

Abstract
Atherosclerotic plaques exhibit high deposition of cholesterol and macrophages. These are not only the main components of the plaques but also key inflammation-triggering sources. However, no existing therapeutics can achieve effective removal of both components within the plaques. Here, we report cargo-switching nanoparticles (CSNP) that are physicochemically designed to bind to cholesterol and release anti-inflammatory drug in the plaque microenvironment. CSNP have a core–shell structure with a core composed of an inclusion complex of methyl-β-cyclodextrin (cyclodextrin) and simvastatin (statin), and a shell of phospholipids. Upon interaction with cholesterol, which has higher affinity to cyclodextrin than statin, CSNP release statin and scavenge cholesterol instead through cargo-switching. CSNP exhibit cholesterol-sensitive multifaceted antiatherogenic functions attributed to statin release and cholesterol depletion in vitro. In mouse models of atherosclerosis, systemically injected CSNP target atherosclerotic plaques and reduce plaque content of cholesterol and macrophages, which synergistically leads to effective prevention of atherogenesis and regression of established plaques. These findings suggest that CSNP provide a therapeutic platform for interfacing with cholesterol-associated inflammatory diseases such as atherosclerosis.
Keywords: atherosclerosis, cargo-switching, nanoparticle, cholesterol, microenvironment
Graphical Abstract
Progression of atherosclerosis is deeply associated with the accumulation of cholesterol and macrophages in the arterial wall, which results in the formation of cholesterol crystals (CC) and foamy macrophages.1–4 The presence of cholesterol crystals and foamy macrophages induces inflammation and promotes further accumulation thereof within plaques,5–8 establishing a vicious positive feedback loop. Therefore, effective removal of both components within plaques would provide significant therapeutic benefits. The advance in nanotechnology has provided chances to improve management of atherosclerosis via efficient delivery of a drug to atherosclerotic plaques, overcoming biological barriers and improving the therapeutic index of a drug.9–12 However, no existing drug delivery system has yet to achieve effective intervention on the key inflammatory sources within plaques.
Statins are widely used as cholesterol-lowering drugs to prevent and manage cardiovascular and coronary heart diseases.13 In addition, the cholesterol-independent or pleiotropic effects of statins on atherosclerotic plaques, including anti-inflammatory and antithrombogenic effects, endothelial function improvement, and plaque stabilization, have been recognized in many studies.14,15 Recently, the systemic injection of simvastatin using reconstituted high-density lipoproteins has shown to be effective for reducing inflammation in plaques of mice by inhibiting macrophage proliferation.16,17 Cyclodextrins are cyclic oligosaccharides that can host small hydrophobic molecules in the inner hydrophobic cavity and form inclusion complexes,18 and can further self-assemble into nanoparticles.19 In addition, cyclodextrins preferentially retain hydrophobic molecules, thus enabling the switching of cargo molecules depending on their affinity.20 Recently, 2-hydroxypropyl-β-cyclodextrin treatment has shown to promote the removal of extracellular and intracellular CC in plaque and reduce atherosclerotic plaque size,21 but its high-dose administration required for the antiatherogenic effect may cause severe hearing loss due to the death of cochlear hair cells,22 posing a therapeutic challenge. Nanoparticle-mediated delivery enables preferential accumulation of multiple drugs in plaque via the disrupted endothelium and macrophage uptake,23,24 thus allowing their therapeutic synergy with reduced side effects.
Herein, we describe the affinity-driven design of cargo-switching nanoparticles (CSNP) that can leverage a cholesterol-rich microenvironment for plaque-targeted drug delivery and scavenge cholesterol for its depletion. We show that potent antiatherogenic agents, cyclodextrin and statin (indicating methyl-β-cyclodextrin and simvastatin in this study, respectively), can form an inclusion complex via self-assembly and then be coated with phospholipids and homogenized into nanoparticles (Figure 1A). We sought to demonstrate that cargo-switching between statin within CSNP and cholesterol takes place based on their physicochemical properties. In mouse models of atherosclerosis, CSNP effectively accumulate within plaques and significantly reduce the content of plaque cholesterol and macrophages though therapeutic synergy of cyclodextrin and statin, which leads to prevention of atherogenesis and regression of established plaques.
Figure 1.
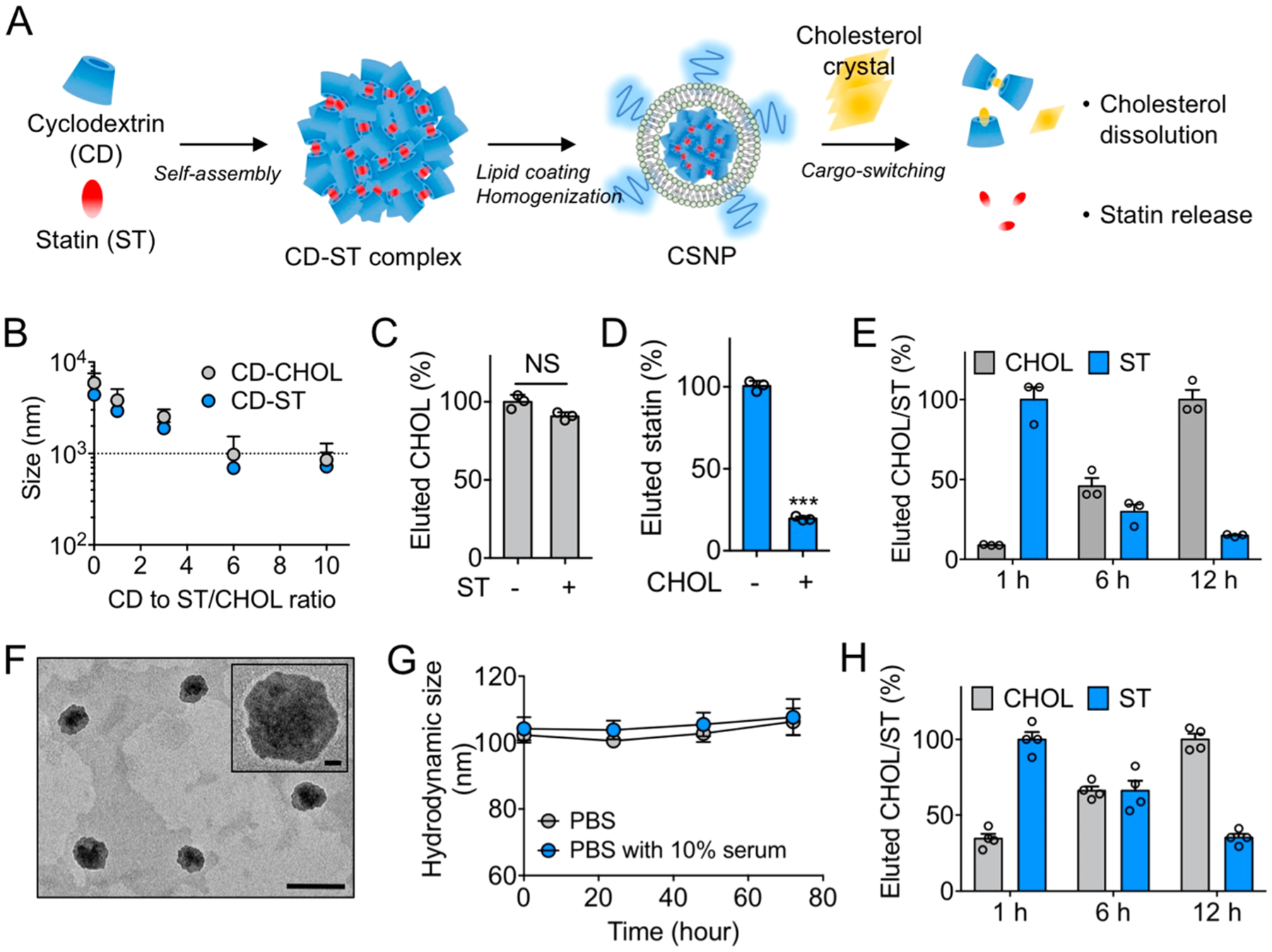
Preparation and physicochemical properties of cargo-switching nanoparticles (CSNP). (A) Schematic of CSNP preparation and cargo-switching. (B) Hydrodynamic sizes of cyclodextrin–statin (CD–ST) and cyclodextrin–cholesterol (CD–CHOL) complexes prepared at different CD to ST/CHOL ratios. (C, D) Competitive binding of (C) CHOL and (D) ST to CD. (E) Release of statin and dissolution of cholesterol by CD–ST complexes via cargo-switching. (F) Representative transmission electron microscopic images of CSNP. The scale bar indicates 200 nm. The inset shows a polymer coating layer on the surface of CSNP. The scale bar in the inset indicates 20 nm. (G) Serum stability of CSNP. Hydrodynamic size of CSNP incubated in PBS and PBS containing 10% serum at 37 °C was monitored over a 72-h period. (H) Cargo-switching property of CSNP. The results are representative of three independent experiments. The results are representative of three independent experiments. Data are means ± s.e.m. [n = 5 for B; n = 3 for C and D; n = 4 for G and H; NS, not significant, ***P < 0.001, unpaired two-tailed Student’s t test for C and D].
RESULTS AND DISCUSSION
Affinity-Driven Cargo-Switching between Statin and Cholesterol.
First, we examined whether cyclodextrin can form inclusion complexes with statin or cholesterol in a controlled manner, and can switch cargo molecules in the complex depending on their affinity. When cyclodextrin was mixed with statin or cholesterol, cyclodextrin–statin or cyclodextrin–cholesterol complexes were formed in an aqueous solution, and their hydrodynamic size decreased as the molar ratio of cyclodextrin to statin or cholesterol increased (Figure 1B). A 6:1 molar ratio of cyclodextrin to statin or cholesterol was used in all subsequent experiments, as this allowed the formation of submicron-sized complexes with minimal concentration of cyclodextrin that can be eluted through a size exclusion column. The complex prepared at the 6:1 molar ratio was easily separated from free statin or cholesterol that self-aggregated without interacting with cyclodextrin (Figure S1A,B). Then we performed a competitive binding assay to examine which molecule binds to cyclodextrin more preferentially. We found that the elution of cholesterol was not significantly altered in the presence of statin, while that of statin was markedly decreased in the presence of cholesterol (Figures 1C,D and S1C). When the total eluted amount was measured, the amount of eluted statin was markedly decreased in contrast to cholesterol, indicating that cholesterol binds competitively with cyclodextrin in place of statin.25,26 These observations reflect that the binding constant of methyl-β-cyclodextrin for cholesterol (7800 ± 110 M−1)27 is higher than that for simvastatin (1700 ± 24 M−1).28 Having observed preferential binding of cyclodextrin to cholesterol, we investigated whether statin preloaded in the complex can be replaced with cholesterol via a cargo-switching process. We prepared cyclodextrin–statin complexes and then incubated with cholesterol at 37 °C. After 1 h of incubation, the elution of statin was dominant, while cholesterol was not eluted effectively (Figures 1E and S1D). However, after 6 h of incubation, the elution of cholesterol increased significantly over time, while that of statin decreased, indicating that the cargo in the complex was switched from preloaded statin to cholesterol. These results suggest that the physicochemical properties of cyclodextrin to form an inclusion complex with statin and to preferentially retain cholesterol by different binding constants can be leveraged to confer a cargo-switching property to the complex.
Preparation of Cargo-Switching Nanoparticles.
Having verified that the cyclodextrin–statin complex can efficiently switch its cargo to cholesterol, we formulated the cyclodextrin–statin complex into homogeneous and stable nanoparticles for systemic injection. A liposomal formulation was exploited to prolong the blood circulation of the cyclodextrin–statin complex and deliver the complex efficiently to the target tissue.29 To optimize a liposomal coating layer that can facilitate cargo-switching between inner cyclodextrin–statin complex and outer cholesterol, we prepared two kinds of liposomal formulations encapsulating free cyclodextrin molecules composed of either 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) with low phase transition temperature (−16.5 °C)30 or hydrogenated soy l-α-phosphatidylcholine (HSPC) with high phase transition temperature (52.5 °C)31 that generated the lipid bilayer in the liquid crystalline or gel phase at body temperature, respectively (Figure S2A). While both DOPC-based and HSPC-based liposomes stably encapsulated free cyclodextrin at 37 °C, the addition of CC induced significant loss of free cyclodextrin only in the DOPC-based liposomes (Figure S2B), which implies that CC can disrupt liquid crystalline structure of DOPC-based liposomal membrane at 37 °C. Consequently, a significant effect of the incorporated cyclodextrin on dissolution of CC was observed in the DOPC-based liposomes, although both empty liposomes slightly dissolved CC due to the function of lipid bilayer as cholesterol sink32,33 (Figure S2C). These results suggest that the fluidic lipid coating layer can allow efficient interaction between encapsulated cyclodextrin and external cholesterol. Therefore, for nanoparticle formulation, the cyclodextrin–statin complex was coated with a lipid bilayer composed of DOPC and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG) at the molar ratio of 95:5 and then homogenized through nanoporous (100 nm) membrane extrusion. The molar ratio of total lipids to complexes was optimized to stabilize the cyclodextrin–statin complexes with the minimum amount of lipids (Figure S3A). In transmission electron microscopy, the nanocomplexes appeared fairly spherical with a lipid coating layer (Figure 1F). The nanocomplexes were stable in the presence of 10% serum at 37 °C over an incubation period of 72 h (Figure 1G). The mean hydrodynamic size and surface charge measured using dynamic light scattering were 104 ± 13 nm and −20 ± 0.8 mV, respectively (Figure S3B,C). 1H NMR spectroscopy revealed that the nanocomplex effectively incorporated cyclodextrin, statin, and lipid with a final molar ratio of approximately 2:1:1.5 (Figure S3D). Importantly, size exclusion chromatography showed that the nanocomplex maintained the pattern of cargo-switching observed by the bare complex after lipid coating (Figure 1H). To further verify the cargo-switching process, we employed a fluorescence resonance energy transfer (FRET) assay where rhodaminylthioureido-conjugated cyclodextrin (RBITC-cyclodextrin) was incorporated into the lipid-coated nanocomplex as the FRET acceptor, and nitrobenzoxadiazole-conjugated cholesterol (NBD-cholesterol) was used as the FRET donor (Figure S4A). When the RBITC-cyclodextrin-incorporated nanocomplexes were incubated with CC containing 2% NBD-cholesterol, the FRET efficiency increased over time (Figure S4B), demonstrating the formation of a cyclodextrin–cholesterol inclusion complex. The pattern of FRET efficiency was similar to that of the cholesterol elution observed in Figure 1H. Taken together, these results suggest that cargo-switching in the lipid-coated nanocomplex can take place via efficient interaction of the incorporated cyclodextrin–statin nanocomplex with external CC through the fluidic lipid layer (Figure S4C). Thus, the nanocomplexes with cargo-switching functions can be prepared through fluidity-based engineering of the surface coating; we named these nanocomplexes cargo-switching nanoparticles (CSNP).
Cholesterol-Scavenging Functions In Vitro.
Next, we studied whether CSNP can bind to and dissolve cholesterol crystals in vitro. Confocal microscopic observations revealed that CSNP actively interacted with CC within 5 min at 37 °C (Figure 2A). For quantitative assessment of cholesterol dissolution, fluorescent CC were incubated with CSNP at different cyclodextrin concentrations for 24 h at 37 °C and then filtered out to quantify only dissolved fluorescent cholesterol. We found that CSNP exhibited effective dose- and time-dependent dissolution of CC (Figure 2B and Figure S5A). It was estimated that 6.5 mg of CSNP could dissolve around 0.2 mg of cholesterol crystals at 24 h post-treatment at 37 °C. Next, we examined whether CSNP can enter macrophages and dissolve intracellular cholesterol crystals. Confocal microscopic observations indicated that CSNP were actively taken up by CC-laden Raw 264.7 murine macrophages and colocalized with intracellular CC (Figure 2C), showing that the negative surface charge of CSNP did not hamper its cellular uptake.34 To evaluate the ability of CSNP to dissolve intracellular CC, Raw 264.7 cells pretreated with fluorescent CC were treated with CSNP for 24 h. CSNP treatment reduced intracellular CC and increased supernatant cholesterol significantly, which was similar to cyclodextrin treatment (Figure 2D,E). These results suggest that CSNP can scavenge both extracellular and intracellular cholesterol effectively via the incorporated cyclodextrin as observed in the previous finding.21
Figure 2.
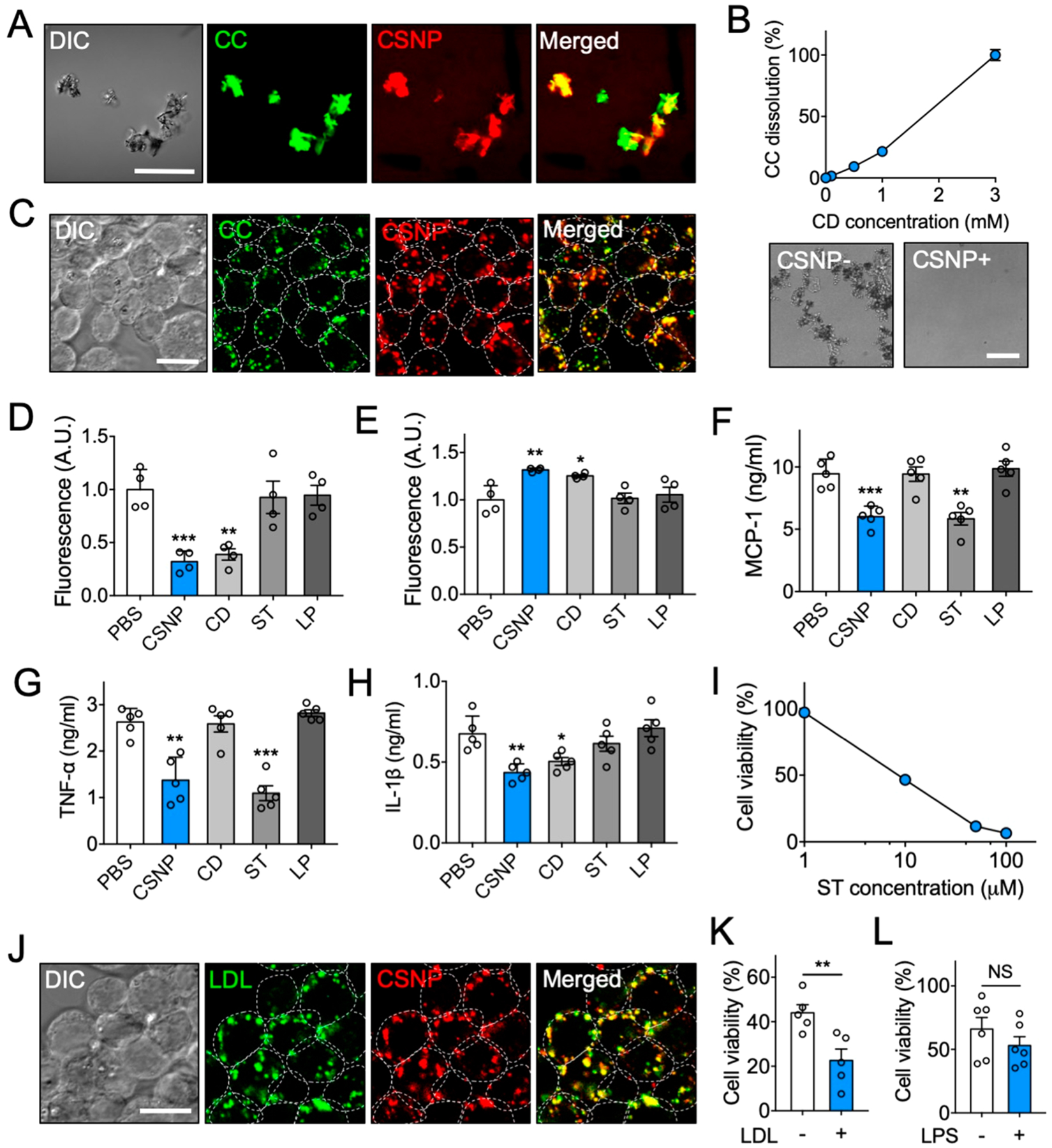
Multiple anti-inflammatory properties of CSNP in vitro. (A) Representative confocal microscopic images of CSNP binding to cholesterol crystals (CC). The scale bar indicates 50 μm. (B) Dose-dependent CC dissolution with CSNP. Bright-field images show complete dissolution of CC 24 h after CSNP treatment at 3 mM CD. (C) Representative confocal microscopic images of CC-laden macrophages after CSNP treatment. The scale bar indicates 10 μm. (D, E) Assessment of cholesterol (D) within cells and (E) in the supernatant after CSNP treatment. (F, G) Secretion of (F) MCP-1 and (G) TNF-α from the LPS-activated macrophages after CSNP treatment. (H) Secretion of IL-1β from the CC-laden LPS-activated macrophages after CSNP treatment. (I) Dose-dependent effects of CSNP on macrophage viability. (J) Representative confocal microscopic images of LDL-laden macrophage after CSNP treatment. The scale bar indicates 10 μm. (K) LDL-dependent effects of CSNP on macrophage viability. (L) LPS-independent effects of CSNP on macrophage viability. The dotted circles indicate individual cells. Data are mean ± s.e.m. [n = 4 for B–E; n = 5 for F–K; n = 6 for L; NS, not significant, *P < 0.01, **P < 0.005, ***P < 0.001, unpaired two-tailed Student’s t test for K and L, compared with the PBS group for D–H].
Cholesterol-Sensitive Anti-Inflammatory Functions In Vitro.
Considering that most of macrophages within advanced plaques exhibit a pro-inflammatory phenotype,35 we evaluated whether CSNP can modulate inflammatory status in the activated macrophages and exert antiproliferative effects on macrophages. For anti-inflammatory study, Raw 264.7 cells pretreated with lipopolysaccharide (LPS) (and CC for inflammasome activation) for 24 h were treated with either PBS, CSNP, free cyclodextrin, free statin, or empty liposomes at the dose where the cell number was minimally affected36,37 (Figure S5B,C). The secretion of MCP-1 and TNF-α was measured to assess anti-inflammatory effects of CSNP and that of IL-1β to assess their inhibitory effects on inflammasome activation. The secretion of MCP-1 and TNF-α was reduced significantly in both CSNP and statin treatments, and that of IL-1β was reduced in both CSNP and cyclodextrin treatments (Figure 2F–H), indicating that CSNP effectively reduced pro-inflammatory status in the activated macrophages via the released statin and inhibited cholesterol-mediated inflammasome activation via the incorporated cyclodextrin. These results suggest that CSNP can exert anti-inflammatory effects of both statin and cyclodextrin. Next, to assess the antiproliferative effects of CSNP, Raw 264.7 cells were treated with either PBS, CSNP, free cyclodextrin, free statin, or empty liposomes. CSNP effectively inhibited macrophage proliferation, which was similar to statin treatment (Figure S5D,E). At higher doses, we also found that CSNP reduced macrophage viability in a dose-dependent manner (Figure 2I). These results suggest that CSNP can exert multiple anti-inflammatory functions attributed to both cyclodextrin and statin.
Since we observed that the presence of cholesterol promotes the release of statin from CSNP, we next studied whether intracellular cholesterol affects the onset of action of CSNP within cells. Macrophages were pretreated with low-density lipoprotein (LDL) for 24 h to induce the accumulation of cholesterol within cells38 (Figure S5F). When treated with CSNP, they showed high colocalization with intracellular LDL (Figure 2J). At 1 day post-treatment, CSNP significantly reduced the viability of LDL-laden macrophages compared to unladen macrophages (Figure 2K). CSNP did not exhibit such effect on macrophages activated with LPS (Figure 2L). These results indicate that the presence of intracellular cholesterol renders macrophages susceptible to CSNP treatment by facilitating the release of statin, although they are still functional without supplementary cholesterol feeding possibly because they naturally go through lysosomal degradation (Figure S5G). Collectively, these results suggest that the antiproliferative effect of CSNP can be amplified within cholesterol-laden cells that are likely to disturb resolution of inflammation within plaques.
Pharmacokinetics, Biodistribution, and Antiatherogenic Functions In Vivo.
Next, we assessed the pharmacokinetics, biodistribution, and plaque accumulation of CSNP to determine their potential for targeted drug delivery to atherosclerotic plaques in vivo. When intravenously injected, the blood half-life of CSNP in wild-type mice was determined to be approximately 9 h (Figure 3A). It was significantly longer than that of free cyclodextrin (39 min) (Figure S6A), which is similar to its reported half-life (31.2 min)39 and that of statin (2 h) reported by another group.40 These results suggest that CSNP can significantly improve the pharmacokinetics of encapsulated drugs. To observe the accumulation of CSNP in plaque and other organs, we used a partial carotid ligation model that forms robust atherosclerosis in the left carotid artery (LCA) of ApoE−/− mice within 2 weeks.41,42 CSNP were injected intravenously on days 8 and 11 after left carotid ligation (Figure 3B). The mice were sacrificed 3 days after the last injection of CSNP to analyze plaque targeting and biodistribution of CSNP. We found that CSNP mainly accumulated in the liver, spleen, and LCA (Figure 3C), showing that CSNP can effectively target atherosclerotic plaques via passive targeting through the disrupted endothelium of the atherosclerotic plaques.23 Plaque accumulation of CSNP was about 10-fold higher than that of free cyclodextrin (Figure S6B,C). Dissected carotid artery images revealed that CSNP accumulated specifically in the plaque lesions of LCA, while their accumulation was negligible in the RCA (Figure 3D). Histological analysis revealed that CSNP effectively colocalized with macrophages and cholesterol within plaques in the LCA (Figures 3E,F and S6D,E). These results suggest that CSNP can effectively deliver the encapsulated drugs to atherosclerotic plaques, especially to macrophages and cholesterol.
Figure 3.
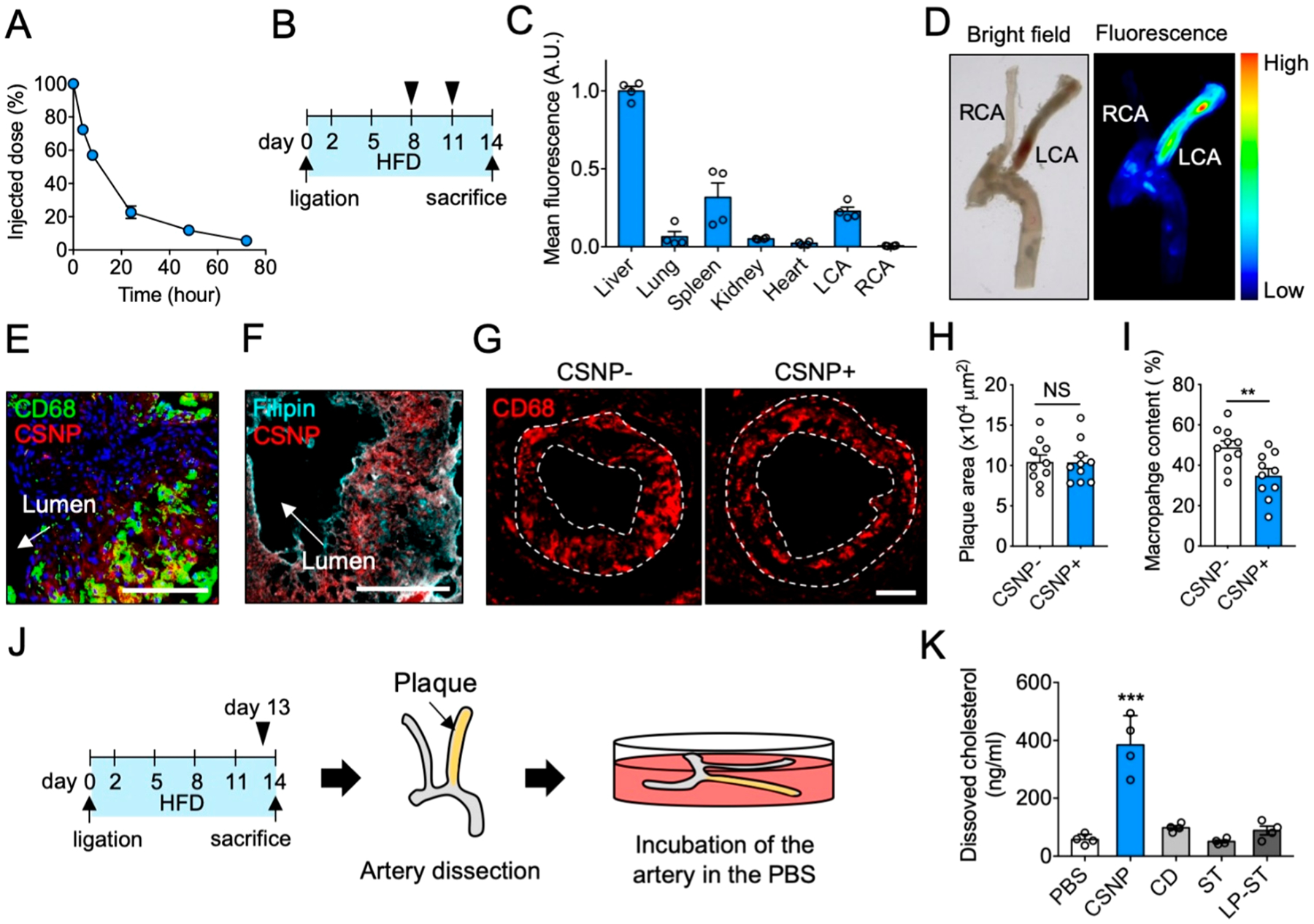
Characterization of CSNP in vivo. (A) Pharmacokinetics of CSNP. (B) Schematic of experimental plan for biodistribution and plaque targeting studies. (C) CSNP accumulation in various organs. (D) Representative ex vivo bright-field and fluorescence images of the dissected carotid artery. (E, F) Representative confocal fluorescence images of the LCA sections after (E) macrophage and (F) cholesterol staining. Anti-CD68 antibody, Filipin, and Hoechst were used to stain macrophage, cholesterol, and nucleus, respectively. The scale bar indicates 100 μm. (G) Representative fluorescence images of the LCA sections showing macrophage content and distribution in plaques. The dotted lines indicate plaque area. (H, I) Quantification of (H) plaque area and (I) macrophage content in panel G. (J) Schematic of experimental plan for ex vivo cholesterol dissolution assay. (K) Quantification of dissolved cholesterol in the PBS. CSNP+ and CSNP− indicate intravenous injection of CSNP and PBS, respectively. Data are mean ± s.e.m. [n = 5 for A; n = 4 for C, H, and I; n = 10 for K; NS, not significant; **P < 0.01, ***P < 0.001, unpaired two-tailed Student’s t test, compared to CSNP− for H and I, and PBS for K].
Next, we investigated whether CSNP can exert therapeutic effects on macrophages and cholesterol in the plaques. To assess antiproliferative effects of CSNP on plaque macrophages, mice were treated with CSNP at day 8 and 11 after partial ligation surgery, and the LCA were collected at day 14. Immunohistochemistry images of the LCA sections revealed that CSNP treatment significantly inhibited proliferation of macrophages in the treatment condition unaffecting plaque size (Figure 3G–I). Considering that the antiproliferative effect of CSNP on macrophages was statin-dependent in vitro (Figure S5D,E) and cyclodextrin did not directly alter macrophage content in vivo,22 it is plausible that statin within CSNP played a significant role in the reduction of plaque macrophages. Then, to assess whether CSNP can dissolve out plaque cholesterol, mice were treated with CSNP at the maximal single dose where the body weight was unaffected (30 mg/kg statin and 200 mg/kg cyclodextrin, Figure S6F) at day 13 after partial ligation surgery and the arteries were collected at day 14 (Figure 3J). The collected arteries were immediately incubated in phosphate-buffered saline (PBS) for 24 h, and the cholesterol dissolved in the PBS was quantified. Ex vivo plaque cholesterol dissolution assay revealed that CSNP induced significant dissolution of cholesterol from the dissected plaques (Figure 3K). Monotherapy with free cyclodextrin at a two-fold higher dose (400 mg/kg) did not induce significant cholesterol dissolution presumably due to its poor pharmacokinetics and plaque targeting efficacy. Monotherapy with free statin or liposomal statin with comparable plaque targeting efficacy to CSNP (Figure S6G,H) at an equivalent dose (30 mg/kg) did not induce significant cholesterol dissolution. These observations demonstrate that CSNP enabled the incorporated cyclodextrin to play a crucial role in the dissolution of plaque cholesterol. Overall, we observed that CSNP exert antiatherogenic functions of both cyclodextrin and statin within plaques.
Atherogenesis Prevention by CSNP.
Having observed the antiatherogenic functions of CSNP within plaques, we assessed whether CSNP can prevent atherogenesis in the ligation model. Since two injections of CNSP at day 8 and 11 after ligation had no significant effect on plaque size (Figure 3G,H), we initiated treatment earlier by injecting CSNP on days 2, 5, 8, and 11 after ligation (Figure 4A). Interestingly, we found that the treatment regimen significantly inhibited lesion formation (Figure 4B,C), plaque growth (Figure 4D,E), and macrophage content (Figure 4F) within plaques. These results show that early treatment is required to prevent atherogenesis in the ligation model that forms atherosclerosis rapidly within 2 weeks.41 However, monotherapy with free cyclodextrin, free statin, and liposomal statin did not show such significant therapeutic effect. Plasma cholesterol level was not altered in all groups (Figure 4G), which is consistent with pervious findings that statins are ineffective in lowering plasma cholesterol level in mice.17,43 Therefore, these results indicate that CSNP exerted local antiatherogenic effects within plaques independent of plasma cholesterol lowering. All treatment groups showed no significant difference in body weight gain (Figure 4H) and enzymatic production by the liver (Figure S7B,C). Furthermore, multiple injections of CSNP along the treatment plan did not affect the weight (Figure S7D,E), size (Figure S7F,G), and histology (Figure S7H,I) of the liver and spleen, where CSNP were mainly cleared.
Figure 4.
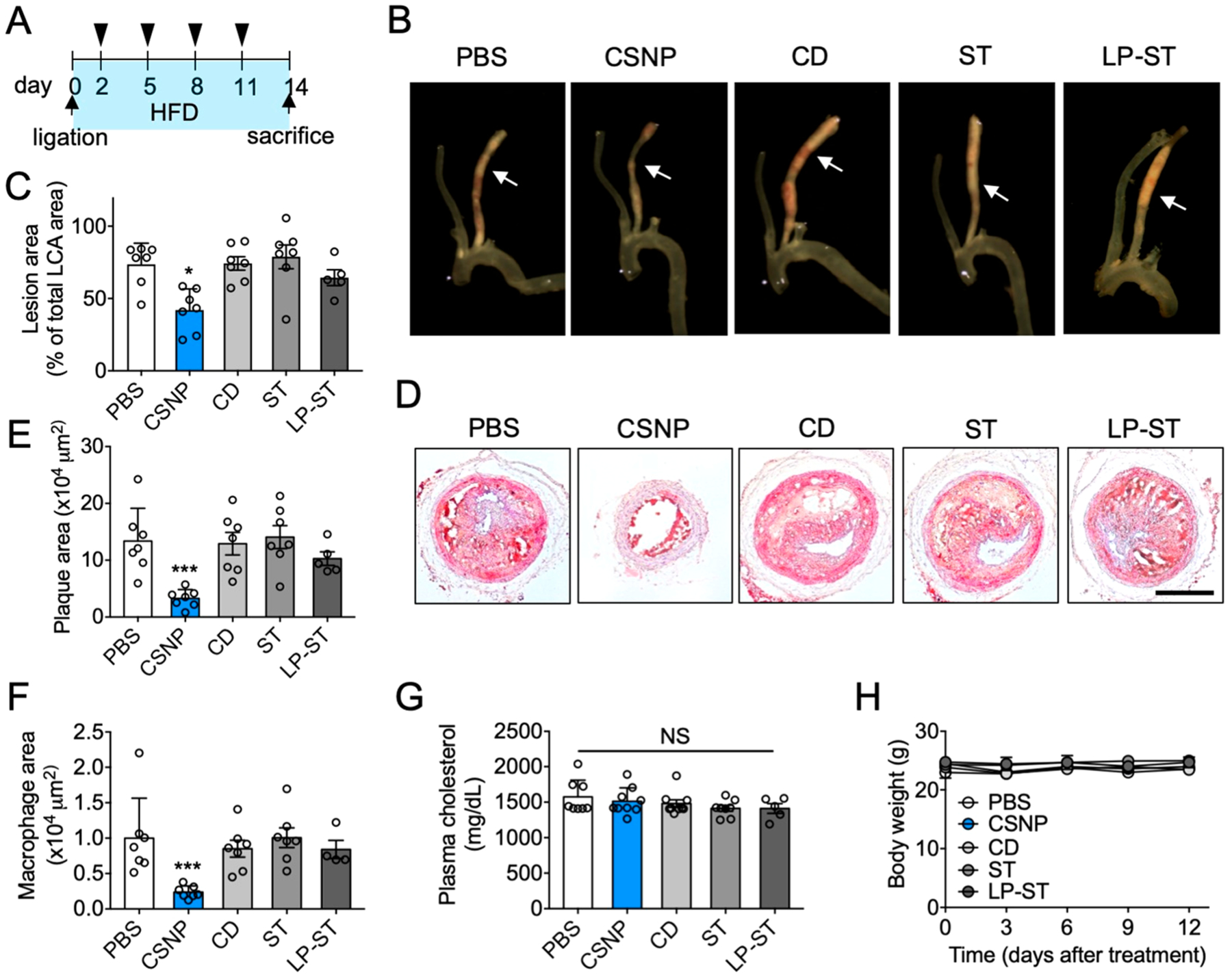
Antiatherogenic effect of CSNP. (A) Schematic of experimental plan for antiatherogenic therapy. Doses per injection were 15 mg/kg ST and 100 mg/kg CD for CSNP group, 400 mg/kg CD for CD group, and 15 mg/kg ST for ST and LP-ST groups. (B) Representative bright field images of dissected carotid arteries. The arrows indicate where cross-sectional images were obtained. (C) Quantification of lesion areas in panel B. (D) Representative histological images of the LCA sections after Oil-Red-O staining. The scale bar indicates 200 μm. (E) Quantification of plaque area in panel D. (F) Quantification of macrophage area in the LCA sections after CD68 staining. (G) Plasma cholesterol level. (H) Body weight during treatment. Data are mean ± s.e.m. [n = 5 (LP-ST) or 7 (PBS, CD, ST, and CSNP) for C, E, F, and G; n = 5 for H; NS, not significant; *P < 0.05, ***P < 0.001, one-way ANOVA and Tukey’s multiple comparison test].
Atherosclerotic Plaque Regression by CSNP.
Next, we examined whether CSNP can regress existing atherosclerotic plaques in the chronic high-fat diet (HFD) mouse model.44 At 12 weeks after HFD, a baseline group was sacrificed. Treatment groups were divided into HFD and normal chow diet (NCD) groups to assess the effect of diet on the therapeutic outcome by CSNP therapy, and each group received intravenous injections of either PBS or CSNP twice a week for another 4 weeks (Figure 5A). Mice in the NCD group exhibited significantly lower systemic cholesterol level compared to those in the HFD group, and CSNP treatments did not change the cholesterol level significantly in both groups (Figure 5B), which shows again that the plasma cholesterol level is dependent on the type of diet, not CSNP treatment. In both HFD and NCD groups, the lesion areas in the aortic root, aortic arch, and thoracic aorta in the CSNP-treated mice were significantly smaller than those in the untreated mice (Figure 5C–F), showing the notable therapeutic effect of CSNP regardless of systemic cholesterol level. These findings have significant clinical implications because some patients who fail to lower systemic cholesterol level with statins could still benefit from local anti-inflammation therapy using nanotherapeutics such as CSNP. Importantly, when compared to baseline, mice in the NCD group showed significant regression of established plaques after CSNP treatments, particularly in the lesion areas of the aortic root and arch, while those in the HFD group showed regression only in the lesion area of the aortic root. As CSNP does not directly affect systemic cholesterol, it is plausible that the accumulation of cholesterol within plaques and its pro-inflammatory functions reduced the anti-inflammatory functions of CSNP in the HFD group. These results show that plaque-targeted anti-inflammation therapy using CSNP can synergistically contribute to plaque regression with systemic cholesterol-lowering regimen. CSNP treatment on advanced atherosclerosis did not affect body weight and liver toxicity (Figure S8A–C), and plaque vulnerability (Figure S8D,E). Overall, these results suggest that CSNP have great potential for the treatment of cholesterol-rich atherosclerotic plaques at least in mice.
Figure 5.
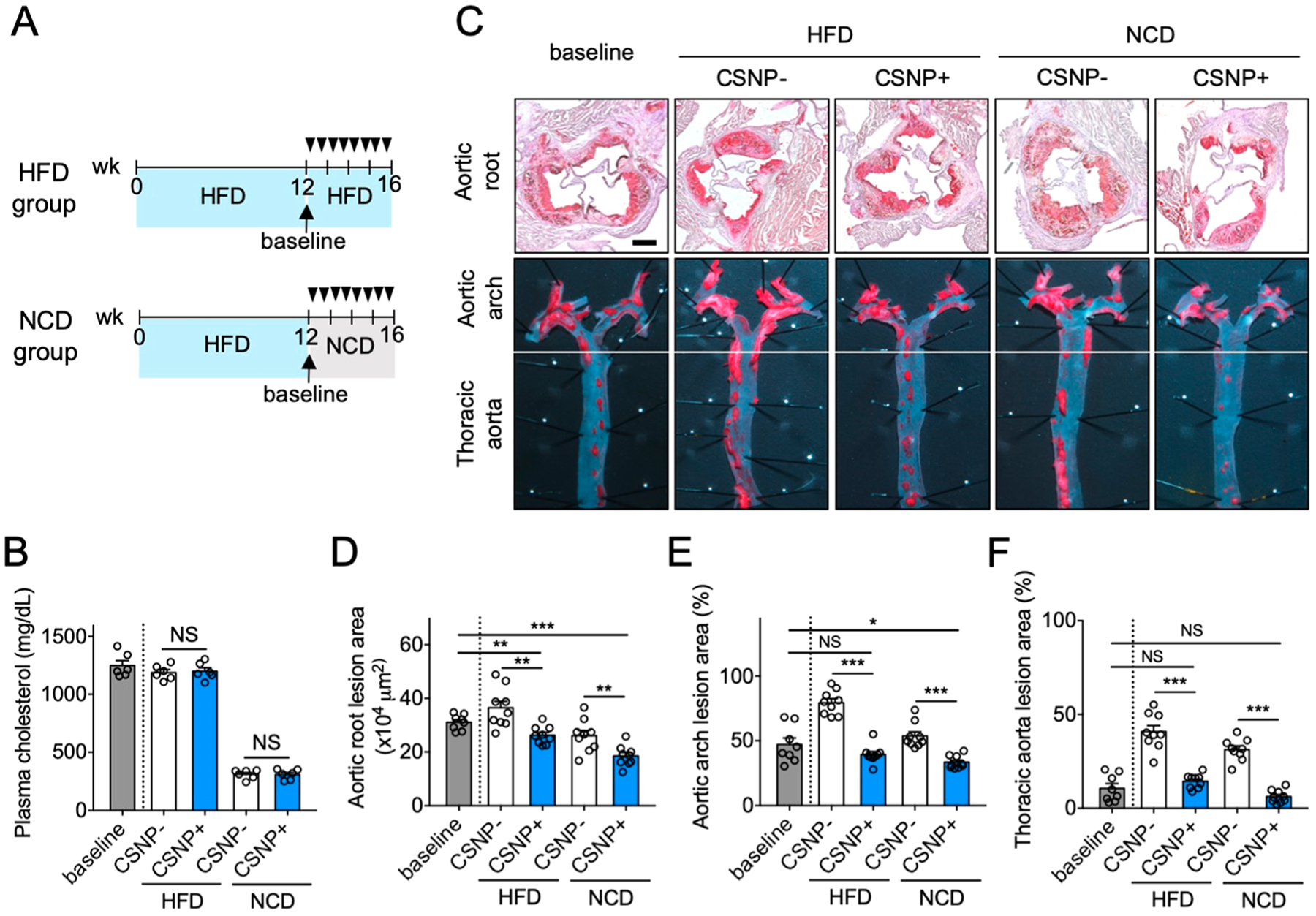
Regression of atherosclerotic plaques using CSNP. (A) Schematic of experimental plan for regression therapy of advanced atherosclerosis. Mice fed with HFD for 12 weeks (baseline) were injected intravenously (black triangles) with PBS (CSNP−) or CSNP (CSNP+) twice a week for 4 weeks, and the diet was either maintained (HFD group) or switched to a normal chow-diet (NCD group). (B) Plasma cholesterol concentrations. Plasma cholesterol concentrations were measured after 4 weeks of atherosclerosis regression therapy. (C) Representative images of aortic root sections and en face aortic arch and thoracic aorta after Oil-Red-O staining. The scale bar indicates 200 μm. (D–F) Quantification of plaque area in (D) aortic root, and lesion areas in (E) aortic arch and (F) thoracic aorta after CSNP treatment. CSNP+ and CSNP− indicate intravenous injection of CSNP and PBS, respectively. Data are means ± s.e.m. [n = 8 (baseline group) or 9 (other groups); NS, not significant; *P < 0.05, **P < 0.01, and ***P < 0.001, unpaired two-tailed Student’s t test].
Hepatic Responses.
Clinically, statins inhibit 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase in the liver after oral administration, which in turn increases the expression of LDL receptors and reduces blood cholesterol level. However, orally taken statins are reported to be ineffective in lowering the blood cholesterol level in mice.43 Similar to the previous findings,16,17 we also observed that intravenous injection of CSNP at a statin dose of 15 mg/kg did not induce significant cholesterol-lowering effects in mice, by which we attributed the effective prevention and regression of atherosclerosis to the local effects of CSNP within the plaque. However, since it has been previously known that orally administered statins increase HMG-CoA reductase activity in humans, rats, and mice regardless of changes in the systemic cholesterol level,45–48 we investigated whether the CSNP accumulated in the liver after systemic administration affects the activity of HMG-CoA reductase. Wild-type mice were injected intravenously with CSNP every 3 days (a total of four times). At 3 days after the last injection, the livers were harvested, and the HMG-CoA reductase activity was measured. CSNP treatment exhibited ~10-fold increase in the HMG-CoA reductase activity in the liver (Figure S9), indicating that systemically administered CSNP can induce hepatic responses similar to orally administered statin. These results indicate that, although ineffective in mice, CSNP may have the potential to lower systemic cholesterol level in humans, which can be synergistically combined with their local therapeutic effects in the plaque for effective treatment of atherosclerosis.
CONCLUSIONS
Therapeutic nanomedicine can be engineered to exploit various microenvironmental factors of a disease such as pH, temperature, reduction/oxidation status, and enzymes to improve disease-specificity and therapeutic efficacy.49,50 Thus, far, no drug delivery system has been developed to exploit a cholesterol-rich microenvironment for effective treatment of a disease. Furthermore, while these disease-associated abnormal microenvironmental factors were exploited simply to stimulate the function of the accumulated nanomedicine, few attempts have been made to normalize the abnormal microenvironment simultaneously. In this study, we described an affinity-driven design of CSNP, formed by the self-assembly of cyclodextrin and a hydrophobic drug, that can effectively exploit the cholesterol-rich microenvironment for the treatment of atherosclerosis. CSNP were physicochemically designed to release low-affinity hydrophobic drug in the cholesterol-rich atherosclerotic microenvironment and scavenge high-affinity cholesterol within the microenvironment. The inherent capacity of CSNP to scavenge and deplete plaque cholesterol in the process of drug release led to superior antiatherogenic efficacy in mouse models of atherosclerosis compared to conventional drug delivery system using liposomes. Although it has been reported that local delivery of statin resolves inflammation and reduces plaque content,16,17 our affinity-driven design of CSNP enabled not only local delivery of statin but also depletion of cholesterol within plaques, which significantly improved atherosclerosis management.
There are various types of cyclodextrins and statins that can form cyclodextrin–statin complexes depending on their physicochemical properties. However, to use the complexes for combination therapy, cyclodextrin and statin should form the complexes at a proper complexation ratio so they can be injected at a dose where both drugs can exert therapeutic effect. In this study, we chose simvastatin among various statins because its excellent local anti-inflammatory effects within plaques has been thoroughly studied.16,17 Although 2-hydroxypropyl-β-cyclodextrin has previously shown to induce regression of atherosclerosis in mice41 and rabbit,51 it requires very high-dose administration. Given that the therapeutic dose of simvastatin (0.036 mmol/kg)17 is much lower than that of 2-hydroxypropyl-β-cyclodextrin (1.3 mmol/kg) in mice,21 we assumed that their binding stoichiometry (1:1 or 1:2)52 would make it hard to prepare CSNP formulation for effective combination therapy. Therefore, we chose methyl-β-cyclodextrin because it exhibits superior efficacy to 2-hydroxypropyl-β-cyclodextrin in dissolving cholesterol and inducing cholesterol efflux from cells.33,53,54 Consequently, CSNP composed of methyl-β-cyclodextrin and simvastatin exerted significant combination therapeutic effects for effective atherosclerosis therapy.
Other than atherosclerosis, there are various diseases associated with defective cholesterol transportation such as NPC155 and multiple sclerosis,56 wherein cyclodextrin treatment and facilitation of cholesterol transport showed significant therapeutic efficacy. However, the unfavorable pharmacokinetics of cyclodextrin requires high-dose administration to exert therapeutic effects and expose subjects to dose-limiting side effects such as hearing loss,22 posing a therapeutic challenge. Nanoparticle formulations of cyclodextrin such as CSNP will help reduce the adverse effects because they can be incorporated together with other potent drugs and engineered to confer tissue-selectivity by conjugation with active-targeting moieties.9,23 Thus, we believe that CSNP provide a therapeutic platform for interfacing with complex diseases featured by cholesterol-rich microenvironment.
METHODS
Preparation and Characterization of Cyclodextrin Inclusion Complexes.
Different amounts of cyclodextrin (methyl-β-cyclodextrin; Sigma-Aldrich, St. Louis, MO) were dissolved in 1 mL of ethanol containing either cholesterol (5 mM; Avanti Polar Lipids, Alabaster, AL) or statin (simvastatin; 5 mM; Sigma-Aldrich, St. Louis, MO) to obtain final cyclodextrin concentrations of 0, 5, 15, 30, and 50 mM. The solutions were dried completely, rehydrated using 1 mL of phosphate buffered saline (PBS), and sonicated until complexes were formed. The size of the aggregates was measured using dynamic light scattering (Zetasizer Nano ZS90; Malvern Instruments, Malvern, UK). To determine whether interaction with cyclodextrin allows the elution of cholesterol and statin through a PD-10 desalting column (GE Healthcare, Little Chalfont, UK), solutions of 100 μL of ethanol containing cholesterol [5 mM, containing 2% nitrobenzoxadiazole (NBD)–cholesterol], statin (5 mM), cholesterol (5 mM, containing 2% NBD-cholesterol) + cyclodextrin (30 mM; Sigma-Aldrich), and statin (5 mM) + cyclodextrin (30 mM) were prepared. Each solution was dried completely, rehydrated using 1 mL of PBS, and sonicated until complexes or aggregates were formed. Then 200 μL of each solution was eluted through the PD-10 column. The eluate was collected in 96-well plates, and elution profiles were obtained by measuring the amounts of cholesterol and statin in the eluted samples. Quantification of cholesterol and statin was performed by measuring the fluorescence of NBD-cholesterol (excitation 480 nm/emission 530 nm) using SpectraMax Gemini XPS Microplate Reader (Molecular Devices, San Jose, CA) and absorbance of statin (240 nm) using SpectraMax Spectra Max plus 384 Microplate Spectrophotometer (Molecular Devices), respectively.
Competitive Binding Assay.
A solution of 100 μL of ethanol containing cholesterol (5 mM, containing 2% NBD-cholesterol), statin (5 mM), and cyclodextrin (30 mM) was prepared. The solution was dried completely and then rehydrated using 1 mL of PBS to allow formation of cyclodextrin inclusion complex with either cholesterol or statin. Then 200 μL of the solution was eluted through the PD-10 column, the eluate containing solubilized cholesterol, and statin was collected in 96-well plates, and elution profiles were obtained by measuring the amounts of cholesterol and statin in each well. Quantification of cholesterol and statin was performed by measuring the fluorescence of NBD-cholesterol (excitation 480 nm/emission 530 nm) and absorbance of statin (240 nm), respectively.
Cargo-Switching Assay.
To form cyclodextrin–statin inclusion complexes, 100 μL of ethanol containing statin (5 mM) and cyclodextrin (30 mM) was prepared. The solution was dried completely and then rehydrated using 1 mL of PBS. Cholesterol aggregates (5 mM, containing 2% NBD-cholesterol) were formed as described above and then added to the cyclodextrin–statin complex. The mixture was incubated for 1, 6, and 12 h at 37 °C to allow interaction and then eluted using the PD-10 column. The eluate containing solubilized cholesterol and statin was collected in 96-well plates, and elution profiles were obtained by measuring the amounts of cholesterol and statin in each well. Quantification of cholesterol and statin was performed by measuring the fluorescence of NBD-cholesterol (excitation 480 nm/emission 530 nm) and absorbance of statin (240 nm), respectively.
Preparation and Characterization of CSNP.
Cyclodextrin and statin were dissolved in ethanol and mixed to achieve final concentrations of 30 mM and 5 mM, respectively. The solution was stirred gently for 30 min, dried completely under a vacuum for 1 h, and then rehydrated using 1 mL of PBS to obtain cyclodextrin–statin complexes. To coat cyclodextrin–statin complexes with phospholipids, a lipid film consisting of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC; Avanti Polar Lipids) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG; Avanti Polar Lipids) at a molar ratio of 95:5 was prepared. To optimize the amount of phospholipid for a coating, lipid films prepared at different phospholipid concentrations (0.5, 1, 2, or 4 mM) were hydrated with cyclodextrin–statin complex solution containing 6 mM cyclodextrin and 1 mM statin, and the hydrodynamic size of phospholipid-coated cyclodextrin–statin complexes was monitored in PBS. A minimal amount of phospholipids that could prevent a size increase was used for CSNP. After hydration, phospholipid-coated cyclodextrin–statin complexes were extruded through a 100 nm polycarbonate membrane (Whatman) at room temperature and then eluted through the PD-10 column to remove free statin and cyclodextrin. The hydrodynamic size and zeta potential of CSNP were measured using dynamic light scattering (Zetasizer Nano ZS90). Amicon ultracentrifugal filters (Milipore) were used to obtain concentrated CSNP for mouse experiments. For preparation of fluorescent CSNP, 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Avanti Polar Lipids) or lipophilic 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DiR; Invitrogen) was added to the lipid film (3 mol % of total phospholipids). For a statin release and cholesterol dissolution assay of CSNP, the cholesterol aggregates and CSNP were incubated for 1, 6, and 12 h at 37 °C and then eluted using the PD-10 column. The eluate containing solubilized cholesterol and statin was collected in 96-well plates, and elution profiles were obtained by measuring the amounts of cholesterol and statin in each well. For assessment of serum stability, fetal bovine serum was diluted in PBS to obtain 10% serum. CSNP were then incubated with either PBS or 10% serum, and hydrodynamic sizes were measured at 0, 24, 48, and 72 h.
1H NMR Analysis of CSNP.
1H NMR spectra of cyclodextrin, statin, phospholipids, and CSNP were obtained using a Bruker Wide-Bore Avance 300 MHz spectrometer (AV300; Bruker, Rheinstetten, Germany). Acquisition of the spectra was carried out using TOPSPIN 2.0 software (Bruker). The acquired 1H NMR spectra were processed with Mnova software (Mestrelab Research) for phase, baseline correction, and quantification.
Transmission Electron Microscopy.
Aliquots of 10 μL of CSNP (0.5 mM lipid) were deposited on Formvar carbon-coated EM grids (Ted Pella, Inc., Redding, CA) for 20 min until dry. The CSNP-coated grids were washed twice with PBS. The grids were then transferred to a 50-μL drop of 1% glutaraldehyde for 5 min before transferring to a 100-μL drops of distilled water for 2 min. This fixation procedure was repeated seven times for a total of eight water washes. The grids were kept wet on the side of the membrane during all steps but dry on the opposite side. For negative staining, the grids were transferred to a 50-μL drops of 2% PTA solution for 2 min, and excess solution was removed with filter paper and then dried. Transmission electron microscopic images were obtained using a JEM-2100F HRTEM operating at 200 kV (JEOL, Tokyo, Japan).
FRET Measurement.
To prepare CSNP incorporating 6-deoxy-6-[(5/6)-rhodaminylthioureido]-β-cyclodextrin (RBITC-cyclodextrin, Cyclolab), cyclodextrin, RBITC-cyclodextrin, and statin were used at a molar ratio of 28.5 mM, 1.5 mM, and 5 mM, respectively. Cholesterol aggregates (5 mM, containing 2% NBD-cholesterol) were formed as described above and then added to CSNP solution. The mixture was incubated for 1, 6, and 12 h at 37 °C to allow interaction. FRET efficiency indicates the fluorescence intensity of RBITC (600 nm) normalized to that of NBD (530 nm) upon excitation at 480 nm. For comparison with bare cyclodextrin–statin complex, the complex solution containing the same concentration of cyclodextrin was used.
Preparation of Cholesterol Crystals.
Ethanol containing cholesterol (2 mg/mL; Avanti Polar Lipids) was prepared. Crystallization was induced by addition of distilled water (30% v/v) to the cholesterol solution and drying the solution completely under a vacuum. The cholesterol crystals (CC) were resuspended in PBS and sonicated for size control. For fluorescent CC, 25-NBD cholesterol (Avanti Polar Lipids) was dissolved together with cholesterol in ethanol at a molar ratio of 1:50.
Preparation of Liposomes.
1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC; Avanti Polar Lipids) or l-α-phosphatidylcholine, hydrogenated (Soy) (HSPC; Avanti Polar Lipids) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG; Avanti Polar Lipids) at a molar ratio of 95:5 were dissolved in chloroform and then dried completely overnight. The lipid film was hydrated with PBS and extruded through a 100 nm polycarbonate membrane. For liposomal cyclodextrin, 30 mM cyclodextrin or 400 μM 6-deoxy-6-[(5/6)-rhodaminylthioureido]-β-cyclodextrin (RBITC-cyclodextrin, Cyclolab) in PBS was used for hydration and free cyclodextrin was removed by size exclusion chromatography. The hydrodynamic size and zeta potential of liposomes were measured using dynamic light scattering.
Cholesterol Crystal Binding and Dissolution.
For binding assay, NBD-labeled CC (50 μM in PBS) were incubated with lissamine rhodamine B-labeled CSNP (50 μM cyclodextrin) for 5 min at room temperature. Then 100 μL of the solution was dropped onto a microscope slide, covered with a coverslip, and imaged under a 60× objective lens on a confocal microscope (Nikon, Tokyo, Japan). For CC dissolution assay, CSNP, cyclodextrin, or cyclodextrin+liposomes were incubated with NBD-labeled CC (500 μM in PBS) at different cyclodextrin concentrations (0, 0.1, 0.5, 1, and 3 mM) for 24 h at 37 °C. The mixtures were then filtrated through 0.22-μm syringe filters and the fluorescence of the filtrates was measured. Bright-field images showing complete dissolution of CC 24 h after CSNP treatment were obtained using a 10× objective lens. To study time-dependent CC dissolution, CSNP (3 mM) were incubated with NBD-labeled CC (500 μM in PBS) for 0, 2, 6, 12, and 24 h at 37 °C. To study CC-mediated liposomal membrane disruption, DOPC or HSPC liposomes loaded with RBITC-cyclodextrin (500 μM) were incubated with or without CC (500 μM) for 24 h at 37 °C. The solution was centrifuged to remove CC, and then the liposomes were isolated using Amicon centrifugal filters with 100 kDa MW cutoff (Millipore). The liposomes were resuspended in PBS, and the RBITC fluorescence was measured for quantification. To study CC dissolution with liposomal cyclodextrin, DOPC and HSPC liposomes (1 mM) loaded with or without cyclodextrin were incubated with NBD-labeled CC (500 μM) for 24 h at 37 °C. The mixtures were then filtrated through 0.22-μm syringe filters, and the fluorescence of dissolved cholesterol in the filtrate was measured for quantification.
Cell Culture.
Raw264.7 and J774A.1 mouse macrophage cells (ATCC TIB-71 and ATCC TIB-67) were maintained in Dulbecco’s modified Eagle’s medium (Hyclone, South Logan, UT). The media were supplemented with 10% fetal bovine serum (Hyclone) and 1% penicillin–streptomycin (Hyclone). All cells were cultured in tissue culture flasks in a humidified incubator at 37 °C in an atmosphere of 95% air and 5% carbon dioxide.
In Vitro Cell Studies.
To observe cellular uptake of CSNP by Raw 264.7 and J774A.1 cells, the cells were seeded in six-well plates for 24 h at a density of 5 × 104 cells per well. The cells were treated with lissamine rhodamine B-labeled CSNP (100 μM statin) for 6 h and observed under a 60× objective lens on a Nikon confocal microscope (Nikon, Tokyo, Japan). Lysosomes were stained with LysoTracker (Thermo Fisher Scientific, Waltham, MA). For an intracellular CC dissolution assay, Raw 264.7 cells were seeded in 48-well plates for 24 h at a density of 2 × 104 cells per well. The cells were treated with NBD-CC (50 μM) for 24 h. Cells were washed thoroughly and then treated with CSNP (10 μM cyclodextrin), cyclodextrin (10 μM), statin (5 μM), and liposome (7.5 μM) for 24 h. The supernatant was collected, and then 200 μL of DMSO was added to lyse cells and dissolve intracellular NBD-cholesterol. The fluorescence of NBD-cholesterol in the supernatant and lysed cell solutions was measured using SpectraMax Gemini XPS Microplate Reader (excitation 480 nm/emission 530 nm). To prepare low-density lipoproteins (LDL)-laden macrophages, Raw 264.7 cells were treated with LDL (50 μg/mL, Catalog Number L35354; Thermo Fisher Scientific) for 24 h. To observe lipid-droplet formation inside the cell, the LDL-laden macrophages were stained with Oil Red O (Sigma-Aldrich). To observe cellular uptake of CSNP by LDL-laden macrophages and its localization, Raw 264.7 cells were treated with Alexa Fluor 488 LDL (50 μg/mL, Acetylated, Catalog Number L23380; Thermo Fisher Scientific) for 24 h and then treated with lissamine rhodamine B-labeled CSNP (100 μM statin) for 4 h and observed by confocal microscopy utilizing 60× objective lens. To assess the effect of CSNP on cell viability, Raw 264.7 and J774A.1 cells were seeded in 48-well plates for 24 h at a density of 2 × 104 cells per well. To assess dose-dependent effects of CSNP on cell viability, each cell line was treated with CSNP at different statin concentrations (1, 10, 50, and 100 μM) for 48 h, and the cell viability was measured using MTT assay. To assess inhibitory effects of CSNP on cell proliferation, Raw 264.7 cells were seeded in a six-well plate at a density of 2 × 105 cells per well. After 24 h, the cells were treated with CelltraceTM CFSE reagents (Catalog Number C34554; Thermo fisher Scientific) at 5 μM for 20 min. The cells were washed with fresh media and then treated with CSNP (10 μM cyclodextrin, 5 μM statin, 7.5 μM lipid), cyclodextrin (10 μM), statin (5 μM), or lipid (7.5 μM) for 48 h. The percentage of proliferating cells of the total treated cells was determined using flow cytometry. To assess the effect of CSNP on the secretion of MCP-1 and TNF-α, Raw264.7 cells were treated with LPS for 24 h and then with CSNP (10 μM cyclodextrin, 5 μM statin, 7.5 μM lipid), cyclodextrin (10 μM), statin (5 μM), or lipid (7.5 μM) for 48 h. The concentration of MCP-1 and TNF-α in the supernatant was measured with ELISA by following the manufacturer’s instructions. To assess the effect of CSNP on the inflammasome activation, Raw264.7 cells were treated with LPS for 24 h and then with CC (50 μM) for 24 h. The cells were then treated with CSNP (10 μM cyclodextrin, 5 μM statin, 7.5 μM lipid), cyclodextrin (10 μM), statin (5 μM), or lipid (7.5 μM) for 48 h. The concentration of IL-1β in the supernatant was measured with ELISA by following the manufacturer’s instructions. To assess the effect of CSNP on the viability of LDL-laden macrophages, the LDL-treated cells were treated with CSNP (100 μM statin) for 24 h, and the cell viability was measured by MTT assay. To assess the effect of CSNP on the viability of proinflammatory macrophage, Raw 264.7 cells were treated with LPS (100 ng/mL; Sigma-Aldrich) for 24 h and then with CSNP (100 μM statin) for 24 h.
Animal Experiments.
Ten-week-old male ApoE−/− mice were purchased from Jackson Laboratory (Bar Harbor, ME). Seven-week old male wild-type C57/BL6 mice were purchased from Koatech (Gyeonggi-do, South Korea). All animal procedures were approved by the Animal Care and Use committees at Emory University and KAIST.
Pharmacokinetics.
For CSNP, aliquots of 200 μL of DiR-labeled CSNP (15 mg/kg statin) were injected into wild-type mice via the tail vein. Retro-orbital bleeding to collect blood was performed at six different time points after injection (0, 4, 8, 24, 48, and 72 h). For free cyclodextrin, 200 μL of 30 μM RBITC-cyclodextrin was injected, and blood was collected at six different time points after injection (0, 2, 4, 8, 12, and 24 h). The fluorescence of blood samples was measured, and the blood half-life was determined by fitting blood fluorescence to a single-exponential equation for a one-compartment open pharmacokinetic model.
Partial Carotid Ligation Model.
Partial carotid ligation surgery was performed as described previously.41 After ligation surgery, ApoE−/− mice were fed with a Paigen’s high-fat diet (HFD, Catalog Number D12336; Research Diets, New Brunswick, NJ) containing 1.25% cholesterol, 16% fat, and 0.5% cholic acid for 2 weeks.
Biodistribution and Plaque Targeting.
In the LCA-ligation model, ApoE−/− mice were injected intravenously with DiR-labeled CSNP (100 mg/kg cyclodextrin and 15 mg/kg statin) every 3 days (total of 2 times) from 8 days after partial ligation surgery. At 3 days after the last injection, the mice were sacrificed and their organs were harvested to analyze the biodistribution and plaque targeting. For biodistribution analysis, CSNP accumulation in liver, spleen, heart, lung, kidney, left carotid artery (LCA), and right carotid artery (RCA) were imaged and quantified using an NIR fluorescent imaging system (LI-COR, Lincoln, NE). For the assessment of plaque delivery efficacy of cyclodextrin, RBITC-cyclodextrin was incorporated into CSNP or dissolved in PBS and then injected intravenously. For immunofluorescence staining, the left and right carotid arteries were collected after CSNP treatment. The samples were fixed with 3.7% formaldehyde, embedded in Tissue-Tek optimal cutting temperature (OCT) compound (Sakura Finetek, Alphen aan den Rijn, The Netherlands), frozen in liquid nitrogen, cut into sections 10 μm thick, and stored at −80 °C until use. For filipin staining, the sections were washed with PBS and incubated with 50 μg/mL filipin (Sigma-Aldrich) for 30 min at room temperature. They were then rinsed with PBS and mounted using aqueous mounting medium. For CD68 staining, the sections were first treated with blocking solution (1% BSA, 5% goat serum, and 0.02% Tween) for 60 min at room temperature. They were incubated with primary rat antimouse CD68 antibody (2 μg/mL in 10% normal goat serum, Catalog Number 137001; Biolegend, San Diego, CA) overnight at 4 °C and then incubated with secondary goat antirat IgG (4 μg/mL in 10% normal goat serum, Catalog Number 405416; Biolegend) for 60 min at room temperature. The sections were washed with PBS and counterstained with Hoechst 33342 (5 μg/mL in PBS; Thermo Fisher Scientific) for 2 min at room temperature. After a final wash with PBS, the sections were mounted using aqueous mounting medium.
Ex Vivo Plaque Cholesterol Dissolution.
To observe cholesterol dissolution within plaques after CSNP treatment in the LCA-ligation model, ApoE−/− mice were injected intravenously with CSNP (200 mg/kg cyclodextrin and 30 mg/kg statin), liposomal statin (30 mg/kg statin), statin (30 mg/kg), cyclodextrin (400 mg/kg), or PBS at 13 days after partial ligation surgery. Twenty-four hours after the injection, the left carotid arteries were collected and put into PBS, respectively. The samples were then incubated at 37 °C in the shaking incubator for 24 h. Dissolved cholesterol in the PBS was measured using a cholesterol assay kit (Catalog Number A12216; Thermo Fisher Scientific) in accordance with the instructions provided by the manufacturer.
Antiatherogenic Therapy In Vivo.
In the LCA-ligation model, ApoE−/− mice were injected intravenously with CSNP (15 mg/kg statin and 100 mg/kg cyclodextrin), liposomal statin (15 mg/kg statin), statin (15 mg/kg), cyclodextrin (400 mg/kg), or PBS at 2, 5, 8, and 11 days after ligation. The mice were maintained on the HFD throughout the 2-week treatment period and sacrificed for analysis at 14 days after ligation.
Plaque Regression Therapy In Vivo.
In the HFD-induced atherosclerosis model, ApoE−/− mice were first fed the HFD for 12 weeks. Then a baseline group was sacrificed, and treatment groups were injected intravenously with either CSNP (100 mg/kg cyclodextrin and 15 mg/kg statin) or PBS twice a week for an additional 4 weeks. During treatment, the diet was either maintained with HFD or switched to normal chow diet. The mice were sacrificed at 16 weeks for analysis.
Plaque Lesion Analysis.
For the LCA-ligation model, the carotid arteries were isolated from the ligated mice after treatment. Left and right carotid arteries were photographed using a charge-coupled device camera (Moticam 2500; Motic, Richmond, BC) attached to a dissection microscope (EMZ-8TRD; Meiji Techno, Saitama, Japan) at 3× magnification, and the opaque area covered by plaque and the total artery area were quantified using NIH ImageJ software. For the HFD-induced advanced atherosclerosis model, aortic arch and thoracic aorta were opened longitudinally and stained with Oil-Red-O. The stained aortic arch and thoracic aorta were photographed using a charge-coupled device camera (Moticam 2500; Motic, Richmond, BC) attached to a dissection microscope (EMZ-8TRD; Meiji Techno, Saitama, Japan) at 3× magnification, and the red area covered by plaque and the total artery area were quantified using NIH ImageJ software.
Histological Analysis of Plaque.
After treatment, the left and right carotid arteries in the partial carotid ligation model, and the aortic roots in the HFD-induced advanced atherosclerosis model were collected, respectively. The samples were fixed with 3.7% formaldehyde. Then they were embedded in Tissue-Tek optimal cutting temperature (OCT) compound (Sakura Finetek), frozen in liquid nitrogen, cut into sections 10 μm thick, and stored at −80 °C until use. For Oil-Red-O staining, the sections were washed with distilled water and rinsed with 60% isopropanol (Sigma-Aldrich). The sections were then stained with 0.3% Oil-Red-O solution for 15 min. Following staining, they were rinsed with isopropanol (60%) and counterstained with hematoxylin (Sigma-Aldrich) for 2 min. After final washing with distilled water, the sections were mounted using aqueous mounting medium. The aortic roots were imaged using a 4× objective lens, and the carotid arteries were imaged using a 20× objective lens on light microscope (Nikon). For the quantification of plaque sizes, at least three cross-section images obtained from Oil-Red-O staining were used for each mouse. For the quantification of plaque sizes in the aortic roots, frozen sections were serially collected when the three leaflets of aortic valve were recognized on unstained sections. To visualize collagen in the plaques, a picrosirius red stain kit (Catalog Number ab150681; Abcam) was used in accordance with the instructions provide by the manufacturer. Necrotic areas were defined as areas that were free of picrosirius red staining (unstained white areas, and yellowish and debris-filled areas). A section in the middle was used for the staining. Photographs of the stained specimens were digitized for data analysis using ImageJ.
Histological Analysis of Liver and Spleen.
After antiatherogenic therapy, liver and spleen were collected, fixed with 3.7% formaldehyde for 24 h, washed with PBS, and put into 30% sucrose solution until they sunk. Then they were embedded in Tissue-Tek optimal cutting temperature (OCT) compound (Sakura Finetek), frozen in the fridge, and cut into sections 10 μm thick. The sections were stained with hematoxylin and eosin.
Blood Tests and Cholesterol Analysis.
Whole blood was collected using heparinized syringes when the mice were sacrificed after treatment. Plasma lipids [total cholesterol, triglycerides, high-density lipoproteins (HDL), and LDL] were analyzed using a CX7 biochemical analyzer (Beckman Coulter, Brea, CA). Blood concentrations of alanine transaminase and aspartate transaminase were analyzed using an AU480 chemistry analyzer (Beckman Coulter).
HMG-CoA Reductase Activity in Liver.
Wild-type mice were injected intravenously with PBS or CSNP (15 mg/kg statin, 100 mg/kg cyclodextrin) every 3 days (total of four times). On day 3 after the last injection, the mice were sacrificed, and the livers were immediately harvested, weighed, and placed in PBS at 4 °C. The livers were then homogenized in ice-cold solution containing 0.3 M sucrose, 10 mM β-mercaptoethanol, and 10 mM sodium EDTA, pH 7.4, for 2 min using a TissueLyserII at 20–30 Hz (QIAGEN, Hilden, Germany). Homogenization was repeated until no tissue debris was visible. At each time of operation, tubes were rearranged to ensure uniform homogenization. After homogenization, cell debris and mitochondria were removed by centrifugation for 15 min at 12 000 × g. The supernatant was centrifuged again under the same conditions. To remove soluble enzymes and concentrate microsomes, the resulting supernatant was transferred to Amicon centrifugal filters with 100 kDa MW cutoff (Millipore) and centrifuged for 30 min at 5000 × g at 4 °C. Centrifugation was performed three times, and the final buffer was changed to a solution containing 0.3 M sucrose, 10 mM β-mercaptoethanol, 10 mM sodium EDTA, and 10 mM dithiothreitol. After isolation of microsomes, BCA assay was performed to measure protein concentration, and the sample concentration was adjusted to 5 mg/mL. HMG-CoA reductase activity in the sample was measured using an HMG-CoA Reductase Assay Kit (Sigma) following the manufacturer’s protocols.
Statistical Analysis.
Data are presented as mean ± s.e.m. The significance of differences was calculated using the paired two-tailed Student’s t test or one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison post hoc test with GraphPad Prism 7.0 (GraphPad Software, San Diego, CA). In all analyses, P < 0.05 was taken to indicate statistical significance (*P < 0.05, **P < 0.01, ***P < 0.001). No statistical methods were used to predetermine sample size for animal or other experiments. For in vivo therapy studies, more than five mice were assigned randomly to each group. All experiments were repeated at least twice and representative data are reported.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Basic Science Research Program through the National Research Foundation (NRF-2017R1E1A1A01074847 to J.-H.P.) funded by the Ministry of Science and ICT, Republic of Korea and the National Institute of Health (HL139757 to H.J.), USA.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.9b08216.
Competitive binding and cargo-switching assay using size exclusion chromatography; CC-mediated disruption of DOPC-based liposomal membrane at 37 °C; preparation and physiochemical properties of CSNP; FRET assay for demonstration of cargo-switching; antiatherogenic effects of CSNP in vitro; pharmacokinetics and plaque targeting of CSNP, cyclodextrin, and liposomal statin; liver and spleen toxicity of CSNP after antiatherogenesis therapy; assessment of systemic side effects and plaque vulnerability after CSNP treatment; change of HMG-CoA reductase (HMGCR) activity after CSNP treatment in wild-type mice (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acsnano.9b08216
The authors declare no competing financial interest.
Contributor Information
Heegon Kim, Department of Bio and Brain Engineering and KAIST Institute for Health Science and Technology, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea.
Hanjoong Jo, Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Division of Cardiology, Department of Medicine, Emory University, Atlanta, Georgia 30322, United States.
Ji-Ho Park, Department of Bio and Brain Engineering and KAIST Institute for Health Science and Technology, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea;.
Data Availability.
The data that support the plots within this paper and other findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- (1).Libby P; Theroux P Pathophysiology of Coronary Artery Disease. Circulation 2005, 111, 3481–3488. [DOI] [PubMed] [Google Scholar]
- (2).Hansson GK; Libby P The Immune Response in Atherosclerosis: A Double-Edged Sword. Nat. Rev. Immunol 2006, 6, 508–519. [DOI] [PubMed] [Google Scholar]
- (3).Duewell P; Kono H; Rayner KJ; Sirois CM; Vladimer G; Bauernfeind FG; Abela GS; Franchi L; Nuñez G; Schnurr M; Espevik T; Lien E; Fitzgerald KA; Rock KL; Moore KJ; Wright SD; Hornung V; Latz E NLRP3 Inflammasomes Are Required for Atherogenesis and Activated by Cholesterol Crystals. Nature 2010, 464, 1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Moore KJ; Sheedy FJ; Fisher EA Macrophages in Atherosclerosis: A Dynamic Balance. Nat. Rev. Immunol 2013, 13, 709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Seimon T; Tabas I Mechanisms and Consequences of Macrophage Apoptosis in Atherosclerosis. J. Lipid Res 2009, 50, S382–S387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Grebe A; Latz E Cholesterol Crystals and Inflammation. Curr. Rheumatol. Rep 2013, 15, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Samstad EO; Niyonzima N; Nymo S; Aune MH; Ryan L; Bakke SS; Lappegard KT; Brekke OL; Lambris JD; Damas JK; Latz E; Mollnes TE; Espevik T Cholesterol Crystals Induce Complement-Dependent Inflammasome Activation and Cytokine Release. J. Immunol 2014, 192, 2837–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Yu XH; Fu YC; Zhang DW; Yin K; Tang CK Foam Cells in Atherosclerosis. Clin. Chim. Acta 2013, 424, 245–252. [DOI] [PubMed] [Google Scholar]
- (9).Flores AM; Ye J; Jarr KU; Hosseini-Nassab N; Smith BR; Leeper NJ Nanoparticle Therapy for Vascular Diseases. Arterioscler., Thromb., Vasc. Biol 2019, 39, 635–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Chan CKW; Zhang L; Cheng CK; Yang HR; Huang Y; Tian XY; Choi CHJ Recent Advances in Managing Atherosclerosis via Nanomedicine. Small 2018, 14, 1702793. [DOI] [PubMed] [Google Scholar]
- (11).Katsuki S; Matoba T; Koga JI; Nakano K; Egashira K Anti-Inflammatory Nanomedicine for Cardiovascular Disease. Front. Cardiovasc. Med 2017, 4, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Gadde S; Rayner KJ Nanomedicine Meets microRNA: Current Advances in RNA-Based Nanotherapies for Atherosclerosis. Arterioscler., Thromb., Vasc. Biol 2016, 36, e73–e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Levine GN; Keaney JF Jr.; Vita JA Cholesterol Reduction in Cardiovascular Disease. Clinical Benefits and Possible Mechanisms. N. Engl. J. Med 1995, 332, 512–521. [DOI] [PubMed] [Google Scholar]
- (14).Jain MK; Ridker PM Anti-Inflammatory Effects of Statins: Clinical Evidence and Basic Mechanisms. Nat. Rev. Drug Discovery 2005, 4, 977–987. [DOI] [PubMed] [Google Scholar]
- (15).Liao JK; Laufs U Pleiotropic Effects of Statins. Annu. Rev. Pharmacol. Toxicol 2005, 45, 89–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Tang J; Lobatto ME; Hassing L; van der Staay S; van Rijs SM; Calcagno C; Braza MS; Baxter S; Fay F; Sanchez-Gaytan BL; Duivenvoorden R; Sager HB; Astudillo YM; Leong W; Ramachandran S; Storm G; Pérez-Medina C; Reiner T; Cormode DP; Strijkers GJ; et al. Inhibiting Macrophage Proliferation Suppresses Atherosclerotic Plaque Inflammation. Sci. Adv 2015, 1, No. e1400223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Duivenvoorden R; Tang J; Cormode DP; Mieszawska AJ; Izquierdo-Garcia D; Ozcan C; Otten MJ; Zaidi N; Lobatto ME; van Rijs SM; Priem B; Kuan EL; Martel C; Hewing B; Sager H; Nahrendorf M; Randolph GJ; Stroes ES; Fuster V; Fisher EA; et al. A Statin-Loaded Reconstituted High-Density Lipoprotein Nanoparticle Inhibits Atherosclerotic Plaque Inflammation. Nat. Commun 2014, 5, 3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Davis ME; Brewster ME Cyclodextrin-Based Pharmaceutics: Past, Present and Future. Nat. Rev. Drug Discovery 2004, 3, 1023–1035. [DOI] [PubMed] [Google Scholar]
- (19).Choisnard L; Geze A; Putaux JL; Wong YS; Wouessidjewe D Nanoparticles of Beta-Cyclodextrin Esters Obtained by Self-Assembling of Biotransesterified Beta-Cyclodextrins. Biomacromolecules 2006, 7, 515–520. [DOI] [PubMed] [Google Scholar]
- (20).Goubet I; Dahout C; Semon E; Guichard E; Le Quere JL; Voilley A Competitive Binding of Aroma Compounds by Beta-Cyclodextrin. J. Agric. Food Chem 2001, 49, 5916–5922. [DOI] [PubMed] [Google Scholar]
- (21).Zimmer S; Grebe A; Bakke SS; Bode N; Halvorsen B; Ulas T; Skjelland M; De Nardo D; Labzin LI; Kerksiek A; Hempel C; Heneka MT; Hawxhurst V; Fitzgerald ML; Trebicka J; Bjorkhem I; Gustafsson JA; Westerterp M; Tall AR; Wright SD; et al. Cyclodextrin Promotes Atherosclerosis Regression via Macrophage Reprogramming. Sci. Transl. Med 2016, 8, 333ra50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Crumling MA; King KA; Duncan RK Cyclodextrins and Iatrogenic Hearing Loss: New Drugs with Significant Risk. Front. Cell. Neurosci 2017, 11, 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lobatto ME; Fuster V; Fayad ZA; Mulder WJM Perspectives and Opportunities for Nanomedicine in the Management of Atherosclerosis. Nat. Rev. Drug Discovery 2011, 10, 845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Mulder WJM; Jaffer FA; Fayad ZA; Nahrendorf M Imaging and Nanomedicine in Inflammatory Atherosclerosis. Sci. Transl. Med 2014, 6, 239sr1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Lopez CA; de Vries AH; Marrink SJ Molecular Mechanism of Cyclodextrin Mediated Cholesterol Extraction. PLoS Comput. Biol 2011, 7, No. e1002020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Sule A; Szente L; Csempesz F Enhancement of Drug Solubility in Supramolecular and Colloidal Systems. J. Pharm. Sci 2009, 98, 484–494. [DOI] [PubMed] [Google Scholar]
- (27).Davidson CD; Fishman YI; Puskas I; Szeman J; Sohajda T; McCauliff LA; Sikora J; Storch J; Vanier MT; Szente L; Walkley SU; Dobrenis K Efficacy and Ototoxicity of Different Cyclodextrins in Niemann-Pick C Disease. Ann. Clin. Transl. Neurol 2016, 3, 366–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Csempesz F; Süle A; Puskás I Induced Surface Activity of Supramolecular Cyclodextrin–Statin Complexes: Relevance in Drug Delivery. Colloids Surf., A 2010, 354, 308–313. [Google Scholar]
- (29).Akbarzadeh A; Rezaei-Sadabady R; Davaran S; Joo SW; Zarghami N; Hanifehpour Y; Samiei M; Kouhi M; Nejati-Koshki K Liposome: Classification, Preparation, and Applications. Nanoscale Res. Lett 2013, 8, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ulrich AS; Sami M; Watts A Hydration of DOPC Bilayers by Differential Scanning Calorimetry. Biochim. Biophys. Acta, Biomembr 1994, 1191, 225–230. [DOI] [PubMed] [Google Scholar]
- (31).Chen J; Cheng D; Li J; Wang Y; Guo JX; Chen ZP; Cai BC; Yang T Influence of Lipid Composition on the Phase Transition Temperature of Liposomes Composed of Both DPPC and HSPC. Drug Dev. Ind. Pharm 2013, 39, 197–204. [DOI] [PubMed] [Google Scholar]
- (32).Brown A; Patel S; Ward C; Lorenz A; Ortiz M; DuRoss A; Wieghardt F; Esch A; Otten EG; Heiser LM; Korolchuk VI; Sun C; Sarkar S; Sahay G PEG-Lipid Micelles Enable Cholesterol Efflux in Niemann-Pick Type C1 Disease-Based Lysosomal Storage Disorder. Sci. Rep 2016, 6, 31750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Atger VM; Moya MD; Stoudt GW; Rodrigueza WV; Phillips MC; Rothblat GH Cyclodextrins as Catalysts for the Removal of Cholesterol from Macrophage Foam Cells. J. Clin. Invest 1997, 99, 773–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Miller CR; Bondurant B; McLean SD; McGovern KA; O’Brien DF Liposome-Cell Interactions In Vitro: Effect of Liposome Surface Charge on the Binding and Endocytosis of Conventional and Sterically Stabilized Liposomes. Biochemistry 1998, 37, 12875–12883. [DOI] [PubMed] [Google Scholar]
- (35).Moore KJ; Sheedy FJ; Fisher EA Macrophages in Atherosclerosis: A Dynamic Balance. Nat. Rev. Immunol 2013, 13, 709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Bagalkot V; Badgeley MA; Kampfrath T; Deiuliis JA; Rajagopalan S; Maiseyeu A Hybrid Nanoparticles Improve Targeting to Inflammatory Macrophages through Phagocytic Signals. J. Controlled Release 2015, 217, 243–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Rajamaki K; Lappalainen J; Oorni K; Valimaki E; Matikainen S; Kovanen PT; Eklund KK Cholesterol Crystals Activate the NLRP3 Inflammasome in Human Macrophages: A Novel Link between Cholesterol Metabolism and Inflammation. PLoS One 2010, 5, No. e11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Zolberg Relevy N; Bechor S; Harari A; Ben-Amotz A; Kamari Y; Harats D; Shaish A The Inhibition of Macrophage Foam Cell Formation by 9-Cis β-Carotene is Driven by BCMO1 Activity. PLoS One 2015, 10, No. e0115272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Grosse PY; Bressolle F; Rouanet P; Joulia JM; Pinguet F Methyl-Beta-Cyclodextrin and Doxorubicin Pharmacokinetics and Tissue Concentrations Following Bolus Injection of These Drugs Alone or Together in the Rabbit. Int. J. Pharm 1999, 180, 215–223. [DOI] [PubMed] [Google Scholar]
- (40).Schachter M Chemical, Pharmacokinetic and Pharmacodynamic Properties of Statins: An Update. Fundam. Clin. Pharmacol 2005, 19, 117–125. [DOI] [PubMed] [Google Scholar]
- (41).Nam D; Ni CW; Rezvan A; Suo J; Budzyn K; Llanos A; Harrison D; Giddens D; Jo H Partial Carotid Ligation is a Model of Acutely Induced Disturbed Flow, Leading to Rapid Endothelial Dysfunction and Atherosclerosis. Am. J. Physiol. Heart Circ. Physiol 2009, 297, H1535–H1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Jo H; Son D; Kumar S The Atypical Mechanosensitive Microrna-712 Derived from Pre-Ribosomal Rna Induces Endothelial Inflammation and Atherosclerosis. Atherosclerosis 2014, 235, E40–E41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Sparrow CP; Burton CA; Hernandez M; Mundt S; Hassing H; Patel S; Rosa R; Hermanowski-Vosatka A; Wang PR; Zhang DH; Peterson L; Detmers PA; Chao YS; Wright SD Simvastatin Has Anti-Inflammatory and Antiatherosclerotic Activities Independent of Plasma Cholesterol Lowering. Arterioscler., Thromb., Vasc. Biol 2001, 21, 115–121. [DOI] [PubMed] [Google Scholar]
- (44).Plump AS; Smith JD; Hayek T; Aalto-Setälä K; Walsh A; Verstuyft JG; Rubin EM; Breslow JL Severe Hyper-cholesterolemia and Atherosclerosis in Apolipoprotein E-Deficient Mice Created by Homologous Recombination in ES Cells. Cell 1992, 71, 343–353. [DOI] [PubMed] [Google Scholar]
- (45).Bergstrom JD; Bostedor RG; Rew DJ; Geissler WM; Wright SD; Chao YS Hepatic Responses to Inhibition of 3-Hydroxy-3-Methylglutaryl-CoA Reductase: A Comparison of Atorvastatin and Simvastatin. Biochim. Biophys. Acta, Lipids Lipid Metab 1998, 1389, 213–221. [DOI] [PubMed] [Google Scholar]
- (46).Yamauchi S; Linscheer WG; Beach DH Increase in Serum and Bile Cholesterol and HMG-CoA Reductase by Lovastatin in Rats. Am. J. Physiol 1991, 260, G625–G630. [DOI] [PubMed] [Google Scholar]
- (47).Reihner E; Rudling M; Stahlberg D; Berglund L; Ewerth S; Bjorkhem I; Einarsson K; Angelin B Influence of Pravastatin, a Specific Inhibitor of HMG-CoA Reductase, on Hepatic Metabolism of Cholesterol. N. Engl. J. Med 1990, 323, 224–228. [DOI] [PubMed] [Google Scholar]
- (48).Schonewille M; de Boer JF; Mele L; Wolters H; Bloks VW; Wolters JC; Kuivenhoven JA; Tietge UJ; Brufau G; Groen AK Statins Increase Hepatic Cholesterol Synthesis and Stimulate Fecal Cholesterol Elimination in Mice. J. Lipid Res 2016, 57, 1455–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Du J; Lane LA; Nie S Stimuli-Responsive Nanoparticles for Targeting the Tumor Microenvironment. J. Controlled Release 2015, 219, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Uthaman S; Huh KM; Park IK Tumor Microenvironment-Responsive Nanoparticles for Cancer Theragnostic Applications. Biomater. Res 2018, 22, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Irie T; Fukunaga K; Garwood MK; Carpenter TO; Pitha J; Pitha J Hydroxypropylcyclodextrins in Parenteral Use 0.2. Effects on Transport and Disposition of Lipids in Rabbit and Humans. J. Pharm. Sci 1992, 81, 524–528. [DOI] [PubMed] [Google Scholar]
- (52).Jun SW; Kim MS; Kim JS; Park HJ; Lee S; Woo JS; Hwang SJ Preparation and Characterization of Simvastatin/Hydroxypropyl-Beta-Cyclodextrin Inclusion Complex Using Super-critical Antisolvent (SAS) Process. Eur. J. Pharm. Biopharm 2007, 66, 413–421. [DOI] [PubMed] [Google Scholar]
- (53).Rosenbaum AI; Zhang GT; Warren JD; Maxfield FR Endocytosis of Beta-Cyclodextrins Is Responsible for Cholesterol Reduction in Niemann-Pick Type C Mutant Cells. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 5477–5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Christian AE; Byun HS; Zhong N; Wanunu M; Marti T; Furer A; Diederich F; Bittman R; Rothblat GH Comparison of the Capacity of Beta-Cyclodextrin Derivatives and Cyclophanes to Shuttle Cholesterol between Cells and Serum Lipoproteins. J. Lipid Res 1999, 40, 1475–1482. [PubMed] [Google Scholar]
- (55).Tanaka Y; Yamada Y; Ishitsuka Y; Matsuo M; Shiraishi K; Wada K; Uchio Y; Kondo Y; Takeo T; Nakagata N; Higashi T; Motoyama K; Arima H; Mochinaga S; Higaki K; Ohno K; Irie T Efficacy of 2-Hydroxypropyl-Beta-Cyclodextrin in Niemann-Pick Disease Type C Model Mice and Its Pharmacokinetic Analysis in a Patient with the Disease. Biol. Pharm. Bull 2015, 38, 844–851. [DOI] [PubMed] [Google Scholar]
- (56).Cantuti-Castelvetri L; Fitzner D; Bosch-Queralt M; Weil MT; Su M; Sen P; Ruhwedel T; Mitkovski M; Trendelenburg G; Lutjohann D; Mobius W; Simons M Defective Cholesterol Clearance Limits Remyelination in the Aged Central Nervous System. Science 2018, 359, 684–688. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the plots within this paper and other findings of this study are available from the corresponding author upon reasonable request.