

Abstract
The heterogeneity associated with glycosylation of the 66 N-glycan sites on the protein trimer making up the spike (S) region of the SARS-CoV-2 virus has been assessed by charge detection mass spectrometry (CDMS). CDMS allows simultaneous measurement of the mass-to-charge ratio and charge of individual ions, so that mass distributions can be determined for highly heterogeneous proteins such as the heavily glycosylated S protein trimer. The CDMS results are compared to recent glycoproteomics studies of the structure and abundance of glycans at specific sites. Interestingly, average glycan masses determined by “top-down” CDMS measurements are 35–47% larger than those obtained from the “bottom-up” glycoproteomics studies, suggesting that the glycoproteomic measurements underestimated the abundances of larger, more-complex glycans. Moreover, the distribution of glycan masses determined by CDMS is much broader than the distribution expected from the glycoproteomics studies, assuming that glycan processing on each trimer is not correlated. The breadth of the glycan mass distribution therefore indicates heterogeneity in the extent of glycan processing of the S protein trimers, with some trimers being much more heavily processed than others. This heterogeneity may have evolved as a way of further confounding the host’s immune system.
Graphical Abstract
INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a novel coronavirus that emerged in Wuhan, China in late 2019 giving rise to the COVID-19 pandemic.1–5 SARS-CoV-2 is an enveloped, positive, single-stranded RNA virus. The viral envelope has two structural glycoproteins called the membrane and spike (S) proteins. The S protein is a large transmembrane protein and trimers of the S protein decorate the surface of the viral envelope, giving the virus its characteristic appearance. The S protein mediates cell entry by fusion of the host and viral membranes. It also plays key roles in neutralizing-antibody and T-cell responses, and consequently it is the primary target for vaccine and therapeutic development.6,7
The S protein consists of two subunits: S1 contains the receptor binding domain (RBD) and S2 is responsible for membrane fusion. S1 binds to the host cell’s angiotensin converting enzyme II (ACE2) receptor. Before ACE2 binding, the prefusion S trimer exists in either an open or closed configuration.8 The closed state shields the RBD of the S protein from immune recognition and ACE2 binding, and the open configuration allows the S trimer to initiate binding.9–11 Once bound to ACE2, S trimers shed their S1 subunits allowing the S2 subunits to fuse to the membranes of host cells.8,12
The S protein is heavily glycosylated and these modifications play key roles in facilitating immune evasion by shielding the underlying protein surface to prevent antibody recognition.13,14 Glycosylation of viral proteins utilizes host-cell machinery, as the viral envelope is developed by budding through the endoplasmic reticulum (ER) or Golgi apparatus. The S protein has 22 potential N-glycosylation sites (66 N-glycan sites on the trimer) and at least 3 sites for O-glycosylation have been predicted.15 To date, glycoproteomics studies of the SARS-CoV-2 S protein using enzymatic digestion and mass spectrometry have been applied to determine the glycan composition at each of these 22 sites N-glycan sites.16–20 N-linked high mannose, hybrid, and complex glycans have been reported.16 However, it appears that the glycan composition and occupancy at each site is different if the S1 and S2 subunits are expressed separately.17 In addition to the 22 N-glycans, O-linked glycans have been detected at two sites on the S1 subunit.17
The number of distinct glycoforms is the product of the numbers of different glycans that can occupy each site on the S protein trimer. If we consider glycans with a population of >1% from previous reports,16 then 8.2 × 1075 glycoforms would be anticipated, assuming that glycosylation at different sites on the trimer is not correlated. Furthermore, the most likely glycoform of the spike trimer has a probability of only 1.9 × 10−34 (see below). Thus, the probability that two spike trimers have the same glycan distribution is vanishingly small. Consequently, every spike trimer present on the surface of a SARS-CoV-2 virus is expected to be different; the question is how different?
To address this question, we have employed a single particle approach, charge detection mass spectrometry (CDMS), to directly determine the mass of individual ions by simultaneously measuring the m/z ratio and charge of each ion.21,22 CDMS has traditionally been used to analyze large molecules and complexes. However, this technique also has great utility for analysis of very heterogeneous mixtures. Herein, we use CDMS to make measurements on thousands of individual trimeric spike protein ions derived from different cell lines to provide information on the glycan mass distribution for the S protein trimer (Scheme 1). A comparison of this CDMS data with previous MS-based glycoproteomics, which identified glycan compositions and abundances at specific sites, reveals a greater average glycan mass determined directly by CDMS. In addition, the distribution of glycan mass determined by CDMS is broader than expected for an uncorrelated glycan distribution (where the processing of glycans at one site is independent of the processing at other sites), indicating that the glycans on some S protein trimers are more heavily processed than on others.
Scheme 1. S Protein samples used for CDMS measurementsa.
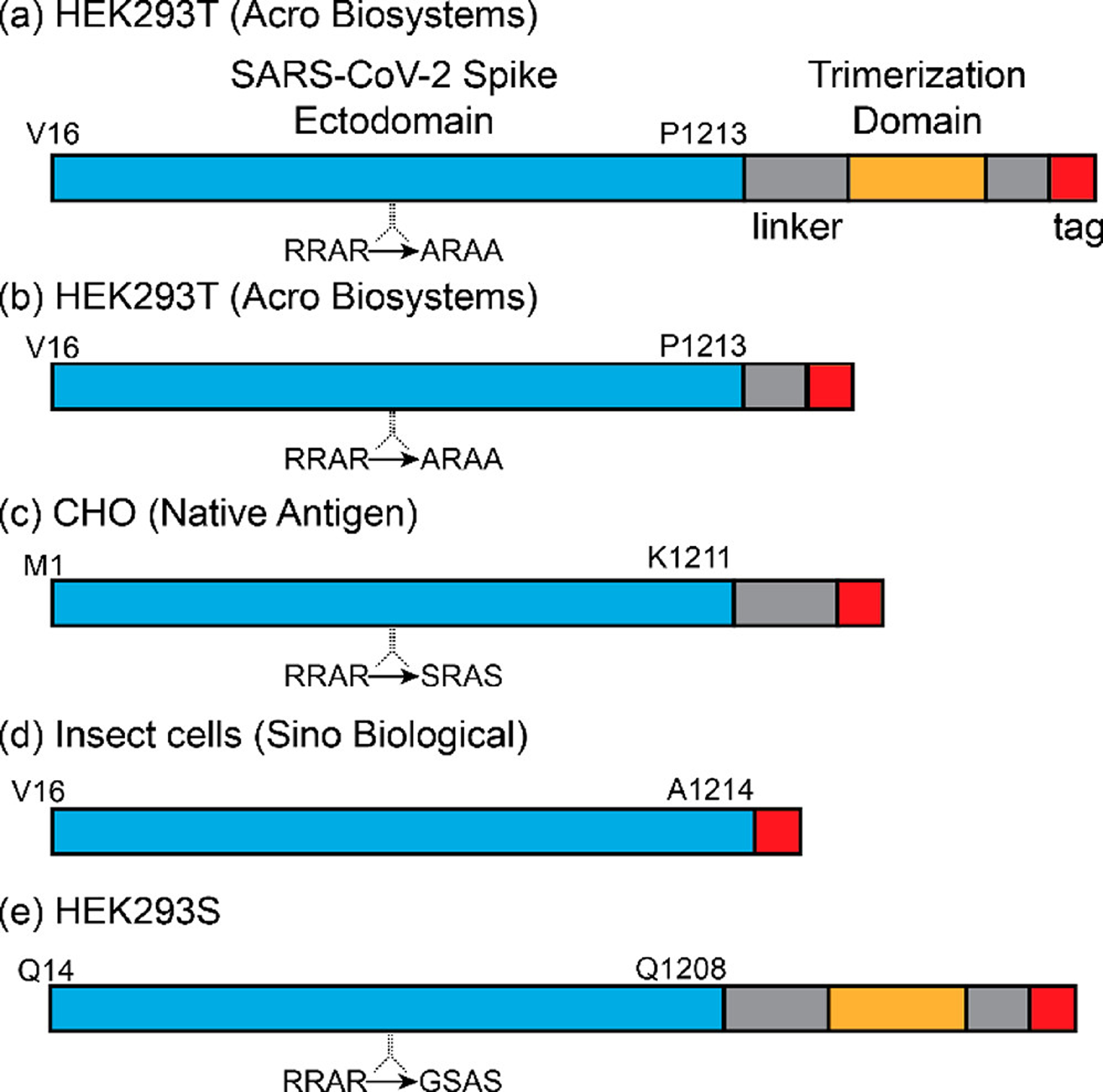
aThe salient features of each sample are (a) An S protein trimer derived from HEK293 cells. It contains V16–P1213, a modified furin cleavage sequence, several linkers, a fibritin trimerization domain23,24 to stabilize the S protein trimer, and a poly-His tag (purchased from Acro Biosystems); (b) S protein identical to part a except it is missing a linker and the fibritin trimerization domain (purchased from Acro Biosystems). (c) S protein derived from CHO cells. It has the predicted sequence M1-K1211, a modified furin cleavage sequence, a linker, and a poly-His tag (purchased from The Native Antigen Company); (d) S protein expressed in insect cells (predicted sequence V16–P1213) containing an unmodified furin cleavage site and a poly-His tag (purchased from Sino Biologics). (e) S protein with fibritin trimerization domain expressed in HEK 293S GnTI- cells to reduce glycan heterogeneity (described in ref 20). It is expected to lead with Q14. The molecular masses of the proteins determined from their sequences (i.e., unglycosylated) are (a) 138068.85 Da, (b) 134642.05 Da, (c) 135607.36, (d) 134366.88 Da, and (e) 140827.76 Da.
RESULTS
Mass Distribution Measured for the S Protein with a Trimerization Domain.
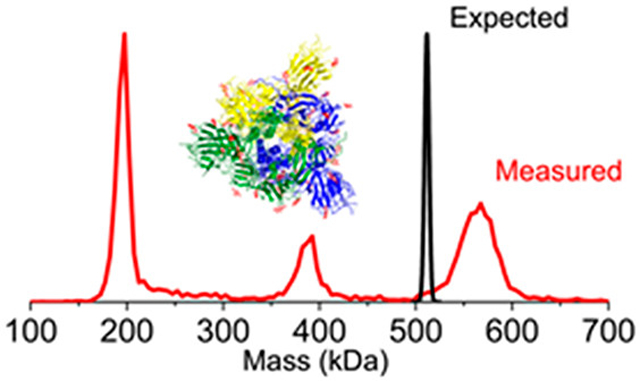
A typical mass distribution recorded following electrospray of the spike protein trimer described in Scheme 1a is shown in Figure 1a (blue). The mass distribution plotted over the mass range of 0–1000 kDa (inset in Figure 1a) reveals a single prominent peak, centered around 560 kDa, attributed to the trimer. This sample incorporates a fibritin trimerization domain23,24 to stabilize the trimer. There are no peaks close to the masses expected for the monomer or dimer in the spectrum. The charge distribution of the spike trimer (Figure 1b) reveals a charge RMSD of 0.191 e (elementary charges). The charge states are almost completely baseline resolved. Therefore, individual ions can be assigned to specific integer charge states with a low error rate and the uncertainty in the charge does not degrade the mass resolution.
Figure 1.
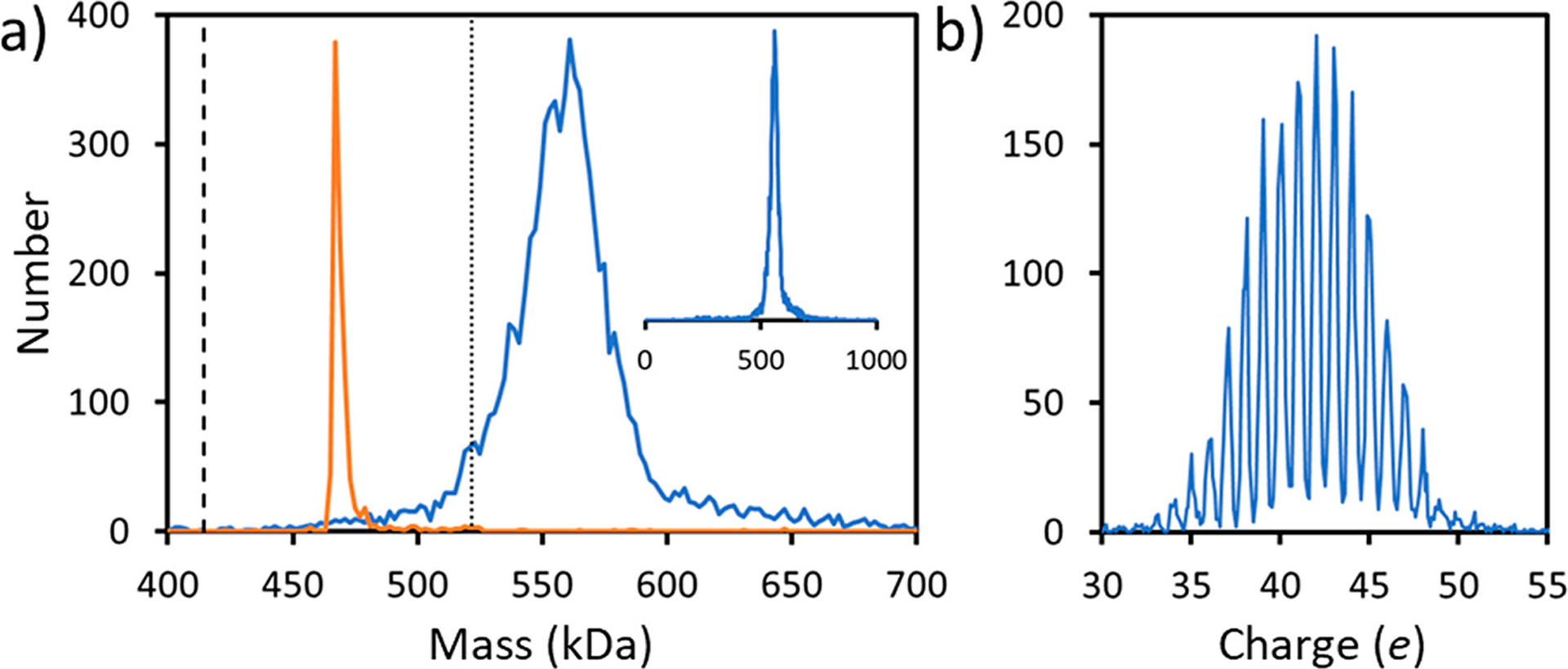
CDMS measurements for the S protein with a trimerization domain (Scheme 1a). (a) Mass spectrum. The blue lines show the measured mass distribution. The inset shows the distribution over the 0–1000 kDa range. (b) Charge spectrum. The charge RMSD is 0.191 e. A bin size of 2 kDa was used for part a and a bin size of 0.1 e was used for part b. The orange line in part a shows the CDMS spectrum measured for β-galactosidase under identical conditions. The dashed line in part a at 414.2 kDa shows the expected mass of the unglycosylated S protein trimer, and the dotted line at 521.7 kDa shows the expected mass of the glycosylated trimer (see text).
The mass distribution for the spike protein trimer is relatively broad, around 35 kDa fwhm. As a comparator, we measured the mass distribution for β-galactosidase using the same experimental conditions (Figure 1a). The peak for β-galactosidase (orange line) is much narrower, and its measured mass (467.6 kDa) is <0.5% larger than the expected mass (465.4 kDa). Masses measured for large protein complexes by MS are usually slightly larger than the expected masses because of residual salt and counterions. This result shows that the broad distribution measured for the spike trimer is not a consequence of the experimental conditions and must therefore arise from heterogeneity.
The expected mass of the glycosylated S protein trimer can be obtained by adding the sequence mass of the unglycosylated trimer (414.2 kDa) to the average mass of the N-glycans (107.5 kDa) from previous glycoproteomics studies.16 The value obtained (521.7 kDa) is shown by the dotted line in Figure 1. The center of the measured mass distribution is ~37 kDa higher than the expected mass. Some of this excess mass could be attributed to residual salt mentioned above. However, this is expected to contribute only 2–3 kDa based on previous studies25,26 and the results for β-galactosidase described above. O-linked glycans could also contribute. Three sites for O-glycosylation have been predicted near the furin cleavage site15 with recent results suggesting that one of the sites (T678) is glycosylated.19 There is also evidence that T323 is glycosylated, and possibly S325.17 However, the extent of O-glycosylation revealed to date is much less than required to account for the 37 kDa additional mass.
Calculation of the Glycan Mass Distribution for the Spike Trimer.
Considering both the additional mass and broad distribution measured for the spike trimer, we calculated the distribution that would arise assuming all 66 sites were populated according to the glycoproteomics study. As noted above, the number of different glycoforms is 8.2 × 1075 if glycans with a population >1% are considered. The probability that a particular glycoform is populated is
| (1) |
where is the probability, from the glycoproteomics study,16 that site 1 is occupied by glycan a, etc. The probability for the most likely glycoform (where all sites are occupied by the most probable glycan) is 1.9 × 10−34. Note that in eq 1, we assume that the glycan sites are uncorrelated. In other words, the glycan at site 2 is completely independent of the nature of the glycan at site 1. Because of the enormous number of possible glycoforms indicated above we calculated the glycan mass distribution using a Monte Carlo (MC) approach with importance sampling, where the probability of sampling a particular glycoform is given by eq 1. The glycan mass distribution calculated in this way is shown in Figure 2a. The overall peak shape is close to Gaussian and the distribution is centered at 107.5 kDa. The distribution in Figure 2a was obtained using 1 Da bins and reveals a series of resonances separated by ~1 kDa. An expanded region shows that the resonances consist of a series of sharp peaks (Figure 2b) while further expansion (Figure 2c) reveals that these peaks are separated by ~8 Da. The results shown by the blue lines in Figure 2 were obtained from 1010 MC samples. To demonstrate that this is sufficient to provide an accurate representation of the mass distribution, the red dashed line in Figure 2c shows results obtained from 109 samples. The results are almost identical, confirming that enough samples were performed.
Figure 2.
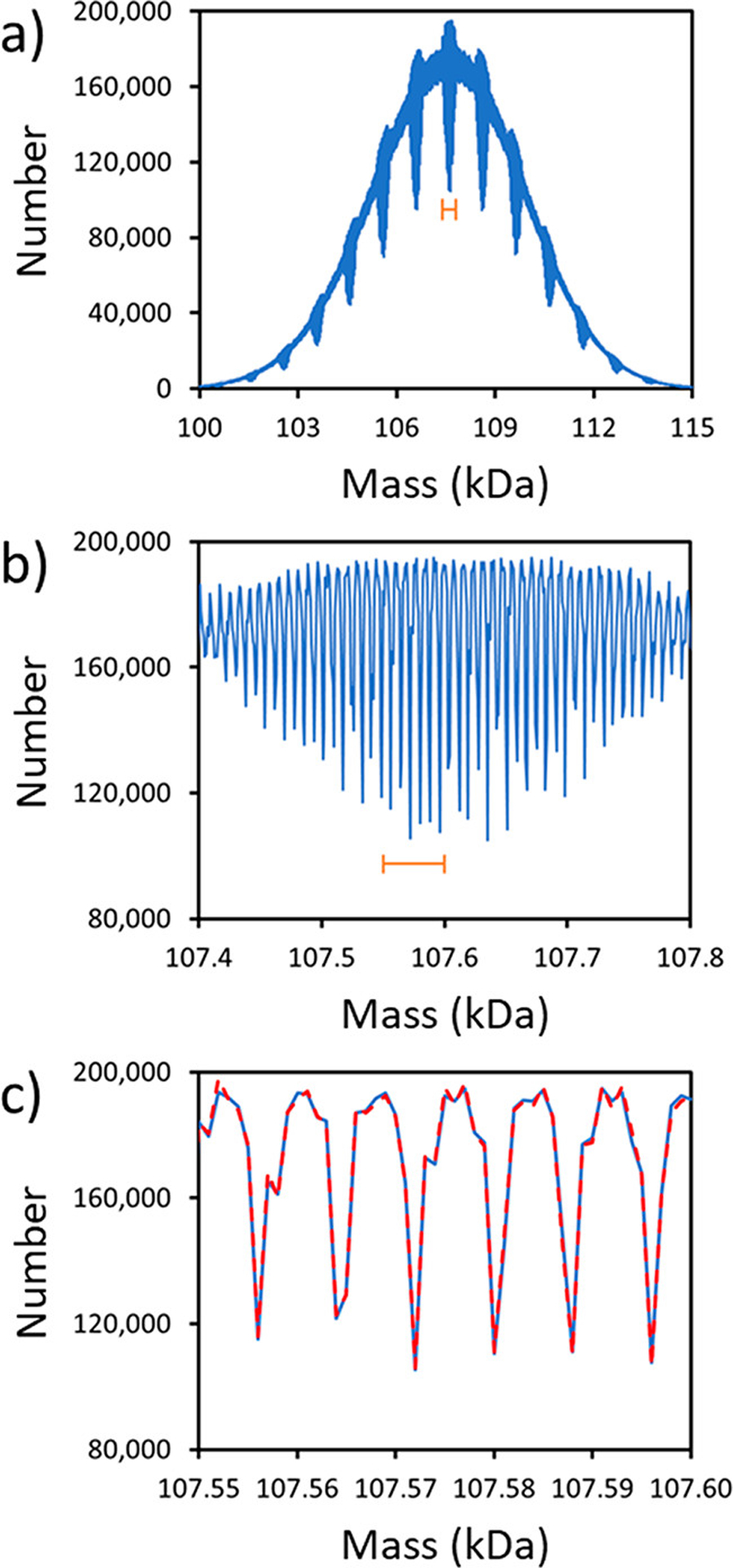
Glycan mass distributions for the S protein trimer from Monte Carlo calculations using probabilities from glycoproteomics17 (see text). (a) The mass distribution calculated from 1010 samples using 1 Da bins. (b) An expanded view of the portion of part a indicated by the orange bar. (c) Expanded view of the portion of part b indicated by the orange bar is shown as the underlying blue line. The glycan mass distribution calculated with 109 samples (scaled up by a factor of 10) is also shown (red dashed line in part c).
The width (fwhm) of the calculated glycan mass distribution (~5.5 kDa) is much narrower than the measured distribution (compare Figure 2a with Figure 1a). If we consider a situation where each site can be occupied by just two glycans with masses of 1200 and 2000 Da, the resulting mass distribution will be Gaussian and extend from 79200 Da (where all glycans are 1200 Da) to 132000 Da (where all glycans are 2000 Da). However, the probability of being at one of the extremes (where all glycans are either 1200 or 2000 Da) is vanishingly small. The fwhm of the distribution is expected to be around Da wide, which is similar to the width from the MC simulations. The calculated distribution is narrow because of the averaging that results from random sampling of the glycans at each site.
Mass Distributions Measured for Other S Proteins Samples.
CDMS measurements were performed for S protein samples from a variety of sources to determine how the cell line or protein sequence influenced the results. A typical mass distribution for the S protein without a trimerization domain expressed in HEK293 cells (Scheme 1b) is shown (Figure 3a). The charge spectrum (red inset) shows well-resolved charge states (charge RMSD 0.175 e). The main peak in the mass spectrum at 196 kDa is assigned to the spike monomer. In addition, there is a small peak at around 388 kDa that we attribute to a dimer and another peak at around 565 kDa attributed to a trimer. The mass of the dimer is slightly less than twice the monomer mass, but the mass of the trimer is substantially less than three times the monomer mass. This trend is observed for all the spectra measured for two different samples of this protein, under a variety of solution conditions. The average dimer mass is ~5 kDa less than twice the monomer mass (392 kDa), and the trimer mass is ~17 kDa less than three times the monomer mass (588 kDa).
Figure 3.
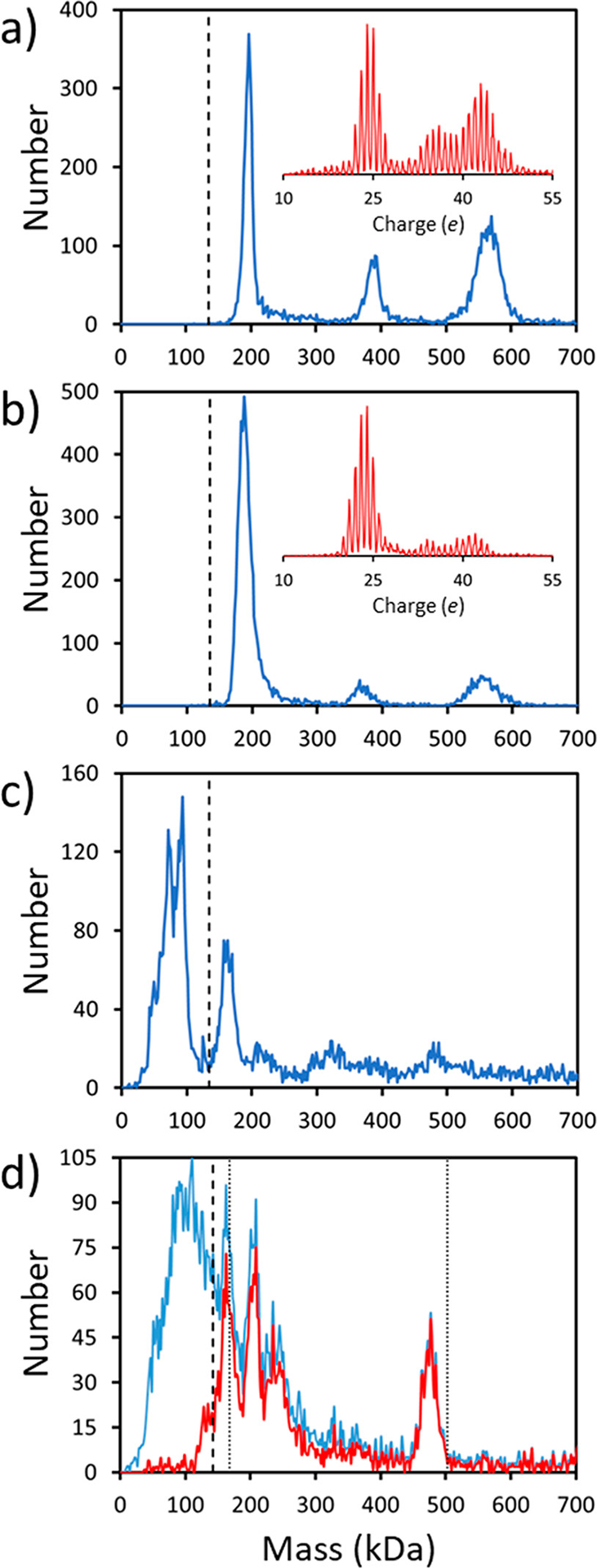
CDMS measurements for other S protein samples: (a) typical mass spectrum (blue) and a charge spectrum (red inset) for the S protein without a trimerization domain from HEK293 cells (Scheme 1b); (b) analogous spectra for the S protein from CHO cells (Scheme 1c); (c) a mass spectrum for the S protein from insect cell expression (Scheme 1d); (d) a mass spectrum for the S protein with a trimerization domain prepared by expression in HEK 293S GnTI- cells for reduced glycan heterogeneity (Scheme 1e). The blue line in part d is the measured spectrum, and the red line shows the spectrum obtained by removing ions with charges <20 e. The dashed vertical lines in parts a–d show the sequence masses of the S protein monomers. The dotted vertical lines in part d shows the expected masses of the fully glycosylated spike monomer and trimer (the sequence mass plus the mass of the glycans assuming that they are all Man5GlcNac2). All mass distributions were generated using 2 kDa bins, and the charge distributions have 0.1 e bins. See text for details.
Figure 3b shows a mass distribution measured for the spike monomer expressed in CHO cells (Scheme 1c). The charge spectrum (red inset) shows well resolved charge states (charge RMSD 0.186 e). The mass distribution is similar to that in Figure 3a, showing a peak at ~188 kDa that we assign as the spike monomer and peaks at ~370 kDa and 555 kDa assigned as the dimer and trimer, respectively. As in Figure 3a, the dimer peak occurs at a mass slightly less than twice the monomer mass (376 kDa), and the trimer peak is at a mass substantially less than three times the monomer mass (564 kDa). Note that the masses of the monomer, dimer, and trimer from the CHO cells are all significantly less than the corresponding masses for HEK293 cells.
Figure 3c shows the mass distribution measured for the spike protein from insect cell expression (Scheme 1d, Sino Biological). We struggled to measure a spectrum for this sample and tried many different solution conditions. The spectrum in Figure 3c was measured with a shorter trapping time (100 ms). Because of the short trapping time, charge states are not resolved, and the mass resolution is significantly lower than that in Figure 3a,b. The data for S derived from insect cell expression shows a high mass tail that extends to beyond 10 MDa. We attribute the peak at 162 kDa to the S protein monomer (sequence mass 136.0 kDa). Unlike the spectra obtained from HEK293 and CHO expression, peaks due to the dimers and trimers are not well-defined here. Instead, there are prominent peaks below the mass of the monomer at around 72.6 and 90.2 kDa. We assign these to the S1 and S2 subdomains of the spike protein because the sum of their masses is 162.8 kDa, near the mass of the monomer. The furin cleavage sequence at the junction between the S1 and S2 subdomains was not modified in this sample (unlike the other samples studied here).
The peak attributed to the spike monomer (at 162 kDa) in Figure 3c is at a significantly lower mass than the monomer peak for the samples in Figure 3a,b that were obtained from mammalian cells. Subtracting the sequence mass (134.4 kDa) from the monomer mass (162 kDa) yields an average glycan mass of 27.6 kDa, which is much smaller than the corresponding glycan masses for the spike monomers from HEK293 cells (61.4 kDa) and CHO cells (52.4 kDa). This observation is consistent with the conclusions of Zhang et al., who found the glycans from the S protein expressed in insect cell lines were mainly of the high mannose type while glycans from mammalian cell lines were mainly complex.18 Thus, the lower mass for the spike protein derived from insect cell expression is a result of different processing of the glycans in insect cells versus mammalian cells.
Figure 3d shows the spectrum measured for the S protein with reduced glycan heterogeneity obtained by expression in HEK 293S GnTI- cells. With this cell line, all N-linked glycans should be occupied by Man5GlcNac2. The blue line in Figure 3d is the measured mass distribution which shows a broad distribution of low mass ions that partially obscure several peaks. The red line shows the distribution obtained after removal of ions with fewer than 20 charges, a process that discriminates against the low mass ions. There are prominent peaks at around 166, 206, and 475 kDa. The sequence mass of this S protein is 140.8 kDa and the mass of the 22 Man5GlcNac2 glycans is 22 × 1216 Da = 26.8 kDa, so the expected mass of the glycosylated spike protein monomer is 167.6 kDa. The peak at 166 kDa is around 1.6 kDa less than the expected mass of the glycosylated spike monomer. The lower-than-expected mass may result from deviations in glycan occupancy (i.e., around one glycan site per monomer is not occupied). The S protein used for the measurement in Figure 3d (Scheme 1e) incorporates a fibritin trimerization domain which may account for the absence of a significant peak attributable to the dimer. The peak at 475 kDa, attributed to the trimer, is at a mass significantly lower than the expected mass (502.8 kDa). Significantly fewer glycan sites must be occupied in the trimer than on the monomer discussed above.
The spectrum in Figure 3d was measured using 100 ms trapping where the diminished accuracy of the charge measurement significantly degrades the mass resolution. Despite the reduced resolution, the trimer peak at 475 kDa in Figure 3d has a fwhm of 25 kDa which is considerably narrower than the measured spike trimer peaks in Figure 1a and Figure 3a,b. The mass resolution for 475 kDa peak in Figure 3d is estimated to be around 23.7 kDa (fwhm) (see Supporting Information). Thus, the width of this peak (25 kDa) is mainly due to instrumental resolution and the underlying peak width is probably in the 5–10 kDa range. This is much narrower than the trimer peaks in Figures 1a and 3a,b where the mass resolution is <2 kDa and the measured peak is representative of the underlying distribution. The narrower peak width is consistent with the reduced glycan heterogeneity expected for this sample.
Figure 4 shows a comparison of the glycan mass distributions determined for the spike trimer from three sources: (a) the S protein with a trimerization domain from HEK293 cells (blue line labeled Tri); (b) the S protein without a trimerization domain from HEK293 cells (orange line labeled HEK); and (c) the S protein without a trimerization domain from CHO cells (yellow line labeled CHO). The distributions in Figure 4 were obtained by subtracting three times the sequence mass of the S proteins (Scheme 1) from the measured masses. The measured glycan mass distributions differ significantly for the different proteins. The average glycan masses are 145.3 kDa for the trimer of the S protein with a trimerization domain from HEK293 cells (Scheme 1a), 158.2 kDa for the trimer of the S-protein without a trimerization domain from HEK293 cells (Scheme 1b), and 150.2 kDa for the trimer of the S protein without a trimerization domain from CHO cells (Scheme 1c). The average glycan masses derived from the CDMS measurements are 35–47% higher than the average glycan mass derived from the glycoproteomics studies.16 The red line in Figure 4 is the calculated N-glycan distribution from the MC simulations described above. In the MC simulations, it is assumed that each glycosylation site is randomly populated with glycans in accordance with their site-specific abundances from glycoproteomics studies.16 The distributions determined from the CDMS measurements are all much broader than the distribution obtained from the MC simulations (red line) and shifted to significantly higher mass. The observation that the measured mass distributions for the spike trimers are much broader than the calculated glycan distribution indicates that glycosylation is not correlated. Some spike trimers appear to have much larger glycans than others, and this is probably related to how the glycans are processed in the cell.
Figure 4.
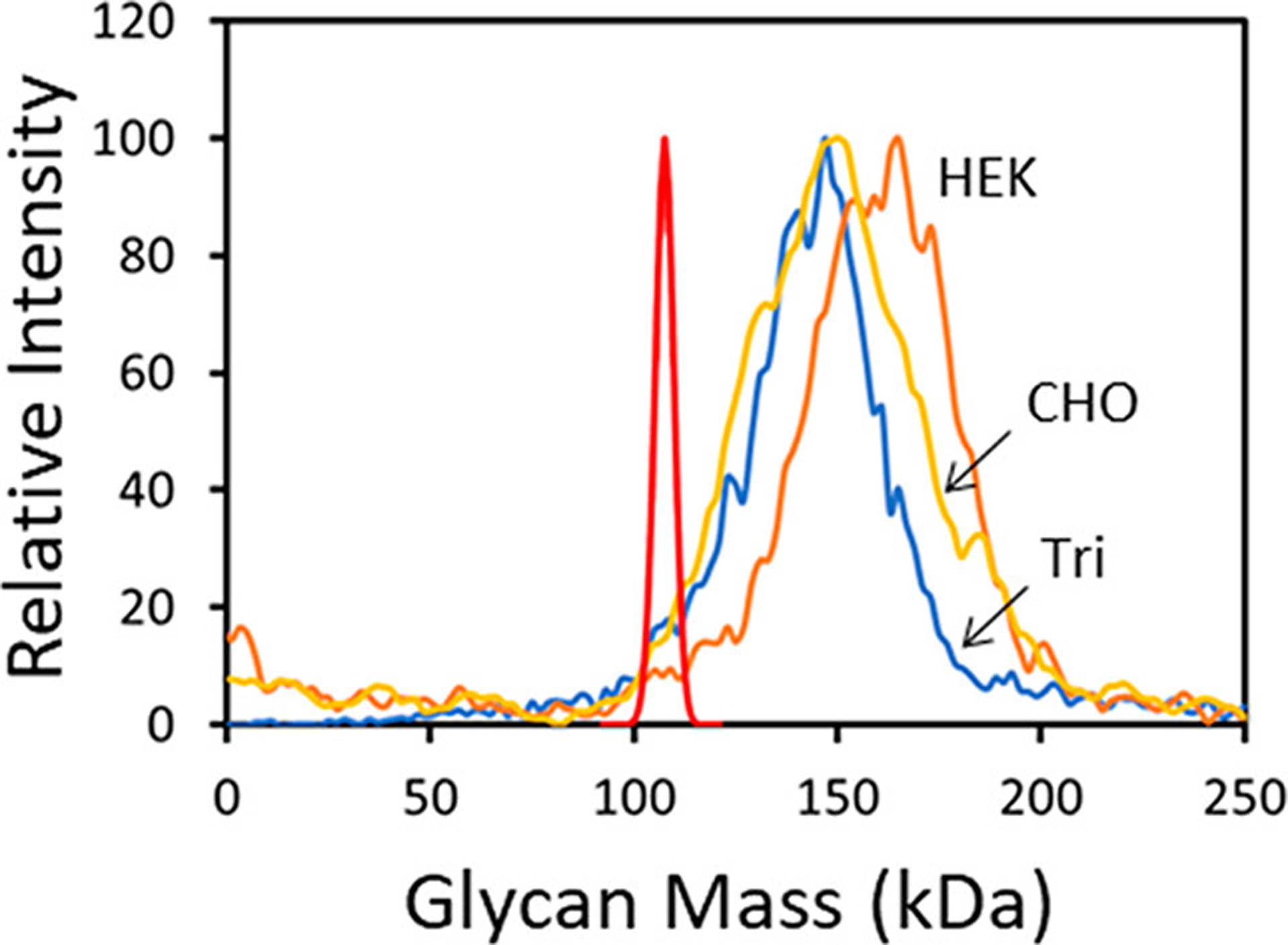
Glycan mass distributions for the S protein trimer from multiple sources. The red line shows the distribution obtained from the Monte Carlo calculation using probabilities from glycoproteomics16 (see text). The other colored lines show experimental results where the glycan distributions were obtained by subtracting the sequence masses for the S protein trimers from the measured masses. The blue line labeled Tri is for the S protein with a trimerization domain (Scheme 1a). The orange and yellow lines are for samples without the trimerization domain. The orange line labeled HEK is for S protein from HEK293 cells (Scheme 1b), and the yellow line labeled CHO is for S protein from CHO cells (Scheme 1c).
DISCUSSION
Protein glycosylation is a complex process.27,28 The coronavirus family of viruses infect a wide range of mammalian species and hijack the host cell glycosylation machinery. For SARS coronaviruses, S protein trimerization and initial N-glycosylation occurs in the ER. Further processing occurs in the Golgi where complex glycans are generated and O-glycans added.29–32 Cleavage of the S1 and S2 subdomains is also thought to occur in the Golgi.33,34 The development of MS-based glycoproteomics methods over the last two decades has enabled the analysis of glycosylation patterns for a wide range of glycoproteins.35–37 The glycopeptides generated by enzymatic digestion of glycoproteins are interrogated by LC-MS, and the abundances of N-glycans at specific sites are inferred from the results. Using this approach, it is possible to determine the identity and relative abundances of the glycans that occupy each site on a glycoprotein. However, this approach cannot provide information about how the glycans at a particular site are correlated with glycans at other sites on individual glycoprotein molecules. In contrast, CDMS analyzes individual intact glycoproteins, and the results can be used to directly assess the heterogeneity within the glycoprotein ensemble. The average glycoprotein mass deduced from the bottom-up MS analysis of glycopeptides should agree with the average mass determined by top-down CDMS analysis. This does not appear to be the case for the S protein studied here. The average glycan masses deduced from the CDMS measurements for S protein trimers from mammalian cells are 35–47% (depending on the trimer) higher than the average glycan mass deduced from glycoproteomic studies16 (107.5 kDa). The deviation for the S protein monomer is even larger (42–72%) because it is systematically more heavily glycosylated than the trimer.
The glycoproteomics studies indicate that all potential sites of N-glycosylation are populated. Thus, a possible explanation for the difference between the average glycan masses obtained from the MS-based glycoproteomics studies and the single molecule CDMS measurements is that the glycoproteomics measurements are skewed in a manner that underestimates the abundances of larger, more complex glycans. This could result for several reasons. There may be differences in the glycopeptide solubilities, rates of enzymatic digestion, efficiency of ionization, and the accuracy of database–based assignments. For a heavily glycosylated protein like the spike protein there are also questions about sensitivity and dynamic range.37,38 A measure of the intact mass by CDMS holistically captures the full distribution because those glycans present in low abundance only contribute to the width of the distribution.
The average mass from the glycoproteomics studies (107.5 kDa) is at the low end of the measured glycan mass distributions (see Figure 4). The upper end of the measured glycan mass distributions is at a mass that is close to twice the glycoproteomics average. The broad width of the glycan mass distribution provides insight about how the spike protein is processed in the cell. It has been reported that the S protein is mannosylated before it assembles into trimers in the ER and acquires complex N-glycans in the Golgi.30 The CDMS results presented here indicate that the degree of processing is highly variable. If the glycosylation sites were processed randomly so that the processing of one site on a trimer is uncorrelated with the processing at another, then the resulting glycan mass distribution would be narrow like the calculated distribution in Figure 2a. The broad glycan mass distribution observed in the experiments suggests that processing is correlated, so that for some trimers, many of the glycan sites are lightly processed, while for others, many of the sites are heavily processed. This additional variance in glycoforms could be another modification that helps the virus escape the host’s immune response.
In addition to the large range of glycoforms found for the S protein, the measured average mass for all S protein monomers was greater than one-third of the measured mass for the S protein trimers. This finding is consistent with complex glycosylation of the trimers occurring after trimerization in the ER, limiting the available sites for complex glycosylation in the trimer. In contrast, all the sites on the unassembled S protein monomer would be fully accessible in the Golgi where they could be processed to obtain complex glycans, leading to the larger measured mass. The differences between the glycosylation of the S protein monomer and the S protein trimer could prove to be important for vaccine approaches that rely on the S protein as the antigen to spur immunity.
CONCLUSIONS
We have used CDMS to investigate the heterogeneity associated with glycosylation of the SARS-CoV-2 spike trimer protein derived from several expression systems, HEK293, CHO, insect cells, and HEK 293S GnTI-. We found that the average glycan mass obtained from these direct “top-down” CDMS measurements is much larger than the average obtained from “bottom-up” MS-based glycoproteomics studies. CDMS is agnostic to the size and nature of the glycans, so it should holistically capture the full mass distribution. Thus, the glycoproteomics studies may have missed some of the larger glycans. To the best of our knowledge, this is the first time that a comparison of top-down and bottom-up glycan mass distributions has been performed. While results presented here are for a single glycoprotein, it is reasonable to expect that this behavior is not restricted to the SARS-CoV-2 S protein. The broad glycan mass distributions measured here indicates that glycan processing is correlated. Thus, most of the glycans on some S protein trimers are heavily processed while on other S protein trimers, most of the glycans are only lightly processed. This study is the first to explore cooperativity in glycan processing. The heterogeneous glycan distribution found in this study may have evolved as a way of further confounding the host’s immune system.
MATERIALS AND METHODS
Sample Preparation.
The variants of the SARS-CoV-2 Spike protein (Scheme 1) were purchased from Acro Biosystems (SPN-C52H8 and SPN-C52H4) for HEK293 expression; The Native Antigen Company (REC31868–100) for CHO expression, and Sino Biological (40589-V08B1) for insect cell expression. The spike protein expressed in HEK293S cells is described below. Prior to CDMS analysis, samples were buffer exchanged into 200 mM ammonium acetate using Zeba microbiospin columns with a 7K MWCO (Thermo Scientific).
Production and Purification of Spike Protein with Limited Glycosylation.
HEK 293S GnTI- cells were obtained from the American Type Culture Collection (CRL-3022) and cultured at 37 °C in 5% CO2. Cells were maintained in DMEM/F12 (Invitrogen) supplemented with nonessential amino acids and 10% FBS. Prior to transfection, the culture media was replaced with Opti-MEM (Invitrogen). Cells were transiently transfected with a plasmid encoding the SARS-CoV-2 S protein ectodomain (a kind gift from Weston Struwe)20 using lipofectamine-2000 (Invitrogen) by following the manufacturers recommended protocol. The supernatant was harvested at 24 and 48 h following transfection and snap frozen at −80 °C until use. The supernatant was thawed on ice and filtered through a 0.45 μm filter before being passed over a 5 mL Ni-NTA prepacked column (GE Healthcare) using an AKTA pure FPLC system preequilibrated with loading buffer (2 mM Tris-HCl (pH 7.4), 200 mM NaCl, 20 mM imidazole). The immobilized protein was washed with five column volumes of wash buffer (2 mM Tris-HCl (pH 7.4), 200 mM NaCl, 80 mM imidazole) before being eluted with the same buffer containing 400 mM imidazole. Peak fractions were concentrated and further purified using a Superdex 200 increase 10/300 GL size exclusion chromatography column equilibrated with 2 mM Tris-HCl (pH 7.4) and 200 mM NaCl. The protein eluted as a single peak which was concentrated to ~1 mg/mL, snap frozen in liquid nitrogen, and stored at −80 °C for further use.
CDMS Measurements.
In CDMS, the masses of individual ions are determined from the simultaneous measurement of each ion’s m/z ratio and charge. The measurements were performed on a home-built CDMS instruments described in detail elsewhere.39–45,26 Briefly, ions generated by nanoelectrospray are carried into the instrument through a capillary. The resulting gas flow and the entrained ions enters the first vacuum chamber which incorporates a FUNPET42 which dissipates the gas jet and transfers the ions through a small aperture into an RF hexapole. The DC potential on the hexapole rods sets the final ion energy. After the hexapole, the ions pass into a segmented RF quadrupole. Ions that exit the quadrupole are focused into an ion beam and then enter a dual hemispherical deflection energy analyzer that is set to pass a narrow band of ion kinetic energies. The transmitted ions are focused into a linear electrostatic ion trap (ELIT).41 The ELIT consists of two end-caps that can be switched between transmission and trapping modes. The trapped ions oscillate back and forth through the detection cylinder that is located between the two end-caps. The detection cylinder is connected to a low-noise charge sensitive amplifier which detects the induced charge from the oscillating ion. The resulting signal is amplified, digitized, and analyzed using fast Fourier transforms.43 The oscillation frequency provides the m/z, and the magnitude provides the charge. Ions that were not trapped for the full trapping time are discarded. The accuracy of the charge measurement depends on the trapping time. Measurements were performed with trapping times of 100 ms and 1.5 s. With 1.5 s trapping, the uncertainty (RMSD) in the charge measurements is around 0.2 e; charge states are well resolved in the charge spectrum, and ions can be assigned to integer charges with a low error rate. The mass resolution is then determined by the m/z resolution. With 100 ms trapping, the uncertainty in the charge is around 1 e, and this becomes the main factor limiting the mass resolution (see Supporting Information).
Supplementary Material
ACKNOWLEDGMENTS
This work is funded in part by grants from the NSF (IIP-2031083) and the NIH (5R01GM121751-04). B.E.D. is funded through an NIH SBIR grant (5R01GM131100-02) awarded to Megadalton Solutions. Work in the CVR laboratory is supported by a Medical Research Council (MRC) program grant (MR/N020413/1), a European Research Council Advanced Grant ENABLE (695511), and a Wellcome Trust Investigator Award (104633/Z/14/Z). C.V.R., C.A.L., and T.J.E. are also a part of the COVID-19 mass spectrometry coalition.46 T.J.E. is supported by the Royal Society as a Royal Society Newton International Fellow. C.A.L. is supported by the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement GPCR-MS 836073. We are grateful for generous support provided by the University of Oxford COVID-19 Research Response fund and its donors (BRD00230).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c00353.
Elucidation of CDMS mass resolution with 100 ms trapping time (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c00353
The authors declare the following competing financial interest(s): BED, DEC, and MFJ are associated with, and are shareholders in, a company that is attempting to commercialize CDMS. The other authors have no conflicts.
REFERENCES
- (1).Huang C; Wang Y; Li X; Ren L; Zhao J; Hu Y; Zhang L; Fan G; Xu J; Gu X; Cheng Z; Yu T; Xia J; Wei Y; Wu W; Xie X; Yin W; Li H; Liu M; Xiao Y; Gao H; Guo L; Xie J; Wang G; Jiang R; Gao Z; Jin Q; Wang J; Cao B Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).WHO Coronavirus Disease (COVID-19) Dashboard https://covid19.who.int/ (accessed 2020-10-10).
- (3).Yang X; Yu Y; Xu J; Shu H; Xia J; Liu H; Wu Y; Zhang L; Yu Z; Fang M; Yu T; Wang Y; Pan S; Zou X; Yuan S; Shang Y Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study, Lancet Resp. Med. Lancet Respir. Med 2020, 8, 475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Zhou P; Yang X-L; Wang X-G; Hu B; Zhang L; Zhang W; Si H-R; Zhu Y; Li B; Huang C-L; Chen H-D; Chen J; Luo Y; Guo H; Jiang R-D; Liu M-Q; Chen Y; Shen X-R; Wang X; Zheng X-S; Zhao K; Chen Q-J; Deng F; Liu L-L; Yan B; Zhan F-X; Wang Y-Y; Xiao G-F; Shi Z-L A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wu A; Peng Y; Huang B; Ding X; Wang X; Niu P; Meng J; Zhu Z; Zhang Z; Wang J; Sheng J; Quan L; Xia Z; Tan W; Cheng G; Jiang T Genome composition and divergence of the novel coronavirus (2019-NCoV) originating in China. Cell Host Microbe 2020, 27, 325–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Amanat F; Krammer F SARS-CoV-2 vaccines: Status report. Immunity 2020, 52, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Tai W; He L; Zhang X; Pu J; Voronin D; Jiang S; Zhou Y; Du L Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol 2020, 17, 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wrapp D; Wang N; Corbett KS; Goldsmith JA; Hsieh C-L; Abiona O; Graham BS; McLellan JS Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Cai Y; Zhang J; Xiao T; Peng H; Sterling SM; Walsh RM; Rawson S; Rits-Volloch S; Chen B Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Turonova B; Sikora M; Schurmann C; Hagen WJH; Welsch S; Blanc FEC; von Bulow S; Gecht M; Bagola K; Horner C; van Zandbergen G; Landry J; de Azevedo NTD; Mosalaganti S; Schwarz A; Covino R; Muhlebach MD; Hummer G; Krijnse Locker J; Beck M In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 2020, 370, 203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Walls AC; Park Y-J; Tortorici MA; Wall A; Mcguire AT; Veesler D Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Walls AC; Tortorici MA; Snijder J; Xiong X; Bosch B-J; Rey FA; Veesler D Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc. Natl. Acad. Sci. U. S. A 2017, 114, 11157–11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Watanabe Y; Bowden TA; Wilson IA; Crispin M Exploitation of glycosylation in enveloped virus pathobiology. Biochim. Biophys. Acta, Gen. Subj 2019, 1863, 1480–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Casalino L; Gaieb Z; Goldsmith JA; Hjorth CK; Dommer AC; Harbison AM; Fogarty CA; Barros EP; Taylor BC; McLellan JS; Fadda E; Amaro RE Beyond shielding: The roles of glycans in the SARS-CoV-2 spike protein. ACS Cent. Sci 2020, 6, 1722–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Andersen KG; Rambaut A; Lipkin WI; Holmes EC; Garry RF The proximal origin of SARS-CoV-2. Nat. Med 2020, 26, 450–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Watanabe Y; Allen JD; Wrapp D; Jason S; McLellan JS; Crispin M Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Shajahan A; Supekar NT; Gleinich AS; Azadi P Deducing the N- and O-glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology 2020, cwaa042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhang Y; Zhao W; Mao Y; Chen Y; Wang S; Zhong Y; Su T; Gong M; Du D; Lu X; Cheng J; Yang H Site-specific N-glycosylation characterization of recombinant SARS-CoV-2 spike proteins. Mol. Cell. Proteomics 2021, 100058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sanda M; Morrison L; Goldman RN and O Glycosylation of the SARS-CoV-2 spike protein. Anal. Chem 2021, 93, 2003–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Brun J; Vasiljevic S; Gangadharan B; Hensen M; Chandran AV; Hill ML; Kiappes JL; Dwek RA; Alonzi DS; Struwe WB; Zitzmann N Analysis of SARS-CoV-2 spike glycosylation reveals shedding of a vaccine candidate. bioRxiv preprint 2020, 384594 DOI: 10.1101/2020.11.16.384594. [DOI] [Google Scholar]
- (21).Fuerstenau SD; Benner HW Molecular weight determination of megadalton DNA electrospray ions using charge detection time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom 1995, 9, 1528–1538. [DOI] [PubMed] [Google Scholar]
- (22).Keifer DZ; Pierson EE; Jarrold MF Charge detection mass spectrometry: Weighing heavier things. Analyst 2017, 142, 1654–1671. [DOI] [PubMed] [Google Scholar]
- (23).Tao Y; Strelkov SV; Mesyanzhinov VV; Rossmann MG Structure of bacteriophage T4 fibritin: a segmented coiled coil and the role of the C-terminal domain. Structure 1997, 5, 789–798. [DOI] [PubMed] [Google Scholar]
- (24).Güthe S; Kapinos L; Möglich A; Meier S; Grzesiek S; Kiefhaber T Very fast folding and association of a trimerization domain from bacteriophage T4 fibritin. J. Mol. Biol 2004, 337, 905–915. [DOI] [PubMed] [Google Scholar]
- (25).Keifer DZ; Pierson EE; Hogan JA; Bedwell GJ; Prevelige PE; Jarrold MF Charge detection mass spectrometry of bacteriophage P22 procapsid distributions above 20 MDa. Rapid Commun. Mass Spectrom 2014, 28, 483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Todd AR; Barnes LF; Young K; Zlotnick A; Jarrold MF Higher resolution charge detection mass spectrometry. Anal. Chem 2020, 92, 11357–11364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Gamblin DP; Scanlan EM; Davis BG Glycoprotein synthesis: An update. Chem. Rev 2009, 109, 131–163. [DOI] [PubMed] [Google Scholar]
- (28).Schjoldager KT; Narimatsu Y; Joshi HJ; Clausen H Global view of human protein glycosylation pathways and functions. Nat. Rev. Mol. Cell Biol 2020, 21, 729. [DOI] [PubMed] [Google Scholar]
- (29).Delmas B; Laude H Assembly of coronavirus spike protein into trimers and its role in epitope expression. J. Virol 1990, 64, 5367–5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Nal B; Chan C; Kien F; Siu L; Tse J; Chu K; Staropoli I; Crescenzo-Chaigne B; Escriou N; van der Werf S; Yuen K-Y; Altmeyer R Differential maturation and subcellular localization of severe acute respiratory syndrome coronavirus surface proteins S, M, and E. J. Gen. Virol 2005, 86, 1423–1434. [DOI] [PubMed] [Google Scholar]
- (31).Stertz S; Reichelt M; Spiegel M; Kuri T; Martínez-Sobrido L; García-Sastre A; Weber F; Kochs G The intracellular sites of early replication and budding of SARS-coronavirus. Virology 2007, 361, 304–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Duan L; Zheng Q; Zhang H; Niu Y; Lou Y; Wang H The SARS-CoV-2 spike glycoprotein biosynthesis, structure, function, and antigenicity: Implications for the design of spike-based vaccine immunogens. Front. Immunol 2020, 11, 576622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Bosshart H; Humphrey J; Deignan E; Davidson J; Drazba J; Yuan L; Oorschot V; Peters PJ; Bonifacino JS The cytoplasmic domain mediates localization of furin to the trans-golgi network en route to the endosomal/lysosomal system. J. Cell Biol 1994, 126, 1157–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hoffmann M; Kleine-Weber H; Pöhlmann S A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol. Cell 2020, 78, 779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wada Y; Azadi P; Costello CE; Dell A; Dwek RA; Geyer H; Geyer R; Kakehi K; Karlsson NG; Kato K; Kawasaki N Comparison of the methods for profiling glycoprotein glycans— HUPO Human Disease Glycomics/Proteome Initiative multi-institutional study. Glycobiology 2007, 17, 411–422. [DOI] [PubMed] [Google Scholar]
- (36).Wada Y; Dell A; Haslam SM; Tissot B; Canis K; Azadi P; Bäckström M; Costello CE; Hansson GC; Hiki Y; Ishihara M Comparison of methods for profiling O-glycosylation: human proteome organisation human disease glycomics/proteome initiative multi-institutional study of IgA1. Mol. Cell Proteomics 2010, 9, 719–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).De Leoz MLA; Duewer DL; Fung A; Liu L; Yau HK; Potter O; Staples GO; Furuki K; Frenkel R; Hu Y; Sosic Z NIST interlaboratory study on glycosylation analysis of monoclonal antibodies: comparison of results from diverse analytical methods. Mol. Cell Proteomics 2020, 19, 11–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hu Y; Shihab T; Zhou S; Wooding K; Mechref Y LC–MS/MS of permethylated N-glycans derived from model and human blood serum glycoproteins. Electrophoresis 2016, 37, 1498–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Contino NC; Jarrold MF Charge detection mass spectrometry for single ions with a limit of detection of 30 charges. Int. J. Mass Spectrom 2013, 345–347, 153–159. [DOI] [PubMed] [Google Scholar]
- (40).Keifer DZ; Shinholt DL; Jarrold MF Charge detection mass spectrometry with almost perfect charge accuracy. Anal. Chem 2015, 87, 10330–10337. [DOI] [PubMed] [Google Scholar]
- (41).Hogan JA; Jarrold MF Optimized electrostatic linear ion trap for charge detection mass spectrometry. J. Am. Soc. Mass Spectrom 2018, 29, 2086–2095. [DOI] [PubMed] [Google Scholar]
- (42).Draper BE; Anthony SN; Jarrold MF The FUNPET- a new hybrid ion funnel-ion carpet atmospheric pressure interface for the simultaneous transmission of a broad mass range. J. Am. Soc. Mass Spectrom 2018, 29, 2160–2172. [DOI] [PubMed] [Google Scholar]
- (43).Draper BE; Jarrold MF Real time analysis and signal optimization for charge detection mass spectrometry. J. Am. Soc. Mass Spectrom 2019, 30, 989–904. [DOI] [PubMed] [Google Scholar]
- (44).Todd AR; Alexander AW; Jarrold MF Implementation of a charge sensitive amplifier without a feedback resistor for charge detection mass spectrometry reduces noise and enables detection of individual ions carrying a single charge. J. Am. Soc. Mass Spectrom 2020, 31, 146–154. [DOI] [PubMed] [Google Scholar]
- (45).Todd AR; Jarrold MF Dynamic calibration enables high accuracy charge measurements on individual ions for charge detection mass spectrometry. J. Am. Soc. Mass Spectrom 2020, 31, 1241–1248. [DOI] [PubMed] [Google Scholar]
- (46).Struwe WB; Emmott E; Bailey M; Sharon M; Sinz A; Corrales FJ; Thalassinos K; Braybrook J; Mills C; Barran P COVID-19 MS Coalition. Lancet 2020, 395, 1761–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.