

ABSTRACT
DNA polymerase kappa (Pol κ) has been well documented thus far for its specialized DNA synthesis activity during translesion replication, progression of replication forks through regions difficult to replicate, restart of stalled forks, and replication checkpoint efficiency. Pol κ is also required for the stabilization of stalled forks, although the mechanisms are poorly understood. In this study, we unveiled an unexpected role for Pol κ in controlling the stability and abundance of checkpoint kinase 1 (Chk1), an important actor for the replication checkpoint and fork stabilization. We found that loss of Pol κ decreased the Chk1 protein level in the nuclei of four human cell lines. Pol κ and not the other Y family polymerase members is required to maintain the Chk1 protein pool all along the cell cycle. We showed that Pol κ depletion affected the protein stability of Chk1 and protected it from proteasome degradation. Importantly, we also observed that the fork restart defects observed in Pol κ-depleted cells could be overcome by the reexpression of Chk1. Strikingly, this new function of Pol κ does not require its catalytic activity. We propose that Pol κ could contribute to the protection of stalled forks through Chk1 stability.
KEYWORDS: DNA polymerase kappa/checkpoint kinase 1/Chk1 stability/fork restart, Chk1, DNA polymerase kappa, replication stress, Y-DNA polymerase
INTRODUCTION
Cells have to continuously cope with a variety of DNA damages induced by exposure to exogenous and endogenous genotoxic agents. Cellular responses such as signaling, repairing, or bypassing the damage are required to deal with DNA damages and avoid the replication fork blockage during S phase. Cells need to limit the accumulation of stalled forks, a process described as replication stress, in order to restrain transmission of DNA damages to daughter cells (1, 2). An important part of the cellular response to replication stress is the induction of the ATR/checkpoint kinase 1 (Chk1) replication checkpoint, which senses stalled replication forks, allows their stabilization and repair, prevents the firing of late replication origins, and inhibits entry into mitosis until the completion of replication (3, 4).
The ATR-Chk1 signaling axis is now described as a tunable brake required during unperturbed cell proliferation to couple replication of the genome and cell cycle progression (5). ATR and Chk1 are essential actors in genome stability maintenance. Indeed, mutations in ATR are responsible for an autosomal recessive disorder called Seckel syndrome (6), and Chk1 heterozygosity leads to defects in cell cycle control and accumulation of DNA damages and predisposes cells to cancer (7). Under physiological conditions, the depletion of Chk1 decreases the global rate of replication (8–10). To ensure correct connections between replication and cell cycle progression, the abundance of Chk1, which relies on its stability, is critical for cellular stress response and checkpoint maintenance (11). One of the best-documented modes of regulation of Chk1 is ubiquitination mediated by the cullin-ring E3-ubiquitin ligases Cul1, Cul4A, and HUWE1, which ubiquitinate the C-terminal degron-like region of Chk1 and target it for proteasomal degradation (12–14). This ubiquitination-dependent regulation of Chk1 can be directly antagonized by the ubiquitin hydrolases USP1, USP3, USP7, and ataxin 3, which deubiquitinate Chk1 (15–18).
Translesion synthesis (TLS) is also an important mechanism to respond to replicative stress. It involves the translesional Y family DNA polymerases (Pol) Pol η (Pol eta), Pol κ (Pol kappa), Pol ι (Pol iota), and Rev1, also called specialized polymerases. They facilitate the bypass of DNA lesions that block the replicative DNA polymerases by insertion of nucleotides opposite DNA lesions. They are devoid of exonuclease activity and show a flexible catalytic site able to adapt to a damaged DNA template (19). Y-DNA polymerases are also implicated in DNA repair mechanisms such nucleotide excision repair and homologous recombination (20–22). In addition to their TLS or repair functions, these specialized DNA polymerases also play critical roles in the replication of non-B-DNA and in the prevention of replication stress induced by oncogenes (23–26). For instance, Pol η contributes to the stability of common fragile sites (CFS), which have the potential to adopt non-B-DNA structures (27). The deoxycytidyl transferase Rev1 is required to replicate DNA sequences prone to form G4 secondary structures or enriched in nucleotide repeats (28). Similarly, Pol κ is required for the replication of structured DNA sequences and rescues the replicative DNA Pol δ when it is stalled at repetitive sequences within CFS (29, 30). Pol κ is also needed to maintain viability upon replication stress induced by oncogene activation (31) since it is required to protect and to restart the replication forks following starvation of deoxynucleoside triphosphates (dNTPs) (32).
Thus far, all these known Pol κ functional roles, i.e., TLS, replication of non-B structured DNA and repetitive sequences, and DNA synthesis on unwound DNA at stalled forks, have been entirely associated with its DNA polymerase catalytic activity. Indeed, mutation of the residues D198 and E199, which belong to the catalytic site of Pol κ (33), abolished the capacity of the polymerase to extend primers in vitro (34), sensitized human cells to benzo[a]pyrene diolepoxide, mitomycin C, and bleomycin (35), decreased the repair of interstrand cross-links (36), and impeded primer synthesis (37) and fork restart (32) at stalled forks.
The multiple functions of Pol κ imply that its cellular level needs to be tightly regulated to avoid perturbation of genome maintenance. In untreated cells, its aberrant recruitment to replication forks in cells depleted of USP1 or p21CDNKN1 leads to a decrease of the fork speed and its overexpression is associated with an instability of CFS, DNA breaks, and tumorigenesis in mice (38–40). In addition, dysregulation of Pol κ expression can affect its normal subcellular localization and can contribute to drug resistance (41, 42). Pol κ depletion in untreated cells induces hallmarks of replication stress with RPA focus formation and γ-H2AX in S phase, which are indicative of endogenous single-stranded DNA (ssDNA) accumulation and DNA breaks, respectively, and common fragile site expression (37, 39) features also observed in the absence of Chk1 (43).
In this study, we unveiled an unexpected role for Pol κ in controlling the stability of Chk1 in mammalian cells. We found that depletion of Pol κ, and not the other Y family polymerase members, induces a decrease of Chk1 protein level in the nuclei of four different human cell lines. Strikingly, this regulation is independent of the catalytic activity of Pol κ and occurs all along the cell cycle. Pol κ depletion does not affect the mRNA expression of Chk1 but favors its degradation through the proteasomal pathway. Finally, we found that the replication defects observed in Pol κ-depleted cells are linked to the low abundance of Chk1. Collectively, our findings highlight a catalytically independent function of Pol κ in genome maintenance.
RESULTS
Chk1 protein level is reduced in mammalian cells depleted of Pol κ.
To better understand the implication of the DNA polymerase kappa (Pol κ) in the maintenance of genome stability, we depleted different human cell lines of Pol κ and analyzed the Chk1 signaling pathway. We found a decrease of Chk1 protein level in the nuclear fraction of MRC5-SV Pol κ-depleted cells (Fig. 1A to C), whereas the levels of other proteins implicated in fork progression and the ATR/Chk1 pathway, such as claspin, TopBP1, Rad18, Rad17, Rad9A, and Pol δ, were not reduced (Fig. 1D). As a control, we treated cells with hydroxyurea (HU), an inhibitor of ribonucleotide reductase which induces the activation of ATR/Chk1 and the phosphorylation of Chk1 before its downregulation, which can be observed later on (44). Analysis of Pol κ and Chk1 protein levels in different nuclear extracts of MRC5-SV cells showed a good correlation (R2 = 0.906 [Fig. 1A]). To confirm this observation, we used immunofluorescence microscopy as a second approach. Chk1 fluorescence intensity was significantly decreased after Chk1 or Pol κ depletion in the nuclei of MRC5-SV cells (Fig. 1B), and importantly, the Chk1 protein level was rescued by ectopic expression of the polymerase (Fig. 1B and C). Thus, we revealed that Pol κ depletion leads reproducibly to a decrease in Chk1 abundance in the nuclei of MRC5-SV cells, an observation that was not reported in previous studies using whole-cell extracts from Pol κ-depleted cells (32, 37, 39). We verified that indeed there was no correlation between Chk1 and Pol κ protein levels in whole-cell extracts (R2 = 0.002 [Fig. 1E]) compared to nuclear extracts (R2 = 0.906 [Fig. 1A]), supporting that the impact on Chk1 level occurs reproducibly in the nuclear compartment. We next checked whether this effect was also observable in additional human cell lines. Fractionation of 293T human cell lines depleted of Pol κ showed a 50% reduction in Chk1 protein level in the nuclear fraction but not in the cytoplasmic fraction, where, in contrast, a slight increase could be observed (Fig. 2A). The reduction of Chk1 in the nucleus of a Pol κ-depleted cell did not rely only on its export from the nucleus to the cytosol, as its level in the cytoplasm did not compensate the decrease observed in the nucleus. This difference between nuclear and cytoplasmic fractions was confirmed in HCT116 cell extracts (Fig. 2B). To confirm with a second approach that Chk1 protein decreases in the nuclear compartment, the fluorescence intensity of Chk1 was monitored in more than 500 nuclei of HCT116 and RKO colon cancer cells after depletion of Pol κ with or without reintroduction of green fluorescent protein (GFP)-Pol κ (Fig. 2C and F). Transfection with small interfering RNA (siRNA) targeting Chk1 was used as a control to decrease the expression of Chk1 in these experiments. The fluorescence intensity of Chk1 decreased by 37% and 28%, respectively, in HCT116 and RKO cells transfected with an siRNA targeting the coding sequence of POLK (Fig. 2D and G) and by 27% in HCT116 cells transfected with siRNA targeting the 3′ untranslated region (UTR) of POLK (Fig. 2D). The Chk1 protein level was rescued by ectopic expression of Pol κ in HCT116 cells (Fig. 2C to E). These results were confirmed in RKO cells by Western blot analysis (Fig. 2H). Collectively, these data support the notion that Pol κ is required to maintain the Chk1 protein level in the nuclei of human cells.
FIG 1.

Chk1 protein level is reduced in the nuclei of MRC5-SV cells depleted of Pol κ. (A) (Left) Western blot analysis of Chk1 and Pol κ in MRC5-SV nuclear extracts 48 h after transfection with a control (Luc), Pol κ (Polκ), or Chk1 (Chk1) siRNA. Cells were left untreated or treated with 1 mM HU for 1 h. ORC4 is shown as a protein-loading control. Quantification of Chk1 is relative to siLuc condition. (Right) Relative Chk1 or Pol κ protein levels in siPolκ nuclear extracts were normalized to the siLuc condition; data from 5 independent experiments in MRC-SV cells are plotted. The regression curve (dashed line) and R-square are shown. (B) MRC5-SV cells were cotransfected with the indicated siRNAs and a vector expressing either GFP-empty or GFP-Pol κ (GFP-Polκ). The fluorescence intensity of Chk1 was quantified in each nucleus. Medians with 25% and 75% interquartile ranges are represented. ***, P = 0.001; ****, P < 0.0001; Mann-Whitney test. A.U, arbitrary units; ns, not significant. (C) Western blot analysis of Pol κ, GFP-Pol κ, and Chk1 in nuclear extracts of MRC5-SV cells 48 h after cotransfection with a control siRNA (Luc) or targeting the 3′ UTR of Pol κ (Polκ3′) with a vector expressing either GFP-empty or GFP-Polκ. MCM7 is shown as a loading control. (D) Western blot analysis of nuclear extracts from MRC5-SV cells transfected with a control (Luc), Polκ, or Chk1 siRNA. Immunodetected proteins are indicated. Fibrillarin and ORC2 are shown as loading controls. (E) Relative Chk1 or Pol κ protein levels in siPol κ whole-cell extracts of MRC5-SV cells were normalized to the siLuc condition. Data from 9 independent experiments are plotted. The regression curve (dashed line) and R-square are shown.
FIG 2.

Chk1 protein level is reduced in the nuclei of 293T, HCT116, and RKO cells depleted of Pol κ. (A and B) Western blot analysis of nuclear (NF) and cytoplasmic (CF) fractions from 293T (A) and HCT116 (B) cells, 48 h after transfection with a control (Luc) or Polκ siRNA. Quantifications of relative Chk1 band intensity from Western blots of 5 independent experiments with means ± SD. (C and F) HCT116 cells were transfected cotransfected with the indicated siRNAs and a vector expressing either GFP-empty or GFP-Polκ (C), and RKO cells were transfected with siRNA only (F). The fluorescence intensity of Chk1 was quantified in each nucleus. Medians with 25% and 75% interquartile ranges are represented. ***, P = 0.001; ****, P < 0.0001; Mann-Whitney test. (D and G) Relative fluorescence intensity of Chk1 in nuclei of HCT116 (D) and RKO (G) cells transfected with the indicated siRNAs (G) or both with a vector expressing either GFP-Polκ or GFP-empty (D). Values are the means (±SEM) of medians of three or four independent experiments. Relative fluorescence intensity was adjusted to the siLuc condition. *, P < 0.05; **, P < 0.01; ****, P < 0.0001; t test. (E) Western blot analysis of Pol κ and GFP-Polκ in nuclear extracts of HCT116 48 h after cotransfection with a control siRNA (Luc) or targeting the 3′ UTR of Pol κ (Polκ3′) with a vector expressing either GFP-empty or GFP-Polκ. MCM7 is shown as a loading control. (H) Western blot analysis of Chk1 and Pol κ in RKO nuclear extracts 48 h after transfection with a control siRNA (Luc) or Polκ siRNA. Fibrillarin is shown as a protein-loading control. Quantification of Chk1 is relative to the siLuc condition.
Among members of the Y-DNA polymerase family, only Pol κ depletion causes a Chk1 nuclear drop.
To check whether the effect on Chk1 is specific to Pol κ, 293T, HCT116, and RKO cells were transiently transfected with siRNA targeting the three other Y family TLS DNA polymerases, Pol η, Pol ι, and Rev1, and the Chk1 fluorescence intensity was measured (Fig. 3). While depletion of Chk1 itself, Pol κ, or USP7, an ubiquitin hydrolase already shown to stabilize Chk1 (18) triggered a Chk1 drop in the nucleus, the depletion of Pol ι, Pol η, or Rev1 did not, supporting that Chk1 is specifically regulated by Pol κ.
FIG 3.

Among members of the Y-DNA polymerase family, only Pol κ depletion causes a Chk1 nuclear drop. Representative images and quantification of Chk1 immunostaining (green) are shown. 293T (A), HCT116 (B) and RKO (C) cells were transfected with the indicated siRNAs, and DNA was stained with DAPI. The fluorescence intensity of Chk1 was quantified in each nucleus. Medians with 25% and 75% interquartile ranges are presented. ****, P < 0.0001; Mann-Whitney test.
The Pol κ-dependent Chk1 downregulation is not due to cell cycle arrest in G1.
The Chk1 protein level in human cells has been shown to accumulate in S and G2 phases and display its lowest level in G1 (45). To exclude the possibility that the Chk1 decrease in Pol κ-depleted cells was the consequence of an enrichment in G1 population, we combined the quantification of Chk1 fluorescent intensity with quantitative image-based cytometry (QIBC) (46) (Fig. 4A). First of all, we confirmed that under the control condition (siLuc) Chk1 accumulates from G1 to G2M and is reduced by Chk1 siRNA transfection. Interestingly, we found that Pol κ depletion impacted the Chk1 protein level in all the phases of the cell cycle (Fig. 4B). Then we compared the cell cycle distribution of Pol κ-depleted cells (siPolκ3′ and siPolκ) with control mocked-depleted cells (siLuc), obtained by QIBC and fluorescence-activated cell sorting (FACS) in HCT116 and RKO cells, respectively (Fig. 4C). We found that 30% to 38% of the Pol κ-depleted cells versus 39% in controls were in G1 in HCT116 cells (Fig. 4C, left) and 40% to 45% of the Pol κ depleted cells versus 43% in controls were in G1 in RKO cells (Fig. 4C, right), demonstrating that no enrichment in G1 cell population occurred in Pol κ-depleted HCT116 and RKO cells. Altogether, these observations indicate that Pol κ-dependent regulation of Chk1 does not result from a cell cycle modification. It is known that upon replication stress the activation of Chk1 can lead to the reduction of its protein level (44). To explore the role of checkpoint activation in the Pol κ-dependent Chk1 regulation, we treated HCT116 cells with VE821, an ATR inhibitor (ATRi) (Fig. 4D). The addition of the inhibitor reduced the phosphorylation of Chk1 on Ser345 induced by HU treatment in control and Pol κ-depleted cells. Moreover, the downregulation of Chk1 induced by the depletion of Pol κ was still observed in the presence of ATRi. Interestingly, when ATRi was added without HU treatment, the reduction of the Chk1 protein level in Pol κ-depleted cells compared to control cells was more important than under untreated conditions. As the depletion of ATR by siRNA has been shown to decrease the Chk1 protein level (11), our result suggests that ATR inhibition and Pol κ depletion lead to additive effects from two independent pathways to induce Chk1 decrease. Collectively, these data show that Chk1 instability in Pol κ-depleted cells is independent of a cell cycle modification or ATR checkpoint pathway activation.
FIG 4.
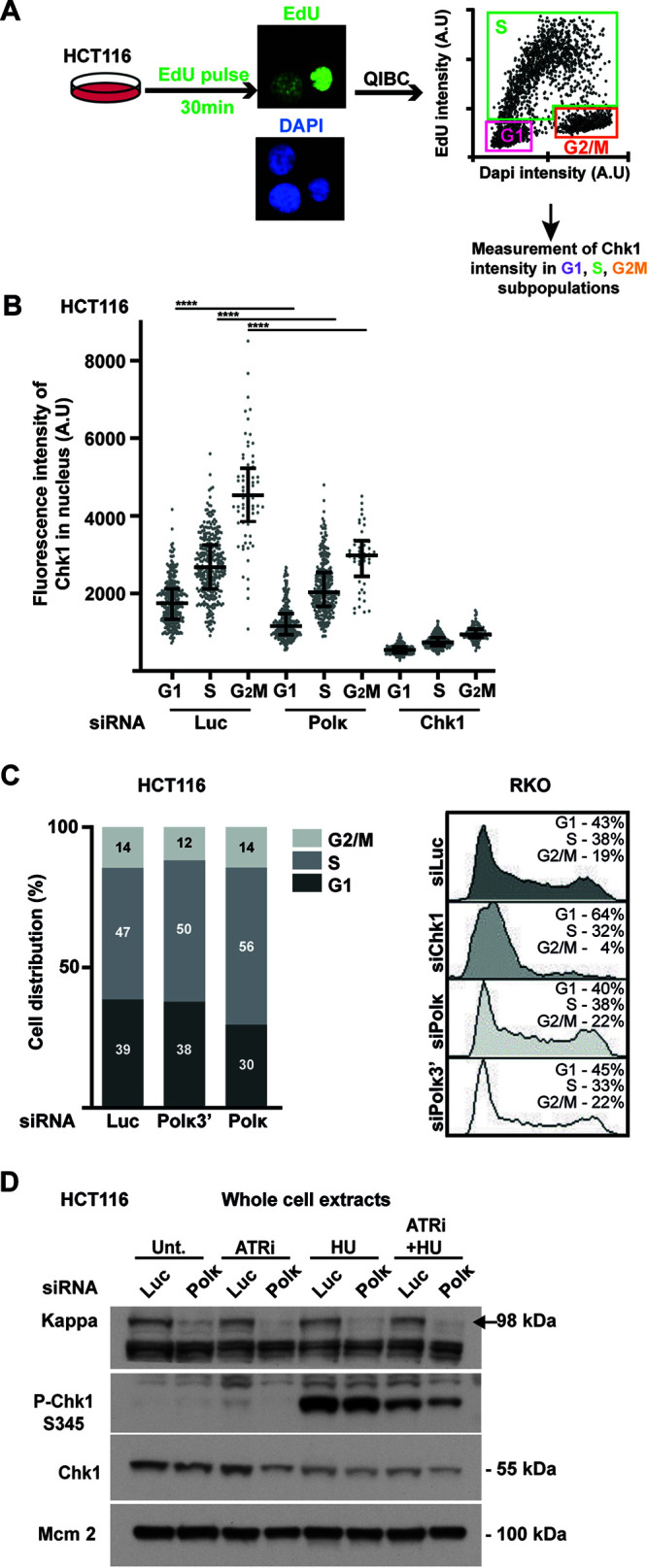
The Pol κ-dependent Chk1 downregulation is not due to cell cycle arrest in G1. (A) Schematic representation of EdU pulse-labeling experiment. HCT116 cells were transfected with control (Luc), Polκ, or Chk1 siRNA. At 48 h after transfection, asynchronous cells were pulse-labeled with 5-ethynyl-2′-deoxyuridine (EdU) (10 μM) for 30 min. Incorporated EdU was coupled to Alexa Fluor 488 in Click-iT reaction, and DNA was stained with DAPI. Quantitative image-based cytometry (QIBC) was performed. The EdU/DAPI dot plot shows the cell cycle distribution. (B) The fluorescence intensity of Chk1 was quantified in each nucleus of each cellular subpopulation transfected with indicated siRNA. Medians with 25% and 75% interquartile ranges are represented. (C) No G1 accumulation in Pol κ-depleted cells. Cell cycle distribution from QIBC analysis in HCT116 cells (left) or from FACS analysis in RKO cells (right) after transfection with the indicated siRNA is shown. The percentage of cells in each phase is indicated in the bar graphs or the FACS profiles. (D) Western blot analysis of Chk1 in whole-cell extracts from HCT116 cells transfected with a control (Luc) or Polκ siRNA. Cells were treated with ATRi (VE821, 10 μM, 3 h), HU (1 mM, 1 h), or ATRi plus HU. MCM2 is shown as a protein-loading control.
Pol κ controls Chk1 abundance independently of its DNA synthesis activity.
For the Pol κ functions such as replication of non-B structured DNA and repetitive sequences, bypass of DNA damages, and full activation of the S-phase checkpoint, the catalytic activity of the polymerase is required. To determine whether this is also true for maintaining the level of Chk1, cells depleted of Pol κ with a 3′ UTR POLK siRNA were transfected with a vector coding for the catalytically inactive form of Pol κ (GFP-Polκ-Dead), and Chk1 level was quantified by immunofluorescence. The results show that similar to the case with wild-type (WT) Pol κ (Fig. 1B and 2C), the Chk1 fluorescence intensity was also restored by expression of the dead Pol κ in HCT116 (Fig. 5A) and RKO (Fig. 5B) cells, demonstrating that the polymerase activity of Pol κ is not required to maintain the Chk1 protein level in human cells.
FIG 5.

Pol κ controls the Chk1 pool independently of its DNA synthesis activity. Quantification of Chk1 immunostaining in HCT116 (A) and RKO (B) cells cotransfected with the indicated siRNAs and with a control vector expressing GFP-empty or GFP-Polκ-Dead. At 48 h after transfection, the fluorescence intensity of Chk1 was quantified in each nucleus. Medians with 25% and 75% interquartile ranges are represented. ****, P < 0.0001; Mann-Whitney test.
Pol κ protects Chk1 from degradation.
To explore how Pol κ affects Chk1 expression, we first carried out reverse transcription and real-time quantitative PCR to analyze the CHEK1 mRNA level in Pol κ-depleted cells with two different siRNAs. We did not find any significant difference from control cells in four different cell lines (Fig. 6A). This observation rules out any potential implication of Pol κ in CHEK1 gene promoter repression, or its binding to the CHEK1 transcript, to explain the downregulation of Chk1. These data are in accordance with the observations presented in Fig. 5 showing that Pol κ reexpression could not restore Chk1 fluorescence induced by Chk1 siRNA-mediated depletion. Next, we monitored Chk1 protein stability by treating 293T cells with cycloheximide (CHX), an inhibitor of protein synthesis, and comparing the dynamics of Chk1 decrease for 7 h in siLuc- versus siPolκ-transfected cells. We found that Chk1 was less stable in Pol κ-depleted cells than in mock-depleted cells, with a shortened half-life (Fig. 6B). The same result was obtained upon the depletion of the deubiquitinylase USP7, which is known to protect Chk1 from proteasome degradation (18). It has been reported that Chk1 can be targeted by ubiquitin ligases to control its stability and its degradation through the proteasome (12–14). To test if Chk1 instability resulted from proteasomal degradation, we treated Pol κ-depleted cells with the proteasome inhibitor MG132 and monitored the level of Chk1 by immunoblotting (Fig. 6C to E). The results clearly showed in whole-cell extracts of three different cell lines and in subcellular fractions of HCT116 cells that proteasome inhibition led to a stabilization of Chk1 under Pol κ-depleted conditions (Fig. 6C and D). Treatment of HCT116 cells with CHX induced the decrease of Chk1 and p53 protein levels under the control condition, and only Chk1 was further affected under Pol κ depletion (Fig. 6E). Addition of MG132 restored Chk1 and p53 levels under CHX-treated conditions. Collectively, these data support that the defective Chk1 pool in Pol κ-depleted cells is not the consequence of a modification of its mRNA abundance but rather the result of its enhanced degradation by the proteasome, and thus, Pol κ seems to be a requisite factor to stabilize the Chk1 protein level in human cells.
FIG 6.

Pol κ protects Chk1 from degradation. (A) Transcript level analysis of Pol κ and Chk1 genes by RT-qPCR in MRC5-SV, HCT116, RKO, and 293T cells transfected with control siRNA (Luc), siRNA targeting the 3′ UTR of Pol κ (Polκ3′) or the coding sequence of Pol κ. Relative expressions were normalized to the siLuc condition. Values are the means (±SEM) of medians of independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; t test). (B) Western Blot analysis of Chk1 in 293T cells transfected with the indicated siRNA for 48 h and treated with 50 μg/ml of cycloheximide (CHX). Lysates were prepared at the indicated times after cycloheximide addition. Quantification of the Chk1 protein level is shown graphically (left). Ponceau is shown as a protein-loading control. (C) Western blot analysis of Chk1 in RKO, HCT116, and 293T cells. At 48 h after transfection with a control siRNA (Luc) or Pol κ siRNA (Polκ), cells were treated or not with MG132 (20 μM) for 4 h just after transfection and then 6 h before to harvest. Actin is shown as a protein-loading control. (D) Western blot analysis of NF and CF fractions from 293T cells. At 48 h after transfection with a control (Luc) or Polκ siRNA, cells were treated with 50 μg/ml of cycloheximide alone (Ch) or in combination with MG132 (20 μM, ch/MG) for 8 h. (E) Western Blot analysis of Chk1 in HCT116 cells extracts. At 48 h after transfection with indicated siRNA, cells were treated with 50 μg/ml of cycloheximide (CHX) in addition or not to MG132 (20 μM) for 5 h. Actin is shown as a protein-loading control.
Fork restart defects associated with Pol κ loss can be restored by Chk1 ectopic expression.
Considering the fact that Chk1 is required for the global genomic replication in the absence of exogenous stress (8, 47), we asked whether the depletion of Pol κ can affect the genome replication to the same extent. We pulse-labeled cells with 5-ethynyl-2′-deoxyuridine (EdU) and we measured the EdU intensity in the S-phase cell population as in Fig. 4A. We found that the depletion of Pol κ decreased the EdU incorporation to a similar extent as the Chk1 depletion (Fig. 7A and B), meaning that Pol κ is required to maintain the global replication in the absence of exogenous stress. Pol κ was shown to be required to restart stalled fork after HU treatment (32). As Chk1 is also implicated in this process (48, 49), we wondered whether the fork restart defect in Pol κ-deficient cells might be explained by low abundance of Chk1 induced by Pol κ depletion. To test this hypothesis, we performed DNA fiber spreading analysis. Cells were labeled with IdU 5′-iodo-2′-deoxyuridine (IdU), treated with HU during 1h at 1 mM, and released in fresh medium containing 5′-chloro-2′-deoxyuridine (CldU) to label the restarting forks (Fig. 7C). The length of the CldU tracts is indicative of the replication recovery efficiency after fork stalling. As shown in Fig. 7D, we found that Pol κ depletion significantly reduced the track length by 44% (2.39 μm in siPolκ versus 4.23 μm in control cells) and reintroduction of GFP-Polκ in siPolκ cells restored the CldU track length. Similarly to the case with Pol κ-depleted cells, we observed that in Chk1-depleted cells the track length is reduced by 46% (2.27 μm in siChk1 cells versus 4.23 μm in control cells). Strikingly, we observed that the inefficient fork recovery in Pol κ-depleted cells was restored by expression of ectopic myc-Chk1, whereas the expression of GFP-Pol κ could not restore the fork recovery in Chk1-depleted cells. Importantly, these data demonstrate that in addition to the role of Pol κ in the fork restart showed by Tonzi et al. (32), the deficiency of the replication stress recovery observed in Pol κ-depleted cells may rely on an insufficient pool of Chk1 in the nucleus.
FIG 7.

The replication defects associated with Pol κ loss can be restored by Chk1 ectopic expression. (A) Cells were transfected with control siRNA (Luc), siRNA targeting the 3′ UTR of Chk1 (Chk1-3′), or Pol κ (Polκ3′). At 48 h after transfection, asynchronous cells were pulse-labeled with EdU (10 μM) for 30 min. Nuclear EdU intensities were quantified in S-phase nuclei. Medians with 25% and 75% interquartile ranges are represented. ****, P < 0.0001; Mann-Whitney test. Number of values (nb values) and median values are indicated. (B) EdU intensity relative to control conditions was determined in 3 independent experiments, and the mean (±SEM) of medians relative to siLuc condition is presented. *, P < 0.05; **, P < 0.01; t test. (C) Schematic representation of experimental DNA fiber labeling. RKO cells were cotransfected with the indicated siRNAs and a vector expressing either GFP-empty, GFP-Polκ, or myc-Chk1. At 48 h after transfection, ongoing DNA replication forks were labeled with IdU (green tracks) for 20 min, treated with HU 1 mM for 1 h, and then labeled with CldU (red tracks) for 30 min. The lengths of CldU tracks were measured and the values of two independent experiments were pooled and plotted. The number of CldU tracks (nb values) measured and the medians of their lengths are indicated. Medians with 25% and 75% interquartile ranges are represented. ***, P = 0.001; ***, P < 0.001; Mann-Whitney test).
Collectively, all these findings highlight a noncatalytic function of Pol κ besides its well-documented TLS function to maintain proper DNA replication and ensure recovery from replication stress through the stability of Chk1.
DISCUSSION
In the absence of exogenous stress, the depletion of Pol κ is associated with increased rates of mutagenesis in mice, DNA breaks, sister-chromatid exchange, expression of common fragile site in human cells, and increased number of 53BP1 nuclear bodies in G1-phase cells (32, 37, 39, 41, 50, 51). These hallmarks of genetic instabilities can reflect a direct role of Pol κ in genomic replication. Indeed, Pol κ is required to copy repetitive sequences known to be fork-stalling sites for replicative DNA polymerase delta (28, 29), tolerate stress induced by oncogenes (31), and promote efficient fork progression under low levels of dNTP (32). We have also previously shown that upon activation of the replication checkpoint with HU, Chk1 phosphorylation is dependent on the presence of Pol κ (37), suggesting that Pol κ could be involved in the activation of the ATR-Chk1 axis. This has been further confirmed in human glioblastoma cell lines treated with the alkylating drug temozolomide (41). All the functions reported thus far have been demonstrated to require the catalytic DNA synthesis activity of the polymerase.
Here, we present evidence supporting a new catalytically independent role for Pol κ in cellular homeostasis through the regulation of Chk1 abundance in human cells. We provide evidence that the depletion of Pol κ in four independent cell lines induces a decrease of the Chk1 protein level. It is specific to Pol κ since this Chk1 reduction is (i) rescued by ectopic GFP-Pol κ and (ii) not shared by other DNA polymerases, consistent with previous studies in which no modification of Chk1 protein level was observed in Pol η-depleted or mutated cells (52). CHK1 knockout is lethal in mice and Chk1 haploinsufficiency leads to carcinogenesis (53). In contrast, the effects of Chk1 instability in Pol κ knockout mice are milder, with increased mutagenesis without affecting viability and with no cancer incidence (51), suggesting that the low level of Chk1 is enough to maintain genome stability and viability. Interestingly, González Besteiro and colleagues reported recently that indeed in Chk1-depleted cells, low levels of exogenous Chk1 were sufficient to rescue origin firing and restore DNA damage signaling (47).
The level of Chk1 in the nucleus depends on a tight equilibrium between protein synthesis, degradation, and nuclear export, and an excess or a lack of Chk1 can be deleterious for genome stability (49). Indeed, a higher abundance of Chk1 protein restricts the replication stress induced by oncogenes or therapeutic treatments and contributes to malignant transformation (54, 55). Conversely, CHK1 deficiency is associated with modifications of replication dynamics, mitotic defects, transmission of underreplicated DNA, and predisposition to cancer (7–10, 37, 56). The tumor suppressor p53 has been shown to regulate the mRNA level of Chk1 and to downregulate its expression in response to stress signals (57). Our data support that the Pol κ effect on Chk1 is independent of p53 since (i) the mRNA of Chk1 is unchanged in Pol κ-depleted cells and (ii) the reduction of Chk1 is observed in p53-proficient cells (RKO and HCT116) as well as p53-deficient cells (293T and MRC5-SV). We also showed that Chk1 downregulation cannot be the consequence of a cell cycle effect induced by the depletion of Pol κ and that Pol κ downregulates Chk1 all along the cell cycle.
Based on our results, we propose that the Chk1 decrease in Pol κ-depleted cells can be the consequence of a lack of protection against its normal degradation. We observed a higher rate of Chk1 disappearance in Pol κ-depleted cells, which can be antagonized by proteasomal inhibition. We propose that Pol κ is a regulator of Chk1 stability as was already observed for Rad17 and ETAA1, members of the ATR-Chk1 signaling pathway (11, 41). However, how Pol κ protects Chk1 from degradation remains to be determined.
Interestingly, we showed that the catalytic activity of Pol κ is not required to maintain the Chk1 protein level, as the GFP-Polκ-Dead also restored the Chk1 protein level in cells depleted of endogenous Pol κ. Noncatalytic function of other polymerases has been previously proposed. Pol η was shown to act as a bridge between PCNA and the ubiquitin-protein ligase Rad18 and to favor the monoubiquitination of PCNA by Rad18 independently of its polymerase activity (58). The C terminus of Rev1, but not the catalytic activity, is necessary to recruit the TLS DNA polymerases Pol η, Pol ι, and Pol κ for DNA damage tolerance (59). The Chk1-regulatory function of Pol κ, which is independent of its catalytic activity, could help in understanding some recent observations from the literature. Human lymphoblastic Nalm6 cells engineered to express a catalytically dead Pol κ mutant showed the same sensitivity to oxidative stress induced by hydrogen peroxide and menadione as Pol κ wild-type cells, while Pol κ knockout counterpart cells were highly sensitive (35). Temprine and colleagues have associated the increased expression of Pol κ to drug resistance but without a high rate of mutagenesis. They propose a noncatalytic function of Pol κ to explain the drug resistance (42). Pol κ can also protect forks against nascent DNA degradation dependent on the fork-remodeler SMARCAL-1 and the nuclease MRE11. The authors showed that the polymerase activity is not essential to perform this task, but they did not demonstrate how Pol κ mediates its fork protection function (32). It has already been demonstrated that the Chk1 activity is required to protect forks from nuclease activities (60–62). Thus, we propose that the stabilization by Pol κ of Chk1 may prevent fork degradation and break accumulation and may explain the catalytically independent function of Pol κ in the fork protection mechanism.
The maintenance of a basal replication checkpoint activity in unchallenged cells is an important concept that is reinforced with recent publications (5, 11). The idea presented by Panagopoulos and Altmeyer is that facing endogenous replication stress due to local impediments (repeats or structured DNA) or dNTP pool decrease, cells adapt instead of arrest their cell cycle progression. They proposed a fine-tuned deceleration and brake release mechanism dependent on the ATR-Chk1 axis (5). Thus, a constant basal activity of Chk1 is prerequisite and Pol κ could be an actor of this regulation, as in response to huge replication stress the dependency on Pol κ to restart stalled forks is less obvious (32).
Thus, Pol κ could act at stalled forks to maintain genome stability in several ways: insertion of nucleotides in front of lesions (TLS function), elongation of primers to allow S-phase checkpoint activation (S-phase checkpoint-associated function), and regulation of the abundance of Chk1 in the nucleus (Chk1 regulator). The first two roles depend on its polymerase activity, whereas the third one does not (Fig. 8).
FIG 8.
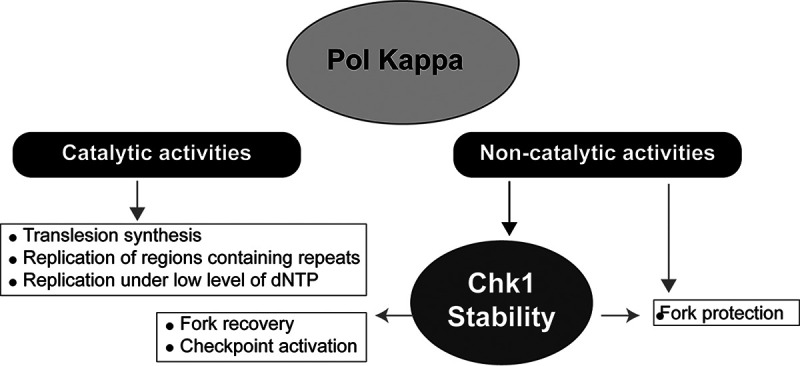
Working model. Under physiological conditions, the DNA polymerase activity of Pol κ is required to perform translesion synthesis, to replicate regions of the genome that contain repetitive sequences or local dNTP imbalance, to overcome fork stalling due to endogenous impediments, and to participate in the S-phase checkpoint activation. In addition, the noncatalytic function of Pol κ may be fundamental to protect forks from degradation and to stabilize Chk1, which, in turn, acts to coordinate the response to fork stalling.
In conclusion, besides the well-documented importance of the catalytic function of the DNA Pol κ for translesion synthesis, restart of stalled forks under low levels of dNTP, and checkpoint activation, our work here unveils an unprecedented described DNA synthesis-independent regulatory function of Pol κ to protect forks from degradation and maintain basal level activity of replication checkpoint.
MATERIALS AND METHODS
Cell culture and cell lines.
RKO, 293T, and HCT116 cells were cultured in Dulbecco modified Eagle medium (DMEM)/GlutaMAX supplemented 10% fetal bovine serum (FBS; GIBCO) and MRC5-SV in alpha-MEM/GlutaMAX with 10% FBS (GIBCO) at 37°C in a humidified incubator in an atmosphere containing 5% CO2 (Panasonic MCO-19AIC-PE). All cells were routinely checked for mycoplasma contamination using PlasmoTest kit (InvivoGen).
Drugs and cell culture supplement.
When indicated, cells were treated with hydroxyurea (Sigma-Aldrich), proteasome inhibitor MG132 (APExBIO), cycloheximide (Biosciences), and ATR inhibitor VE821 (Sigma-Aldrich) at doses and for times indicated in the figure legends. The dNTP analogs CldU and IdU (Sigma-Aldrich) were used as indicated in figures and in “DNA fiber assay” below.
siRNA and plasmid transfections.
A total of 1.5 × 106 to 2 × 106 cells were seeded 24 h before transfection with 45 nM siRNA using Lipofectamine RNAiMAX (Life Technologies). The following siRNA molecules used targeting the coding sequence of Pol κ (Polκ), 5′CCAAUAGACAAGCUGUGAU3′ from Sigma-Aldrich; the 3′ UTR of Pol κ (Polκ3′), 5′ACUCCAGCCUGAAGAGCGA3′ from Sigma-Aldrich; the coding sequence of USP7 (USP7), 5′CCCAAAUUAUUCCGCGGCAAA3′ from Sigma-Aldrich; the coding sequence of Chk1 (Chk1), 5′GAAGCAGUCGCAGUGAAGA3′ from Sigma-Aldrich; the 3′ UTR of Chk1 (Chk1-3′), 5′CUGGUGAAUAUAGUGCUGCUA3′ from Sigma-Aldrich; the coding sequence of Polι (iota), SMART pool (5′CCACAGUUGGUAUUAGUUA3′, 5′GCACUAUGGUCGUGAGAGU3′, 5′CGGGUCAUGUAUACAAUAA3′, and 5′GAACAUCAGGCUUUAAUAG3′ from Dharmacon); the 3′ UTR of Polη (eta), 5′GCAAUGAGGGCCUUGAACA3′ from Sigma-Aldrich; and the coding sequence of Rev1 (Rev1), 5′CAGCGCAUCUGUGCCAAAGAA3′ from Sigma-Aldrich. Control siRNA against luciferase and the catalytically inactive mutant of human Pol κ (Dead Pol κ; D198A and E199A) were previously described by Bétous et al. (37). The wild-type human Pol κ coding sequence was cloned into the peGFP vector (peGFP-Kappa_WT). Transfections of 4 to 8 μg of plasmids were carried out using JetPrime (Polyplus transfection) following the manufacturer’s recommendations.
Cell extracts, Western blotting, and quantification.
Nuclear extracts were obtained by using the NE-PER kit and nuclear and cytoplasmic extracts were obtained by using a subcellular protein fractionation kit (Thermo Scientific) according to the manufacturer’s recommendations. For whole-cell extracts, cells were lysed in a buffer containing 50 mM Tris (pH 7.5), 300 mM NaCl, 1% Triton, 5 mM EDTA, and 1 mM dithiothreitol (DTT), supplemented with inhibitors. Cell lysates were cleared by centrifugation for 10 min at 10,000 rpm. Cell extracts were boiled in loading buffer (Bio-Rad; 4× Laemmli sample buffer) with 0.1 M DTT.
Proteins were dosed by the Bradford method (Bio-Rad) and separated by SDS-PAGE (Life Technologies; NUPAGE, 4 to 12% or 3 to 8%). For immunoblotting, primary antibodies were incubated overnight at 4°C in 1× Tris-buffered saline (TBS) with 0.1% Tween. Secondary peroxidase-coupled antibodies (Jackson ImmunoResearch, Life Technologies) were incubated at room temperature for 1 h. Blots were detected by ECL plus Western blotting substrate (Pierce) or ECL Bright Quantum (Diagomix) on Blue Devil autoradiography film (Genesee Scientific) or with the ChemiDoc imaging system (Bio-Rad). Primary antibodies were used at the following dilutions: Pol κ (from T. Nohmi), 1/1,000; Pol κ (from Sigma; HPA012035), 1/1,000; Chk1 (Abcam; ab32531), 1/1,000; Chk1-pS345 (Cell Signaling; 2348), 1/1,000; H2AX (Abcam; ab11175), 1/4,000; ORC2 (MBL; M055-3), 1/1,000; ORC4 (Transduction Laboratories; H83120), 1/500; MCM7 (Santa Cruz; sc22782), 1/1,000; MCM2 (Abcam; ab4461), 1/2,000; actin (Millipore; MAB1501), 1/10,000; actinin (Millipore; 05-384), 1/2,000; fibrillarin (Sigma-Aldrich; SAB4300633), 1/1,000; and p53 (BD Sciences; 554294), 1/2,000. Where indicated, proteins were quantified using ImageJ software.
RNA extraction and quantitative PCR analysis.
Total RNA was extracted from cell using the RNeasy kit (Qiagen) according to the supplier’s instructions, and then 1 μg of total RNA was reverse transcribed into cDNA using Superscript II (Invitrogen) according to the manufacturer’s recommendations. Duplicate quantitative PCR assays were run on the StepOne real-time system from Applied Biosystems with TaqMan Universal Master Mix II and specific probes from the assay on demand (Applied Biosystems). Three housekeeping genes (ACTIN B, YWHAZ, and GAPDH) were also amplified and used as references. The relative amount of each mRNA level was normalized to the control condition and calculated using the threshold cycle (ΔΔCT) method.
EdU incorporation, QIBC, and Chk1 immunofluorescence.
For immunostaining, transfected cell lines used for immunofluorescence were plated on an 8-well chamber slide (Lab-Tek). When EdU incorporation was performed, EdU was added 15 or 30 min at a final concentration of 10 μM in the culture media of exponentially growing cells. Fixation was done with a 4% paraformaldehyde (PFA)/phosphate-buffered saline (PBS) solution for 10 min, and then a permeabilization and saturation step was performed in 1× PBS containing 3% bovine serum albumin (BSA) and 0.1× Triton for 30 min (PBS-Triton/BSA). Detection of EdU was performed prior to incubation with the primary antibodies using the Click-iT Plus EdU–Alexa Fluor 488 imaging kit according to the manufacturer’s instructions (Thermo Fisher Scientific). Chk1 primary antibody (Abcam; ab32531, 1/1,000) was diluted in PBS-Triton/BSA and incubated for 1 h at room temperature. The 8-well chamber slides were washed three times with 1× PBS and incubated in PBS-Triton/BSA containing fluorescent secondary antibodies at 1/800 (Alexa fluorophores; Life Technologies) for 30 min. Three washes were performed and nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). For mounting the slides, Prolong Diamond antifade reagent (Thermo Fisher Scientific) was used. Ten to 20 images were acquired randomly. After acquisition, the images were processed for automated analysis with Cell Profiler image analysis software. DAPI signal was used for segmentation of the nuclei according to intensity threshold, generating a mask which identified each individual nucleus as an individual object. This mask was applied to quantify pixel intensities in the different channels for each individual cell/object. The values quantified for EdU and DAPI staining per cell were graph plotted by dual-parameter (EdU versus DNA)-generating diagrams in a flow cytometry-like fashion (quantitative-based image cytometry [QIBC]) for each cell condition. This approach allows the assignment of cells to G1, S, or G2/M phase.
Microscope image acquisition and analysis.
Image acquisition of multiple random fields was carried out on a wide-field Nikon Eclipse Ni-E microscope equipped with a 63× oil immersion objective Nikon Plan Apo 1.4 λ using a C-mos DsQi2 camera driven by NIS-Elements AR software. Fluorescence quantifications were performed with Cell Profiler 2.1.1. Images were assembled with Adobe Photoshop and Adobe Illustrator.
DNA fiber assay.
Cells were pulse-labeled with 50 μM ldU for 20 min, washed, and then treated with HU (1 mM) for 1 h. After removal of HU and washing, ongoing DNA fibers were labeled in medium containing CldU at a 100 μM final concentration for 30 min. The cells were harvested and lysed in 200 mM Tris-HCl (pH 7.4), 0.5% SDS, and 50 mM EDTA, and the DNA fibers were spread on glass slides. The slides were incubated with 0.5 mg/ml of pepsin in 30 mM HCl at 37°C for 20 min, and the DNA was denatured in 2.5 M HCl for 1 h and blocked with 1% BSA containing 0.1% Tween 20 in 1× PBS. The nucleotide analogues were detected with primary antibodies against CldU (Novus) and IdU (BD Biosciences) and the secondary antibodies anti-rat antibody–Alexa Fluor 555 and anti-mouse antibody–Alexa Fluor 488 (Thermo Fisher Scientific). Coverslips were mounted on slides using the Prolong Diamond antifade reagent (Thermo Fisher Scientific). Images were captured with a Nikon Ni-E microscope and a DS-Qi2 camera equipped with a 20× objective, and lengths of CldU tracks were measured with NIS-Elements AR imaging software.
Statistical analysis.
GraphPad Prism version 5.03 for Windows was used for statistical analysis. Differences were considered statistically significant at a P value of <0.05. Data are reported as the medians with 25% to 75% interquartile ranges or as the means ± standard errors of the means (SEM). Results were compared by 2-tailed Student’s t test for two groups or a Mann-Whitney nonparametric test as indicated in figure legends.
ACKNOWLEDGMENTS
We thank V. Bergoglio and S. Manenti from the Cancer Research Center of Toulouse for critical reading of the manuscript and helpful discussions. We thank Takehiko Nohmi (National Institute of Health Sciences, Japan) and Masami Yamada (National Institute of Health Sciences, Japan) for the Rabbit anti-Polκ antibodies. We thank S. Britton (IPBS, CNRS Toulouse) for technical support.
M. Dall’Osto was supported by the Region Languedoc Roussillon Midi Pyrénées/INSERM fellowship (U1037-R16068BB-Region). This work was supported by funding from INCa-PLBIO 2016, ANR PRC 2016, Labex Toucan, and La Ligue contre le Cancer (Equipe labelisée) to J.-S.H., as well as Ligue Régionale grant RAB17004BBA to M.-J.P.
M.-J.P., M.D., and L.P. performed experiments. N.V. performed the rescue experiments with the GFP-Pol κ vector. M.D. and M.-J.P. conceived the study design. M.D. carried out data analysis with the supervision of M.-J.P. M.D., M.-J.P., and J.-S.H. wrote the manuscript. J.-S.H. and M.-J.P. acquired the funding.
We have no financial or other conflict of interest to declare.
Contributor Information
Jean-Sébastien Hoffmann, Email: jean-sebastien.hoffmann@inserm.fr.
Marie-Jeanne Pillaire, Email: marie-jeanne.pillaire@inserm.fr.
REFERENCES
- 1.Zeman MK, Cimprich KA. 2014. Causes and consequences of replication stress. Nat Cell Biol 16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Técher H, Koundrioukoff S, Nicolas A, Debatisse M. 2017. The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat Rev Genet 18:535–550. doi: 10.1038/nrg.2017.46. [DOI] [PubMed] [Google Scholar]
- 3.González Besteiro MA, Gottifredi V. 2015. The fork and the kinase: a DNA replication tale from a CHK1 perspective. Mutat Res Rev Mutat Res 763:168–180. doi: 10.1016/j.mrrev.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y, Hunter T. 2014. Roles of Chk1 in cell biology and cancer therapy. Int J Cancer 134:1013–1023. doi: 10.1002/ijc.28226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Panagopoulos A, Altmeyer M. 2 December 2020. The hammer and the dance of cell cycle control. Trends Biochem Sci doi: 10.1016/j.tibs.2020.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Lecona E, Fernandez-Capetillo O. 2018. Targeting ATR in cancer. Nat Rev Cancer 18:586–595. doi: 10.1038/s41568-018-0034-3. [DOI] [PubMed] [Google Scholar]
- 7.Lam MH, Liu Q, Elledge SJ, Rosen JM. 2004. Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell 6:45–59. doi: 10.1016/j.ccr.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 8.Petermann E, Maya-Mendoza A, Zachos G, Gillespie DAF, Jackson DA, Caldecott KW. 2006. Chk1 requirement for high global rates of replication fork progression during normal vertebrate S phase. Mol Cell Biol 26:3319–3326. doi: 10.1128/MCB.26.8.3319-3326.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petermann E, Woodcock M, Helleday T. 2010. Chk1 promotes replication fork progression by controlling replication initiation. Proc Natl Acad Sci USA 107:16090–16095. doi: 10.1073/pnas.1005031107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Técher H, Koundrioukoff S, Carignon S, Wilhelm T, Millot GA, Lopez BS, Brison O, Debatisse M. 2016. Signaling from Mus81-Eme2-dependent DNA damage elicited by Chk1 deficiency modulates replication fork speed and origin usage. Cell Rep 14:1114–1127. doi: 10.1016/j.celrep.2015.12.093. [DOI] [PubMed] [Google Scholar]
- 11.Michelena J, Gatti M, Teloni F, Imhof R, Altmeyer M. 2019. Basal CHK1 activity safeguards its stability to maintain intrinsic S-phase checkpoint functions. J Cell Biol 218:2865–2875. doi: 10.1083/jcb.201902085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y-W, Brognard J, Coughlin C, You Z, Dolled-Filhart M, Aslanian A, Manning G, Abraham RT, Hunter T. 2009. The F box protein Fbx6 regulates Chk1 stability and cellular sensitivity to replication stress. Mol Cell 35:442–453. doi: 10.1016/j.molcel.2009.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y-W, Otterness DM, Chiang GG, Xie W, Liu Y-C, Mercurio F, Abraham RT. 2005. Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol Cell 19:607–618. doi: 10.1016/j.molcel.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 14.Cassidy KB, Bang S, Kurokawa M, Gerber SA. 2020. Direct regulation of Chk1 protein stability by E3 ubiquitin ligase HUWE1. FEBS J 287:1985–1999. doi: 10.1111/febs.15132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng Y-C, Shieh S-Y. 2018. Deubiquitinating enzyme USP3 controls CHK1 chromatin association and activation. Proc Natl Acad Sci USA 115:5546–5551. doi: 10.1073/pnas.1719856115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guervilly J-H, Renaud E, Takata M, Rosselli F. 2011. USP1 deubiquitinase maintains phosphorylated CHK1 by limiting its DDB1-dependent degradation. Hum Mol Genet 20:2171–2181. doi: 10.1093/hmg/ddr103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tu Y, Liu H, Zhu X, Shen H, Ma X, Wang F, Huang M, Gong J, Li X, Wang Y, Guo C, Tang T-S. 2017. Ataxin-3 promotes genome integrity by stabilizing Chk1. Nucleic Acids Res 45:4532–4549. doi: 10.1093/nar/gkx095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alonso-de Vega I, Martín Y, Smits VAJ. 2014. USP7 controls Chk1 protein stability by direct deubiquitination. Cell Cycle 13:3921–3926. doi: 10.4161/15384101.2014.973324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo C, Kosarek-Stancel JN, Tang T-S, Friedberg EC. 2009. Y-family DNA polymerases in mammalian cells. Cell Mol Life Sci 66:2363–2381. doi: 10.1007/s00018-009-0024-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McVey M, Khodaverdian VY, Meyer D, Cerqueira PG, Heyer W-D. 2016. Eukaryotic DNA polymerases in homologous recombination. Annu Rev Genet 50:393–421. doi: 10.1146/annurev-genet-120215-035243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ogi T, Limsirichaikul S, Overmeer RM, Volker M, Takenaka K, Cloney R, Nakazawa Y, Niimi A, Miki Y, Jaspers NG, Mullenders LHF, Yamashita S, Fousteri MI, Lehmann AR. 2010. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol Cell 37:714–727. doi: 10.1016/j.molcel.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 22.Lange SS, Takata K, Wood RD. 2011. DNA polymerases and cancer. Nat Rev Cancer 11:96–110. doi: 10.1038/nrc2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bournique E, Dall’Osto M, Hoffmann J-S, Bergoglio V. 2018. Role of specialized DNA polymerases in the limitation of replicative stress and DNA damage transmission. Mutat Res 808:62–73. doi: 10.1016/j.mrfmmm.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 24.Tsao W-C, Eckert KA. 2018. Detours to replication: functions of specialized dna polymerases during oncogene-induced replication stress. Int J Mol Sci 19:3255. doi: 10.3390/ijms19103255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pillaire M-J, Bétous R, Hoffmann J-S. 2014. Role of DNA polymerase κ in the maintenance of genomic stability. Mol Cell Oncol 1:e29902. doi: 10.4161/mco.29902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tonzi P, Huang TT. 2019. Role of Y-family translesion DNA polymerases in replication stress: implications for new cancer therapeutic targets. DNA Repair (Amst) 78:20–26. doi: 10.1016/j.dnarep.2019.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bergoglio V, Boyer A-S, Walsh E, Naim V, Legube G, Lee MYWT, Rey L, Rosselli F, Cazaux C, Eckert KA, Hoffmann J-S. 2013. DNA synthesis by Pol η promotes fragile site stability by preventing under-replicated DNA in mitosis. J Cell Biol 201:395–408. doi: 10.1083/jcb.201207066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarkies P, Reams C, Simpson LJ, Sale JE. 2010. Epigenetic instability due to defective replication of structured DNA. Mol Cell 40:703–713. doi: 10.1016/j.molcel.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barnes RP, Hile SE, Lee MY, Eckert KA. 2017. DNA polymerases eta and kappa exchange with the polymerase delta holoenzyme to complete common fragile site synthesis. DNA Repair (Amst) 57:1–11. doi: 10.1016/j.dnarep.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walsh E, Wang X, Lee MY, Eckert KA. 2013. Mechanism of replicative DNA polymerase delta pausing and a potential role for DNA polymerase kappa in common fragile site replication. J Mol Biol 425:232–243. doi: 10.1016/j.jmb.2012.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Y, Gao Y, Mutter-Rottmayer L, Zlatanou A, Durando M, Ding W, Wyatt D, Ramsden D, Tanoue Y, Tateishi S, Vaziri C. 2017. DNA repair factor RAD18 and DNA polymerase Polκ confer tolerance of oncogenic DNA replication stress. J Cell Biol 216:3097–3115. doi: 10.1083/jcb.201702006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tonzi P, Yin Y, Lee CWT, Rothenberg E, Huang TT. 2018. Translesion polymerase kappa-dependent DNA synthesis underlies replication fork recovery. Elife 7:e41426. doi: 10.7554/eLife.41426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lone S, Townson SA, Uljon SN, Johnson RE, Brahma A, Nair DT, Prakash S, Prakash L, Aggarwal AK. 2007. Human DNA polymerase kappa encircles DNA: implications for mismatch extension and lesion bypass. Mol Cell 25:601–614. doi: 10.1016/j.molcel.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 34.Gerlach VL, Feaver WJ, Fischhaber PL, Friedberg EC. 2001. Purification and characterization of polκ, a DNA polymerase encoded by the human DINB1 gene. J Biol Chem 276:92–98. doi: 10.1074/jbc.M004413200. [DOI] [PubMed] [Google Scholar]
- 35.Kanemaru Y, Suzuki T, Niimi N, Grúz P, Matsumoto K, Adachi N, Honma M, Nohmi T. 2015. Catalytic and non-catalytic roles of DNA polymerase κ in the protection of human cells against genotoxic stresses. Environ Mol Mutagen 56:650–662. doi: 10.1002/em.21961. [DOI] [PubMed] [Google Scholar]
- 36.Williams HL, Gottesman ME, Gautier J. 2012. Replication-independent repair of DNA interstrand crosslinks. Mol Cell 47:140–147. doi: 10.1016/j.molcel.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bétous R, Pillaire M-J, Pierini L, van der Laan S, Recolin B, Ohl-Séguy E, Guo C, Niimi N, Grúz P, Nohmi T, Friedberg E, Cazaux C, Maiorano D, Hoffmann J-S. 2013. DNA polymerase κ-dependent DNA synthesis at stalled replication forks is important for CHK1 activation. EMBO J 32:2172–2185. doi: 10.1038/emboj.2013.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bavoux C, Leopoldino AM, Bergoglio V, O-Wang J, Ogi T, Bieth A, Judde J-G, Pena SDJ, Poupon M-F, Helleday T, Tagawa M, Machado C, Hoffmann J-S, Cazaux C. 2005. Up-regulation of the error-prone DNA polymerase κ promotes pleiotropic genetic alterations and tumorigenesis. Cancer Res 65:325–330. [PubMed] [Google Scholar]
- 39.Mansilla SF, Bertolin AP, Bergoglio V, Pillaire M-J, González Besteiro MA, Luzzani C, Miriuka SG, Cazaux C, Hoffmann J-S, Gottifredi V. 2016. Cyclin kinase-independent role of p21CDKN1A in the promotion of nascent DNA elongation in unstressed cells. Elife 5:e18020. doi: 10.7554/eLife.18020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pillaire M-J, Betous R, Conti C, Czaplicki J, Pasero P, Bensimon A, Cazaux C, Hoffmann J-S. 2007. Upregulation of error-prone DNA polymerases beta and kappa slows down fork progression without activating the replication checkpoint. Cell Cycle 6:471–477. doi: 10.4161/cc.6.4.3857. [DOI] [PubMed] [Google Scholar]
- 41.Peng C, Chen Z, Wang S, Wang H-W, Qiu W, Zhao L, Xu R, Luo H, Chen Y, Chen D, You Y, Liu N, Wang H. 2016. The error-prone DNA polymerase κ promotes temozolomide resistance in glioblastoma through Rad17-dependent activation of ATR-Chk1 signaling. Cancer Res 76:2340–2353. doi: 10.1158/0008-5472.CAN-15-1884. [DOI] [PubMed] [Google Scholar]
- 42.Temprine K, Campbell NR, Huang R, Langdon EM, Simon-Vermot T, Mehta K, Clapp A, Chipman M, White RM. 2020. Regulation of the error-prone DNA polymerase Polκ by oncogenic signaling and its contribution to drug resistance. Sci Signal 13:eaau1453. doi: 10.1126/scisignal.aau1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gagou ME, Zuazua-Villar P, Meuth M. 2010. Enhanced H2AX phosphorylation, DNA replication fork arrest, and cell death in the absence of Chk1. Mol Biol Cell 21:739–752. doi: 10.1091/mbc.e09-07-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leung-Pineda V, Huh J, Piwnica-Worms H. 2009. DDB1 targets Chk1 to the Cul4 E3 ligase complex in normal cycling cells and in cells experiencing replication stress. Cancer Res 69:2630–2637. doi: 10.1158/0008-5472.CAN-08-3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaneko YS, Watanabe N, Morisaki H, Akita H, Fujimoto A, Tominaga K, Terasawa M, Tachibana A, Ikeda K, Nakanishi M, Kaneko Y. 1999. Cell-cycle-dependent and ATM-independent expression of human Chk1 kinase. Oncogene 18:3673–3681. doi: 10.1038/sj.onc.1202706. [DOI] [PubMed] [Google Scholar]
- 46.Toledo LI, Altmeyer M, Rask M-B, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, Lukas J. 2013. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155:1088–1103. doi: 10.1016/j.cell.2013.10.043. [DOI] [PubMed] [Google Scholar]
- 47.González Besteiro MA, Calzetta NL, Loureiro SM, Habif M, Bétous R, Pillaire M-J, Maffia A, Sabbioneda S, Hoffmann J-S, Gottifredi V. 2019. Chk1 loss creates replication barriers that compromise cell survival independently of excess origin firing. EMBO J 38:e101284. doi: 10.15252/embj.2018101284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hromas R, Williamson EA, Fnu S, Lee Y-J, Park S-J, Beck BD, You J-S, Leitao A, Laitao A, Nickoloff JA, Lee S-H. 2012. Chk1 phosphorylation of Metnase enhances DNA repair but inhibits replication fork restart. Oncogene 31:4245–4254. doi: 10.1038/onc.2011.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smits VAJ, Gillespie DA. 2015. DNA damage control: regulation and functions of checkpoint kinase 1. FEBS J 282:3681–3692. doi: 10.1111/febs.13387. [DOI] [PubMed] [Google Scholar]
- 50.Hakura A, Sui H, Sonoda J, Matsuda T, Nohmi T. 2019. DNA polymerase kappa counteracts inflammation-induced mutagenesis in multiple organs of mice. Environ Mol Mutagen 60:320–330. doi: 10.1002/em.22272. [DOI] [PubMed] [Google Scholar]
- 51.Stancel JNK, McDaniel LD, Velasco S, Richardson J, Guo C, Friedberg EC. 2009. Polk mutant mice have a spontaneous mutator phenotype. DNA Repair (Amst) 8:1355–1362. doi: 10.1016/j.dnarep.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Despras E, Daboussi F, Hyrien O, Marheineke K, Kannouche PL. 2010. ATR/Chk1 pathway is essential for resumption of DNA synthesis and cell survival in UV-irradiated XP variant cells. Hum Mol Genet 19:1690–1701. doi: 10.1093/hmg/ddq046. [DOI] [PubMed] [Google Scholar]
- 53.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ. 2000. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 54.David L, Fernandez-Vidal A, Bertoli S, Grgurevic S, Lepage B, Deshaies D, Prade N, Cartel M, Larrue C, Sarry J-E, Delabesse E, Cazaux C, Didier C, Récher C, Manenti S, Hoffmann J-S. 2016. CHK1 as a therapeutic target to bypass chemoresistance in AML. Sci Signal 9:ra90. doi: 10.1126/scisignal.aac9704. [DOI] [PubMed] [Google Scholar]
- 55.López-Contreras AJ, Gutierrez-Martinez P, Specks J, Rodrigo-Perez S, Fernandez-Capetillo O. 2012. An extra allele of Chk1 limits oncogene-induced replicative stress and promotes transformation. J Exp Med 209:455–461. doi: 10.1084/jem.20112147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Speroni J, Federico MB, Mansilla SF, Soria G, Gottifredi V. 2012. Kinase-independent function of checkpoint kinase 1 (Chk1) in the replication of damaged DNA. Proc Natl Acad Sci USA 109:7344–7349. doi: 10.1073/pnas.1116345109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gottifredi V, Karni-Schmidt O, Shieh SS, Prives C. 2001. p53 down-regulates CHK1 through p21 and the retinoblastoma protein. Mol Cell Biol 21:1066–1076. doi: 10.1128/MCB.21.4.1066-1076.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Durando M, Tateishi S, Vaziri C. 2013. A non-catalytic role of DNA polymerase η in recruiting Rad18 and promoting PCNA monoubiquitination at stalled replication forks. Nucleic Acids Res 41:3079–3093. doi: 10.1093/nar/gkt016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tissier A, Kannouche P, Reck M-P, Lehmann AR, Fuchs RPP, Cordonnier A. 2004. Co-localization in replication foci and interaction of human Y-family members, DNA polymerase pol eta and REVl protein. DNA Repair (Amst) 3:1503–1514. doi: 10.1016/j.dnarep.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 60.Forment JV, Blasius M, Guerini I, Jackson SP. 2011. Structure-specific DNA endonuclease Mus81/Eme1 generates DNA damage caused by Chk1 inactivation. PLoS One 6:e23517. doi: 10.1371/journal.pone.0023517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Murfuni I, Basile G, Subramanyam S, Malacaria E, Bignami M, Spies M, Franchitto A, Pichierri P. 2013. Survival of the replication checkpoint deficient cells requires MUS81-RAD52 function. PLoS Genet 9:e1003910. doi: 10.1371/journal.pgen.1003910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thompson R, Montano R, Eastman A. 2012. The Mre11 nuclease is critical for the sensitivity of cells to Chk1 inhibition. PLoS One 7:e44021. doi: 10.1371/journal.pone.0044021. [DOI] [PMC free article] [PubMed] [Google Scholar]