: primary and secondary active transport (4). Fig. 1B shows that primary active transport requires the hydrolysis of ATP at specific protein domains, which are known as nucleotide-binding domains (NBDs) or ATPase domains, to provide energy for transport. Secondary active transport shown in Figs. 1C–1E uses a concentration or electrochemical gradient. The electrochemical gradient can be established by an ion gradient or through an H+ gradient from an ATPase such as the F-Type ATPases (14).
Figure 1. Transport-mediated translocation of molecules across membranes.

A) Passive diffusion. B) Primary active transport involving ATP hydrolysis, which is denoted on the figure. Secondary active transport through C) antiporters, D) symporters (cotransporters), and E) uniporters.
Secondary active transport can be divided into three categories depending on the direction that species (i.e., molecules or ions) are transported across the membrane. For antiporters (Fig. 1C), two or more molecules or ions move across the membrane in opposite directions. Symporters (a.k.a. cotransporters, Fig. 1D) can transport two or more molecules, or ions move across the membrane in the same direction. The third type of secondary active transport is a uniporter (Fig. 1E), which is where a single species of the molecule or ion transverse the membrane in a single direction. In cases of secondary active transport, uniporters function by facilitated diffusion. Both passive and facilitated diffusion is driven by a concentration gradient. Facilitated diffusion is different than passive diffusion in that it involves a protein and is saturable.
1.2. Drug Transporter Superfamilies
Drug transporters belong to three major superfamilies (2, 5, 6). The ABC transporters are primary active uniporters (15–17). The ABC transporter superfamily has 7 members that are divided into ABCA-ABCG subfamilies (17). Except for the ABCE and ABCF subfamilies, the remaining transporter members of the superfamily use the hydrolysis of ATP to move molecules unidirectionally across the membrane (15–18). The SLC superfamily has 65 families, excluding the SLC21 family that is now part of the SLCO superfamily, that can transport a diverse range of substrates and ions (19–21). The members of SLC superfamily are examples of secondary active transporters that are driven by electrochemical or concentration gradients (13, 19, 22). They can also indirectly use energy from ion gradients produced from ATP-dependent pumps (22). The third superfamily of drug transporters that was formerly part of the SLC21 subfamily, and is now known as the SLCO or the organic anion transporting polypeptide (OATP) superfamily (20). The SLCO superfamily is divided into 6 families with a range of functions (20, 23, 24). They are responsible for transporting a range of large molecules over 300 Da, such as bile salts, prostaglandins, and xenobiotics, but their transport mechanisms are not well understood (20, 23, 24). No significant X-ray or cryo-EM structures have been solved for this superfamily (25). This book chapter will focus on the X-ray crystal and cryo-EM structures of promiscuous drug transporters because they have a strong effect on drug disposition, they are prone to drug-drug interactions, and their substrate and inhibitor preference is difficult to define. Tables are also provided within this chapter with additional structural information about these and other drug transporters.
1.3. Drug Transport Mechanisms
A variety of transport mechanisms have been proposed for drug transporters and reviewed in (10–12). For ABC transporter superfamily, “alternating access,” “ATP-switch,” and the “constant contact” transport models have been proposed (10). For the SLC transporter superfamily, transport mechanisms are mostly variations of the “alternating access” model (11, 26, 27). Because little is known about the structure of transporters within the SLCO superfamily, the structural processes involved in transport within this superfamily are not well understood, although investigations suggest that the transport processes may be quite complex (20, 23, 24). In general, several studies have indicated that they may act as electroneutral exchangers, while others indicate they are driven by pH or electrochemical gradients (20, 23, 24).
1.3.1. “Alternating Access” Transport Models.
The “alternating access” model has been proposed for both transporters in the ABC and the SLC superfamilies (10–12, 28). In general, the “alternating access” model involves conformational changes of the transporter where the transporter exposes itself to opposing sides of the membrane, while delivering the substrate (10, 12, 27). For the SLC superfamily, there are variations in the “alternating access” model for transport (11, 26). There is a “rocker-switch” variant of the mechanism where the conformational changes between similar domains occur around a bound substrate that are exposed to different sides of the membrane (11, 26). In the “rocking bundle” or “gate-pore” mechanism of the transporter, two asymmetric domains rearrange to allow substrate access to different sides of the membrane with the involvement of a protein gate (11). There is an “elevator” mechanism variant of the mechanism where the substrate is vertically translated across the membrane and released to different sides of the membrane by a “gate” (11, 26).
1.3.2. Other ABC Transporter Mechanisms.
In addition to the “alternating access” model for ABC transporters, additional models have been proposed, including the “ATP switch” (29), “sequential binding” (30), and “constant contact” (31, 32) models. Each of these models has distinct characteristics, but they are not necessarily mutually exclusive (10).
The “ATP switch” (a.k.a. “alternating switch” or “switch”) model only applies to the ABC transporter superfamily (10, 29). The binding of two molecules of ATP shifts the NBDs of the ABC transporter together while opening the transmembrane helices (TMs) on the extracellular (EC)-side of the membrane (29). Hydrolysis of those ATP molecules causes the NBDs of ABC transporter to separate, while the EC-side coalesces together (29). This model is supported by a recent molecular dynamics (MD) study for unidirectional transport of a bacterial ABC transporter (33).
Another model that has been proposed for ABC transporters is called the “sequential binding” (a.k.a. “alternating site”) transport model (30). In the model, one NBD undergoes ATP hydrolysis after the binding of ATP at the other NBD (30). A model called the “constant contact” model was proposed where nucleotides in different states of hydrolysis continually occupy the NBDs during transport (31, 32). A “nucleotide occlusion” model was proposed where ATP hydrolysis alternates between the NBDs as a result of ATP being sequentially “occluded” at one of the NBDs (10). A “processive clamp” model was proposed where ATP binding to the NBDs leads to NBD dimer formation followed by successive ATP hydrolysis at each NBD and followed by NBD separation (10). A distinct transport model was proposed for the bacterial ABC transporter maltose transporter MalFGK2 (10). This model is referred to as a “tweezer-like” model where the NBDs play a direct role in mediating substrate (i.e., maltose) transport across the membrane (10).
2.1. ABC Transporter Topology
The functional unit of an ABC transporter has two TM domains (TMDs) and two NBDs (10). Fig. 2 shows the general topology of ABC transporters. Some ABC transporters such as the ABCB1 transporter possess all four domains and are fully functional as monomers and are known as full transporters (10). Full transporters in the ABCA, ABCB, and ABCC transporter subfamilies possess two 6 helix TMDs and 2 NBDs and have a topology order of TMD-NBD-TMD-NBD (Fig. 2A–C) (34, 35). Some of ABCC subfamily isoforms, ABCC1–3,6,8,9, have an additional five helix TMD at the N-terminus that is referred to as TMD0 (Fig. 2B) (34, 35). The ABCA subfamily, such as ABCA1 and ABCA4, have significant extracellular (EC) domains (Fig. 2C) (34, 36, 37). Other ABC transporters are half transporters and are found in the ABCB and ABCD subfamilies, such as ABCB2/ABCB3 and ABCB10 that form heterodimers and homodimers, respectively (10, 17, 38). These half transporters have a topology order of a 6 helix TMD followed by an NBD (Fig. 2D) (34, 35). The members of the ABCG family, such as ABCG2 transporter, are “reverse” half transporters with a topology order of NBD-TMD (Fig. 2E) (10). The ABCE and the ABCF subfamilies only possess two NBDs connected together like pearls on a string (39–41).
Figure 2. The topology of ABC Transporters.

A) Full transporters in the ABCB and ABCC transporter subfamilies. B) “Long” full transporters with some members of the ABCC subfamily. C) Full transporters in the ABCA transporter subfamilies with EC domains. D) Half transporters in the ABCB and ABCD subfamilies. E) “Reverse” half transporters in the ABCG subfamily. F) Non-transporters in the ABCE and ABCF subfamilies. Abbreviations: EC, Extracellular Domain; NBD, Nucleotide Binding Domain; TM0, Transmembrane Domain 0 of ABCC family transporters.
The NBDs within ABC transporters contain conserved sequence motifs. They possess characteristic amino acid sequences that are known as Walker A and B motifs (42–45). The Walker sequences are flanked by characteristic “C,” “LSGGQ,” or “signature” motifs, which are not found in P-loop NTPases (43, 45–47). In ABC transporters, except the ABCG family (48), residues upstream of the Walker A sequence are a group of conserved aromatic residues called the “A-loop” that are critical for ATP binding (49). In addition, the ABCG transporter family has a conserved NPXDF motif within the NBDs that play an important role in cholesterol transport (50).
The NBDs of ABC transporters form a characteristic fold and have several conserved structures and sequences within them (10, 18, 51, 52). A Q-loop region, which lies between the Walker A and Walker B sequences and is defined by a conserved glutamine, has a variable conformation in ABC transporters (18, 51). The Q-loop has been proposed to be coupled to the TMs during transport and ATP hydrolysis (18, 45, 53). Upstream of the Walker signature sequences are the D-loop and the H-loop, which is also known as the switch region (51). A conserved aspartate in the D-loop has been proposed to stabilize an asparagine in the Walker A signature sequence to promote ATP hydrolysis (54). Interestingly, when the aspartate is mutated to histidine in the cystic fibrosis conductance regulator (CFTR), the mutation leads to cystic fibrosis (54, 55). The H-loop possesses a highly conserved histidine that interacts with glutamate in the Walker B sequence, which has been proposed to facilitate ATP hydrolysis (45). In a bacterial ABC transporter, the H-loop histidine has been proposed to shuttle protons required for ATP hydrolysis (56).
2.3. ATP Hydrolysis Mechanisms of ABC Transporters
The mechanism by which ABC transporters undergo ATP hydrolysis by their NBDs has not been completely resolved. Investigations support two types of ATP hydrolysis mechanisms: the “general-base catalysis” (10, 57), and the “substrate-assisted catalysis” (10, 58). For the “general-base catalysis” mechanism, the reaction rate will be sensitive to pH and D2O (57). In the “substrate-assisted catalysis” mechanism, a functional group of the substrate contributes to catalysis by the enzyme (58). An essential factor in both of these mechanisms is the coordination of a Mg2+ to the β and γ phosphates of ATP, and a hydrolytic water molecule (10).
The “general-base catalysis” mechanism was proposed for ATP hydrolysis by ABC transporters with catalysis occurring through a conserved glutamate that lies adjacent to the Walker B motif (59). The importance of the glutamate in catalysis was demonstrated with mutagenesis of the glutamate in the bacterial half transporter BmrA (60). Several X-ray crystal structures of the Escherichia (E.) coli ABC transporter maltose transporter, which is comprised of four subunits, with various nucleotide analogs, further support that ATP hydrolysis with ABC transporters occurs by a “general-base catalysis” mechanism (61). However, because there was no apparent decrease in the reaction velocity in the presence of D2O and other effects, a “substrate-assisted catalysis” (SAC) mechanism was proposed for the bacterial ABC half transporter HlyB (62, 63).
From work with the HlyB transporter, a structural model for ATP hydrolysis was proposed called the “linchpin” model (62, 63). In the model, the conserved glutamate adjacent to the Walker B motif forms a hydrogen bond with a conserved histidine on the H-loop, while the imidazole ring forms a hydrogen bond with the γ-phosphate of ATP allowing access to a catalytic water molecule (62, 63).
2.4. ABC Transporters and their Bacterial Homologs
Fig. 3 shows cartoon and molecular representations of select drug ABC transporters and their bacterial homologs that will be discussed in more detail. In the left column, the ABC transporters are front-facing, while in the remaining columns, they are side facing. Fig. 3A are general categories of ABC transporter conformations in a cartoon format (64–68). The NBDs are located on the cytosolic (C) side of the membrane, and substrates are transported unidirectionally to the EC-side of the membrane (69). These cartoon representations are based on structural work from X-ray crystal structures of bacterial ABC transporters that are shown in Fig. 3B. As explained below, the ABC transporter conformations can be divided into three general categories: inside-open (left), occluded (middle), and outside-open (right). The next row (Fig. 3C) shows the structures of the ABCB family transporter member ABCB1 (P-gp) (70, 71). The ABCC family transporter ABCC1 (MRP1) is shown in Fig. 3D (72, 73), and the ABCG family transporter ABCG2 (BCRP) is shown in Fig 3E (48, 74).
Figure 3. Structures of Select ABC Transporters in Different Conformations.
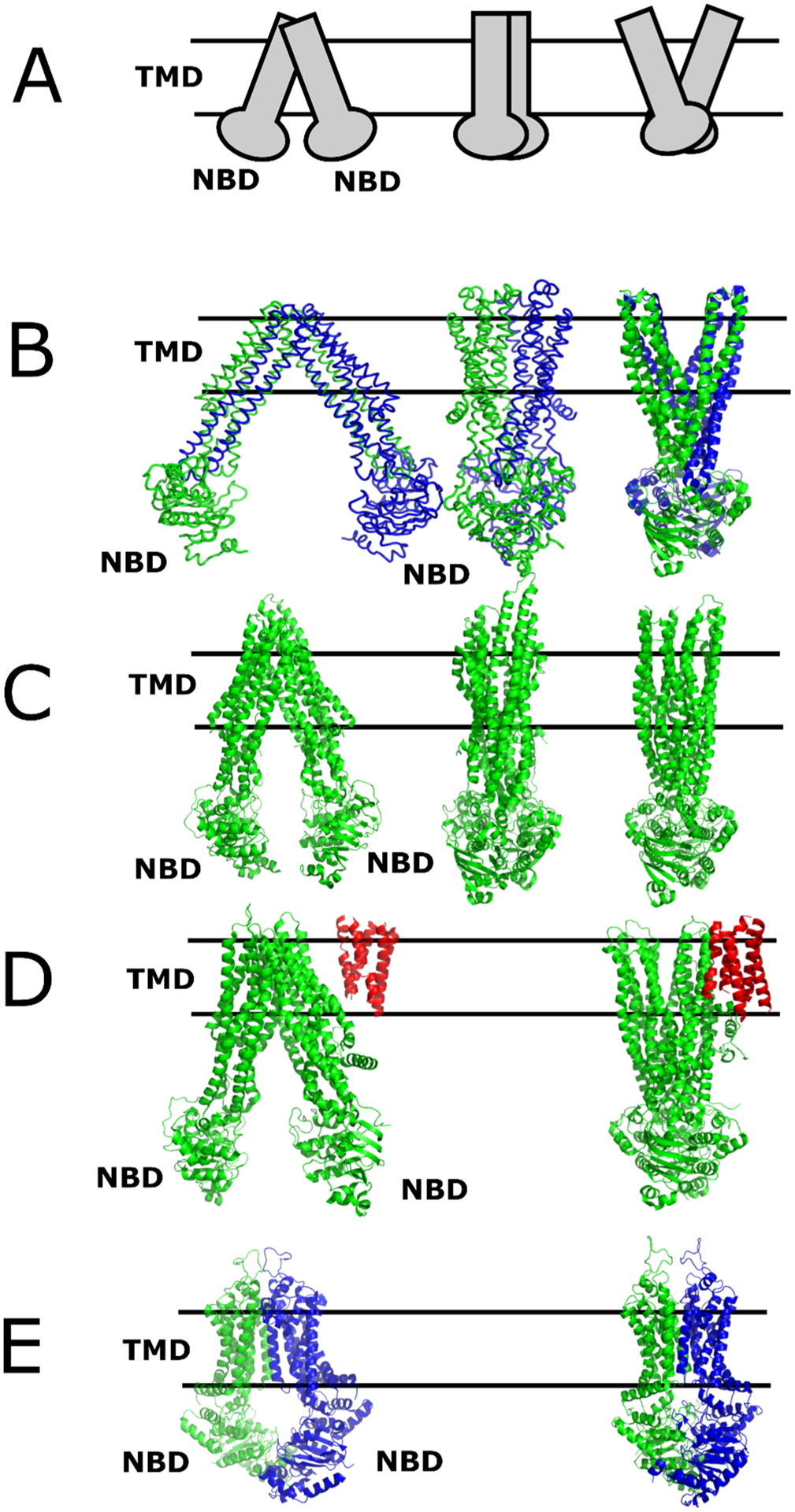
A) Cartoon of a generic ABC transporter, B) the bacterial MsbA ABC transporter (PDB IDs: 3B5W (left), 3B5X (middle) and 3B60 (right) (65)), C) P-gp; (ABCB1; PDB IDs: 5KPI (left) (70), 6FN4 (middle) (109), 6C0V (right) (71), D) MRP1 (ABCC1; PDB IDs: 5UJ9 (left) (72), 6BHU (right) (73)) and E) BCRP (ABCG2; 5NJ3 (left) (74), 6HBU (right) (48)) in inside-open (left) in front-facing view, and the occluded (middle) and outside-open conformation (right) in side view. The proteins are colored green or blue to differentiate the monomers. TMD0 of ABCC1 is colored red. The parallel lines denote the position of the membrane with the top being the EC-side and the bottom being the C-side. The TMDs and NBDs of the structures on the left are labeled.
2.4.1. Bacterial ABC Transporters.
X-ray crystal structures of bacterial ABC transporters were the first to identify key conformations in the transport process. Bacterial ABC half transporters were solved of the MsbA from E. coli that functions as a flippase for lipopolysaccharide amphiphilic cations and gram-positive Sav1866 from Staphylococcus (S.) aureus (65–68). The half transporters have six TM helices and an NBD (Fig. 2D) and form functional dimers (65–67). In the presence of nucleotides and nucleotide analogs, these transporters were in inside-open, occluded and outside-open conformations (Fig. 3B) (65–67). An “alternating access” mechanism was proposed with these transporters with an additional twisting motion proposed for the MsbA transporter (65–67). Additional details of the bacterial X-ray crystal structures are presented in Table 1.
Table 1.
X-ray crystal structures of key bacterial ABC transporters.
| Gene | Name | Species | X/Ca | PDB | Res.b | ~c | Comments | Ref.c |
|---|---|---|---|---|---|---|---|---|
| msbA | MsbA | E. coli | X | 3B5W | 5.30 | I | Apo | (65) |
| X | 3B5X | 5.50 | C | Apo | ||||
| X | 3B5Y | 4.50 | O | with AMPPNP (low resolution) | ||||
| X | 3B5Z | 4.20 | O | MsbA with ADP and OVO3 | ||||
| X | 3B60 | 3.70 | O | with AMPPNP (high resolution) | ||||
| SAV1866 | Sav1866 | S. aureus | X | 2HYD | 3.00 | O | with ADP | (67) |
| SAV1866 | Sav1866 | S. aureus | X | 2ONJ | 3.40 | O | with AMPPNP | (68) |
X-ray crystal (X) or Cryo-em (C) structure.
Resolution in Å.
Conformation: I, inside‐open conformation; O, outside‐Open conformation; C, occluded conformation; -, not applicable.
Reference. Abbreviations: AMPPNP, adenylyl-imidodiphosphate; NBD, Nucleotide-binding domain.
2.4.2. ABC Transporters in Human Drug Transport
The human ABC transporter superfamily is divided into seven subfamilies: ABCA-ABCG (17). The ABCA subfamily is primarily associated with lipid transport and trafficking, but it has also been implicated in drug transport and human disease (37). The ABCA2 and ABCA3 isoforms have been implicated in anti-cancer drug resistance in leukemia (75–78). The ABCB subfamily has a diverse range of substrates, including drugs, endogenous molecules, heme, lipids, and bile (38, 56, 79–83). The ABCB subfamily also has diverse functions such as multidrug resistance (MDR), antigen processing, and heme processing (38, 56, 79–83). The ABCB1 transporter isoform (a.k.a. P-glycoprotein (P-gp) ) mediates multi-drug resistance (MDR) and transports a range of clinically relevant hydrophobic xenobiotics (56). The ABCC subfamily has transporters that function in toxin mediation and MDR as well as ion channels (17). Within the ABCC subfamily are multidrug resistance proteins (MRPs) (i.e., ABCC1-ABCC6 and ABCC10-ABC12 isoforms) that transport a range of xenobiotics (78, 84). The ABCD subfamily is responsible for transporting long and very long fatty acids and these transporters are found in the lysosomes and peroxisomes, and is not associated with drug transport (78, 85). The ABCE subfamily, which consists of a single member (i.e., ABCE1) and lacks a TMD, is associated with interferon activity and translation (17, 86). The ABCF subfamily appears to be involved in antiviral, antibacterial and translational processes (17, 39, 40, 87). Due to a lack of a TMD, the ABCE and ABCF subfamilies are unlikely to be involved in drug transport (17, 78). The ABCG subfamily plays an important role in cholesterol/lipid homeostasis, MDR, peptide transport and sterol excretion (17). The ABCG2 isoform, which is also known as the breast cancer resistance protein (BCRP), transports a range of substrates such as organic anions and steroids (88). Structural details of the most promiscuous members of the ABC transporter family are described below. Additional structural information is provided in Table 2 of ABC transporters solved by X-ray crystallography and cryo-EM.
Table 2.
X-ray crystal or Cryo-em structures of eukaryotic and bacterial homologs of ABC superfamily transporters.
| Gene | Name | Species | X/Ca | PDB | Res.b | ~c | Comments | Ref.d |
|---|---|---|---|---|---|---|---|---|
| ABCB1 | ||||||||
| Abcb1a | P-gp | mouse | X | 3G5U | 3.80 | I | Apo | (104) |
| X | 3G60 | 4.40 | I | with QZ59-RRR | ||||
| X | 3G61 | 4.35 | I | with QZ59-SSS | ||||
| Pgp-1 | P-gp | C. elegans | X | 4F4C | 3.40 | I | Apo | (107) |
| Abcb1a | P-gp | mouse | X | 4LSG | 3.80 | I | Apo | NP |
| Abcb1a | P-gp | mouse | X | 4KSB | 3.80 | I | (106) | |
| X | 4KSC | 4.00 | I | |||||
| X | 4KSD | 4.10 | I | with nanobody | ||||
| Abcb1a | P-gp | mouse | X | 4M1M | 3.80 | I | Apo | (105) |
| X | 4M2S | 4.40 | I | with QZ59-RRR | ||||
| X | 4M2T | 4.35 | I | with QZ59-SSS | ||||
| Abcb1a | P-gp | mouse | X | 4Q9H | 3.40 | I | Apo | (110) |
| X | 4Q9I | 3.78 | I | with QZ-Ala | ||||
| X | 4Q9J | 3.60 | I | with QZ-Val | ||||
| X | 4Q9K | 3.80 | I | with QZ-Leu | ||||
| X | 4Q9L | 3.80 | I | with QZ-Phe | ||||
| Abcb1a | P-gp | mouse | X | 4XWK | 3.50 | I | with PBDE-100 | (216) |
| Abcb1a | P-gp | mouse | X | 5KPI | 4.01 | I | Apo | (70) |
| X | 5KPJ | 3.50 | I | methylated | ||||
| X | 5KO2 | 3.30 | I | 34 residue linker deleted with Hg2+ | ||||
| X | 5KOY | 3.85 | I | E552Q, E1197Q, 34 residue linker deleted with ATP | ||||
| X | 5KPD | 3.35 | I | E552Q, E1197Q, 34 residue linker deleted | ||||
| ABCB1 | P-gp | human | C | 6C0V | 3.40 | O | E566Q, E1201Q with ATP, Mg2+ | (71) |
| Abcb1a, ABCB1 | P-gp | mouse/humane | C | 6FN1 | 3.58 | C | LEVLFQGP replaced residues 668–675 and cross-linked with Zosuquidar and UIC2 Fab | (109) |
| C | 6FN4 | 4.14 | C | LEVLFQGP replaced residues 668–675 and cross-linked with UIC2 antibody and Fab | ||||
| Abcb1a | P-gp | mouse | C | 6GDI | 7.91 | I | Apo | (108) |
| C | 6Q81 | 7.90 | I | with ADP | ||||
| Abcb1a, ABCB1 | P-gp | mouse/humane | C | 6QEE | 3.90 | I | E555Q, E1200Qf with Zosuquidar, Fab, cholesterol and lipid | (111) |
| C | 6QEX | 3.60 | I | with Paclitaxel, Fab, cholesterol and lipid | ||||
| Abcb1a | P-gp | mouse | X | 6UTT | 4.17 | I | Y303A C952A with PBDE-100 | (217) |
| X | 6UJS | 4.17 | I | F728A C952A with PBDE-100 | ||||
| X | 6UJW | 4.15 | I | Y306A C952A with PBDE-100 | ||||
| X | 6UJN | 3.98 | I | C952A with PBDE-100 | ||||
| X | 6UJP | 3.98 | I | E979A C952A with PBDE-100 | ||||
| X | 6UJR | 4.10 | I | E724A C952A with PBDE-100 | ||||
| ABCC1 | ||||||||
| ABCC1 | MRP1 | human | X | 2CBZ | 1.50 | - | NBD1 with ATP and Mg2+ | (117) |
| ABCC1 | MRP1 | human | X | 4C3Z | 2.10 | - | NBD1 | (116) |
| ABCC1 | MRP1 | bovine | C | 5UJ9 | 3.49 | I | Apo | (72) |
| C | 5UJA | 3.34 | I | with leukotriene C4 | ||||
| ABCC1 | MRP1 | bovine | C | 6BHU | 3.24 | O | with ATP and Mg2+ | (73) |
| ABCC6 | ||||||||
| ABCC6 | MRP6 | human | X | 6BZS | 2.30 | - | NBD1 | (218) |
| MRP6 | human | X | 6NLO | 2.85 | - | NBD1 H812A | NP | |
| ABCG2 | ||||||||
| ABCG2 | BCRP | human | C | 5NJ3 | 3.78 | Ig | with antibody | (74) |
| C | 5NJG | 3.78 | - | TM region with antibody | ||||
| ABCG2 | BCRP | human | C | 6FEQ | 3.60 | Ig | with antibody and Zosuquidar inhibitor derivative MB136 | (129) |
| C | 6ETI | 3.10 | Ig | with antibody and Ko143 inhibitor derivative MZ29 | ||||
| C | 6FFC | 3.56 | Ig | with Ko143 inhibitor derivative MZ29 | ||||
| C | 6HIJ | 3.56 | Ig | with Ko143 inhibitor derivative MZ29, cholesterol, and PE lipid | ||||
| ABCG2 | BCRP | human | C | 6HBU | 3.09 | O | E211Q with ATP and Mg2+ | (48) |
| C | 6HCO | 3.58 | Ig | E211Q with antibody and estrone 3-sulfate | ||||
X-ray (X) or Cryo-em (C) structure.
Resolution in Å,
Conformation: I, inside-open conformation; O, outside-open conformation; C, occluded conformation; -, not applicable.;
Reference;
N-terminal half is mouse P-gp, and the C-terminal half is human P-gp;
PDB ID: 6QEE numbering;
NBDs are in contact. Abbrevations: aCAP, CmABCB1-binding cyclic peptide; AMPPCP, phosphomethylphosphonic acid adenylate ester; AMPPNP, phosphoaminophosphonic acid-adenylate ester; NBD, Nucleotide binding domain; NBD1, Nucleotide binding domain 1; NP, not published; GSH, glutathione; GSSG, glutathione disulfide; GS-Hg-SG, mercury derivative of glutathione disulfide; PBDE-100, 2,4-dibromophenyl 2,4,6-tribromophenyl ether.
P-glycoprotein (P-gp).
The P-gp transporter is a ubiquitously-expressed member of the ABCB transporter family that plays a large clinical role by effluxing a wide range of clinically relevant hydrophobic drugs through ATP hydrolysis at the NBDs (56). The transporter has several crucial physiological functions in drug disposition (56). P-gp is highly expressed on the apical side of enterocytes of the small intestine, where it mediates the oral absorption by effluxing drugs back into the intestinal lumen (56). It is also expressed at several important biological barriers, including the blood-brain-barrier, the blood-testes-barrier, and the blood-retinal-barrier (3, 89, 90). P-gp within these barriers protect against toxic insults but may also block potentially useful therapeutics (3, 89, 90). P-gp is concentrated on the apical side (urine side) of proximal convoluted tubule kidney cells and the bile canaliculi side of hepatocytes, where it contributes to excretion (2). In the placenta, the transporter is found at the trophoblast layer, but not the fetal capillary endothelial cells, to protect the developing fetus (91, 92). In addition to these physiological functions, P-gp reduces the efficacy of anti-cancer drugs by being highly expressed in cancerous tumors (93).
A number of structure-function studies have been carried out on mammalian and eukaryotic P-gp. P-gp is predicted to have the topology of a full transporter with 2 TMDs and 2 NBDs (Fig. 2A) (34). A low-resolution cryo-EM study of human P-gp and an atomic force microscope (AFM) study of mouse P-gp, under physiological conditions in a lipid bilayer, demonstrated that the transporter does transition between many conformations including those shown in Fig. 3A (94, 95). A differentiating feature of P-gp versus the bacterial half transporters is a linker between NBD1 and TMD2 (TM7), which is essential for activity (96). There is also strong evidence that ATP hydrolysis between the NBDs is asymmetric (e.g., 32, 97, 98). Also, lipid and cholesterol are known to modulate P-gp mediated ATP hydrolysis (99–103)
X-ray crystal structures of mouse and C. elegans P-gp were in an inside-open conformation with the NBDs separated (Fig. 3A (left)) (104–107). Large diffferences in the NBD separation between the P-gp X-ray structures suggested that P-gp has a lot of conformational flexibility and provides an explanation for its broad substrate specificity (104–107). An X-ray crystallographic study of P-gp with mutations of the linker region between that NBD1 and TM7 suggested that it controls the degree of NBD separation (70). In the presence Mg2+ ATP and mutations of the NBDs to prevent ATP hydrolysis, a cryo-EM structure of human P-gp was in an outside-open conformation (Fig. 3C, right) (71). Two Mg2+ ATP were bound in the structure suggesting that ATP binding rather than hydrolysis promotes substrate release (71). To examine the structure of P-gp after ATP hydrolysis, a cryo-EM structure was solved of mouse P-gp in the presence of ADP and OVO3 as a surrogate for inorganic phosphate (108). The P-gp conformation shifted back to the inside-open conformation with a partially resolved linker that was proposed to serve as a “steric blocker” to keep the NBDs separated (108). An occluded conformation of a mouse/human P-gp chimera (Fig. 3C, middle) was achieved by chemical cross-linking of the NBDs and an antigen-binding fragment (Fab) (109). The structure had an internal pocket spanning the length of the transporter (109).
Several structural studies were also undertaken to identify inhibitor and substrate binding sites on the P-gp transporter as well as the conformational changes that underly undirectional substrate efflux and modulation. The locations of the customized P-gp cyclic peptide inhibitors, QZ59-RRR and QZ59-SSS, were the first to be indentified in the X-ray crystal structures of mouse P-gp (104, 105). Despite their structural similiarity, these inhibitors bound at distinct hydrophobic niches near the EC-side of the cavity between the TMDs, while P-gp was in the inside-open conformation (Fig. 3A (left) (104, 105). Additional cyclic peptide ligands, with varying binding affinities and effects on P-gp-mediated ATP hydrolysis, were investigated by X-ray crystallography with mouse P-gp (110). All the ligands singly and doubly occupied the EC-side cavity between the TMDs as in the previous P-gp structures (104, 105, 110). One of the ligands occupied an additional niche formed by a bend in the middle of TM2, which the investigators proposed as a C-side ligand-binding site for substrate entry through the inner leaflet (110). These ligand-bound structures also revealed differences in TM4, which the investigators correlated to previous involvment of the helix in substrate binding and coupling of ATP hydrolysis with the NBDs (110). Further insights into the conformational changes involved in substrate binding and transport came from the cryo-EM mouse/human chimeric P-gp structure of paclitaxel and the inhibitor zosuquidar (111). Like the other P-gp structures, both zosuquidar and paclitaxel occupy a cavity on the EC-side of the transporter between the TMDs with the transporter in the inside-open conformation (111). In the presence of the ligands, the TM 4 and TM 10 helices were bent and TM 6 and TM 12 were significantly shifted, when compared to ligand-free P-gp (111). A cryo-EM structure of the mouse/human chimeric P-gp in the occluded conformation with zosuquidar showed the molecule trapped on the EC-side by TM 4 and TM 10 helices that are bent in the center (109). The ligand-induced bend observed in TM4 and TM10 helices in the cryo-EM P-gp structures suggested a potential mechanism for unidirectional substrate transport (8, 109, 111). Furthermore, paclitaxel and zosuquidar induced conformational changes in P-gp also created binding sites for cholesterol and phospholipids (111). This observation provides a structural explanation for lipid and cholesterol modulation of P-gp mediated ATP hydrolysis (99–103).
Multidrug Resistance-associated Protein 1 (MRP1)).
MRP1, which is also known as the ABCC1 transporter, is a promiscuous transporter that is known to efflux a wide range of drugs and is known to play a significant role in anti-cancer drug resistance (Fig. 3D) (112). Additionally, the transporter plays an important physiological function effluxing anions, the antioxidant GSH, and the pro-inflammatory cysteinyl leukotriene C4 (112). Genetic polymorphisms have been associated with increased drug resistance and disease susceptibility (113). MRP1 has 17 TMs, which form three TMDs, and two NBDs (112). Two of the TMDs, TMD1 and TMD2, form the transport channel, while the function of the third amino-terminal TMD0 remains to be clarified (112, 114, 115). The first molecular structures of MRP1 were of its first NBD in the presence and absence of Mg2+ ATP (116, 117). The overall NBD1 conformation was similar to other solved NBD structure with Mg2+ ATP interactions and conformational changes that support a “general-base catalysis” mechanism for ATP hydrolysis (116, 117). Complete structures of bovine MRP1 were solved by cryo-EM without substrate and with leukotriene C4 (72). The structures showed that the TMD1 and TMD2 form two pockets (72). There is a positively charged region called the P-pocket and a hydrophobic region called the H-pocket (72). The TMD0 was not well defined in the structure, and the removal of TMD0 did not have significant effects on the ATPase activity (73, 118). In the leukotriene C4 bound structure, the NBDs were closer together with the leukotriene C4 molecule occupying both binding pockets (72). A cryo-EM structure of MRP1 was also solved with Mg2+ ATP bound, which shifted the NBDs together and caused the EC-side to open up into an outside-open conformation (73). Like P-gp (71), these ATP-induced conformational shifts suggested that substrate release occurs before ATP hydrolysis (73).
Breast Cancer Resistance Protein (BCRP).
BCRP, which is also known as ABCG2, transporter name comes from the breast cancer cell line that it originated from and has broad substrate specificity (119). The transporter has significant effects on drug disposition being expressed in most normal tissues, including relatively high levels in placental syncytiotrophoblasts (119, 120). Despite the BCRP name, high expression levels of the transporter have been associated more with acute myeloid leukemia than breast cancer (119–121). BCRP transporter activity is modulated by sterols such as cholesterol, which is an essential activator, and steroids (122–124). Many transporters in the ABCG subfamily are known to transport sterols (125), but the role of BCRP in vivo remains to be clarified (123).
BCRP is a reverse half transporter (Fig. 2E) with 6 TM helices and an NBD that functions as a homodimer (126). On the EC-side of the transporter is an asparagine (N596) that is glycosylated, but the functional role remains unresolved (127, 128). The glycosylation-deficient N596Q ABCG2 mutant produced in a virus expression system showed comparable transport activity to the wild type suggesting that asparagine is not essential (127). However, the expression of the mutant was significantly reduced in a human embryonic kidney cell line, which the authors interpreted as reduced transporter stability and enhanced ubiquitin-mediated degradation (128).
A cryo-EM structure of the BCRP was solved with the extracellular loops complexed with a fragment antigen-binding (Fab) and cholesterol (74). The overall fold of the homodimer is similar to P-gp, but the vertical span of the transporter is shorter with the NBDs and the EC domain closer to the membrane surface (74). The structure was in an inside-open conformation with the NBDs contacting (Fig. 3E, left) (74). Within the EC domain, the N596 was glycosylated. There are two intracellular disulfides and one intercellular disulfide bond holding the homodimer together (74). In the structure, two cholesterol molecules were found symmetrically bound on opposite sides within the central cavity formed by the dimer, which is consistent with their role in modulating transporter activity (74, 122). A couple of cryo-EM structures of BCRP were solved with a Ko143 derivative (i.e., MZ29) and a tariquidar-derivative (i.e., MB136) (129). Ko143 is a novel analog of the fungal toxin fumitremorgin C and a potent inhibitor of BCRP (130). While being a potent P-gp inhibitor (131), tariquidar is both an inhibitor and a substrate for BCRP (132). Two molecules of the Ko143 derivative and BCRP inhibitor were found symmetrically bound within the same niches occupied by the cholesterol molecules (129). In contrast, a single molecule of the tariquidar-derivative was found spanning both cholesterol binding sites in BCRP (129). The overall conformation of BCRP in the presence of these inhibitors was similar to the cholesterol-bound BCRP structure (74, 129). A cryo-EM structure was solved of BCRP in two conformations bound with the substrate estrone-3-sulfate and Mg2+ ATP, providing insight into BCRP-mediated ATP hydrolysis and transport (48). The estrone-3-sulfate bound BCRP structure was in an inside-open conformation with the NBDs separated with the ligand modeled in two different orientations (48). In the Mg2+ ATP-bound structure, the NBDs coalesced into an outside-open conformation (Fig. 3E, right), while the TMs on the EC-side opened up with respect to the nucleotide-free structure (48). The positions and orientations of the two ATP molecules within the NBDs were similiar to other ABC transporters, except for some distinct interactions with the adenine functional drug due to the absence of a conserved A-loop (48). The ATP-induced opening up of the EC-side and the presence of two bound ATP molecules in the structure imply that substrate release occurs prior to ATP hydrolysis in BCRP like other ABC transporters (48, 71, 73).
3. SLC transporter superfamily members that are involved in drug transport
Out of 65 families of transporters within the SLC superfamily many are known to transport drugs or to be potential drug targets (21, 22). The SLC1 transporter family move neurotransmitters and amino acids across membranes and are associated with a range of neurodegenerative disorders and cancer (133–135). Dysfunctions and mutations in the SLC1A3 transporter are associated with a range of neurological and mental health disorders which makes this transporter a potential neurotherapeutic drug target (136, 137). The SLC1A5 transporter is overexpressed in several cancers, so it is a target for anti-cancer drugs (138, 139). The SLC6 family is comprised of Na+/neurotransmitter antiporters that are found in the brain and are inhibited by a range of drugs (140, 141). The SLC6A4 transporter is a serotonin transporter (SERT) associated with a range of neurological disorders and the target of serotonin reuptake inhibitors (SSRIs) (142–144). The SLC6A19 transporter is a Na+/neutral amino acid antiporter that is responsible for amino acid absorption in the intestine and may be a potential drug target for type 2 diabetes and protein restriction treatment (142, 145, 146). The SLC7 and SLC3 transporter families form a heterodimer that functions as a Na+/amino acid antiporter (147). The SLC7A5-SLC3A2 heterodimer (a.k.a. LAT1-4F2hc) can exchange amino acid drugs and is a target for anticancer drugs (147–150). The SLC10 transporter family is a group of Na+/bile acid symporters that can translocate conjugated bile acids, steroids, and drugs (151–153). The SLC15 transporter family is comprised of H+/oligonucleotide cotransporters, which are known to transport a diverse range of clinically relevant drugs (154). Although no structures have been solved in this transporter family, the SLC22 transporter family is important to mention because the transporter family includes Organic Anionic Transporters (OATs) and Organic Cationic Transporters (OCTs) that can translocate drugs, metabolites, and toxins (155). The equilibrative nucleoside transporters (ENTs) and concentrative nucleoside transporters (CNTs) are part of the SLC28 and SLC29 transporter families, respectively, and are known to translocate nucleoside-derived therapeutics (156–159). The members of the SLC35 transporter family are nucleotide sugar antiporters, but SLC35F2 is the only member known to transport any cancer drugs, and there is no structural information on the transporter (160, 161). Finally, the SLC47 family is known as the multidrug and toxic compound extrusion (MATE) family (162, 163). Within the family, the MATE1 (SLC47A1) isoform has the broadest substrate specificity within the SLC superfamily (162, 163). Table 3 provides information on the X-ray and cryo-EM structures of drug transporters within the SLC superfamily. Additional structural details are provided below for the MATE transporter family, which possesses the most promiscuous isoform within the SLC superfamily.
Table 3.
X-ray Crystal, Cryo-em and Solution NMR structures of eukaryotic and bacterial homologs of SLC superfamily of transporters.
| Gene | Name | Species | X/Ca | PDB | Res.b | ~c | Comments | Ref.c |
|---|---|---|---|---|---|---|---|---|
| SLC1A3 | ||||||||
| SLC1A3 | EAAT1 | human | X | 5MJU |
3.71 |
O | thermostable mutant with TBOATFB and UCPH101 inhibitors | (136) |
| X | 5LLM | 3.25 | O | thermostable mutant with aspartate and UCPH101 inhibitor | ||||
| X | 5LM4 | 3.10 | O | thermostable mutant with aspartate UCPH101 inhibitor | ||||
| X | 5LLV | 3.32 | O | thermostable mutant with aspartate | ||||
| SLC1A5 | ||||||||
| SLC1A5 | ASCT2 | human | C | 6GCT | 3.85 | C | with glutamine | (168) |
| SLC1A5 | ASCT2 | human | C | 6RVY | 4.13 | I | C467R | (219) |
| C | 6RVX | 3.61 | I | C467R in presence of TBOA | ||||
| SLC1A5 | ASCT2 | human | C | 6MP6 | 3.54 | O | Apo | (214) |
| C | 6MPB | 3.84 | O | with glutamine | ||||
| SLC6 | ||||||||
| dDAT | dDAT | D. melanogaster | X | 4M48 | 2.96 | O | V74A, V275A, V311A, L415A, G538L with nortriptyline | (202) |
| dDAT | dDAT | D. melanogaster | X | 4XPF | 3.27 | O | V275A, V311A, G538L, D121G, S426M with cocaine analog RTI-55 and Fab | (220) |
| X | 4XPG | 3.21 | O | V275A, V311A, G538L, D121G, S426M with β-CFT or Win35428 | ||||
| X | 4XPH | 2.90 | O | V275A, V311A, G538L, D121G, S426M with 3,4-dichlorophenethyla mine and Fab | ||||
| X | 4XP4 | 2.80 | O | V275A, V311A, G538L, D121G, S426M with cocaine | ||||
| X | 4XP5 | 3.30 | O | V275A, V311A, G538L with 3,4-dichlorophenethyla mine and Fab | ||||
| X | 4XP9 | 2.80 | O | V275A, V311A, G538L with D-amphetamine and Fab | ||||
| X | 4XP1 | 2.89 | O | V275A, V311A, G538L with dopamine and Fab | ||||
| X | 4XPT | 3.36 | O | V275A, V311A, G538L, Δ162–201, D121G, S426M with 3,4-dichlorophenethyla mine and Fab | ||||
| X | 4XNX | 3.00 | O | with reboxetine and Fab | ||||
| X | 4XPB | 3.05 | O | D121G, S426M with cocaine and Fab | ||||
| X | 4XP6 | 3.10 | O | with methamphetamine | ||||
| X | 4XPA | 2.90 | O | with 3,4-dichloro phenethylamine | ||||
| SLC6A4 | ||||||||
| SLC6A4 | SERT | human | X | 5I6Z | 4.53 | O | I291A, T439S with 8B6 Fab | (171) |
| X | 5I74 | 3.40 | O | I291A, T439S with 8B6 Fab and Br-citalopram | ||||
| X | 5I73 | 3.24 | O | I291A, T439S with 8B6 Fab and s-citalopram at 2 sites | ||||
| X | 5I75 | 3.49 | O | I291A, T439S with 8B6 Fab, s-citalopram and Br-citalopram | ||||
| X | 5I6X | 3.14 | O | I291A, T439S with 8B6 Fab and paroxetine | ||||
| X | 5I71 | 3.15 | O | I291A, T439S with 8B6 Fab and s-citalopram | ||||
| SLC6A4 | SERT | human | X | 6AWO | 3.53 | O | I291A with 8B6 Fab and sertraline | (221) |
| X | 6AWN | 3.62 | O | I291A with 8B6 Fab and paroxetine | ||||
| X | 6AWQ | 4.05 | O | I291A with 8B6 Fab and sertraline | ||||
| X | 6AWP | 3.80 | O | I291A with 8B6 Fab and fluvoxamine | ||||
| SLC6A4 | SERT | human | C | 6DZW | 4.30 | O | S277C with 15B8 Fab, 8B6 Fv, and paraoxetine | (222) |
| C | 6DZV | 4.20 | C | N- and C-terminal truncated (ΔN72/C13) S277C with 15B8 Fab and ibogaine | ||||
| C | 6DZY | 4.10 | O | S277C with 15B8 Fab, 8B6 Fv and ibogaine | ||||
| C | 6DZZ | 3.60 | I | S277C with 15B8 Fab and ibogaine | ||||
| SLC6A4 | SERT | human | C | 6VRK | 4.10 | - | with 8B6 Fab and Br-paroxetine | NP |
| C | 6VRL | 3.80 | - | with 8B6 Fab and I-paroxetine | ||||
| C | 6VRH | 3.30 | - | with 8B6 Fab and paroxetine | ||||
| SLC6A4 | SERT | human | X | 6W2B | 4.70 | - | with 8B6 Fab and Br-paroxetine | NP |
| X | 6W2C | 6.30 | - | with 8B6 Fab and I-paroxetine | ||||
| SLC6A19 | ||||||||
| SLC6A19 | B0AT1 | human | C | 6M17 | 2.90 | I | with ACE2 and COVID-19 RBD | (203) |
| C | 6M18 | 2.90 | O | with ACE2 | ||||
| C | 6M1D | 4.50 | O | with ACE2 | ||||
| SLC7 | ||||||||
| ApcT | GkApcT | G. kaustophilus | X | 5OQT | 2.86 | C | with alanine cholesterol and lipid | (205) |
| X | 6F34 | 3.13 | C | with arginine, cholesterol and lipid | ||||
| CAT7_03719 | BasC | Carnobacterium sp. AT7 | X | 6F2G | 2.92 | I | with nanobody | (223) |
| X | 6F2W | 3.40 | I | with nanobody and α-aminoisobutyric acid | ||||
| SLC7A5, SLC3A2 | ||||||||
| SLC3A2 | 4F2hc | human | X | 2DH2 | 2.10 | - | Apo | (224) |
| 2DH3 | 2.80 | - | with Zn2+ | |||||
| Slc3a2 | CD98hc | mouse | X | 6I9Q | 2.10 | - | Apo | |
| SLC7A5, SLC3A2 | LAT1-4F2hc | human | C | 6IRS | 3.30 | I | with JPH203 inhibitor | (172) |
| C | 6IRT | 3.50 | I | with BCH | ||||
| SLC7A5, SLC3A2 | LAT1-4F2hc | human | C | 6JMQ | 3.31 | I | with MEM-108 Fab and cholesterol | (204) |
| C | 6JMR | 4.10 | I | with HBJ127 and MEM-108 | ||||
| SLC10 | ||||||||
| not available | ASBTNM | N. meningitidis | X | 3ZUX | 2.20 | I | with taurocholic acid (332 residues) | (176) |
| X | 3ZUY | 2.20 | I | with taurochlolic acid (323 residues) | ||||
| yfred0001_28600 | ASBTYf | Y. frederiksenii | X | 4N7W | 1.95 | I | Apo | (225) |
| X | 4N7W | 2.50 | O | E254A | ||||
| SLC15 | ||||||||
| BL313_09825 | PepT1 | S. hominis | X | 6EXS | 2.50 | I | with malodour precursor | (215) |
| BL313_09825 | PepT1 | S. hominis | X | 6GZ9 | 3.10 | I | with Valacyclovir | (226) |
| X | 6HZU | 2.80 | I | with 5ALA | ||||
| X | 6HZP | 2.50 | I | with 5ALA | ||||
| dtpA | DtpA | E. coli | X | 6GS4 | 2.65 | I | with nanobody and valganciclovir | (206) |
| X | 6GS7 | 3.30 | I | with nanobody and glycine buffer | ||||
| X | 6GS1 | 3.29 | I | with nanobody and MES buffer | ||||
| SLC15A1 | ||||||||
| Slc15a1 | PepT1 | mouse | X | 5A9D | 2.10 | - | EC domain | (207) |
| SLC15A2 | ||||||||
| Slc15a2 | PepT2 | rat | X | 5A9H | 2.06 | - | EC domain | (207) |
| X | 5A9I | 2.84 | - | EC domain | ||||
| SLC28 | ||||||||
| VC_2352 | vcCNT | V. cholerae | X | 3TIJ | 2..44 | I | with uridine and Na+ | (167) |
| VC_2352 | vcCNT | V. cholerae | X | 4PDA | 2.61 | I | with cytidine and Na+ | (227) |
| X | 4PD8 | 2.75 | I | with pyrrolo-cytidine and Na+ | ||||
| X | 4PD9 | 3.10 | I | with adenosine and Na+ | ||||
| X | 4PD5 | 2.91 | I | with gemcitabine and Na+ | ||||
| X | 4PD6 | 2.08 | I | with uridine and Na+ | ||||
| X | 4PD7 | 2.91 | I | with zebularine and Na+ | ||||
| X | 4PB1 | 2.80 | I | L7C, I8C with ribavirin and Na+ | ||||
| X | 4PB2 | 2.30 | I | L7C, I8C with 5-fluoridine and Na+ | ||||
| SLC29A1 | ||||||||
| SLC29A1 | ENT1 | human | X | 6OB6 | 2.90 | O | with S-(4-nitrobenzyl)-6-thioinosine | (175) |
| X | 6OB7 | 2.30 | O | with dilazep | ||||
| SLC47 | ||||||||
| NorM | ||||||||
| Norm | NorM-VC | V. cholerae | X | 3MKT | 3.65 | O | Apo | (174) |
| X | 3MKU | 4.20 | O | Rb+ | ||||
| NGTW08_0430 | NorM-NG | N. gonorrhoeae | X | 4HUK | 3.59 | O | with TPP and monobody | (190) |
| X | 4HUL | 3.81 | O | with Cs+ and monobody | ||||
| X | 4HUM | 3.49 | O | with ethidium and monobody | ||||
| X | 4HUN | 3.59 | O | with R6G and monobody | ||||
| NGO0395 | NorM-NG | N. gonorrhoeae | X | 5C6P | 3.00 | O | with monobody and verapamil | (196) |
| clbM | ClbM | E. coli | X | 4Z3N | 2.70 | O | Apo | NP |
| X | 4Z3P | 3.30 | O | with Rb+ | ||||
| DinF | ||||||||
| PF0708 | pfMATE | P. furiosus | X | 3VVN | 2.40 | O | basic TM1 “straight” form | (191) |
| X | 3VVO | 2.50 | O | acidic TM1 “bent” form | ||||
| X | 3VVP | 2.91 | O | with Br-NRF | ||||
| X | 3VVR | 3.00 | O | with MaD5 cyclic peptide | ||||
| X | 3VVS | 2.60 | O | with MaD35 cyclic peptide | ||||
| X | 3WBN | 2.45 | O | with MaL6 cyclic peptide | ||||
| X | 3W4T | 2.10 | O | P26A acidic TM1 “straight form” | ||||
| BH2163 | DinF-BH | B. halodurans | X | 4LZ6 | 3.20 | O | Apo | (192) |
| X | 4LZ9 | 3.70 | O | with R6G | ||||
| BH2163 | DinF-BH | B. halodurans | X | 5C6O | 3.00 | O | with verapamil | (196) |
| X | 5C6N | 3.00 | O | D40N | ||||
| CGT79_05080 | VcmN | V. cholerae | X | 6IDR | 2.50 | O | acidic TM1 “bent” form | (200) |
| X | 6IDP | 2.21 | O | basic TM1 “straight” form | ||||
| X | 6IDS | 2.79 | O | D35N | ||||
| PF0708 | pfMATE | P. furiosus | X | 4MLB | 2.35 | O | with Cl− | (198) |
| X | 6FHZ | 2.80 | I | Apo | ||||
| X | 6HFB | 3.50 | O | Apo | ||||
| X | 6GWH | 2.80 | O | Apo | ||||
| eMATE | ||||||||
| DTX14 | AtDTX14 | A. thaliana | X | 5Y50 | 2.60 | O | Apo | (228) |
| false flax | CasMATE | C. sativa | X | 5YCK | 2.30 | O | with Rb+ | (229) |
X-ray (X), Cryo-em (C) or NMR (N) structures.
Resolution in Å,
Conformation O, outside-open conformation; I, inside-open conformation; C, Intermediate or occluded conformation, or – not applicable/not determined.;
Reference. Abbrevations: 5ALA, 5-aminolevulinic acid; Br-NRF, norfloxacin-derivative; EC, extracellular; eMATE, eukaryotic multidrug and toxin extrusion transporter; NP, not published; R6G, rhodamine 6G; TBOA, DL-threo-β-benzyloxyaspartic acid; TBOATFB, (2S,3S)-3-[3-[4-(trifluoromethyl) benzoylamino] benzyloxy] aspartate; TPP, tetraphenylphosphonium.
3.1. SLC Transporter Folds
There is considerably more conformational diversity for the SLC superfamily than the ABC transporter family. SLC superfamily has at least six distinct folds that are described below and reviewed in detail in (160, 163–167). Fig. 4 organizes structures of SLC drug transporters according to six distinct fold types: GltPh (Fig. 4A), LeuT (Fig. 4B), MATE (Fig. 4C), MFS (Fig. 4D), NhaA (Fig. 4E) and vcCNT folds (Fig. 4F).
Figure 4. Structures of Select SLC transporters with Major Features Highlighted.

GltPh fold: A) EAAT1 (SLC1A5; PDB ID: 6MP6 (214)) with ScaD (green), TranD (blue). LeuT fold: B, left) SERT (SLC6A4; PDB ID: 5I6Z (171)) with NTD (green), CTD (blue) and TM11-12 (red). B, right) LAT1-4F2hc (SLC7A5/SLC3A2; PDB ID: 6IRS (172)) with TM1-5 (N-terminal domain, green), TM6-10 (C-terminal domain, blue), TM11-12 (red) and the ectodomain 4F2hc (SLC3A2, magenta). MATE fold: C) Bacterial MATE Norm-VC (SLC47 analog PDB ID: 3MKT (174)) with NTD (green) and CTD (blue). MFS fold: D) Bacterial PepTsh (SLC15A1 analog, PDB ID: 6EXS (215)) with NTD (green) and CTD (blue). NhaA fold: E) Bacterial ASBTNM (SLC10A2 analog, PDB ID: 3ZUX (176)) with the core (green) panel (domains). vcCNT fold: F) vcCNT (SLC28 analog, PDB ID: 3TIJ (167)) with ScaD (green), TranD (blue).The parallel lines denote the position of the membrane with the top being the EC-side and the bottom being the C-side.
3.1.1. GltPh fold.
The SLC1 transporter family has a conformational fold that is similar to the bacterial glutamate transporter from Pyrococcus (P.) horikoshii (GltPh) (136, 168). GltPh fold (Fig. 4A) has a scaffold domain (ScaD), transport domain (TranD), and two hairpins (HP1 and HP2) (136, 164, 169, 170). HP1 and HP2 have been proposed to act as intracellular (IC) intracellular and EC gates of the transporter (164).
3.1.2. LeuT fold.
The fold is common among SLC transporters, and it is based on the bacterial leucine transporter (LeuT) (166). The LeuT fold (Fig. 4B) is characterized by two inverted structural 5 TM repeats or inverted 5 + 5 TM repeats (166). SLC transporters in this group include SERT (SLC6A4, Fig. 4B, left (171)) and the Na+/amino acid antiporter LAT1-4F2hc (SLC7A5/SLC3A2, Fig. 4B, right (172))
3.1.3. MATE fold.
The MATE fold (Fig. 4C) is distinct from the other transporter folds (163, 173). Bacterial MATE transporters are known to have 12 TM helices, while mammalian MATEs are predicted to an additional TM13 and an EC domain on the C-terminus (163, 173). Like the MFS fold, the 12 TM domains of bacterial MATEs split into two six-helix N-terminal (NTD) and C-terminal (CTD) domains (174). Two pairs of TM helices, TM1/TM8, and TM2/TM7 also create a distinct opening into the transporter for substrates (174).
3.1.4. MFS fold.
The major facilitator superfamily (MFS) includes the SLC15 transporter family and has a characteristic topology and fold (Fig. 4D) with TM 12 helices (154, 166). They are divided into NTD and CTD that have 6 TM helices each, and each domain has pairs of inverted structural repeats (166). The equilibrative nucleoside transporter 1 (ENT1) (SLC29A1) has an MFS-like fold that has 11 helices that follow the topology of the MSF fold with pseudo-symmetric 6 + 5 TM topology and inverted structural repeats (175).
3.1.5. NhaA fold.
One of the bacterial homologs of an SLC10 transporter homolog has an unusual fold found in the Na+/H+ antiporter (NhaA) (165, 166, 176). The NhaA fold (Fig. 4E) has a panel (a.k.a. dimer interface domain) and a core domain with antiparallel inverted crossing TM helices with connecting loops (165, 166)
3.1.6. vcCNT fold
The bacterial concentrative nucleoside transporter from Vibrio (V.) cholerae (vcCNT) of the SLC28 transporter family has a distinct fold (Fig. 4F) formed from 8 TM helices (167). The TM helices form a ScaD (TM1-TM3) and a TranD (TM5-TM8) comprised of two hairpins, HP1 and HP2, that flank a long TM6 that was proposed to play a major role in transport (167).
3.2. Multidrug And Toxin Extrusion (MATE) family
The MATE transporter family, which is also known as SLC47, exchange organic cations through electroneutral exchange with protons and are found in both prokaryotes and eukaryotes (177). Within the MATE transporter family, there is MATE1, which is also known as SLC47A1, and there are several variants of the SLC47A2 isoform, including MATE2, MATE2-B, and MATE2-K (177–181). MATE1 has broad substrate specificity, including many clinically-relevant drugs (162, 163, 182). MATE1 transported substrates and drugs usually have a low molecular weight and are cationic, but zwitterionic and ionic molecules are known substrates (163). The MATE1 transporter can even transport the toxic heavy metal cadmium (183). The substrate specificity for the MATE2 isoform remains to be elucidated (162, 163). The only known substrate for MATE2 is tetraethylammonium (TEA), but MATE2 expresssion has been correlated to the resistance of antiproliferative drug metformin in cancer cells (184). MATE transporters have species-dependent differences in expression levels and tissue localization (177–181). For human MATE1 (hMATE1), significant expression occurs in the liver, skeletal muscle, and kidneys with minor expression in the heart (178, 179). Mouse MATE1 (mMATE1) localization has some similarities to hMATE1, but was not found in skeletal muscle (178, 179). MATE2 transporter expresses in the kidneys in humans and the testes of mice and rats (178, 181). MATE2-B was originally isolated from the human brain but lacks transport activity (180). MATE2-K is highly expressed in the kidneys in humans and the testes of mice and rodents (179, 181).
3.2.2. Bacterial MATE Transporters as Models.
Members in the MATE transporter family share a 40% sequence similarity with each other (174, 185). Therefore, eukaryotic and bacterial MATE transporters likely have similarities in their structures and their transport mechanisms (174, 185). However, the bacterial MATE transporters are not perfect models for their mammalian counterparts (173). The bacterial MATE transporters are known to have 12 TM helices, while cysteine scanning and epitope tagging suggest a 13th C-terminal TM helix with an EC-domain for mammalian MATEs (163, 173). The putative TM13 has an unknown transport function since the deletion of the helix did not disrupt transport (173). Bacterial MATE transporters can be divided into the NorM or the DNA damage-inducible protein F (DinF) subfamilies, which can couple with Na+ ions and/or H+ to substrate transport (186–189). In contrast, eukaryotic MATE (eMATE) transporters only couple with H+ to substrate transport (177). Several X-ray crystal solved structures of MATE transporters have improved our understanding of the substrate binding and the transport process (e.g., 174, 189–191). Although the proposed MATE transport models fit within an “alternating access” mechanism, significant differences exist between them (e.g., 174, 192). Therefore, details of substrate binding and their proposed transport mechanisms for several bacterial MATEs are presented in this review.
3.2.3. Bacterial NorM MATE transporter subfamily.
Bacterial NorM MATE transporters are typically Na+/substrate antiporters (186, 190, 193), although some may couple with both Na+ and H+ (194, 195).
NorM MATE transporter from Vibrio (V.) cholerae (NorM-VC).
The NorM-VC couple both Na+ and H+ to substrate transport (194, 195). X-ray crystal structures were solved for the MATE transporter with and without Na+ analogs, rubidium (Rb+) and cesium (Cs+) (174). NorM-VC had the distinctive MATE fold with NTD and CTD, and intersecting helices (174). There is also a characteristic asymmetric pairing of TM1/TM8 and TM2/TM7 that provides an opening for ligands into the transporter (174). The NorM-VC structure was found in the outside-open conformation toward the EC-side with a large cavity volume of approximately 4,300 Å3 (174). The monovalent Na+ analogs, Rb+ and a Cs+, were sequestered by several residues, including E255 and F288, which was presumed to be the putative cation binding site (174). From the X-ray crystal structure, they proposed the “rocker-switch” variant of the “alternating access” mechanism for transport (174). They proposed that the outside-open conformation was part of the transport cycle with high affinity for monovalent cations and lower affinity for substrates (174). In the model, the high-affinity monovalent cation shifts the conformation of NorM-VC to an inside-open conformation where substrate binding is higher affinity (174). The release of the cation or binding of the substrate shifts the conformation of NorM-VC back to the outside-open conformation (174). During the proposed transport cycle, the substrate is pumped to the EC-side of the membrane, while the cation is pumped into the cytosol (174).
NorM MATE transporter from Neisseria (N.) gonorrhoeae (NorM-NG).
NorM-NG is a typical NorM MATE Na+/antiporter (192, 193). To understand the transport process, X-ray crystal structures of NorM-NG were solved with the several substrates, and with the Na+ analog cesium (Cs+) (190, 196). Despite some conformational differences, NorM-NG X-ray crystal structures had an overall fold and conformation that was similar to NorM-VC (174, 190). All the ligand-bound X-ray crystal structures of NorM-NG were in the outside-open conformation with minor differences in the ligand-binding sites within the central cavity (190, 196). The Cs+ binding site of NorM-NG was found just outside the ligand-binding site with E261 and Y294 coordinating it (190). In the Cs+-bound NorM-NG structure, the TM7 and TM8 helices showed the largest differences from the other structures suggesting an important role in the transport cycle (190). With additional information provided by the ligand and cation bound structures, a more detailed “alternating access” mechanism for the antiporter was proposed involving conformational changes of TM7 and TM8 with the involvement of TM1 and TM2 during the cation and substrate antiport (190).
3.2.4. DNA damage-inducible protein F (DinF) MATE subfamily.
The DNA damage-inducible protein F (DinF) MATE subfamily are H+/substrate antiporters (191, 192, 197), although recent structural and computational investigations with the DinF transporter from Pyrococcus (P.) furiosus (pfMATE) suggested that Na+ binding may be involved in the transport process as well (198, 199)
DinF MATE transporter from Pyrococcus (P.) furiosus (pfMATE).
The DinF transporter pfMATE is an H+/substrate antiporter (191, 197), but may serve additional transport functions (198). Several X-ray crystal structures of pfMATE were solved at different pH’s, with a norfloxacin-derivative and with several different inhibitory cyclic peptides (191, 198). The overall fold of pfMATE was similar to NorM-VC and Norm-NG MATE structures (174, 190, 191). Ligand-free pfMATE crystals grown under acidic conditions had a kinked TM1, while crystals ground under basic conditions had a straight TM1 (191). This pH-dependent kinking effect on TM1 was ultimately concluded to be a crystallization artifact, but several experimental observations still suggested that TM1 was important for transport (191, 198). Mutagenesis of P26 within TM1 virtually eliminated transport activity, but had little effect on the transporter structure (191). The outside-open and inside-open pfMATE structures pivoted around residue G30 of TM1 (191, 198). The importance of TM1 in substrate transport by pfMATE was further reinforced by MD simulations of the pfMATE with a “bent” TM1 showing a modeled Na+ ion stabilizing the kink in the helix (191, 198, 199). Based on these computational results, the residues near D41 on TM1 were designated as a cation (i.e., Na+/H+) binding site (198, 199). The MD simulations also revealed a chloride (Cl−) binding site (198). From this, the authors proposed that pfMATE might also function as a Cl−/Na+ antiporter (198). In addition, the substrate binding site of pfMATE was found within the NTD (191). In contrast, the inhibitor binding site was found in a central cavity between the NTD and CTD (191). Inhibition was proposed to restrict movement of the NTD and CTD domains of pfMATE during the transport cycle (191). The pfMATE transport cycle is likely a variant of the “alternating access” transport model, but details of the proton coupling process still need be resolved including the potential involvement of Na+ and Cl− ions (191, 198).
DinF MATE transporter from Bacillus (B.) halodurans (DinF-BH).
The DinF MATE DinF-BH is a H+-coupled antiporter that can translocate a diverse range of molecules, including ciprofloxacin, ethidium, and R6G (192). Several X-ray crystal structure investigations of DinF-BH were undertaken to unravel the transporter process (192, 196). All the DinF MATE structures were virtually identical with an outside-open MATE conformation (192). Notable structural features were a bend between TM7 and TM8 and a “straight” conformation between TM1 and TM2, which produces a larger pocket for the CTD than NTD (192). The central pocket between these domains were occupied by ligands in the X-ray crystal structures (192). The TM7 and TM8 helices produce a crevasse that was proposed to serve as a potential lateral entry point for the substrate and H+ (192). Mutagenesis and ligand affinity experiments with a highly conserved aspartate (D40) that overlaps with the substrate binding site suggested that D40 was a likely cation (proton) of the transporter (192, 196). The proposed transport model for DinF-BH resembles a “rocker-switch” variant of an “alternating access” transport mechanism but differs from other MATE transport models in that it involves direct competition between the substrate and cation binding (protonation) at D40 of DinF-BH, and the conformational transitions between outside-open and inside-open DinF-BH conformations (192).
DinF MATE Transporter from Vibrio (V.) cholerae (VcmN).
The DinF MATE transporter VcmN is an H+/substrate antiporter (200). X-ray crystallographic studies of the H+/substrate antiporters with pfMATE and DinF-BH were unable to provide a unifying molecular mechanism of H+-coupled substrate transport (e.g., 192, 198). To resolve a possible mechanism for H+-coupled substrate transport, several X-ray crystal structures were solved of VcmN (200). Like other MATE transporters, they all had “V-shaped” MATE folds with NTD and CTD (200). Like the original pfMATE X-ray crystal structures, the TM1 helix was “bent” under acidic crystallization conditions, and “straight” under basic crystallization conditions (191, 200). A conserved D35 is presumed to be the proton acceptor for VcmN, as suggested with other DinF MATE transporters (191, 192, 200). Based on electron density from monoolein, which is used in crystallization, the substrate binding site was proposed to reside within the NTD like pfMATE (191, 200).
Based on their results, they propose a model that is similar to the original transport model for pfMATE (191, 200). A substrate binds to VcmN with the TM1 in a “straight” conformation (200). Cation (H+) binding through protonation of D35 causes the bending of TM1 at P20, leading to the release of the substrate into the EC space (200). Without a substrate, the protonation of D35 induces a conformational change in the VcmN transporter from an outside-open to an inside-open conformation (200). While in the inside-open conformation, the proton is released into the cytosol (200). With the D35 of VcmN deprotonated and in an inside-open conformation, a substrate from the cytosol binds and shifts VcmN back to an outside-open conformation, and the process is repeated (200).
4. Summary and Future Prospects
Our structural understanding of drug transporters has increased dramatically over the past two decades with all the solved X-ray and cryo-EM structures within the PDB (201). In the beginning, the first structural information of ABC transporters came from the soluble NBDs (64). Later, more complete structures were solved of bacterial homologs of ABC transporters (65–68). Using nucleotide analogs, these structures were solved in different stages in the transport cycle (65–68). Soon after, analogous structures were solved of the mammalian and eukaryotic ABC transporters (72, 74, 104). The bacterial and mammalian structures were found in several distinct conformations suggesting conformational flexibility (65, 106). However, the structures reflect molecular snapshots of the transporters. Investigations using low-resolution cryo-EM and atomic force microscopy (AFM) of mammalian ABC transporters suggest that they are very conformationally dynamic and have many conformations (94, 95). This inherent dynamic behavior might explain why many ABC transporter structures in the PDB are relatively low resolution (>3 Å) (201). Increasing the resolution and capturing the transporter in a range in conformations will improve our molecular understanding of the drug transport process. X-ray and cryo-EM structures of drug transporters have been solved in the ABCB and ABC subfamilies. The only remaining subfamily that still have no drug transporting members is the ABCA subfamily (i.e., ABCA2 and ABCA3). The lipid exporter ABCA1 was recently solved by cryo-EM (36), so high-resolution structures of the drug transporting ABCA2 and ABCA3 are likely to be forthcoming.
We have made great progress in understanding the transport mechanism of members within the SLC superfamily, but many gaps in our knowledge still remain. All models of SLC transport involve some variant of the “alternating access” transport model (11, 12), despite significant differences in their protein folds (Fig. 4). The SLC1 family plays a critical role in transporting neurotransmitters and amino acids in the brain (133, 134). Unfortunately, X-ray crystallography of human EAAT1 (SLC1A3) required significant mutations for crystallization (136), so future structural studies with wild-type may be necessary to resolve its substrate specificity. Our current structural understanding of the SLC6 neurotransmitter antiporter family is incomplete. Members of the SLC6 neurotransmitter antiporter family have been solved from the AT, DAT and SERT subfamilies (e.g., 171, 202, 203), but the transporters in the CHT and NET subfamilies remain to be elucidated. For the SLC7 amino acid antiporter family, bacterial homologs of the CAT subfamily have been determined, and a human structure from the LAT subfamily has been solved (e.g., 204, 205). The mammalian CAT transporters are predicted to have two more TM helices and have soluble external domains that are absent in the bacterial transporters (147, 205). Solving mammalian CAT structures will aid in determining the function of these mammalian-specific features. In addition, solving the LAT1-4F2hc (SLC7A5-SLC3A2) heterodimer in additional conformations and in the absence of the 4F2hc (SLC3A2) will be helpful in defining its molecular transport mechanism and the role of the 4F2hc ectodomain in transport. Within the SLC10 bile acid symporter family, only bacterial homologs of the ASBT subfamily have been solved to date (e.g., 176), which have more TM helices than their mammalian counterparts. Future structural investigations of mammalian SLC10 transporters are needed to resolve these differences. Structural information of the SLC15 family comes from the bacterial PepT transporters and the murine EC-domains of PepT1 and PeptT2 (e.g., 206, 207), but the role of the mammalian EC-domains with respect to the TMD remains unclear. For the CNTs in the SLC28 transporter family, only the structure of the bacterial homolog is known, and it contains between 4 and 7 helices less than the mammalian CNTs, and they possess no outside soluble domains (60, 167, 208). Despite several conserved residues between the transporters (167), the mammalian CNT structures may have considerably different transport mechanisms than their bacterial counterparts. The hENT1 structure was the first mammalian structure solved from the SLC29 family (175) but has a <50% sequence identity with its members (156). Thus, structural studies of other ENT members may be necessary to resolve differences in their substrate specificity within the family. The bacterial MATE and the plant eMATE transporters have increased our understanding of the SLC47 family, but no mammalian transporters have been solved in this family, which may possess an extra TM helix and an EC domain (173). For the DinF H+/substrate antiporters of the SLC47 MATE transporter family, structural investigations lead to different conclusions about their transport mechanisms (191, 192, 198, 200). Additional structural investigations of the transporters at different pH’s may help clarify their transport mechanisms. A large gap in our knowledge of SLC transporters exists in the SLC22 transporter family that is comprised of OATs and OCTs because no structures have been solved within this group.
SLCO superfamily (formerly known as the SLC21 family) was not discussed in this book chapter because it is the only superfamily member that lacks a significant X-ray or a cryo-EM structure (25). This absence may end soon because of the exponential increase of X-ray crystal and cryo-EM structures (201). Although high-resolution X-ray crystal and cryo-EM structures have been extremely valuable and informative in elucidating transport mechanisms, a limitation of these techniques is that they often need to be stabilized in unnatural ways such as reductive methylation (209), antibody fragments (210, 211) and mutations (212) to produce high-resolution structures. To overcome these limitations, many recent structural studies also include biochemical or structural information from other techniques such as DEER (213). A multidisciplinary approach to structural studies is key to unraveling the complex molecular processes involved in drug transport.
5. Questions
-
5.1.
What family of ABC transporters has the topology of the NBD and the TMD in the reverse order from the ABC transporters? Hint: See Section 2.1
Short Answer: The ABCG family of transporters have topology in reverse order with the NBD first followed by the TMD, which includes BCRP.
5.2.The catalytic transport cycle of ABC transporters involves coupling of ATP hydrolysis to unidirectional transport of substrates. The ABC transporter structures solved in the presence of Mg2+ ATP suggest that substrate release occurs at what stage of the catalytic cycle? Hint: Review the conclusions for the transporters in Section 2.4.2
Short Answer: The Mg2+ ATP bound cryo-EM structures suggest that substrate release occurs before ATP hydrolysis.
5.3.What is the main transport mechanism for the SLC transporter superfamily? Hint: See Section 1.3
Short Answer: All the SLC transporters that have been solved to date are variants of the “alternating access” transport model.
6. Reference List
- 1.Giacomini KM (1997) Membrane transporters in drug disposition. J Pharmacokinet Biopharm 25:731–741 [DOI] [PubMed] [Google Scholar]
- 2.Giacomini KM, Huang S-M, and Tweedie DJ (2010) Membrane transporters in drug development. Nat Rev Drug Discov 9:215–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Girardin F (2006) Membrane transporter proteins: a challenge for CNS drug development. Dialogues Clin Neurosci 8:311–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petzinger E and Geyer J (2006) Drug transporters in pharmacokinetics. Naunyn Schmiedebergs Arch Pharmacol 372:465–475 [DOI] [PubMed] [Google Scholar]
- 5.Nigam SK (2015) What do drug transporters really do? Nat Rev Drug Discov 14:29–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liang Y, Li S, and Chen L (2015) The physiological role of drug transporters. Protein Cell 6:334–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vinothkumar KR and Henderson R (2010) Structures of membrane proteins. Q Rev Biophys 43:65–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lusvarghi S, Robey RW, Gottesman MM, et al. (2020) Multidrug transporters: recent insights from cryo-electron microscopy-derived atomic structures and animal models. F1000Research 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng H, Handing KB, Zimmerman MD, et al. (2015) X-ray crystallography over the past decade for novel drug discovery – where are we heading next? Expert Opin Drug Discov 10:975–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Szöllősi D, Rose-Sperling D, Hellmich UA, et al. (2018) Comparison of mechanistic transport cycle models of ABC exporters. Biochim Biophys Acta BBA - Biomembr 1860:818–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drew D and Boudker O (2016) Shared Molecular Mechanisms of Membrane Transporters. Annu Rev Biochem 85:543–572 [DOI] [PubMed] [Google Scholar]
- 12.Jardetzky O (1966) Simple Allosteric Model for Membrane Pumps. Nature 211:969–970 [DOI] [PubMed] [Google Scholar]
- 13.Colas C, Ung PM-U, and Schlessinger A (2016) SLC Transporters: Structure, Function, and Drug Discovery. MedChemComm 7:1069–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kühlbrandt W (2019) Structure and Mechanisms of F-Type ATP Synthases. Annu Rev Biochem 88:515–549 [DOI] [PubMed] [Google Scholar]
- 15.Dean M, Rzhetsky A, and Allikmets R (2001) The human ATP-binding cassette (ABC) transporter superfamily. Genome Res 11:1156–1166 [DOI] [PubMed] [Google Scholar]
- 16.Xiong J, Feng J, Yuan D, et al. (2015) Tracing the structural evolution of eukaryotic ATP binding cassette transporter superfamily. Sci Rep 5:16724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vasiliou V, Vasiliou K, and Nebert DW (2009) Human ATP-binding cassette (ABC) transporter family. Hum Genomics 3:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rees DC, Johnson E, and Lewinson O (2009) ABC transporters: The power to change. Nat Rev Mol Cell Biol 10:218–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He L, Vasiliou K, and Nebert DW (2009) Analysis and update of the human solute carrier (SLC) gene superfamily. Hum Genomics 3:195–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hagenbuch B and Stieger B (2013) The SLCO (former SLC21) superfamily of transporters. Mol Aspects Med 34:396–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schaller L and Lauschke VM (2019) The genetic landscape of the human solute carrier (SLC) transporter superfamily. Hum Genet 138:1359–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin L, Yee SW, Kim RB, et al. (2015) SLC Transporters as Therapeutic Targets: Emerging Opportunities. Nat Rev Drug Discov 14:543–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roth M, Obaidat A, and Hagenbuch B (2012) OATPs, OATs and OCTs: the organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br J Pharmacol 165:1260–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stieger B and Hagenbuch B (2014) Organic Anion Transporting Polypeptides. Curr Top Membr 73:205–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reiser J-B, Legoux F, Gras S, et al. (2014) Analysis of Relationships between Peptide/MHC Structural Features and Naive T Cell Frequency in Humans. J Immunol 193:5816–5826 [DOI] [PubMed] [Google Scholar]
- 26.Ryan RM and Vandenberg RJ (2016) Elevating the alternating-access model. Nat Struct Mol Biol 23:187–189 [DOI] [PubMed] [Google Scholar]
- 27.Kazmier K, Claxton DP, and Mchaourab HS (2017) Alternating access mechanisms of LeuT-Fold transporters: trailblazing towards the promised energy landscapes. Curr Opin Struct Biol 45:100–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Locher KP (2016) Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat Struct Mol Biol 23:487–493 [DOI] [PubMed] [Google Scholar]
- 29.Higgins CF and Linton KJ (2004) The ATP switch model for ABC transporters. Nat Struct Mol Biol 11:918–926 [DOI] [PubMed] [Google Scholar]
- 30.Senior AE, Shawi MK al-, and Urbatsch IL (1995) The catalytic cycle of P-glycoprotein. FEBS Lett 377:285–289 [DOI] [PubMed] [Google Scholar]
- 31.Sauna ZE and Ambudkar SV (2001) Characterization of the catalytic cycle of ATP hydrolysis by human P-glycoprotein. The two ATP hydrolysis events in a single catalytic cycle are kinetically similar but affect different functional outcomes. J Biol Chem 276:11653–11661 [DOI] [PubMed] [Google Scholar]
- 32.Siarheyeva A, Liu R, and Sharom FJ (2010) Characterization of an asymmetric occluded state of P-glycoprotein with two bound nucleotides: implications for catalysis. J Biol Chem 285:7575–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu Y, Seelig A, and Bernèche S (2017) Unidirectional Transport Mechanism in an ATP Dependent Exporter. ACS Cent Sci 3:250–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tusnády GE, Sarkadi B, Simon I, et al. (2006) Membrane topology of human ABC proteins. FEBS Lett 580:1017–1022 [DOI] [PubMed] [Google Scholar]
- 35.Klein I, Sarkadi B, and Váradi A (1999) An inventory of the human ABC proteins. Biochim Biophys Acta BBA - Biomembr 1461:237–262 [DOI] [PubMed] [Google Scholar]
- 36.Qian H, Zhao X, Cao P, et al. (2017) Structure of the Human Lipid Exporter ABCA1. Cell 169:1228–1239.e10 [DOI] [PubMed] [Google Scholar]
- 37.Albrecht C and Viturro E (2007) The ABCA subfamily—gene and protein structures, functions and associated hereditary diseases. Pflüg Arch - Eur J Physiol 453:581–589 [DOI] [PubMed] [Google Scholar]
- 38.Seguin A and Ward DM (2018) Mitochondrial ABC Transporters and Iron Metabolism. J Clin Exp Pathol 8:1–5 [Google Scholar]
- 39.Qu L, Jiang Y, Cheng C, et al. (2018) Crystal Structure of ATP-Bound Human ABCF1 Demonstrates a Unique Conformation of ABC Proteins. Structure 26:1259–1265.e3 [DOI] [PubMed] [Google Scholar]
- 40.Peterson E, Shippee E, Brinton MA, et al. (2019) Biochemical characterization of the mouse ABCF3 protein, a partner of the flavivirus-resistance protein OAS1B. J Biol Chem jbc.RA119.008477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kovalchuk A and Driessen AJ (2010) Phylogenetic analysis of fungal ABC transporters. BMC Genomics 11:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walker JE, Saraste M, Runswick MJ, et al. (1982) Distantly related sequences in the alpha-and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J 1:945–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rea PA (1999) MRP subfamily ABC transporters from plants and yeast. J Exp Bot 50:895–913 [Google Scholar]
- 44.Higgins CF (1992) ABC Transporters: From Microorganisms to Man. Annu Rev Cell Biol 8:67–113 [DOI] [PubMed] [Google Scholar]
- 45.Beek J ter, Guskov A, and Slotboom DJ (2014) Structural diversity of ABC transporters. J Gen Physiol 143:419–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Loo TW, Bartlett MC, and Clarke DM (2002) The “LSGGQ” motif in each nucleotide-binding domain of human P-glycoprotein is adjacent to the opposing walker A sequence. J Biol Chem 277:41303–6 [DOI] [PubMed] [Google Scholar]
- 47.Prasad R, Sharma M, and Rawal MK (2011) Functionally Relevant Residues of Cdr1p: A Multidrug ABC Transporter of Human Pathogenic Candida albicans. J Amino Acids 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Manolaridis I, Jackson SM, Taylor NMI, et al. (2018) Cryo-EM structures of a human ABCG2 mutant trapped in ATP-bound and substrate-bound states. Nature 563:426–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ambudkar SV, Kim I-W, Xia D, et al. (2006) The A-loop, a novel conserved aromatic acid subdomain upstream of the Walker A motif in ABC transporters, is critical for ATP binding. FEBS Lett 580:1049–1055 [DOI] [PubMed] [Google Scholar]
- 50.Wang F, Li G, Gu H, et al. (2013) Characterization of the Role of a Highly Conserved Sequence in ATP Binding Cassette Transporter G (ABCG) Family in ABCG1 Stability, Oligomerization, and Trafficking. Biochemistry 52:9497–9509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davidson AL, Dassa E, Orelle C, et al. (2008) Structure, Function, and Evolution of Bacterial ATP-Binding Cassette Systems. Microbiol Mol Biol Rev MMBR 72:317–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oswald C, Holland IB, and Schmitt L (2006) The motor domains of ABC-transporters. Naunyn Schmiedebergs Arch Pharmacol 372:385–399 [DOI] [PubMed] [Google Scholar]
- 53.Jones PM and George AM (2002) Mechanism of ABC transporters: A molecular dynamics simulation of a well characterized nucleotide-binding subunit. Proc Natl Acad Sci 99:12639–12644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De la Rosa MB and Nelson SW (2011) An Interaction between the Walker A and D-loop Motifs Is Critical to ATP Hydrolysis and Cooperativity in Bacteriophage T4 Rad50. J Biol Chem 286:26258–26266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bareil C, Thèze C, Béroud C, et al. (2010) UMD-CFTR: A database dedicated to CF and CFTR-related disorders. Hum Mutat 31:1011–1019 [DOI] [PubMed] [Google Scholar]
- 56.Zhou S-F (2008) Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition. Xenobiotica 38:802–832 [DOI] [PubMed] [Google Scholar]
- 57.Fersht A (2018) Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding, Kaissa Publications [Google Scholar]
- 58.Dall’Acqua W and Carter P (2000) Substrate-assisted catalysis: molecular basis and biological significance. Protein Sci Publ Protein Soc 9:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Geourjon C, Orelle C, Steinfels E, et al. (2001) A common mechanism for ATP hydrolysis in ABC transporter and helicase superfamilies. Trends Biochem Sci 26:539–544 [DOI] [PubMed] [Google Scholar]
- 60.Orelle C, Dalmas O, Gros P, et al. (2003) The Conserved Glutamate Residue Adjacent to the Walker-B Motif Is the Catalytic Base for ATP Hydrolysis in the ATP-binding Cassette Transporter BmrA. J Biol Chem 278:47002–47008 [DOI] [PubMed] [Google Scholar]
- 61.Oldham ML and Chen J (2011) Snapshots of the maltose transporter during ATP hydrolysis. Proc Natl Acad Sci 108:15152–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hanekop N, Zaitseva J, Jenewein S, et al. (2006) Molecular insights into the mechanism of ATP-hydrolysis by the NBD of the ABC-transporter HlyB. FEBS Lett 580:1036–1041 [DOI] [PubMed] [Google Scholar]
- 63.Zaitseva J, Jenewein S, Jumpertz T, et al. (2005) H662 is the linchpin of ATP hydrolysis in the nucleotide-binding domain of the ABC transporter HlyB. EMBO J 24:1901–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hung L-W, Wang IX, Nikaido K, et al. (1998) Crystal structure of the ATP-binding subunit of an ABC transporter. Nature 396:703–707 [DOI] [PubMed] [Google Scholar]
- 65.Ward A, Reyes CL, Yu J, et al. (2007) Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc Natl Acad Sci 104:19005–19010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dawson RJP, Hollenstein K, and Locher KP (2007) Uptake or extrusion: crystal structures of full ABC transporters suggest a common mechanism. Mol Microbiol 65:250–257 [DOI] [PubMed] [Google Scholar]
- 67.Dawson RJP and Locher KP (2006) Structure of a bacterial multidrug ABC transporter. Nature 443:180–185 [DOI] [PubMed] [Google Scholar]
- 68.Dawson RJP and Locher KP (2007) Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett 581:935–938 [DOI] [PubMed] [Google Scholar]
- 69.Sharom FJ (2014) Complex interplay between the P-glycoprotein multidrug efflux pump and the membrane: Its role in modulating protein function. Front Oncol 4:1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Esser L, Zhou F, Pluchino KM, et al. (2016) Structures of the multidrug transporter P-glycoprotein reveal asymmetric ATP binding and the mechanism of polyspecificity. J Biol Chem 292:446–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim Y and Chen J (2018) Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 359:915–919 [DOI] [PubMed] [Google Scholar]
- 72.Johnson ZL and Chen J (2017) Structural Basis of Substrate Recognition by the Multidrug Resistance Protein MRP1. Cell 168:1075–1085.e9 [DOI] [PubMed] [Google Scholar]
- 73.Johnson ZL and Chen J (2018) ATP Binding Enables Substrate Release from Multidrug Resistance Protein 1. Cell 172:81–89.e10 [DOI] [PubMed] [Google Scholar]
- 74.Taylor NMI, Manolaridis I, Jackson SM, et al. (2017) Structure of the human multidrug transporter ABCG2. Nature 546:504–509 [DOI] [PubMed] [Google Scholar]
- 75.Aberuyi N, Rahgozar S, Khosravi Dehaghi Z, et al. (2017) The translational expression of ABCA2 and ABCA3 is a strong prognostic biomarker for multidrug resistance in pediatric acute lymphoblastic leukemia. OncoTargets Ther 10:3373–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Steinbach D, Gillet J-P, Sauerbrey A, et al. (2006) ABCA3 as a Possible Cause of Drug Resistance in Childhood Acute Myeloid Leukemia. Clin Cancer Res 12:4357–4363 [DOI] [PubMed] [Google Scholar]
- 77.Chapuy B, Koch R, Radunski U, et al. (2008) Intracellular ABC transporter A3 confers multidrug resistance in leukemia cells by lysosomal drug sequestration. Leukemia 22:1576–1586 [DOI] [PubMed] [Google Scholar]
- 78.Glavinas H, Krajcsi P, Cserepes J, et al. (2004) The role of ABC transporters in drug resistance, metabolism and toxicity. Curr Drug Deliv 1:27–42 [DOI] [PubMed] [Google Scholar]
- 79.Lehnert E and Tampé R (2017) Structure and Dynamics of Antigenic Peptides in Complex with TAP. Front Immunol 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moitra K, Scally M, McGee K, et al. (2011) Molecular Evolutionary Analysis of ABCB5: The Ancestral Gene Is a Full Transporter with Potentially Deleterious Single Nucleotide Polymorphisms. PLoS One 6:e16318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Oude Elferink RPJ and Paulusma CC (2007) Function and pathophysiological importance of ABCB4 (MDR3 P-glycoprotein). Pflüg Arch - Eur J Physiol 453:601–610 [DOI] [PubMed] [Google Scholar]
- 82.Zhao C, Tampé R, and Abele R (2006) TAP and TAP-like — Brothers in arms? Naunyn Schmiedebergs Arch Pharmacol 372:444–450 [DOI] [PubMed] [Google Scholar]
- 83.Przybylla S, Stindt J, Kleinschrodt D, et al. (2016) Analysis of the Bile Salt Export Pump (ABCB11) Interactome Employing Complementary Approaches. PLoS One 11:e0159778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Roy U, Barber P, Tse Dinh Y ching, et al. (2015) Role of MRP transporters in regulating antimicrobial drug inefficacy and oxidative stress-induced pathogenesis during HIV-1 and TB infections. Front Microbiol 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kawaguchi K and Morita M (2016) ABC Transporter Subfamily D: Distinct Differences in Behavior between ABCD1–3 and ABCD4 in Subcellular Localization, Function, and Human Disease. BioMed Res Int 2016:e6786245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen Z, Dong J, Ishimura A, et al. (2006) The Essential Vertebrate ABCE1 Protein Interacts with Eukaryotic Initiation Factors. J Biol Chem 281:7452–7457 [DOI] [PubMed] [Google Scholar]
- 87.Murina V, Kasari M, Takada H, et al. (2019) ABCF ATPases Involved in Protein Synthesis, Ribosome Assembly and Antibiotic Resistance: Structural and Functional Diversification across the Tree of Life. J Mol Biol 431:3568–3590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Moitra K, Silverton L, Limpert K, et al. (2011) Moving out: from sterol transport to drug resistance – the ABCG subfamily of efflux pumps. Drug Metab Pers Ther 26:105–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Melaine N, Liénard M-O, Dorval I, et al. (2002) Multidrug Resistance Genes and P-Glycoprotein in the Testis of the Rat, Mouse, Guinea Pig, and Human. Biol Reprod 67:1699–1707 [DOI] [PubMed] [Google Scholar]
- 90.Chapy H, Saubaméa B, Tournier N, et al. (2016) Blood–brain and retinal barriers show dissimilar ABC transporter impacts and concealed effect of P-glycoprotein on a novel verapamil influx carrier. Br J Pharmacol 173:497–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mölsä M, Heikkinen T, Hakkola J, et al. (2005) Functional role of P-glycoprotein in the human blood-placental barrier. Clin Pharmacol Ther 78:123–131 [DOI] [PubMed] [Google Scholar]
- 92.Ceckova-Novotna M, Pavek P, and Staud F (2006) P-glycoprotein in the placenta: expression, localization, regulation and function. Reprod Toxicol 22:400–10 [DOI] [PubMed] [Google Scholar]
- 93.Robinson K and Tiriveedhi V (2020) Perplexing Role of P-Glycoprotein in Tumor Microenvironment. Front Oncol 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Frank GA, Shukla S, Rao P, et al. (2016) Cryo-EM analysis of the conformational landscape of human P-glycoprotein (ABCB1) during its catalytic cycle. Mol Pharmacol 90:35–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sigdel KP, Wilt LA, Marsh BP, et al. (2018) The conformation and dynamics of P-glycoprotein in a lipid bilayer investigated by atomic force microscopy. Biochem Pharmacol 156:302–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hrycyna CA, Airan LE, Germann UA, et al. (1998) Structural flexibility of the linker region of human P-glycoprotein permits ATP hydrolysis and drug transport. Biochemistry 37:13660–73 [DOI] [PubMed] [Google Scholar]
- 97.Tombline G, Bartholomew LA, Urbatsch IL, et al. (2004) Combined mutation of catalytic glutamate residues in the two nucleotide binding domains of P-glycoprotein generates a conformation that binds ATP and ADP tightly. J Biol Chem 279:31212–20 [DOI] [PubMed] [Google Scholar]
- 98.Sauna ZE, Kim I-W, Nandigama K, et al. (2007) Catalytic Cycle of ATP Hydrolysis by P-Glycoprotein: Evidence for Formation of the E·S Reaction Intermediate with ATP-γ-S, a Nonhydrolyzable Analogue of ATP. Biochemistry 46:13787–13799 [DOI] [PubMed] [Google Scholar]
- 99.Kimura Y, Kioka N, Kato H, et al. (2007) Modulation of drug-stimulated ATPase activity of human MDR1/P-glycoprotein by cholesterol. Biochem J 401:597–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Clay AT and Sharom FJ (2013) Lipid bilayer properties control membrane partitioning, binding, and transport of P-Glycoprotein substrates. Biochemistry 52:343–354 [DOI] [PubMed] [Google Scholar]
- 101.Clay AT, Lu P, and Sharom FJ (2015) Interaction of the P-Glycoprotein Multidrug Transporter with Sterols. Biochemistry 54:6586–6597 [DOI] [PubMed] [Google Scholar]
- 102.Loo TW and Clarke DM (2016) P-glycoprotein ATPase activity requires lipids to activate a switch at the first transmission interface. Biochem Biophys Res Commun 472:379–383 [DOI] [PubMed] [Google Scholar]
- 103.Marcoux J, Wang SC, Politis A, et al. (2013) Mass spectrometry reveals synergistic effects of nucleotides, lipids, and drugs binding to a multidrug resistance efflux pump. Proc Natl Acad Sci 110:9704–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Aller SG, Yu J, Ward A, et al. (2009) Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323:1718–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Li J, Jaimes KF, and Aller SG (2014) Refined structures of mouse P-glycoprotein. Protein Sci 23:34–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ward AB, Szewczyk P, Grimard V, et al. (2013) Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc Natl Acad Sci 110:13386–13391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jin MS, Oldham ML, Zhang Q, et al. (2012) Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature 490:566–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Thonghin N, Collins RF, Barbieri A, et al. (2018) Novel features in the structure of P-glycoprotein (ABCB1) in the post-hydrolytic state as determined at 7.9 Å resolution. BMC Struct Biol 18:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Alam A, Küng R, Kowal J, et al. (2018) Structure of a zosuquidar and UIC2-bound human-mouse chimeric ABCB1. Proc Natl Acad Sci 115:E1973–E1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Szewczyk P, Tao H, McGrath AP, et al. (2015) Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr D 71:732–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Alam A, Kowal J, Broude E, et al. (2019) Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 363:753–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cole SPC (2014) Multidrug Resistance Protein 1 (MRP1, ABCC1), a “Multitasking” ATP-binding Cassette (ABC) Transporter. J Biol Chem 289:30880–30888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jiye Y and Jianting Z (2011) Multidrug resistance-associated protein 1 (MRP1/ABCC1) polymorphism: from discovery to clinical application. Zhong Nan Da Xue Xue Bao Yi Xue Ban 36:927–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bakos E, Evers R, Szakacs G, et al. (1998) Functional multidrug resistance protein (MRP1) lacking the N-terminal transmembrane domain. J Biol Chem 273:32167–75 [DOI] [PubMed] [Google Scholar]
- 115.Westlake CJ, Cole SPC, and Deeley RG (2005) Role of the NH2-terminal Membrane Spanning Domain of Multidrug Resistance Protein 1/ABCC1 in Protein Processing and Trafficking. Mol Biol Cell 16:2483–2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chaptal V,S, Magnard R, et al. (2014) Nucleotide-Free Crystal Structure of Nucleotide-Binding Domain 1 from Human Abcc1 Supports a “General-Base Catalysis” Mechanism for ATP Hydrolysis. Biochem Pharmacol Open Access 3:1–4 [Google Scholar]
- 117.Ramaen O, Leulliot N, Sizun C, et al. (2006) Structure of the Human Multidrug Resistance Protein 1 Nucleotide Binding Domain 1 bound to Mg2+/ATP Reveals a Non-productive Catalytic Site. J Mol Biol 359:940–949 [DOI] [PubMed] [Google Scholar]
- 118.Rosenberg MF, Oleschuk CJ, Wu P, et al. (2010) Structure of a human multidrug transporter in an inward-facing conformation. J Struct Biol 170:540–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mao Q and Unadkat JD (2014) Role of the Breast Cancer Resistance Protein (BCRP/ABCG2) in Drug Transport—an Update. AAPS J 17:65–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Faneyte IF, Kristel PMP, Maliepaard M, et al. (2002) Expression of the Breast Cancer Resistance Protein in Breast Cancer. Clin Cancer Res 8:1068–1074 [PubMed] [Google Scholar]
- 121.Natarajan K, Xie Y, Baer MR, et al. (2012) Role of Breast Cancer Resistance Protein (BCRP/ABCG2) in Cancer Drug Resistance. Biochem Pharmacol 83:1084–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Telbisz Á, Hegedüs C, Váradi A, et al. (2014) Regulation of the Function of the Human ABCG2 Multidrug Transporter by Cholesterol and Bile Acids: Effects of Mutations in Potential Substrate and Steroid Binding Sites. Drug Metab Dispos 42:575–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Janvilisri T, Venter H, Shahi S, et al. (2003) Sterol Transport by the Human Breast Cancer Resistance Protein (ABCG2) Expressed in Lactococcus lactis. J Biol Chem 278:20645–20651 [DOI] [PubMed] [Google Scholar]
- 124.Pál Á, Méhn D, Molnár É, et al. (2007) Cholesterol Potentiates ABCG2 Activity in a Heterologous Expression System: Improved in Vitro Model to Study Function of Human ABCG2. J Pharmacol Exp Ther 321:1085–1094 [DOI] [PubMed] [Google Scholar]
- 125.Baldán Á, Bojanic DD, and Edwards PA (2009) The ABCs of sterol transport. J Lipid Res 50:S80–S85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Horsey AJ, Cox MH, Sarwat S, et al. (2016) The multidrug transporter ABCG2: still more questions than answers. Biochem Soc Trans 44:824–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Diop NK and Hrycyna CA (2005) N-Linked Glycosylation of the Human ABC Transporter ABCG2 on Asparagine 596 Is Not Essential for Expression, Transport Activity, or Trafficking to the Plasma Membrane. Biochemistry 44:5420–5429 [DOI] [PubMed] [Google Scholar]
- 128.Nakagawa H, Wakabayashi-Nakao K, Tamura A, et al. (2009) Disruption of N-linked glycosylation enhances ubiquitin-mediated proteasomal degradation of the human ATP-binding cassette transporter ABCG2. FEBS J 276:7237–7252 [DOI] [PubMed] [Google Scholar]
- 129.Jackson SM, Manolaridis I, Kowal J, et al. (2018) Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat Struct Mol Biol 25:333–340 [DOI] [PubMed] [Google Scholar]
- 130.Allen JD, Loevezijn A van, Lakhai JM, et al. (2002) Potent and Specific Inhibition of the Breast Cancer Resistance Protein Multidrug Transporter in Vitro and in Mouse Intestine by a Novel Analogue of Fumitremorgin C 1 This work was supported in part by grant NKI 97–1433 from the Dutch Cancer Society (to A. H. S.). Synthesis investigations by A. v. L. and G-J. K. were supported by the Netherlands Research Council for Chemical Sciences (NWO/CW) and the Netherlands Technology Foundation (STW).1. Mol Cancer Ther 1:417–425 [PubMed] [Google Scholar]
- 131.Fox E and Bates SE (2007) Tariquidar (XR9576): a P-glycoprotein drug efflux pump inhibitor. Expert Rev Anticancer Ther 7:447–59 [DOI] [PubMed] [Google Scholar]
- 132.Kannan P, Telu S, Shukla S, et al. (2011) The “specific” P-glycoprotein inhibitor Tariquidar is also a substrate and an inhibitor for breast cancer resistance protein (BCRP/ABCG2). ACS Chem Neurosci 2:82–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Grewer C, Gameiro A, and Rauen T (2014) SLC1 Glutamate Transporters. Pflugers Arch 466:3–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gether U, Andersen PH, Larsson OM, et al. (2006) Neurotransmitter transporters: molecular function of important drug targets. Trends Pharmacol Sci 27:375–383 [DOI] [PubMed] [Google Scholar]
- 135.Cha YJ, Kim E-S, and Koo JS (2018) Amino Acid Transporters and Glutamine Metabolism in Breast Cancer. Int J Mol Sci 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Canul-Tec JC, Assal R, Cirri E, et al. (2017) Structure and allosteric inhibition of excitatory amino acid transporter 1. Nature 544:446–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hu C, Tao L, Cao X, et al. (2020) The solute carrier transporters and the brain: Physiological and pharmacological implications. Asian J Pharm Sci 15:131–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wahi K and Holst J (2019) ASCT2: a potential cancer drug target. Expert Opin Ther Targets 23:555–558 [DOI] [PubMed] [Google Scholar]
- 139.Liu Y, Zhao T, Li Z, et al. (2018) The role of ASCT2 in cancer: A review. Eur J Pharmacol 837:81–87 [DOI] [PubMed] [Google Scholar]
- 140.Rudnick G, Krämer R, Blakely RD, et al. (2014) The SLC6 transporters: perspectives on structure, functions, regulation, and models for transporter dysfunction. Pflugers Arch 466:25–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bröer S and Gether U (2012) The solute carrier 6 family of transporters. Br J Pharmacol 167:256–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Pramod AB, Foster J, Carvelli L, et al. (2013) SLC6 transporters: Structure, function, regulation, disease association and therapeutics. Mol Aspects Med 34:197–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Murphy DL, Fox MA, Timpano KR, et al. (2008) How the Serotonin Story is Being Rewritten By New Gene-Based Discoveries Principally Related to SLC6A4, the Serotonin Transporter Gene, Which Functions To Influence All Cellular Serotonin Systems. Neuropharmacology 55:932–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhu J, Klein-Fedyshin M, and Stevenson JM (2017) Serotonin Transporter Gene Polymorphisms and Selective Serotonin Reuptake Inhibitor Tolerability: Review of Pharmacogenetic Evidence. Pharmacother J Hum Pharmacol Drug Ther 37:1089–1104 [DOI] [PubMed] [Google Scholar]
- 145.Scalise M, Pochini L, Galluccio M, et al. (2020) Glutamine transporters as pharmacological targets: From function to drug design. Asian J Pharm Sci 15:207–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Cheng Q, Shah N, Bröer A, et al. (2017) Identification of novel inhibitors of the amino acid transporter B0AT1 (SLC6A19), a potential target to induce protein restriction and to treat type 2 diabetes. Br J Pharmacol 174:468–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Fotiadis D, Kanai Y, and Palacín M (2013) The SLC3 and SLC7 families of amino acid transporters. Mol Aspects Med 34:139–158 [DOI] [PubMed] [Google Scholar]
- 148.Puris E, Gynther M, Auriola S, et al. (2020) L-Type amino acid transporter 1 as a target for drug delivery. Pharm Res 37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Uchino H, Kanai Y, Kim DK, et al. (2002) Transport of Amino Acid-Related Compounds Mediated by L-Type Amino Acid Transporter 1 (LAT1): Insights Into the Mechanisms of Substrate Recognition. Mol Pharmacol 61:729–737 [DOI] [PubMed] [Google Scholar]
- 150.Dickens D, Webb SD, Antonyuk S, et al. (2013) Transport of gabapentin by LAT1 (SLC7A5). Biochem Pharmacol 85:1672–1683 [DOI] [PubMed] [Google Scholar]
- 151.Geyer J, Wilke T, and Petzinger E (2006) The solute carrier family SLC10: more than a family of bile acid transporters regarding function and phylogenetic relationships. Naunyn Schmiedebergs Arch Pharmacol 372:413–431 [DOI] [PubMed] [Google Scholar]
- 152.Silva TC da Polli JE, and Swaan PW (2013) The solute carrier family 10 (SLC10): beyond bile acid transport. Mol Aspects Med 34:252–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Döring B, Lütteke T, Geyer J, et al. (2012) Chapter Four - The SLC10 Carrier Family: Transport Functions and Molecular Structure, In: Bevensee MO (ed.) Current Topics in Membranes, pp. 105–168 Academic Press; [DOI] [PubMed] [Google Scholar]
- 154.Smith DE, Clémençon B, and Hediger MA (2013) Proton-coupled oligopeptide transporter family SLC15: Physiological, pharmacological and pathological implications. Mol Aspects Med 34:323–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Nigam SK (2018) The SLC22 Transporter Family: A Paradigm for the Impact of Drug Transporters on Metabolic Pathways, Signaling, and Disease. Annu Rev Pharmacol Toxicol 58:663–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Baldwin SA, Beal PR, Yao SYM, et al. (2004) The equilibrative nucleoside transporter family, SLC29. Pflüg Arch 447:735–743 [DOI] [PubMed] [Google Scholar]
- 157.Boswell-Casteel RC and Hays FA (2017) Equilibrative Nucleoside Transporters – A Review. Nucleosides Nucleotides Nucleic Acids 36:7–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Young JD, Yao SYM, Sun L, et al. (2008) Human equilibrative nucleoside transporter (ENT) family of nucleoside and nucleobase transporter proteins. Xenobiotica 38:995–1021 [DOI] [PubMed] [Google Scholar]
- 159.Pastor-Anglada M and Pérez-Torras S (2018) Emerging Roles of Nucleoside Transporters. Front Pharmacol 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Parker JL and Newstead S (2019) Gateway to the Golgi: Molecular mechanisms of nucleotide sugar transporters. Curr Opin Struct Biol 57:127–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hadley B, Litfin T, Day CJ, et al. (2019) Nucleotide Sugar Transporter SLC35 Family Structure and Function. Comput Struct Biotechnol J 17:1123–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tanihara Y, Masuda S, Sato T, et al. (2007) Substrate specificity of MATE1 and MATE2-K, human multidrug and toxin extrusions/H+-organic cation antiporters. Biochem Pharmacol 74:359–371 [DOI] [PubMed] [Google Scholar]
- 163.Nies AT, Damme K, Kruck S, et al. (2016) Structure and function of multidrug and toxin extrusion proteins (MATEs) and their relevance to drug therapy and personalized medicine. Arch Toxicol 90:1555–1584 [DOI] [PubMed] [Google Scholar]
- 164.Jiang J and Amara SG (2011) New views of glutamate transporter structure and function: advances and challenges. Neuropharmacology 60:172–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Padan E and Michel H (2015) NhaA: A Unique Structural Fold of Secondary Active Transporters. Isr J Chem 55:1233–1239 [Google Scholar]
- 166.Shi Y (2013) Common Folds and Transport Mechanisms of Secondary Active Transporters. Annu Rev Biophys 42:51–72 [DOI] [PubMed] [Google Scholar]
- 167.Johnson ZL, Cheong C-G, and Lee S-Y (2012) Crystal structure of a concentrative nucleoside transporter from Vibrio cholerae at 2.4 Å. Nature 483:489–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Garaeva AA, Oostergetel GT, Gati C, et al. (2018) Cryo-EM structure of the human neutral amino acid transporter ASCT2. Nat Struct Mol Biol 25:515–521 [DOI] [PubMed] [Google Scholar]
- 169.Reyes N, Ginter C, and Boudker O (2009) Transport mechanism of a bacterial homologue of glutamate transporters. Nature 462:880–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Yernool D, Boudker O, Jin Y, et al. (2004) Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature 431:811–818 [DOI] [PubMed] [Google Scholar]
- 171.Coleman JA, Green EM, and Gouaux E (2016) X-ray structures and mechanism of the human serotonin transporter. Nature 532:334–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Yan R, Zhao X, Lei J, et al. (2019) Structure of the human LAT1-4F2hc heteromeric amino acid transporter complex. Nature 568:127–130 [DOI] [PubMed] [Google Scholar]
- 173.Zhang X and Wright SH (2009) MATE1 has an external COOH terminus, consistent with a 13-helix topology. Am J Physiol-Ren Physiol 297:F263–F271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.He X, Szewczyk P, Karyakin A, et al. (2010) Structure of a cation-bound multidrug and toxic compound extrusion transporter. Nature 467:991–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wright NJ and Lee S-Y (2019) Structures of human ENT1 in complex with adenosine reuptake inhibitors. Nat Struct Mol Biol 26:599–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hu N-J, Iwata S, Cameron AD, et al. (2011) Crystal structure of a bacterial homologue of the bile acid sodium symporter ASBT. Nature 478:408–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Pelis RM and Wright SH (2014) Chapter Six - SLC22, SLC44, and SLC47 Transporters—Organic Anion and Cation Transporters: Molecular and Cellular Properties, In: Bevensee MO (ed.) Current Topics in Membranes, pp. 233–261 Academic Press; [DOI] [PubMed] [Google Scholar]
- 178.Otsuka M, Matsumoto T, Morimoto R, et al. (2005) A human transporter protein that mediates the final excretion step for toxic organic cations. Proc Natl Acad Sci 102:17923–17928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Motohashi H, Nakao Y, Masuda S, et al. (2013) Precise comparison of protein localization among OCT, OAT, and MATE in human kidney. J Pharm Sci 102:3302–3308 [DOI] [PubMed] [Google Scholar]
- 180.Masuda S, Terada T, Yonezawa A, et al. (2006) Identification and functional characterization of a new human kidney-specific H+/organic cation antiporter, kidney-specific multidrug and toxin extrusion 2. J Am Soc Nephrol JASN 17:2127–2135 [DOI] [PubMed] [Google Scholar]
- 181.A Y and K I (2011) Importance of the multidrug and toxin extrusion MATE/SLC47A family to pharmacokinetics, pharmacodynamics/toxicodynamics and pharmacogenomics. Br J Pharmacol 164:1817–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Li Q and Shu Y (2014) Role of solute carriers in response to anticancer drugs. Mol Cell Ther 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Yang H, Guo D, Obianom ON, et al. (2017) Multidrug and Toxin Extrusion Proteins Mediate Cellular Transport of Cadmium. Toxicol Appl Pharmacol 314:55–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Chowdhury S, Yung E, Pintilie M, et al. (2016) MATE2 Expression Is Associated with Cancer Cell Response to Metformin. PLoS One 11:e0165214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Omote H, Hiasa M, Matsumoto T, et al. (2006) The MATE proteins as fundamental transporters of metabolic and xenobiotic organic cations. Trends Pharmacol Sci 27:587–593 [DOI] [PubMed] [Google Scholar]
- 186.Morita Y, Kataoka A, Shiota S, et al. (2000) NorM of Vibrio parahaemolyticus Is an Na+-Driven Multidrug Efflux Pump. J Bacteriol 182:6694–6697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Brown MH, Paulsen IT, and Skurray RA (1999) The multidrug efflux protein NorM is a prototype of a new family of transporters. Mol Microbiol 31:394–395 [DOI] [PubMed] [Google Scholar]
- 188.Kuroda T and Tsuchiya T (2009) Multidrug efflux transporters in the MATE family. Biochim Biophys Acta BBA - Proteins Proteomics 1794:763–768 [DOI] [PubMed] [Google Scholar]
- 189.Kusakizako T, Miyauchi H, Ishitani R, et al. (2019) Structural biology of the multidrug and toxic compound extrusion superfamily transporters. Biochim Biophys Acta BBA -Biomembr 183154 [DOI] [PubMed] [Google Scholar]
- 190.Lu M, Symersky J, Radchenko M, et al. (2013) Structures of a Na+-coupled, substrate-bound MATE multidrug transporter. Proc Natl Acad Sci 110:2099–2104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Tanaka Y, Hipolito CJ, Maturana AD, et al. (2013) Structural basis for the drug extrusion mechanism by a MATE multidrug transporter. Nature 496:247–251 [DOI] [PubMed] [Google Scholar]
- 192.Lu M, Radchenko M, Symersky J, et al. (2013) Structural insights into H+-coupled multidrug extrusion by a MATE transporter. Nat Struct Mol Biol 20:1310–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Long F, Rouquette-Loughlin C, Shafer WM, et al. (2008) Functional Cloning and Characterization of the Multidrug Efflux Pumps NorM from Neisseria gonorrhoeae and YdhE from Escherichia coli. Antimicrob Agents Chemother 52:3052–3060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Claxton DP, Jagessar KL, Steed PR, et al. (2018) Sodium and proton coupling in the conformational cycle of a MATE antiporter from Vibrio cholerae. Proc Natl Acad Sci U S A 115:E6182–E6190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Jin Y, Nair A, and Veen HW van (2014) Multidrug Transport Protein NorM from Vibrio cholerae Simultaneously Couples to Sodium- and Proton-Motive Force. J Biol Chem 289:14624–14632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Radchenko M, Symersky J, Nie R, et al. (2015) Structural basis for the blockade of MATE multidrug efflux pumps. Nat Commun 6:7995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Jagessar KL, Mchaourab HS, and Claxton DP (2019) The N-terminal domain of an archaeal multidrug and toxin extrusion (MATE) transporter mediates proton coupling required for prokaryotic drug resistance. J Biol Chem jbc.RA119.009195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Zakrzewska S, Mehdipour AR, Malviya VN, et al. (2019) Inward-facing conformation of a multidrug resistance MATE family transporter. Proc Natl Acad Sci 116:12275–12284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ficici E, Zhou W, Castellano S, et al. (2018) Broadly conserved Na+-binding site in the N-lobe of prokaryotic multidrug MATE transporters. Proc Natl Acad Sci U S A 115:E6172–E6181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kusakizako T, Claxton DP, Tanaka Y, et al. (2019) Structural Basis of H+-Dependent Conformational Change in a Bacterial MATE Transporter. Structure 27:293–301.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Berman HM, Westbrook J, Feng Z, et al. (2000) The protein data bank. Nucleic Acids Res 28:235–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Penmatsa A, Wang KH, and Gouaux E (2013) X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 503:85–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Yan R, Zhang Y, Li Y, et al. (2020) Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367:1444–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lee Y, Wiriyasermkul P, Jin C, et al. (2019) Cryo-EM structure of the human L-type amino acid transporter 1 in complex with glycoprotein CD98hc. Nat Struct Mol Biol 26:510–517 [DOI] [PubMed] [Google Scholar]
- 205.Jungnickel KEJ, Parker JL, and Newstead S (2018) Structural basis for amino acid transport by the CAT family of SLC7 transporters. Nat Commun 9:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Ural-Blimke Y, Flayhan A, Strauss J, et al. (2019) Structure of Prototypic Peptide Transporter DtpA from E. coli in Complex with Valganciclovir Provides Insights into Drug Binding of Human PepT1. J Am Chem Soc 141:2404–2412 [DOI] [PubMed] [Google Scholar]
- 207.Beale JH, Parker JL, Samsudin F, et al. (2015) Crystal Structures of the Extracellular Domain from PepT1 and PepT2 Provide Novel Insights into Mammalian Peptide Transport. Structure 23:1889–1899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hamilton SR, Yao SYM, Ingram JC, et al. (2001) Subcellular Distribution and Membrane Topology of the Mammalian Concentrative Na+-Nucleoside Cotransporter rCNT1. J Biol Chem 276:27981–27988 [DOI] [PubMed] [Google Scholar]
- 209.Rypniewski WR, Holden HM, and Rayment I (1993) Structural consequences of reductive methylation of lysine residues in hen egg white lysozyme: An x-ray analysis at 1.8-.ANG. resolution. Biochemistry 32:9851–9858 [DOI] [PubMed] [Google Scholar]
- 210.Griffin L and Lawson A (2011) Antibody fragments as tools in crystallography. Clin Exp Immunol 165:285–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Brown ZP and Takagi J (2019) Advances in domain and subunit localization technology for electron microscopy. Biophys Rev 11:149–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Price SR and Nagai K (1995) Protein engineering as a tool for crystallography. Curr Opin Biotechnol 6:425–430 [DOI] [PubMed] [Google Scholar]
- 213.Alonso-García N, García-Rubio I, Manso JA, et al. (2015) Combination of X-ray crystallography, SAXS and DEER to obtain the structure of the FnIII-3,4 domains of integrin α6β4. Acta Crystallogr D 71:969–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Yu X, Plotnikova O, Bonin PD, et al. (2019) Cryo-EM structures of the human glutamine transporter SLC1A5 (ASCT2) in the outward-facing conformation. eLife 8:e48120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Minhas GS, Bawdon D, Herman R, et al. (2018) Structural basis of malodour precursor transport in the human axilla. eLife 7:e34995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Nicklisch SC, Rees SD, McGrath AP, et al. (2016) Global marine pollutants inhibit P-glycoprotein: Environmental levels, inhibitory effects, and cocrystal structure. Sci Adv 2:e1600001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Le CA, Harvey DS, and Aller SG Structural definition of polyspecific compensatory ligand recognition by P-glycoprotein. IUCrJ In Press; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Ran Y, Zheng A, and Thibodeau PH (2018) Structural analysis reveals pathomechanisms associated with pseudoxanthoma elasticum–causing mutations in the ABCC6 transporter. J Biol Chem 293:15855–15866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Garaeva AA, Guskov A, Slotboom DJ, et al. (2019) A one-gate elevator mechanism for the human neutral amino acid transporter ASCT2. Nat Commun 10:3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Wang KH, Penmatsa A, and Gouaux E (2015) Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature 521:322–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Coleman JA and Gouaux E (2018) Structural basis for recognition of diverse antidepressants by the human serotonin transporter. Nat Struct Mol Biol 25:170–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Coleman JA, Yang D, Zhao Z, et al. (2019) Serotonin transporter–ibogaine complexes illuminate mechanisms of inhibition and transport. Nature 569:141–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Errasti-Murugarren E, Fort J, Bartoccioni P, et al. (2019) L amino acid transporter structure and molecular bases for the asymmetry of substrate interaction. Nat Commun 10:1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Fort J, Ballina LR de la, Burghardt HE, et al. (2007) The Structure of Human 4F2hc Ectodomain Provides a Model for Homodimerization and Electrostatic Interaction with Plasma Membrane. J Biol Chem 282:31444–31452 [DOI] [PubMed] [Google Scholar]
- 225.Zhou X, Levin EJ, Pan Y, et al. (2014) Structural basis of the alternating-access mechanism in a bile acid transporter. Nature 505:569–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Minhas GS and Newstead S (2019) Structural basis for prodrug recognition by the SLC15 family of proton-coupled peptide transporters. Proc Natl Acad Sci 116:804–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Johnson ZL, Lee J-H, Lee K, et al. (2014) Structural basis of nucleoside and nucleoside drug selectivity by concentrative nucleoside transporters. eLife 3:e03604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Miyauchi H, Moriyama S, Kusakizako T, et al. (2017) Structural basis for xenobiotic extrusion by eukaryotic MATE transporter. Nat Commun 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Tanaka Y, Iwaki S, and Tsukazaki T (2017) Crystal Structure of a Plant Multidrug and Toxic Compound Extrusion Family Protein. Structure 25:1455–1460.e2 [DOI] [PubMed] [Google Scholar]