

Abstract
Background:
Alcoholic chronic pancreatitis (ACP) is a serious inflammatory disorder of exocrine pancreatic gland. A previous study from this laboratory has reported that ethanol (EtOH) causes cytotoxicity, dysregulates AMPKα, ER/oxidative stress signaling and induces inflammatory responses in primary human pancreatic acinar cells (hPACs). Therefore, a differential cytotoxicity of EtOH and its oxidative (acetaldehyde), and nonoxidative (fatty acid ethyl esters; FAEEs) metabolites in hPACs was examined to understand the metabolic basis and mechanism of ACP.
Methods:
Concentration-dependent cytotoxicity, AMPKα inactivation, ER/oxidative stress and inflammatory responses were evaluated in hPACs incubated with EtOH, acetaldehyde, or FAEEs for 6 hr at clinically relevant concentrations reported in alcoholic subjects using conventional methods. Cellular bioenergetics (mitochondrial stress and a real-time ATP production rate) were determined using Seahorse XFp analyzer in AR42J cells treated with acetaldehyde or FAEEs.
Results:
Concentration-dependent increases for LDH release, inactivation of AMPKα along with upregulation of ACC1 and FAS (key lipogenic proteins), downregulation of p-LKB1 (an oxidative stress-sensitive upstream kinase regulating AMPKα), and CPT1A (involved in β-oxidation of fatty acids) were observed in hPACs treated with EtOH, acetaldehyde or FAEEs. An oxidative stress and ER stress as measured by GRP78, unspliced XBP1, p-eIF2α, and CHOP along with activation of p-JNK1/2, p-ERK1/2, and p-P38MAPK were found in cells treated with EtOH, acetaldehyde or FAEEs were increased concentration dependently. Furthermore, a significant decrease for total ATP production rate with subsequent mitochondrial stress were observed in AR42J cells treated with acetaldehyde and FAEEs.
Conclusions:
EtOH and its metabolites, acetaldehyde and FAEEs; caused cytotoxicity, ER/oxidative and mitochondrial stress and dysregulated AMPKα signaling suggesting a key role of EtOH metabolism in etiopathogenesis of ACP. Since oxidative EtOH metabolism is negligible in the exocrine pancreas, pathogenesis of ACP could be associated with the formation of FAEEs and related pancreatic acinar cell injury.
Keywords: Alcoholic pancreatitis, Acetaldehyde, AMPKα, ER stress, Fatty acid ethyl esters, Human pancreatic acinar cells
Introduction
Chronic alcohol consumption is the major cause of chronic pancreatitis (CP), a serious fibro-inflammatory disorder of the exocrine pancreas, and may result in multiple dysfunctions / insufficiencies of the exocrine and endocrine pancreatic gland including diabetes and pancreatic cancer (Rasineni et al., 2020). The risk of developing alcoholic chronic pancreatitis (ACP) is shown to be directly associated with the amount and duration of alcohol intake. However, effective preventive measures and therapies could not be developed for ACP due to poor understanding regarding its mechanism(s) and metabolic basis.
Several putative mechanisms of ethanol (EtOH)-induced pancreatic damage have been reviewed (Kaphalia and Ansari, 2001, Apte et al., 2010, Pandol et al., 2011, Takahashi et al., 2020). Both oxidative as well as nonoxidative metabolism of EtOH to acetaldehyde and fatty acid ethyl esters (FAEEs), respectively, and their toxicity to the pancreas have been reported (Kaphalia and Ansari, 2001, Vonlaufen et al., 2007, Apte et al., 2010, Rasineni et al., 2020). While acetaldehyde has been shown to activate pancreatic stellate cells (McCarroll et al., 2003, Apte et al., 2010), FAEEs cause uncoupling of oxidative phosphorylation and induce inflammatory pathways in the pancreatic acinar cells (Gukovskaya et al., 2002, Haber et al., 2004, Criddle et al., 2006).
Of note, a majority of the ingested EtOH is metabolized primarily by oxidative metabolism to acetaldehyde by hepatic alcohol dehydrogenase (ADH), which is reduced during chronic alcohol consumption (Nuutinen et al., 1983, Sharkawi, 1984, Panes et al., 1993, Kaphalia et al., 1996). Furthermore, the hepatic ADH deficiency in deer mice fed chronic EtOH is shown to elevate body burden of EtOH despite the induction of hepatic cytochrome P450 2E1 (CYP2E1) (Srinivasan et al., 2019), and facilitates formation of FAEEs by several folds in the pancreas causing pancreatitis. (Bhopale et al., 2006, Kaphalia et al., 2010, Amer et al., 2018). During chronic EtOH ingestion, EtOH is predominantly metabolized via nonoxidative metabolism in the pancreas, catalyzed by FAEE synthase (Laposata and Lange, 1986, Werner et al., 1997, Werner et al., 2001, Werner et al., 2002, Kaphalia and Ansari, 2003). It is now apparent that blood alcohol levels and concentration of pancreatic FAEEs are key determining factors for the development of ACP (Werner et al., 2001, Kaphalia et al., 2004, Kaphalia et al., 2010).Thus, EtOH metabolism itself might play a crucial role in pathogenesis of ACP.
Experimental evidence suggests that dysregulation of unfolded protein response (UPR) induced by endoplasmic reticulum (ER) stress in acinar cells after chronic alcohol consumption promotes the development of pancreatitis (Pandol et al., 2010, Lugea et al., 2017a). On the other hand, dysregulation of AMP-Activated Protein Kinase (AMPKα), a major regulator of cellular metabolism and ER/oxidative stress (Steinberg and Kemp, 2009, Kim et al., 2015), plays a key role in the pathogenesis of various diseases, including EtOH-related disorders (Chen et al., 2010, Liangpunsakul et al., 2010, Shearn et al., 2014, Kaphalia et al., 2019, Srinivasan et al., 2019). An underlying link between EtOH-induced AMPKα inactivation and ER/oxidative stress in relation to pancreatic acinar cell injury, and its inflammatory responses and cellular bioenergetics could be key factors for the initiation of ACP (Srinivasan et al., 2020). Therefore, a differential cytotoxicity, AMPKα signaling, and ER/oxidative and mitochondrial stress were examined in human pancreatic acinar cells treated with EtOH, acetaldehyde and FAEEs to understand the mechanism(s) and metabolic basis of ACP.
Materials and Methods
All the chemicals, reagents, kits, antibodies, and techniques used in this study are detailed previously (Srinivasan et al., 2020) and provided in the Supplementary Section.
Human pancreatic acinar cells (hPACs) were obtained from Prodo Laboratories (Cat # HAR-001, Irvine, CA) and Department of Surgery, University of Louisville, Kentucky These cells have been shown to maintain most of their physiological functions and phenotype in culture for several hours and are suitable for the mechanistic studies (Coutts et al., 2007, Lugea et al., 2017b, Srinivasan et al., 2020).
The pancreatic acinar cells were harvested during islet isolation procedure from brain-dead donors (Ricordi et al., 1988, Balamurugan et al., 2010), [male (n=2) and female (n=2) Caucasians] who were non-diabetic, average age 35 years, with no history of chronic alcohol use, smoking, drug abuse, and gastrointestinal disorders. Upon receipt, hPACs were washed, distributed into cell culture flasks (0.5 × 106 cells/flask), and incubated at 37°C supplied with purified air containing 5% CO2 as described earlier (Srinivasan et al., 2020).
The reported blood acetaldehyde concentration in chronic alcoholics has ranged from 0–250 μM (Korsten et al., 1975, Nuutinen et al., 1983). Further, 50 μM FAEEs has shown to be sufficient to cause pancreatic injury (Haber et al., 1993). Previous studies from this laboratory have reported the plasma concentration of FAEEs in chronic alcoholic subjects to be in the range of 1–50 μg/ml and ~ 50 μg / g (wet weight) in the pancreas of deer mouse model fed chronic EtOH (Kaphalia et al., 2004, Kaphalia et al., 2010). Based on the findings by us and others, a concentration-dependent study was performed by incubating hPACs with 1, 2.5, 5 and 10 μg/ml (~ 23, 57, 113 and 227 μM) acetaldehyde (Cat # 402788, Sigma-Aldrich, St. Louis, MO) or 10, 25, 50, 100, and 200 μg/ml (~ 32, 80, 160, 320 and 640 μM) FAEEs mixture for a period of 6 hrs. In addition, the hPACs were also incubated with 1, 3 and 6 mg/ml EtOH (Cat # 2716, Decon Laboratories, Inc, King of Prussia, PA) under similar experimental conditions used for acetaldehyde or FAEEs, as described previously (Srinivasan et al., 2020). The FAEEs mixture was prepared in the ratio of 1: 2.2: 4.7: 2.8 wt/wt for ethyl palmitate (Cat # P9009, Sigma-Aldrich, St. Louis, MO), ethyl oleate (Cat # 268011, Sigma-Aldrich, St. Louis, MO), ethyl linoleate (Cat # L1751, Sigma-Aldrich, St. Louis, MO), and ethyl arachidonate (Cat # A9135, Sigma-Aldrich, St. Louis, MO), respectively, based upon their levels reported in the plasma of chronic alcoholic subjects (Kaphalia et al., 2004).
FAEEs were dissolved in EtOH (1 mg/ml) as a vehicle, because, at this concentration of EtOH, the formation of FAEEs was comparable to the control levels without any significant pancreatic acinar cell injury (Wu et al., 2008, Srinivasan et al., 2020). Therefore, hPACs were also incubated with an equivalent amount of EtOH to serve as control for FAEEs treated cells.
Importantly, all the cell culture studies with primary hPACs were completed within 30hr of their isolation from the donor (Srinivasan et al., 2020).
Pancreatic acinar cell viability and injury, dysregulated AMPKα and ER/oxidative stress signaling and inflammatory responses by EtOH, acetaldehyde and FAEEs
The methods used to evaluate cytotoxicity, AMPKα and ER/oxidative stress signaling, lipogenesis and inflammatory responses in hPACs treated with EtOH, acetaldehyde, or FAEEs herein are detailed previously (Srinivasan et al., 2020), and provided in the Supplementary Section.
Mitochondrial stress test and real-time ATP production rate
Due to a short lifespan and inherent clumping of freshly isolated hPACs, cellular bioenergetics studies were carried out using rat pancreatic tumor (AR42J) cells obtained from ATCC® (CRL-14092™, Rockville, MD). We previously determined mitochondrial stress and a real-time ATP production rate in AR42J cells treated with EtOH using Seahorse XFp extracellular flux analyzer (Agilent, Santa Clara, CA) (Srinivasan et al., 2020). Therefore, in this study, we measured both the parameters in AR42J cells treated with acetaldehyde and FAEEs, only.
Based on the concentration-dependent responses observed for acetaldehyde and FAEEs treated hPACs, a concentration of 5 μg/ml (~113 μM) for acetaldehyde and 100 μg/ml (320 μM) for FAEEs was preferred to determine mitochondrial stress and real-time ATP production rate.
Statistical analysis
The data sets were analyzed using One Way ANOVA followed by the Tukey Multiple Comparison test for the statistical significance. The results are expressed as Mean ± SEM (standard error of mean). Statistical analysis was performed using GraphPad Prism (v 7.0, San Diego, CA) and p-Value ≤0.05 was considered statistically significant.
Results
Viability and markers of cell injury
To begin with, the cell viability as a percent of the total number of cells used in all the experiments was found to be ~95%. EtOH, acetaldehyde, or FAEEs treated hPACs showed a concentration-dependent cell death (Fig. 1A–D). Amylase and lipase activities assayed in culture medium from hPACs treated either with EtOH, acetaldehyde, or FAEEs were not altered, significantly (Fig. 2A, B). Furthermore, a significant concentration-dependent increases for the lactate dehydrogenase (LDH) activity were found in the culture medium from hPACs treated with EtOH (3 and 6 mg/ml), acetaldehyde (5 and 10 μg/ml), or FAEEs (100 and 200 μg/ml), respectively, indicating their cytotoxic effects (Fig. 2C).
Fig. 1.
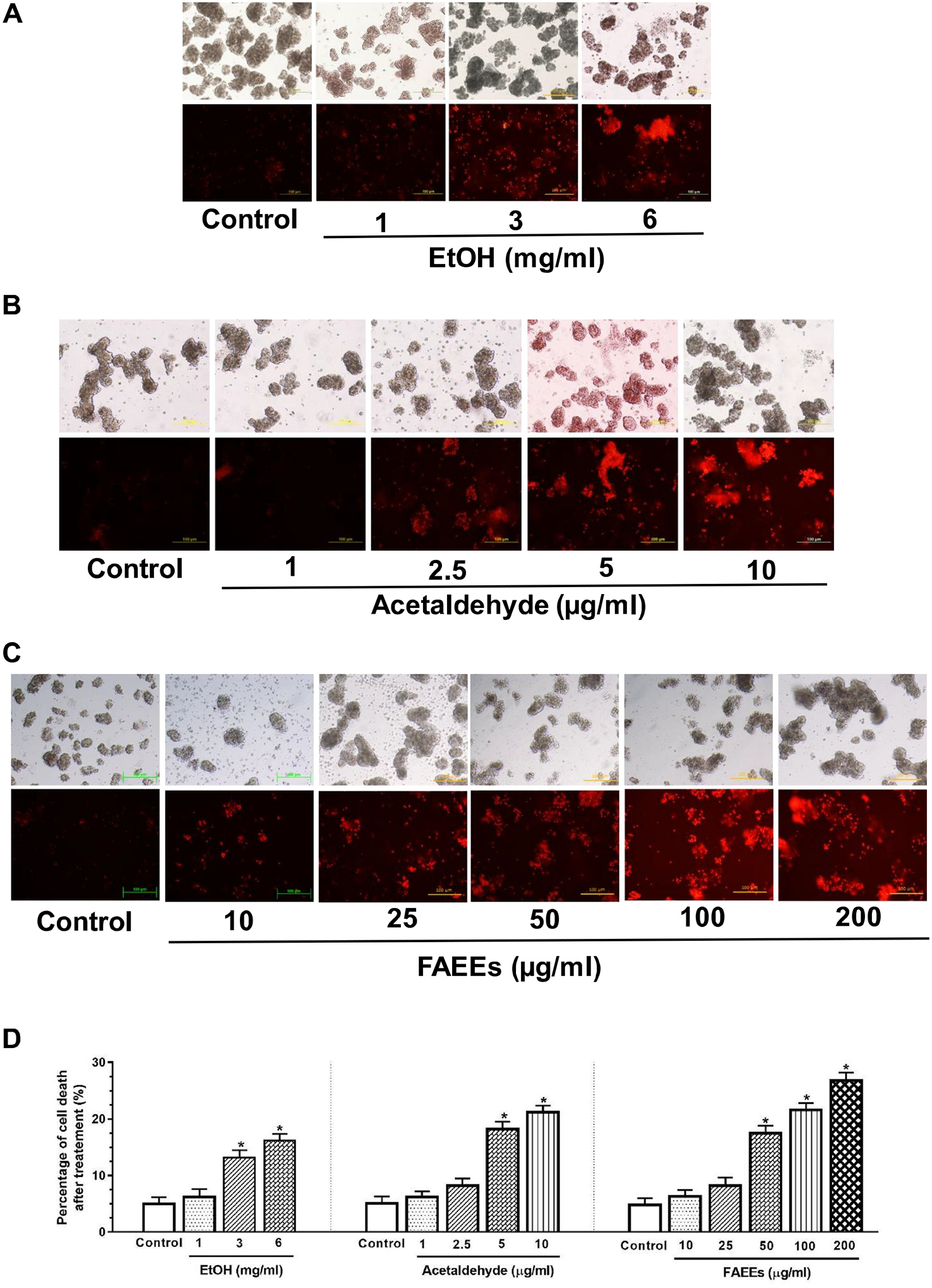
Viability/cell death in hPACs treated with EtOH (A), acetaldehyde (B) or FAEEs (C) for 6 hr using ethidium bromide (EB) staining. Red fluorescence intensity as a measure of dead cells was quantified using Image J (D) (original magnification × 20). Values are expressed as Mean ± SEM (n = 6 replicates).
Fig. 2.
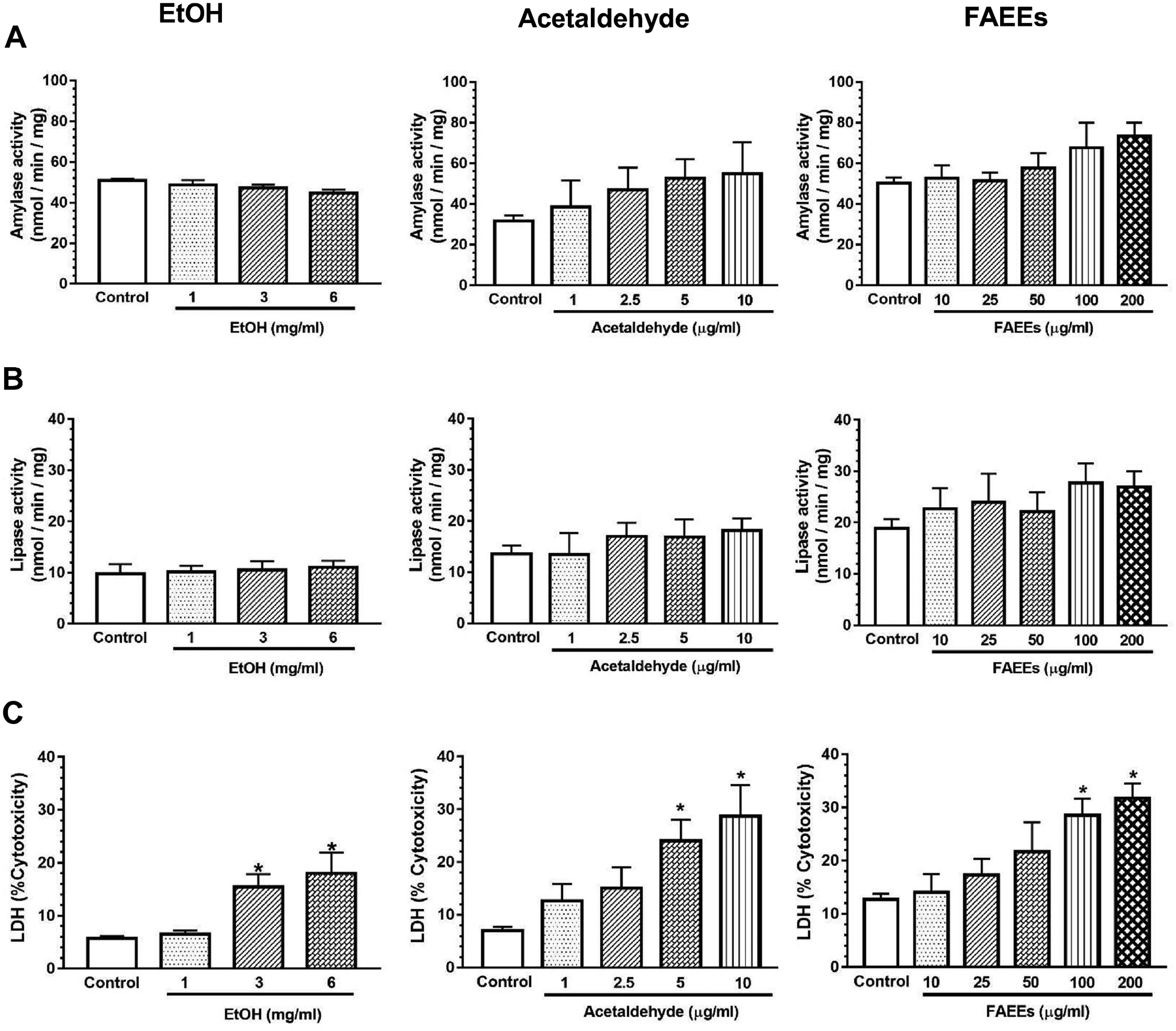
Markers for hPACs injury. Amylase (A), lipase (B) and lactate dehydrogenase (C) activities (markers of injury) in the culture medium of hPACs incubated for 6 hr with EtOH (left panel) acetaldehyde (middle panel) and FAEEs (right panel). Values are expressed as Mean ± SEM (n =12 replicates). * p value ≤ 0.05 with respect to controls.
Dysregulated AMPKα signaling
Concentration-dependent decreases for phospho (p)-AMPKα levels were observed in the hPACs treated with EtOH, acetaldehyde, or FAEEs as compared to their respective controls (Fig. 3A). The levels of p-AMPKα were significantly reduced by 2 to 2.5 fold in hPACs treated with 3 and 6 mg/ml EtOH, ~2.5 and 3-fold with 5 and 10 μg/ml of acetaldehyde, and 2.5 to 5-fold with 50, 100, or 200 μg/ml of FAEEs, respectively.
Fig. 3.
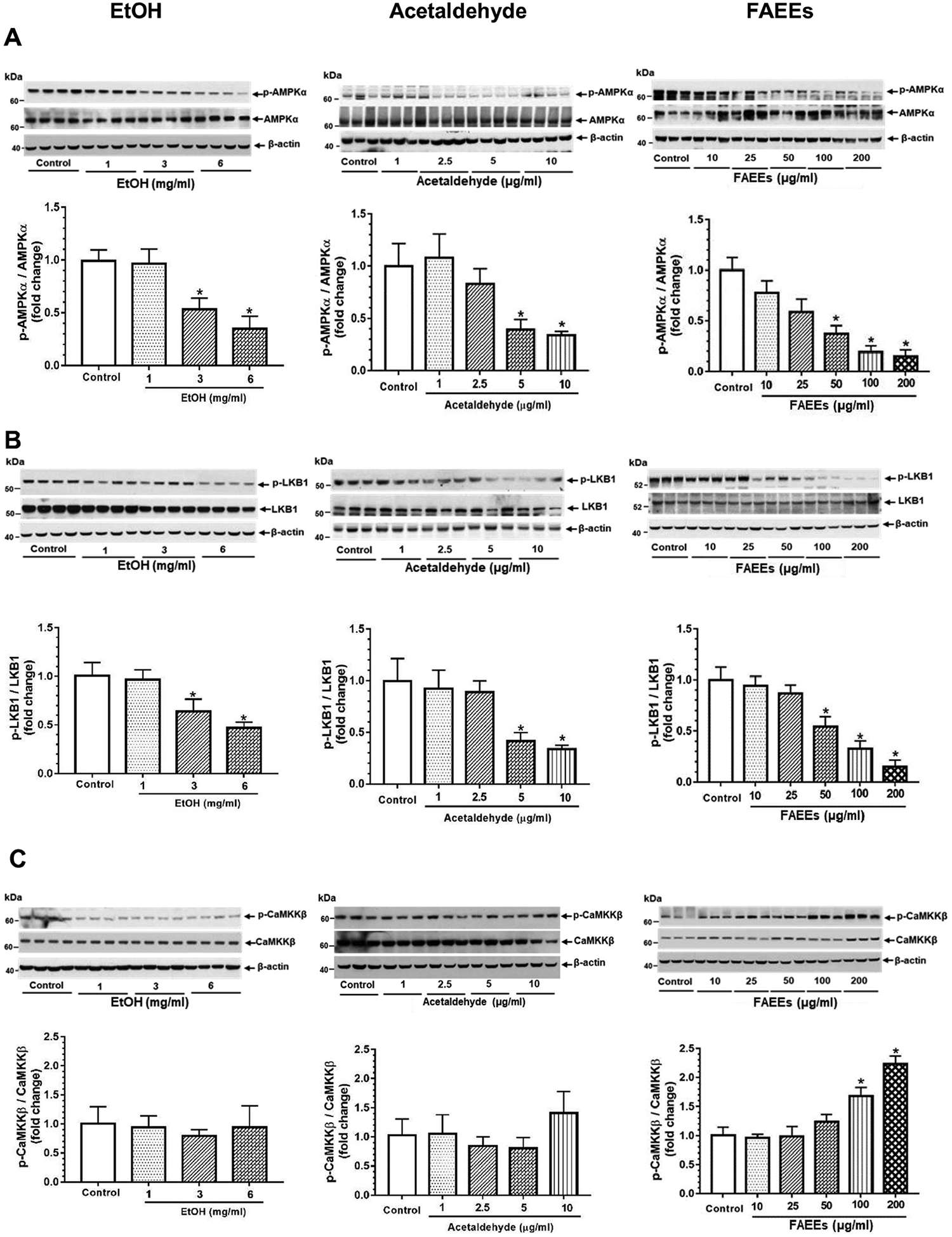
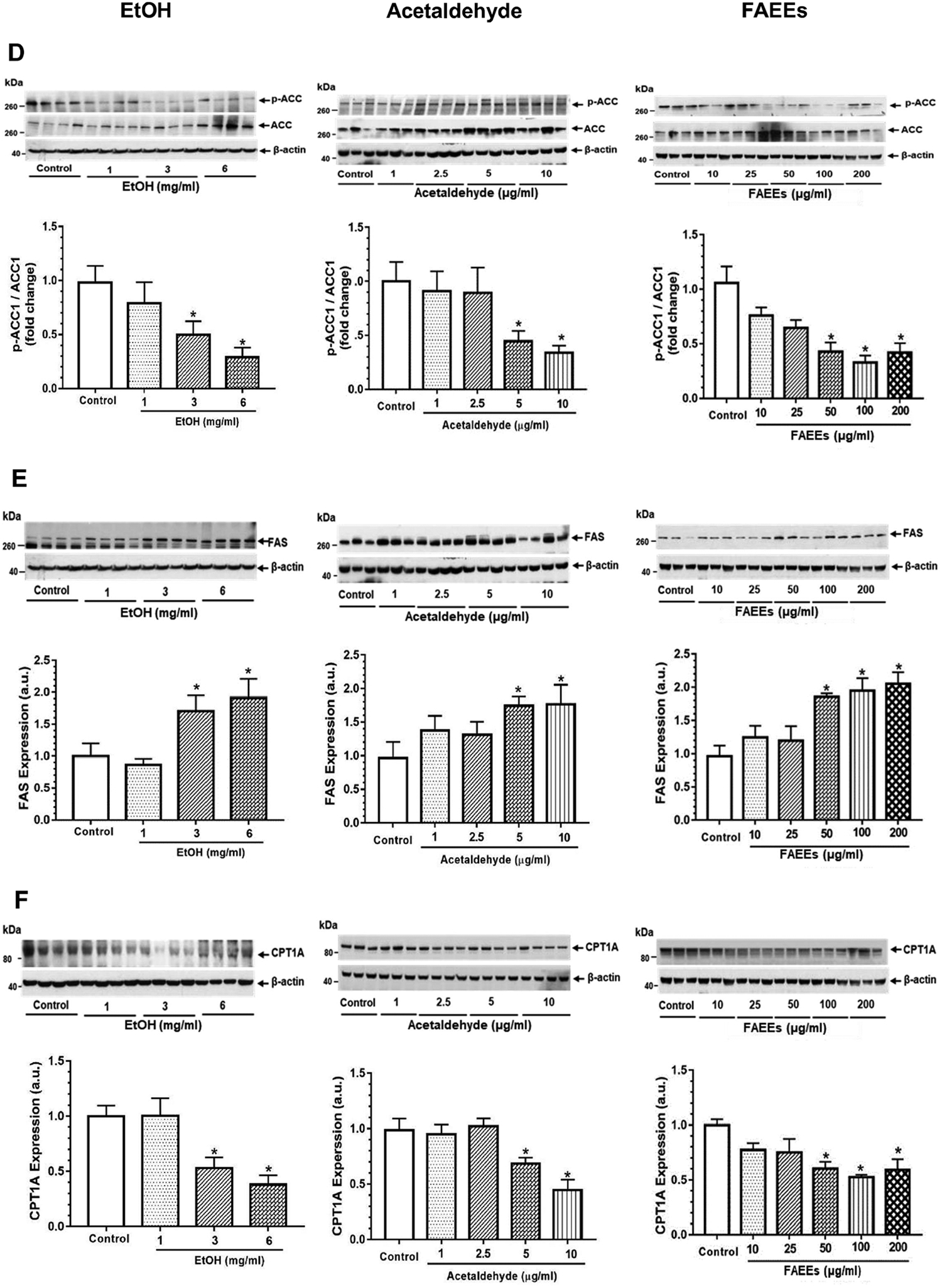
Dysregulation of AMPKα signaling in hPACs treated with EtOH (left panel), acetaldehyde (middle panel) and FAEEs (right panel) for 6hr. Representative immunoblots along with respective bar diagrams show relative intensities for p-AMPKα/AMPKα (A), p-LKB1/LKB1 (B), p-CaMKKβ/CaMKKβ (C), p-ACC1/ACC1 (D), FAS (E), and CPT1A (F). Intensities were normalized to β-actin (loading control). Values are expressed as Mean ± SEM (n =12 replicates). * p value ≤ 0.05 with respect to controls.
Of importance, significant concentration-dependent decreased levels of p- Liver kinase B1 (p-LKB1) (Fig. 3B), an oxidative stress-sensitive upstream kinase regulating AMPKα, were observed in hPACs treated with EtOH, acetaldehyde or FAEEs. On the other hand, p-Ca2+/CaM-dependent protein kinase kinase (p-CaMKKβ) (Fig. 3C), an upstream ER stress-sensitive kinase regulating AMPKα, was significantly upregulated in a concentration-dependent manner in hPACs treated with FAEEs. However, no significant changes for p-CaMKKβ were observed in hPACs treated with EtOH or acetaldehyde (Fig. 3C).
Further, concentration-dependent increases for the key proteins involved in lipogenesis [acetyl CoA carboxylase 1 (ACC1) (Fig. 3D), fatty acid synthase (FAS) (Fig. 3E)], were noted in hPACs treated with EtOH, acetaldehyde, or FAEEs. Of note, a significant concentration-dependent decrease was observed for p-ACC1 (Fig. 3D, a downstream signaling protein regulated by AMPKα) and carnitine palmitoyltransferase 1A (CPT1A), a key protein involved in β-oxidation of fatty acids, (Fig. 3F) in hPACs treated with EtOH, acetaldehyde or FAEEs.
ER/oxidative stress
ER stress as evaluated by increased expression of glucose regulated protein 78 (GRP78) and various such unfolded protein responses as p-Inositol-requiring enzyme 1α (p-IRE 1α), unspliced X-Box binding protein 1 (uXBP1), p- eukaryotic translation initiation factor 2α (p-eIF2α), and CCAAT-enhancer-binding protein homologous protein (CHOP) in hPACs incubated with EtOH, acetaldehyde, or FAEEs was found to be concentration-dependent (Fig. 4 A–F). A significant change was noted in the cells treated with EtOH (3 and 6 mg/ml), acetaldehyde (5 and 10 μg/ml) or FAEEs (100 and 200 μg/ml). Of note, the expression of protein kinase RNA-like ER kinase (PERK) was significantly decreased in hPACs treated with EtOH (3 and 6 mg/ml) (Fig. 4D), in contrast to its increased expression in the cells treated with acetaldehyde (5 and 10 μg/ml) and FAEEs (100 and 200 μg/ml) (Fig. 4D). However, the expression of ATF6 was not altered in hPACs treated with either EtOH, acetaldehyde, or FAEEs (data not shown).
Fig. 4.
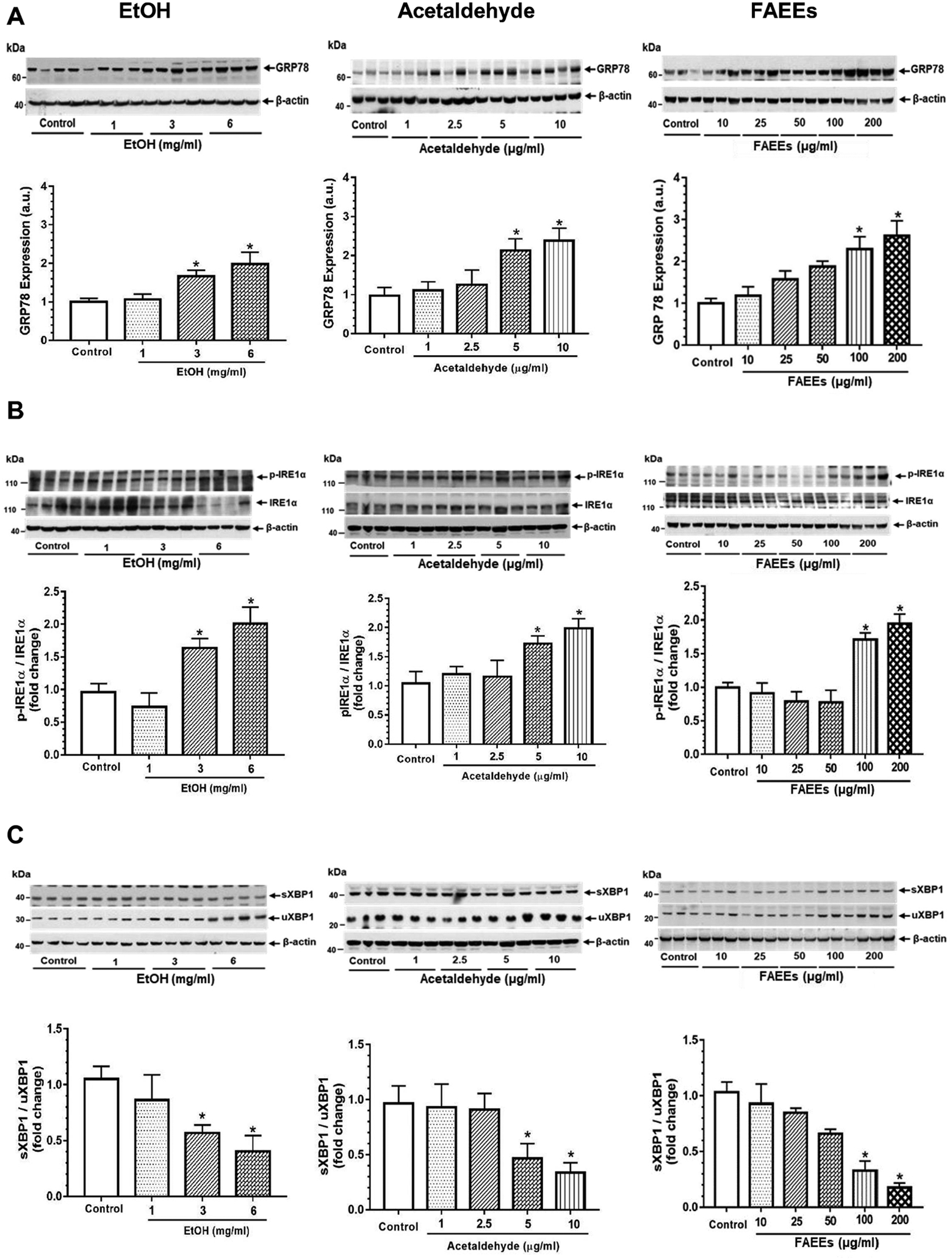
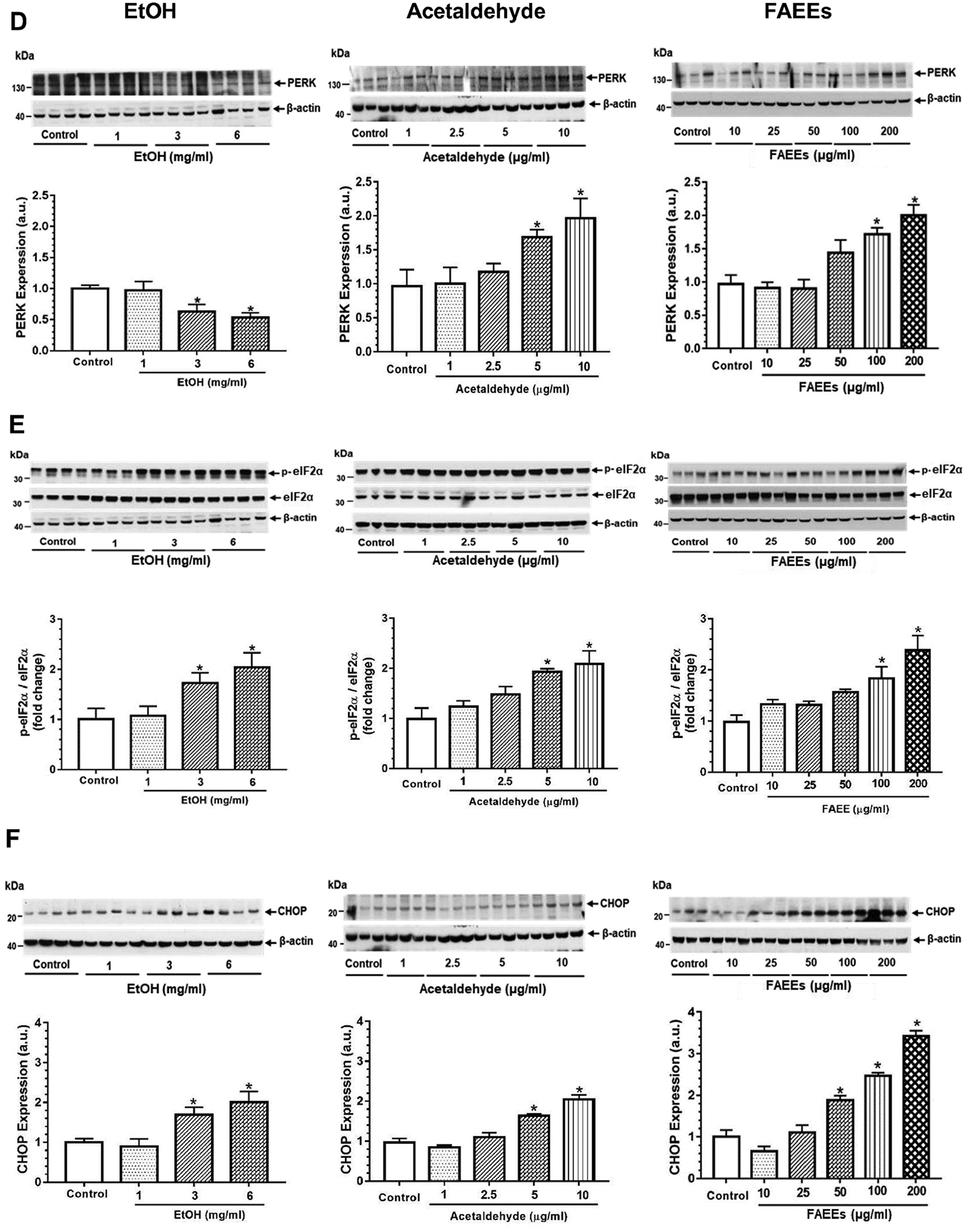
ER stress signaling in hPACs treated with EtOH (left panel), acetaldehyde (middle panel) and FAEEs (right panel) for 6hr. Representative immunoblots along with respective bar diagram show expression of GRP78 (A), p-IRE1α/ IRE1α (B), sXBP1/uXBP1 (C), PERK (D), p-eIF2α/eIF2α (E) and CHOP (F). Intensities were normalized to β-actin (loading control). Values are expressed as Mean ± SEM (n =12 replicates). * p value ≤ 0.05 with respect to controls.
As shown in Fig. 5A, the levels of 4-hydroxynonenal (4-HNE) modified protein adducts were significantly increased in hPACs treated with EtOH (3 and 6 mg/ml), acetaldehyde (5 and 10 μg/ml) or FAEEs (100 and 200 μg/ml), respectively.
Fig. 5.
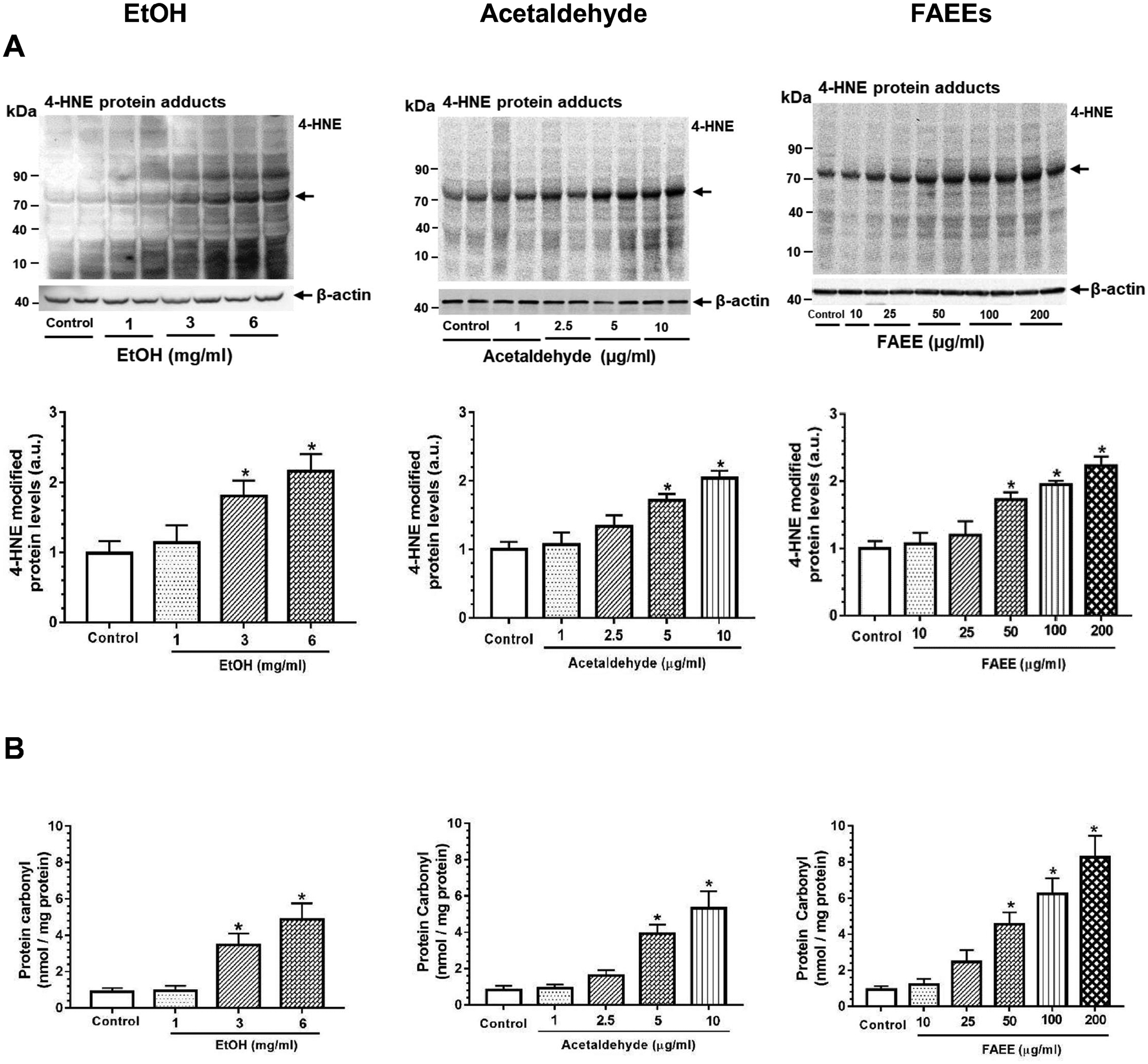
Levels of 4-hydroxynonenal (4-HNE, marker of Oxidative stress) protein adducts in hPACs incubated with EtOH (left panel), acetaldehyde (middle panel) and FAEEs (right panel) for 6hr (A). Intensities were normalized to β-actin (loading control). Values are expressed as Mean ± SEM (n = 4 replicates). * p value ≤ 0.05 with respect to controls.
Levels of protein carbonyl in hPACs incubated with EtOH (left panel), acetaldehyde (middle panel) and FAEEs (right panel) for 6hr (B). Values are expressed as Mean ± SEM (n = 4 replicates). * p value ≤ 0.05 with respect to controls.
In addition, the levels of protein carbonyl, a maker for protein oxidation and oxidative stress were significantly elevated in hPACs treated with EtOH (3 and 6 mg/ml), acetaldehyde (5 and 10 μg/ml) or FAEEs (100 and 200 μg/ml), respectively (Fig. 5B). Overall, EtOH as well as its metabolites caused a concentration-dependent oxidative stress in hPACs.
Activation of inflammatory pathways and cytokine secretion
Mitogen activated protein kinases (MAPKs), a key pathway activated during oxidative stress/inflammation, were significantly upregulated in hPACs treated with EtOH, acetaldehyde, or FAEEs as evidenced by a concentration-dependent increased expression of c-Jun N-terminal kinase 1/2 (JNK1/2), extracellular signal-regulated kinases 1/2 (ERK1/2), and p38 mitogen-activated protein kinases (P38MAPK) (Fig. 6A–C).
Fig. 6.
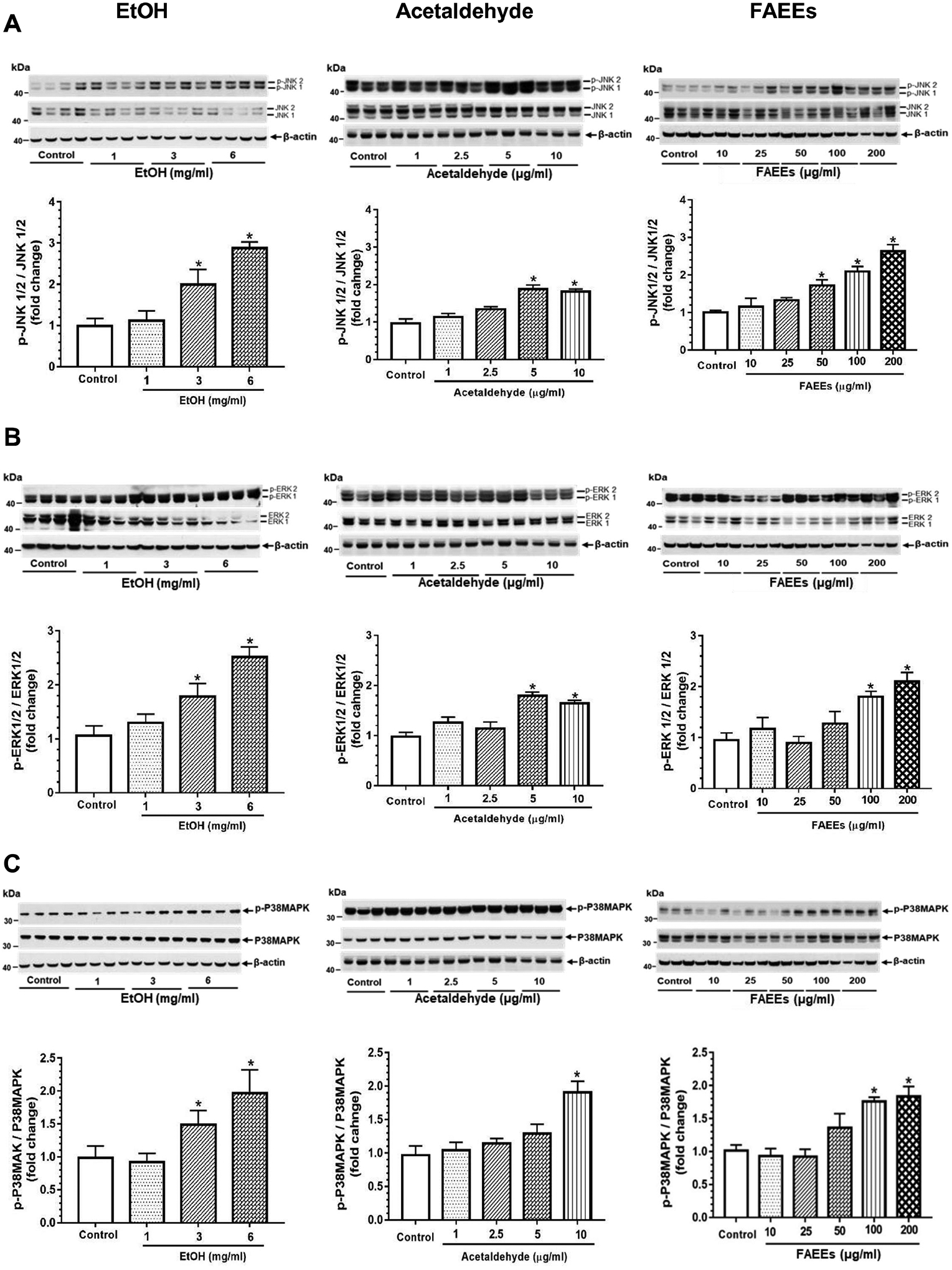
Activation of mitogen activated protein kinases (MAPKs) in hPACs treated with EtOH (left panel), acetaldehyde (middle panel) and FAEEs (right panel) for 6hr. Representative immunoblots along with respective bar diagram show the levels of p-JNK1/2/JNK1/2 (A), p-ERK1/2/ ERK1/2(B) and p-P38MAPK/P38MAPK (C), key proteins upregulated during oxidative stress and activate inflammatory pathway in hPACs. Intensities were normalized to β-actin (loading control). Values are expressed as Mean ± SEM (n =12 replicates). * p value ≤ 0.05 with respect to controls.
Of importance, an increased expression of tumor necrosis factor α (TNFα, an inflammatory cytokine) was noted in the hPACs treated with EtOH (3 and 6 mg/ml), acetaldehyde (5 and 10 μg/ml) or FAEEs (100 and 200 μg/ml) (Fig. 7A). Out of 23 human cytokines and chemokines assayed in the culture medium of acinar cells treated with acetaldehyde or FAEEs, only 11 (5 inflammatory cytokines and 6 chemokines) were secreted and detected (Fig. 7B). The expression levels of secreted inflammatory cytokines [interleukin 1α (IL-1α), IL-6, IL-8, TNFα, and TNFβ] and chemokines [monocyte chemoattractant protein-1 (MCP-1), MCP-2, MCP-3, growth-regulated oncogene (GRO), GROα, and regulated upon activation, normal T cell expressed and presumably secreted (RANTES)] were found to be significantly higher in the cells treated with acetaldehyde (5 and 10 μg/ml) and FAEEs (100 and 200 μg/ml) as compared to the respective controls.
Fig. 7.
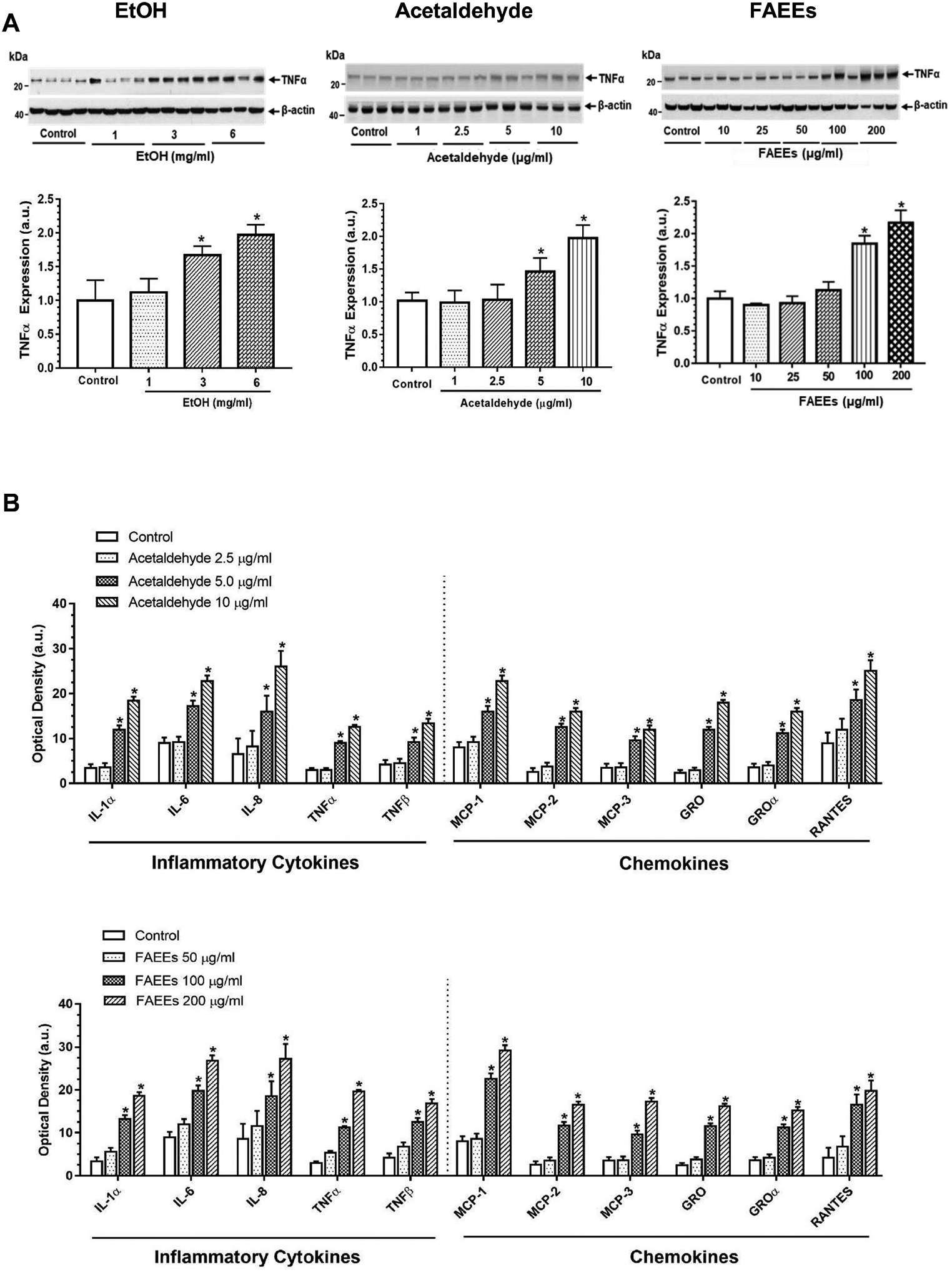
Markers of inflammation. Levels of tumor necrosis factor α (TNFα, a key inflammatory cytokine) in hPACs treated with EtOH (left panel), acetaldehyde (middle panel) and FAEEs (right panel) for 6hr (A). Intensities were normalized to β-actin (loading control). Values are expressed as Mean ± SEM (n =12 replicates). * p value ≤ 0.05 with respect to controls.
Levels of cytokines and chemokines secreted into culture medium from hPACs incubated with different concentrations of acetaldehyde (upper panel) and FAEEs (lower panel) for 6hr showing optical density of 11 detected cytokines (out of 23) (B). Values are expressed as Mean ± SEM (n =4). * p value ≤ 0.05 with respect to controls. IL-, Interleukin; TNF, tumor necrosis factor; MCP, Monocyte Chemoattractant Protein. Following cytokines/chemokines were tested but not detected in culture medium. IL-2; IL-3, IL-5; IL-7; IL-10; IL-13; IL-15; IFN-γ; MIG; G-CSF, GM-CSF and TGF-β1.
Dysregulated cellular bioenergetics in AR42J cells
As shown in Fig. 8A, AR42J cells treated with acetaldehyde (5 μg/ml) exhibited a significant reduction in basal oxygen consumption rate (OCR) from ~96 pmol/min to 74 pmol/min, along with ~2-fold decrease in spare respiratory capacity (SRC, an index of mitochondrial function). Further, an impaired mitochondrial function was found to be associated with a concomitant decrease in ATP production rate from ~75 pmol/min to 62 pmol/min. Similarly, as shown in Fig. 8A, AR42J cells treated with FAEEs (100 μg/ml) showed a significant decrease in basal OCR (from ~109 pmol/min to 54 pmol/min) and SRC (~ 1.5 fold) as compared to the respective control cells treated with 1 mg/ml EtOH. Of note, the ATP production rate was reduced significantly from ~102 pmol/min to 48 pmol/min.
Fig. 8.
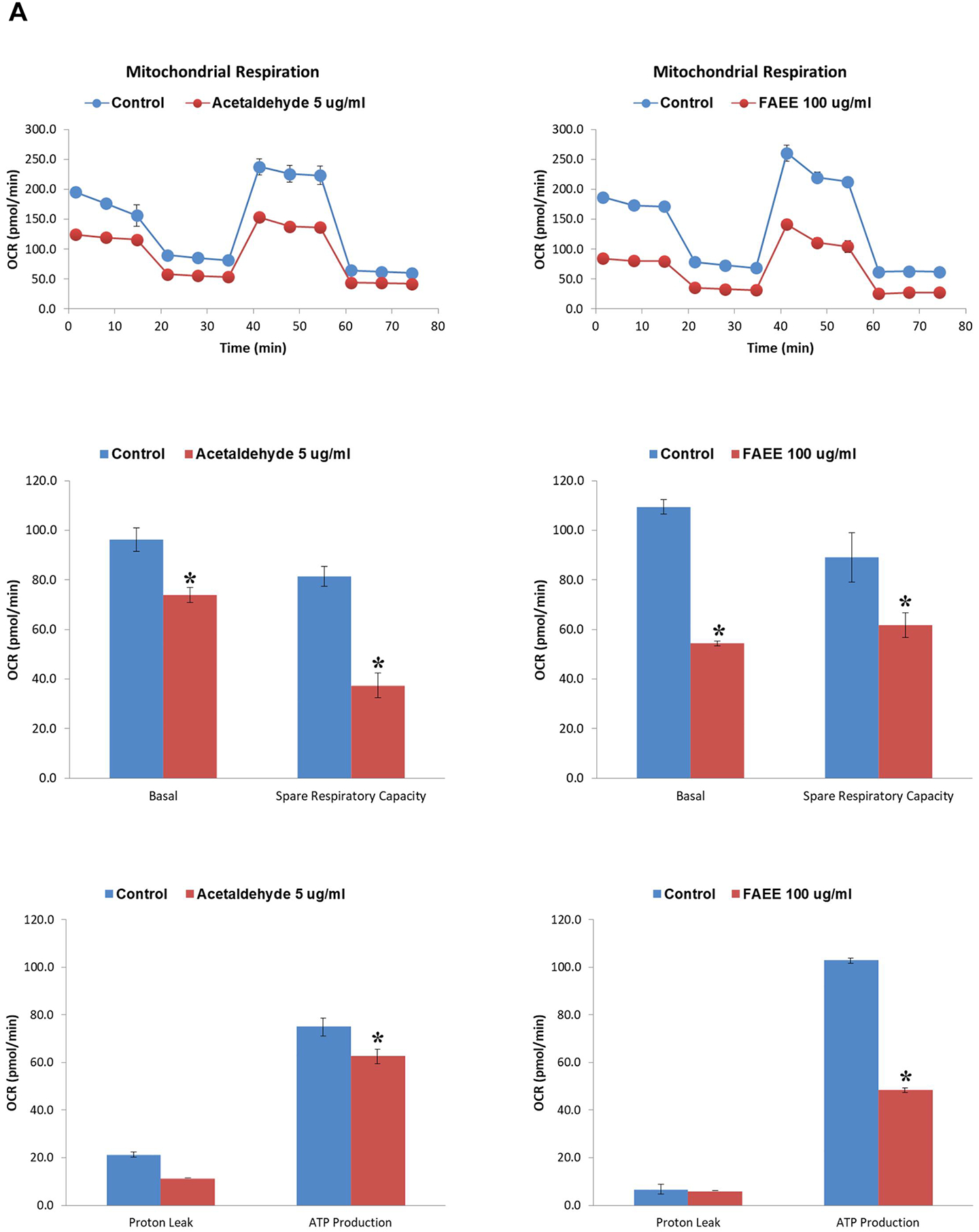
Cellular biogenetics in AR42J cells treated with acetaldehyde and FAEEs. Acute effects of acetaldehyde (left panel) and FAEE (right panel) on mitochondrial bioenergetics and real-time ATP production rate in AR42J cells were determined using Seahorse extracellular flux analyzer. Mito stress test showing the oxygen consumption rate, spare respiratory capacity and ATP production rate in AR42J cells (A). Data are shown as the Mean ± SEM (n = 3 independent experiments). * p value ≤ 0.05 with respect to controls.
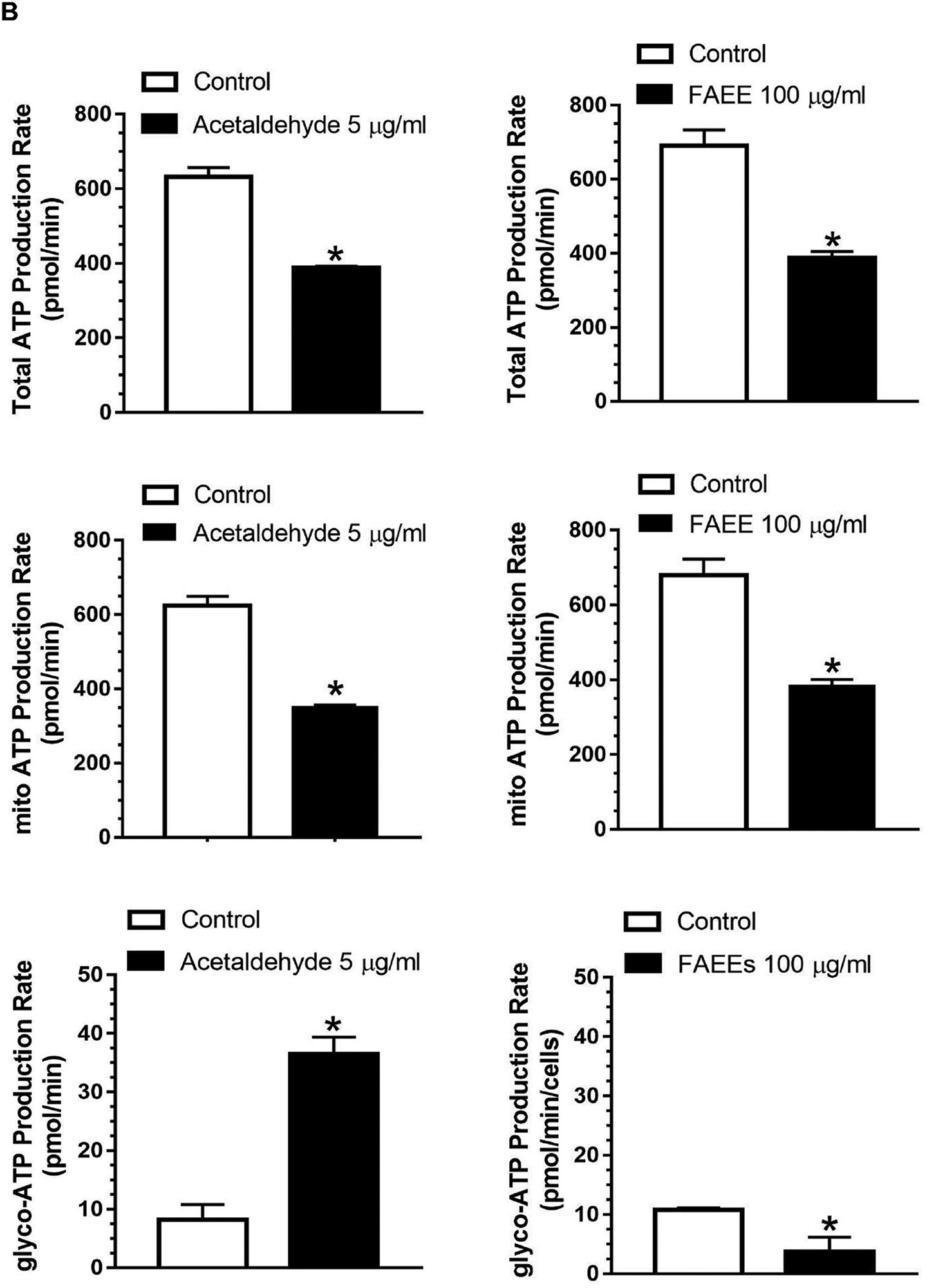
Real-time ATP production rate assay showing total ATP production rate, mitochondrial and Glycolytic ATP production rate in AR42J cells (B). Data are shown as the Mean ± SEM (n = 3 independent experiments). * p value ≤ 0.05 with respect to controls.
As shown in Fig. 8B, real-time total ATP production rate in the AR42J cells treated with acetaldehyde (5 μg/ml) decreased from ~641 pmol/min to 388 pmol/min. Furthermore, the treated cells exhibited a significant reduction in the mitochondrial ATP production rate (from ~ 623 pmol/min to 351 pmol/min) with a concomitant increase in glycolytic ATP production rate (from 17.9 pmol/min to 36.5 pmol/min). Similarly, as shown in Fig.8B, the AR42J cells treated with FAEEs (100 μg/ml) showed a significant reduction in real-time total ATP production rate from ~ 690 pmol/min to 388 pmol/min with a subsequent reduction of mitochondrial ATP production rate (from ~ 679 pmol/min to 384 pmol/min) as found for the cells treated with acetaldehyde, but the glycolytic ATP production rate was also reduced from ~ 11 pmol/min to 3 pmol/min in contrast to that by acetaldehyde.
Discussion
Although pancreatic acinar cells express both oxidative and nonoxidative metabolism of EtOH, the nonoxidative metabolism to fatty acid ethyl esters (FAEEs, catalyzed by FAEE synthase) is prevalent than oxidative metabolism to acetaldehyde [catalyzed by alcohol dehydrogenase (ADH) and cytochrome P450 2E1 (CYP2E1)] in the pancreas (Laposata and Lange, 1986, Gukovskaya et al., 2002, Werner et al., 2002, Wilson and Apte, 2003, Amer et al., 2018). Pancreatic ADH and CYP2E1 are shown to be relatively very low and are not induced by chronic EtOH exposure (Werner et al., 2002, Amer et al., 2018). Therefore, an increased expression of FAEE synthase in the pancreas after chronic EtOH exposure could significantly contribute to pancreatic EtOH disposition via nonoxidative metabolism.
Of note, FAEEs can be detected in systemic circulation and tissues after chronic alcohol consumption and that pancreatic FAEE synthase is significantly induced in alcohol-related pancreatitis (Laposata and Lange, 1986, Doyle et al., 1994, Kaphalia et al., 2004, Miyasaka et al., 2005). Furthermore, concentration-dependent increased expression of carboxyl ester lipase (CEL, the major FAEE synthase present in the pancreatic acinar cells) and subsequent formation of FAEEs in hPACs treated with EtOH has been reported earlier by us (Srinivasan et al., 2020). Thus, FAEEs formed during chronic alcohol abuse, itself could be responsible for pancreatic injury.
Nevertheless, exogenous acetaldehyde infusion / injection has been shown to alter the pancreatic morphology and exocrine dysfunction in some isolated pancreas models (Majumdar et al., 1986, Nordback et al., 1991). Rat pancreatic acini treated with very high concentrations of acetaldehyde (1000 μM) can cause perturbation in exocytosis (Dolai et al., 2012), as compared to 0–250 μM blood acetaldehyde concentration commonly reported in chronic alcoholics (Korsten et al., 1975, Nuutinen et al., 1983), but, endogenously produced acetaldehyde has failed to induce pancreatitis (He et al., 2001). Therefore, this is the first study to evaluate differential cytotoxicity of EtOH, acetaldehyde, and FAEEs in primary hPACs at concentrations reported in chronic alcoholic subjects.
AMPKα is a serine/threonine-protein kinase, a sensor of cellular energy, which regulates basal pancreatic acinar cell functions, but its inactivation could be one of the key underlying mechanisms in EtOH-mediated pancreatic acinar cell injury (Srinivasan et al., 2020). A concentration dependent inactivation of AMPKα by acetaldehyde or FAEEs in hPACs as observed in this study suggests that EtOH metabolism itself could be a determining factor for the inactivation of AMPKα and related ER/oxidative stress. However, this conclusion needs to be further validated by modulating oxidative and nonoxidative metabolism of EtOH (Bhopale et al., 2014). Upregulation of lipogenesis and downregulation of fatty acid oxidation as found in this study could also contribute to oxidative stress (Hauck and Bernlohr, 2016). Therefore, dysregulated AMPKα signaling by EtOH and its metabolites could play a key role in EtOH-induced pancreatic acinar cell dysfunction. Amelioration of EtOH-induced AMPKα inactivation and ER/oxidative stress including the formation of FAEEs by AMPKα activator (5-Aminoimidazole-4-carboxamide ribonucleotide, AICAR) suggests an interrelationship among AMPKα and ER/oxidative signaling and formation of FAEEs (Srinivasan et al., 2020). However, a similar beneficial role of antioxidants could enable develop a much simpler and economically viable therapeutic strategy for ACP.
Upstream kinases, LKB1 and/or CaMKKβ, related to oxidative stress and ER stress, respectively, can regulate activation of AMPKα (Carling et al., 2008, Willows et al., 2017). Thus, inhibition of LKB1 in hPACs treated with EtOH, acetaldehyde and FAEEs as found in this study could also contribute towards the inactivation of AMPKα. On the other hand, an upregulation of CaMKKβ as observed only in hPACs treated with FAEEs is a key finding and warrants further investigations in the context of FAEE-induced ER stress (Ozcan and Tabas, 2010) and calcium metabolism (Tokumitsu et al., 2000, Tokumitsu et al., 2001). Of importance, FAEEs have been shown to cause calcium toxicity linked to the sequestration of calcium from the ER membrane in pancreatic acinar cells (Criddle et al., 2004, Criddle et al., 2006). Thus, an increased expression of CaMKKβ in hPACs treated with FAEEs, only, could be caused by an elevated cytosolic calcium and ER stress suggesting a differential regulation of AMPKα by EtOH and its metabolites.
Since a putative link has been found between AMPKα inactivation and ER/oxidative stress in relation to EtOH-induced pancreatic acinar cell injury (Srinivasan et al., 2020), a systemic response to ER stress observed in hPACs treated with EtOH, acetaldehyde and FAEEs could be interrelated. Moreover, a reduced expression of sXBP1 together with increased expression for PERK/CHOP arm of UPR in hPACs treated with EtOH, acetaldehyde and FAEEs suggests a lack of adaptive UPR to maintain ER homeostasis commonly observed in subjects with alcoholic chronic pancreatitis (Sah et al., 2014, Lugea et al., 2017a).
Furthermore, despite increased phosphorylation of IRE1α, downregulation of sXBP1 in hPACs treated with EtOH, acetaldehyde / FAEEs is supported by increased levels of uXBP1. sXBP1 is an important translational and transcriptional regulator involved in homeostasis of ER membrane, but a precise molecular mechanism underlying EtOH and its metabolites induced downregulation of sXBP1 is largely unknown. Several factors including decreased zymogen granules, degradation of sXBP1, and inhibition of IRE1α-RNASE activity and post translational regulatory mechanism of XBP1 could contribute towards downregulation of sXBP1 in cells undergoing prolonged ER stress (Yoshida et al., 2006, Lugea et al., 2017a, Sun et al., 2019). Therefore, further studies to understand the underlying mechanism of EtOH-induced gene regulation of XBP1 can open new avenues for therapeutics of ACP.
Increased levels of inflammatory cytokines and chemokines have been reported in patients with severe acute pancreatitis and animal models of acute pancreatitis (Gukovskaya et al., 1997, Brivet et al., 1999, Hirota et al., 2000, Yang et al., 2000, Regner et al., 2008, Aoun et al., 2009). However, EtOH and its metabolites can regulate transcription factors and cytokines either positively or negatively depending on the effect of oxidative or nonoxidative metabolic pathways (Gukovskaya et al., 2002). A concentration-dependent activation of three major classes of MAPKs as found in hPACs treated with EtOH, acetaldehyde, or FAEEs can also mediate the production of pro-inflammatory cytokines and chemokines involved in the development of pancreatitis (Dabrowski et al., 2000, Irrera et al., 2014) and support our findings of an increased expression of inflammatory cytokines (TNFα along with IL-1α, IL-6, IL-8) and chemokines (MCP-1, GRO, RANTES).The activation of MAPKs in hPACs by EtOH and its metabolites could also be linked to oxidative stress and/or ER stress (Pearson et al., 2001, Aroor and Shukla, 2004, Apte et al., 2005). These seminal findings in hPACs further strengthen a case for the metabolic basis of alcoholic pancreatitis.
Oxidative stress is a pathological condition caused by an imbalance between production and accumulation of reactive oxygen species (ROS) in cells and tissues. Alcohol promotes the production of ROS and causes oxidative stress in many tissues including the pancreas (Das and Vasudevan, 2007). In addition to activation of pro-inflammatory pathways, alcohol-induced oxidative stress plays a key role in pathogenesis of ACP and could be linked to EtOH metabolism, itself (Apte et al., 2010, Perez et al., 2015). Lipid peroxidation and protein oxidation are very common reactions of ROS generated during EtOH oxidative metabolism and can result in cell death and inflammation (Barrera et al., 2015, Gaschler and Stockwell, 2017). 4-Hydroxynonenal (4-HNE, a reactive aldehyde and marker for oxidative stress) is a product of lipid peroxidation cascade and forms protein adducts resulting in protein oxidation (Barrera et al., 2015).Therefore, 4-HNE levels and protein carbonyl contents, alone or together indicate oxidative stress in hPACs treated with EtOH, acetaldehyde and FAEEs. Since oxidative metabolism of EtOH in the pancreas is negligible, we hypothesize that EtOH-induced pancreatic oxidative stress is associated with formation of FAEEs and lipid peroxidation.
FAEE-induced mitochondrial damage is one such putative mechanism which can explain several mechanism(s) related to pancreatic acinar cell injury (Criddle et al., 2006). In this regard, a diminished spare respiratory capacity/ impaired mitochondrial reserve as found in AR42J cells by acetaldehyde and FAEEs supports mitochondrial toxicity contributing to pancreatic acinar cell injury. Besides, a decreased mitochondrial ATP production rate as found in this study, together with a loss of secretory capacity of acinar cells, could shift the pancreatic cell death from apoptosis to necrosis (Armstrong et al., 2018).
Interestingly, a robust increase in ATP turnover from glycolysis concomitantly with inhibition of mitochondrial ATP turnover in response to acetaldehyde exposure, suggest presence of an in situ efficient operative compensatory metabolic shift towards glycolysis under impaired oxidative phosphorylation. However, despite potential boost of ATP production by this route, the glycolytic ATP production rate appear to be not sufficient to restore the cellular functions and homeostasis in the cells treated with acetaldehyde. In contrast, an inhibition of both glycolytic and oxidative phosphorylation pathways by FAEEs suggests incompetency of cells to meet cellular energy demands under elevated stress conditions, which could be linked to an array of acinar cell dysfunctions including lack of execution of apoptosis and autophagy (Mukherjee et al., 2016). It is likely that a decreased release for amylase and lipase as observed in hPACs treated with EtOH, acetaldehyde, or FAEEs could be linked to diminished mitochondrial ATP production rate. Of note, it has been shown that EtOH, acetaldehyde, or FAEEs could inhibit the secretory capacity of acinar cells by dysregulating the process of exocytosis (Sankaran et al., 1985, Dolai et al., 2012). Furthermore, an increased LDH release in contrast to impaired amylase and lipase secretion could contribute to cytotoxic effects of EtOH, acetaldehyde or FAEEs. Overall, contribution of oxidative versus nonoxidative EtOH metabolism is very different for the pancreas than that in the liver, indicating a greater role of nonoxidative metabolism of EtOH in the etiology of alcoholic pancreatitis.
In summary, EtOH and its metabolites, acetaldehyde and FAEEs cause acinar cell injury via dysregulated AMPKα signaling and related ER/oxidative stress, which could be linked to etiopathogenesis of ACP. Of importance, AMPKα activity can be reduced not only under the condition of oxidative stress but also during decreased ATP (Shearn et al., 2014). An increased ER/oxidative stress could be one of the factors for the inactivation of AMPKα or vice versa as observed in hPACs treated with EtOH or its metabolites. However, the pathogenesis of ACP likely to be associated with the formation of FAEEs and related pancreatic acinar cell injury in view of negligible oxidative EtOH metabolism reported in the exocrine pancreas. Further studies to examine the effect of individual FAEEs, modulation of EtOH metabolism and role of various modulators of ER/oxidative stress in acinar cell injury could identify potential etiologic agent(s).
Supplementary Material
Acknowledgments:
This work is supported by grants AA24699 and AA25850 from the National Institute on Alcohol Abuse and Alcoholism (NIAAA), National Institutes of Health (NIH), and its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIAAA/NIH. The authors sincerely thank Kentucky Organ Donor Affiliates (KODA) for the supply of human pancreas. We also thank the islet isolation team members of Dr. A.N. Balamurugan.
Abbreviations:
- 4-HNE
4-Hydroxynonenal
- ACC1
Acetyl CoA carboxylase 1
- ACP
Alcoholic chronic pancreatitis
- ADH1
Alcohol dehydrogenase 1
- ADP
Adenosine diphosphate
- AMP
Adenosine monophosphate
- AMPKα
AMP activated protein kinase α
- AP
Acute pancreatitis
- ATF6
Activating transcription factor 6
- ATP
Adenosine triphosphate
- CaMKK β
Ca2+/CaM-dependent protein kinase kinase β
- CEL
Carboxyl ester lipase
- CHOP
CCAAT-enhancer-binding protein homologous protein
- CP
Chronic pancreatitis
- CPT1A
Carnitine palmitoyltransferase 1A
- CYP2E1
Cytochrome P450 2E1
- ECAR
Extracellular acidification rate
- eIF2α
Eukaryotic translation initiation factor 2α
- EtOH
Ethanol
- ER
Endoplasmic reticulum
- ERK1/2
Extracellular signal-regulated kinases 1/2
- FAEEs
Fatty acid ethyl esters
- FAS
Fatty acid synthase
- GRP78
Glucose regulated protein 78
- hPACs
Human Pancreatic acinar cells
- IRE1α
Inositol-requiring enzyme 1α
- JNK1/2
c-Jun N-terminal kinase ½
- LDH
Lactate dehydrogenase
- LKB1
Liver kinase 1
- MAPK
Mitogen activated protein kinase
- OCR
Oxygen consumption rate
- P38MAPK
p38 mitogen-activated protein kinases
- PERK
Protein kinase RNA-like ER kinase
- SREBP1c
Sterol regulatory element-binding protein 1c
- TNFα
Tumor necrosis factor α
- XBP1
X-Box binding protein 1
- UPR
Unfolded protein response
Footnotes
Conflict of Interest
The authors declare no conflicts of interest.
References
- Amer SM, Bhopale KK, Kakumanu RD, Popov VL, Rampy BA, El-Mehallawi IH, Ashmawy MM, Shakeel Ansari GA, Kaphalia BS (2018) Hepatic alcohol dehydrogenase deficiency induces pancreatic injury in chronic ethanol feeding model of deer mice. Exp Mol Pathol 104:89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoun E, Chen J, Reighard D, Gleeson FC, Whitcomb DC, Papachristou GI (2009) Diagnostic accuracy of interleukin-6 and interleukin-8 in predicting severe acute pancreatitis: a meta-analysis. Pancreatology 9:777–785. [DOI] [PubMed] [Google Scholar]
- Apte MV, Pirola RC, Wilson JS (2010) Mechanisms of alcoholic pancreatitis. J Gastroenterol Hepatol 25:1816–1826. [DOI] [PubMed] [Google Scholar]
- Apte MV, Zima T, Dooley S, Siegmund SV, Pandol SJ, Singer MV (2005) Signal transduction in alcohol-related diseases. Alcohol Clin Exp Res 29:1299–1309. [DOI] [PubMed] [Google Scholar]
- Armstrong JA, Cash NJ, Ouyang Y, Morton JC, Chvanov M, Latawiec D, Awais M, Tepikin AV, Sutton R, Criddle DN (2018) Oxidative stress alters mitochondrial bioenergetics and modifies pancreatic cell death independently of cyclophilin D, resulting in an apoptosis-to-necrosis shift. J Biol Chem 293:8032–8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroor AR, Shukla SD (2004) MAP kinase signaling in diverse effects of ethanol. Life Sci 74:2339–2364. [DOI] [PubMed] [Google Scholar]
- Balamurugan AN, Breite AG, Anazawa T, Loganathan G, Wilhelm JJ, Papas KK, Dwulet FE, McCarthy RC, Hering BJ (2010) Successful human islet isolation and transplantation indicating the importance of class 1 collagenase and collagen degradation activity assay. Transplantation 89:954–961. [DOI] [PubMed] [Google Scholar]
- Barrera G, Pizzimenti S, Ciamporcero ES, Daga M, Ullio C, Arcaro A, Cetrangolo GP, Ferretti C, Dianzani C, Lepore A, Gentile F (2015) Role of 4-hydroxynonenal-protein adducts in human diseases. Antioxid Redox Signal 22:1681–1702. [DOI] [PubMed] [Google Scholar]
- Bhopale KK, Falzon M, Ansari GA, Kaphalia BS (2014) Alcohol oxidizing enzymes and ethanol-induced cytotoxicity in rat pancreatic acinar AR42J cells. In Vitro Cell Dev Biol Anim 50:373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhopale KK, Wu H, Boor PJ, Popov VL, Ansari GA, Kaphalia BS (2006) Metabolic basis of ethanol-induced hepatic and pancreatic injury in hepatic alcohol dehydrogenase deficient deer mice. Alcohol 39:179–188. [DOI] [PubMed] [Google Scholar]
- Brivet FG, Emilie D, Galanaud P (1999) Pro- and anti-inflammatory cytokines during acute severe pancreatitis: an early and sustained response, although unpredictable of death. Parisian Study Group on Acute Pancreatitis. Crit Care Med 27:749–755. [DOI] [PubMed] [Google Scholar]
- Carling D, Sanders MJ, Woods A (2008) The regulation of AMP-activated protein kinase by upstream kinases. Int J Obes (Lond) 32 Suppl 4:S55–59. [DOI] [PubMed] [Google Scholar]
- Chen L, Wang F, Sun X, Zhou J, Gao L, Jiao Y, Hou X, Qin CY, Zhao J (2010) Chronic ethanol feeding impairs AMPK and MEF2 expression and is associated with GLUT4 decrease in rat myocardium. Exp Mol Med 42:205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutts M, Hinton S, Zheng J, Scharp DW (2007) Hypothermic storage and preservation of human pancreatic acinar tissue. In Vitro Cell Dev Biol Anim 43:2–6. [DOI] [PubMed] [Google Scholar]
- Criddle DN, Murphy J, Fistetto G, Barrow S, Tepikin AV, Neoptolemos JP, Sutton R, Petersen OH (2006) Fatty acid ethyl esters cause pancreatic calcium toxicity via inositol trisphosphate receptors and loss of ATP synthesis. Gastroenterology 130:781–793. [DOI] [PubMed] [Google Scholar]
- Criddle DN, Raraty MG, Neoptolemos JP, Tepikin AV, Petersen OH, Sutton R (2004) Ethanol toxicity in pancreatic acinar cells: mediation by nonoxidative fatty acid metabolites. Proc Natl Acad Sci U S A 101:10738–10743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowski A, Boguslowicz C, Dabrowska M, Tribillo I, Gabryelewicz A (2000) Reactive oxygen species activate mitogen-activated protein kinases in pancreatic acinar cells. Pancreas 21:376–384. [DOI] [PubMed] [Google Scholar]
- Das SK, Vasudevan DM (2007) Alcohol-induced oxidative stress. Life Sci 81:177–187. [DOI] [PubMed] [Google Scholar]
- Dolai S, Liang T, Lam PPL, Fernandez NA, Chidambaram S, Gaisano HY (2012) Effects of ethanol metabolites on exocytosis of pancreatic acinar cells in rats. Gastroenterology 143:832–843 e837. [DOI] [PubMed] [Google Scholar]
- Doyle KM, Bird DA, al-Salihi S, Hallaq Y, Cluette-Brown JE, Goss KA, Laposata M (1994) Fatty acid ethyl esters are present in human serum after ethanol ingestion. J Lipid Res 35:428–437. [PubMed] [Google Scholar]
- Gaschler MM, Stockwell BR (2017) Lipid peroxidation in cell death. Biochem Biophys Res Commun 482:419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gukovskaya AS, Gukovsky I, Zaninovic V, Song M, Sandoval D, Gukovsky S, Pandol SJ (1997) Pancreatic acinar cells produce, release, and respond to tumor necrosis factor-alpha. Role in regulating cell death and pancreatitis. J Clin Invest 100:1853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gukovskaya AS, Mouria M, Gukovsky I, Reyes CN, Kasho VN, Faller LD, Pandol SJ (2002) Ethanol metabolism and transcription factor activation in pancreatic acinar cells in rats. Gastroenterology 122:106–118. [DOI] [PubMed] [Google Scholar]
- Haber PS, Apte MV, Moran C, Applegate TL, Pirola RC, Korsten MA, McCaughan GW, Wilson JS (2004) Non-oxidative metabolism of ethanol by rat pancreatic acini. Pancreatology 4:82–89. [DOI] [PubMed] [Google Scholar]
- Haber PS, Wilson JS, Apte MV, Pirola RC (1993) Fatty acid ethyl esters increase rat pancreatic lysosomal fragility. J Lab Clin Med 121:759–764. [PubMed] [Google Scholar]
- Hauck AK, Bernlohr DA (2016) Oxidative stress and lipotoxicity. J Lipid Res 57:1976–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He ZJ, Ericksson P, Alho H, Harmoinen A, Nordback I (2001) Effects on endogenous acetaldehyde production by disulfiram and ethanol feeding on rat pancreas. J Gastrointest Surg 5:531–539. [DOI] [PubMed] [Google Scholar]
- Hirota M, Nozawa F, Okabe A, Shibata M, Beppu T, Shimada S, Egami H, Yamaguchi Y, Ikei S, Okajima T, Okamoto K, Ogawa M (2000) Relationship between plasma cytokine concentration and multiple organ failure in patients with acute pancreatitis. Pancreas 21:141–146. [DOI] [PubMed] [Google Scholar]
- Irrera N, Bitto A, Interdonato M, Squadrito F, Altavilla D (2014) Evidence for a role of mitogen-activated protein kinases in the treatment of experimental acute pancreatitis. World J Gastroenterol 20:16535–16543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaphalia BS, Ansari GA (2001) Fatty acid ethyl esters and ethanol-induced pancreatitis. Cell Mol Biol (Noisy-le-grand) 47 Online Pub:OL173–179. [PubMed] [Google Scholar]
- Kaphalia BS, Ansari GA (2003) Purification and characterization of rat pancreatic fatty acid ethyl ester synthase and its structural and functional relationship to pancreatic cholesterol esterase. J Biochem Mol Toxicol 17:338–345. [DOI] [PubMed] [Google Scholar]
- Kaphalia BS, Bhopale KK, Kondraganti S, Wu H, Boor PJ, Ansari GA (2010) Pancreatic injury in hepatic alcohol dehydrogenase-deficient deer mice after subchronic exposure to ethanol. Toxicol Appl Pharmacol 246:154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaphalia BS, Cai P, Khan MF, Okorodudu AO, Ansari GA (2004) Fatty acid ethyl esters: markers of alcohol abuse and alcoholism. Alcohol 34:151–158. [DOI] [PubMed] [Google Scholar]
- Kaphalia BS; Khan MF; Caroll RM; Aronson J; Ansari GA Subchronic toxicity of 2-chloroethanol and 2-bromoethanol in rats. Res. Commun. Pharmacol. Toxicol 1996, 1, 173–186. [Google Scholar]
- Kaphalia L, Srinivasan MP, Kakumanu RD, Kaphalia BS, Calhoun WJ (2019) Ethanol Exposure Impairs AMPK Signaling and Phagocytosis in Human Alveolar Macrophages: Role of Ethanol Metabolism. Alcohol Clin Exp Res 43:1682–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Moon SY, Kim JS, Baek CH, Kim M, Min JY, Lee SK (2015) Activation of AMP-activated protein kinase inhibits ER stress and renal fibrosis. Am J Physiol Renal Physiol 308:F226–236. [DOI] [PubMed] [Google Scholar]
- Korsten MA, Matsuzaki S, Feinman L, Lieber CS (1975) High blood acetaldehyde levels after ethanol administration. Difference between alcoholic and nonalcoholic subjects. N Engl J Med 292:386–389. [DOI] [PubMed] [Google Scholar]
- Laposata EA, Lange LG (1986) Presence of nonoxidative ethanol metabolism in human organs commonly damaged by ethanol abuse. Science 231:497–499. [DOI] [PubMed] [Google Scholar]
- Liangpunsakul S, Sozio MS, Shin E, Zhao Z, Xu Y, Ross RA, Zeng Y, Crabb DW (2010) Inhibitory effect of ethanol on AMPK phosphorylation is mediated in part through elevated ceramide levels. Am J Physiol Gastrointest Liver Physiol 298:G1004–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugea A, Gerloff A, Su HY, Xu Z, Go A, Hu C, French SW, Wilson JS, Apte MV, Waldron RT, Pandol SJ (2017a) The Combination of Alcohol and Cigarette Smoke Induces Endoplasmic Reticulum Stress and Cell Death in Pancreatic Acinar Cells. Gastroenterology 153:1674–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugea A, Waldron RT, Mareninova OA, Shalbueva N, Deng N, Su HY, Thomas DD, Jones EK, Messenger SW, Yang J, Hu C, Gukovsky I, Liu Z, Groblewski GE, Gukovskaya AS, Gorelick FS, Pandol SJ (2017b) Human Pancreatic Acinar Cells: Proteomic Characterization, Physiologic Responses, and Organellar Disorders in ex Vivo Pancreatitis. Am J Pathol 187:2726–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar AP, Vesenka GD, Dubick MA, Yu GS, DeMorrow JM, Geokas MC (1986) Morphological and biochemical changes of the pancreas in rats treated with acetaldehyde. Am J Physiol 250:G598–606. [DOI] [PubMed] [Google Scholar]
- McCarroll JA, Phillips PA, Park S, Doherty E, Pirola RC, Wilson JS, Apte MV (2003) Pancreatic stellate cell activation by ethanol and acetaldehyde: is it mediated by the mitogen-activated protein kinase signaling pathway? Pancreas 27:150–160. [DOI] [PubMed] [Google Scholar]
- Miyasaka K, Ohta M, Takano S, Hayashi H, Higuchi S, Maruyama K, Tando Y, Nakamura T, Takata Y, Funakoshi A (2005) Carboxylester lipase gene polymorphism as a risk of alcohol-induced pancreatitis. Pancreas 30:e87–91. [DOI] [PubMed] [Google Scholar]
- Mukherjee R, Mareninova OA, Odinokova IV, Huang W, Murphy J, Chvanov M, Javed MA, Wen L, Booth DM, Cane MC, Awais M, Gavillet B, Pruss RM, Schaller S, Molkentin JD, Tepikin AV, Petersen OH, Pandol SJ, Gukovsky I, Criddle DN, Gukovskaya AS, Sutton R, Unit NPBR (2016) Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: inhibition prevents acute pancreatitis by protecting production of ATP. Gut 65:1333–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordback IH, MacGowan S, Potter JJ, Cameron JL (1991) The role of acetaldehyde in the pathogenesis of acute alcoholic pancreatitis. Ann Surg 214:671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuutinen H, Lindros KO, Salaspuro M (1983) Determinants of blood acetaldehyde level during ethanol oxidation in chronic alcoholics. Alcohol Clin Exp Res 7:163–168. [DOI] [PubMed] [Google Scholar]
- Ozcan L, Tabas I (2010) Pivotal role of calcium/calmodulin-dependent protein kinase II in ER stress-induced apoptosis. Cell cycle 9:223–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandol SJ, Gorelick FS, Gerloff A, Lugea A (2010) Alcohol abuse, endoplasmic reticulum stress and pancreatitis. Dig Dis 28:776–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandol SJ, Lugea A, Mareninova OA, Smoot D, Gorelick FS, Gukovskaya AS, Gukovsky I (2011) Investigating the pathobiology of alcoholic pancreatitis. Alcohol Clin Exp Res 35:830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panes J, Caballeria J, Guitart R, Pares A, Soler X, Rodamilans M, Navasa M, Pares X, Bosch J, Rodes J (1993) Determinants of ethanol and acetaldehyde metabolism in chronic alcoholics. Alcohol Clin Exp Res 17:48–53. [DOI] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH (2001) Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 22:153–183. [DOI] [PubMed] [Google Scholar]
- Perez S, Pereda J, Sabater L, Sastre J (2015) Redox signaling in acute pancreatitis. Redox Biol 5:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasineni K, Srinivasan MP, Balamurugan AN, Kaphalia BS, Wang S, Ding WX, Pandol SJ, Lugea A, Simon L, Molina PE, Gao P, Casey CA, Osna NA, Kharbanda KK (2020) Recent Advances in Understanding the Complexity of Alcohol-Induced Pancreatic Dysfunction and Pancreatitis Development. Biomolecules 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regner S, Appelros S, Hjalmarsson C, Manjer J, Sadic J, Borgstrom A (2008) Monocyte chemoattractant protein 1, active carboxypeptidase B and CAPAP at hospital admission are predictive markers for severe acute pancreatitis. Pancreatology 8:42–49. [DOI] [PubMed] [Google Scholar]
- Ricordi C, Lacy PE, Finke EH, Olack BJ, Scharp DW (1988) Automated method for isolation of human pancreatic islets. Diabetes 37:413–420. [DOI] [PubMed] [Google Scholar]
- Sah RP, Garg SK, Dixit AK, Dudeja V, Dawra RK, Saluja AK (2014) Endoplasmic reticulum stress is chronically activated in chronic pancreatitis. J Biol Chem 289:27551–27561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran H, Lewin MB, Wong A, Deveney CW, Wendland MF, Leimgruber RM, Geokas MC (1985) Irreversible inhibition by acetaldehyde of cholecystokinin-induced amylase secretion from isolated rat pancreatic acini. Biochem Pharmacol 34:2859–2863. [DOI] [PubMed] [Google Scholar]
- Sharkawi M (1984) In vivo inhibition of liver alcohol dehydrogenase by ethanol administration. Life Sci 35:2353–2357. [DOI] [PubMed] [Google Scholar]
- Shearn CT, Backos DS, Orlicky DJ, Smathers-McCullough RL, Petersen DR (2014) Identification of 5’ AMP-activated kinase as a target of reactive aldehydes during chronic ingestion of high concentrations of ethanol. J Biol Chem 289:15449–15462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan MP, Bhopale KK, Amer SM, Wan J, Kaphalia L, Ansari GS, Kaphalia BS (2019) Linking Dysregulated AMPK Signaling and ER Stress in Ethanol-Induced Liver Injury in Hepatic Alcohol Dehydrogenase Deficient Deer Mice. Biomolecules 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan MP, Bhopale KK, Caracheo AA, Amer SM, Khan S, Kaphalia L, Loganathan G, Balamurugan AN, Kaphalia BS (2020) Activation of AMP-activated protein kinase attenuates ethanol-induced ER/oxidative stress and lipid phenotype in human pancreatic acinar cells. Biochem Pharmacol:114174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg GR, Kemp BE (2009) AMPK in Health and Disease. Physiol Rev 89:1025–1078. [DOI] [PubMed] [Google Scholar]
- Sun S, Kelekar S, Kliewer SA, Mangelsdorf DJ (2019) The orphan nuclear receptor SHP regulates ER stress response by inhibiting XBP1s degradation. Genes Dev 33:1083–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Miao Y, Kang F, Dolai S, Gaisano HY (2020) Susceptibility Factors and Cellular Mechanisms Underlying Alcoholic Pancreatitis. Alcohol Clin Exp Res. [DOI] [PubMed] [Google Scholar]
- Tokumitsu H, Iwabu M, Ishikawa Y, Kobayashi R (2001) Differential regulatory mechanism of Ca2+/calmodulin-dependent protein kinase kinase isoforms. Biochemistry 40:13925–13932. [DOI] [PubMed] [Google Scholar]
- Tokumitsu H, Muramatsu M, Ikura M, Kobayashi R (2000) Regulatory mechanism of Ca2+/calmodulin-dependent protein kinase kinase. J Biol Chem 275:20090–20095. [DOI] [PubMed] [Google Scholar]
- Vonlaufen A, Wilson JS, Pirola RC, Apte MV (2007) Role of alcohol metabolism in chronic pancreatitis. Alcohol Res Health 30:48–54. [PMC free article] [PubMed] [Google Scholar]
- Werner J, Laposata M, Fernandez-del Castillo C, Saghir M, Iozzo RV, Lewandrowski KB, Warshaw AL (1997) Pancreatic injury in rats induced by fatty acid ethyl ester, a nonoxidative metabolite of alcohol. Gastroenterology 113:286–294. [DOI] [PubMed] [Google Scholar]
- Werner J, Saghir M, Fernandez-del Castillo C, Warshaw AL, Laposata M (2001) Linkage of oxidative and nonoxidative ethanol metabolism in the pancreas and toxicity of nonoxidative ethanol metabolites for pancreatic acinar cells. Surgery 129:736–744. [DOI] [PubMed] [Google Scholar]
- Werner J, Saghir M, Warshaw AL, Lewandrowski KB, Laposata M, Iozzo RV, Carter EA, Schatz RJ, Fernandez-Del Castillo C (2002) Alcoholic pancreatitis in rats: injury from nonoxidative metabolites of ethanol. Am J Physiol Gastrointest Liver Physiol 283:G65–73. [DOI] [PubMed] [Google Scholar]
- Willows R, Sanders MJ, Xiao B, Patel BR, Martin SR, Read J, Wilson JR, Hubbard J, Gamblin SJ, Carling D (2017) Phosphorylation of AMPK by upstream kinases is required for activity in mammalian cells. Biochem J 474:3059–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JS, Apte MV (2003) Role of alcohol metabolism in alcoholic pancreatitis. Pancreas 27:311–315. [DOI] [PubMed] [Google Scholar]
- Wu H, Bhopale KK, Ansari GA, Kaphalia BS (2008) Ethanol-induced cytotoxicity in rat pancreatic acinar AR42J cells: role of fatty acid ethyl esters. Alcohol Alcohol 43:1–8. [DOI] [PubMed] [Google Scholar]
- Yang BM, Demaine AG, Kingsnorth A (2000) Chemokines MCP-1 and RANTES in isolated rat pancreatic acinar cells treated with CCK and ethanol in vitro. Pancreas 21:22–31. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Oku M, Suzuki M, Mori K (2006) pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J Cell Biol 172:565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.