

Summary
Membrane proteins are central to many pathophysiological processes, yet remain very difficult to analyze structurally. Moreover, high-throughput structure-based drug discovery has not yet been exploited for membrane proteins because of lack of automation. Here, we present a facile and versatile platform for in meso membrane protein crystallization, enabling rapid atomic structure determination at both cryogenic and room temperatures. We apply this approach to human integral membrane proteins, which allowed us to identify different conformational states of intramembrane enzyme-product complexes and analyze by molecular dynamics simulations the structural dynamics of the ADIPOR2 integral membrane protein. Finally, we demonstrate an automated pipeline combining high-throughput microcrystal soaking, automated laser-based harvesting, and serial crystallography, enabling screening of small-molecule libraries with membrane protein crystals grown in meso. This approach brings needed automation to this important class of drug targets and enables high-throughput structure-based ligand discovery with membrane proteins.
Keywords: LCP crystallization, automated crystal harvesting, drug discovery, CrystalDirect, high throughput ligand screening, CRIMS, data management, automation, SSX, LIMS
Graphical abstract
Highlights
-
•
A platform for rapid in meso structures by serial crystallography (SSX)
-
•
Insights into ADIPOR2 receptor-ligand dynamic interactions
-
•
A web-based application for remote user-guided experimental design and execution
-
•
An automated SSX-based ligand discovery pipeline for membrane proteins is introduced
Motivation
Membrane proteins are key regulators of most physiological processes and represent attractive targets for drug discovery programs. One of the most successful methods to obtain high-resolution structures of membrane proteins relies on in meso crystallization in combination with serial synchrotron crystallography. However, this remains a difficult process, with challenges at every step, including complex manual sample recovery protocols leading to limited throughput and sample loss and the difficulty in carrying out high-throughput ligand screening experiments. We have developed a new approach enabling rapid, automated structural analysis of membrane proteins in meso based on the CrystalDirect technology that addresses these issues, enabling high-throughput drug discovery with membrane proteins.
Membrane proteins control many biological processes and represent attractive targets for drug discovery, but are difficult to study structurally. Healey et al. present an automated approach, combining the CrystalDirect technology and serial crystallography, for rapid structural analysis of membrane proteins and opening new opportunities for high-throughput drug discovery.
Introduction
Membrane proteins are key regulators of most physiological processes and represent attractive targets for drug discovery programs (Andrews et al., 2014; Congreve et al., 2020). Their structure determination remains a powerful strategy to gain insight into their function and accelerates drug development (Andrews et al., 2014; Ishchenko et al., 2019). One of the most successful methods to obtain high-resolution structures from membrane proteins relies on in meso crystallization (i.e., lipidic cubic phase, LCP) and X-ray diffraction (Caffrey, 2015; Landau and Rosenbusch, 1996; Landau et al., 1997; Liu and Cherezov, 2011; Pebay-Peyroula, 1997). This strategy has been particularly successful at solving structures of the G-protein-coupled receptor (GPCR) family and other integral membrane proteins (Caffrey, 2015; Rasmussen et al., 2011; Vasiliauskaité-Brooks et al., 2017, 2018). In meso crystallization remains a long process with challenges at every step. This might justify the underrepresentation of membrane protein structures in the worldwide Protein Data Bank (wwPDB) (Berman, 2000). Structure-based ligand discovery efforts targeting membrane proteins, such as fragment screening, remain inadequate due to technical barriers in experimental setups.
In meso crystallization trials are typically performed with sandwich plates, where the sample mesophase is squeezed between two glass plates (Caffrey and Cherezov, 2009; Cherezov et al., 2004). Membrane protein crystals grown in meso are often of micrometer-sized dimensions and decay rapidly because of radiation damage. Thus, analysis by X-ray diffraction is usually performed through the so-called serial synchrotron crystallography (SSX) approach, applied at both ambient and cryogenic temperatures (Gati et al., 2014; Huang et al., 2015, 2016; Weierstall et al., 2014). For room-temperature experiments, delivery of LCP membrane protein crystals to an X-ray beam for SSX can be achieved by using sample jets (Botha et al., 2015; Nogly et al., 2015, 2018; Weierstall et al., 2014; Weinert et al., 2017). The LCP injection method has many benefits for time-resolved experiments. However, it requires large quantities of protein, extensive optimization of sample preparation, manual delivery protocols, and long times for data collection at the beamline. For cryogenic SSX analysis, crystals grown in sandwich plates are manually harvested, for which one of the glass plates must be removed, or broken, and the crystals manually recovered and flash-frozen in a matter of seconds before the crystals are damaged (Caffrey and Cherezov, 2009; Li et al., 2012). Even in experienced laboratories, manual LCP crystal harvesting is a difficult, time-consuming, and inefficient process. Post-crystallization treatments, such as ligand or fragment soaking, done in an automated and straightforward manner are not possible. A promising approach to allow soaking of in meso crystals has been suggested, in which crystallization occurs between two plastic films (Huang et al., 2015, 2016). However, this method poses technical challenges, entailing manual handling and low throughput (Huang et al., 2018). Currently, the field lacks automation that could support higher-throughput approaches (Rucktooa et al., 2018), a necessity to expedite structure-based ligand discovery for membrane proteins.
The CrystalDirect method was initially developed to enable automated recovery of crystals from soluble proteins grown through vapor diffusion (Cipriani et al., 2012; Márquez and Cipriani, 2014; Zander et al., 2016). In this approach, crystals are grown on thin films in a commercially available 96-well crystallization plate (a CrystalDirect plate; Figure 1) and harvested directly on X-ray data collection pins by a robotic laser-based excision system (Zander et al., 2016). This approach has thus far been applied to processing crystals from soluble proteins (Aragón et al., 2019; Bezerra et al., 2017; Martin-Malpartida et al., 2017). However, its application to membrane proteins was limited by incompatibility of standard LCP sandwich plates with the CrystalDirect laser-excision technology. To the best of our knowledge, automated harvesting of crystals grown in meso has not been described to date, and manually harvesting crystals remains a major technical challenge, limiting high-throughput applications.
Figure 1.

In meso crystallization and SSX analysis of membrane proteins in CrystalDirect plates
On the left is a CrystalDirect (CD) plate with LCP crystallization experiments (inset). Once LCP crystals have grown, two different workflows for structural analysis can be applied under the same crystallization conditions. Room-temperature in situ X-ray diffraction data can be collected directly from the CrystalDirect plate (middle top). For cryogenic structures (middle bottom), a workflow including automated laser-based harvesting and cryo-cooling can be utilized. Room-temperature and cryogenic structures of ADIPOR2 (right, top and bottom, respectively) obtained with these approaches are shown.
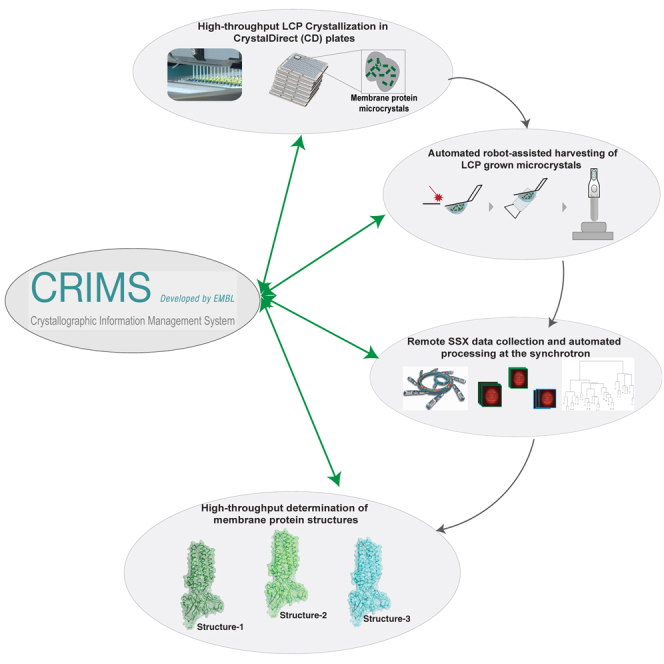
Here, we introduce a platform to grow and monitor membrane protein crystals in CrystalDirect plates by using two pharmacologically important integral membrane protein targets: human alkaline ceramidase 3 (ACER3) and adiponectin receptor 2 (ADIPOR2). Using CrystalDirect as a support for LCP crystallization allows SSX diffraction to be monitored in situ at ambient and cryogenic temperatures, enabling the direct comparison of ADIPOR2 structures solved at different temperatures and revealing insights into ADIPOR2-lipid interactions and conformational states (Figure 2). Moreover, we demonstrate how this technology can be applied to high-throughput ligand screening with membrane proteins. To facilitate automated experimental workflows, we have developed the web-based Crystallographic Information Management System (CRIMS). This information system allows automated tracking of crystallographic experiments and web-based access to experimental design and evaluation tools encompassing protein sample registration, crystallization setup and monitoring, and crystal harvesting, as well as shipment to and recovery of processed data from synchrotrons (Figure 3). Through the combination of the CrystalDirect technology and CRIMS software, the HTX facility in Grenoble provides access to a resource for the rapid structural analysis of membrane proteins in meso, removing major hurdles and allowing rapid structural analysis of membrane proteins. This resource is open to scientists worldwide through different access programs and could be replicated elsewhere.
Figure 2.

ADIPOR2-lipidic product dynamics
(A) Overlay of CrystalDirect structures: cryogenic (magenta) and room temperature (cyan) with molecular dynamics (MD) simulation (yellow).
(B) MD trajectory plots overlaying oleic acid (OLA)-bound (yellow) and OLA-depleted (black) simulations, measuring the ADIPOR2 transmembrane helix 5 (TM5) opening of 4 Å (distance: R275™5–H348TM7) with the density plot of the trajectory shown on the right.
(C) MD trajectory plot of OLA-bound simulation measuring displacement of OLA from the active site by 3.9 Å (distance: catalytic zinc atom − carboxylic head of OLA). Spearman's correlation coefficient between TM5 opening and OLA movement is 0.74.
Figure 3.

A pipeline for high-throughput ligand discovery with membrane protein crystals
After LCP crystallization in CrystalDirect plates, crystals can be directly accessed by removal of the top plastic seal on the plate (top row). Soaking solutions are introduced into the experiment by a pipetting robot, after which the plate is resealed and incubated for the necessary time (top row). After soaking, LCP crystals are auto-harvested with the CrystalDirect robot into pins and stored in UniPucks for shipment to the synchrotron (middle row, “Experiment”). Serial diffraction data are collected for each uniquely soaked bolus and merged after established protocols before structure refinement and ligand fitting (bottom row, “Results”). The automated data flow in the pipeline, shown by double-arrowed dashed lines, is maintained through the CRIMS, through which users can access, monitor, and analyze data via web interfaces.
Results and discussion
Automated harvesting of membrane protein crystals
We have developed a new method for LCP crystallization compatible with the CrystalDirect technology, thereby enabling automated harvesting, soaking, and cryo-cooling of crystals grown in meso (STAR Methods). In this method, LCP crystallization experiments are set inside the vapor diffusion chambers of a CrystalDirect plate, instead of on a sandwich plate. To avoid evaporation, the reservoir side of the vapor diffusion well is filled with an excess volume of crystallization solution and the plate is sealed from the top with a standard film (Figures 1, 3, and S5). Under these conditions, experiments are stable for several weeks. Once crystals develop, the plates are transferred to the CrystalDirect harvester robot, where film pieces containing the crystal-laden LCP bolus or parts of it are automatically excised, mounted onto a standard data collection pin, and transferred to a cryogenic sample storage device (Figure 4).
Figure 4.

Automated in meso crystal harvesting and cryo-cooling
(A) Schematic representation of the CrystalDirect harvesting process: the laser cuts an aperture in the drop and the robot gently removes the excess precipitant solution by aspiration, then the laser-based film excision is applied and the film piece containing the sample is automatically glued to a data collection pin and cryo-cooled. Samples are then transferred to a high-capacity cryogenic storage system (not shown).
(B–E) Examples of ADIPOR2 and ACER3 microcrystals (pink arrows) grown and harvested from CrystalDirect plates. Different scenarios involving harvesting of full boluses or sections of them are presented. (B) Full LCP drop; (C) high-density area; (D) single crystals; (E) single crystals.
The overall management of crystallization experiments presented here is made possible through the CRIMS, a web application for the design, evaluation, and analysis of crystallographic experiments (https://htxlab.embl.fr/) (STAR Methods). Using CRIMS, scientists can register protein samples, design crystallization screens, monitor crystal growth, and define automated harvesting (Figure S1). Once crystals have been identified, harvesting is performed by the CrystalDirect harvester robot (Figure S1). The CrystalDirect robot then transfers the sample pin to a standard sample holder in a cryogenic storage device, whereas CRIMS records sample locations. The entire process is shown in Video S1. A single harvesting operation takes approximately 1 min, and the dewar has capacity for up to 312 samples (pins).
This workflow is based on the combination of the CrystalDirect technology and serial X-ray diffraction data analysis, and can be controlled over the web through the Crystallographic Information Management System (CRIMS)
For this study we chose the seven-transmembrane protein ADIPOR2, a receptor activated by the protein hormone adiponectin. Adiponectin exerts a pleiotropic action in mammals and is implicated in metabolic and cardiovascular-related disorders (Straub and Scherer, 2019); small molecules replicating adiponectin effects have long been sought after for clinical applications in the treatment of metabolic and cardiovascular-related diseases (Okada-Iwabu et al., 2013). The CrystalDirect LCP method produced crystals under the same precipitant conditions and with similar size and morphology compared with those previously reported by using the classical sandwich glass plate approach (Vasiliauskaité-Brooks et al., 2017). We collected cryogenic SSX diffraction data on ADIPOR2 crystals harvested from single in meso boluses. A laser-cut area containing multiple ADIPOR2 microcrystals was defined in CRIMS as single pin and harvested by the CrystalDirect harvester such that a single pin contained 10–100 crystals (Figure S2). Diffraction data were collected at the Swiss Light Source (SLS) on PXI (X06SA) from each microcrystal as 10° wedges of minisets as previously described (Basu et al., 2019; Wojdyla et al., 2016) (STAR Methods). A complete ADIPOR2 dataset was obtained from a single harvested pin, requiring 300 ng of protein. The structure of ADIPOR2 harvested automatically from a CrystalDirect plate was resolved at 2.4 Å (Figures 1 and S6C; STAR Methods); this resolution is equivalent to that previously reported from manual harvesting from glass plates (Vasiliauskaité-Brooks et al., 2017), indicating that our automated approach does not lead to any reduction in data quality.
We also crystallized an intramembrane sphingolipid hydrolase, ACER3, using this method (STAR Methods). In glass sandwich plates, ACER3 crystals tend to grow in the sponge phase and are particularly challenging to harvest manually. Using the CrystalDirect harvester, all attempted ACER3 crystal harvests were successful. Nine harvested pins were required to collect a complete dataset for ACER3 employing the same SSX methodology as for ADIPOR2. The data collection required 36 min of synchrotron beam time at the Swiss Light Source (STAR Methods). A high-quality structure of ACER3 at 2.6 Å resolution was resolved, requiring 1.6 μg of protein (Figure S3; STAR Methods). This resolution was equivalent to that previously reported by using classical sandwich glass plates (Vasiliauskaité-Brooks et al., 2018), demonstrating that in meso crystallization in CrystalDirect plates does not compromise data quality.
In situ membrane protein serial crystallography
The CrystalDirect plates can also be directly used for in situ X-ray experiments (Figure 1). We used this approach for room-temperature analysis of ADIPOR2 through in situ diffraction (STAR Methods). We collected in situ datasets from ADIPOR2 crystals by mounting the plate directly on an SBS-plate goniometer at the P14 beamline of the Petra III synchrotron (Hamburg, Germany). The crystals were exposed to the X-ray beam and diffraction was recorded at room temperature with a continuously oscillating raster-scan across each LCP bolus (Gati et al., 2014) (Video S2 and Figure S6A). A collection strategy that optimized beam transmission and exposure time while minimizing radiation damage was applied (STAR Methods). A complete dataset was acquired from five boluses in different wells in approximately 20 min of beam time, representing a total of 1.5 μg of protein. At room temperature, ADIPOR2 crystals diffracted to 2.9 Å resolution (Figure 1 and Figure S6B; STAR Methods). An added benefit of using the CrystalDirect plate for in meso crystallization is that structural analysis can be carried out at cryogenic and room temperature in the same support and under the exact same conditions in a straightforward manner. Unlike existing sample-to-beam delivery methods, such as LCP jets or microchips (Botha et al., 2015; Nogly et al., 2015; Weierstall et al., 2014), our approach requires essentially no optimization. In CrystalDirect plates, crystal growth conditions are easily translated from glass sandwich plates. CrystalDirect plates can also be used for primary LCP crystallization screening.
Structural analysis of ADIPOR2 and its interaction with endogenous lipid at different temperatures
Cooling protein crystals to cryogenic temperatures allows for a reduction in radiation sensitivity, but can also decrease the heterogeneity of protein conformations (Russi et al., 2017). By collecting diffraction data at ambient and cryogenic temperatures, alternative conformations relevant to protein function can be revealed (Fraser et al., 2009, 2011).
Adiponectin receptors belong to a larger family of transmembrane receptors putatively assigned with hydrolase activity because of a conserved active motif of three histidines coordinating a zinc ion (Pei et al., 2011). The adiponectin receptors 1 and 2 (ADIPOR1/2) function as ceramide hydrolases, utilizing the bioactive lipid ceramide as a substrate and producing sphingosine and free fatty acids, such as oleic acid (OLA) (Vasiliauskaité-Brooks et al., 2017). The structures of ADIPOR1 and ADIPOR2 reveal an internal cavity containing the active site, enclosed by seven-transmembrane helices (7TMs). The mechanism by which substrate enters the cavity to be processed into products is not yet characterized. Recent structural data have revealed the existence of distinct conformations of transmembrane helix 5 (TM5) for ADIPOR1 and ADIPOR2, so-called open and closed, respectively (Tanabe et al., 2020; Vasiliauskaité-Brooks et al., 2017, 2019).
In the ADIPOR2 structures determined here, TM5 is in a closed conformation, and a free fatty acid product, OLA, is inside the catalytic cavity (Figures 2A and 5G, 5H, and 5K). Overall, the room-temperature and cryogenic structures showed similar backbone conformations (root-mean-square deviation [RMSD] = 0.3 Å). However, we did observe a conformational difference in the bound OLA molecule (Figures 2 and 5). To explore the relevance of this enzymatic product conformational difference, we performed replica-exchange molecular dynamics simulations of ADIPOR2 at room temperature, using OLA-depleted and OLA-bound cryogenic structures as starting models (STAR Methods). During the OLA-bound simulation, OLA rapidly adopted a conformation corresponding to that of our room temperature structure, and initially continued to oscillate around this position (Figure 2C). As the OLA-bound simulation proceeded, the intracellular terminal of ADIPOR2 TM5 transitioned into a more open position, coinciding with a displacement of OLA away from the active-site zinc and toward the lipid bilayer (Figures 2B and 2C). This simulation captures the conformational changes required for ADIPOR2 to release its enzymatic product, OLA. The open TM5 conformation observed in the molecular dynamics (MD) simulations resembles that of the open structure reported in the ADIPOR1 crystal structures (Tanabe et al., 2020; Vasiliauskaité-Brooks et al., 2017), suggesting conserved conformational transition in the ADIPOR family associated with product release and necessary for enzymatic activity. In simulations performed in the absence of OLA, TM5 was observed to gradually close (Figure 2B). The versatility of CrystalDirect plates as in meso crystallization supports allowed us to capture different conformational states of intramembrane enzyme-lipid complexes. To maximize comparability, we were able to use identical crystallization conditions for both cryogenic and in situ experiments with protein prepared from a single purification batch, enabling a true comparison between the resulting protein structures. Our results highlight the potential of this technique, when combined with computational approaches, to analyze the structural dynamics of membrane proteins. This information is key to provide a mechanistic understanding of protein function and can be exploited for drug design.
Figure 5.

High-throughput structural analysis of integral membrane proteins ADIPOR2 and ACER3
Structures of the ACER3 and ADIPOR2 seven-transmembrane enzyme receptors were obtained with the workflows presented in Figure 1. Protein crystal structures are shown with the membrane plane indicated as a gray zone.
(A–C) (A) ACER3 cryogenic structure, shown as a cartoon in gray and (2Fo − Fc) map in blue; (B) ADIPOR2-scFv cryogenic structure, shown as cartoon in salmon with (2Fo − Fc) map; and (C) ADIPOR2-scFv room-temperature (RT) structure, shown as cyan cartoon with orange (2Fo − Fc) map.
(D and E) ADIPOR2-scFv structures shown as surface structures, highlighting (D) Tb complex and (E) Gd complex as soaked clusters with (2Fo − Fc) map in blue. All (2Fo − Fc) maps are contoured at 1.0 σ.
(F–H) Inset views of active sites of (A), (B), and (C), respectively, showing Zn2+ atoms as magenta spheres, coordinated by His residues shown as sticks, with the respective (2Fo − Fc) maps, matching the color.
(I and J) Inset views of the (I) Tb complex and (J) Gd complex, shown in magenta sticks with metal atoms shown as gray spheres, along with the (2Fo − Fc) map in blue and anomalous maps in green (contoured at 7.0 σ). PDB codes are ACER3 cryo: 6YXH; ADIPOR2 cryo: 6YX9; ADIPOR2 RT: 6YXD; ADIPOR2-Tb-XO4: 6YXG; ADIPOR2-Gd-DO3: 6YXF.
(K) Superposition showing distinct conformations of the oleic acid molecule at the catalytic site of ADIPOR2 in the cryogenic (green) and room-temperature structures (yellow).
A pipeline for automated ligand soaking in meso
To expand the possibilities of high-throughput structure-based ligand discovery on membrane protein targets, we have developed an automated pipeline. This pipeline incorporates LCP crystal production, ligand soaking, crystal harvesting, cryogenic SSX data collection, and structure determination of microcrystals grown in meso (Figure 3). Automation of crystal manipulation is a key consideration for any type of screening, as it enables high throughput and reduces the possibility of human error. Soaking of LCP crystals in traditional plates requires manual separation of one of the two sandwich layers holding the LCP bolus, which is a delicate operation and can result in sample damage. This has limited the application of high-throughput ligand screening approaches for membrane proteins. The resealable CrystalDirect plate is an optimal support for in meso experiments, allowing direct access to experiments for post-crystallization manipulation without damaging the bolus or the crystals. To soak ADIPOR2 crystals, the top sealing film of the plate was removed, ligand solutions were added with a mosquito robot, and the plate was resealed for the desired soaking time (Figure 3, top row, and STAR Methods). Addition of 96 soaking solutions to an entire microplate takes <2 min in total, indicating the rapidity and high throughput of our automated soaking pipeline. After addition of the ligands, experiments were incubated inside a crystal imaging robot and images of soaked crystals were used to define harvesting plans. After soaking, crystals were harvested and prepared for synchrotron shipment as outlined for ADIPOR2 and ACER3 (Figure 3, middle row).
Processing of SSX diffraction data for a large number of samples remains a challenging computational task. Efficient detection of well-diffracting microcrystals via raster scanning and automatic triggering of multiple dataset collection with small wedges are imperative for high-throughput SSX data collection (Basu et al., 2019). On-the-fly data processing and feedback together with efficient selection of useful datasets is also necessary to enable high-throughput screening of samples. In this work, cryogenic SSX measurements were carried out at the Swiss Light Source, where data collection and real-time data processing, selection, and merging are available through the sxdm package (Basu et al., 2019).
To confirm that in meso crystals grown in CrystalDirect plates could be soaked with small molecules, we soaked ADIPOR2 crystals with heavy-metal-containing organic caged molecules, typically used as phasing agents (Engilberge et al., 2017; Girard et al., 2003). After incubation with ligand, automated harvesting was performed on regions of interest containing dozens of microcrystals. For initial soaking experiments, we soaked several in meso boluses for each ligand and then collected cryogenic SSX diffraction data. The electron-density maps show two distinct binding sites for Tb-XO4, in clefts on the surface of the antibody fragment portion of the ADIPOR2-scFv complex (Figures 5D and 5I). The structure of ADIPOR2 soaked with Gd-DO3 revealed binding of the compound to transmembrane helix 3 (Figures 5E and 5J). Both ADIPOR2 metal complexes were solved at 3 Å resolution (Table 1).
Table 1.
Crystallography data collection and refinement statistics
| Protein |
|||||
|---|---|---|---|---|---|
| ACER3-cryo | ADIPOR2-cryo | ADIPOR2-RT | ADIPOR2-Tb-XO4 | ADIPOR2-Gd-DO3 | |
| PDB ID | 6YXH | 6YX9 | 6YXD | 6YXG | 6YXF |
| Data collection | |||||
| Photon energy (keV) | 12.4 | 12.4 | 12.4 | 7.5 | 7.2 |
| Beam size (μm2) | 20 × 10 | 20 × 10 | 10 × 5 | 20 × 10 | 20 × 10 |
| Synchrotron | Swiss Light Source | Swiss Light Source | P14–PETRA III, DESY | Swiss Light Source | Swiss Light Source |
| Detector | EIGER-16M | EIGER-16M | EIGER-16M (4M ROI) | EIGER-16M | EIGER-16M |
| Photon flux (ph/s) | 6.1 × 1011 | 6.1 × 1011 | 1.17 × 1013 | 6.1 × 1011 | 6.1 × 1011 |
| Exposure time (ms)/frame | 100 | 100 | 1.3 | 100 | 100 |
| Oscillation range/crystal | 10° | 10° | 0.6° | 10° | 10° |
| Collection strategy | 10° wedge/crystal | 10° wedge/crystal | helical line scan on each drop; ∼400 lines; 2- and 10-μm vertical and horizontal steps, respectively | 10° wedge/crystal | 10° wedge/crystal |
| Total no. of frames | NA | NA | 382,552 | NA | NA |
| No. of hits (datasets collected) | 21,250 (425) | 15,750 (315) | 28,949 | 28,500 (570) | 20,500 (410) |
| No. of indexed framesa | 18,550 (371) | 5,200 (104) | 25,297 | 23,350 (467) | 17,800 (356) |
| No. of datasets merged | 101 | 100 | N/A | 210 | 96 |
| No. of CD boluses | 9 | 1 | 5 | 16 | 7 |
| No. of crystals/bolus | ∼17 | ∼33 | ∼100 | ∼27 | ∼22 |
| Effective collection timeb (min) | 8.4 (30.9) | 8.3 (26.2) | 20 (76) | 38.9 (47.5) | 29.6 (35) |
| Protein consumption (μL) | 1.6 | 0.3 | 1.5 | 4.8 | 2.1 |
| Crystal size (μm3, approx.) | 20 × 10 × 5 | 30 × 20 × 5 | 30 × 20 × 5 | 30 × 20 × 5 | 30 × 20 × 5 |
| Space group | C2221 | P21221 | P21221 | P21221 | P21221 |
| Unit cell: a, b, c (Å) | 61.5, 69.9, 259.5 | 74.7, 100.6, 109.7 | 75.4, 100.8, 114.9 | 74.6, 100.7, 110.5 | 74.6, 100.6, 111.2 |
| Structure refinement | |||||
| Resolution rangec (Å) | 46.2–2.6 (2.62–2.6) | 22–2.4 (2.49–2.40) | 50–2.9 (2.98–2.9) | 48.0–3.0 (3.1–3.0) | 48.7–3.0 (3.1–3.0) |
| No. of unique reflectionsa | 17,701 (1,275) | 30,556 (2,326) | 15,292 (1,971) | 32,012 (2,150) | 31,468 (2,336) |
| Multiplicitya | 36.01 (35.28) | 37.12 (33.69) | 130.57 (4.24) | 34.82 (13.14) | 14.98 (6.18) |
| CC1/2a | 0.99 (0.330) | 0.99 (0.561) | 0.98 (0.737) | 0.99 (0.3) | 0.98 (0.3) |
| I/σ(I)a | 8.4 (0.98) | 8.3 (1.02) | 8.6 (1.4) | 7.33 (1.5) | 5.06 (1.1) |
| Completeness (%)a | 100 (100) | 99.4 (100) | 100 (75.7) | 99.9 (100) | 98.8 (99.7) |
| Wilson B factor (Å2) | 65.9 | 60.7 | 42.3 | 79.1 | 78.9 |
| R-work/R-free (%)a | 23.2 (32.2)/28.0 (34.9) | 20.7 (38.0)/24.8 (40.6) | 19.9 (23.4)/25.3 (39.3) | 21.5 (34.6)/25.2 (50.3) | 21.4 (30.6)/25.9 (40.8) |
| Clashscore | 9.2 | 8.1 | 6.4 | 10.9 | 11.1 |
| RMS bond (Å) | 0.01 | 0.005 | 0.003 | 0.01 | 0.01 |
| RMS angle (°) | 1.06 | 0.710 | 0.659 | 1.10 | 1.15 |
| Ramachandran favored/outlier (%) | 93.97/1.44 | 98.22/0.0 | 98.02/0.0 | 93.76/0.97 | 92.48/0.99 |
| Average B factor (Å2) | |||||
| Macromolecule | 77.0 | 79.7 | 47.8 | 79.5 | 78.8 |
| Ligand | – | 100.3 | 68.5 | 91.5 | 90.5 |
| Solvent | 65.6 | 72.5 | 57.4 | 79.3 | 82.8 |
| Metal ions | Zn2+: 99.0 | Zn2+: 72.0 | Zn2+: 33.0 | Zn2+: 66.0 | Zn2+: 62.0 |
| Ca2+:116.0 | |||||
| Na+: 84.0 | |||||
| Mg2+: 71.5 | |||||
| SO42–: 123.1 | |||||
Values in parenthesis represent number of 10˚ wedges used.
Values in parenthesis indicate total time in minutes, which includes the screening of many CrystalDirect pins or drops in plate-containing microcrystals.
Values in parentheses represent high-resolution shells.
To demonstrate that our pipeline can also be applied to ligand discovery, we soaked ADIPOR2 crystals with a library composed of 60 small-molecule fragments. We soaked each fragment into two in meso boluses containing ADIPOR2 microcrystals in order to ensure collection of a complete dataset. Soaked crystals were harvested and subjected to SSX diffraction data collection, data processing, and ligand detection (STAR Methods). Structures were refined against the data by using the fully automated Pipedream pipeline from Global Phasing, including structure refinement with autoBuster and automated ligand fitting with Rhofit (Figure 3). This led to the identification of two fragment hits, named B08 and F04 (Figure S4), demonstrating the feasibility of this automated pipeline for ligand screening.
Conclusions
The combination of in meso crystallization with CrystalDirect technology enables facile in situ data collection and automation of in meso crystal soaking, harvesting, and processing, producing high-resolution membrane protein structures (Figure 5). This approach removes critical bottlenecks in sample handling and streamlines SSX diffraction analysis of membrane proteins. In combination with CRIMS, providing web access to experimental design, analysis, and crystal mounting interfaces, as well as an automated SSX data processing pipeline, we provide for the first time an automated, protein-to-structure pipeline for the crystallographic analysis of membrane proteins remotely operated over the web. The method is versatile, enabling structure determination both at room temperature and under cryogenic conditions from a single crystallization support. All workflows described are currently available at the HTX platform at EMBL Grenoble (https://htxlab.embl.fr/) and can be reproduced at other laboratories, as the technology is readily available. The technique is compatible with standard beamline setups, thereby eliminating the need to adapt crystal production protocols to the type of experiment or sample delivery system. This approach facilitates the systematic analysis of membrane proteins at different temperatures, helping to identify conformational flexibility in membrane proteins and contributing to the study of receptor dynamics (Fraser et al., 2011). We have demonstrated this by capturing aspects of intramembrane enzyme-product dynamics. Finally, in meso ligand soaking experiments are key to support structure-guided drug design against membrane proteins (Ishchenko et al., 2019; Rucktooa et al., 2018). The approach presented here uniquely combines a web-based CRIMS database and the CrystalDirect platform, providing a complete, automated workflow for high-throughput membrane protein ligand discovery.
Limitations of the study
The proposed method has some limitations. Naturally, samples should be available in sufficient amount to carry out crystallographic screening and should be of good enough quality to produce crystals with sufficient diffracting power. Optimization of protein expression and purification methods is often required for successful crystallization of membrane proteins.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E. coli cells DH5α | NEB | C2987 |
| E. coli cells DH10Bac | Invitrogen | 10361012 |
| Chemicals, peptides, and recombinant proteins | ||
| pFastBac vector | Thermofisher | 10712024 |
| pCDNA3.1 vector | Thermofisher | V79020 |
| pMT vector | ThermoFisher | V413020 |
| Iodoacetamide | Sigma | I6125 |
| Leupetin | Euromedex | SP-04-2217 |
| Benzamidine | Sigma | B6506 |
| Phenylmethanesulfonyl fluoride (PMSF) | Sigma | P7626 |
| n-dodecyl-beta-D-maltopyranoside (DDM) | Anatrace | D310 |
| Lauryl maltose neopentyl glycol (MNG) | Anatrace | NG310 |
| Cholesterol hemisucinate (CHS) | Sigma | C6512 |
| ANTI-FLAG® M2 Agarose Affinity Gel | Sigma-Aldrich | A2220 |
| FLAG peptide | Covalab | https://www.covalab.com/peptide-synthesis |
| EX-CELL420 Serum-Free Medium | Sigma-Aldrich | 14420 |
| Strep-Tactin® Superflow® high capacity resin | IBA | 2-1206 |
| Desthiobiotin | IBA | 2-1000 |
| Puromycin dihydrochloride | ThermoFisher | A1113803 |
| Transfection reagent Sf9 (Transfectin) | BioRad | 1703350 |
| Transfection reagent S2 (Effectene) | Qiagen | 301425 |
| Cadmium chloride hydrate | Sigma | 208299 |
| Experimental models: cell lines | ||
| insect cell line Sf9 | Life Technologies | 11496015 |
| insect cell line S2 | ThermoFisher | R69007 |
| Recombinant DNA | ||
| pMT-scFv-ADIPOR | Vasiliauskaité-Brooks et al., 2017 | N/A |
| pFastBac-ADIPOR2(100-386) | Vasiliauskaité-Brooks et al., 2017 | N/A |
| pFastBac-ACER3(1-244)-BRIL | Vasiliauskaité-Brooks et al., 2018 | N/A |
| Deposited data | ||
| Cryogenic structure of ADIPOR2 at 2.4 Å ligand determined by SSX crystallography using CrystalDirect | This paper | PDB: 6YX9 |
| Cryogenic structure of ACER3 at 2.6 Å determined by SSX crystallography using CrystalDirect | This paper | PDB: 6YXH |
| Room temperature structure of ADIPOR2 at 2.9 Å ligand determined by SSX crystallography using CrystalDirect | This paper | PDB: 6YXD |
| Cryogenic structure of ADIPOR2 with Gd-DO3 ligand determined by SSX crystallography using CrystalDirect | This paper | PDB: 6YXF |
| Cryogenic structure of ADIPOR2 with Tb-XO4 ligand determined by SSX crystallography using CrystalDirect | This paper | PDB: 6YXG |
| Software and algorithms | ||
| CRIMS | This paper | https://git.embl.de/grp-marquez/crims_lift |
| PyMOL | Schrödinger | https://pymol.org/2/ |
| XDS | Kabsch, 2010 | https://xds.mr.mpg.de/ |
| CrystFEL | White et al., 2012 | https://www.desy.de/∼twhite/crystfel/ |
| Sxdm | Basu et al., 2019 | git@github.com:shibom/sxdm.git |
| CCP4 | https://doi.org/10.1107/S0907444910045749 | http://www.ccp4.ac.uk/ |
| COOT | Emsley and Cowtan, 2004 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Buster | Vonrhein et al., 2006 | http://www.globalphasing.com/buster/ |
| Pipedream | Sharff et al., 2011 | https://www.globalphasing.com/buster/manual/pipedream/manual/index.html#_pipedream |
| H++ server | Gordon et al., 2005 | http://biophysics.cs.vt.edu/ |
| GROMACS 5.1 | Van Der Spoel et al., 2005 | https://www.gromacs.org |
| PLUMED 2.3 | Tribello et al., 2014 | https://www.plumed.org/ |
| PACKMOL-Memgen | Schott-Verdugo and Gohlke, 2019 | https://github.com/alanwilter/acpype/blob/master/amber19-0_linux/bin/packmol-memgen |
Resource availability
Lead contacts
Further information and requests for reagents should be directed to and will be fulfilled by the lead contacts, Sébastien Granier (sebastien.granier@igf.cnrs.fr) and José Antonio Márquez (marquez@embl.fr).
Materials availability
DNA constructs generated by the authors can be obtained upon request from the Lead Contact, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Experimental model and subject details
Human ADIPOR2 and ACER3 were expressed in Sf9 cells infected with recombinant baculovirus (pFastBac, Invitrogen). Synthetic anti-ADIPOR scFv was expressed in S2 cells transfected with plasmid DNA (pMT, ThermoFisher).
Quantification and statistical analysis
Quantification and statistical analyses of data are described in Method details and Figure legends.
Method details
scFv Anti-ADIPOR2 expression and purification
The DNA sequence of anti-ADIPOR was subcloned into a pMT expression vector (Invitrogen) containing an N-terminal BiP secretion signal and C-terminal twin-strep tag. S2 Schneider cells (ThermoFisher) expressing the scFv were induced for 10 days with cadmium chloride and supernatant containing scFv was harvested. The supernatant was concentrated and loaded onto a StrepTactin column (IBA) in scFv wash buffer (100 mM Tris pH 8, 150 mM NaCl, 1 mM EDTA), the column was washed then scFv was eluted using scFv wash buffer containing 2.5 mM desthiobiotin (IBA). The eluted scFv was concentrated and injected on a Superdex S200 size exclusion chromatography column in SEC scFv buffer (10 mM HEPES pH 7.5, 150 mM NaCl). Eluted fractions were concentrated to 15 mg/mL, snap-frozen in liquid nitrogen prior to complexation with purified receptor.
ADIPOR2(100-386) expression, purification and complexation with scFv
We utilized an N-terminally truncated human ADIPOR2 construct containing an N-terminal Flag-tag for affinity purification. Recombinant baculoviruses were generated using the pFastBac baculovirus system (ThermoFisher). High titer baculovirus encoding ADIPOR2 gene were used to infect Sf9 cells at a cell density of 4 × 106 cells per ml in suspension in EX-Cell 420 serum free media (Expression System). Cells were harvested by centrifugation 48 h post-infection and stored at -80°C until purification.
Cell pellets were resuspended in lysis buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA buffer containing 2 mg·mL-1 iodoacetamide and protease inhibitors) without salt to lyse the cells by hypotonic lysis. Lysed cells were centrifuged (38,420 g) and the membranes were solubilized for 1 hour at 4 degrees in solubilization buffer (20 mM HEPES pH 7.5, 100mM NaCl, 1% (w/v) n-dodecyl-β-D-maltoside (DDM), 0.1% (w/v) cholesteryl hemisuccinate (CHS), 2 mg·mL-1 iodoacetamide and protease inhibitors). The solubilized receptor was recovered by centrifugation (38,420 g), loaded onto anti-Flag M2 column (Sigma) and washed thoroughly with DDM wash buffer (20 mM HEPES pH 7.5, 200 mM NaCl, 0.025% (w/v) DDM and 0.0001% (w/v) CHS). While on the M2 antibody resin, the detergent was exchanged to lauryl maltose neopentyl glycol (MNG, Anatrace) using MNG exchange buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 0.5% (w/v) MNG). The detergent exchange was performed by washing the column with a series of seven buffers (3 CV each) made up of the following ratios (%/%) DDM wash buffer and MNG exchange buffer: 100:0, 50:50, 25:75, 10:90, 5:95, 1:99, 0:100. The column was then washed with MNG wash buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 0.02% (w/v) MNG) and the bound receptor was eluted in the same buffer supplemented with 0.2 mg·mL-1 Flag peptide (Covalab).
Eluted ADIPOR2 was incubated with purified antiADIPOR-scFv at a molar ratio of 2:1, ADIPOR2:scFv on ice for 30 minutes to form a complex, then loaded onto a StrepTactin column (IBA). The complex was washed with MNG wash buffer and the bound complex was eluted in the same buffer supplemented with 2.5 mM desthiobiotin. Eluted complex was concentrated to 500 μL and injected on a Superdex S200 size exclusion chromatography column in SEC MNG buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 0.002% (w/v) MNG). Purified complex was concentrated to 15 mg/mL and snap-frozen in liquid nitrogen prior to crystallization.
ACER3(1-244) expression and purification
We utilized a C-terminally truncated human ACER3 construct containing an N-terminal Flag-tag for affinity purification. Recombinant baculoviruses were generated using the pFastBac baculovirus system (ThermoFisher). High titer baculovirus encoding ACER3 gene were used to infect Sf9 cells at a cell density of 4 × 106 cells per ml in suspension in EX-Cell 420 serum free media (Expression System). Cells were harvested by centrifugation 48 h post-infection and stored at -80°C until purification.
Cell pellets were resuspended in lysis buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA buffer containing 2 mg·mL-1 iodoacetamide and protease inhibitors) without salt to lyse the cells by hypotonic lysis. Lysed cells were centrifuged (38,420 g) and the membranes were solubilized for 1 hour at 4 degrees in solubilization buffer (20 mM HEPES pH 7.5, 100mM NaCl, 1% (w/v) n-dodecyl-β-D-maltoside (DDM), 0.1% (w/v) cholesteryl hemisuccinate (CHS), 2 mg·mL-1 iodoacetamide and protease inhibitors). The solubilized enzyme was recovered by centrifugation (38,420 g), loaded onto anti-Flag M2 column (Sigma) and washed thoroughly with DDM wash buffer (20 mM HEPES pH 7.5, 200 mM NaCl, 0.025% (w/v) DDM and 0.0001% (w/v) CHS). While on the M2 antibody resin, the detergent was exchanged to lauryl-maltose-neopentyl-glycol (MNG, Anatrace) using MNG exchange buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 0.5% (w/v) MNG). The detergent exchange was performed by washing the column with a series of seven buffers (3 CV each) made up of the following ratios (%/%) DDM wash buffer and MNG exchange buffer: 100:0, 50:50, 25:75, 10:90, 5:95, 1:99, 0:100. The column was then washed with MNG wash buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 0.02% (w/v) MNG) and the bound enzyme was eluted in the same buffer supplemented with 0.2 mg·mL-1 Flag peptide (Covalab). Eluted enzyme was concentrated to 500 μL and injected on a Superdex S200 size exclusion chromatography column in SEC MNG buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 0.002% (w/v) MNG). Purified ACER3 was concentrated to 18 mg/mL and snap-frozen in liquid nitrogen prior to crystallization.
LCP crystallization in CrystalDirect plates
Protein constructs, expression and purification were as previously described for ADIPOR2 (Vasiliauskaité-Brooks et al., 2017) and ACER3(Vasiliauskaité-Brooks et al., 2018). Snap-frozen protein aliquots were sent on dry ice to the high-throughput crystallization lab (HTX Lab) at EMBL Grenoble for crystallization set-up (Dimasi et al., 2007). Sample mesophases of ADIPOR2 and ACER3 were generated using the LCP method (Caffrey, 2015; Landau and Rosenbusch, 1996; Liu and Cherezov, 2011). Concentrated protein solutions (ADIPOR2: 15 mg/mL, ACER3: 18 mg/mL) were thawed and reconstituted with a 10:1 monoolein:cholesterol (Sigma) mixture in a ratio of 2:3 protein:lipid (v/v). The mesophase was formed by using the coupled two-syringe method (Caffrey, 2015; Caffrey and Cherezov, 2009; Liu and Cherezov, 2011). The mesophase was dispensed onto the crystallization film of CrystalDirect plates (MiTeGen, ref. M-XDIR-96-2) in 30 or 50 nl bolus and overlaid with 600 nl precipitant solution using either a Mosquito LCP robot (SPT Labtech) equipped with humidifier or a Gryphon LCP robot (Art Robbins Instruments). Reservoir wells next to each mesophase bolus contained 45 μL of precipitant solution to maintain drop humidity during crystallization (Figure 3). Precipitant solutions contained 42.3% PEG 400, 110 mM potassium citrate, 100 mM HEPES pH 7.0 for ADIPOR2 and 39.5–41% PEG 400, 100 mM HEPES pH 7.5, 75 mM magnesium sulphate and 5% DMSO for ACER3. Under these conditions ADIPOR2 crystals were stable for several weeks.
The plates were incubated at 20˚C and monitored with a Rock Imager-1000 robot (Formulatrix). In our experience the surface of the bolus in contact with the precipitant solution presents a rough structure, which interferes with crystal imaging. To overcome this, and facilitate crystal detection, a plate adaptor was developed to invert the plate inside the imager (Figure S5). This adaptor consists of a plastic frame compatible with the SBS-plate format into which the crystallization plate is inserted and turned upside-down (crystallization film facing up) whilst guaranteeing safe handling by the imaging robot. Thanks to this adaptor; crystallization experiments are imaged through the film in which the LCP bolus is resting, therefore presenting a smooth and flattened surface to the camera, notably improving crystal imaging.
Design and follow-up of crystallization experiments was carried out remotely using CRIMS. Crystals were observed within one day and reached full size within five days: ∼30 x 20 x 5 μm3 for ADIPOR2 in cubic phase and ∼20 x 10 x 5 μm3 for ACER3 in sponge phase (Figure S3).
CrystalDirect plates are compatible with standard crystallization robotics, which makes setting LCP crystallization experiments very straightforward. They can be used both for crystallization screening and optimization. In our experience it was straightforward to transfer LCP crystallization from sandwich to CrystalDirect plates and vice versa by minor adjustment of the precipitant concentration.
Room temperature serial crystallography data collection
In situ diffraction experiments were carried out at the P14 beam line operated by EMBL Hamburg at the PETRA III storage ring (Gati et al., 2014) (DESY, Hamburg, Germany). While the crystallization film used in CrystalDirect plates is extremely thin (20 μm), the plate is typically sealed on the top side with a standard film (c.a. 100 μm thick). In order to reduce the scattering background, the top sealing film was removed from the plates and replaced with a CrystalDirect (Zander et al., 2016) in situ plate seal (Mitegen, ML-CDSF1-10, with the same thickness as the crystallization film) immediately before data collection. The CrystalDirect plates were mounted on a SBS-plate goniometer. Data collection was carried out by rastering each LCP bolus applying multiple vertical “helical-lines” separated by 10 μm in the horizontal direction and applying a continuous oscillation with the plate goniometer from the bottom to the top of the line (Video S2). A 10 x 5 μm2 beam operated at 12.4 KeV and 100% transmission at a photon flux of 1.17 x 1013 was used, which delivered roughly 100 kGy per frame (Gati et al., 2014). The length and number of lines as well as the oscillation were adjusted to match the size of each bolus. Typically, between 300 and 400 helical lines about 1.8 mm in length were necessary to sample a full LCP bolus with an oscillation of between 0.3 and 0.4 degrees per frame. At the P14 beam line optimal data collection patterns can be easily selected by simply entering the number of lines, separation and oscillation range per line in a dedicated user interface. Images were collected using an Eiger X 16M operated in 4M mode compatible with a fast exposure time of 1.3 msec. A typical data collection session took four minutes per bolus (including focusing and centering the bolus into the beam). For ADIPOR2 five LCP boluses and about 20 minutes of data collection were sufficient to obtain a full dataset. This consisted of 382,552 diffraction images, out of which 28,949 were crystal hits and 25,297 diffraction images were indexable (Table 1).
Automated, laser-based LCP-crystal harvesting and cryocooling
Automated harvesting of crystals grown in mesophase media was carried out with the help of the CRIMS software and the CrystalDirect harvester robot (Cipriani et al., 2012; Márquez and Cipriani, 2014; Zander et al., 2016) (Video S1). Briefly, crystallization images taken by the crystal imaging farm are presented through a CRIMS web interface (Figure S1). This interface enables scientists to select from a collection of different shapes that can then be placed onto the crystallization image and rotated for optimal crystal recovery. The scientist has a number of choices concerning sample mounting. For example, it is possible to mount multiple crystals in a single pin or use the laser to isolate single crystals (Figure 4). It is also possible to perform multiple crystal harvests from a single drop. All harvesting operations are recorded by the CRIMS system.
To harvest the samples, the crystallization plate is transferred to the CrystalDirect harvester from which point all harvesting operations are automatically performed (Video S1). The harvester uses a femto-second laser beam operating in the photoablation regime enabling precise excision of the crystallization film while avoiding heat transfer or mechanical damage to the sample (Aragón et al., 2019; Bezerra et al., 2017; Martin-Malpartida et al., 2017). The harvester robot uses the plate barcode to recover all harvesting operations stored by CRIMS. A camera system along with automated image recognition algorithms allows the harvester to automatically locate crystallization experiments and precisely position the harvesting shapes according to the design specified through the CRIMS harvesting interfaces. After laser excision, the film piece containing the crystals is automatically attached to a SPINE standard X-ray data collection pin and transferred to a nitrogen gas stream at 100 K. Cryo-cooled samples are then recovered by a robotic arm that transfers them to a cryogenic sample storage dewar. The system can work both with SPINE and UNIPUCK puck formats that are compatible with most synchrotron beam lines. Multiple harvests can be performed consecutively in batch-mode. The sample storage dewar has capacity for 312 pins. All data, including sample, pin and puck locations for each sample along with harvesting images automatically generated during the harvesting processes is transferred back and stored in CRIMS.
Soaking of ADIPOR2 crystals
The CrystalDirect LCP crystallization method facilitates post-crystallization treatments, including soaking, by enabling the removal of the top sealing film without disturbing the bolus. Two commercially available caged-metal complexes (Engilberge et al., 2017; Girard et al., 2003) were used for soaking experiments, lanthanide phasing compound Gd-HPDO3A (Jena Bioscience PK-CSM002-0002A) at a concentration of 500 mM and polyvalan Crystallophore kit Tb-XO4 (Molecular Dimensions ref: MD2-80) at 100 mM. For soaking experiments, the top sealing film of the CrystalDirect plate was removed and 30–150 nl of Tb-XO4 or 60–120 nl of Gd-DO3 ligand solutions were delivered directly to the LCP crystallization drops with a SPT-Labtech crystallization robot, after which the plate was resealed. This process takes a few seconds for a drop and just a couple of minutes for the whole plate, avoiding dehydration of the LCP drops. Plates were resealed and crystals were soaked for 90 minutes at 20°C before crystal harvesting. The final concentrations of ligands were approximately 5–25 mM Tb-XO4 and 10–25 mM Gd-DO3. The same protocol was used for fragment soaking, adding approximately 32nl of DMSO stock solutions at 500 mM to the boluses and final experimental concentrations of about 25 mM.
Cryogenic serial crystallography data collection
Diffraction data from samples vitrified using the CrystalDirect technology were collected remotely at the X06SA beamline of the Swiss Light Source (SLS), Villigen, Switzerland. After sample transfer to the goniometer, pins containing multiple crystals were rapidly raster-scanned (Figure S2) with the X-ray beam to identify positions corresponding to diffracting crystals (Basu et al., 2019; Wojdyla et al., 2016). In a second step 10° wedges were collected with 0.1 sec exposure time from each of the identified positions at a speed of 2°/sec. If dose permitted, 2-3 sweeps of 10° wedges were collected from each of those identified crystals. Each 10° wedge takes typically 5 sec. Effective data collection time required to obtain a complete dataset from each target is summarized in Table 1. Diffraction images were collected using an EIGER X 16M detector (Dectris) with a sample-to-detector distance of 30 cm, beam energy was 12.4 KeV for apo crystals, and 7.2 KeV for soaked crystals in order to exploit the anomalous signals from the heavy elements. The beam size was 10x20 μm2 and 100% transmission.
Data processing and structure refinement
Initial diffraction patterns, collected under cryogenic condition at the SLS, were processed using XDS (Kabsch, 2010), followed by real-time automated selection and merging of statistically equivalent datasets with XSCALE using the sxdm tool (Basu et al., 2019) as deployed at the beamline. Datasets were selected using a criterion or a combination of multiple criteria, including ISa cut-off of 3.0, unit-cell, pair-CC based hierarchical clustering as well as Δ-CC1/2 iteratively (Assmann et al., 2020). Overall, pre-processed data was manually transferred from SLS and refined with BUSTER program in Global Phasing. In case of fragment screening datasets, we further processed unmerged scaled MTZs with Global-Phasing Pipedream software pipeline. For in situ datasets collected at PetraIII P14 beamline, we used an in-house script to run CrystFEL (White et al., 2012) version 0.8.0 automatically as the data had arrived. This allowed us to quickly identify hits and reduce data volume and produce a mtz file ready for structural analysis. All the structures were solved by molecular replacement using PHASER and previously determined structures of ACER3 and ADIPOR2 as search models (PDB IDs 6G7O, 5LWY respectively). The resolution cut-offs for all structures were determined by a combined metric of CC1/2 of 0.30 and I/σ(I) of 1.0 (Karplus and Diederichs, 2012). Structure refinement was carried out iteratively with BUSTER (Vonrhein et al., 2006) version 2.10.3 and Coot(Emsley and Cowtan, 2004). At late stages of refinement, translation liberation screw-motion parameters generated within BUSTER (Vonrhein et al., 2006) were used. MolProbity (Williams et al., 2018) was used to assess the quality of the structure and the data collection and refinement statistics are summarized in Table 1. The data refinement and analysis software were compiled and supported by the SBGrid consortium (https://sbgrid.org/).
ADIPOR2-RT
About 3.8 million diffraction patterns were collected from 19 drops in 1 hour and 16 minutes approximately from 19 LCP boluses in one 96-well plate, out of which 170,944 images correspond to crystal hits. However, a complete room-temperature dataset was obtained from only 5 drops in a 96 well-plate, which yielded 25,297 indexed diffraction patterns. The room-temperature structure was refined at 2.9-Å resolution using AUTOBUSTER with Rwork of 19.9 % and Rfree of 25.3 %. The relative lower resolution for ADIPOR2-RT over ADIPOR2-Cryo is owing to the 10-fold less deposited dose at room-temperature. The structure was determined from images obtained from 5 drops, requiring 20 min of collection time.
ADIPOR2-Cryo
315 sweeps of 10° wedges were collected from only 1 laser-cut solid support , containing about 33 microcrystals. 104 datasets were successfully indexed and processed using XDS, out of which 100 datasets were selected based on ISa value cutoff of 3.0, followed by merging with XSCALE. The final mtz file, obtained from automated pipeline, was used for structure determination and refined at 2.4 Å resolution using AUTOBUSTER with Rwork of 20.7 % and Rfree of 24.8 %. The structure was determined from 100 datasets, obtained from 8.3 min of collection time.
ACER3-Cryo
425 sweeps of 10° wedges were collected from 9 CrystalDirect pins, out of which 375 datasets were successfully processed. 101 datasets were selected based on Δ-CC1/2(Assmann et al., 2020) and ISa values as implemented in sxdm tool (Basu et al., 2019), followed by merging with XSCALE. The final mtz file was used for structure determination in Molecular replacement method. ACER3 structure was refined at 2.6 Å resolution using AUTOBUSTER with Rwork of 23.5 % and Rfree of 28.9 %. The structure was determined from 101 datasets, obtained from 8.4 min of collection time.
ADIPOR2-TbXO4
570 sweeps of 10° wedges were collected from 16 pins, out of which 467 datasets were successfully processed in XDS, followed by 210 datasets being selected and merged with and sxdm, at X06SA beamline of the SLS. The Tb atoms from the soaked cluster produced strong anomalous signals. The minimum datasets required to extract sufficient anomalous signals to validate the presence of soaked ligand, were obtained in 17.5 min of collection time. The structure was refined at 3.0- Å resolution using AUTOBUSTER with R-work of 21.5% and Rfree of 25.2%.
ADIPOR2-GdDO3
410 x 10° wedges were collected from 7 pins, out of which 356 datasets were processed in XDS, followed by 96 datasets, being selected and merged with XSCALE automatically by sxdm tool, as installed at X06SA beamline of the SLS. The soaked heavy metal Gd-cluster produced a strong anomalous signal. The minimum datasets required to obtain sufficient anomalous signals to validate the presence of soaked ligand, were collected in 8 min. The structure was refined at 3.0 Å resolution using AUTOBUSTER with R-work of 21.4% and Rfree of 25.9%.
Molecular dynamics simulations
The initial receptor models were based on the X-ray crystal structures of ADIPOR2 in complex with an oleic acid. The protonation states of titratable residues were predicted at pH 6.5 using the H++ server (Gordon et al., 2005). The receptor systems were embedded in explicit POPC membrane and aqueous solvent neutralized with Cl- ions. Effective point charges of the ligands were obtained by RESP fitting (Wang et al., 2000) of the electrostatic potentials calculated with the HF/6-31G∗ basis set using Gaussian 09 (Frisch et al., 2016). The Amber 99SB-ildn (Wang et al., 2004) and GAFF (Lindorff-Larsen et al., 2010) force fields were used for the protein and the ligands, respectively. Parameters for the active site were adopted from the Zinc AMBER Force Field (Peters et al., 2010). The TIP3P (Jorgensen et al., 1983) and the Joung-Cheatham models (Joung and Cheatham, 2008) were used for the water and the ions, respectively. After energy minimization, all-atom MD simulations were carried out using Gromacs 2018 (Van Der Spoel et al., 2005) patched with the PLUMED 2.3 plugin(Tribello et al., 2014). Each system was gradually heated to 298 K and pre-equilibrated during 10 ns of brute-force MD in the NPT-ensemble. The replica exchange with solute scaling (REST2) (Wang et al., 2011) technique was then employed to enhance the sampling with 48 replicas in the NVT ensemble. The protein and the ligands were considered as “solute” in the REST2 scheme–force constants of their van der Waals, electrostatic and dihedral terms were subject to scaling. The effective temperatures used for generating the REST2 scaling factors ranged from 298 K to 700 K, following a distribution calculated with the Patriksson-van der Spoel approach (Patriksson and van der Spoel, 2008). Exchange between replicas was attempted every 1000 simulation steps. This setup resulted in an average exchange probability of ∼40% during 40 ns (× 48 replicas) of simulations. The first 10 ns were discarded for equilibration. Only the original unscaled replica (at 298 K effective temperature) was collected and analyzed.
Acknowledgments
We want to thank Thomas Schneider and the EMBL Hamburg Team for excellent support with data collection at P14 of the PETRA III synchrotron (DESY, Hamburg, Germany), the staff at the Swiss Light Source (Paul Scherrer Institut, Villigen, Switzerland), and the staff at the ID23-2 microfocus beamline at the European Synchrotron Radiation Facility (ESRF, Grenoble, France). In addition, we thank Frank Felisaz and the EMBL Grenoble Diffraction Instrumentation Team for support during the design of the plate adaptor. This work benefited from access to the HTX Lab at EMBL Grenoble. This project was supported by funding from the European Union’s Horizon 2020 Programme under the projects iNEXT (grant 653706) and iNEXT Discovery (grant 871037), as well as the Région Auvergne-Rhône-Alpes through the Booster program. This research has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement 730872, project CALIPSOplus. This project has also received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement 647687).
Author contributions
R.H., S.B., and C.L. produced the samples and carried out serial data collection and structural analysis. A.S.H., F.D., and A.P. carried out crystallization experiments, adapted the CrystalDirect technology to LCP, and contributed to serial data collection experiments. X.C. and J.B. performed molecular dynamics simulations. J.A.M. and S.G. supervised the work and wrote the manuscript with R.H. and S.B. All authors contributed to the final version of the manuscript.
Declaration of interests
J.A.M. is a co-author of a patent on the CrystalDirect technology and co-founder of ALPX s.a.s.
Published: October 25, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2021.100102.
Contributor Information
Sébastien Granier, Email: sebastien.granier@igf.cnrs.fr.
José Antonio Márquez, Email: marquez@embl.fr.
Supplemental information
Data and code availability
The article includes all datasets generated or analyzed during this study. Macromolecular coordinates for the structure displayed can be accessed through the wwPDB under accession codes are listed in the Key resources table.
All original code has been deposited as indicated in the key resources table at and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Andrews S.P., Brown G.A., Christopher J.A. Structure-based and fragment-based GPCR drug discovery. ChemMedChem. 2014;9:256–275. doi: 10.1002/cmdc.201300382. [DOI] [PubMed] [Google Scholar]
- Aragón E., Wang Q., Zou Y., Morgani S.M., Ruiz L., Kaczmarska Z., Su J., Torner C., Tian L., Hu J., et al. Structural basis for distinct roles of SMAD2 and SMAD3 in FOXH1 pioneer-directed TGF-β signaling. Genes Dev. 2019;33:1506–1524. doi: 10.1101/gad.330837.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assmann G.M., Wang M., Diederichs K. Making a difference in multi-data-set crystallography: simple and deterministic data-scaling/selection methods. Acta Crystallogr. Sect. Struct. Biol. 2020;76:636–652. doi: 10.1107/S2059798320006348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S., Kaminski J.W., Panepucci E., Huang C.-Y., Warshamanage R., Wang M., Wojdyla J.A. Automated data collection and real-time data analysis suite for serial synchrotron crystallography. J. Synchrotron Radiat. 2019;26:244–252. doi: 10.1107/S1600577518016570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H.M. The protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezerra G.A., Ohara-Nemoto Y., Cornaciu I., Fedosyuk S., Hoffmann G., Round A., Márquez J.A., Nemoto T.K., Djinović-Carugo K. Bacterial protease uses distinct thermodynamic signatures for substrate recognition. Sci. Rep. 2017;7:2848. doi: 10.1038/s41598-017-03220-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botha S., Nass K., Barends T.R.M., Kabsch W., Latz B., Dworkowski F., Foucar L., Panepucci E., Wang M., Shoeman R.L., et al. Room-temperature serial crystallography at synchrotron X-ray sources using slowly flowing free-standing high-viscosity microstreams. Acta Crystallogr. D Biol. Crystallogr. 2015;71:387–397. doi: 10.1107/S1399004714026327. [DOI] [PubMed] [Google Scholar]
- Caffrey M. A comprehensive review of the lipid cubic phase or in meso method for crystallizing membrane and soluble proteins and complexes. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2015;71:3–18. doi: 10.1107/S2053230X14026843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffrey M., Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat. Protoc. 2009;4:706–731. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V., Peddi A., Muthusubramaniam L., Zheng Y.F., Caffrey M. A robotic system for crystallizing membrane and soluble proteins in lipidic mesophases. Acta Crystallogr. D Biol. Crystallogr. 2004;60:1795–1807. doi: 10.1107/S0907444904019109. [DOI] [PubMed] [Google Scholar]
- Cipriani F., Röwer M., Landret C., Zander U., Felisaz F., Márquez J.A. CrystalDirect: a new method for automated crystal harvesting based on laser-induced photoablation of thin films. Acta Crystallogr. D Biol. Crystallogr. 2012;68:1393–1399. doi: 10.1107/S0907444912031459. [DOI] [PubMed] [Google Scholar]
- Congreve M., de Graaf C., Swain N.A., Tate C.G. Impact of GPCR structures on drug discovery. Cell. 2020;181:81–91. doi: 10.1016/j.cell.2020.03.003. [DOI] [PubMed] [Google Scholar]
- Dimasi N., Flot D., Dupeux F., Márquez J.A. Expression, crystallization and X-ray data collection from microcrystals of the extracellular domain of the human inhibitory receptor expressed on myeloid cells IREM-1. Acta Crystallograph. Sect. F Struct. Biol. Cryst. Commun. 2007;63:204–208. doi: 10.1107/S1744309107004903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Engilberge S., Riobé F., Di Pietro S., Lassalle L., Coquelle N., Arnaud C.-A., Pitrat D., Mulatier J.-C., Madern D., Breyton C., et al. Crystallophore: a versatile lanthanide complex for protein crystallography combining nucleating effects, phasing properties, and luminescence. Chem. Sci. 2017;8:5909–5917. doi: 10.1039/c7sc00758b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser J.S., Clarkson M.W., Degnan S.C., Erion R., Kern D., Alber T. Hidden alternative structures of proline isomerase essential for catalysis. Nature. 2009;462:669–673. doi: 10.1038/nature08615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser J.S., van den Bedem H., Samelson A.J., Lang P.T., Holton J.M., Echols N., Alber T. Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proc. Natl. Acad. Sci. 2011;108:16247–16252. doi: 10.1073/pnas.1111325108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Cheeseman R.J.R., Scalmani G., Barone V., Peterson G.A., Nakatsuji H., Li X., et al. Gaussian Inc.; 2016. Gaussian 09, Revision A.01. [Google Scholar]
- Gati C., Bourenkov G., Klinge M., Rehders D., Stellato F., Oberthür D., Yefanov O., Sommer B.P., Mogk S., Duszenko M., et al. Serial crystallography on in vivo grown microcrystals using synchrotron radiation. IUCrJ. 2014;1:87–94. doi: 10.1107/S2052252513033939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard É., Stelter M., Vicat J., Kahn R. A new class of lanthanide complexes to obtain high-phasing-power heavy-atom derivatives for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2003;59:1914–1922. doi: 10.1107/s0907444903020511. [DOI] [PubMed] [Google Scholar]
- Gordon J.C., Myers J.B., Folta T., Shoja V., Heath L.S., Onufriev A. H++: a server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005;33:W368–W371. doi: 10.1093/nar/gki464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.-Y., Olieric V., Ma P., Panepucci E., Diederichs K., Wang M., Caffrey M. In meso in situ serial X-ray crystallography of soluble and membrane proteins. Acta Crystallogr. D Biol. Crystallogr. 2015;71:1238–1256. doi: 10.1107/S1399004715005210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.-Y., Olieric V., Ma P., Howe N., Vogeley L., Liu X., Warshamanage R., Weinert T., Panepucci E., Kobilka B., et al. In meso in situ serial X-ray crystallography of soluble and membrane proteins at cryogenic temperatures. Acta Crystallogr. Sect. Struct. Biol. 2016;72:93–112. doi: 10.1107/S2059798315021683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.-Y., Olieric V., Howe N., Warshamanage R., Weinert T., Panepucci E., Vogeley L., Basu S., Diederichs K., Caffrey M., et al. In situ serial crystallography for rapid de novo membrane protein structure determination. Commun. Biol. 2018;1:124. doi: 10.1038/s42003-018-0123-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishchenko A., Stauch B., Han G.W., Batyuk A., Shiriaeva A., Li C., Zatsepin N., Weierstall U., Liu W., Nango E., et al. Toward G protein-coupled receptor structure-based drug design using X-ray lasers. IUCrJ. 2019;6:1106–1119. doi: 10.1107/S2052252519013137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- Joung I.S., Cheatham T.E. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B. 2008;112:9020–9041. doi: 10.1021/jp8001614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus P.A., Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau E.M., Rosenbusch J.P. Lipidic cubic phases: a novel concept for the crystallization of membrane proteins. Proc. Natl. Acad. Sci. 1996;93:14532–14535. doi: 10.1073/pnas.93.25.14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau E.M., Rummel G., Cowan-Jacob S.W., Rosenbusch J.P. Crystallization of a polar protein and small molecules from the aqueous compartment of lipidic cubic phases †. J. Phys. Chem. B. 1997;101:1935–1937. [Google Scholar]
- Li D., Boland C., Aragao D., Walsh K., Caffrey M. Harvesting and cryo-cooling crystals of membrane proteins grown in lipidic mesophases for structure determination by macromolecular crystallography. J. Vis. Exp. 2012;4001 doi: 10.3791/4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J.L., Dror R.O., Shaw D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field: improved Protein Side-Chain Potentials. Proteins Struct. Funct. Bioinforma. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Cherezov V. Crystallization of membrane proteins in lipidic mesophases. J. Vis. Exp. 2011;2501 doi: 10.3791/2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Márquez J.A., Cipriani F. In: Structural Genomics. Chen Y.W., editor. Humana Press; 2014. CrystalDirect™: a novel approach for automated crystal harvesting based on photoablation of thin films; pp. 197–203. [DOI] [PubMed] [Google Scholar]
- Martin-Malpartida P., Batet M., Kaczmarska Z., Freier R., Gomes T., Aragón E., Zou Y., Wang Q., Xi Q., Ruiz L., et al. Structural basis for genome wide recognition of 5-bp GC motifs by SMAD transcription factors. Nat. Commun. 2017;8:2070. doi: 10.1038/s41467-017-02054-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogly P., James D., Wang D., White T.A., Zatsepin N., Shilova A., Nelson G., Liu H., Johansson L., Heymann M., et al. Lipidic cubic phase serial millisecond crystallography using synchrotron radiation. IUCrJ. 2015;2:168–176. doi: 10.1107/S2052252514026487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogly P., Weinert T., James D., Carbajo S., Ozerov D., Furrer A., Gashi D., Borin V., Skopintsev P., Jaeger K., et al. Retinal isomerization in bacteriorhodopsin captured by a femtosecond x-ray laser. Science. 2018:eaat0094. doi: 10.1126/science.aat0094. [DOI] [PubMed] [Google Scholar]
- Okada-Iwabu M., Yamauchi T., Iwabu M., Honma T., Hamagami K., Matsuda K., Yamaguchi M., Tanabe H., Kimura-Someya T., Shirouzu M., et al. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature. 2013;503:493–499. doi: 10.1038/nature12656. [DOI] [PubMed] [Google Scholar]
- Patriksson A., van der Spoel D. A temperature predictor for parallel tempering simulations. Phys. Chem. Chem. Phys. 2008;10:2073. doi: 10.1039/b716554d. [DOI] [PubMed] [Google Scholar]
- Pebay-Peyroula E. X-ray structure of bacteriorhodopsin at 2.5 angstroms from microcrystals grown in lipidic cubic phases. Science. 1997;277:1676–1681. doi: 10.1126/science.277.5332.1676. [DOI] [PubMed] [Google Scholar]
- Pei J., Millay D.P., Olson E.N., Grishin N.V. Crest - a large and diverse superfamily of putative transmembrane hydrolases. Biol. Direct. 2011;6:37. doi: 10.1186/1745-6150-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters M.B., Yang Y., Wang B., Füsti-Molnár L., Weaver M.N., Merz K.M. Structural survey of zinc-containing proteins and development of the zinc AMBER force field (ZAFF) J. Chem. Theor. Comput. 2010;6:2935–2947. doi: 10.1021/ct1002626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen S.G.F., DeVree B.T., Zou Y., Kruse A.C., Chung K.Y., Kobilka T.S., Thian F.S., Chae P.S., Pardon E., Calinski D., et al. Crystal structure of the β2 adrenergic receptor–Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rucktooa P., Cheng R.K.Y., Segala E., Geng T., Errey J.C., Brown G.A., Cooke R.M., Marshall F.H., Doré A.S. Towards high throughput GPCR crystallography: in Meso soaking of Adenosine A2A Receptor crystals. Sci. Rep. 2018;8:41. doi: 10.1038/s41598-017-18570-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russi S., González A., Kenner L.R., Keedy D.A., Fraser J.S., van den Bedem H. Conformational variation of proteins at room temperature is not dominated by radiation damage. J. Synchrotron Radiat. 2017;24:73–82. doi: 10.1107/S1600577516017343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott-Verdugo S., Gohlke H. PACKMOL-Memgen: A Simple-To-Use, Generalized Workflow for Membrane-Protein–Lipid-Bilayer System Building. J. Chem. Inf. Model. 2019;6:2522–2528. doi: 10.1021/acs.jcim.9b00269. [DOI] [PubMed] [Google Scholar]
- Sharff A., Keller P., Vonrhein C., Smart O., Womack T., Flensburg C., Paciorek C., Bricogne G. Global Phasing Ltd; 2011. Pipedream, Version 1.3.0. [Google Scholar]
- Straub L.G., Scherer P.E. Metabolic messengers: adiponectin. Nat. Metab. 2019;1:334–339. doi: 10.1038/s42255-019-0041-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe H., Fujii Y., Okada-Iwabu M., Iwabu M., Kano K., Kawana H., Hato M., Nakamura Y., Terada T., Kimura-Someya T., et al. Human adiponectin receptor AdipoR1 assumes closed and open structures. Commun. Biol. 2020;3:446. doi: 10.1038/s42003-020-01160-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tribello G.A., Bonomi M., Branduardi D., Camilloni C., Bussi G. Plumed 2: new feathers for an old bird. Comput. Phys. Commun. 2014;185:604–613. [Google Scholar]
- Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A.E., Berendsen H.J.C. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Vasiliauskaité-Brooks I., Sounier R., Rochaix P., Bellot G., Fortier M., Hoh F., De Colibus L., Bechara C., Saied E.M., Arenz C., et al. Structural insights into adiponectin receptors suggest ceramidase activity. Nature. 2017;544:120–123. doi: 10.1038/nature21714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiliauskaité-Brooks I., Healey R.D., Rochaix P., Saint-Paul J., Sounier R., Grison C., Waltrich-Augusto T., Fortier M., Hoh F., Saied E.M., et al. Structure of a human intramembrane ceramidase explains enzymatic dysfunction found in leukodystrophy. Nat. Commun. 2018;9:5437. doi: 10.1038/s41467-018-07864-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiliauskaité-Brooks I., Healey R.D., Granier S. 7TM proteins are not necessarily GPCRs. Mol. Cell. Endocrinol. 2019;491:110397. doi: 10.1016/j.mce.2019.02.009. [DOI] [PubMed] [Google Scholar]
- Vonrhein C., Blanc E., Roversi P., Bricogne G. Volume 2. Humana Press; 2006. Automated structure solution with autoSHARP; pp. 215–230. (Macromolecular Crystallography Protocols). [Google Scholar]
- Wang J., Cieplak P., Kollman P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000;21:1049–1074. [Google Scholar]
- Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Wang L., Friesner R.A., Berne B.J. Replica exchange with solute scaling: a more efficient version of replica exchange with solute tempering (REST2) J. Phys. Chem. B. 2011;115:9431–9438. doi: 10.1021/jp204407d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weierstall U., James D., Wang C., White T.A., Wang D., Liu W., Spence J.C.H., Bruce Doak R., Nelson G., Fromme P., et al. Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat. Commun. 2014;5:3309. doi: 10.1038/ncomms4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert T., Olieric N., Cheng R., Brünle S., James D., Ozerov D., Gashi D., Vera L., Marsh M., Jaeger K., et al. Serial millisecond crystallography for routine room-temperature structure determination at synchrotrons. Nat. Commun. 2017;8:542. doi: 10.1038/s41467-017-00630-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White T.A., Kirian R.A., Martin A.V., Aquila A., Nass K., Barty A., Chapman H.N. CrystFEL : a software suite for snapshot serial crystallography. J. Appl. Crystallogr. 2012;45:335–341. [Google Scholar]
- Williams C.J., Headd J.J., Moriarty N.W., Prisant M.G., Videau L.L., Deis L.N., Verma V., Keedy D.A., Hintze B.J., Chen V.B., et al. MolProbity: more and better reference data for improved all-atom structure validation: protein science. ORG. Protein Sci. 2018;27:293–315. doi: 10.1002/pro.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojdyla J.A., Panepucci E., Martiel I., Ebner S., Huang C.-Y., Caffrey M., Bunk O., Wang M. Fast two-dimensional grid and transmission X-ray microscopy scanning methods for visualizing and characterizing protein crystals. J. Appl. Crystallogr. 2016;49:944–952. doi: 10.1107/S1600576716006233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zander U., Hoffmann G., Cornaciu I., Marquette J.-P., Papp G., Landret C., Seroul G., Sinoir J., Röwer M., Felisaz F., et al. Automated harvesting and processing of protein crystals through laser photoablation. Acta Crystallogr. Sect. Struct. Biol. 2016;72:454–466. doi: 10.1107/S2059798316000954. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This workflow is based on the combination of the CrystalDirect technology and serial X-ray diffraction data analysis, and can be controlled over the web through the Crystallographic Information Management System (CRIMS)
Data Availability Statement
The article includes all datasets generated or analyzed during this study. Macromolecular coordinates for the structure displayed can be accessed through the wwPDB under accession codes are listed in the Key resources table.
All original code has been deposited as indicated in the key resources table at and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.