

Abstract
The protein folding problem was first articulated as question of how order arose from disorder in proteins: How did the various native structures of proteins arise from interatomic driving forces encoded within their amino acid sequences, and how did they fold so fast? These matters have now been largely resolved by theory and statistical mechanics combined with experiments. There are general principles. Chain randomness is overcome by solvation-based codes. And in the needle-in-a-haystack metaphor, native states are found efficiently because protein haystacks (conformational ensembles) are funnel-shaped. Order-disorder theory has now grown to encompass a large swath of protein physical science across biology.
Keywords: protein folding theory, statistical mechanics, disordered proteins, protein aggregation, coarse-grained modeling
The History of the Folding Problem
What is (or was) the protein folding problem1–4? And, why does it matter? First of all, among classes of molecules, proteins occupy a niche of special importance. Not only are they components of all living things, and not only do they matter for health and disease, but also their material powers are extraordinary: they are the working components of beings that walk, talk and think. The folding problem was viewed as a grand challenge of high order, natural catnip for scientists. It was a basic science problem that attracted great interest.
A central question in the folding problem arose from the first experimental determinations of globular protein structures around 1957 by Perutz, Kendrew, Levinthal, Anfinsen, Kauzmann, Baldwin, Tanford, Zimm, Creighton, Englander and others. The chain trace was not random. The native structure was unique, yet without apparent symmetry5–10 (Fig. 1). Proteins were neither disordered like polymers nor ordered like sodium chloride salts. What explained the shape of the chain trace in a protein structure? What forces, encoded within the sequence, drove these structures? What was the sequence-to-structure folding code? As knowledge grew, proteins increasingly appeared to be unlike ordinary materials. Unlike other polymers, proteins fold into unique structures. They have a sequence-to-structure code: different folds are encoded within different sequences. And different folds perform different actions.
Fig. 1.

The first view of a protein structure. Myoglobin at 6 Å resolution, published in 1958 by Kendrew and coworkers.6 Its basic chain trace (white) did not have the simple symmetries or regularities previously seen in crystals of smaller molecules. [With permission from the Medical Research Council Laboratory of Molecular Biology].
Second, what could explain how proteins fold so quickly2,4,11,3? Despite their large conformational spaces, proteins often fold in the microsecond or millisecond time range. What are the rates and routes (pathways) by which the astronomically large conformational spaces of a protein are searched so efficiently, by random processes, to find these uniquely structured native states? How could order arise from disorder so fast?
Third, it was clear there was not just one story to be told here, but a whole library-full. In addition to the most stable native structure of huge numbers of sequences,12 the many non-native structures would be crucial for understanding human health and disease. Proteins differ in a universe of shapes, of jobs that they do, how they do them, and their roles in organisms. There are huge numbers of stories to tell: for about 20,000 human proteins, 9 million different organisms, many different mutations, and shapes that depend on conditions, environments and binding partners.
The community sought a framework of logic for physical reasoning that would be applicable across all proteins. We needed to find universal principles, to progress from descriptions applicable to individual proteins or sets of conditions. We confronted questions about proteins as a general state of matter, so very different from other materials, including other polymeric materials. These fundamental aspects of the protein folding problem boiled down to questions about how order arises from disorder, often cooperatively, in sequence-patterned chain molecules.
How could theory and computation address these issues? In the early days, modeling aimed to test whether the inter-atomic potentials of the day could explain native structures.13–16 Over time, three roles emerged: (1) Database models. Using the growing database of protein structures (Protein DataBank, PDB), could we determine unknown structures using sequence homologies to known structures? (2) Computational molecular physics. As Molecular Dynamics (MD) with semi-empirical force fields improved, they joined with experiments to tell protein microstories, i.e. the details of motions, binding and other actions, roughly one protein at a time, one condition at a time. (3) Theory & coarse-grained simulations. Statistical mechanics and polymer theories were developed to tell macrostories, i.e. to address more global questions of principle, of commonalities and differences among proteins.
Think about these three steps in solving a crime mystery: First, you get static snapshots of the suspects. Second, you trace his/her steps and actions. Third, you aim to learn their motives. (1) Database modeling gives snapshots of native structure culprits. Database inference approaches have dominated the 14 Critical Assessment of protein Structure Prediction (CASP) events over 27 years of community-wide structure prediction competitions. These underwent a major advance this year with Google’s AlphaFold deep learning algorithm.17 (2) MD gives the step-by-step physical actions that we call the microstories. MD-based protein storytelling is reviewed elsewhere.12 (3) Theory seeks to learn the macrostories, the universal drives and principles. While all three approaches give insights into proteins, a grounding in physical truths is important for modern protein storytelling. In the present work, we summarize the role of theory that, in conjunction with experiments, has explained disorder-to-order processes in proteins.
Seeking Principles from Polymer Theories
Starting in the 1980’s, we and others looked to polymer statistical mechanics for insights into protein folding.18–23 The polymer perspective went against the grain at the time because protein science was generally seen by protein experimentalists to be about atomistic detail and structural specifics. In contrast, synthetic polymer properties were understood through their disorder – their chain length and conformational distributions, often modeled in random-flight and lattice solution theories, and by Flory length distributions.24
What questions could polymer physics address about how proteins found native states (needles in haystacks) among all possible conformations (the haystacks)? Could polymer physics describe the forces that drive stabilities of certain states (native) relative to others, and the speed of the search? The questions were about the nature of the seemingly astronomically large conformational spaces of non-native states of proteins, the equilibrium structures they attained, and the kinetics of searching those spaces and finding native states.
In the 19th century, research on proteins and polymers had tracked together. JJ Berzelius coined both terms, polymer in 1832 and protein in 1839. The idea that small molecules could form chains through covalent linkages was controversial until Hermann Staudinger’s macromolecule hypothesis of the late 1920’s (Nobel Prize in Chemistry in 1953) was proven true by parallel work on proteins – from both Theo Svedberg’s centrifugation experiments on hemoglobin in the 1920’s (Nobel Prize in Chemistry 1926) and Wallace Carothers’ syntheses in the 1930s of polymers such as neoprene and nylon. The protein and polymer fields then diverged for years as Flory and others showed that the properties of synthetic macromolecules were explainable by disorder – distributions of chain lengths and conformations, while Perutz and Kendrew and structural biologists in the 1950s and ‘60s showed that proteins had atomistically detailed structures, different for different proteins, and that those differences matter for function.
On the one hand, polymer theories at that time were mostly focused on the simplest polymers – homopolymers and random or block co-polymers. Modeling had not previously explored sequence-structure relationships because it had not been possible to make specific sequences of non-biological synthetic polymers. Nor was there work on polymers that had unique folded structures like the native states of proteins. On the other hand, polymer theory was a good starting point for several reasons.
First, astronomical conformational spaces are the daily business of polymer statistical mechanics. Once you take the logarithm of a count of conformations and turn it into an entropy, the numbers are all quite measurable and ordinary. Unlike thinking about small molecules, which is often more about energies than entropies, thinking about polymers means confronting large conformational spaces, disorder-to-order transitions (like protein folding), and how various simple interactions can direct fast searches of conformational spaces to reach ordered states.
Second, traditional polymer theory had provided many good simplifications that clearly added value to models. For example, chains were often approximated as a string of beads, each representing roughly a monomer unit. Bonds were approximated as fixed-length vectors. Bond pairs were represented with simple orientational freedom. On the one hand, that random-flight model and its variants was sufficient to model some size properties of denatured states. On the other hand, Flory-Huggins theory had successfully modeled more compact states in polymer liquids with averaged solvent-mediated contact interactions among non-neighboring beads.24,25 In these ways, polymers in complex situations could be reduced to smaller sub-problems, either of rotational freedom in short segments, or of contact interactions of the isolated beads, measurable in simple solvation transfer experiments. Combined with small-molecule model-compound experiments, you could get coarse-grained insights into conformational shapes, chain entropies and enthalpies.
In the early days of harnessing such ideas from polymer science, there was push-back from some quarters, where it was felt that the details should matter. However, important lessons from polymer science were (i) that entropies are often large, sometimes dominant (as in elastic solids, viscoelastic liquids, and in polymer blends, mixtures and melts); and (ii) that getting right the entropies and disorder is more about the general nature of whole conformational spaces, through the largescale sampling and lack of bias in coarse-grained models, even at the sacrifice of atomic detail.
What is the nature of disorder in denatured proteins?
Characterizing a disordered protein is less straightforward than describing a folded protein.26–28 A useful, comprehensible, and general description of conformational ensembles of large numbers of microscopic states is needed. Polymer theory has provided crucial concepts and tools for this. First, it defines certain ensemble averages, such as the radius of gyration (Rg) or end-to-end distance (Re), that are good descriptors of the sizes of conformational ensembles.
Second, polymer theories explain the huge importance the solvent plays in polymer conformations. A key organizing principle here is of the Flory theta solvent or Θ point. Imagine a fictitious dial with which you could control an energetic property of solvent molecules, ranging from preferentially interacting with other solvent molecules to preferentially interacting with monomers of a polymer chain. When monomer-solvent interactions are strongly favorable (a “good solvent”), a polymer chain will expand extensively. When monomer-solvent interactions are very unfavorable (“poor solvent”), the chain will collapse to avoid the solvent. When the dial is in the middle, that balance point is a theta solvent. At the theta solvent point, a polymer chain will obey ideal random-flight statistics. It will be neither more collapsed nor more expanded than a random-flight chain. Proteins generally fold in poor solvents (like pure water) and unfold in good solvents (denaturants). But unlike homopolymers, proteins can have more complex sequence-dependent behaviors in solvents. Water may act as good or poor solvent for unfolded states of proteins.29,30
Third, these ideas about solvation give ways to learn key features of protein conformational ensembles. The radius of gyration Rg of a chain will depend on its number of chain monomers N, as Rg ≈ Nv,33–38,29,39 where v, called the “Flory exponent” depends on the solvent. In a random-flight chain, v = 1/2, and for denatured proteins ingood solvents, v ≈ 0.6 (Fig. 2). Unfolded proteins in high denaturant are in Flory good solvents. Unfolded proteins in low denaturant have varying compactness, and depend on hydrophobicity and charge. These simple homopolymer theory concepts have been much more powerful than we had any right to expect for protein conformational distributions.31,32,40–42,39,43,44
Fig. 2.
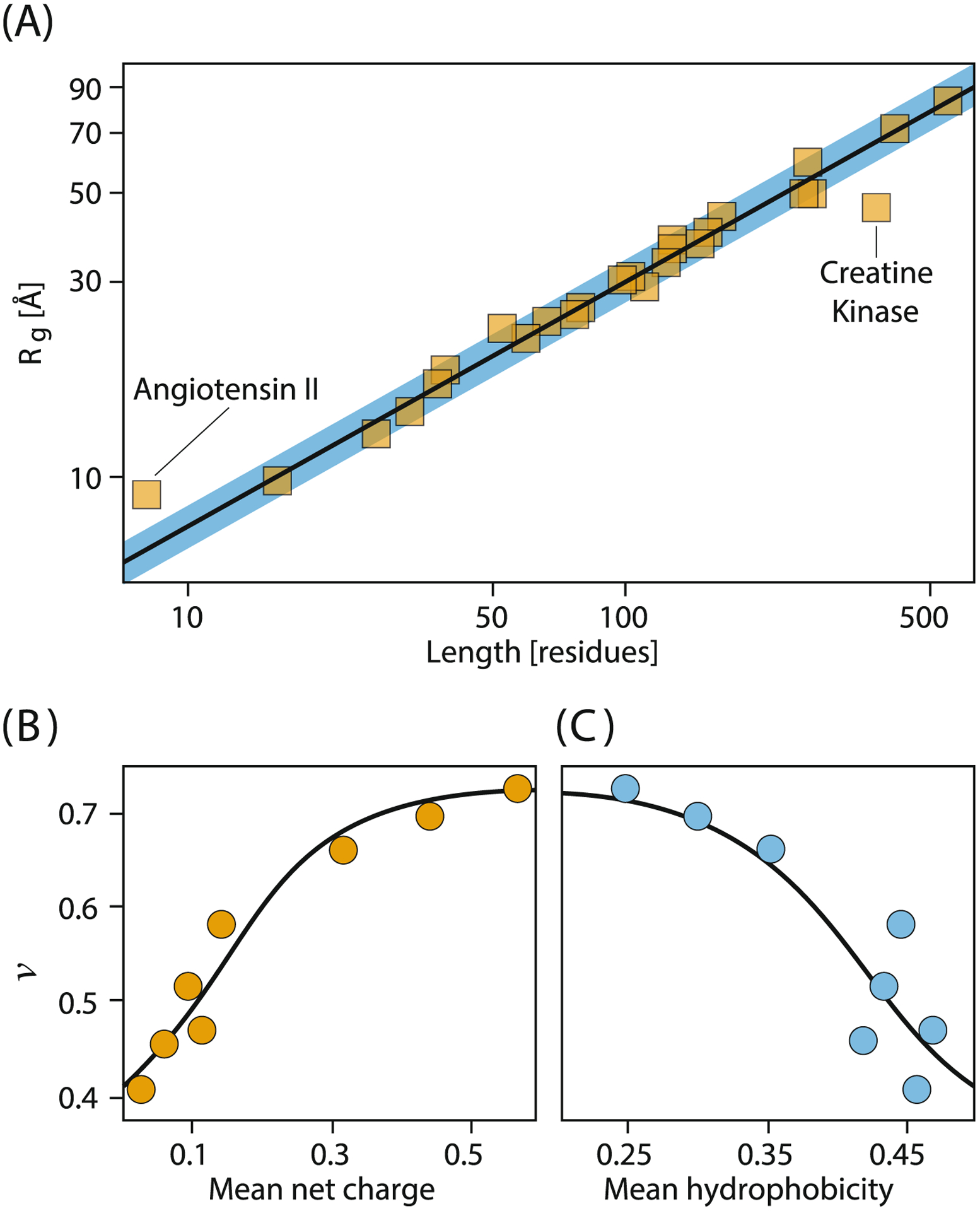
Unfolded-state radii depend on chain length, charge and hydrophobicity. (A) Unfolded proteins in concentrated denaturant are highly expanded like polymers in Flory good solvents (Rg ~ Nv, where N is number of monomers and v ≈ 0.6). (B,C) In low denaturant, the compactnesses of unfolded proteins depend on hydrophobicity and charge. Data from Refs. 31,32
Fourth, ensembles can sometimes seem chameleon-like, where different experiments give different weightings to ensemble sub-components. This is where theory and modeling can be useful, in calculating these different facets properly, resolving apparent differences from different experiments.41,45–47,44
How Does a Protein Reach its Ordered Folded State from its Unfolded Ensemble?
What is the nature of the ordering in a folded native protein, and how does that ordering arise from its highly disordered denatured state? The basic ideas are expressed through statistical mechanics. The relative stabilities of states depend on their free energies. At equilibrium, the probability of occupying a state depends on its Boltzmann’s weight, exp(−ΔG/kBT), where ΔG is the difference in free energies of the states, native and unfolded in this case, kB is Boltzmann’s constant and T is temperature.48,49 Small proteins typically fold cooperatively, i.e. through relatively sharp transitions between the disordered and ordered states.50,51
Disorder can be overcome by energetic preferences for certain states over others. For proteins, there was early focus on hydrogen bonding. In 1951, Linus Pauling deduced that hydrogen bonding would favor α-helical structures in peptides.56,57 The early evidence for α-helices in proteins raised the possibility that short model peptide chains might undergo helix-coil transitions in solution, in the absence of the other parts of the protein, and that such model systems might give insights into the bigger questions of protein folding. Consistent with that speculation, Perutz and Kendrew observed considerable amounts of α-helix in the first protein structures that were learned at atomic resolution, myoglobin in 1957 and hemoglobin in 1959 (Fig. 1 shows an early low-resolution structure).58,6 For this discovery, they were awarded the Nobel Prize in 1962.59
Helix-coil transition: order from disorder in peptides.
Among the first successes in modeling protein conformational changes were the helix-coil theories of Schellman,60 Zimm and Bragg,61 and Lifson and Roig62 (reviewed in Ref.51). How is a helical chain conformation stable inside a protein, in light of the large entropic favorability of the much larger number of the chain’s disordered conformational states (“coil”)? The answer, in the language of the Zimm-Bragg theory, was that the entropic challenge of so many open configurations could be balanced against factors for (i) the difficulty of forming the first turn of a helix (expressed as an equilibrium constant σ), and (ii) the energetic gain of hydrogen bonding of additional turns (expressed as an equilibrium constant s). These theories, in conjunction with an associated experimental enterprise,63,64 showed that although nucleation is entropically disfavored [10−4 < σ < 10−2], a small energetic preference for propagation [s slightly greater than 1] favors cooperative formation of helices, provided the chains are long enough, and σ is sufficiently small (Fig. 3). It was later shown by Baldwin and coworkers63,65 that short peptides (<20 amino acids) comprising certain amino acid sequences can display substantial helical character. Helix-coil propensities of peptides are summarized in database models.64
Fig. 3.
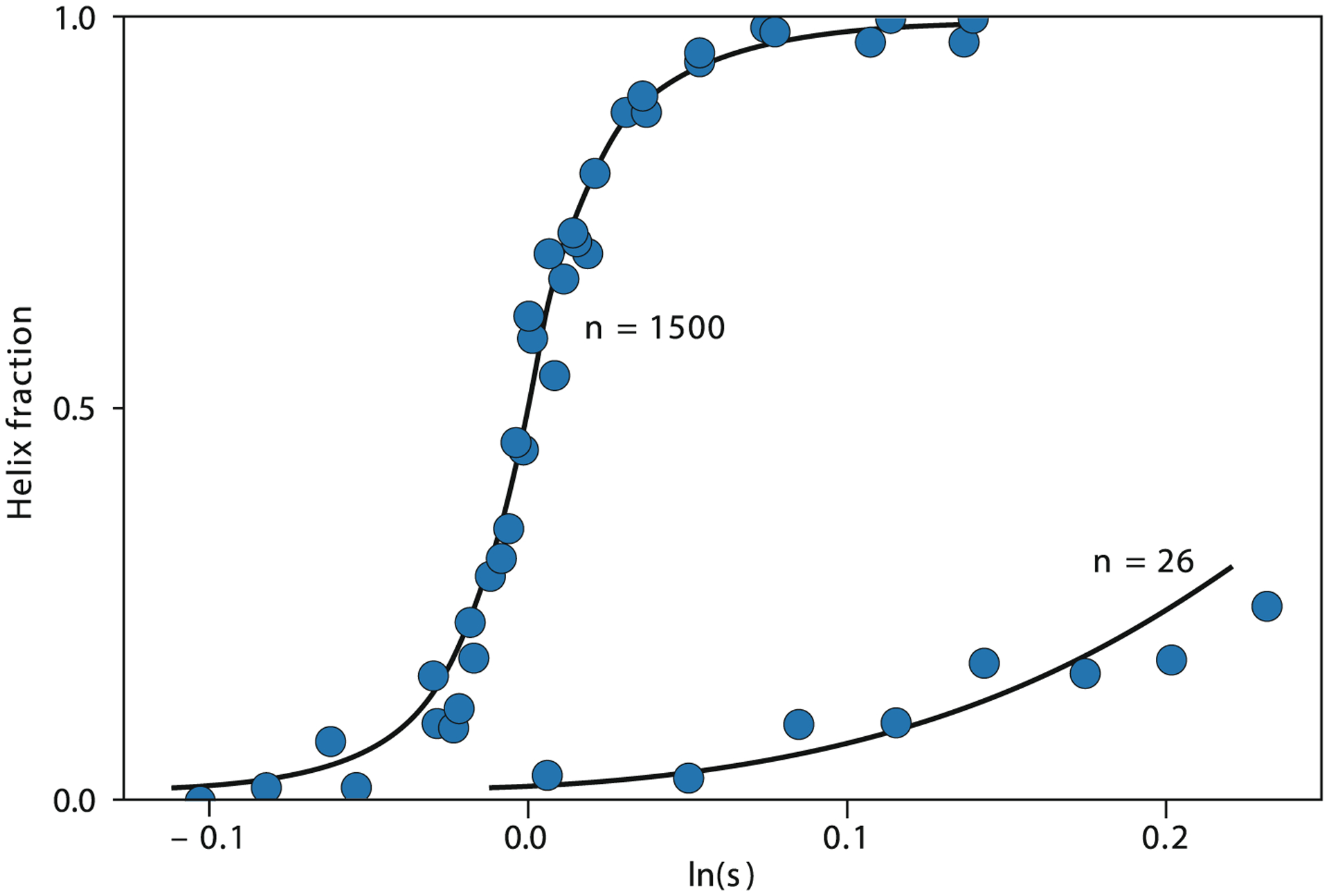
The helix-coil transition (Zimm and Bragg, 1959).61 The model was among the first explanations of disorder-order in helices, which are a key component of folded proteins. It’s not a full explanation for general folding, however, because β-sheets also fold cooperatively, and because these helix forces are weak (here, 1500-mer helices are required to show the same level of folding cooperativity as typical 100-mer proteins).
Thus, simple theories that elucidated the organizing principle of helical ordering shone light on hydrogen bonding as an important driver, and showed how to ground our understanding in experimental physical chemistry.
Protein folding: collapse, the folding code & cooperativity.
Like helix-coil processes, much of protein folding is also highly cooperative.50 However, protein folding was more challenging to understand than the coil-to-helix transition. Proteins are much longer chains, and their native structures are very well packed with various internal organizations of helices and coils, β-sheets and loops. There was found to be a folding code where different native structures are encoded by different sequences, but the physical basis of that code was not clear. Partly based on successes of helix-coil theory, a prominent view at the time was that the folding code that determined how one protein differed from another was predominantly due to hydrogen bonding interactions. Hydrophobic interactions were seen as a sort of nonspecific glue that just made proteins compact in some oily glob-like way.
Our group took an alternative view. We supposed that the folding code must reside within the sequence of side chains, not the backbone, because each protein has a different side-chain ordering, but all proteins have the same backbone. We assumed that hydrophobicity was the dominant interaction. We supposed that the folding code was mostly a solvation code in the hydrophobic interactions, and was not from hydrogen bonding. To explore the folding transition, polymer theory was recruited and included in an approximate analytical theory18 and in a so-called “exact” lattice model.21 We explored the ideas that the hydrophobic interactions embedded in the protein sequence determined why each protein folded to a different structure, and that the hydrogen bonding was just a generic “glue” that stabilized compact states. And, we supposed that the atomistic details of the tight van der Waals packing was more of a consequence than a driver.66,67
Proteins fold on funnel-shaped energy landscapes.
Lattice polymer models indicated how a binary code with two types of monomers, hydrophobic (H) and polar (P), could lead many different HP sequences to have different native folds. Even with only HH interactions, which are weak, nondiscriminatory and combinatoric – any one H can interact with any other H (except its nearest neighbors in the sequence) – the models predicted sharp cooperative transitions from the large unfolded ensemble to a single native state that maximizes the number of HH contacts (i.e., spatial neighbors on the lattice that are not neighbors in the sequence), depending on the HP sequence.68–70,21,71 These models demonstrated that the folding code was predominantly a solvation-based binary code of relatively nonspecific interactions, driving a chain to a native state encoded in its sequence of hydrophobic and polar monomers. Thus, a protein in a poor solvent (see above for definition), like water, collapses to a particular native fold due to its particular HP sequence. HP models also showed a funnel-like shape in the folding energy landscape, describing the transition from many states at the top having few HH contacts to very few conformations at the bottom having many HH contacts.72,73
The seeds of the folding-funnel concept came from insights of polymer theory about how chain conformational space reduces as chain density increases, as when proteins fold from random configurations to a compact folded state.18,74 In modeling polymer liquids, PJ Flory predicted a factor of (1/e)N as the reduction in numbers of conformations available to each chain having N beads, in bulk liquid polymer relative to its random flight state.75 The effect was called excluded volume. Put into folding conditions, we inferred that a protein chain forms increasing numbers of hydrophobic contacts, some native and some not. As the chain becomes increasingly compact, the space of remaining viable conformations diminishes sharply.
Further analytical modeling based on defining protein free energy as the summation of pairwise contact energies quantified the number of sequences expected to fold to a given native structure.76–79 Rather than studying how protein sequences fold into a given structure, this inverse computation provided insight into how certain protein structures can be more amenable to encoding by several different sequences.80,81
Experimental validation of the binary HP code hypothesis.
Proofs of principle followed. First, experiments tested the binary HP code hypothesis. Sauer et al. demonstrated that protein structures are highly tolerant to extensive mutations, provided that the core hydrophobic residues are preserved.82,83 And, Kamtekar et al. randomized the amino acid sequences of a 4-helix bundle protein, keeping only the hydrophobic and polar code intact and fixing given loops. They found that the large preponderance of these non-biological sequences still fold to the correct native structure.84 Similar results were recently observed by Koga et al. for designed proteins.85 Moreover, the thermodynamics of hydrophobic and polar interactions were found to give enthalpies, entropies and heat capacities of folding consistent with experiments.86–88 A key prediction was that a large excluded-volume entropy opposed folding. Subsequently, a useful minimal alphabet for folding for a broader range of native folds was found to be IKEAG – a hydrophobe, one positive and one negative amino acid, alanine and glycine.89,90 Recent refined models also incorporate helix-coil cooperativity into the folding collapse cooperativity.51,64
From the folding code to non-biological foldamers.
These notions about folding codes were useful in developing new polymeric materials called foldamers.91–93 The implication from modeling that the backbone was not critical in creating sequence-dependent foldable polymers led to explorations in non-biological types of materials, such as peptoids.94,95
How is protein folding so fast and so directed?
The protein folding problem has also been regarded as two questions of folding speeds and paths: (a) How is folding so fast, from unfolded to folded states? (b) Is there a folding mechanism, a particular route or pathway that is both specific enough to explain how a given amino-acid sequence reaches its native state, and general enough to say why most proteins have such routes?
(a). The Levinthal paradox of folding speeds.
At a meeting in 1969,96 Cyrus Levinthal noted that the number of atomistic level conformations of a protein would be huge, of the order of 10300, and that a random search of them would require essentially forever for the protein to find its native state; see also DB Wetlaufer97 and TE Creighton.98 The implications were that even though folding is stochastic, nevertheless it must also involve some physical basis for not exploring the vastness of entire conformational spaces. Folding funnels gave an explanation, both from equilibrium modeling of densities of states18,72,73 and kinetic models.74,99 When jumped to folding conditions, the chain randomly forms increasingly many HH contacts, lowering the free energy, at the same time reducing the remaining space of searchable conformations. Folding is not a random search; it is strongly pruned by the exponentially diminishing numbers of conformations available to search as the protein becomes increasingly compact. Even so, it raised the question of how to compute folding rates across proteins.
The Thirumalai glassy kinetics model presciently predicted folding rates. In 1995, Dave Thirumalai formulated the relationship between protein folding rates kf and protein chain length, N.100 His insight originated from the resemblance of protein ensemble behavior at low energies and entropies to those of glassy dynamics, where the transitions from the many compact near-native ensembles to the presumably unique native state are separated by a free energy barrier ΔG‡ having a Gaussian distribution, where the relevant property is the size of the average fluctuation (standard deviation) . Consequently, the Arrhenius folding rates are given by at a given temperature. This prediction long preceded its experimental validation (Fig. 4). Thirumalai and coworkers also predicted that folding occurs most efficiently when the chain is put into a temperature TΘ, close to the folding midpoint temperature Tf, at which the polypeptide chain behaves as an ideal polymer chain.100–103 This prediction was confirmed by Hofmann et al.,32 and later others,42,40 using single-molecule spectroscopy. Those groups measured the Flory scaling exponent v of unfolded proteins under varying conditions of denaturant. Under native conditions (low-denaturant), they found that denatured chains are near the theta state (v = 0.5), i.e. more compact than highly denatured chains are.104,105 The next puzzle, described below, was to understand how different folding pathways were physically encoded in different protein amino-acid sequences.
Fig. 4.
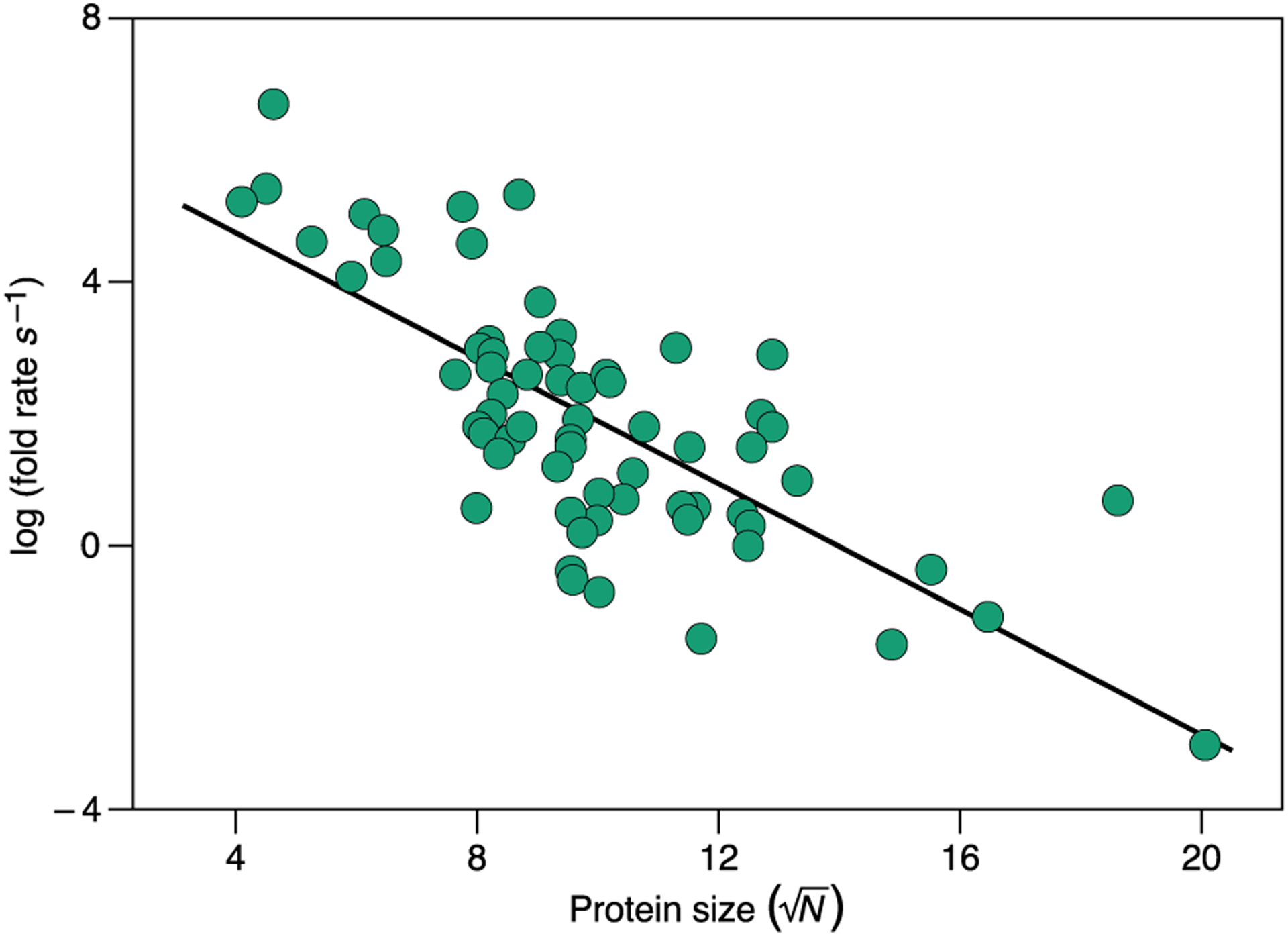
Experimental validation of the Thirumalai glassy folding kinetics model. Protein folding rates as a function of size. The folding rates were computed as the inverse of folding times from the data in Ref. 106 confirming the theoretically derived relation log kf ~ N1/2 over 10 orders of magnitude. The black line is a linear fit to the data.
(b). The folding mechanism: the conformational routes.
What is the mechanism of protein folding? The term “mechanism” has meant a narrative of a protein’s pathways of folding at a level that is sufficiently general and coarse-grained to apply to all proteins, and yet sufficiently granular to account for the differences across proteins. This would not be an atom-by-atom nanosecond-by-nanosecond sequence of events, for that would surely apply only to one protein. Nor is the funnel concept alone a sufficient descriptor of mechanism because it is too generic to tell us differences in folding routes of different proteins.
Early on, experimental kineticists had observed that a protein’s secondary structures would often form early in its folding pathway. These kinetic building block units were termed “foldons”.107–111 But, the physics was not clear. Secondary structures were not stable by themselves. How did the protein know where to start? How were the folding pathways encoded in the amino-acid sequence? How did the protein avoid searching the vast majority of its conformational space to follow its pathway? What was sought was a general theory that would be grounded, like helix-coil theory is, in known physical chemistry experimental model systems. Among the first efforts to embody the secondary-structure-first idea was diffusion-collision theory, where secondary structures formed early then diffused and collided ultimately into a native structure.52,53 A more microscopic physical treatment was the Ising-like model of Eaton and coworkers.54,55 Each residue could adopt one of two states, native or non-native, and the model formulated a partition sum over them, successfully describing generic folding kinetics with foldons for the simplest proteins. But, it remained to understand how residues knew native from non-native.
In the Foldon Funnel Model,112,51 the first kinetic steps of folding are those based on local conformational preferences in the chain, such as helices and turns, that populate fractionally and transiently, but not at sufficiently high levels to be stable by themselves. Consider a helix bundle protein. Next along the folding route, two local helical pieces then assemble together to persist as a pair over longer time scales. Those pair units then accrete additional helices to persist even longer, and so on. Each step is more persistent than the last, even though none is yet fully stable. The barrier to folding here comes from the substantial conformational entropy loss of forming the ordered helices, with the gain of native contacts not yet sufficient to offset it. Full stability is only achieved at the end when all of the helices are in their native structure. Such landscapes look like volcanoes: uphill at first, then the downhill funnel only for the last steps; see Fig. 5.
Fig. 5.
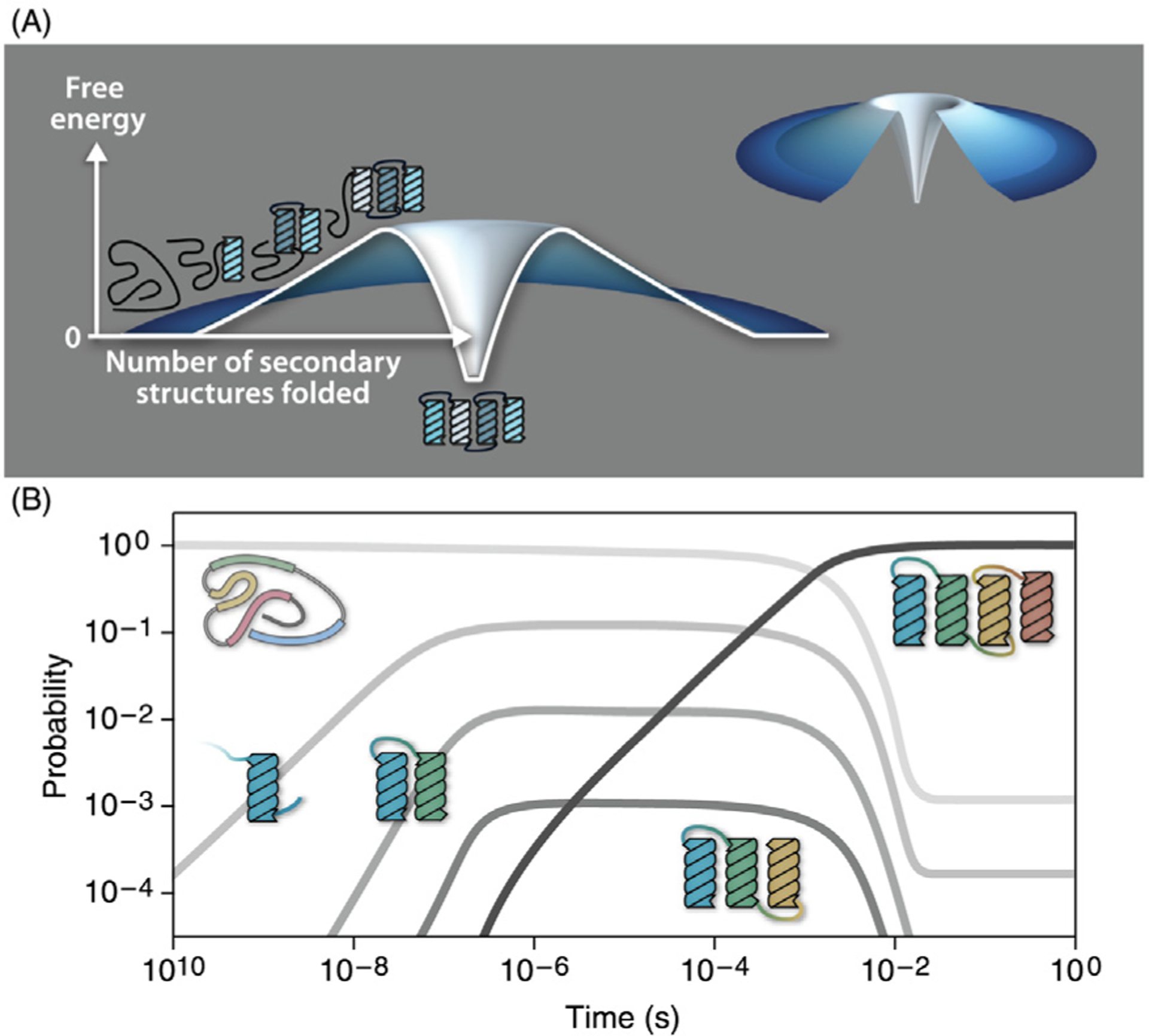
The Foldon Funnel Model describes the thermodynamics and kinetics of folding. (A) The energy landscape of folding is volcano-shaped for a 4-helix-bundle. As the protein progresses along the folding pathway it accumulates local secondary structures that are not stable by themselves (uphill landscape). Tertiary folds result from the cooperative associations of secondary structural elements that ultimately confer the protein its stable native fold at the last step (downhill landscape). (B) The Foldon Funnel Model describes state populations versus time. A single helix is meta-stable long enough to form helix dimers, then trimers, and so on. The black line shows the native state rising late and becoming fully populated with time as dimers and trimers progress on the folding landscape. Reproduced from Ref. 112
This model explains how “local-first, global later” results simply from the nature of the time scales of polymer molecules sampling their conformations.113 Two residues i and j close to each other in sequence have fewer degrees of freedom between them than two residues that are further apart. And sampling speed of smaller spaces is faster than of larger spaces. Of course, if wrong local structures, or competing native contacts, are formed early, then some backtracking is required later.114,115 However, exact lattice model studies show that there are almost always routes where local-first, global-later can achieve native structures without backtracking.116 Hue Sun Chan and coworkers used lattice models of a small protein with varying levels of cooperativity within and between the helices (i.e. local and nonlocal interactions) to reason about the ordering of folding events and shed light on observations from hydrogen exchange experiments.117,118
Key points of such modeling are: to give quantitative relationships between macro observables, such as folding rates over many different proteins, the general two-state thermodynamics across simple proteins, and the observation of folding by foldons; and to give micro properties that can be measured in physical chemical model experiments, such as helix-coil properties σ and s, and a hydrophobic interaction equilibrium constant among amino acids. A principal prediction from folding kinetics theories was that individual chains would fold stochastically – each following a different microscopic process, but with averaged behavior as described by this mechanism. This predicted folding route heterogeneity came to be called the “new view”,119 and has been confirmed by experiments120,121 and simulations.122,123
Chaperones help other proteins to fold by unfolding them.
Some proteins are able to fold into their native structure in isolation, while many proteins could use a little help to overcome the thermodynamic and kinetic barriers to folding.124–127 Proteins called chaperones can generically help other proteins reach their unique folded state.128–130 Although originally thought to function by a lock and key mechanism, chaperones are now known to help perform the complicated act of folding by instead performing the uncomplicated act of randomly unfolding proteins through iterative annealing.131–135 Many proteins, especially large ones, can get stuck in kinetic traps, i.e. misfolded states that are not able to proceed to the folded state. By ‘iteratively’ unfolding misfolded proteins powered by ATP hydrolysis, chaperones allow proteins with complex funnel landscapes to successfully traverse the landscape and ‘anneal’ to the native state.132
This mechanism of action elucidates how one chaperone is capable of assisting hundreds of different proteins in folding.124,136,129 Chaperone activity is important in the context of protein homeostasis in the cell. If chaperones do not properly operate, the number of copies of functional proteins can be too low for the cell to remain healthy.137 In addition, the number of misfolded proteins can overwhelm the proteostasis machinery, causing cell collapse.138
Intrinsic Disorder: leveraging disorder for function.
There has been growing interest in protein conformations that are unfolded or disordered.139,140 Some proteins are entirely or partially disordered. These intrinsically disordered proteins (IDPs) and intrinsically disordered regions (IDRs) themselves often have biological functions.141,142 Conformational heterogeneity is often found to play a critical role in biological processes such as regulation by post-translational modification, transient binding interactions, autoinhibition, and assembly into non-stoichiometric cellular assemblies.143–145,28,146 These proteins may function while fully disordered, or may sample ordered conformations that encode function.147,148
Averages, such as Rg and Re, over polymer conformational ensembles often usefully describe how IDP properties depend on concentrations of denaturants32,40,149,45 or temperature.150–152 Protein conformations are often well modeled by simple classical homopolymer theories.33 But sometimes more subtle insights are sought. To fill that need, more granular metrics can help differentiate the properties of different IDPs, their conformational ensembles, interactions, and functions. For instance, one can use the full ensemble-averaged distance matrix of all residue pairs, 〈Rij〉.153–155 This level of modeling gives deeper insights about distinct IDP conformations,43,156,157 and can give insight into functional relationships between IDPs where sequence conservation cannot be used as with folded proteins.157 Experimental validation of these metrics remains a forefront challenge.
Why do the sizes of IDPs matter? Intrinsic disorder can have biological relevance; it can be modulated through covalent modifications to amino acid side chains, or through post-translational modifications (PTMs).143,158 PTMs can shift conformational ensembles by altering their electrostatic charge compositions159 or their specific charge sequence patterns.160 The consequence is that ensemble radii or shape distributions can depend differently on external stimuli, such as salt concentration.159,161,162
For explaining sequence effects on IDP conformational properties, statistical mechanical modeling and coarse-grained simulations have been critical. Sequence patterning effects occur through the local clustering of like and unlike charges, expressed through Pappu’s κ parameter161 or on the pairwise distances between like and unlike charges in Ghosh’s sequence charge decoration (SCD) parameter.166 These parameters stem from earlier polymer physics theories.167–169 Both of these quantities correlate well with the average properties (i.e. Rg, Re, v) of IDPs.170 Theory involved in development of SCD has successfully predicted the size of an IDP and its phosphorylated variants (Fig. 6B,C). The variants were designed to minimize or maximize SCD and to observe the greatest degree of collapse or expansion in the IDP, demonstrating the role of theory in determining PTM hotspots that would be difficult to identify otherwise (Fig. 6). In addition to charge patterning in amino-acid sequences, hydrophobic and polar (HP) patterning can also shift ensembles of disordered protein chains.171,172 More advanced recent theory treats more granular patterns in which certain amino acids distribute as blocks or clusters within the sequences (Fig. 6A).165 Together with charge patterning metrics, one can predict experimentally-determined IDP dimensions with good accuracy (Fig. 6D,E). In short, improved theory and modeling of disordered protein states, which should help to understand biological control and regulation, is ever more accurate in capturing their compactness, exposure, and interactions with other biomolecules.173–175
Fig. 6.
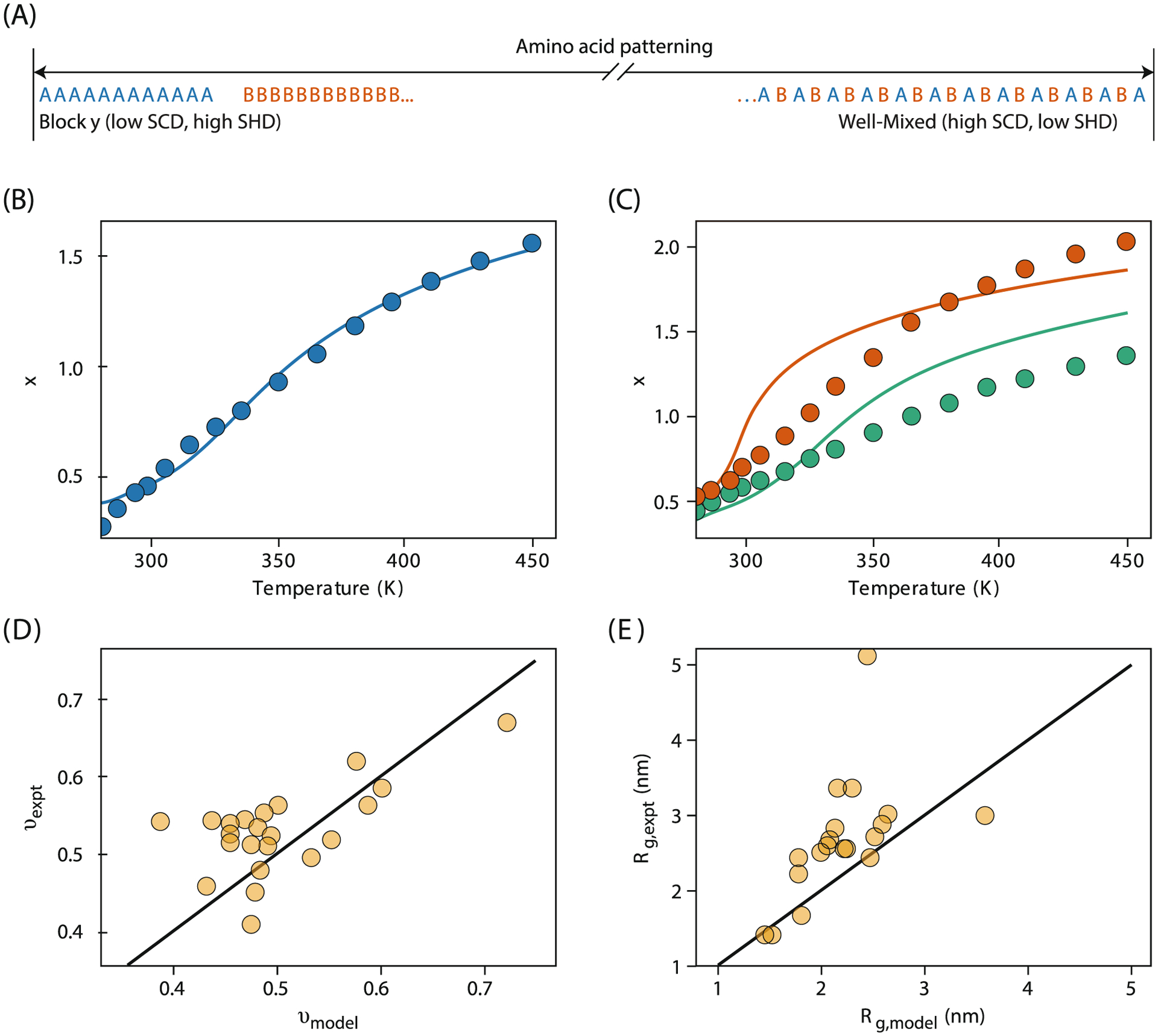
IDP conformations depend on sequence patterns, as explained by models and simulations. (A) Two sequence patterns: blocky (left) and strictly alternating (right). SCD measures the blockiness of anionic and cationic residues, while SHD measures the blockiness of amino acids of differing hydropathy values. (B,C) Analytical theory of SCD predicts IDP conformations (x) (where ) for (B) wild type and (C) its two phosphorylated variants. Theory (lines) agrees well with all-atom simulations (points) (Ref. 159). (D,E) Modeling predicts the effects of charge and hydropathy patterning on the scaling exponent and Rg from experiments32,40,45,41,152,163,164 (Ref. 165).
Order-disorder Processes Also Contribute to Multi-protein Associations
While folding is a single-chain disorder-to-order transition, agglomeration and complex formation are multi-chain disorder-to-order transitions. Agglomeration can occur in the formation of amyloid assemblies and fibrils in neurodegenerative diseases176,177 or in functional cellular processes178,179; in unwanted states of biotech formulations of proteins and antibody biologics180; in the assembly of membraneless organelles, which may be highly organized or completely disordered181,182; and in protein crystallization, a major step in determining native structures.183 These order-disorder transitions, which are predominantly along a protein concentration coordinate rather than an intramolecular folding coordinate, are appropriately treated using the statistical physics of solutions and polymers.
Modeling liquid phase equilibria dates back more than a century for simple liquids, and to Flory-Huggins theory for polymers in the 1940’s.34 But proteins posed new challenges in their heterogeneity of intra- and inter-molecular structures and in their sequence-structure relationships. How do different proteins associate into different morphologies with different properties? And, how can we explain the rates of association, including those that are extremely slow, such as the growth of amyloid in neurodegeneration that takes place over individual lifetimes? The cooperativity of protein aggregation is best studied by statistical mechanical theory. Atomistic molecular simulations are challenged to treat multiple proteins, or the big state changes with density from dilute to dense, or the need to compute free energies and populations in addition to the structures.
Proteins can assemble into native-like aggregates.
Proteins at sufficiently high concentrations in solution can agglomerate. Examples include formulations of antibodies as biological drugs, which can form reversible or irreversible aggregates184,185; liquid-like droplets formed through a liquid-liquid phase separation186,187; and small reversible clusters that give rise to very high solution viscosities in the absence of macroscopic aggregates.188,189 Collagens are another well-known example of proteins that self-associate and aggregate.190,191 They are the most abundant proteins in mammals, having major structural roles in bone, skin and muscle tissue. Crystallins are proteins kept at high concentrations in the lens of the eye, whose aggregation results in cataract disease.192–194 Such assemblies have translational ordering as a function of protein concentration – a sharp change to high local density, like the freezing of solids or the crystallization of salts in solution. The process is generic; many different proteins do it.
Simplifications are needed to model large systems of many associating proteins. Early modeling of native-state association treated proteins as spheres with radial like-charge repulsions and sticky van der Waals interactions distributed uniformly across their surfaces.195–201 In the past decade, more microscopically realistic structures and energetics have become possible due to advances in theory and computing power. Successful coarse-grained models have been developed based on population balance equations that quantitatively reproduce experimental measurements of aggregate concentrations increasing with time.202–206 A recent development is the adaptation of the Wertheim statistical mechanics theory of strongly associating liquids to describe liquid solutions of proteins such as antibodies, which have complex shapes.207–212 Here, the antibodies are represented as 7-bead molecular structures held together by strong sticking forces, which associate with each other in aqueous solution by weaker forces (Fig. 7).209,213 These models show how protein shapes and protein-protein interactions predict the formation of dimers, clusters and higher associations, reflected in solution properties of phase equilibria and viscosities.
Fig. 7.
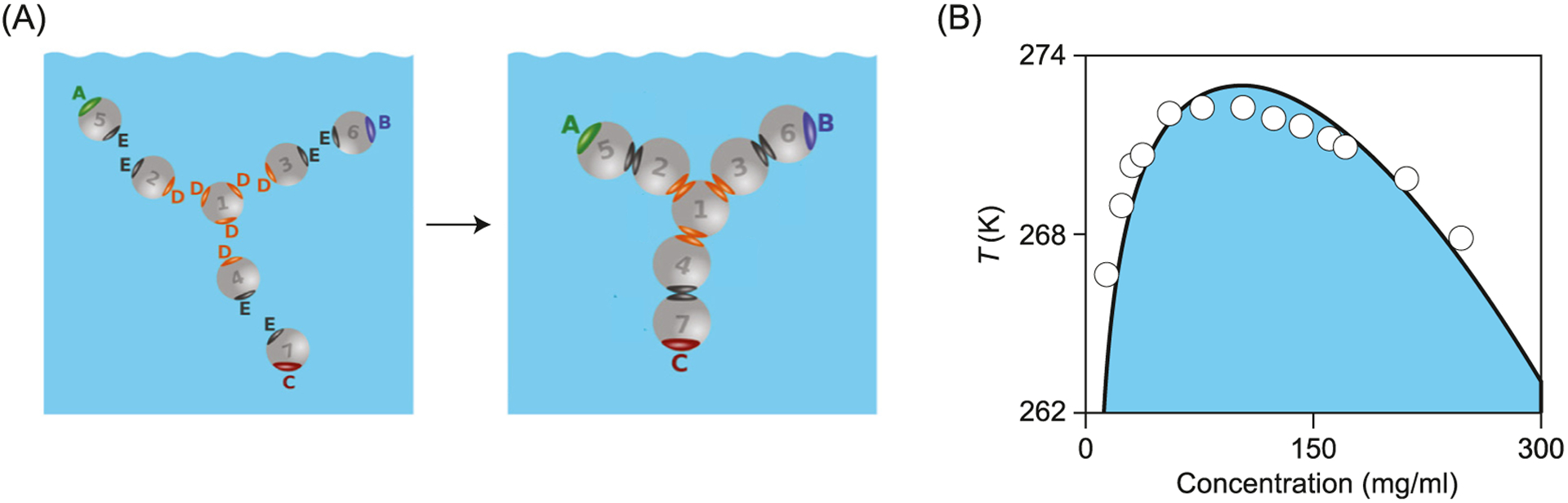
Antibody liquid phase equilibria computed from Wertheim liquid theory. (A) Isolated beads first stick together as 7-bead Y-shaped (covalent) molecules. These can then come together into (noncovalent) assemblies in solution. (B) Phase diagram of temperature T versus antibody concentration from experiments (points) and theory (line). Adapted from Ref. 213
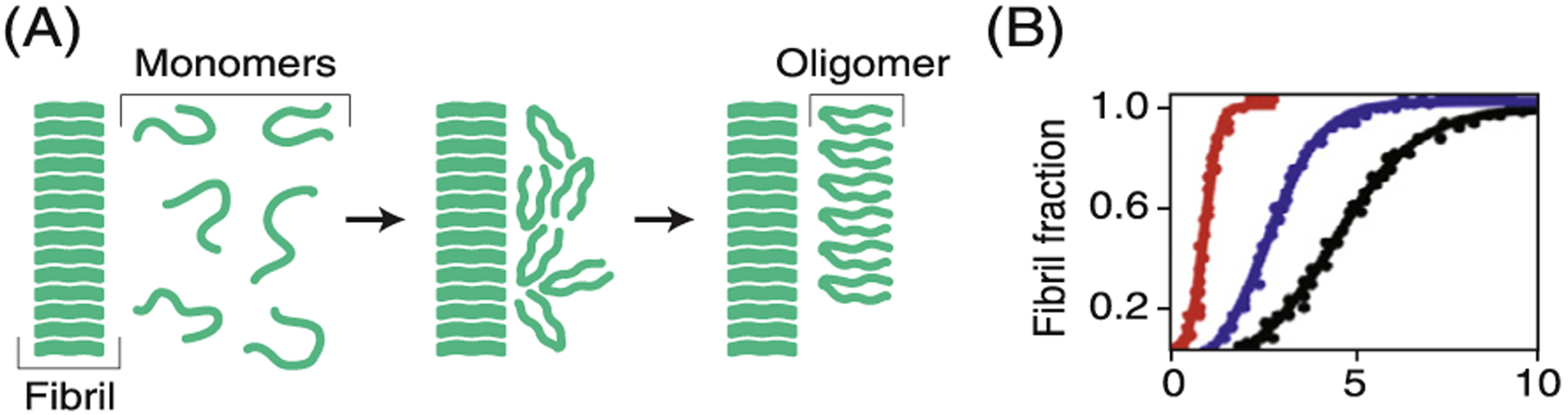
Proteins can assemble into amyloid oligomers and fibrils.
Remarkably, many different proteins are capable of forming fibrils. At high concentrations, peptides and proteins can form organized multi-molecular β-sheet fibril structures.176,177,214 They often have a second stable, less ordered oligomeric state. Theory describes the equilibrium states of the oligomers as micelle-like: relatively disordered assemblies of a few protein molecules, driven to form hydrophobic cores. The fibril state is more ordered, with a further loss of entropy of the aligned chains, but a further gain of stability from the hydrogen bonding and tight packing of the multiple aligned chains in the fibrils, bunched together like dry spaghetti in a box.215–218 Indeed, combined experimental and computational studies have explained the remarkable nanomechanical stabilities of amyloid fibrils in terms of hydrogen bonding densities and side chain packing efficiencies.219–223 Intriguingly, studies have shown that fibrils are more thermodynamically stable than the isolated native states, but kinetically separated from them on the energy landscape.224
Prions are misfolded aggregated states of proteins that can further promote the misfolding and recruitment of normal variants of the protein into the aggregate, hence, showing a protein-based infectious character, often leading to incurable pathologies such as the Creutzfeldt-Jacob disease in humans and mad cow disease in cattle.225 HP lattice modeling shows the existence of stable non-native states in small model prion proteins.226,227 Two or more proteins can achieve greater stability by non-native assembly in dimers or trimers. Often the assemblies have β sheet-like character, and the potential to incorporate more monomers into the assembly, resembling amyloid forms.
Theory has also been developed for the kinetics of fibril formation.228,229 Fibrils form on very slow time scales – tens of years in human amyloid diseases, much too slowly for modeling by molecular dynamics simulations. Schmit et al. modeled fibril formation as the zippering of a landing chain onto an existing partial fibril. This is a 1-dimensional random walk with sticking and unsticking steps representing hydrogen bonds (Fig. 8). Each added bond holds the landing chain in place longer than the last one did, as each bond adds stability to the zippered chain. Overall, fibril growth is slow because it is so rare that a landing chain will randomly align in the proper register. But, when it does, zipping is fast. Fig. 8 shows that the model captures well the growth rates of insulin fibers depending on temperature and denaturant.
Fig. 8.
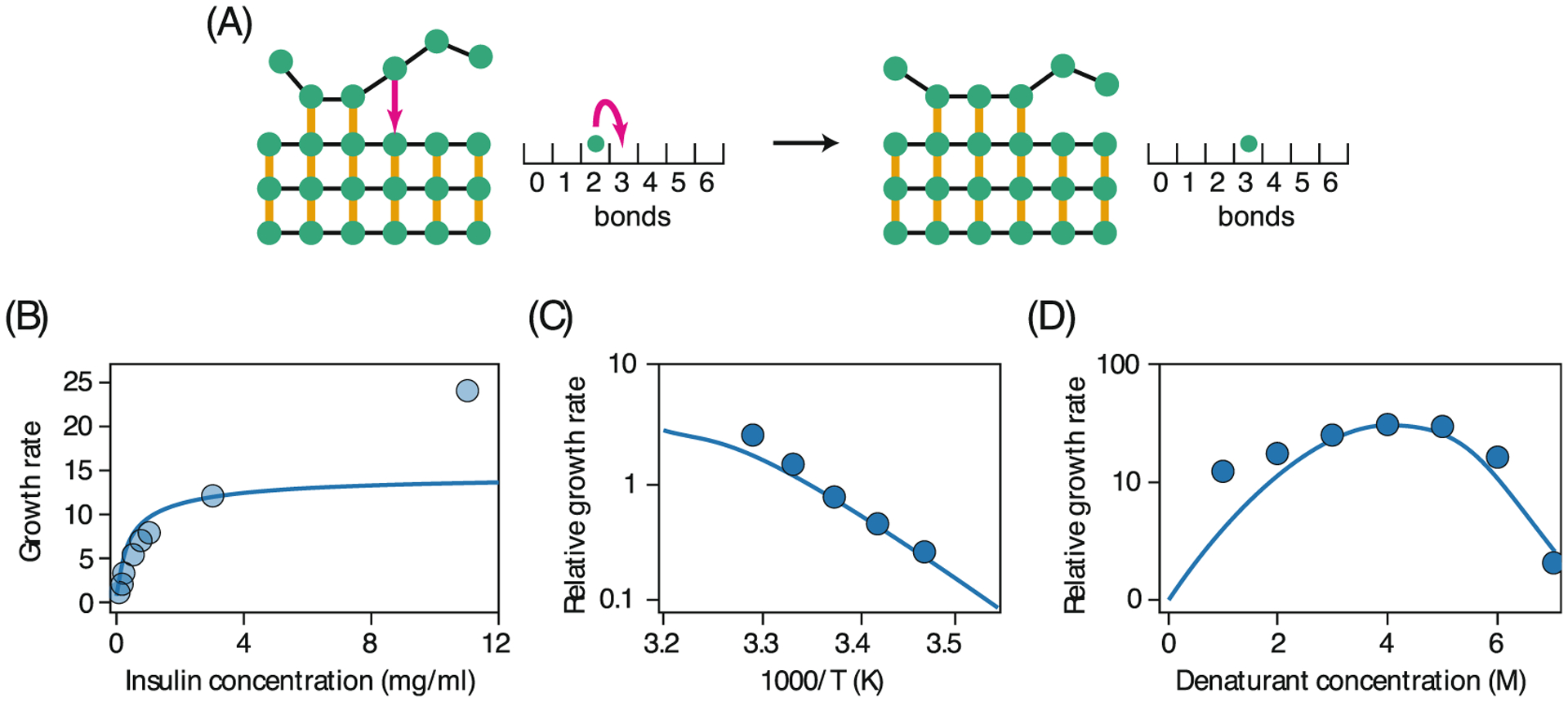
Kinetic theory for amyloid fibril formation. (A) The number of hydrogen bonds made at the templating fibril is a quantity that undergoes a random walk in the range [0,N] where N = 6 is number of monomers in the incoming peptide. The time for a chain to fully attach (or release) from the fibril is computed from the rates of forming (or breaking) all hydrogen bonds in this simplified scheme. The growth rate for insulin fibrils computed by this theory qualitatively matches that measured by experiments as a function of (B) peptide concentration, (C) temperature and (D) denaturant concentration (Ref. 228).
Rates of fibril formation have been modeled with mass-action kinetics by Knowles and Dobson. They capture the temperature effects by assuming the Arrhenius rate equation k = A exp(−ΔG‡/RT), with standard thermodynamic relations, ΔG‡ = ΔH‡ − TΔS‡ and ΔH‡/R = − (∂logk)/(∂1/T). Cohen and coworkers231,232 recently used integrated kinetic rate laws to quantitatively determine the size of activation energy barriers (ΔG‡) and the corresponding enthalpy (ΔH‡) and entropy (ΔS‡) contributions for describing experimental fibril growth curves for Aβ42, a peptide whose aggregation is implicated in Alzheimer’s disease.
The model of Knowles and Dobson233,234,230,232 that best agrees with experiments describes amyloid oligomer (nuclei) formation via three competing pathways: fibril fragmentation (breakage), primary nucleation through direct monomer associations, and secondary nucleation through fibril surface auto-catalysis (Fig. 9). These microscopic pathways have distinct kinetic and thermodynamic hallmarks, and quantitatively characterizing them revealed that the dominant pathway for Aβ42 oligomer formation is through the secondary nucleation mechanism.231 Saric et al.235 developed theory for this self-replication behavior of amyloid fibrils. They varied the monomer-monomer and monomer-fibril interaction energies, showing that fibril self-replication occurs only under very specific combinations of the interaction strengths, as observed in experiments. Nucleation can also be accelerated by adsorption to surfaces, such as lipid membranes.236 A fuller review is given in Ref. 237
Fig. 9.
The dominant mechanism of forming amyloid oligomers. (A) Free monomers attach to an existing fibril surface and rearrange into new oligomers with rate k2. This is known as secondary nucleation. (B) By solving the population balance equations under different scenarios, the likely mechanism dominating fibril nucleation, the secondary pathway shown here, can be inferred by fitting to experimental data at different monomer concentrations (colored lines). The kinetic data were obtained from Ref. 230.
Proteins undergo phase separations inside the cell.
Inside the cell, proteins (and other biomolecules) can become concentrated, driving a phase transition,238,181 like the crystallization of folded proteins183 or the phase equilibria in disordered protein polymers.239 These intracellular phase transitions have been linked to decision-making in cell division, signalling, stress responses, and other biological actions.238,181,240–242 They’ve also been linked to disease pathologies,243 including the formation of amyloids.244–246 Through phase separation, proteins and other biomolecules create compartments in the absence of lipid membranes, and are thus called membraneless organelles (MLOs) or more generally, biomolecular condensates.247 The first MLO to be observed was likely the nucleolus, first described in the 1830s by Wagner and by Valentin.248–250 Although the phase separation of proteins in vitro has long been known, it was only recognized in the formation of cellular inclusion bodies in 1995251 and demonstrated experimentally in 2009.238
The basic process of general nonspecific protein-protein association arises from a balance of attractions and repulsions. The attractions come from interchain “sticky” interactions. The repulsions come from steric interactions, from like charges in the sequences, and from the entropy losses of translational and rotational freedom when free chains join together. Polymer theories, such as the Flory-Huggins theory of homopolymer liquids and mixing,24,25 have proven useful for describing the phase separations that occur in biology.146,252,253,174,254,153 A key prediction of Flory-Huggins theory is that longer polymers will phase separate at lower concentrations. Similar length effects are also observed for proteins.181,252,255,256
A second key result from classical theories of homopolymers is a relationship between a single-chain property of collapse and a multi-chain property of dense liquid solutions. A classic polymer theory result is that a solvent that is dialed to be at the Θ point for the collapse of a single polymer chain will also cause multi-chain systems to be at their liquid-liquid phase equilibrium critical point.35,257,170 Remarkably, this basic principle of uncharged homopolymers is also true for many proteins, even though they are sequence-specific heteropolymers.258 This is found in computer simulations of HP-like chains255,259 (Fig. 10) and evidenced by experiments.260,241,152,261
Fig. 10.

A single-chain collapse property predicts a multi-chain phase equilibrium property of IDPs. (A) Collapse transition of an IDP (B) The critical temperature and Θ temperature of an IDP were determined in coarse-grained simulations, and show good agreement. (C) The agreement is consistent over a wide range of IDPs. Reproduced from Ref. 255 (D) Information from single chain simulations can be used to predict coexistence concentrations (+ symbols) and show agreement with concentrations obtained with coarse-grained phase-separation simulations (o symbols). Temperatures are in reduced units. Reproduced from Ref. 259.
Even more remarkably, this single-chain/multiple-chain correspondence principle often holds even for proteins having sequence-dependent charges,262,170 and multicomponent phase separation.254 Major advances in theory and modeling relating liquid protein phase equilibria to sequence dependent collapse and folding of individual chains derive from: the HPS theory of Jeetain Mittal’s lab;153,175 the stickers-and-spacers framework from Rohit Pappu’s lab263,264,152 originating from Rubin-stein’s associative polymer models;265 the analytical random phase approximation (RPA) theory of Hue Sun Chan’s lab;266 and others267–269 (reviewed in Refs. 270–272).
Protein self-assembly into virus capsids.
The self-assembled capsids of virus particles are beautiful examples of protein ordering and organization. A capsid, which is a protective shell around a virus, is a symmetrical construct of hundreds of protein subunits. Theory has made two major contributions to understanding how viral capsids form: (i) a general formalism for explaining the various capsid architectures of different viruses and (ii) thermodynamic and kinetic models that describe the process of capsid assembly.
Early geometrical analysis by Caspar and Klug led to mathematical principles of symmetry for the common icosahedral capsids. They showed how capsid symmetries arise from the symmetries of their pentameric and hexameric protein subunits.273 They described capsids as icosahedra whose size varies based on the number of hexamers that are placed in between the 12 pentamers occupying the vertices of the icosahedron (Fig. 11). The number of hexamers in the capsid is 10(T-1), where T is the triangulation number given by discrete integers T = 1,3,4,7, etc. The pioneering work of Caspar and Klug has since led to insights into design laws, protein subunit mosaics and layout deviations in capsid structures of various viruses.274–276 Only two types of protein-protein interaction – a rigid-body repulsion and an effective hydrophobic interaction – are needed to explain the stable states for these diverse icosahedral symmetries, and to give the design rules noted above. Such minimal statistical mechanical models also allow prediction of pH and salt effects on capsid formation.277
Fig. 11.
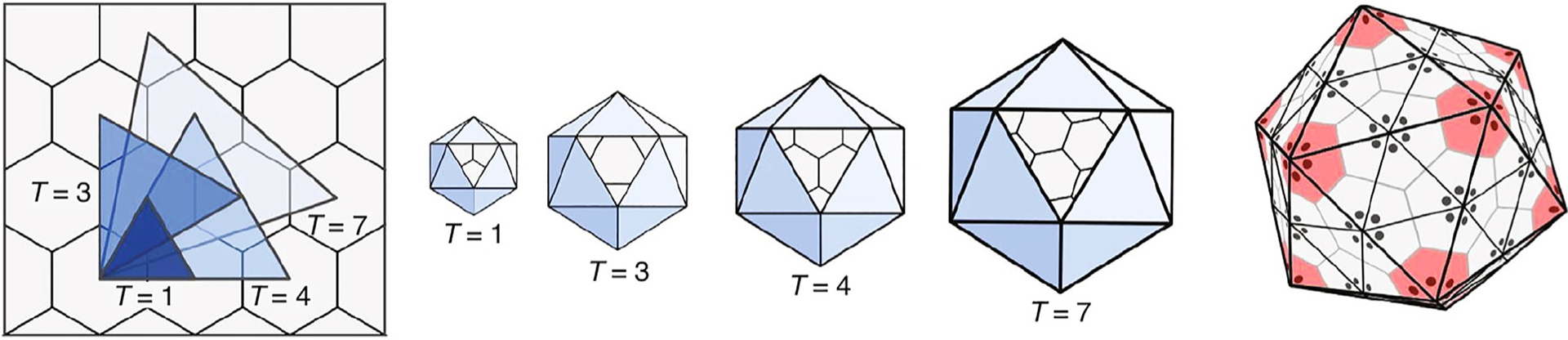
Viral capsids result from simple geometry rules. (Left) Different capsid sizes come from different geometric triangulations T, between the centers of polyhedra on a hexagonal lattice. (Middle) Stable capsids result by placing pentamers on the 12 icosahedral vertices and hexamers in between. (Right) The individual pentameric and hexameric protein complexes of the capsid are formed from the individual proteins shown as dot bundles at the center of each pentamer/hexamer. Reproduced from Ref. 276
What is the assembly process? The Zlotnick and Rudnick groups developed kinetic models for an assembly pathway from free monomers to partially formed intermediates to full capsids. They consider the growth of objects having n subunits through rates of addition and removal.278,279 They express the ratio of on and off rates through the intermediate state free energy ΔG as (k+/k−) ~ exp(−βΔG). They find that early stage oligomers are unstable and transient due to the small number of favorable contacts any protein subunit can make, and that full stability is achieved only at the last step of assembling the entire capsid. Substantial additional insights are described elsewhere.280,281 This unstable till the end strategy resembles that of the foldon assembly model of protein folding described above.
What are tomorrow’s protein order-disorder grand challenges? Here are just a few. (1) Proteome actions in cell behaviors. How do cells maintain proteostasis (homeostasis of proteome folding)?282,283 How is that balance lost in cells that are sick or old?138,284 (2) Understanding cotranslational folding: How do folding rates and mechanisms differ for proteins that emerge from ribosomes?285–288 (3) Rates of evolution. What protein properties determine rates of cell evolution?289,290 For some enzymes at certain timescales, evolution depends on the rates of product formation,291–295 which are relevant to the efficacies of antibiotics, for example.296,297 In other cases, cell evolution rates depend on folding stabilities, misfolding, aggregation and chaperoning.298–309 Insofar as living systems are seemingly endless hierarchies of organization and complexity, there is no shortage of protein disorder-to-order challenges to explicate.
Conclusions
Proteins constitute an extraordinary state of matter. Unlike other polymers, their power arises from their many physical and chemical actions encoded within their monomer sequences. The protein folding problem was a set of questions about disorder-to-order equilibria, rates, forces and the folding code that specify these unique compact non-symmetrical native structures. In the needle-in-a-haystack metaphor, these were questions not just about needles, but also importantly about their haystacks, the conformational ensembles in which they were embedded and how they were searched.
These problems have now largely been solved by statistical mechanics, polymer theory and coarse-grained computer simulations, in conjunction with experiments. In short, polymer chains having irregular sequences and a solvation code have funnel-shaped haystacks. While proteins have polymeric conformational disorder, as well as steric and charge repulsions, they also have attractions from hydrophobic interactions, solvation and unlike-charge attractions that can overcome the disorder. Across scales of size and time, these forces drive coils into helices, disordered chains into compact unique native states, peptides into amyloid and prion assemblies, proteins into solid and liquid agglomerates, chaperones to fold proteins by unfolding them, and viral capsids to self-assemble. Those few early questions of basic science have now mushroomed into an important component of protein physical chemistry.
Acknowledgments
We thank Hue Sun Chan, Kingshuk Ghosh, Jeetain Mittal, Rohit Pappu, Jeremy Schmit, Barbara Hribar-Lee, Miha Luksic and Dave Thirumalai for deeply insightful comments. We are grateful to Sarina Bromberg for help with the figures and for valuable comments on the manuscript. We are also grateful for the support from NIH, grant RM1GM135136, and the Laufer Center for Physical and Quantitative Biology at Stony Brook University.
Footnotes
Exact doesn’t mean exactly true or exactly right; it just refers to a model that leaves no discretion for the modeler – no free parameters, no choices about sampling, no approximations. It is perfectly faithful to some underlying prescription.
DECLARATION OF COMPETING INTEREST
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Dill Ken A., Banu Ozkan, S., Weikl Thomas R., Chodera John D., Voelz Vincent A., (2007). The protein folding problem: when will it be solved?. Curr. Opin. Struct. Biol, 17 (3), 342–346. [DOI] [PubMed] [Google Scholar]
- 2.Dill Ken A., Banu Ozkan S, Scott Shell M, Weikl Thomas R., (2008). The protein folding problem. Annual Review. Biophysics, 37, 289–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dill Ken A., MacCallum Justin L., (2012). The protein-folding problem, 50 years on. Science, 338 (6110), 1042–1046. [DOI] [PubMed] [Google Scholar]
- 4.Service Robert F., (2008). Problem solved*(* sort of). Science, 321 (5890), 784–786. [DOI] [PubMed] [Google Scholar]
- 5.Perutz MF, Rossmann MG, Cullis Ann F., Muirhead H, Will Georg, (1960). North ACT. Structure of haemoglobin. Nature, 185 (4711), 416–422. [DOI] [PubMed] [Google Scholar]
- 6.Kendrew John C., Bodo G, Dintzis Howard M., Parrish RG, Wyckoff Harold, Phillips David C., (1958). A three-dimensional model of the myoglobin molecule obtained by x-ray analysis. Nature, 181 (4610), 662–666. [DOI] [PubMed] [Google Scholar]
- 7.Levinthal Cyrus, (1968). Are there pathways for protein folding?. J. Chim. Phys, 65, 44–45. [Google Scholar]
- 8.Taniuchi Hiroshi, Anfinsen Christian B., (1969). An experimental approach to the study of the folding of staphylococcal nuclease. J. Biol. Chem, 244 (14), 3864–3875. [PubMed] [Google Scholar]
- 9.Kauzmann Walter, (1964). The three dimensional structures of proteins. Biophys. J, 4 (1), 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanford Charles, (1962). Contribution of hydrophobic interactions to the stability of the globular conformation of proteins. J. Am. Chem. Soc, 84 (22), 4240–4247. [Google Scholar]
- 11.Dill Ken A., (1990). Dominant forces in protein folding. Biochemistry, 29 (31), 7133–7155. [DOI] [PubMed] [Google Scholar]
- 12.Brini Emiliano, Simmerling Carlos, Dill Ken, (2020). Protein storytelling through physics. Science, 370 (6520) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuntz ID, (1972). Protein folding. J. Am. Chem. Soc, 94 (11), 4009–4012. [DOI] [PubMed] [Google Scholar]
- 14.Levitt Michael, Warshel Arieh, (1975). Computer simulation of protein folding. Nature, 253 (5494), 694–698. [DOI] [PubMed] [Google Scholar]
- 15.Kuntz ID, Crippen GM, Kollman PA, Kimelman D, (1976). Calculation of protein tertiary structure. J. Mol. Biol, 106 (4), 983–994. [DOI] [PubMed] [Google Scholar]
- 16.Andrew McCammon J, Gelin Bruce R., Karplus Martin, (1977). Dynamics of folded proteins. Nature, 267 (5612), 585–590. [DOI] [PubMed] [Google Scholar]
- 17.Senior Andrew W., Evans Richard, Jumper John, Kirkpatrick James, Sifre Laurent, Green Tim, Qin Chongli, Žídek Augustin, et al. , (2020). Improved protein structure prediction using potentials from deep learning. Nature, 577 (7792), 706–710. [DOI] [PubMed] [Google Scholar]
- 18.Dill Ken A., (1985). Theory for the folding and stability of globular proteins. Biochemistry, 24 (6), 1501–1509. [DOI] [PubMed] [Google Scholar]
- 19.Bryngelson Joseph D., Wolynes Peter G., (1987). Spin glasses and the statistical mechanics of protein folding. Proc. Natl. Acad. Sci. USA, 84 (21), 7524–7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dill Ken A., Alonso Darwin O.V., Hutchinson Karen, (1989). Thermal stabilities of globular proteins. Biochemistry, 28 (13), 5439–5449. [DOI] [PubMed] [Google Scholar]
- 21.Lau Kit Fun, Dill Ken A., (1989). A lattice statistical mechanics model of the conformational and sequence spaces of proteins. Macromolecules, 22 (10), 3986–3997. [Google Scholar]
- 22.Chan Hue Sun, Dill Ken A., (1990). The effects of internal constraints on the configurations of chain molecules. J. Chem. Phys, 92 (5), 3118–3135. [Google Scholar]
- 23.Shakhnovich Eugene I., Gutin Alexander M., (1990). Implications of thermodynamics of protein folding for evolution of primary sequences. Nature, 346 (6286), 773–775. [DOI] [PubMed] [Google Scholar]
- 24.Flory Paul J., (1942). Thermodynamics of high polymer solutions. J. Chem. Phys, 10 (1), 51–61. [Google Scholar]
- 25.Huggins Maurice L., (1942). Some properties of solutions of long-chain compounds. J. Phys. Chem, 46 (1), 151–158. [Google Scholar]
- 26.Jane Dyson H, Wright Peter E., (1998). Equilibrium NMR studies of unfolded and partially folded proteins. Nature Struct. Biol, 5 (7), 499–503. [DOI] [PubMed] [Google Scholar]
- 27.Mukhopadhyay Samrat, Krishnan Rajaraman, Lemke Edward A., Lindquist Susan, Deniz Ashok A., (2007). A natively unfolded yeast prion monomer adopts an ensemble of collapsed and rapidly fluctuating structures. Proc. Natl. Acad. Sci. USA, 104 (8), 2649–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Der Lee Robin, Buljan Marija, Lang Benjamin, Weatheritt Robert J., Daughdrill Gary W., Keith Dunker A, Fuxreiter Monika, Gough Julian, et al. , (2014). Classification of intrinsically disordered regions and proteins. Chem. Rev, 114 (13), 6589–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holehouse Alex S., Pappu Rohit V., (2018). Collapse transitions of proteins and the interplay among backbone, sidechain, and solvent interactions. Ann. Rev. Biophys, 47, 19–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clark Patricia L., Plaxco Kevin W., Sosnick Tobin R., (2020). Water as a good solvent for unfolded proteins: Folding and collapse are fundamentally different. J. Mol. Biol, 432 (9), 2882–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kohn Jonathan E., Millett Ian S., Jacob Jaby, Zagrovic Bojan, Dillon Thomas M., Cingel Nikolina, Dothager Robin S., Seifert Soenke, et al. , (2004). Random-coil behavior and the dimensions of chemically unfolded proteins. Proc. Natl. Acad. Sci. USA, 101 (34), 12491–12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hofmann Hagen, Soranno Andrea, Borgia Alessandro, Gast Klaus, Nettels Daniel, Schuler Benjamin, (2012). Polymer scaling laws of unfolded and intrinsically disordered proteins quantified with single-molecule spectroscopy. Proc. Natl. Acad. Sci. USA, 109 (40), 16155–16160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flory Paul J., (1949). The configuration of real polymer chains. J. Chem. Phys, 17 (3), 303–310. [Google Scholar]
- 34.Flory Paul J., (1953). Principles of polymer chemistry. Cornell University Press. [Google Scholar]
- 35.De Gennes Pierre-Gilles, (1979). Scaling concepts in polymer physics. Cornell University Press. [Google Scholar]
- 36.Sanchez Isaac C, (1979). Phase transition behavior of the isolated polymer chain. Macromolecules, 12 (5), 980–988. [Google Scholar]
- 37.Chan Hue Sun, Dill Ken A., (1991). Polymer principles in protein structure and stability. Ann. Rev. Biophys. Biophys. Chem, 20 (1), 447–490. [DOI] [PubMed] [Google Scholar]
- 38.Rubinstein Michael, Colby Ralph H., et al. , (2003). Polymer Physics,, vol. 23 Oxford University Press, New York. [Google Scholar]
- 39.Thirumalai Dave, Samanta Himadri S., Maity Hiranmay, Reddy Govardhan, (2019). Universal nature of collapsibility in the context of protein folding and evolution. Trends Biochem. Sci, 44 (8), 675–687. [DOI] [PubMed] [Google Scholar]
- 40.Borgia Alessandro, Zheng Wenwei, Buholzer Karin, Borgia Madeleine B., Schüler Anja, Hofmann Hagen, Soranno Andrea, Nettels Daniel, et al. , (2016). Consistent view of polypeptide chain expansion in chemical denaturants from multiple experimental methods. J. Am. Chem. Soc, 138 (36), 11714–11726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riback Joshua A., Bowman Micayla A., Zmyslowski Adam M., Knoverek Catherine R., Jumper John M., Hinshaw James R., Kaye Emily B., Freed Karl F., et al. , (2017). Innovative scattering analysis shows that hydrophobic disordered proteins are expanded in water. Science, 358 (6360), 238–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peran Ivan, Holehouse Alex S., Carrico Isaac S., Pappu Rohit V., Bilsel Osman, Raleigh Daniel P., (2019). Unfolded states under folding conditions accommodate sequence-specific conformational preferences with random coil-like dimensions. Proc. Natl. Acad. Sci. USA, 116 (25), 12301–12310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baul Upayan, Chakraborty Debayan, Mugnai Mauro L., Straub John E., Thirumalai Dave, (2019). Sequence effects on size, shape, and structural heterogeneity in intrinsically disordered proteins. J. Phys. Chem. B, 123 (16), 3462–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Best Robert B., (2020). Emerging consensus on the collapse of unfolded and intrinsically disordered proteins in water. Curr. Opin. Struct. Biol, 60, 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fuertes Gustavo, Banterle Niccolò, Ruff Kiersten M., Chowdhury Aritra, Mercadante Davide, Koehler Christine, Kachala Michael, Girona Gemma Estrada, et al. , (2017). Decoupling of size and shape fluctuations in heteropolymeric sequences reconciles discrepancies in SAXS vs. FRET measurements. Proc. Natl. Acad. Sci. USA, 114 (31), E6342–E6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng Wenwei, Zerze Gül H., Borgia Alessandro, Mittal Jeetain, Schuler Benjamin, Best Robert B., (2018). Inferring properties of disordered chains from FRET transfer efficiencies. J. Chem. Phys, 148 (12), 123329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zheng Wenwei, Best Robert B., (2018). An extended Guinier analysis for intrinsically disordered proteins. J. Mol. Biol, 430 (16), 2540–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dill Ken, Bromberg Sarina, (2012). Molecular driving forces: statistical thermodynamics in biology, chemistry, physics, and nanoscience. Garland Science. [Google Scholar]
- 49.Chandler D, (1987). Introduction to modern statistical mechanics. Oxford University Press. [Google Scholar]
- 50.Privalov PL, Khechinashvili NN, (1974). A thermodynamic approach to the problem of stabilization of globular protein structure: a calorimetric study. J. Mol. Biol, 86 (3), 665–684. [DOI] [PubMed] [Google Scholar]
- 51.Bahar Ivet, Jernigan Robert L., Dill Ken A., (2017). Protein actions: principles and modeling. Garland Science. [Google Scholar]
- 52.Karplus Martin, Weaver David L., (1976). Protein-folding dynamics. Nature, 260, 404–406. [DOI] [PubMed] [Google Scholar]
- 53.Karplus Martin, Weaver David L., (1994). Protein folding dynamics: The diffusion-collision model and experimental data. Protein Sci, 3 (4), 650–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Muñoz Victor, Eaton William A., (1999). A simple model for calculating the kinetics of protein folding from three-dimensional structures. Proc. Natl. Acad. Sci. USA, 96 (20), 11311–11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Henry Eric R., Best Robert B., Eaton William A., (2013). Comparing a simple theoretical model for protein folding with all-atom molecular dynamics simulations. Proc. Natl. Acad. Sci. USA, 110 (44), 17880–17885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pauling Linus, Corey Robert B., Branson Herman R., (1951). The structure of proteins: two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. USA, 37 (4), 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pauling Linus, Corey Robert B., (1951). Atomic coordinates and structure factors for two helical configurations of polypeptide chains. Proc. Natl. Acad. Sci. USA, 37 (5), 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kendrew JC, Perutz MF, (1957). X-ray studies of compounds of biological interest. Annu. Rev. Biochem, 26 (1), 327–372. [DOI] [PubMed] [Google Scholar]
- 59.Kendrew John C., (1964). Myoglobin and the structure of proteins. Nobel Lectures. Chemistry, 1942–1962, 676–698. [Google Scholar]
- 60.Schellman John A., (1958). The factors affecting the stability of hydrogen-bonded polypeptide structures in solution. J. Phys. Chem, 62 (12), 1485–1494. [Google Scholar]
- 61.Zimm Bruno H., Bragg JK, (1959). Theory of the phase transition between helix and random coil in polypeptide chains. J. Chem. Phys, 31 (2), 526–535. [Google Scholar]
- 62.Lifson Shneior, Roig A, (1961). On the theory of helix–coil transition in polypeptides. J. Chem. Phys, 34 (6), 1963–1974. [Google Scholar]
- 63.Martin Scholtz J, Baldwin Robert L., (1992). The mechanism of alpha-helix formation by peptides. Annu. Rev. Biophys. Biomol. Struct, 21 (1), 95–118. [DOI] [PubMed] [Google Scholar]
- 64.Munoz Victor, Serrano Luis, (1997). Development of the multiple sequence approximation within the AGADIR model of α-helix formation: Comparison with Zimm-Bragg and Lifson-Roig formalisms. Biopolym. Original Res. Biomol, 41 (5), 495–509. [DOI] [PubMed] [Google Scholar]
- 65.Marqusee Susan, Robbins Virginia H., Baldwin Robert L., (1989). Unusually stable helix formation in short alanine-based peptides. Proc. Natl. Acad. Sci. USA, 86 (14), 5286–5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bromberg Sarina, Dill Ken A., (1994). Side-chain entropy and packing in proteins. Protein Sci, 3 (7), 997–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liang Jie, Dill Ken A., (2001). Are proteins well-packed? Biophys. J, 81 (2), 751–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stigter Dirk, Alonso DO, Dill Ken A., (1991). Protein stability: electrostatics and compact denatured states. Proc. Natl. Acad. Sci. USA, 88 (10), 4176–4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dill Ken A., Stigter Dirk, (1995). Modeling protein stability as heteropolymer collapse. Adv. Protein Chem, 46, 59–104. [DOI] [PubMed] [Google Scholar]
- 70.Dill Ken A., Bromberg Sarina, Yue Kaizhi, Fiebig Klaus M., Yee David P., Thomas Paul D., Chan Hue Sun, (1995). Principles of protein folding–a perspective from simple exact models. Protein Sci, 4 (4), 561–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Camacho Carlos J., Thirumalai D, (1993). Minimum energy compact structures of random sequences of heteropolymers. Phys. Rev. Letters, 71 (15), 2505. [DOI] [PubMed] [Google Scholar]
- 72.Dill Ken A., (1987). The stabilities of globular proteins. Protein Eng,, 187–192. [Google Scholar]
- 73.Dill Ken A., Chan Hue Sun, (1997). From Levinthal to pathways to funnels. Nature Struct. Biol, 4 (1), 10–19. [DOI] [PubMed] [Google Scholar]
- 74.Leopold Peter E., Montal Mauricio, Onuchic José N., (1992). Protein folding funnels: a kinetic approach to the sequence-structure relationship. Proc. Natl. Acad. Sci. USA, 89 (18), 8721–8725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Flory Paul J., (1941). Thermodynamics of high polymer solutions. J. Chem. Phys, 9 (8), 660. [Google Scholar]
- 76.Wolynes Peter G., (1996). Symmetry and the energy landscapes of biomolecules. Proc. Natl. Acad. Sci. USA, 93, 14249–14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.England Jeremy L., Shakhnovich Eugene I., (2003). Structural determinant of protein designability. Phys. Rev. Letters, 90 (21), 218101. [DOI] [PubMed] [Google Scholar]
- 78.Choi Jeong-Mo, Gilson Amy I., Shakhnovich Eugene I., (2017). Graph’s topology and free energy of a spin model on the graph. Phys. Rev. Letters, 118 (088302), 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bialek William, (2012). Sequence Ensembles. Biophysics: Searching for Principals,. Princeton University Press, pp. 262–263 (Chapter 5). [Google Scholar]
- 80.Li Hao, Helling Robert, Tang Chao, Wingreen Ned, (1996). Emergence of preferred structures in a simple model of protein folding. Science, 273 (5275), 666–669. [DOI] [PubMed] [Google Scholar]
- 81.England Jeremy L., Shakhnovich Boris E., Shakhnovich Eugene I., (2003). Natural selection of more designable folds: a mechanism for thermophilic adaptation. Proc. Natl. Acad. Sci. USA, 100 (15), 8727–8731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lim Wendell A., Sauer Robert T., (1989). Alternative packing arrangements in the hydrophobic core of λ repressor. Nature, 339 (6219), 31–36. [DOI] [PubMed] [Google Scholar]
- 83.Bowie James U., Reidhaar-Olson John F., Lim Wendell A., Sauer Robert T., (1990). Deciphering the message in protein sequences: tolerance to amino acid substitutions. Science, 247 (4948), 1306–1310. [DOI] [PubMed] [Google Scholar]
- 84.Kamtekar Satwik, Schiffer Jarad M., Xiong Huayu, Babik Jennifer M., Hecht Michael H., (1993). Protein design by binary patterning of polar and nonpolar amino acids. Science, 262 (5140), 1680–1685. [DOI] [PubMed] [Google Scholar]
- 85.Koga Rie, Yamamoto Mami, Kosugi Takahiro, Kobayashi Naohiro, Sugiki Toshihiko, Fujiwara Toshimichi, Koga Nobuyasu, (2020). Robust folding of a de novo designed ideal protein even with most of the core mutated to valine. Proc. Natl. Acad. Sci. USA, 117 (49), 31149–31156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Robertson Andrew D., Murphy Kenneth P., (1997). Protein structure and the energetics of protein stability. Chem. Rev, 97 (5), 1251–1268. [DOI] [PubMed] [Google Scholar]
- 87.Ghosh Kingshuk, Dill Ken A., (2009). Computing protein stabilities from their chain lengths. Proc. Natl. Acad. Sci. USA, 106 (26), 10649–10654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ghosh Kingshuk, Dill Ken, (2010). Cellular proteomes have broad distributions of protein stability. Biophys. J, 99 (12), 3996–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chan Hue Sun, (1999). Folding alphabets. Nature Struct. Biol, 6 (11), 994–996. [DOI] [PubMed] [Google Scholar]
- 90.Riddle David S., Santiago Jed V., Bray-Hall Susan T., Doshi Nikunj, Grantcharova Viara P., Yi Qian, Baker David, (1997). Functional rapidly folding proteins from simplified amino acid sequences. Nature Struct. Biol, 4 (10), 805–809. [DOI] [PubMed] [Google Scholar]
- 91.Gellman Samuel H., (1998). Foldamers: a manifesto. Acc. Chem. Res, 31 (4), 173–180. [Google Scholar]
- 92.Kirshenbaum Kent, Zuckermann Ronald N., Dill Ken A., (1999). Designing polymers that mimic biomolecules. Curr. Opin. Struct. Biol, 9 (4), 530–535. [DOI] [PubMed] [Google Scholar]
- 93.Guseva Elizaveta, Zuckermann Ronald N., Dill Ken A., (2017). Foldamer hypothesis for the growth and sequence differentiation of prebiotic polymers. Proc. Natl. Acad. Sci. USA, 114 (36), E7460–E7468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee Byoung-Chul, Zuckermann Ronald N., Dill Ken A., (2005). Folding a nonbiological polymer into a compact multihelical structure. J. Am. Chem. Soc, 127 (31), 10999–11009. [DOI] [PubMed] [Google Scholar]
- 95.Sun Jing, Zuckermann Ronald N., (2013). Peptoid polymers: a highly designable bioinspired material. ACS Nano, 7 (6), 4715–4732. [DOI] [PubMed] [Google Scholar]
- 96.Levinthal Cyrus, (1969). How to fold graciously. Mossbauer Spectrosc. Biol. Syst, 67, 22–24. [Google Scholar]
- 97.Wetlaufer Donald B., (1973). Nucleation, rapid folding, and globular intrachain regions in proteins. Proc. Natl. Acad. Sci. USA, 70 (3), 697–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Creighton Thomas E., (1979). Experimental studies of protein folding and unfolding. Prog. Biophys. Mol. Biol, 33, 231–297. [DOI] [PubMed] [Google Scholar]
- 99.Wolynes Peter G., Onuchic Jose N., Thirumalai D, (1995). Navigating the folding routes. Science, 267 (5204), 1619–1620. [DOI] [PubMed] [Google Scholar]
- 100.Thirumalai D, (1995). From minimal models to real proteins: time scales for protein folding kinetics. J. Phys. I, 5 (11), 1457–1467. [Google Scholar]
- 101.Thirumalai D, O’Brien Edward P., Morrison Greg, Hyeon Changbong, (2010). Theoretical perspectives on protein folding. Ann. Rev. Biophys, 39, 159–183. [DOI] [PubMed] [Google Scholar]
- 102.Li Mai Suan, Klimov Dmitri K., Thirumalai D, (2004). Finite size effects on thermal denaturation of globular proteins. Phys. Rev. Letters, 93 (26), 268107. [DOI] [PubMed] [Google Scholar]
- 103.Kremer K, Baumgartner A, Binder K, (1982). Collapse transition and crossover scaling for self-avoiding walks on the diamond lattice. J. Phys. A: Math. Gen, 15 (9), 2879. [Google Scholar]
- 104.Klimov DK, Thirumalai D, (1996). Criterion that determines the foldability of proteins. Phys. Rev. Letters, 76 (21), 4070. [DOI] [PubMed] [Google Scholar]
- 105.Klimov DK, Thirumalai D, (1996). Factors governing the foldability of proteins. Proteins: Struct., Funct., Bioinf, 26 (4), 411–441. [DOI] [PubMed] [Google Scholar]
- 106.Naganathan Athi N., Muñoz Victor, (2005). Scaling of folding times with protein size. J. Am. Chem. Soc, 127 (2), 480–481. [DOI] [PubMed] [Google Scholar]
- 107.Kim Peter S., Baldwin Robert L., (1982). Specific intermediates in the folding reactions of small proteins and the mechanism of protein folding. Annu. Rev. Biochem, 51 (1), 459–489. [DOI] [PubMed] [Google Scholar]
- 108.Kim Peter S., Baldwin Robert L., (1990). Intermediates in the folding reactions of small proteins. Annu. Rev. Biochem, 59 (1), 631–660. [DOI] [PubMed] [Google Scholar]
- 109.Englander S. Walter, (2000). Protein folding intermediates and pathways studied by hydrogen exchange. Annu. Rev. Biophys. Biomol. Struct, 29 (1), 213–238. [DOI] [PubMed] [Google Scholar]
- 110.Maity Haripada, Maity Mita, Krishna Mallela M.G., Mayne Leland, Walter Englander S, (2005). Protein folding: the stepwise assembly of foldon units. Proc. Natl. Acad. Sci. USA, 102 (13), 4741–4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Walter Englander S, Mayne Leland, (2014). The nature of protein folding pathways. Proc. Natl. Acad. Sci. USA, 111 (45), 15873–15880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rollins Geoffrey C., Dill Ken A., (2014). General mechanism of two-state protein folding kinetics. J. Am. Chem. Soc, 136 (32), 11420–11427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Weikl Thomas R., Dill Ken A., (2003). Folding rates and low-entropy-loss routes of two-state proteins. J. Mol. Biol, 329 (3), 585–598. [DOI] [PubMed] [Google Scholar]
- 114.Capraro Dominique T., Roy Melinda, Onuchic José N., Jennings Patricia A., (2008). Backtracking on the folding landscape of the β-trefoil protein interleukin-1β? Proc. Natl. Acad. Sci. USA, 105 (39), 14844–14848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chavez Leslie L., Gosavi Shachi, Jennings Patricia A., Onuchic José N., (2006). Multiple routes lead to the native state in the energy landscape of the β-trefoil family. Proc. Natl. Acad. Sci. USA, 103 (27), 10254–10258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Voelz Vincent A., Dill Ken A., (2007). Exploring zipping and assembly as a protein folding principle. Proteins: Struct., Funct., Bioinf, 66 (4), 877–888. [DOI] [PubMed] [Google Scholar]
- 117.Kaya Hüseyin, Chan Hue Sun, (2005). Explicit-chain model of native-state hydrogen exchange: Implications for event ordering and cooperativity in protein folding. Proteins: Struct. Funct. Bioinformatics, 58 (1), 31–44. [DOI] [PubMed] [Google Scholar]
- 118.Chan Hue Sun, Zhang Zhuqing, Wallin Stefan, Liu Zhirong, (2011). Cooperativity, local-nonlocal coupling, and nonnative interactions: principles of protein folding from coarse-grained models. Ann. Rev. Phys. Chem, 62 [DOI] [PubMed] [Google Scholar]
- 119.Baldwin Robert L., (1995). The nature of protein folding pathways: the classical versus the new view. J. Biomol. NMR, 5 (2), 103–109. [DOI] [PubMed] [Google Scholar]
- 120.Cecconi Ciro, Shank Elizabeth A., Bustamante Carlos, Marqusee Susan, (2005). Direct observation of the three-state folding of a single protein molecule. Science, 309 (5743), 2057–2060. [DOI] [PubMed] [Google Scholar]
- 121.Bhatia Sandhya, Krishnamoorthy Guruswamy, Udgaonkar Jayant B., (2021). Mapping distinct sequences of structure formation differentiating multiple folding pathways of a small protein. J. Am. Chem. Soc, 143 (3), 1447–1457. [DOI] [PubMed] [Google Scholar]
- 122.Noé Frank, Schütte Christof, Vanden-Eijnden Eric, Reich Lothar, Weikl Thomas R., (2009). Constructing the equilibrium ensemble of folding pathways from short off-equilibrium simulations. Proc. Natl. Acad. Sci. USA, 106 (45), 19011–19016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Voelz Vincent A., Bowman Gregory R., Beauchamp Kyle, Pande Vijay S., (2010). Molecular simulation of ab initio protein folding for a millisecond folder NTL9(1–39). J. Am. Chem. Soc, 132 (5), 1526–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kerner Michael J., Naylor Dean J., Ishihama Yasushi, Maier Tobias, Chang Hung-Chun, Stines Anna P., Georgopoulos Costa, Frishman Dmitrij, et al. , (2005). Proteome-wide analysis of chaperonin-dependent protein folding in Escherichia coli. Cell, 122 (2), 209–220. [DOI] [PubMed] [Google Scholar]
- 125.Gong Yunchen, Kakihara Yoshito, Krogan Nevan, Greenblatt Jack, Emili Andrew, Zhang Zhaolei, Houry Walid A., (2009). An atlas of chaperone–protein interactions in Saccharomyces cerevisiae: implications to protein folding pathways in the cell. Mol. Syst. Biol, 5 (1), 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Calloni Giulia, Chen Taotao, Schermann Sonya M., Chang Hung-chun, Genevaux Pierre, Agostini Federico, Tartaglia Gian Gaetano, Hayer-Hartl Manajit, et al. , (2012). DnaK functions as a central hub in the E. coli chaperone network. Cell Rep, 1 (3), 251–264. [DOI] [PubMed] [Google Scholar]
- 127.Sala Ambre J., Bott Laura C., Morimoto Richard I., (2017). Shaping proteostasis at the cellular, tissue, and organismal level. J. Cell Biol, 216 (5), 1231–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Young Jason C., Agashe Vishwas R., Siegers Katja, Ulrich Hartl F, (2004). Pathways of chaperone-mediated protein folding in the cytosol. Nature Rev. Mol. Cell Biol, 5 (10), 781–791. [DOI] [PubMed] [Google Scholar]
- 129.Ulrich Hartl F, Bracher Andreas, Hayer-Hartl Manajit, (2011). Molecular chaperones in protein folding and proteostasis. Nature, 475 (7356), 324–332. [DOI] [PubMed] [Google Scholar]
- 130.Kim Yujin E., Hipp Mark S., Bracher Andreas, Hayer-Hartl Manajit, Ulrich Hartl F, (2013). Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem, 82, 323–355. [DOI] [PubMed] [Google Scholar]
- 131.Chan Hue Sun, Dill Ken A., (1996). A simple model of chaperonin-mediated protein folding. Proteins: Struct., Funct., Bioinf, 24 (3), 345–351. [DOI] [PubMed] [Google Scholar]
- 132.Todd Matthew J., Lorimer George H., Thirumalai D, (1996). Chaperonin-facilitated protein folding: optimization of rate and yield by an iterative annealing mechanism. Proc. Natl. Acad. Sci. USA, 93 (9), 4030–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Thirumalai Devarajan, Lorimer George H., (2001). Chaperonin-mediated protein folding. Annu. Rev. Biophys. Biomol. Struct, 30, 245–269. [DOI] [PubMed] [Google Scholar]
- 134.Thirumalai D, Lorimer George H., Hyeon Changbong, (2020). Iterative annealing mechanism explains the functions of the GroEL and RNA chaperones. Protein Sci, 29 (2), 360–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chakrabarti Shaon, Hyeon Changbong, Ye Xiang, Lorimer George H., Thirumalai D, (2017). Molecular chaperones maximize the native state yield on biological times by driving substrates out of equilibrium. Proc. Natl. Acad. Sci. USA, 114 (51), E10919–E10927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Taipale Mikko, Jarosz Daniel F., Lindquist Susan, (2010). HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nature Rev. Mol. Cell Biol, 11 (7), 515–528. [DOI] [PubMed] [Google Scholar]
- 137.Bershtein Shimon, Mu Wanmeng, Serohijos Adrian W. R., Zhou Jingwen, Shakhnovich Eugene I., (2013). Protein quality control acts on folding intermediates to shape the effects of mutations on organismal fitness. Mol. Cell, 49 (1), 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Santra Mantu, Dill Ken A., Graff De, Adam MR, (2019). Proteostasis collapse is a driver of cell aging and death. Proc. Natl. Acad. Sci. USA, 116 (44), 22173–22178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Keith Dunker A, David Lawson J, Brown Celeste J., Williams Ryan M., Romero Pedro, Oh Jeong S., Oldfield Christopher J., Campen Andrew M., et al. , (2001). Intrinsically disordered protein. J. Mol. Graph. Modell, 19 (1), 26–59. [DOI] [PubMed] [Google Scholar]
- 140.Keith Dunker A, Brown Celeste J., David Lawson, J., Iakoucheva Lilia M., Obradović Zoran, (2002). Intrinsic disorder and protein function. Biochemistry, 41 (21), 6573–6582. [DOI] [PubMed] [Google Scholar]
- 141.Uversky Vladimir N., Oldfield Christopher J., Keith Dunker A, (2005). Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signalling. J. Mol. Recog. Interdiscipl. J, 18 (5), 343–384. [DOI] [PubMed] [Google Scholar]
- 142.Tompa Peter, Fersht Alan, (2009). Structure and function of intrinsically disordered proteins. CRC Press. [Google Scholar]
- 143.Galea Charles A., Wang Yuefeng, Sivakolundu Sivashankar G., Kriwacki Richard W., (2008). Regulation of cell division by intrinsically unstructured proteins: intrinsic flexibility, modularity, and signaling conduits. Biochemistry, 47 (29), 7598–7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Trudeau Travis, Nassar Roy, Cumberworth Alexander, Wong Eric T.C., Woollard Geoffrey, Gsponer Jörg, (2013). Structure and intrinsic disorder in protein autoinhibition. Structure, 21 (3), 332–341. [DOI] [PubMed] [Google Scholar]
- 145.Cumberworth Alexander, Guillaume Lamour M, Babu Madan, Gsponer Jörg, (2013). Promiscuity as a functional trait: intrinsically disordered regions as central players of interactomes. Biochem. J, 454 (3), 361–369. [DOI] [PubMed] [Google Scholar]
- 146.Brangwynne Clifford P., Tompa Peter, Pappu Rohit V., (2015). Polymer physics of intracellular phase transitions. Nature Phys, 11 (11), 899–904. [Google Scholar]
- 147.Mohan Amrita, Oldfield Christopher J., Radivojac Predrag, Vacic Vladimir, Cortese Marc S., Keith Dunker A, Uversky Vladimir N., (2006). Analysis of molecular recognition features (MoRFs). J. Mol. Biol, 362 (5), 1043–1059. [DOI] [PubMed] [Google Scholar]
- 148.Borgia Alessandro, Borgia Madeleine B., Bugge Katrine, Kissling Vera M., Heidarsson Pétur O., Fernandes Catarina B., Sottini Andrea, Soranno Andrea, et al. , (2018). Extreme disorder in an ultrahigh-affinity protein complex. Nature, 555 (7694), 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zheng Wenwei, Borgia Alessandro, Buholzer Karin, Grishaev Alexander, Schuler Benjamin, Best Robert B., (2016). Probing the action of chemical denaturant on an intrinsically disordered protein by simulation and experiment. J. Am. Chem. Soc, 138 (36), 11702–11713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wuttke René, Hofmann Hagen, Nettels Daniel, Borgia Madeleine B., Mittal Jeetain, Best Robert B., Schuler Benjamin, (2014). Temperature-dependent solvation modulates the dimensions of disordered proteins. Proc. Natl. Acad. Sci. USA, 111 (14), 5213–5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zerze Gül H., Best Robert B., Mittal Jeetain, (2015). Sequence-and temperature-dependent properties of unfolded and disordered proteins from atomistic simulations. J. Phys. Chem. B, 119 (46), 14622–14630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Martin Erik W., Holehouse Alex S., Peran Ivan, Farag Mina, Jeremias Incicco J, Bremer Anne, Grace Christy R., Soranno Andrea, et al. , (2020). Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science, 367 (6478), 694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Dignon Gregory L., Zheng Wenwei, Kim Young C., Best Robert B., Mittal Jeetain, (2018). Sequence determinants of protein phase behavior from a coarse-grained model. PLoS Comput. Biol, 14 (1), e1005941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Huihui Jonathan, Ghosh Kingshuk, (2020). An analytical theory to describe sequence-specific inter-residue distance profiles for polyampholytes and intrinsically disordered proteins. J. Chem. Phys, 152 (16), 161102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gomes Gregory-Neal W., Krzeminski Mickaël, Namini Ashley, Martin Erik W., Mittag Tanja, Head-Gordon Teresa, Forman-Kay Julie D., Gradinaru Claudiu C., (2020). Conformational ensembles of an intrinsically disordered protein consistent with NMR, SAXS, and single-molecule FRET. J. Am. Chem. Soc, 142 (37), 15697–15710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Samanta Himadri S., Chakraborty Debayan, Thirumalai D, (2018). Charge fluctuation effects on the shape of flexible polyampholytes with applications to intrinsically disordered proteins. J. Chem. Phys, 149 (16), 163323. [DOI] [PubMed] [Google Scholar]
- 157.Huihui J, Ghosh K, (2021). Intra-chain interaction topology can identify functionally similar intrinsically disordered proteins. Biophys. J,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bah Alaji, Vernon Robert M., Siddiqui Zeba, Krzeminski Mickaël, Muhandiram Ranjith, Zhao Charlie, Sonenberg Nahum, Kay Lewis E., et al. , (2015). Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature, 519 (7541), 106–109. [DOI] [PubMed] [Google Scholar]
- 159.Firman Taylor, Ghosh Kingshuk, (2018). Sequence charge decoration dictates coil-globule transition in intrinsically disordered proteins. J. Chem. Phys, 148 (12), 123305. [DOI] [PubMed] [Google Scholar]
- 160.Perdikari Theodora Myrto, Jovic Nina, Dignon Gregory L., Kim Young C., Fawzi Nicolas L., Mittal Jeetain, (2021). A coarse-grained model for position-specific effects of post-translational modifications on disordered protein phase separation. Biophys. J,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Das Rahul K., Pappu Rohit V., (2013). Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. USA, 110 (33), 13392–13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Huihui Jonathan, Firman Taylor, Ghosh Kingshuk, (2018). Modulating charge patterning and ionic strength as a strategy to induce conformational changes in intrinsically disordered proteins. J. Chem. Phys, 149 (8), 085101. [DOI] [PubMed] [Google Scholar]
- 163.Rieloff Ellen, Skepö Marie, (2020). Phosphorylation of a disordered peptide–structural effects and force field inconsistencies. J. Chem. Theory Comput, 16 (3), 1924–1935. [DOI] [PubMed] [Google Scholar]
- 164.Cragnell Carolina, Durand Dominique, Cabane Bernard, Skepö Marie, (2016). Coarse-grained modeling of the intrinsically disordered protein Histatin 5 in solution: Monte Carlo simulations in combination with SAXS. Proteins: Struct., Funct., Bioinf, 84 (6), 777–791. [DOI] [PubMed] [Google Scholar]
- 165.Zheng Wenwei, Dignon Gregory, Brown Matthew, Kim Young C., Mittal Jeetain, (2020). Hydropathy patterning complements charge patterning to describe conformational preferences of disordered proteins. J. Phys. Chem. Letters, 11 (9), 3408–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Sawle Lucas, Ghosh Kingshuk, (2015). A theoretical method to compute sequence dependent configurational properties in charged polymers and proteins. J. Chem. Phys, 143 (8), 08B615_1. [DOI] [PubMed] [Google Scholar]
- 167.Dobrynin Andrey V., Rubinstein Michael, (1995). Flory theory of a polyampholyte chain. J. Phys. II, 5 (5), 677–695. [Google Scholar]
- 168.Edwards Sam F., Singh Pooran, (1979). Size of a polymer molecule in solution. Part 1. – Excluded volume problem. J. Chem. Soc. Faraday Trans. Mol. Chem. Phys, 75, 1001–1019. [Google Scholar]
- 169.Edwards Sam F., Jeffers Eamon F., (1979). Size of a polymer molecule in solution. Part 2. –Semi-dilute solutions. J. Chem. Soc. Faraday Trans. Mol. Chem. Phys., 75, 1020–1029. [Google Scholar]
- 170.Lin Yi-Hsuan, Chan Hue Sun, (2017). Phase separation and single-chain compactness of charged disordered proteins are strongly correlated. Biophys. J, 112 (10), 2043–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Khokhlov Alexei R., Khalatur Pavel G., (1999). Conformation-dependent sequence design (engineering) of AB copolymers. Phys. Rev. Letters, 82 (17), 3456. [Google Scholar]
- 172.Ashbaugh Henry S., (2009). Tuning the globular assembly of hydrophobic/hydrophilic heteropolymer sequences. J. Phys. Chem. B, 113 (43), 14043–14046. [DOI] [PubMed] [Google Scholar]
- 173.Amin Alan N., Lin Yi-Hsuan, Das Suman, Chan Hue Sun, (2020). Analytical theory for sequence-specific binary fuzzy complexes of charged intrinsically disordered proteins. J. Phys. Chem. B, 124 (31), 6709–6720. [DOI] [PubMed] [Google Scholar]
- 174.Brady Jacob P., Farber Patrick J., Sekhar Ashok, Lin Yi-Hsuan, Huang Rui, Bah Alaji, Nott Timothy J., Chan Hue Sun, et al. , (2017). Structural and hydrodynamic properties of an intrinsically disordered region of a germ cell-specific protein on phase separation. Proc. Natl. Acad. Sci. USA, 114 (39), E8194–E8203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Schuster Benjamin S., Dignon Gregory L., Tang Wai Shing, Kelley Fleurie M., Ranganath Aishwarya Kanchi, Jahnke Craig N., Simpkins Alison G., Regy Roshan Mammen, et al. , (2020). Identifying sequence perturbations to an intrinsically disordered protein that determine its phase-separation behavior. Proc. Natl. Acad. Sci. USA, 117 (21), 11421–11431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Chiti Fabrizio, Webster Paul, Taddei Niccolò, Clark Anne, Stefani Massimo, Ramponi Giampietro, Dobson Christopher M., (1999). Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc. Natl. Acad. Sci. USA, 96 (7), 3590–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Chiti Fabrizio, Dobson Christopher M., (2006). Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem, 75, 333–366. [DOI] [PubMed] [Google Scholar]
- 178.Otzen Daniel, Riek Roland, (2019). Functional amyloids. Cold Spring Harbor Perspect. Biol, 11 (12), a033860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Maji Samir K., Perrin Marilyn H., Sawaya Michael R., Jessberger Sebastian, Vadodaria Krishna, Rissman Robert A., Singru Praful S., Peter K, Nilsson R, Simon Rozalyn, Schubert David, et al. , (2009). Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science, 325 (5938), 328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Roberts Christopher J., (2014). Therapeutic protein aggregation: mechanisms, design, and control. Trends Biotechnol, 32 (7), 372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Li Pilong, Banjade Sudeep, Cheng Hui-Chun, Kim Soyeon, Chen Baoyu, Guo Liang, Llaguno Marc, Hollingsworth Javoris V., et al. , (2012). Phase transitions in the assembly of multivalent signalling proteins. Nature, 483 (7389), 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Murthy Anastasia C., Dignon Gregory L., Kan Yelena, Zerze Gül H., Parekh Sapun H., Mittal Jeetain, Fawzi Nicolas L., (2019). Molecular interactions underlying liquid-liquid phase separation of the FUS low-complexity domain. Nature Struct. Mol. Biol, 26 (7), 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Asherie Neer, (2004). Protein crystallization and phase diagrams. Methods, 34 (3), 266–272. [DOI] [PubMed] [Google Scholar]
- 184.Shire Steven J., Shahrokh Zahra, Liu Jun, (2004). Challenges in the development of high protein concentration formulations. J. Pharm. Sci, 93 (6), 1390–1402. [DOI] [PubMed] [Google Scholar]
- 185.Woldeyes Mahlet A., Qi Wei, Razinkov Vladimir I., Furst Eric M., Roberts Christopher J., (2019). How well do low-and high-concentration protein interactions predict solution viscosities of monoclonal antibodies?. J. Pharm. Sci, 108 (1), 142–154. [DOI] [PubMed] [Google Scholar]
- 186.Mason Bruce D., Enk Jian Zhang-van, Zhang Le, Remmele Richard L. Jr., Zhang Jifeng, (2010). Liquid-liquid phase separation of a monoclonal antibody and nonmonotonic influence of Hofmeister anions. Biophys. J, 99 (11), 3792–3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wang Ying, Lomakin Aleksey, Latypov Ramil F., Benedek George B., (2011). Phase separation in solutions of monoclonal antibodies and the effect of human serum albumin. Proc. Natl. Acad. Sci. USA, 108 (40), 16606–16611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Arora Jayant, Hickey John M., Majumdar Ranajoy, Esfandiary Reza, Bishop Steven M., Samra Hardeep S., Russell Middaugh, C., Weis David D., et al. , (2015). Hydrogen exchange mass spectrometry reveals protein interfaces and distant dynamic coupling effects during the reversible self-association of an IgG1 monoclonal antibody. MAbs, 7 (3), 525–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Arora Jayant, Hu Yue, Esfandiary Reza, Sathish Hasige A., Bishop Steven M., Joshi Sangeeta B., Russell Middaugh, C., Volkin David B., et al. , (2016). and elevated viscosity. MAbs, 8 (8), 1561–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Shoulders Matthew D., Raines Ronald T., (2009). Collagen structure and stability. Annu. Rev. Biochem, 78, 929–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Silver Frederick H., Freeman Joseph W., Seehra Gurinder P., (2003). Collagen self-assembly and the development of tendon mechanical properties. J. Biomech, 36 (10), 1529–1553. [DOI] [PubMed] [Google Scholar]
- 192.Boatz Jennifer C., Whitley Matthew J., Li Mingyue, Gronenborn Angela M., van der Wel Patrick C.A., (2017). Cataract-associated P23T γD-crystallin retains a native-like fold in amorphous-looking aggregates formed at physiological pH. Nature Commun, 8 (1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Bloemendal Hans, de Jong Wilfried, Jaenicke Rainer, Lubsen Nicolette H., Slingsby Christine, Tardieu Annette, (2004). Ageing and vision: structure, stability and function of lens crystallins. Prog. Biophys. Mol. Biol, 86 (3), 407–485. [DOI] [PubMed] [Google Scholar]
- 194.Serebryany Eugene, King Jonathan A., (2014). The bccrystallins: native state stability and pathways to aggregation. Prog. Biophys. Mol. Biol, 115 (1), 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Vlachy Vojko, Prausnitz John M., (1992). Donnan equilibrium: hypernetted-chain study of one-component and multicomponent models for aqueous polyelectrolyte solutions. J. Phys. Chem, 96 (15), 6465–6469. [Google Scholar]
- 196.Vlachy Vojko, Blanch HW, Prausnitz John M., (1993). Liquid-liquid phase separations in aqueous solutions of globular proteins. AIChE J, 39 (2), 215–223. [Google Scholar]
- 197.Curtis RA, Prausnitz JM, Blanch HW, (1998). Protein-protein and protein-salt interactions in aqueous protein solutions containing concentrated electrolytes. Biotechnol. Bioeng, 57 (1), 11–21. [DOI] [PubMed] [Google Scholar]
- 198.Wu JZ, Bratko D, Blanch HW, Prausnitz JM, (1999). Monte Carlo simulation for the potential of mean force between ionic colloids in solutions of asymmetric salts. J. Chem. Phys, 111 (15), 7084–7094. [Google Scholar]
- 199.Neal BL, Asthagiri D, Lenhoff AM, (1998). Molecular origins of osmotic second virial coefficients of proteins. Biophys. J, 75 (5), 2469–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Roth Charles M., Lenhoff Abraham M., (1993). Electrostatic and van der Waals contributions to protein adsorption: computation of equilibrium constants. Langmuir, 9 (4), 962–972. [Google Scholar]
- 201.Lin MY, Lindsay H_M, Weitz DA, Ball RC, Klein R, Meakin P, (1989). Universality in colloid aggregation. Nature, 339 (6223), 360–362. [Google Scholar]
- 202.Nicoud Lucrece, Arosio Paolo, Sozo Margaux, Yates Andrew, Norrant Edith, Morbidelli Massimo, (2014). Kinetic analysis of the multistep aggregation mechanism of monoclonal antibodies. J. Phys. Chem. B, 118 (36), 10595–10606. [DOI] [PubMed] [Google Scholar]
- 203.Nicoud Lucrèce, Owczarz Marta, Arosio Paolo, Morbidelli Massimo, (2015). A multiscale view of therapeutic protein aggregation: a colloid science perspective. Biotechnol. J, 10 (3), 367–378. [DOI] [PubMed] [Google Scholar]
- 204.Borgia Madeleine B., Nickson Adrian A., Clarke Jane, Hounslow Michael J., (2013). A mechanistic model for amorphous protein aggregation of immunoglobulin-like domains. J. Am. Chem. Soc, 135 (17), 6456–6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Calero-Rubio Cesar, Saluja Atul, Roberts Christopher J., (2016). Coarse-grained antibody models for weak protein–protein interactions from low to high concentrations. J. Phys. Chem. B, 120 (27), 6592–6605. [DOI] [PubMed] [Google Scholar]
- 206.Chowdhury Amjad, Bollinger Jonathan A., Dear Barton J., Cheung Jason K., Johnston Keith P., Truskett Thomas M., (2020). Coarse-grained molecular dynamics simulations for understanding the impact of short-range anisotropic attractions on structure and viscosity of concentrated monoclonal antibody solutions. Mol. Pharm, 17 (5), 1748–1756. [DOI] [PubMed] [Google Scholar]
- 207.Zhou Huan-Xiang, (2003). Quantitative account of the enhanced affinity of two linked scfvs specific for different epitopes on the same antigen. J. Mol. Biol, 329 (1), 1–8. [DOI] [PubMed] [Google Scholar]
- 208.Schmit Jeremy D., He Feng, Mishra Shradha, Ketchem Randal R., Woods Christopher E., Kerwin Bruce A., (2014). Entanglement model of antibody viscosity. J. Phys. Chem. B, 118 (19), 5044–5049. [DOI] [PubMed] [Google Scholar]
- 209.Kastelic Miha, Dill Ken A., Kalyuzhnyi Yura V., Vlachy Vojko, (2018). Controlling the viscosities of antibody solutions through control of their binding sites. J. Mol. Liq, 270, 234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Ramallo Nelson, Paudel Subhash, Schmit Jeremy, (2019). Cluster formation and entanglement in the rheology of antibody solutions. J. Phys. Chem. B, 123 (18), 3916–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kastelic Miha, Kalyuzhnyi Yurij V., Hribar-Lee Barbara, Dill Ken A., Vlachy Vojko, (2015). Protein aggregation in salt solutions. Proc. Natl. Acad. Sci. USA, 112 (21), 6766–6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Kastelic Miha, Kalyuzhnyi Yurij V., Vlachy Vojko, (2016). Modeling phase transitions in mixtures of β–γlens crystallins. Soft Matter, 12 (35), 7289–7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Kastelic Miha, Vlachy Vojko, (2018). Theory for the liquid–liquid phase separation in aqueous antibody solutions. J. Phys. Chem. B, 122 (21), 5400–5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Chiti Fabrizio, Dobson Christopher M., (2017). Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu. Rev. Biochem, 86, 27–68. [DOI] [PubMed] [Google Scholar]
- 215.Nelson Rebecca, Sawaya Michael R., Balbirnie Melinda, Madsen Anders Ø., Riekel Christian, Grothe Robert, Eisenberg David, (2005). Structure of the cross-β spine of amyloid-like fibrils. Nature, 435 (7043), 773–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Sawaya Michael R., Sambashivan Shilpa, Nelson Rebecca, Ivanova Magdalena I., Sievers Stuart A., Apostol Marcin I., Thompson Michael J., Balbirnie Melinda, et al. , (2007). Atomic structures of amyloid cross-b spines reveal varied steric zippers. Nature, 447 (7143), 453–457. [DOI] [PubMed] [Google Scholar]
- 217.Schmit Jeremy D., Ghosh Kingshuk, Dill Ken, (2011). What drives amyloid molecules to assemble into oligomers and fibrils?. Biophys. J, 100 (2), 450–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Phan Tam T.M., Schmit Jeremy D., (2020). Thermodynamics of Huntingtin aggregation. Biophys. J, 118 (12), 2989–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Knowles Tuomas P., Fitzpatrick Anthony W., Meehan Sarah, Mott Helen R., Vendruscolo Michele, Dobson Christopher M., Welland Mark E., (2007). Role of intermolecular forces in defining material properties of protein nanofibrils. Science, 318 (5858), 1900–1903. [DOI] [PubMed] [Google Scholar]
- 220.Knowles Tuomas P.J., Buehler Markus J., (2011). Nanomechanics of functional and pathological amyloid materials. Nature Nanotechnol, 6 (8), 469–479. [DOI] [PubMed] [Google Scholar]
- 221.Lamour Guillaume, Nassar Roy, Chan Patrick H.W., Bozkurt Gunes, Li Jixi, Bui Jennifer M., Yip Calvin K., Mayor Thibault, et al. , (2017). Mapping the broad structural and mechanical properties of amyloid fibrils. Biophys. J, 112 (4), 584–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Nassar Roy, Wong Eric, Bui Jennifer M., Yip Calvin K., Li Hongbin, Gsponer Jörg, Lamour Guillaume, (2018). Mechanical anisotropy in GNNQQNY amyloid crystals. J. Phys. Chem. Letters, 9 (17), 4901–4909. [DOI] [PubMed] [Google Scholar]
- 223.Nassar Roy, Wong Eric, Gsponer Jörg, Lamour Guillaume, (2018). Inverse correlation between amyloid stiffness and size. J. Am. Chem. Soc, 141 (1), 58–61. [DOI] [PubMed] [Google Scholar]
- 224.Baldwin Andrew J., Knowles Tuomas P.J., Tartaglia Gian Gaetano, Fitzpatrick Anthony W., Devlin Glyn L., Shammas Sarah Lucy, Waudby Christopher A., Mossuto Maria F., et al. , (2011). Metastability of native proteins and the phenomenon of amyloid formation. J. Am. Chem. Soc, 133 (36), 14160–14163. [DOI] [PubMed] [Google Scholar]
- 225.Prusiner Stanley B., (1998). Prions. Proc. Natl. Acad. Sci. USA, 95 (23), 13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Harrison Paul M., Chan Hue Sun, Prusiner Stanley B., Cohen Fred E., (1999). Thermodynamics of model prions and its implications for the problem of prion protein folding. J. Mol. Biol, 286 (2), 593–606. [DOI] [PubMed] [Google Scholar]
- 227.Harrison Paul M., Chan Hue Sun, Prusiner Stanley B., Cohen Fred E., (2001). Conformational propagation with prion-like characteristics in a simple model of protein folding. Protein Sci, 10 (4), 819–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Schmit Jeremy D., (2013). Kinetic theory of amyloid fibril templating. J. Chem. Phys, 138 (18), 05B611_1. [DOI] [PubMed] [Google Scholar]
- 229.Huang Caleb, Ghanati Elaheh, Schmit Jeremy D., (2018). Theory of sequence effects in amyloid aggregation. J. Phys. Chem. B, 122 (21), 5567–5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Knowles Tuomas P.J., Vendruscolo Michele, Dobson Christopher M., (2014). The amyloid state and its association with protein misfolding diseases. Nature Rev. Mol. Cell Biol, 15 (6), 384–396. [DOI] [PubMed] [Google Scholar]
- 231.Cohen Samuel I.A., Cukalevski Risto, Michaels Thomas C.T., Saric Andela, Törnquist Mattias, Vendruscolo Michele, Dobson Christopher M., Buell Alexander K., Knowles Tuomas P.J., Linse Sara, (2018). Distinct thermodynamic signatures of oligomer generation in the aggregation of the amyloid-bpeptide. Nature Chem, 10 (5), 523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Michaels Thomas C.T., Saric Andela, Habchi Johnny, Chia Sean, Meisl Georg, Vendruscolo Michele, Dobson Christopher M., Knowles Tuomas P.J., (2018). Chemical kinetics for bridging molecular mechanisms and macroscopic measurements of amyloid fibril formation. Ann. Rev. Phys. Chem, 69, 273–298. [DOI] [PubMed] [Google Scholar]
- 233.Knowles Tuomas P.J., Waudby Christopher A., Devlin Glyn L., Cohen Samuel I.A., Aguzzi Adriano, Vendruscolo Michele, Terentjev Eugene M., Welland Mark E., et al. , (2009). An analytical solution to the kinetics of breakable filament assembly. Science, 326 (5959), 1533–1537. [DOI] [PubMed] [Google Scholar]
- 234.Cohen Samuel I.A., Linse Sara, Luheshi Leila M., Hellstrand Erik, White Duncan A., Rajah Luke, Otzen Daniel E., Vendruscolo Michele, et al. , (2013). Proliferation of amyloid-b 42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA, 110 (24), 9758–9763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Saric Andela, Buell Alexander K., Meisl Georg, Michaels Thomas C.T., Dobson Christopher M., Linse Sara, Knowles Tuomas P.J., Frenkel Daan, (2016). Physical determinants of the self-replication of protein fibrils. Nature Phys, 12 (9), 874–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Krausser Johannes, Knowles Tuomas P.J., Saric Andela, (2020). Physical mechanisms of amyloid nucleation on fluid membranes. Proc. Natl. Acad. Sci. USA, 117 (52), 33090–33098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Nguyen Phuong H., Ramamoorthy Ayyalusamy, Sahoo Bikash R., Zheng Jie, Faller Peter, Straub John E., Dominguez Laura, Shea Joan-Emma, et al. , (2021). and Amyotrophic Lateral Sclerosis. Chem. Rev,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Brangwynne Clifford P., Eckmann Christian R., Courson David S., Rybarska Agata, Hoege Carsten, Gharakhani Jöbin, Jülicher Frank, Hyman Anthony A., (2009). Germline p granules are liquid droplets that localize by controlled dissolution/condensation. Science, 324 (5935), 1729–1732. [DOI] [PubMed] [Google Scholar]
- 239.Dzuricky Michael, Roberts Stefan, Chilkoti Ashutosh, (2018). Convergence of artificial protein polymers and intrinsically disordered proteins. Biochemistry, 57 (17), 2405–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Hyman Anthony A., Weber Christoph A., Jülicher Frank, (2014). Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol, 30, 39–58. [DOI] [PubMed] [Google Scholar]
- 241.Riback Joshua A., Katanski Christopher D., Kear-Scott Jamie L., Pilipenko Evgeny V., Rojek Alexandra E., Sosnick Tobin R., Allan Drummond D, (2017). Stress-triggered phase separation is an adaptive, evolutionarily tuned response. Cell, 168 (6), 1028–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Frottin F, Schueder F, Tiwary S, Gupta R, Körner R, Schlichthaerle T, Cox J, Jungmann R, Hartl FU, Hipp MS, (2019). The nucleolus functions as a phase-separated protein quality control compartment. Science, 365 (6451), 342–347. [DOI] [PubMed] [Google Scholar]
- 243.Heinkel Florian, Abraham Libin, Ko Mary, Chao Joseph, Bach Horacio, Hui Lok Tin, Li Haoran, Zhu Mang, et al. , (2019). Phase separation and clustering of an ABC transporter in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA, 116 (33), 16326–16331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Patel Avinash, Lee Hyun O., Jawerth Louise, Maharana Shovamayee, Jahnel Marcus, Hein Marco Y., Stoynov Stoyno, Mahamid Julia, et al. , (2015). A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell, 162 (5), 1066–1077. [DOI] [PubMed] [Google Scholar]
- 245.Forman-Kay Julie D., Kriwacki Richard W., Seydoux Geraldine, (2018). Phase separation in biology and disease. J. Mol. Biol, 430 (23), 4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Mathieu Cécile, Pappu Rohit V., Paul Taylor J, (2020). Beyond aggregation: pathological phase transitions in neurodegenerative disease. Science, 370 (6512), 56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Shin Yongdae, Brangwynne Clifford P., (2017). Liquid phase condensation in cell physiology and disease. Science, 357 (6357) [DOI] [PubMed] [Google Scholar]
- 248.Wagner Rudolph, (1835). Einige bemerkungen und fragen über das keimbläschen (vesicular germinativa). Müller Archiv für Anatomie Physiologie und Wissenschaftliche Medicin, 268, 373–377. [Google Scholar]
- 249.Valentin Gabriel, (1837). Repertorium für Anatomie und Physiologie. Veit und comp,. [Google Scholar]
- 250.Pederson Thoru, (2011). The nucleolus. Cold Spring Harbor Perspect. Biol, 3 (3), a000638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Walter Harry, Brooks Donald E., (1995). Phase separation in cytoplasm, due to macromolecular crowding, is the basis for microcompartmentation. FEBS Letters, 361 (2–3), 135–139. [DOI] [PubMed] [Google Scholar]
- 252.Uversky Vladimir N., Kuznetsova Irina M., Turoverov Konstantin K., Zaslavsky Boris, (2015). Intrinsically disordered proteins as crucial constituents of cellular aqueous two phase systems and coacervates. FEBS Letters, 589 (1), 15–22. [DOI] [PubMed] [Google Scholar]
- 253.Zhou Huan-Xiang, Nguemaha Valery, Mazarakos Konstantinos, Qin Sanbo, (2018). Why do disordered and structured proteins behave differently in phase separation?. Trends Biochem. Sci, 43 (7), 499–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Lin Yi-Hsuan, Brady Jacob P., Forman-Kay Julie D., Chan Hue Sun, (2017). Charge pattern matching as a fuzzy mode of molecular recognition for the functional phase separations of intrinsically disordered proteins. New J. Phys, 19 (11), 115003. [Google Scholar]
- 255.Dignon Gregory L., Zheng Wenwei, Best Robert B., Kim Young C., Mittal Jeetain, (2018). Relation between single-molecule properties and phase behavior of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA, 115 (40), 9929–9934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Schuster Benjamin S., Reed Ellen H., Parthasarathy Ranganath, Jahnke Craig N., Caldwell Reese M., Bermudez Jessica G., Ramage Holly, Good Matthew C., Hammer Daniel A., (2018). Controllable protein phase separation and modular recruitment to form responsive membraneless organelles. Nature Commun, 9 (1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Sheng Yu Jane, Panagiotopoulos Athanassios Z., Kumar Sanat K., Szleifer Igal, (1994). Monte Carlo calculation of phase equilibria for a bead-spring polymeric model. Macromolecules, 27 (2), 400–406. [Google Scholar]
- 258.Fields Gregg B., Alonso Darwin O.V., Stigter Dirk, Dill Ken A., (1992). Theory for the aggregation of proteins and copolymers. J. Phys. Chem, 96 (10), 3974–3981. [Google Scholar]
- 259.Zeng Xiangze, Holehouse Alex S., Chilkoti Ashutosh, Mittag Tanja, Pappu Rohit V., (2020). Connecting coil-to-globule transitions to full phase diagrams for intrinsically disordered proteins. Biophys. J, 119 (2), 402–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Monahan Zachary, Ryan Veronica H., Janke Abigail M., Burke Kathleen A., Rhoads Shannon N., Zerze Gül H., O’Meally Robert, Dignon Gregory L., Alexander Conicella E, Zheng Wenwei, et al. , (2017). Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J, 36 (20), 2951–2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Martin Erik W., Hopkins Jesse B., Mittag Tanja, (2020). Small-angle X-ray scattering experiments of monodisperse intrinsically disordered protein samples close to the solubility limit. Methods Enzymol, 646, 185–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Lin Yi-Hsuan, Forman-Kay Julie D., Chan Hue Sun, (2016). Sequence-specific polyampholyte phase separation in membraneless organelles. Phys. Rev. Letters, 117 (17), 178101. [DOI] [PubMed] [Google Scholar]
- 263.Wang Jie, Choi Jeong-Mo, Holehouse Alex S., Lee Hyun O., Zhang Xiaojie, Jahnel Marcus, Maharana Shovamayee, Lemaitre Régis, et al. , (2018). A molecular grammar governing the driving forces for phase separation of prion-like RNA binding proteins. Cell, 174 (3), 688–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Choi Jeong-Mo, Holehouse Alex S., Pappu Rohit V., (2020). Physical principles underlying the complex biology of intracellular phase transitions. Ann. Rev. Biophys, 49, 107–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Rubinstein Michael, Dobrynin Andrey V., (1997). Solutions of associative polymers. Trends in Polymer. Science, 5 (6), 181–186. [Google Scholar]
- 266.Lin Yi-Hsuan, Song Jianhui, Forman-Kay Julie D., Chan Hue Sun, (2017). Random-phase-approximation theory for sequence-dependent, biologically functional liquid-liquid phase separation of intrinsically disordered proteins. J. Mol. Liq, 228, 176–193. [Google Scholar]
- 267.Lin Yi-Hsuan, Brady Jacob P., Chan Hue Sun, Ghosh Kingshuk, (2020). A unified analytical theory of heteropolymers for sequence-specific phase behaviors of polyelectrolytes and polyampholytes. J. Chem. Phys, 152 (4), 045102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.McCarty James, Delaney Kris T., Danielsen Scott P.O., Fredrickson Glenn H., Shea Joan-Emma, (2019). Complete phase diagram for liquid–liquid phase separation of intrinsically disordered proteins. J. Phys. Chem. Letters, 10 (8), 1644–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Danielsen Scott P.O., McCarty James, Shea Joan-Emma, Delaney Kris T., Fredrickson Glenn H., (2019). Molecular design of self-coacervation phenomena in block polyampholytes. Proc. Natl. Acad. Sci. USA, 116 (17), 8224–8232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Dignon Gregory L., Zheng Wenwei, Mittal Jeetain, (2019). Simulation methods for liquid–liquid phase separation of disordered proteins. Curr. Opin. Chem. Eng, 23, 92–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Ruff Kiersten M., Pappu Rohit V., Holehouse Alex S., (2019). Conformational preferences and phase behavior of intrinsically disordered low complexity sequences: insights from multiscale simulations. Curr. Opin. Struct. Biol, 56, 1–10. [DOI] [PubMed] [Google Scholar]
- 272.Shea Joan-Emma, Best Robert B., Mittal Jeetain, (2021). Physics-based computational and theoretical approaches to intrinsically disordered proteins. Curr. Opin. Struct. Biol, 67, 219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Caspar Donald L.D., Klug Aaron, (1962). Physical principles in the construction of regular viruses. Cold Spring Harb. Symp. Quant. Biol,, volume 27 Cold Spring Harbor Laboratory Press, pp. 1–24. [DOI] [PubMed] [Google Scholar]
- 274.Berger Bonnie, Shor Peter W., Tucker-Kellogg Lisa, King Jonathan, (1994). Local rule-based theory of virus shell assembly. Proc. Natl. Acad. Sci. USA, 91 (16), 7732–7736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Zandi Roya, Reguera David, Bruinsma Robijn F., Gelbart William M., Rudnick Joseph, (2004). Origin of icosahedral symmetry in viruses. Proc. Natl. Acad. Sci. USA, 101 (44), 15556–15560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Twarock Reidun, Luque Antoni, (2019). Structural puzzles in virology solved with an overarching icosahedral design principle. Nature Commun, 10 (1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Kegel Willem K., van der Schoot Paul, (2006). Physical regulation of the self–assembly of tobacco mosaic virus coat protein. Biophys. J, 91 (4), 1501–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Zlotnick Adam, (2007). Distinguishing reversible from irreversible virus capsid assembly. J. Mol. Biol, 366 (1), 14–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Morozov Alexander Yu, Bruinsma Robijn F., Rudnick Joseph, (2009). Assembly of viruses and the pseudo-law of mass action. J. Chem. Phys, 131 (15), 10B607. [DOI] [PubMed] [Google Scholar]
- 280.Perlmutter Jason D., Hagan Michael F., (2015). Mechanisms of virus assembly. Annu. Rev. Phys. Chem, 66, 217–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Bruinsma Robijn F., Wuite Gijs J.L., Roos Wouter H., (2021). Physics of viral dynamics. Nature Rev. Phys,, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Santra Mantu, Farrell Daniel W., Dill Ken A., (2017). Bacterial proteostasis balances energy and chaperone utilization efficiently. Proc. Natl. Acad. Sci. USA, 114 (13), E2654–E2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Santra Mantu, Dill Ken A., de Graff Adam M.R., (2018). How do chaperones protect a cell’s proteins from oxidative damage?. Cell Syst, 6 (6), 743–751.e3. [DOI] [PubMed] [Google Scholar]
- 284.De Graff Adam M.R., Mosedale David E., Sharp Tilly, Dill Ken A., Grainger David J., (2020). Proteostasis is adaptive: balancing chaperone holdases against foldases. PLoS Comput. Biol, 16 (12), 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Holtkamp Wolf, Kokic Goran, Jäger Marcus, Mittelstaet Joerg, Komar Anton A., Rodnina Marina V., (2015). Cotranslational protein folding on the ribosome monitored in real time. Science, 350 (6264), 1104–1107. [DOI] [PubMed] [Google Scholar]
- 286.Bitran Amir, Jacobs William M., Zhai Xiadi, Shakhnovich Eugene, (2020). Cotranslational folding allows misfolding-prone proteins to circumvent deep kinetic traps. Proc. Natl. Acad. Sci. USA, 117 (3), 1485–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Liutkute Marija, Samatova Ekaterina, Rodnina Marina V., (2020). Cotranslational folding of proteins on the ribosome. Biomolecules, 10 (1), 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Zhao Victor, Jacobs William M., Shakhnovich Eugene I., (2020). Effect of protein structure on evolution of cotranslational folding. Biophys. J, 119 (6), 1123–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Sella Guy, Hirsh Aaron E., (2005). The application of statistical physics to evolutionary biology. Proc. Natl. Acad. Sci. USA, 102 (27), 9541–9546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Agozzino Luca, Balázsi Gábor, Wang Jin, Dill Ken A., (2020). How do cells adapt? Stories told in landscapes. Ann. Rev. Chem. Biomol. Eng, 11, 155–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Dykhuizen Daniel E., Dean Antony M., Hartl Daniel L., (1987). Metabolic flux and fitness. Genetics, 115 (1), 25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Bershtein Shimon, Serohijos Adrian W.R., Bhattacharyya Sanchari, Manhart Michael, Choi Jeong-Mo, Mu Wanmeng, Zhou Jingwen, Shakhnovich Eugene I., (2015). Protein homeostasis imposes a barrier on functional integration of horizontally transferred genes in bacteria. PLoS Genet, 11 (10), e1005612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Adkar Bharat V., Manhart Michael, Bhattacharyya Sanchari, Tian Jian, Musharbash Michael, Shakhnovich Eugene I., (2017). Optimization of lag phase shapes the evolution of a bacterial enzyme. Nature Ecol. Evol, 1 (6), 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Socha Raymond D., Chen John, Tokuriki Nobuhiko, (2019). The molecular mechanisms underlying hidden phenotypic variation among metallo-β-lactamases. J. Mol. Biol, 431 (6), 1172–1185. [DOI] [PubMed] [Google Scholar]
- 295.Adkar Bharat V., Bhattacharyya Sanchari, Gilson Amy I., Zhang Wenli, Shakhnovich Eugene I., (2019). Substrate inhibition imposes fitness penalty at high protein stability. Proc. Natl. Acad. Sci. USA, 116 (23), 11265–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Rodrigues João V., Bershtein Shimon, Li Anna, Lozovsky Elena R., Hartl Daniel L., Shakhnovich Eugene I., (2016). Biophysical principles predict fitness landscapes of drug resistance. Proc. Natl. Acad. Sci. USA, 113 (11), E1470–E1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Knies Jennifer L., Cai Fei, Weinreich Daniel M., (2017). Enzyme efficiency but not thermostability drives cefotaxime resistance evolution in TEM-1 b-lactamase. Mol. Biol. Evol, 34 (5), 1040–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Pál Csaba, Papp Baláz, Hurst Laurence D., (2001). Highly expressed genes in yeast evolve slowly. Genetics, 158, 927–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Wang Zhi, Zhang Jianzhi, (2009). Why is the correlation between gene importance and gene evolutionary rate so weak?. PLoS Genet., 5 (1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Drummond DA, Bloom JD, Adami C, Wilke CO, Arnold FH, (2005). Why highly expressed proteins evolve slowly. Proc. Natl. Acad. Sci. USA, 102 (40), 14338–14343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Allan Drummond D, Wilke Claus O., (2008). Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell, 134 (2), 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Yang Jian Rong, Zhuang Shi Mei, Zhang Jianzhi, (2010). Impact of translational error-induced and error-free misfolding on the rate of protein evolution. Mol. Syst. Biol, 6 (421), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Serohijos Adrian W.R., Rimas Zilvinas, Shakhnovich Eugene I., (2012). Protein biophysics explains why highly abundant proteins evolve slowly. Cell Rep, 2 (2), 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Agozzino Luca, Dill Ken A., (2018). Protein evolution speed depends on its stability and abundance and on chaperone concentrations. Proc. Natl. Acad. Sci. USA, 115 (37), 9092–9097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Yang J-R, Liao B-Y, Zhuang S-M, Zhang J, (2012). Protein misinteraction avoidance causes highly expressed proteins to evolve slowly. Proc. Natl. Acad. Sci. USA, 109 (14), E831–E840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Zhang Jianzhi, Yang Jian-Rong, (2015). Determinants of the rate of protein sequence evolution. Nature Rev. Genet, 16 (7), 409–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Tartaglia Gian Gaetano, Pechmann Sebastian, Dobson Christopher M., Vendruscolo Michele, (2007). Life on the edge: a link between gene expression levels and aggregation rates of human proteins. Trends Biochem. Sci, 32 (5), 204–206. [DOI] [PubMed] [Google Scholar]
- 308.Vecchi Giulia, Sormanni Pietro, Mannini Benedetta, Vandelli Andrea, Tartaglia Gian Gaetano, Dobson Christopher M., Ulrich Hartl F, Vendruscolo Michele, (2020). Proteome-wide observation of the phenomenon of life on the edge of solubility. Proc. Natl. Acad. Sci. USA, 117 (2), 1015–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Razban Rostam M, Dasmeh Pouria, Serohijos Adrian W.R., Shakhnovich Eugene I., (2021). Avoidance of protein unfolding constrains protein stability in long-term evolution. Biophys. J, 120 (12), 2413–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]