

Abstract
Motivation
Access to large-scale genomics and transcriptomics data from various tissues and cell lines allowed the discovery of wide-spread alternative splicing events and alternative promoter usage in mammalians. Between human and mouse, gene-level orthology is currently present for nearly 16k protein-coding genes spanning a diverse repertoire of over 200k total transcript isoforms.
Results
Here, we describe a novel method, ExTraMapper, which leverages sequence conservation between exons of a pair of organisms and identifies a fine-scale orthology mapping at the exon and then transcript level. ExTraMapper identifies more than 350k exon mappings, as well as 30k transcript mappings between human and mouse using only sequence and gene annotation information. We demonstrate that ExTraMapper identifies a larger number of exon and transcript mappings compared to previous methods. Further, it identifies exon fusions, splits and losses due to splice site mutations, and finds mappings between microexons that are previously missed. By reanalysis of RNA-seq data from 13 matched human and mouse tissues, we show that ExTraMapper improves the correlation of transcript-specific expression levels suggesting a more accurate mapping of human and mouse transcripts. We also applied the method to detect conserved exon and transcript pairs between human and rhesus macaque genomes to highlight the point that ExTraMapper is applicable to any pair of organisms that have orthologous gene pairs.
Availability and implementation
The source code and the results are available at https://github.com/ay-lab/ExTraMapper and http://ay-lab-tools.lji.org/extramapper.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The fundamental process of gene expression in mammalian genomes is a highly complex procedure that is regulated at multiple different levels (Lelli et al., 2012). An important part of this regulation involves the transcription of alternative gene products from a single gene through the use of alternative promoters and alternative splicing (Ayoubi and Van De Ven, 1996; Davuluri et al., 2008; Matlin et al., 2005; Schibler and Sierra, 1987). Alternative transcription allows an organism to produce multiple protein isoforms that may substantially differ in their structures and functions (Lerch et al., 2012; Murray-Zmijewski et al., 2006; Seijffers et al., 2007). Recent advances in sequencing technology and the analysis of sequencing data now allow researchers to explore ‘one gene → multiple isoforms → multiple functions’ paradigm instead of still commonly used ‘one gene → one protein → one function’ approach (Pal et al., 2014; Shiraki et al., 2003; Wang et al., 2009). While there already are methods aimed at overcoming the challenges of transcript/isoform level gene expression analysis (Anders and Huber, 2010; Kim et al., 2011; Robinson et al., 2010; Trapnell et al., 2009), only a limited number of studies attempt to comparatively analyze transcripts to find transcript-level orthology relationship among different organisms (Pavesi et al., 2008; Zambelli et al., 2010).
One important intermediate step in revealing transcript-level relationships between a pair of organisms is to identify their conserved exons. To date, there is only a limited number of published studies that aim to find exon-level relationships among different organisms (Blekhman, 2012; Douzery et al., 2014; Fu and Lin, 2012; Modrek and Lee, 2003; Zhang et al., 2009). These methods use either sequence alignment tools, such as BlastN and tBlastX, or reverse engineer the protein level similarity between isoforms to find conserved exons between two organisms. Often, only the fully coding exons are considered by these methods completely leaving out the partially coding and the non-coding exons such as first and last exons of a transcript. At the transcript level, Exalign, described in Pavesi et al. (2008) and used for transcript mappings in Zambelli et al. (2010), computes similarities between two given transcripts by utilizing only the exon-intron structure and the coding lengths of exons. However, Exalign does not utilize any sequence similarity or orthology information between the exons or the transcripts and is biased toward assigning higher similarity scores for transcripts with large number of exons.
Here, we describe a new method, ExTraMapper, which extracts fine-scale mappings between the exons and transcripts of a given pair of ortholog genes between two organisms using sequence conservation between the two genomes and their gene annotations(Fig. 1). To demonstrate the use of our method, we first find exon and transcript mappings for nearly 16k protein coding genes with orthology defined between human and mouse. Compared to previous methods, ExTraMapper identifies a significantly larger number of exon mappings, as well as transcript mappings with differing stringencies for conservation. Notably, our exon mappings include microexons (<21 bp), which are largely missed by previous approaches. Using similarity both in coding domain and in overall sequence, ExTraMapper better distinguishes transcript pair similarities leading to resolution of many-to-many relationships between transcripts into one-to-one when possible. Furthermore, transcript similarity scores computed by ExTraMapper show less dependency to the number of exons of a transcript compared to an existing method that uses only exon length information for scoring transcript pairs. We also compared and found that RNA-seq expression profiles (Yue et al., 2014) of orthologous transcripts from human and mouse tissues identified by ExTraMapper show a higher degree of correlation and more similar extent of expression level then Exalign specific isoforms. Our results on multiple important human-mouse ortholog gene pairs and comparing their respective isoform-level expression profiles across different tissues from human and mouse show that ExTraMapper finds biologically relevant transcript-level mappings. The results also provide examples of exon fusions, splits and losses due to splice site mutations. In addition to human-mouse mapping, we also apply ExTraMapper to find exon and transcript mappings between human and rhesus macaque (Macaca mulatta) genomes. ExTraMapper reports over 442k exon mappings at a similarity threshold of 0.8 involving ∼359k human and ∼207k rhesus macaque exons. For transcripts, similar to human-mouse mappings, ExTraMapper identifies ∼30k transcript pairs. However, over 14k of human-rhesus transcript mappings are perfect mappings (similarity score of 1) compared to 8k for human-mouse comparison reflecting their evolutionary distance. These results highlight the suitability of ExTraMapper in analyzing different pairs of genomes with a varying degree of conservation.
Fig. 1.
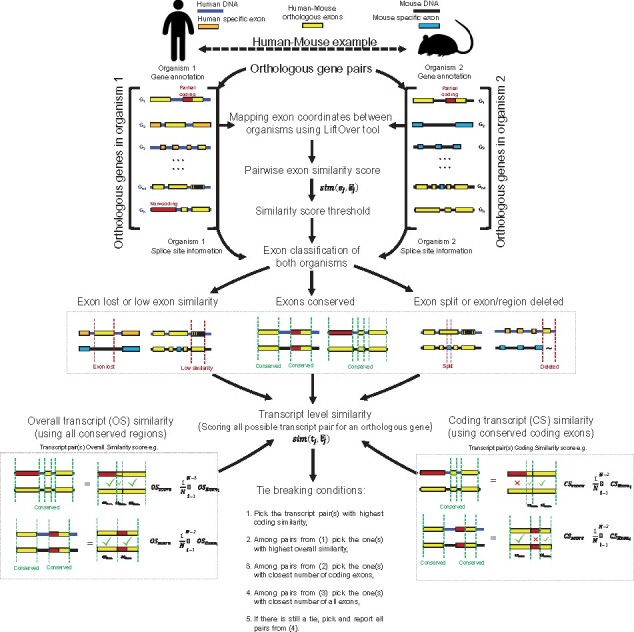
Overview of ExTraMapper. For each orthologous gene pair between the two organisms, ExTraMapper first computes exon-level similarities using gene annotations and the liftover tool that maps regions from one organism to the other. Each exon is then categorized according to its conservation status using the computed similarity scores and splice site information from acceptor and donor sites. The exon-level similarity scores are used to compute transcript-level similarities through an exon pairing algorithm. The transcript-level mappings for each gene are then determined using a greedy method that applies multiple tie break conditions to ensure mostly one-to-one mappings
2 Materials and methods
2.1 Publicly available datasets and methods
2.1.1. Gene annotations and orthologous pairs
We download and use gene annotations and orthology information from Ensembl database release 81 (Flicek et al., 2014). For human genome, we use the GTF file Homo_sapiens.GRCh38.81.gtf.gz and the corresponding reference genome build of hg38. For mouse genome, we use the GTF file Mus_musculus.GRCm38.81.gtf.gz and the genome build mm10. For the orthology information of gene pairs between human and mouse genomes, we use ensembl_mart_81/hsapiens_gene_ensembl__homolog_mmus__dm.txt.gz and ensembl_mart_81/mmusculus_gene_ensembl__homolog_hsap__dm.txt.gz files. These files provide orthology pairings for 16 711 genes, 15 846 of which are annotated as protein coding genes in both organisms. Almost all the remaining orthologous gene pairs code for various types of RNA and are between single-exon, single-transcript genes which are trivial cases for our interest. Summary of exon and transcript-level annotations for the 15 846 orthologous gene pairs are presented in Supplementary Table S1.
For the repeat of the same human-mouse analysis with Ensembl release 102 (November 2020), we use GTF files Homo_sapiens.GRCh38.102.gtf.gz and Mus_musculus.GRCm38.102.gtf.gz for gene annotations and hsapiens_gene_ensembl__homolog_mmusculus__dm.txt.gz and mmusculus_gene_ensembl__homolog_hsapiens__dm.txt.gz for orthology, which provide 15 177 protein coding genes that are orthologous. For the exon and transcript mapping of human and rhesus macaque genomes (Ensembl release 102), we use the GTF files Homo_sapiens.GRCh38.102.gtf.gz, Macaca_mulatta.Mmul_10.102.gtf.gz for gene annotations as well as files hsapiens_gene_ensembl__homolog_mmulatta__dm.txt.gz, mmulatta_gene_ensembl__homolog_hsapiens__dm.txt.gz providing us with 16 150 protein coding genes that are orthologous.
2.1.2. Conservation scores between human and mouse
We use Multiz alignments provided as MAF (Multiple Alignment Format) files and PhastCons to compute a conservation score for each human and mouse exon (Blanchette et al., 2004; Siepel et al., 2005; Siepel and Haussler, 2004). We filter the MAF files downloaded from http://hgdownload.cse.ucsc.edu/goldenPath/hg38/multiz7way/hg38.7way.maf.gz for human genome and from http://hgdownload.cse.ucsc.edu/goldenPath/mm10/multiz60way/maf/ for mouse genome by using maf_parse to only keep human to mouse and mouse to human alignments. We then use phastCons twice, once to obtain tree models and once to generate wig files with conservation scores at single base pair resolution. Using these scores, for each exon in either organism, we compute average conservation scores (i) for the whole exon body, (ii) for the coding portion of the exon and, (iii) for the acceptor and donor sites of the exon.
2.1.3. Exon mappings from OrthoExon
We download mappings between united exons described in Fu and Lin (2012) from http://tdl.ibms.sinica.edu.tw/OrthoExon/download.html. Since the exon coordinates in these mappings belong to earlier reference genome versions (hg18 for human and mm9 for mouse), we first use liftOver to convert these coordinates and then use bedtools intersect to find corresponding exons in hg38 and mm10 genome builds.
2.1.4. Exon mappings from protein isoforms using Inparanoid
We download mappings between orthologous exons described in Zhang et al. from http://genomebiology.com/content/supplementary/gb-2009-10-11-r120-s5.zip. Since the exon coordinates in the mappings provided in Dataset_s2_Human_and_mouse_orthologous_exons_list.tsv belong to earlier reference genome versions (hg18 for human and mm8 for mouse), we first use liftOver to convert these coordinates and then use bedtools intersect to find corresponding exons in hg38 and mm10 genome builds.
2.1.5. Transcript mappings from Exalign
We parse the transcript information for only protein coding transcripts from Ensembl database (release 81) for human and mouse in the input format desired by Exalign. We use ‘exalign -O <organism>.rf –freq’ to compute exon length frequency files required by Exalign for each organism using all their genes. Then, for each orthologous gene pair, we create a query/database file pair using only the transcripts for that gene pair. We then run exalign with default parameters to find transcript mappings from human to mouse and from mouse to human using exon length frequencies generated from all genes.
2.1.6. Tissue specific RNA-seq data and expression profile analysis
We download the human and mouse tissue specific RNA-seq data (Lin et al., 2014) from ENCODE project site in FASTQ format (Yue et al., 2014). For our analysis we first map the human and mouse tissue specific RNA-seq reads on GRCh38 and GRCm38 genome assembly, respectively, using Hisat2 (Kim et al., 2015). We then use featureCounts (Liao et al., 2014) to generate the initial read counts for gene, transcript and exon-wise Ensembl genomic annotations (Yates et al., 2016) for each tissue per organism from the mapped reads. For correlation calculation, we use expression values from 13 281 protein coding genes that: (i) have at least one reported transcript pair from both ExTraMapper and Exalign mappings, (ii) are among the 15 846 orthologous genes between human and mouse. We next follow a similar protocol as described by Gilad and Mizrahi-Man (2015) to account for the sequencing study design batch effect using ‘ComBat’ function from ‘sva’ package (Leek et al., 2012) to get the final gene, transcript and exon-wise expression values for downstream analysis.
2.2 Detailed description of ExTraMapper
2.2.1. Computation of pairwise exon similarities
From the orthologous gene pairs, we first extract all exons from all annotated transcripts for both genomes. We use liftOver chains between the two organisms to find, for each exon coordinate in one organism, the corresponding genomic coordinates in the other organism. To allow for non-perfect exon mappings, we use multiple settings for the ‘-minMatch’ parameter (0.9, 0.95 and 1.0) of liftOver which sets the minimum ratio of bases that must remap to the target organism (default value is 0.95). Also, to capture potential one-to-many mappings, we use the ‘-multiple’ option of liftOver which allows outputting multiple target regions per input region. For each gene pair , for each exon of the gene in one organism, we find the exons of the gene from the other organism that intersect with the lifted over coordinates of . For each possible pairing of the from and from , we compute a similarity score using:
the amount of overlap between the lifted over coordinates and the original coordinates,
the ratio between the original length and the lifted over length of .
We compute the same score for the pair by reversing the order of the two organisms and take the maximum score between the two orderings. More formally, let denote the lifted over coordinates and denote the length of the exon . Then, we define the similarity for the exon pair as:
| (1) |
This score is zero for any two exons that do not overlap after both ways of liftOver and it is proportional to the percentage of overlap for overlapping exon pairs. A similarity score of 1 corresponds to perfect conservation of length and overlap for an exon between the two genomes.
We calculate the above similarity over the whole exon body regardless of whether the exon is fully coding, partially coding or non-coding. To compute a coding similarity counterpart of this overall exon similarity, we analyze three cases of exon coding type separately. For the case of fully coding exons, the overall similarity equals coding similarity. For non-coding exons the coding similarity is trivially undefined and is set to zero. For the case of partially coding exons, we compute coding similarity score using only the coding portion of an exon. We use the coding portions of each partially coding exon for the liftOver between the organisms and then for the length and overlap calculations. We then use these values in the same formulation as above to compute the coding similarity score.
2.2.2. Computation of pairwise transcript similarities
For each orthologous gene pair , for each pair of transcripts such that , and , we compute two similarity scores (i.e. overall and coding) using the similarities of the conserved exons between the two organisms. Computation of these similarity scores in non-trivial since an exon of one organism sometimes overlaps with several other exons of the other. For this we choose a greedy approach that selects the ‘most’ similar exon pair between the two transcripts, match the two exons to each other, remove them from the set of exons to be paired and repeat this process until there is no exon pair with a similarity score of at least a given threshold. When determining the ‘most’ similar exon pair at each step requires tie break, we first look at the coding similarity of each exon pair then compare the overall exon similarity. If there are still ties, we pick one exon pair and report it and then repeat the process.
Once a set of one-to-one and non-contradicting exons mappings are found for a transcript pair , we can then define the similarity score as:
| (2) |
Note that for partially coding exons, we take as the maximum of overall exon similarity and coding exon similarity. We use all exons, including non-coding exons, for the overall pairwise transcript similarity. For the coding transcript similarity, we use only the exons that are either fully coding or partially coding for both exon similarity and transcript length calculations in the above equation. These transcript similarity scores range between 0 and 1, with a similarity score of 1 indicating perfect conservation of a transcript between human and mouse genomes including the transcript length and exon identity.
2.2.3. Extraction of transcript-level mappings
Once we compute pairwise transcript similarities for each gene pair, we get a transcript similarity matrix that is where and correspond to number of transcripts of the two genes in the orthologous gene pair. Each transcript from one organism potentially has similarity to multiple transcripts from the other organism leading to one-to-many or many-to-many relationships among the transcripts of the two organisms. We use a greedy method to extract a set of transcript mappings which mostly includes one-to-one mappings that do not conflict with each other. Note that, it is possible to extract such set of mappings with highest total similarity score by solving the maximum weight bipartite matching (MWBM) which can be done in polynomial time. However, MWBM prefers multiple suboptimal mappings (e.g. two mappings with score of 0.6 and 0.5) to a perfect mapping of score 1 which conflicts with the two suboptimal mappings. This feature, even though desirable in some scenarios, is undesirable for our purpose which is to find a set of transcript mappings, even though it may be a small set, that are highly conserved between the two organisms. Therefore, we greedily select the transcript pair with the highest similarity score first, accept this transcript mapping into our results set and then set the corresponding row and column in matrix to zero to ensure no conflicting mappings will selected in the later iterations. We repeat this process until no more non-zero entries are left in . We also post-process the set resulting set of mappings and discard those transcript pairs with a similarity score below a given threshold (at least 0.8) to ensure that only highly conserved pairs are reported.
Since the above described method of extracting transcript mappings can include ties, such as two pairs with the same exact score in , in the process, we break these ties in the best way possible to favor pairs that are highly similar. We list the criteria for breaking these ties and their ordering in Figure 1. Briefly, we first look at the coding similarity between transcripts to pick a single transcript pair. In case of a tie for this criterion, we resort to overall transcript similarity to break the tie among the tied pairs from the previous step. If there is another tie, we pick among the tied pairs the transcript pair that has the smallest difference between the numbers of coding exons of the two transcripts. If there are still tied pairs after all the previous criteria, we try to break it by the difference between the numbers of overall exons of the two transcripts. Lastly, we pick and report all transcript pairs that are tied in all the four criteria we checked because all these pairs have equivalent value for our purposes.
3 Results
3.1 ExTraMapper utilizes exons with different coding and positional types
An important step in expanding orthology from gene level to transcript isoform level is to establish an orthology relationship between the exons, which are the building blocks of transcripts. To define such a relationship, we gather all exons from 15 846 gene pairs that are (i) protein coding in both organisms, (ii) one-to-one mapped in human-mouse orthology on Ensembl release 81 (Section 2). This provides us with nearly 500k and 345k exons for human and mouse, respectively (407k and 289k unique ones after removing duplicated coordinates). The median numbers of exons and coding exons per gene were 22 and 14 for human, and 15 and 11 for mouse (Supplementary Fig. S1a, b). Supplementary Table S1 reports further grouping of these unique coordinates with respect to their coding types (fully coding, partially coding, non-coding) and their positions (first, middle, last) within each transcript. For each classification; we report a fourth category for exons that have multiple different types in different transcripts (e.g. an exon with the exact same coordinates that is labelled as coding in one transcript and non-coding in another one). Our further analysis of different exon types reported in using evolutionary conservation scores demonstrate that coding exons are more conserved compared to non-coding exons as expected (Supplementary Fig. S1c, d). Also, exons that are exclusively in the middle of each transcript they participate are highly conserved (Supplementary Fig. S1e, f) and are mainly fully coding (43.7% and 60.5% for human and mouse). In comparison, full coding exons make up only 10.7% and 8.7% among first or last exons of human and mouse genomes, respectively. These statistics highlight the importance of utilizing all exon types in computing transcript similarities and mappings.
3.2 ExTraMapper identifies conserved exons between human and mouse genomes
Without any assumption on the gene or transcript structure, ExTraMapper computes exon-level similarity scores and classifies exons with respect to their conservation between human and mouse (Section 2). Supplementary Figure S2 summarizes the fraction of exons for each gene that ExTraMapper mapped from human to mouse (a–c) and from mouse to human (d–f) with different stringencies. With an exon similarity threshold of 0.9, more than half the genes had at least 83% (human) and 96% (mouse) of all their coding exons mapped (Supplementary Fig. S2). These percentages are 59% (human) and 75% (mouse) when all exons are considered (Supplementary Fig. S2). Notably, a larger fraction of mouse exons is classified as mapped for each threshold choice reflecting the smaller number of total exons for mouse compared to human.
We also compute the inclusion level for an exon as the fraction of all protein coding transcripts that include a given exon (Alekseyenko et al., 2007; Modrek and Lee, 2003), and ask whether the inclusion level is a determinant of exon conservation. Similar to Modrek and Lee (2003), we observe that very large percent (95.9% for human and 95.7% for mouse) of constitutive and major-form exons (i.e. inclusion level >0.5) are mapped between the two organisms by ExTraMapper. For minor-form exons (i.e. inclusion level <0.5) the percentages of mapped ones are 52.6% and 58.8% for human and mouse, respectively. These results are in conjunction with Modrek et al. which suggests that around 98% of constitutive and major-form exons and only around 28% of minor-form exons existed before the divergence of the mouse and human genomes (Modrek and Lee, 2003). The difference in the percentages between Modrek et al. in 2003 (∼40k total exons) and our work here (>840k total exons) is likely due to improvement in the exon annotations of both organisms within the past decade.
3.3 ExTraMapper reports additional exon mappings compared to existing methods
We compare ExTraMapper with two previous methods that find exon-level mappings between human and mouse. For an orthologous gene pair, OrthoExon uses a two-step Blast search to find orthology exon pairs (Fu and Lin, 2012). OrthoExon first performs BlastN alignment from all human exons to mouse, and vice versa, to find significant hits (Fu and Lin, 2012). In the second step, OrthoExon realigns the exons with no significant hits from one organism to similar exons of the other using tBlastX to allow for sequence divergence. Another exon mapping method by Zhang et al. uses protein level information from Inparanoid database to find ortholog and in-paralog exons between human and mouse using (Zhang et al., 2009). This approach excludes first and last exons and requires exact length match between the two exons to deem that they are orthologs. OrthoExon suffers from dependency of sequence alignment scores to exon lengths, whereas Zhang et al. approach inherently limits the number of exons considered for mapping.
For comparison with our exon mapping results, we download and process the exon mappings from these two methods (Section 2). Figure 2a highlights that ExTraMapper reports a significantly larger number of exons, which are conserved for both organisms even for the most stringent threshold of 1.0 (i.e. 100% overlap between the two exons after liftOver). One main reason for this improvement is that ExTraMapper does not discard part-coding exons or first and last exons and uses both coding similarity and overall similarity to find mappings. Indeed, ExTraMapper finds perfect mappings (threshold of 1.0) for more than three times as many part-coding exons compared to two previous methods.
Fig. 2.

Summary of exon-level mapping results from three differ-ent computational approaches for human and mouse genome (a) for all exons, (b) for only microexons (<21bp). The results for the number of human (left) and mouse (right) microexons are shown separately. ExTraMapper exon similarity threshold is indicated for each Venn diagram
Another important advantage of ExTraMapper is that, since it uses sequence conservation information at the exact coordinate level, it allows identification of mappings between very short exons, termed microexons (Scheckel and Darnell, 2015), between human and mouse. Microexons are known to be highly conserved and have recently been shown to modulate and encode for protein domains involved in protein–protein interactions in the context of neurogenesis and autism (Irimia et al., 2014; Porter et al., 2018; Quesnel-Vallieres et al., 2016). To assess the ability of different tools in capturing orthologous mappings of microexons, we compared ExTraMapper, OrthoExon and Inparanoid. When we define microexons as exons that are shorter than 21 bps, ExTraMapper mapped 939 human and 829 mouse microexons, whereas OrthoExon and Inparanoid reported <100 and <350 exons, respectively (Fig. 2b). Similar trends hold when we use a more liberal threshold of <51 bps for microexon definition with ExTraMapper reporting ∼4–6k additional exon mappings while capturing nearly all mappings reported by the other two tools (Supplementary Fig. S3).
3.4 ExTraMapper computes transcript-level similarities using exon-level conservation
Now that we compute exon similarity scores and classify human and mouse exons according to their conservation between the two organisms, the next step is to compute similarity scores and establish mappings at the transcript level. Ensembl annotations (release 81) for the 15 846 gene pairs we analyze for human and mouse genomes provide nearly 126k and 69k transcripts out of which more than 68k and 40k are protein coding for human and mouse, respectively. For these genes, the human orthologs have a median number of six transcripts and three coding transcripts per gene, whereas these numbers were three and two, respectively, for the mouse genome (Supplementary Fig. S4). The smaller number of transcripts for mouse in comparison with human is in agreement with the smaller number of exons per gene for mouse. Whether these numbers reflect differences in biological diversity of transcripts or differences between the annotation qualities of the two organisms, it is clear that the number of mouse transcripts will be the bottleneck for transcript mappings that will be identified by ExTraMapper or any other algorithm.
We compute transcript-level similarity scores using a greedy method to find non-overlapping and non-contradicting set of exon pairings for a transcript with that maximizes the sum of exon similarity scores, either overall or only coding (Section 2). Using this approach, we compute an overall similarity score and a coding similarity score for a total of 730 440 pairs of transcripts generated from the 15 846 orthologous gene pairs. There are only 13 701 and 70 737 transcript pairs (1.9% and 9.7% of all possible pairs) giving a coding or overall similarity score of 1 and 0.9, respectively. Such transcript pairs span in total 36 464 and 29 389 different human and mouse transcripts out of which 16 585 45.5% and 16 937 57.6% appeared in more than one perfect pair, suggesting one-to-many mappings exist even within perfectly conserved sets of transcripts of the two organisms. This is partly due to multiple transcripts in Ensembl with the same exact set of coordinates and partly because a perfect conservation in coding similarity still allows for difference in non-coding regions such as untranslated regions (UTRs).
3.5 ExTraMapper identifies mainly one-to-one transcript mappings between human and mouse
To reduce the number of one-to-many transcript mappings, we employ a set of tie breakers as outlined in Figure 1 and described in detail in Section 2. Briefly, we consider first the coding similarity, then overall transcript similarity, then difference between the numbers of coding exons and lastly the difference between the numbers of overall exons. If tie breaks still persist, we then report all tied transcript pairs. In practice, nearly all ties are resolved with the first two tie break conditions. Between these two conditions, we choose coding similarity as the first tie breaker to prioritize similarity at the level of protein sequence. Similar to the computation of transcript similarity scores from exon similarities, we choose to use a greedy approach because we want to favor a smaller number of highly conserved transcript pairs instead of a larger number of moderately conserved ones. After this greedy approach with tie breaks, we get 8 007, 25 146 and 30 388 transcript pairs with either coding or overall transcript similarity score of 1, greater than 0.9 and greater than 0.8, respectively. These transcripts span a total of 30 190 unique human and 30 150 unique mouse transcripts. Out of these, only 177 human and 195 mouse transcripts are reported as one-to-many mappings suggesting that ExTraMapper eliminates a very large portion of one-to-many mappings when there is enough information to distinguishing between multiple pairs with equal coding similarity.
We next compare the transcript mappings from ExTraMapper with those from a previous method, namely Exalign, which uses fully coding exon lengths to compute transcript similarities (Pavesi et al., 2008; Zambelli et al., 2010). The total number of human transcripts that are exactly mapped to only one mouse transcript is 30 013 for ExTraMapper (score >0.8) while this number was only 10 405 for Exalign (Supplementary Fig. S5a). Accordingly, ExTraMapper only reports 177 human transcripts that map to more than one mouse isoform, whereas Exalign reports 13 462 such human transcript. A similar trend can also be seen when numbers are computed with respect to mouse transcripts (Supplementary Fig. S5b). The fraction of one-to-one mappings is obviously dependent on the score threshold for ExTraMapper, however, even for a more stringent threshold of 0.9, ExTraMapper still reports over 25k such human transcripts suggesting its utility for breaking ties among potential one-to-many or many-to-many mappings. To study this on a specific example, we analyze the retinoic acid receptor alpha (RARa) gene, which is a critical nuclear receptor expressing specific isoforms that are found either in nucleus or cytoplasm and perform distinct functions depending on their cellular compartment (Larange and Cheroutre, 2016; Leroy et al., 1991). Even though six possible pairs among two human and three mouse isoforms for this gene result in perfect coding similarity scores (both from ExTraMapper and Exalign), ExTraMapper breaks ties using the similarity between non-coding portions of these transcripts to report two one-to-one transcript mappings (Supplementary Fig. S6). We also compare ExTraMapper and Exalign transcript mappings for three other important genes (TP53, TP63 and TP73; Supplementary Table S2), which are known to play multiple critical roles in cell differentiation and response to stress through their rich repertoires of isoforms (Murray-Zmijewski et al., 2006). For instance, for the 24 protein-coding human transcripts of TP53 and 4 protein-coding mouse transcripts of Trp53, Exalign reports 16 pairs with identical scores (among 4 human and 4 mouse transcripts). In contrast, ExTraMapper, using information from other exons including those that are part-coding, reports four different one-to-one mappings (Supplementary Table S2). Such differences between the two methods in distinguishing transcript pairs from each other is observed for other genes where multiple different transcripts have the same number of exons such (e.g. 11 exons for the case of TP53). For TP63-Trp63 and TP73-Trp73, since most isoforms vary in exon length both methods successfully capture transcript mappings (Supplementary Table S2).
3.6 ExTraMapper eliminates biases in transcript similarity score computation
In order to compare ExTraMapper the transcript similarity with Exalign, we download and apply Exalign to the same set of gene annotations used for ExTraMapper (Section 2). First, we compare the best Exalign alignment score and the best ExTraMapper similarity score for the top scoring transcript pair for each gene with Exalign E-value <0.001 and ExTraMapper score ≥0.8 (either coding or overall transcript similarity). Figure 3a shows that there is very strong correlation between the best Exalign score and the number of exons a human transcript has (Pearson’s r = 0.94). On the other hand, ExTraMapper scores have substantially lower dependence on the transcript length (Fig. 3b, Pearson’s r = 0.07). Both figures are in agreement that human transcripts which code longer proteins (over 50 coding exons) have very similar ortholog transcripts in mouse, suggesting they are evolutionarily conserved. However, a near-linear relationship between the transcript length and Exalign scores across the whole range of lengths suggest that the transcript length biases the Blast-like score used by Exalign. Similar plots with respect to mouse transcript lengths exhibit the same trend (Fig. 3c, d). We repeat the same analysis using Exalign and ExTraMapper transcript similarity scores for all transcript pairs (Supplementary Fig. S7; rather than the best scoring one for each gene as in Fig. 3) again, highlighting a stronger dependency of Exalign scores to transcript length compared to ExTraMapper.
Fig. 3.
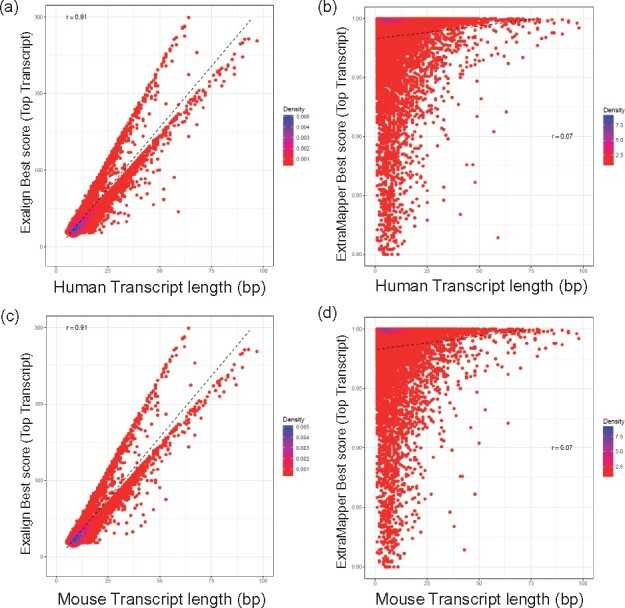
The length bias of the transcript-level similarity scores computed by Exalign. The density plots of the best Exalign alignment score and the best ExTraMapper similarity score for the top scoring transcript pair for each gene with Exalign E-value < 0.001 and ExTraMapper score > 0.8 (either coding or overall transcript similarity) are plotted for human transcripts using (a) Exalign, (b) Ex-TraMapper, mouse transcripts using (c) Exalign, (d) ExTraMapper. Transcript length indicates the number of coding (either fully or partly) exons of a transcript. r = Pearson correlation
3.7 Expression profile comparison of orthologous transcript pairs
Previous work has shown that expression profiles of the conserved genes between human and mouse are highly similar between matched tissues (Gilad and Mizrahi-Man, 2015; Yue et al., 2014) by reanalyzing tissue-specific RNA-seq profiles from a study that suggested otherwise (Lin et al., 2014). Here we use the same dataset (Supplementary Table S3) to compare transcript-level expression estimates for pairs of transcripts identified by either ExTraMapper or Exalign. For reference, for each of the 13 tissues, we first compute the correlation between expression values at the gene level as was done by Gilad and Mizrahi-Man (2015) resulting in a median R of 0.83 (Pearson correlation; Fig. 4a). We then repeat the correlation calculation using top-scoring orthologous transcript pairs obtained either from ExTraMapper, from Exalign or by both methods (i.e. common pairs). In the case of ties for the top-scoring pair, we take all transcript pairs with the highest score for each method leading to 2441 pairs exclusive to ExTraMapper, 7335 pairs exclusive to Exalign and 10 517 pairs that are in common pairs from 13 281 genes we considered for correlation calculation (Section 2). We observe that common transcript pairs exhibit the highest correlation followed by pairs predicted by ExTraMapper (Fig. 4a). Exalign reported transcript pairs slightly lower correlation as well as higher quantile rank difference of individual transcripts based on their expression distribution in every human and mouse tissue compared to common pairs and pairs from ExTraMapper (Fig. 4b). To better understand the source of higher expression correlation and smaller rank difference for ExTraMapper pairs compared to Exalign, we next compare the same entities using a subset of 562 human and 469 mouse transcripts where ExTraMapper and Exalign both identified a single orthologous isoform in mouse and human respectively, but the identified orthologous partners are different by both programs. For these 562 human and 460 mouse transcripts with their respective ExTraMapper and Exalign identified orthologous partners in the other organism, the transcript-level expression correlations also show marginally higher values for ExTraMapper mappings compared to Exalign (Fig. 4c, d).
Fig. 4.
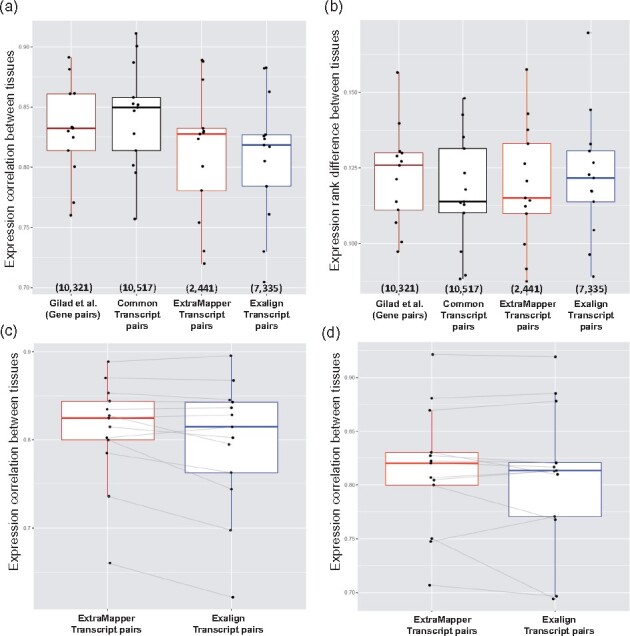
Reanalysis of tissue-matched human and mouse RNA-seq gene expression profiles. (a) Gene- and transcript-level expression correlation. (b) Gene- and transcript-level expression rank difference. (c) Gene- and transcript-level expression correlation for human tran-scripts where ExTraMapper and Exalign both report a single match-ing mouse transcript but their reported pairs are different from each other. (d) Similar plot to (c) but for mouse transcripts with a single matching human transcript. Each dot represents a specific tissue type
3.8 ExTraMapper identifies exon and transcript mappings between human and rhesus macaque genomes
In order to demonstrate the utility of ExTraMapper beyond human-mouse comparison, we also include the rhesus macaque (Macaca mulatta), a genome with relatively poor gene annotations compared to human and mouse, in our analysis. Compared to human genome, rhesus macaque has nearly three times smaller number of exons and four times smaller number of transcripts annotated (Supplementary Table S4; Supplementary Fig. S8). The number of rhesus genes with orthology to human (16 150) is, however, slightly higher than that of mouse. For these 16 150 gene pairs, ExTraMapper reports 442 165 mappings (exon similarity score ≥ 0.8) for 359 403 unique human exons and 207 441 unique rhesus macaque exons (Supplementary Fig. S8). The number of exon mappings with the perfect score of 1, either from coding or overall similarity, is 338 144. Using these exon mappings, we identify 29 634 transcript mappings (transcript similarity score ≥ 0.8) between 29 486 unique human and 29 384 unique rhesus macaque transcripts (Supplementary Fig. S8). Even though the number of transcript mappings from human-mouse and human-rhesus comparisons are comparable at similarity thresholds of 0.8 (30 388 versus 29 634) and 0.9 (25 146 versus 25 137), the number of perfect mappings (similarity score of 1) are substantially higher for human-rhesus (8007 versus 14 343) comparison.
3.9 A case study: finding transcript-level mappings of the BRAF–Braf gene pair
Human BRAF gene is a proto-oncogene on chromosome 7 that encodes a RAF kinase (BRAF), which participates in the MAP kinase/ERK signaling pathway. Mutations in BRAF gene are shown to play important roles in multiple cancers including melanoma, long and colon cancers as well as in developmental disorders (Davies et al., 2002; Hussain et al., 2015; Tidyman and Rauen, 2009). Braf, located on chromosome 6, is the mouse ortholog of human BRAF gene. According to Ensembl annotations (release 81), BRAF has 33 exons and Braf has 42 exons, and each have 5 transcript isoforms with 2 that are protein coding.
In Ensembl database, the orthology between BRAF and Braf genes is reported as a one-to-one relationship between two protein isoforms ENSP00000288602 and ENSMUSP00000002487 that are encoded by the transcripts BRAF-001 and Braf-001, respectively (Fig. 5a). However, Braf-001 has 4 more exons and codes for 38 more amino acids (aa) compared to BRAF-001, the third exon of BRAF-001 corresponds to a fusion of two exons in Braf-001 plus the retention of the 100 bp intron between them and several exons of Braf-001 are lost in human (35, 119 bp and the last exon). Despite these considerable differences, Ensembl orthology is defined through BRAF-001 and Braf-001 isoforms, likely due to default use of longest protein isoforms for orthology relationships.
Fig. 5.
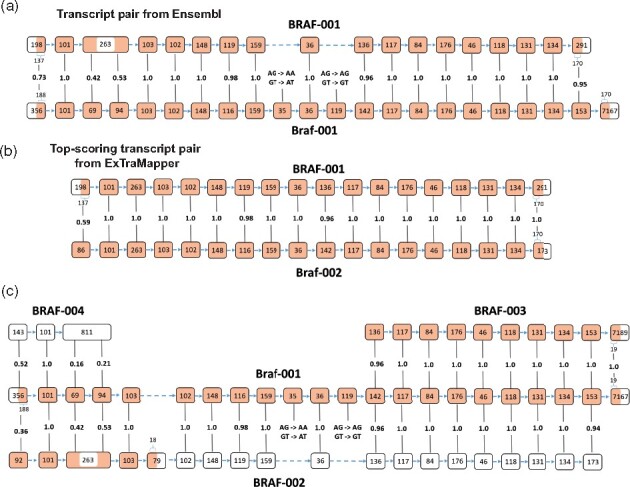
Transcript-level mappings for the BRAF–Braf gene pair. (a) The transcript mapping reported as the basis for orthology between the two genes by Ensembl (coding similarity = 0.86). (b) The highest scoring transcript pair reported by ExTraMapper (coding similarity = 0.97). (c) Partial mappings from multiple human transcripts to the longest mouse transcript Braf-001
Figure 5b illustrates the best transcript-level mapping identified with ExTraMapper, which links protein isoforms ENSP00000288602 and ENSMUSP00000099036 that are encoded by the transcripts BRAF-001 and Braf-002, respectively. These two transcripts have the same number of exons and an almost perfect mapping between their exons except the first ones. ExTraMapper coding transcript similarity score for this pair is 0.97 out of 1 whereas it was 0.86 for the transcript pair reported by Ensembl gene orthology illustrating the need for systematic pairing at the transcript level. Figure 5c demonstrates other suboptimal mappings found between the longest mouse transcript Braf-001 (protein coding) and three human transcripts BRAF-002 (non-sense mediated decay), BRAF-003 (protein coding) and BRAF-004 (retained intron). Overall, this case study provides us examples of complex exonic and splicing events that lead to the different transcripts with varying biotypes in human and mouse. Furthermore, it demonstrates the use of ExTraMapper in identifying such events as well as maximum similarity transcript pairings that are not readily available in any public dataset to the best of our knowledge.
4 Discussion
The ∼16k gene pairs that are orthologs between human and mouse (a similar number holds for human—rhesus macaque comparison) span a diverse repertoire of over 200k total transcript isoforms more than 125k of which are protein coding. Current tools, including HomoloGene (http://www.ncbi.nlm.nih.gov/homologene) do not provide any information on which of these transcripts from mouse correspond to those in human in terms of their DNA sequence, protein sequence and function. Here, we developed a novel method, ExTraMapper, which leverages sequence conservation between exons of a pair of organisms and identifies a fine-scale orthology mapping at the exon and then transcript level. We demonstrated that ExTraMapper mappings cover a large fraction of exons and transcripts when human gene annotations are compared to those from mouse or to rhesus macaque. Our comparative results show that ExTraMapper identified two to three times more perfect exon mappings (100% conservation) compared to two existing methods as well as a larger number of transcript mappings compared to Exalign, a method that suffers from biases in its similarity score calculations. As an orthogonal validation, we showed that transcript pairs identified by ExTraMapper have more correlated expression patterns compared to Exalign identified pairs. Aside from genome-wide comparisons, we also presented specific cases where ExTraMapper transcript mappings capture expected biology for genes with known isoform-specific functions (RARA, TP53, TP63 and TP73). We created genome browser tracks for visualizing such mappings from either one of the compared genomes (UCSC visualizations for the FOXP3-Foxp3 gene pair; Supplementary Fig. S9).
In order to assess the variation in ExTraMapper results caused by gene annotation differences, we compared human-mouse exon and transcript mappings using Ensembl release 81 (Jul. 2015) versus release 102 (Nov. 2020). This analysis highlighted a small change in the number of orthologous gene pairs (15 846 versus 15 177; Supplementary Fig. S10a, d; Supplementary Table S5) but a substantial increase in the number of annotated exons (Supplementary Fig. S10 b, e) and transcripts (Supplementary Fig. S10c, f) for these genes especially for the mouse genome (Supplementary Fig. S10e, f). This was also reflected in the number of mappings exclusively found from the recent release for both exons (Supplementary Fig. S11) and transcripts (Supplementary Fig. S12). The comparison of release-specific mappings to those that are common between releases showed that the two groups were very similar in their exon similarity score distributions (Supplementary Fig. S11), whereas release-specific transcript pairs had a shift toward lower similarity scores likely pertaining to differences in mappings beyond the top scoring pairs (Supplementary Fig. S12).
We believe that ExTraMapper will have a great impact for translational sciences as it provides a dictionary for translating transcript-level information about gene expression and gene regulation from one organism to another. For example, one direction this translation can be done is from mouse models to human genome. This will be specifically useful for work with certain tissues or samples that are difficult to obtain from human donors or come in very limited quantities. Another important aspect of our work is that it allows us to identify which genes have highly conserved exon-intron structures and transcript repertoires between two organisms such as human and mouse. This information is important for understanding the extent of evolutionary differences with respect to specific gene functions and biological pathways. For instance, our GO term enrichment analyses showed that human genes that have all their protein coding transcripts map perfectly to a corresponding mouse transcript are enriched in certain developmental processes such pattern specification process and regionalization. On the other hand, genes that have none of their coding transcripts map to any mouse transcript are enriched in response related processes such as defense response to other organism suggesting higher level of divergence between human and mouse in their immune system related transcripts. Further research directions include better annotation of transcript mappings found by ExTraMapper as well as its extension to finding mappings across multiple mammalian genomes simultaneously.
Supplementary Material
Acknowledgements
The authors thank Wen-chang Lin for making their OrthoExon database available for batch download.
Funding
The work was supported in part by the National Institutes of Health (NIH) under award numbers R01-LM011297 (R.V.D.) and R35-GM128938 (F.A.).
Conflict of Interest: none declared.
Contributor Information
Abhijit Chakraborty, Centers for Cancer Immunotherapy and Autoimmunity, La Jolla Institute for Immunology, La Jolla, CA 92037, USA.
Ferhat Ay, Centers for Cancer Immunotherapy and Autoimmunity, La Jolla Institute for Immunology, La Jolla, CA 92037, USA; Department of Pediatrics, UC San Diego—School of Medicine, La Jolla, CA 92093, USA.
Ramana V Davuluri, Department of Biomedical Informatics, Stony Brook University, Stony Brook, NY 11794, USA.
References
- Alekseyenko A.V. et al. (2007) Global analysis of exon creation versus loss and the role of alternative splicing in 17 vertebrate genomes. RNA, 13, 661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., Huber W. (2010) Differential expression analysis for sequence count data. Genome Biol., 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoubi T.A., Van De Ven W.J. (1996) Regulation of gene expression by alternative promoters. FASEB J, 10, 453–460. [PubMed] [Google Scholar]
- Blanchette M. et al. (2004) Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res., 14, 708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blekhman R.A. (2012) A database of orthologous exons in primates for comparative analysis of RNA-seq data. Nat Prec. 10.1038/npre.2012.7054.1. [DOI]
- Davies H. et al. (2002) Mutations of the BRAF gene in human cancer. Nature, 417, 949–954. [DOI] [PubMed] [Google Scholar]
- Davuluri R.V. et al. (2008) The functional consequences of alternative promoter use in mammalian genomes. Trends Genet., 24, 167–177. [DOI] [PubMed] [Google Scholar]
- Douzery E.J. et al. (2014) OrthoMaM v8: a database of orthologous exons and coding sequences for comparative genomics in mammals. Mol. Biol. Evol., 31, 1923–1928. [DOI] [PubMed] [Google Scholar]
- Flicek P. et al. (2014) Ensembl 2014. Nucleic Acids Res., 42, D749–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu G.C., Lin W.C. (2012) Identification of gene-oriented exon orthology between human and mouse. BMC Genomics, 13, S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad Y., Mizrahi-Man O. (2015) A reanalysis of mouse ENCODE comparative gene expression data. F1000Res, 4, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain M.R. et al. (2015) BRAF gene: from human cancers to developmental syndromes. Saudi J. Biol. Sci., 22, 359–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia M. et al. (2014) A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell, 159, 1511–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. et al. (2015) HISAT: a fast spliced aligner with low memory requirements. Nat. Methods, 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. et al. (2011) IsoformEx: isoform level gene expression estimation using weighted non-negative least squares from mRNA-Seq data. BMC Bioinformatics, 12, 305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larange A., Cheroutre H. (2016) Retinoic acid and retinoic acid receptors as pleiotropic modulators of the immune system. Annu. Rev. Immunol., 34, 369–394. [DOI] [PubMed] [Google Scholar]
- Leek J.T. et al. (2012) The SVA package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics, 28, 882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelli K.M. et al. (2012) Disentangling the many layers of eukaryotic transcriptional regulation. Annu. Rev. Genet., 46, 43–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerch J.K. et al. (2012) Isoform diversity and regulation in peripheral and central neurons revealed through RNA-Seq. PLoS One, 7, e30417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy P. et al. (1991) Multiple isoforms of the mouse retinoic acid receptor alpha are generated by alternative splicing and differential induction by retinoic acid. EMBO J, 10, 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y. et al. (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lin S. et al. (2014) Comparison of the transcriptional landscapes between human and mouse tissues. Proc. Natl. Acad. Sci. USA, 111, 17224–17229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlin A.J. et al. (2005) Understanding alternative splicing: towards a cellular code. Nat. Rev. Mol. Cell Biol., 6, 386–398. [DOI] [PubMed] [Google Scholar]
- Modrek B., Lee C.J. (2003) Alternative splicing in the human, mouse and rat genomes is associated with an increased frequency of exon creation and/or loss. Nat. Genet., 34, 177–180. [DOI] [PubMed] [Google Scholar]
- Murray-Zmijewski F. et al. (2006) p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ., 13, 962–972. [DOI] [PubMed] [Google Scholar]
- Pal S. et al. (2014) Genome-wide mapping of RNA Pol-II promoter usage in mouse tissues by ChIP-seq. Methods Mol. Biol., 1176, 1–9. [DOI] [PubMed] [Google Scholar]
- Pavesi G. et al. (2008) Exalign: a new method for comparative analysis of exon-intron gene structures. Nucleic Acids Res., 36, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter R.S. et al. (2018) Neuron-specific alternative splicing of transcriptional machineries: implications for neurodevelopmental disorders. Mol. Cell Neurosci., 87, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesnel-Vallieres M. et al. (2016) Misregulation of an activity-dependent splicing network as a common mechanism underlying autism spectrum disorders. Mol. Cell, 64, 1023–1034. [DOI] [PubMed] [Google Scholar]
- Robinson M.D. et al. (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheckel C., Darnell R.B. (2015) Microexons–tiny but mighty. EMBO J., 34, 273–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schibler U., Sierra F. (1987) Alternative promoters in developmental gene expression. Annu. Rev. Genet., 21, 237–257. [DOI] [PubMed] [Google Scholar]
- Seijffers R. et al. (2007) ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J. Neurosci., 27, 7911–7920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraki T. et al. (2003) Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc. Natl. Acad. Sci. USA, 100, 15776–15781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A. et al. (2005) Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res., 15, 1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A., Haussler D. (2004) Combining phylogenetic and hidden Markov models in biosequence analysis. J. Comput. Biol., 11, 413–428. [DOI] [PubMed] [Google Scholar]
- Tidyman W.E., Rauen K.A. (2009) The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr. Opin. Genet. Dev., 19, 230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C. et al. (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics, 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. et al. (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet., 10, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates A. et al. (2016) Ensembl 2016. Nucleic Acids Res., 44, D710–D716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue F. et al. ; Mouse ENCODE Consortium. (2014) A comparative encyclopedia of DNA elements in the mouse genome. Nature, 515, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambelli F. et al. (2010) Assessment of orthologous splicing isoforms in human and mouse orthologous genes. BMC Genomics, 11, 534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. et al. (2009) Divergence of exonic splicing elements after gene duplication and the impact on gene structures. Genome Biol., 10, R120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.