

ABSTRACT
Staphylococcus aureus is an important human pathogen that can cause a variety of diseases ranging from mild superficial skin infections to life-threatening conditions like necrotizing pneumonia, endocarditis, and septicemia. Polymorphonuclear leukocytes (PMNs; neutrophils in particular herein) are essential for host defense against S. aureus infections, and the microbe is phagocytosed readily. Most ingested bacteria are killed, but some S. aureus strains—such as the epidemic USA300 strain—have an enhanced ability to cause PMN lysis after phagocytosis. Although progress has been made, the mechanism for lysis after phagocytosis of S. aureus remains incompletely determined. Here, we tested the hypothesis that disruption of phagosome integrity and escape of S. aureus from the PMN phagosome into the cytoplasm precedes PMN lysis. We used USA300 wild-type and isogenic deletion strains to evaluate and/or verify the role of selected S. aureus molecules in this cytolytic process. Compared to the wild-type USA300 strain, Δagr, Δhla, ΔlukGH, and Δpsm strains each caused significantly less lysis of human PMNs 3 h and/or 6 h after phagocytosis, consistent with previous studies. Most notably, confocal microscopy coupled with selective permeabilization assays demonstrated that phagosome membrane integrity is largely maintained prior to PMN lysis after S. aureus phagocytosis. We conclude that PMN lysis does not require escape of S. aureus from the phagosome to the cytoplasm and that these are independent phenomena. The findings are consistent with the ability of S. aureus (via selected molecules) to trigger lysis of human PMNs by an undetermined signaling mechanism.
IMPORTANCE S. aureus strain USA300 has the ability to cause rapid lysis of human neutrophils after phagocytosis. Although this phenomenon likely contributes to the success of USA300 as a human pathogen, our knowledge of the mechanism remains incomplete. Here, we used a selective permeabilization assay coupled with confocal microscopy to demonstrate that USA300 is contained within human neutrophil phagosomes until the point of host cell lysis. Thus, consistent with a process in macrophages, S. aureus fails to escape into the neutrophil cytoplasm prior to cytolysis.
KEYWORDS: PMN, MRSA, USA300, phagocytosis, cytolysis, cell death, Staphylococcus aureus, neutrophils
INTRODUCTION
Staphylococcus aureus is a human commensal bacterium and opportunistic pathogen (1). Inasmuch as all humans have antibody specific for S. aureus and ∼60% are either permanently or intermittently colonized, the level of exposure is high (2). In addition, S. aureus is notorious for its ability to acquire resistance to multiple classes of antibiotics, and antibiotic resistance—most notably resistance to beta-lactam antibiotics—has been a major problem for treatment of S. aureus infections for decades (3, 4). Indeed, methicillin-resistant S. aureus (MRSA) is considered a serious public health threat by the U.S. Centers for Disease Control and Prevention (https://www.cdc.gov/mrsa/healthcare/index.html). MRSA strains classified as USA300 by molecular typing are prominent in community (community-associated MRSA [CA-MRSA]) and health care settings in the United States. (5–10). CA-MRSA strains, such as USA300, are notable because they can cause infections in otherwise healthy individuals. The pathogen produces numerous molecules that have been shown to contribute to virulence in animal infection models, and many promote evasion of killing by neutrophils in vitro.
Polymorphonuclear leukocytes (PMNs; neutrophils in particular herein) are essential for defense against bacterial and fungal infections (11, 12). Individuals with low PMN counts or congenital PMN disorders are susceptible to recurrent and/or severe infections, especially those caused by S. aureus (13). Thus, the enhanced ability of USA300 to circumvent killing by PMNs is consistent with its ability to cause infections in healthy individuals. This attribute is not unusual for S. aureus. For example, landmark studies in the 1950s by Rogers and colleagues revealed that some S. aureus strains can avoid killing by PMNs in vitro and in vivo in rabbits and can ultimately cause neutrophil lysis after phagocytosis (14–16). The authors proposed a model whereby S. aureus is ingested rapidly by PMNs, which then migrate into tissues and lyse, thereby releasing any viable microbes (16). Similar work using a mouse infection model was performed by Gresham et al. decades later (17). More recently, work with human PMNs demonstrated that USA300 promotes rapid (within 3 h) lysis of PMNs after phagocytosis (18–20). Subsequent studies have shown that the process is distinct from apoptosis and has characteristics of programmed necrosis or necroptosis (19, 21). However, the mechanism for PMN lysis after phagocytosis of S. aureus remains incompletely determined.
A number of S. aureus molecules and gene regulators have been proposed to be involved in this cytolytic process, including alpha-hemolysin (Hla), leukocidin GH (LukGH) (also known as leukocidin AB [LukAB]), alpha-type phenol-soluble modulins (PSMα), accessory gene regulator (Agr), and SaeRS (22–27). Here, we used USA300 wild-type and mutant strains combined with confocal laser scanning microscopy to gain additional insight into the mechanism that underlies this cytolytic process in human PMNs.
RESULTS
Verification of the phenotype associated with PMN lysis after phagocytosis of S. aureus.
As a step toward gaining insight into the mechanism of neutrophil lysis after phagocytosis of S. aureus, we verified the ability of USA300 strain LAC (representative of the USA300 epidemic clone of the early 2000s) to cause rapid lysis of human PMNs as reported previously (Fig. 1A) (19). Consistent with previous work, PMN lysis after phagocytosis of S. aureus was time dependent and increased with higher bacterial cell-to-PMN ratios (Fig. 1A). Inasmuch as the S. aureus growth phase has a significant impact on the production of cytolytic toxins in vitro (28–30), we tested whether culture of USA300 to exponential or stationary phases of growth alters PMN lysis after phagocytosis (Fig. 1B). There was no significant difference in the rates of PMN lysis after phagocytosis at 3 h and 6 h using USA300 cultured to exponential- (optical density at 600 nm [OD600] = 0.75) or stationary- (OD600 = 1.75) growth phases (Fig. 1B). These findings indicate that the culture conditions tested here had no bearing on lysis after PMN phagocytosis. PMN lysis was completely prevented by killing of USA300 with heat or UV light, as described previously (Fig. 1C) (19). We next used lysostaphin (LS) to kill extracellular S. aureus, and there was a significant reduction in PMN lysis if lysostaphin was added at the start of the assay, but not when it was added at later time points (Fig. 1D). These results are consistent with the known rapid PMN phagocytosis of S. aureus, as intracellular S. aureus are protected from killing by lysostaphin. To test this notion more directly, we treated PMNs with cytochalasin D (to inhibit phagocytosis) or added lysostaphin concurrently (prior to the start of the assay) (Fig. 1E). Cytochalasin D alone failed to prevent PMN lysis by 6 h after culture with USA300, although there were many bacteria associated with (surface bound) or in close proximity to all PMNs. This finding merits further investigation. On the other hand, the combination of cytochalasin D and lysostaphin prevented PMN lysis almost completely (PMN lysis was 2.0% ± 3.4% [mean ± standard deviation], versus 50.1% ± 10.6% for treated and untreated cultures; P < 0.0001) (Fig. 1E). In addition, supernatant from S. aureus cultured alone for 6 h in RPMI assay medium—an estimate of the maximum potential cytolytic capacity of the medium in 6-h PMN assays if bacteria are not phagocytosed—caused limited PMN lysis (7.8% ± 7.0%) (Fig. 1E). Taken together, these results provide further strong support to the idea that PMN lysis in these assays is caused largely by ingested USA300 cells and not toxins secreted into the culture medium.
FIG 1.

Human PMN lysis after phagocytosis of USA300 strain LAC. (A) PMN lysis is dependent on time and bacterial cell-to-PMN ratio (n = 4). (B) PMN lysis is not influenced by bacterial growth phase (n ≥ 4). (C) PMN lysis after phagocytosis requires live S. aureus. Bacteria were heat killed (LAC HK) at 95°C for 5 min or UV irradiated (LAC UV) at 4,000 × 100 μJ/cm2 (n = 3). (D) Phagocytosed S. aureus are protected from killing by lysostaphin (LS). LS (6.5 U/107 CFU) was added to assays at the times indicated, and USA300-mediated PMN lysis was determined as described in Materials and Methods. (E) PMN lysis can be caused by intracellular and/or extracellular staphylococci. Cytochalasin D (CD; 10 μg/ml) was added to PMNs 15 min prior to addition of bacteria. LS was added 30 min after the start of the experiment when added alone (LAC+LS) or 15 min prior to addition of bacteria when added concurrently with CD (LAC+LS+CD) (n ≥ 3). To generate culture supernatant (SN), overnight cultures of LAC were diluted 1:100 into prewarmed RPMI/H and grown for 6 h at 37°C as described in Materials and Methods. Data are presented as mean values ± standard deviations (SD), and assays whose results are shown in panels B to E were performed using a 10:1 bacterial cell/PMN ratio. (A) Data were analyzed using repeated-measures ANOVA and Tukey’s posttest. Asterisks marking data colored blue and red indicate a P value of <0.05 versus all other bacterial cell/PMN ratios at 4 h and versus 1:1 and 2:1 ratios at 6 h. (B) Results were analyzed using an unpaired two-tailed t test. ns, not significant. (C, D) Data were analyzed using repeated-measures ANOVA and Tukey’s posttest. **, P < 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001. (E) Data were analyzed with mixed-effects analysis and Dunnett’s posttest. ****, P ≤ 0.0001.
Multiple S. aureus cytolytic toxins contribute to PMN lysis after phagocytosis.
Inasmuch as multiple S. aureus cytolytic toxins effect leukocyte lysis in vitro, we next evaluated the ability of USA300 wild-type and isogenic mutant strains to cause PMN lysis after phagocytosis (Fig. 2). In general, S. aureus is known to be ingested readily by human neutrophils (16). Consistent with previous studies (18, 31), USA300 was phagocytosed readily by human PMNs (phagocytosis was ≥80.2% ± 0.01% within 40 min for all strains tested), and there were no differences in uptake between wild-type and mutant strains. In contrast, PMN lysis at 3 h was decreased significantly following phagocytosis of ΔlukGH (P < 0.0001), Δpsmα (P = 0.02), Δhla (P = 0.03), and Δagr (P < 0.0001) strains compared to the phagocytosis of the USA300 wild-type strain (Fig. 2A). The findings with ΔlukGH, Δpsmα, and Δagr strains were also observed at 6 h, although differences compared to the wild-type strain were reduced as the level of PMN cytolysis increased over time (Fig. 2B). We selected ΔlukGH as a representative strain with which to confirm specificity. Complementation of the ΔlukGH strain with lukGH on a plasmid (ΔlukGH::plukGH) restored cytolytic capacity in full, thereby verifying that LukGH alone can contribute to PMN lysis (Fig. 2C). The addition of IgG specific for LukG to these assays also blocked PMN lysis significantly, providing further support to the idea that LukGH promotes lysis after phagocytosis (Fig. 2C). Collectively, these results confirm previous work with selected S. aureus molecules and, additionally, provide data that bears on the relative contribution of each to PMN lysis after phagocytosis (Fig. 2A and B) (22–27).
FIG 2.

Lysis of human PMNs following phagocytic interaction with LAC wild-type and isogenic mutant strains. (A, B) PMN lysis 3 h (A) or 6 h (B) after incubation with the indicated strains. Each red circle indicates the results for a separate experiment (n = 10 to 16 [A] and n = 6 to 13 [B]). (C) PMN lysis was determined following phagocytosis of wild-type, ΔlukGH, or complemented ΔlukGH strain (left) or with or without anti-LukG antibody (αLukG) or control IgG as indicated (right) (n ≥ 5). Lysostaphin (LS) was added 30 min after the start of the assay to kill extracellular S. aureus (right) (n ≥ 6). Assays were performed using a 10:1 bacterial cell/PMN ratio, and results are presented as the mean values ± SD. (A, B) Data were analyzed using a mixed-effects analysis and Dunnett’s posttest. (C) Data were analyzed using a mixed-effects analysis and Tukey’s (left) or Dunnett’s (right) posttest. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
S. aureus-containing phagosomes remain intact during PMN lysis.
We next tested whether escape of S. aureus from the phagosome to the cytoplasm precedes PMN lysis after phagocytosis. Digitonin can be used to selectively permeabilize the plasma membrane of eukaryotic cells (after fixation) without damaging intracellular membranes and has been used to measure vacuolar escape of intracellular pathogens like Francisella tularensis and Salmonella enterica serotype Typhimurium (32–34). Therefore, we developed a selective-permeabilization assay with digitonin that permeabilizes the PMN plasma membrane but not the phagosome (Fig. 3A to C). S. aureus was visualized by direct labeling with fluorescein isothiocyanate (FITC) and with IgG specific for S. aureus (anti-S. aureus antibody), which was used to determine the accessibility of S. aureus following PMN phagocytosis, chemical fixation, and permeabilization with digitonin or Triton X-100 (Fig. 3B and C). Notably, Triton X-100 permeabilized both plasma membranes and phagosome membranes of fixed PMNs, thereby permitting cytoplasmic-actin staining with phalloidin (Fig. 3A, green), staining of intraphagosomal S. aureus with anti-S. aureus antibody (Fig. 3C, red S. aureus), and labeling of S. aureus-containing phagosomes with LAMP1 (Fig. 4). In comparison, digitonin permeabilizes plasma membranes (Fig. 3A, green) but not phagosome membranes. Therefore, fixed PMNs with ingested FITC-labeled strain LAC failed to stain with anti-S. aureus antibody after treatment with digitonin (Fig. 3B, green S. aureus only—no red antibody stain). Selective permeabilization of the plasma membrane by digitonin was then used to estimate phagosome integrity in subsequent experiments. We used CD16 as a PMN plasma membrane marker, and staining of PMNs with anti-CD16 antibody revealed a circumscribed staining pattern consistent with the presence of plasma membrane (Fig. 4A and 5; Fig. S2 in the supplemental material). PMNs containing USA300 at 30 min had circumscribed staining with CD16, whereas it was absent in some of the PMNs associated with/containing bacteria at 3 h (a time point at which PMN lysis is typically first detected) (Fig. 5). Using this methodology, we were able to estimate phagosome integrity during the process of PMN lysis, including the specific sequence of events (i.e., whether loss of phagosome integrity occurred in the presence or absence of an intact plasma membrane). In accordance with the PMN lysis assays, the percentage of S. aureus-associated PMNs (FITC positive [FITC+]) that retained plasma membrane staining (CD16+) and contained intact phagosomes (anti-S. aureus antibody negative [αSa−]) decreased over time (from 88% ± 2.8% at 30 min to 27.8% ± 11.3% at 3 h) (Fig. 5A, B, and F, green squares). Consistent with this finding, the percentage of PMNs that lost both plasma membrane staining (CD16−) and phagosome integrity (FITC+ anti-S. aureus antibody positive [αSa+]) increased in parallel, but only to a limited extent over the time period tested (from 0% to 14.5% ± 12.8% at 3 h) (Fig. 5D and F, red circles). This phenotype represents fully lysed PMNs (Fig. 5F, red circles). In comparison, there was a significant increase in the number of PMNs that lacked plasma membrane staining but contained intact S. aureus phagosomes (from 0% at 30 min to 38.2% ± 8.3% at 3 h) (Fig. 5C and F, green circles; Table S1). Moreover, the percentage of PMNs that retained CD16 staining at the plasma membrane and contained disrupted S. aureus phagosomes—a phenotype consistent with escape of S. aureus to the cytoplasm prior to lysis—was low (5.5% ± 6.2% at 3 h) and did not change significantly over time (Fig. 5E and F, red squares; Table S1). Taken together, these findings indicate that the plasma membrane is likely compromised prior to disruption of the phagosome membrane. The results are compatible with a cytolytic process that does not require escape of S. aureus from the PMN phagosome.
FIG 3.
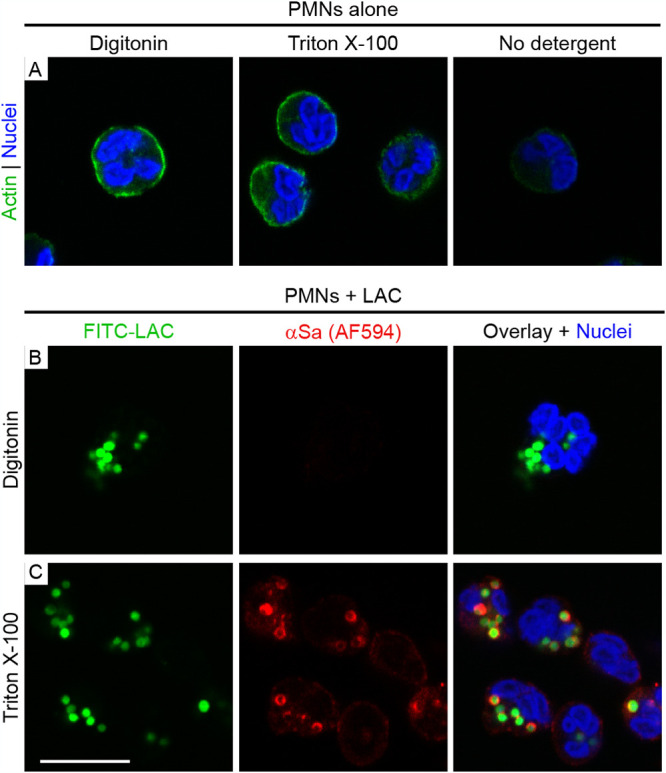
Selective permeabilization of the PMN plasma membrane by digitonin. (A) Digitonin and Triton X-100 each permeabilize the plasma membrane of fixed PMNs, as demonstrated by cytoplasmic-actin staining with AF488-phalloidin. Nuclei were stained with NucBlue. (B, C) LAC was labeled with FITC (FITC-LAC) prior to the start of the experiment and is pseudocolored green in all panels. The digitonin treatment conditions used did not permit staining of intraphagosomal bacteria with anti-S. aureus antibody (αSa) coupled to AF594 (B), whereas intraphagosomal bacteria are stained readily after treatment of PMN with Triton X-100 (C). Scale bar = 10 μm.
FIG 4.
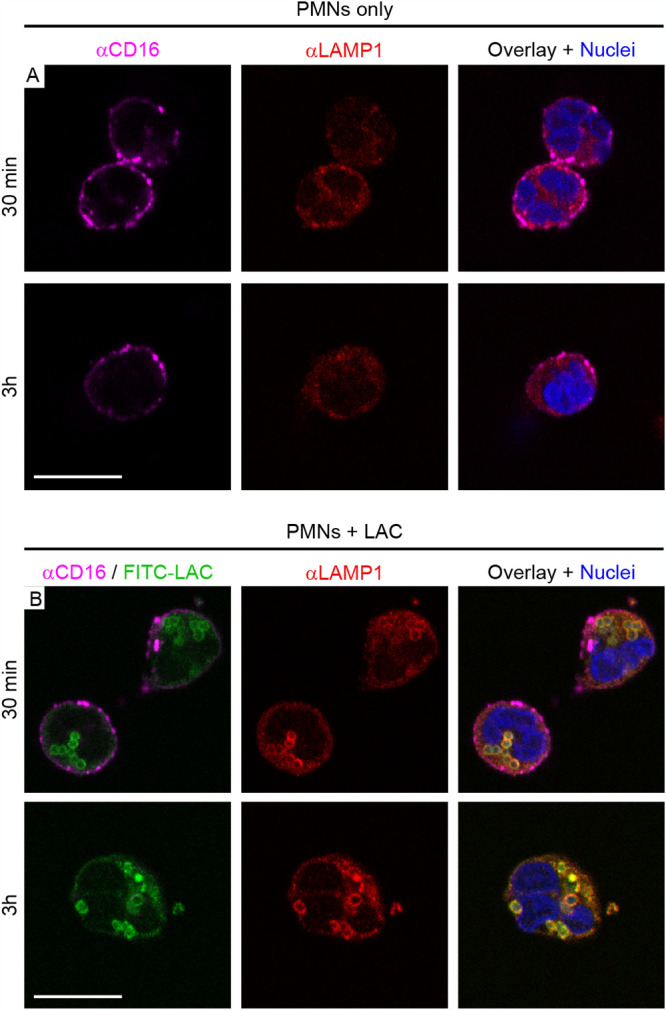
Subcellular distribution of CD16 and LAMP1 in human PMNs following phagocytosis of S. aureus (strain LAC). (A) Staining patterns of control PMNs. At the indicated time points, control PMNs (no bacteria) were labeled with antibody specific for CD16 (αCD16), fixed with paraformaldehyde, permeabilized with Triton X-100, and subsequently labeled with antibody specific for LAMP1 (αLAMP1) as described in Materials and Methods. Nuclei were stained with NucBlue. (B) Staining patterns in PMNs following phagocytosis of S. aureus. Strain LAC was labeled with FITC (FITC-LAC) prior to the start of the phagocytosis experiment and is pseudocolored green in all panels. At the indicated time points, PMNs were labeled with αCD16 and αLAMP1 antibody as described for panel A. Scale bar = 10 μm.
FIG 5.

PMN-S. aureus phenotypes as observed by confocal microscopy. For all panels, PMNs were combined with S. aureus for 30 min or 3 h, labeled with anti-CD16 antibody (αCD16), fixed with paraformaldehyde, permeabilized selectively with digitonin, and stained as indicated. (A, B) CD16-positive PMNs with intact S. aureus phagosomes (CD16+ FITC+ αSa− [anti-S. aureus negative]). (C) CD16-negative PMNs with intact S. aureus phagosomes (CD16− FITC+ αSa−) and a juxtaposed CD16+ PMN lacking S. aureus. (D) CD16-negative PMNs with disrupted S. aureus phagosomes (CD16− FITC+ αSa+) next to a CD16-positive PMN with intact phagosomes. (E) CD16-positive PMN with a single disrupted S. aureus phagosome (CD16+ FITC+ αSa+). Arrows in all panels indicate representative staining patterns for PMNs (and associated phagosomes) that were used to quantitate each phenotype as labeled on the right. Scale bar = 10 μm. (F) Quantification of the PMN-S. aureus phenotypes described for panels A to E. Green symbols and solid lines indicate intact phagosomes; red symbols and dashed lines indicate loss of phagosome integrity. Results are presented as the mean values ± SD from 4 separate experiments. Statistics were performed using repeated-measures nonparametric ANOVA (Friedman test) with Dunn’s posttest to correct for multiple comparisons. *, P < 0.05 for 3 h versus 0.5 h; †, P < 0.05 versus red square (2 h, 3 h) or red circle (1 h).
DISCUSSION
Although most S. aureus strains are readily ingested by PMNs and killed, some strains, such as USA300, have the ability to circumvent killing by PMNs and cause rapid lysis of these important immune cells (18–21). Inasmuch as neutrophils comprise the major cellular component of host defense against S. aureus infections, this cytolytic phenomenon has the potential to influence the virulence of a strain and its success as a human pathogen. Although progress has been made in understanding this phenomenon, the cellular and molecular events that culminate with PMN lysis after phagocytosis of S. aureus remain incompletely determined.
Work with nonprofessional phagocytic cells (nonphagocytes) has provided intriguing insights into host cell-S. aureus interactions, including host cell death mechanisms. Numerous studies have demonstrated that S. aureus can be ingested by nonphagocytes in vitro and that the microbes reside within a membrane-bound vacuole often characterized as a phagolysosome (35–44). Some of the sequestered bacteria then escape into the cytoplasm and cause host cell death and/or lysis, albeit the process is strain dependent and can differ among types of host cells (35–37, 45, 46). The ability of S. aureus to escape from the phagolysosome of nonphagocytes is dependent on a functional accessory gene regulator (Agr) system and may involve PSMs and β-toxin (38, 46–49). Recently, Stelzner et al. reported that S. aureus ingested by nonphagocytes disrupts intracellular calcium homeostasis, which in turn leads to cell death by apoptosis and/or necrosis (50). These results provide support to the idea that intracellular S. aureus triggers cytolysis/cell death by altering metabolic processes of host cells rather than targeting the plasma membrane via cytolytic toxins.
The cell death process in professional phagocytes following phagocytosis of S. aureus appears distinct from that of nonphagocytes. For example, Kubica et al. reported that S. aureus ingested by human monocyte-derived macrophages survived for several days within an intracellular vacuole (51). This intracellular persistence required S. aureus alpha-toxin and was followed by macrophage lysis and release of the pathogen (51). More recent studies have also shown that S. aureus is contained within phagosomes of macrophages and macrophage-like cell lines until the point of cell death and lysis (reviewed by Moldovan and Fraunholz [52]). Although previous work by Grosz et al. found limited (∼5%) escape of USA300 strain LAC from THP-1 phagosomes (53), studies by Tranchemontagne et al., Jubrail et al., and Flannagan et al. demonstrate that S. aureus is retained fully in macrophage phagosomes (54–57). Jubrail et al. found that as the ratio of bacteria per macrophage increases, bacteria are continually phagocytosed but the efficiency of intracellular killing is decreased (54). These viable intracellular S. aureus can then replicate within the phagosome and ultimately cause host cell death (55, 57). This macrophage death process has features of apoptosis and necrosis, which suggests the host cell undergoes programmed cell death (55). This finding, combined with the observation that S. aureus is contained within an intact phagolysosome until the point of macrophage cytolysis, indicates that any contribution of S. aureus toxins to the process is indirect. It is noteworthy that the replication of USA300 within spent macrophages is optimal within (or requires) an acidic intracellular compartment, such as the phagolysosome (57). Such a process has the potential to promote a continuous cycle of phagocytosis, intracellular replication, and lysis, thereby leading to persistence of S. aureus in tissues.
PMN lysis after S. aureus phagocytosis is also accompanied by characteristics of apoptosis and necrosis, although the mechanism remains incompletely understood (19–21). Our previous studies used ultrastructural analyses to provide evidence that S. aureus is retained within the PMN phagosome until cytolysis (19). Here, we used a selective permeabilization assay to demonstrate that the integrity of the S. aureus phagosome is largely maintained until the PMN undergoes cytolysis. These data provide strong support to the idea that there is little or no escape of ingested S. aureus from the phagosome to the cytoplasm of human PMNs. To our knowledge, no study to date has demonstrated the escape of S. aureus from PMN phagosomes. Our results are thus consistent with findings generated using macrophages and macrophage cell lines (55–57).
Previous studies identified S. aureus molecules, including cytolytic toxins, that contribute to PMN lysis after phagocytosis (22–25). Here, we compared the ability of USA300 wild-type and isogenic mutant strains to cause lysis of human PMNs after phagocytosis. Among the toxin mutant strains tested, the reduction in cytolysis (versus the cytolysis caused by the wild-type strain) was greatest following uptake of the ΔlukGH mutant strain (Fig. 2). Interestingly, lukGH is upregulated in human blood and the promoter is preferentially activated following exposure to PMNs (58, 59). Moreover, a genome-wide screen identified LukGH as a prominent effector of PMN lysis caused by USA300 (26). DuMont et al. identified CD11b as the cell surface receptor for LukAB (also known as LukGH) and showed that cytolysis of host cells by this leukocidin occurs through binding of CD11b on the cell surface (59). CD11b would also be accessible to intraphagosomal S. aureus, based on the topology of the phagosomal membrane. It is possible that binding of LukGH to its receptor (or receptors) present in the phagosome membrane initiates a signaling cascade that ultimately leads to PMN lysis.
In summary, we show that PMN lysis after phagocytosis of S. aureus does not require escape of the pathogen into the cytoplasm. Based on these and previous studies, the contribution of S. aureus toxins to this cytolytic process is likely indirect and involves signaling or triggering of PMN death from within. More work is needed to identify the signaling pathways involved in this process and thereby elucidate the specific mechanism for cytolysis.
MATERIALS AND METHODS
Statement of ethics.
Venous blood was obtained from healthy human volunteers in accordance with a protocol (01IN055) approved by the Institutional Review Board for human subjects, National Institutes of Health (Bethesda, MD, USA), and the research follows the principles of the Declaration of Helsinki. All volunteers gave written informed consent prior to participation.
Strains and culture conditions.
S. aureus strain LAC (and pulsed-field type USA300 in general [60, 61]) has been well characterized (18, 19, 31, 60–63). All mutant strains used in this study were derived from the LAC wild-type strain. The ΔlukGH, Δhla, Δhlg, Δpvl, and Δpsmα strains were described previously (23, 58, 64–66). A new LACΔlukGH::plukGH strain was constructed as described previously (63), and LACΔlukED was constructed as described below. All strains were grown overnight in Trypticase soy broth (TSB; BD Biosciences) with shaking at 250 rpm or on Trypticase soy agar (TSA; BD Biosciences) at 37°C. Overnight cultures of the ΔlukGH::plukGH strain were supplemented with 12.5 μg/ml tetracycline to ensure retention of the complementation plasmid. For PMN lysis assays, the ΔlukGH::plukGH strain was grown in the presence of 0.5% xylose to an OD600 of 0.75 to induce production of LukGH. E. coli One Shot OmniMAX 2 T1 phage-resistant cells (Invitrogen) used during the construction of the ΔlukED strain were grown at 37°C with shaking at 250 rpm in the presence of 100 μg/ml ampicillin in Luria Bertani broth or on Luria Bertani agar (LB; BD Biosciences).
Construction of USA300 LACΔlukED.
Isogenic deletion of lukED from the LAC chromosome was conducted according to Schneewind and Missiakas (67). PCRs were performed using Phusion high-fidelity DNA polymerase (NEB), and chromosomal DNA was purified from LAC wild type using the DNeasy blood and tissue kit (Qiagen). Gateway cloning was performed using the Gateway BP Clonase II enzyme mix (Invitrogen) according to the manufacturer’s instructions. The primer pairs used for amplification of 1-kbp regions upstream from lukD and downstream from lukE were as follows: upstream fragment, attB1_LukD_up_for (GGGGACAAGTTTGTACAAAAAAGCAGGCTGTTGAAGTTAAGGCCTACTTC) and LukD_up_rev_BamHI (GGATCCCTGTTAGAAATAGAGGAAGTTAAACCCGAAATTTAAGAGTTAAAC), and downstream fragment, LukE_down_for_BamHI (GGATCCAAGTTTCACTTTCTTTCTATATAAATTTTTAATACAAATTTATTCT) and attB2_LukE_down_rev (GGGGACCACTTTGTACAAGAAAGCTGGGTCCATCTGGATTATCGTGAGT). Amplicons were digested using BamHI-HF (NEB) and subsequently ligated with T4 DNA ligase (NEB). The plasmid pKOR1ΔlukED was passaged via S. aureus strain RN4220 and electroporated into strain LAC. Integration and excision of the plasmid into and from the chromosome of LAC were achieved by temperature shifts as described previously (67). Deletion of lukED from the LAC chromosome and maintenance of virulence plasmids pUSA01 and pUSA03 were verified by PCR and whole-genome sequencing.
Isolation of human PMNs.
Human PMNs were isolated from heparinized venous blood of healthy volunteers by using dextran sedimentation and Ficoll-Hypaque gradient centrifugation as described previously (68). The purity and viability of PMNs were determined by flow cytometry (FACSCalibur; BD Biosciences) as described previously (68). PMN preparations typically contain 98 to 99% granulocytes, and the viability was typically greater than 98%.
PMN lysis.
Lysis of human PMNs was measured by the release of lactate dehydrogenase (LDH) using the Roche cytotoxicity detection kit plus (LDH) (Roche Applied Sciences) and a microplate spectrophotometer (SpectraMax plus; Molecular Devices) according to the manufacturer’s instructions and as described previously (19). In summary, bacteria were cultured to early-exponential (OD600 of 0.75) or early-stationary (OD600 of 1.75) phases of growth, washed with Dulbecco’s phosphate-buffered saline (DPBS), and opsonized with 50% normal human serum (NHS) for 30 min at 37°C with shaking. Opsonized bacteria were then resuspended at the desired concentration in RPMI 1640 medium (Invitrogen) buffered with 10 mM HEPES (RPMI/H, pH 7.2). Bacteria were diluted in DPBS and plated on TSA in triplicates to verify CFU. For assays comparing live and dead bacteria, staphylococci were heated at 95°C for 5 min or exposed to UV light (4,000 × 100 μJ/cm2 count down mode) using a Hoefer UV cross-linker (Thermo Fisher) and plated on TSA to ensure no viable bacteria remained. All assays were conducted in 96-well flat-bottom plates precoated with 20% NHS for 30 min at 37°C. Unless indicated otherwise, 1 × 106 PMNs were incubated with 1 × 107 bacteria in a total volume of 0.2 ml. Plates were centrifuged at 524 × g for 8 min at 4°C to synchronize phagocytosis and then incubated at 37°C with 5% CO2. In some assays, lysostaphin (LS) (product number L7386, 6.5 U/107 CFU; Sigma-Aldrich) was added to the PMN assays to kill extracellular bacteria. LS killed strain LAC efficiently using our assay conditions, and wells containing PMNs plus LS alone (no bacteria) were used as controls. Where indicated, cytochalasin D (CD) (product number C8273; Sigma-Aldrich) was used at 10 μg/ml to inhibit phagocytosis. PMNs were incubated with CD or the combination of CD and LS for 15 min at 37°C with 5% CO2 prior to the addition of bacteria. The ability of CD to inhibit phagocytosis was verified for each experiment by light microscopy (Zeiss Axioscope 5 and Zeiss EC Plan-Neofluar 100× oil objective lens; Carl Zeiss Microscopy, LLC). Images were captured by using a Zeiss Axiocam 208 color camera (Carl Zeiss Microscopy, LLC) (Fig. S1). CD failed to induce PMN lysis in control assays. Bacterial supernatant was generated for use in neutrophil lysis assays as follows. Overnight cultures of LAC were diluted 1:100 into RPMI/H prewarmed at 37°C and grown for 3 h (∼OD600 of 0.4) or 6 h (∼OD600 of 0.8). Cultures were then centrifuged at ambient temperature for 10 min at 3,000 × g, supernatant was sterile filtered, and a 0.1-ml aliquot was added to wells containing PMNs. Microplates were incubated at 37°C for the indicated times, and PMN lysis was determined using the assay kit as described above. Untreated PMNs and bacteria alone were used as negative controls, and PMNs treated with 3% Triton X-100 were used as positive controls for PMN lysis. Lysis caused by Triton X-100 was set at 100%, and the percentage of lysis in samples was calculated by the following formula: (ODsample − ODbacteria alone − ODPMN alone)/(ODPMN+Triton X-100 − ODPMN alone) × 100. Any negative values were set to zero for subsequent analyses (minimum lysis is 0%).
Alternatively, neutrophil lysis assays were performed in the presence of an anti-LukG antibody. Purified rabbit anti-LukG IgG (23) and rabbit control IgG (catalog number 31235; Invitrogen) were incubated with purified S. aureus protein A (product number 539202; Calbiochem) overnight at 4°C using a ratio of 1 μg IgG/1 μg protein A. Anti-LukG and rabbit control IgG (189 μg/ml final concentration) were added to 1 × 106 PMNs prior to the addition of the LAC or ΔlukGH strain (each at 1 × 107 CFU and grown to an OD600 of 0.75). LS (6.5 U/107 CFU) was added 30 min after the start of the assay (after phagocytosis) to kill extracellular bacteria. Samples were incubated at 37°C for 3 h, and LDH release was measured as described above.
Phagocytosis assays.
Synchronized phagocytosis assays were performed as described previously (31, 69). Briefly, LAC and isogenic mutant strains were cultured to the early-exponential phase of growth (OD600 of 0.75), washed with DPBS, and opsonized with 50% NHS. Opsonized bacteria (108 CFU) were labeled with fluorescein isothiocyanate (FITC) in DPBS (0.5 ml FITC solution at 50 μg/ml) for 30 min at ambient temperature in the dark with rotation and resuspended at the desired concentration in RPMI/H. Assays with human PMNs (3 × 106 CFU/106 PMNs) were conducted in 24-well flat-bottom tissue culture plates containing serum-coated coverslips as described previously (18) but with a few modifications. Plates were centrifuged at 524 × g for 8 min at 4°C to synchronize phagocytosis and incubated for 40 min at 37°C with 5% CO2. Samples were washed with DPBS, fixed with 4% paraformaldehyde for 15 min at ambient temperature, and blocked with 5% goat serum for 30 min in the dark. Staining was performed with rabbit anti-S. aureus antibody (S. aureus-spcific IgG) at a 1:100 dilution (31), followed by Alexa Fluor 594-conjugated goat anti-rabbit IgG (catalog number A-11012; Thermo Fisher) at a 1:1,500 dilution for 30 min at ambient temperature. Nuclei were counterstained with NucBlue live ReadyProbes reagent (catalog number R37605; Thermo Fisher) for 10 min at ambient temperature in the dark, and samples were mounted with ProLong gold antifade mountant (catalog number P36934; Thermo Fisher). Extracellular and intracellular bacteria were quantitated by fluorescence microscopy using 50 PMNs in at least 5 random fields of view (Zeiss Axioskop 2 plus). The percentage (%) of internalized bacteria was calculated according to the following formula: [(bacteriagreen − bacteriared)/bacteriagreen] × 100 = bacteriainternalized (31).
Differential permeabilization assay with digitonin.
A differential permeabilization assay using digitonin was adapted from Checroun et al. (32). In brief, LAC was cultured to the early-exponential-growth phase and opsonized with 50% NHS for 30 min at 37°C with shaking. Opsonized bacteria were washed with DPBS, labeled with FITC, washed again, and resuspended in RPMI/H. FITC-labeled LAC (2 × 106 CFU) was then added to wells containing 1 × 106 PMNs (2:1 bacterial cell/PMN ratio) on coverslips precoated with NHS in sterile 24-well plates. Plates were centrifuged at 524 × g for 8 min at 4°C to synchronize phagocytosis and subsequently incubated at 37°C with 5% CO2 for 0.5 to 3 h. At the indicated time points, supernatants were aspirated and samples washed once with RPMI/H. To visualize plasma membranes, live cells were stained with 0.3 ml of a 1:100 dilution of mouse anti-human CD16-AF647 monoclonal antibody (no. 239275; Abcam) for 30 min in the dark at ambient temperature. Samples were then fixed with 4% paraformaldehyde in DPBS (15 min at ambient temperature), washed three times with DPBS, and permeabilized at ambient temperature with either 0.3 ml 0.3% Triton X-100 for 10 min or 0.2 ml digitonin diluted in RPMI/H to 10 μg/ml (2 μg/106 PMNs) for 1 min. After permeabilization, samples were washed 3 times with DPBS, and those containing bacteria were blocked with 0.25 ml 5% goat serum (30 min at ambient temperature in the dark). Samples were labeled with anti-S. aureus primary antibody and goat anti-rabbit IgG-AF594 secondary antibody as described for the phagocytosis assays above. In some assays, PMNs permeabilized with 0.3% Triton X-100 were stained with a 1:2,000 dilution of rabbit anti-LAMP1 antibody conjugated to Cy3 (product number L0419; Sigma-Aldrich) to visualize phagosomes. Nuclei were stained with NucBlue live ReadyProbes reagent (Thermo Fisher) for 10 min at ambient temperature in the dark, and samples were mounted with ProLong gold antifade mountant (Thermo Fisher). In preliminary experiments, we determined the digitonin treatment conditions needed to selectively permeabilize the PMN plasma membrane. These treatment conditions do not permeabilize the phagosome membrane. For this process, we first titrated digitonin on fixed (4% paraformaldehyde) uninfected PMNs and stained the PMNs for cytoplasmatic actin using phalloidin-AF488 (catalog number A12379; Invitrogen). We then selected the lowest concentrations and shortest durations of digitonin treatment that yielded consistent phalloidin (actin) staining in human PMNs. We performed a similar series of experiments to determine the conditions for staining intracellular LAC using the combination of anti-S. aureus antibody and goat anti-rabbit IgG-AF594 antibody. We determined that 2 μg digitonin/106 PMNs (in 0.2 ml final volume) for 1 min at ambient temperature permitted cytoplasmic staining of actin but did not permeabilize the phagosome membrane. For positive controls, LAC-containing PMNs were treated with 0.3% Triton X-100 in DPBS to permeabilize the plasma- and phagosome membranes. Samples were stained with anti-S. aureus antibody/goat anti-rabbit IgG-AF594. Uninfected, CD16-stained PMNs served as controls for quality of the CD16 staining at all times tested. Images were acquired using a Zeiss laser scanning confocal microscope (LSM 880) driven by ZEN version 2.3 software (Carl Zeiss Microscopy, LLC). Data were generated in four independent experiments, and 50 PMNs/sample were used for quantification purposes. PMNs were quantified as CD16+ when the cell shape was clearly visible by CD16 stain alone, even if the staining was punctate, and the transmitted light channel (T-PMT) was used to confirm PMN morphology. PMNs were quantified as CD16− when no signal was visible and there was no clear cell outline. Laser power and gain settings for the CD16 signal were initially set to that of the uninfected PMNs at 30 min or, in case of the anti-S. aureus antibody/AF594 signal, to LAC-containing PMNs permeabilized with Triton X-100 and then applied to all samples. The signal intensities for all channels were not adjusted manually prior to scoring or assembly of the figures.
Statistical analysis.
Statistics were determined using GraphPad Prism (version 8.4.4; GraphPad Software, LLC). Data were assessed for normal distribution by the Shapiro-Wilk test. A paired t test was used for comparisons of two data sets only. A one-way or repeated-measures analysis of variance (ANOVA) with posttest was used to compare three or more data sets. The influence of bacterial cell/PMN ratio and time on PMN lysis was determined by two-way ANOVA with Geisser-Greenhouse correction and Tukey’s posttest. Where sample matching was not possible, we used a mixed-effects analysis and appropriate posttest. Data are presented as the mean value ± standard deviation. Significance values are indicated as follows: *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; and ****, P ≤ 0.0001.
ACKNOWLEDGMENTS
We are grateful to Anita Mora (Visual Medical Arts, NIAID) for assistance with preparation of figures.
This work was supported by the Intramural Research Program of the National Institutes of Allergy and Infectious Diseases, National Institutes of Health (F.R.D. and M.O.), and NIH grant 1R01 AI149491 (J.M.V).
We declare no conflicts of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Frank R. DeLeo, Email: fdeleo@niaid.nih.gov.
Joanna B. Goldberg, Emory University School of Medicine
REFERENCES
- 1.Lowy FD. 1998. Staphylococcus aureus infections. N Engl J Med 339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 2.Albrecht VS, Limbago BM, Moran GJ, Krishnadasan A, Gorwitz RJ, McDougal LK, Talan DA, EMERGEncy ID NET Study Group. 2015. Staphylococcus aureus colonization and strain type at various body sites among patients with a closed abscess and uninfected controls at U.S. emergency departments. J Clin Microbiol 53:3478–3484. doi: 10.1128/JCM.01371-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chambers HF, DeLeo FR. 2009. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Rev Microbiol 7:629–641. doi: 10.1038/nrmicro2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cassini A, Högberg LD, Plachouras D, Quattrocchi A, Hoxha A, Simonsen GS, Colomb-Cotinat M, Kretzschmar ME, Devleesschauwer B, Cecchini M, Ouakrim DA, Oliveira TC, Struelens MJ, Suetens C, Monnet DL, Strauss R, Mertens K, Struyf T, Catry B, Latour K, Ivanov IN, Dobreva EG, Tambic Andraševic A, Soprek S, Budimir A, Paphitou N, Žemlicková H, Schytte Olsen S, Wolff Sönksen U, Märtin P, Ivanova M, Lyytikäinen O, Jalava J, Coignard B, Eckmanns T, Abu Sin M, Haller S, Daikos GL, Gikas A, Tsiodras S, Kontopidou F, Tóth Á, Hajdu Á, Guólaugsson Ó, Kristinsson KG, Murchan S, Burns K, Pezzotti P, Gagliotti C, Dumpis U, Burden of AMR Collaborative Group, et al. 2019. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: a population-level modelling analysis. Lancet Infect Dis 19:56–66. doi: 10.1016/S1473-3099(18)30605-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Diep BA, Sensabaugh GF, Somboona NS, Carleton HA, Perdreau-Remington Foise. 2004. Widespread skin and soft-tissue infections due to two methicillin-resistant Staphylococcus aureus strains harboring the genes for Panton-Valentine leucocidin. J Clin Microbiol 42:2080–2084. doi: 10.1128/JCM.42.5.2080-2084.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moran GJ, Krishnadasan A, Gorwitz RJ, Fosheim GE, McDougal LK, Carey RB, Talan DA, EMERGEncy ID Net Study Group. 2006. Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med 355:666–674. doi: 10.1056/NEJMoa055356. [DOI] [PubMed] [Google Scholar]
- 7.McDougal LK, Steward CD, Killgore GE, Chaitram JM, McAllister SK, Tenover FC. 2003. Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: establishing a national database. J Clin Microbiol 41:5113–5120. doi: 10.1128/JCM.41.11.5113-5120.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tenover FC, McDougal LK, Goering RV, Killgore G, Projan SJ, Patel JB, Dunman PM. 2006. Characterization of a strain of community-associated methicillin-resistant Staphylococcus aureus widely disseminated in the United States. J Clin Microbiol 44:108–118. doi: 10.1128/JCM.44.1.108-118.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Popovich KJ, Snitkin ES, Hota B, Green SJ, Pirani A, Aroutcheva A, Weinstein RA. 2017. Genomic and epidemiological evidence for community origins of hospital-onset methicillin-resistant Staphylococcus aureus bloodstream infections. J Infect Dis 215:1640–1647. doi: 10.1093/infdis/jiw647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.David MZ, Cadilla A, Boyle-Vavra S, Daum RS. 2014. Replacement of HA-MRSA by CA-MRSA infections at an academic medical center in the midwestern United States, 2004-5 to 2008. PLoS One 9:e92760. doi: 10.1371/journal.pone.0092760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kobayashi SD, Malachowa N, DeLeo FR. 2018. Neutrophils and bacterial immune evasion. J Innate Immun 10:432–441. doi: 10.1159/000487756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. 2012. Neutrophil function: from mechanisms to disease. Annu Rev Immunol 30:459–489. doi: 10.1146/annurev-immunol-020711-074942. [DOI] [PubMed] [Google Scholar]
- 13.Bennett JE, Dolin R, Blaser MJ. 2020. Mandell, Douglas, and Bennett’s principles and practice of infectious diseases, 9th ed. Elsevier, Philadelphia, PA. [Google Scholar]
- 14.Melly MA, Thomison JB, Rogers DE. 1960. Fate of staphylococci within human leukocytes. J Exp Med 112:1121–1130. doi: 10.1084/jem.112.6.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rogers DE, Melly MA. 1960. Further observations on the behavior of staphylococci within human leukocytes. J Exp Med 111:533–558. doi: 10.1084/jem.111.4.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rogers DE, Tompsett R. 1952. The survival of staphylococci within human leukocytes. J Exp Med 95:209–230. doi: 10.1084/jem.95.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gresham HD, Lowrance JH, Caver TE, Wilson BS, Cheung AL, Lindberg FP. 2000. Survival of Staphylococcus aureus inside neutrophils contributes to infection. J Immunol 164:3713–3722. doi: 10.4049/jimmunol.164.7.3713. [DOI] [PubMed] [Google Scholar]
- 18.Voyich JM, Braughton KR, Sturdevant DE, Whitney AR, Said-Salim B, Porcella SF, Long RD, Dorward DW, Gardner DJ, Kreiswirth BN, Musser JM, DeLeo FR. 2005. Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J Immunol 175:3907–3919. doi: 10.4049/jimmunol.175.6.3907. [DOI] [PubMed] [Google Scholar]
- 19.Kobayashi SD, Braughton KR, Palazzolo-Ballance AM, Kennedy AD, Sampaio E, Kristosturyan E, Whitney AR, Sturdevant DE, Dorward DW, Holland SM, Kreiswirth BN, Musser JM, DeLeo FR. 2010. Rapid neutrophil destruction following phagocytosis of Staphylococcus aureus. J Innate Immun 2:560–575. doi: 10.1159/000317134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenlee-Wacker MC, Rigby KM, Kobayashi SD, Porter AR, DeLeo FR, Nauseef WM. 2014. Phagocytosis of Staphylococcus aureus by human neutrophils prevents macrophage efferocytosis and induces programmed necrosis. J Immunol 192:4709–4717. doi: 10.4049/jimmunol.1302692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greenlee-Wacker MC, Kremserova S, Nauseef WM. 2017. Lysis of human neutrophils by community-associated methicillin-resistant Staphylococcus aureus. Blood 129:3237–3244. doi: 10.1182/blood-2017-02-766253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pang YY, Schwartz J, Thoendel M, Ackermann LW, Horswill AR, Nauseef WM. 2010. agr-dependent interactions of Staphylococcus aureus USA300 with human polymorphonuclear neutrophils. J Innate Immun 2:546–559. doi: 10.1159/000319855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ventura CL, Malachowa N, Hammer CH, Nardone GA, Robinson MA, Kobayashi SD, DeLeo FR. 2010. Identification of a novel Staphylococcus aureus two-component leukotoxin using cell surface proteomics. PLoS One 5:e11634. doi: 10.1371/journal.pone.0011634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DuMont AL, Yoong P, Surewaard BG, Benson MA, Nijland R, van Strijp JA, Torres VJ. 2013. Staphylococcus aureus elaborates leukocidin AB to mediate escape from within human neutrophils. Infect Immun 81:1830–1841. doi: 10.1128/IAI.00095-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Surewaard BG, de Haas CJ, Vervoort F, Rigby KM, DeLeo FR, Otto M, van Strijp JA, Nijland R. 2013. Staphylococcal alpha-phenol soluble modulins contribute to neutrophil lysis after phagocytosis. Cell Microbiol 15:1427–1437. doi: 10.1111/cmi.12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang D, Ho YX, Cowell LM, Jilani I, Foster SJ, Prince LR. 2019. A genome-wide screen identifies factors involved in S. aureus-induced human neutrophil cell death and pathogenesis. Front Immunol 10:45. doi: 10.3389/fimmu.2019.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Voyich JM, Vuong C, DeWald M, Nygaard TK, Kocianova S, Griffith S, Jones J, Iverson C, Sturdevant DE, Braughton KR, Whitney AR, Otto M, DeLeo FR. 2009. The SaeR/S gene regulatory system is essential for innate immune evasion by Staphylococcus aureus. J Infect Dis 199:1698–1706. doi: 10.1086/598967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kong C, Neoh HM, Nathan S. 2016. Targeting Staphylococcus aureus toxins: a potential form of anti-virulence therapy. Toxins (Basel) 8:72. doi: 10.3390/toxins8030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oogai Y, Matsuo M, Hashimoto M, Kato F, Sugai M, Komatsuzawa H. 2011. Expression of virulence factors by Staphylococcus aureus grown in serum. Appl Environ Microbiol 77:8097–8105. doi: 10.1128/AEM.05316-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheung AL, Bayer AS, Zhang G, Gresham H, Xiong YQ. 2004. Regulation of virulence determinants in vitro and in vivo in Staphylococcus aureus. FEMS Immunol Med Microbiol 40:1–9. doi: 10.1016/S0928-8244(03)00309-2. [DOI] [PubMed] [Google Scholar]
- 31.Lu T, Porter AR, Kennedy AD, Kobayashi SD, DeLeo FR. 2014. Phagocytosis and killing of Staphylococcus aureus by human neutrophils. J Innate Immun 6:639–649. doi: 10.1159/000360478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Checroun C, Wehrly TD, Fischer ER, Hayes SF, Celli J. 2006. Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc Natl Acad Sci USA 103:14578–14583. doi: 10.1073/pnas.0601838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chong A, Wehrly TD, Nair V, Fischer ER, Barker JR, Klose KE, Celli J. 2008. The early phagosomal stage of Francisella tularensis determines optimal phagosomal escape and Francisella pathogenicity island protein expression. Infect Immun 76:5488–5499. doi: 10.1128/IAI.00682-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meunier E, Broz P. 2015. Quantification of cytosolic vs. vacuolar Salmonella in primary macrophages by differential permeabilization. J Vis Exp 2015:e52960. doi: 10.3791/52960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bayles KW, Wesson CA, Liou LE, Fox LK, Bohach GA, Trumble WR. 1998. Intracellular Staphylococcus aureus escapes the endosome and induces apoptosis in epithelial cells. Infect Immun 66:336–342. doi: 10.1128/IAI.66.1.336-342.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lam TT, Giese B, Chikkaballi D, Kuhn A, Wolber W, Pane-Farre J, Schafer D, Engelmann S, Fraunholz M, Sinha B. 2010. Phagolysosomal integrity is generally maintained after Staphylococcus aureus invasion of nonprofessional phagocytes but is modulated by strain 6850. Infect Immun 78:3392–3403. doi: 10.1128/IAI.00012-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jarry TM, Cheung AL. 2006. Staphylococcus aureus escapes more efficiently from the phagosome of a cystic fibrosis bronchial epithelial cell line than from its normal counterpart. Infect Immun 74:2568–2577. doi: 10.1128/IAI.74.5.2568-2577.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jarry TM, Memmi G, Cheung AL. 2008. The expression of alpha-haemolysin is required for Staphylococcus aureus phagosomal escape after internalization in CFT-1 cells. Cell Microbiol 10:1801–1814. doi: 10.1111/j.1462-5822.2008.01166.x. [DOI] [PubMed] [Google Scholar]
- 39.Hess DJ, Henry-Stanley MJ, Erickson EA, Wells CL. 2003. Intracellular survival of Staphylococcus aureus within cultured enterocytes. J Surg Res 114:42–49. doi: 10.1016/s0022-4804(03)00314-7. [DOI] [PubMed] [Google Scholar]
- 40.Kahl BC, Goulian M, van Wamel W, Herrmann M, Simon SM, Kaplan G, Peters G, Cheung AL. 2000. Staphylococcus aureus RN6390 replicates and induces apoptosis in a pulmonary epithelial cell line. Infect Immun 68:5385–5392. doi: 10.1128/IAI.68.9.5385-5392.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lowy FD, Fant J, Higgins LL, Ogawa SK, Hatcher VB. 1988. Staphylococcus aureus–human endothelial cell interactions. J Ultrastruct Mol Struct Res 98:137–146. doi: 10.1016/s0889-1605(88)80906-6. [DOI] [PubMed] [Google Scholar]
- 42.Hamill RJ, Vann JM, Proctor RA. 1986. Phagocytosis of Staphylococcus aureus by cultured bovine aortic endothelial cells: model for postadherence events in endovascular infections. Infect Immun 54:833–836. doi: 10.1128/iai.54.3.833-836.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ogawa SK, Yurberg ER, Hatcher VB, Levitt MA, Lowy FD. 1985. Bacterial adherence to human endothelial cells in vitro. Infect Immun 50:218–224. doi: 10.1128/iai.50.1.218-224.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sinha B, Francois P, Que YA, Hussain M, Heilmann C, Moreillon P, Lew D, Krause KH, Peters G, Herrmann M. 2000. Heterologously expressed Staphylococcus aureus fibronectin-binding proteins are sufficient for invasion of host cells. Infect Immun 68:6871–6878. doi: 10.1128/IAI.68.12.6871-6878.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Strobel M, Pfortner H, Tuchscherr L, Volker U, Schmidt F, Kramko N, Schnittler HJ, Fraunholz MJ, Loffler B, Peters G, Niemann S. 2016. Post-invasion events after infection with Staphylococcus aureus are strongly dependent on both the host cell type and the infecting S. aureus strain. Clin Microbiol Infect 22:799–809. doi: 10.1016/j.cmi.2016.06.020. [DOI] [PubMed] [Google Scholar]
- 46.Giese B, Glowinski F, Paprotka K, Dittmann S, Steiner T, Sinha B, Fraunholz MJ. 2011. Expression of delta-toxin by Staphylococcus aureus mediates escape from phago-endosomes of human epithelial and endothelial cells in the presence of beta-toxin. Cell Microbiol 13:316–329. doi: 10.1111/j.1462-5822.2010.01538.x. [DOI] [PubMed] [Google Scholar]
- 47.Wesson CA, Liou LE, Todd KM, Bohach GA, Trumble WR, Bayles KW. 1998. Staphylococcus aureus Agr and Sar global regulators influence internalization and induction of apoptosis. Infect Immun 66:5238–5243. doi: 10.1128/IAI.66.11.5238-5243.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shompole S, Henon KT, Liou LE, Dziewanowska K, Bohach GA, Bayles KW. 2003. Biphasic intracellular expression of Staphylococcus aureus virulence factors and evidence for Agr-mediated diffusion sensing. Mol Microbiol 49:919–927. doi: 10.1046/j.1365-2958.2003.03618.x. [DOI] [PubMed] [Google Scholar]
- 49.Qazi SN, Counil E, Morrissey J, Rees CE, Cockayne A, Winzer K, Chan WC, Williams P, Hill PJ. 2001. agr expression precedes escape of internalized Staphylococcus aureus from the host endosome. Infect Immun 69:7074–7082. doi: 10.1128/IAI.69.11.7074-7082.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stelzner K, Winkler AC, Liang C, Boyny A, Ade CP, Dandekar T, Fraunholz MJ, Rudel T. 2020. Intracellular Staphylococcus aureus perturbs the host cell Ca(2+) homeostasis to promote cell death. mBio 11:e02250-20. doi: 10.1128/mBio.02250-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kubica M, Guzik K, Koziel J, Zarebski M, Richter W, Gajkowska B, Golda A, Maciag-Gudowska A, Brix K, Shaw L, Foster T, Potempa J. 2008. A potential new pathway for Staphylococcus aureus dissemination: the silent survival of S. aureus phagocytosed by human monocyte-derived macrophages. PLoS One 3:e1409. doi: 10.1371/journal.pone.0001409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moldovan A, Fraunholz MJ. 2019. In or out: phagosomal escape of Staphylococcus aureus. Cell Microbiol 21:e12997. doi: 10.1111/cmi.12997. [DOI] [PubMed] [Google Scholar]
- 53.Grosz M, Kolter J, Paprotka K, Winkler AC, Schafer D, Chatterjee SS, Geiger T, Wolz C, Ohlsen K, Otto M, Rudel T, Sinha B, Fraunholz M. 2014. Cytoplasmic replication of Staphylococcus aureus upon phagosomal escape triggered by phenol-soluble modulin alpha. Cell Microbiol 16:451–465. doi: 10.1111/cmi.12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jubrail J, Morris P, Bewley MA, Stoneham S, Johnston SA, Foster SJ, Peden AA, Read RC, Marriott HM, Dockrell DH. 2016. Inability to sustain intraphagolysosomal killing of Staphylococcus aureus predisposes to bacterial persistence in macrophages. Cell Microbiol 18:80–96. doi: 10.1111/cmi.12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Flannagan RS, Heit B, Heinrichs DE. 2016. Intracellular replication of Staphylococcus aureus in mature phagolysosomes in macrophages precedes host cell death, and bacterial escape and dissemination. Cell Microbiol 18:514–535. doi: 10.1111/cmi.12527. [DOI] [PubMed] [Google Scholar]
- 56.Flannagan RS, Kuiack RC, McGavin MJ, Heinrichs DE. 2018. Staphylococcus aureus uses the GraXRS regulatory system to sense and adapt to the acidified phagolysosome in macrophages. mBio 9:e01143-18. doi: 10.1128/mBio.01143-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tranchemontagne ZR, Camire RB, O’Donnell VJ, Baugh J, Burkholder KM. 2016. Staphylococcus aureus strain USA300 perturbs acquisition of lysosomal enzymes and requires phagosomal acidification for survival inside macrophages. Infect Immun 84:241–253. doi: 10.1128/IAI.00704-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Malachowa N, Whitney AR, Kobayashi SD, Sturdevant DE, Kennedy AD, Braughton KR, Shabb DW, Diep BA, Chambers HF, Otto M, DeLeo FR. 2011. Global changes in Staphylococcus aureus gene expression in human blood. PLoS One 6:e18617. doi: 10.1371/journal.pone.0018617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.DuMont AL, Yoong P, Day CJ, Alonzo F, III, McDonald WH, Jennings MP, Torres VJ. 2013. Staphylococcus aureus LukAB cytotoxin kills human neutrophils by targeting the CD11b subunit of the integrin Mac-1. Proc Natl Acad Sci USA 110:10794–10799. doi: 10.1073/pnas.1305121110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kennedy AD, Otto M, Braughton KR, Whitney AR, Chen L, Mathema B, Mediavilla JR, Byrne KA, Parkins LD, Tenover FC, Kreiswirth BN, Musser JM, DeLeo FR. 2008. Epidemic community-associated methicillin-resistant Staphylococcus aureus: recent clonal expansion and diversification. Proc Natl Acad Sci USA 105:1327–1332. doi: 10.1073/pnas.0710217105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miller LG, Perdreau-Remington F, Rieg G, Mehdi S, Perlroth J, Bayer AS, Tang AW, Phung TO, Spellberg B. 2005. Necrotizing fasciitis caused by community-associated methicillin-resistant Staphylococcus aureus in Los Angeles. N Engl J Med 352:1445–1453. doi: 10.1056/NEJMoa042683. [DOI] [PubMed] [Google Scholar]
- 62.Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, Lin F, Lin J, Carleton HA, Mongodin EF, Sensabaugh GF, Perdreau-Remington F. 2006. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367:731–739. doi: 10.1016/S0140-6736(06)68231-7. [DOI] [PubMed] [Google Scholar]
- 63.Malachowa N, Kobayashi SD, Braughton KR, Whitney AR, Parnell MJ, Gardner DJ, Deleo FR. 2012. Staphylococcus aureus leukotoxin GH promotes inflammation. J Infect Dis 206:1185–1193. doi: 10.1093/infdis/jis495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Voyich JM, Otto M, Mathema B, Braughton KR, Whitney AR, Welty D, Long RD, Dorward DW, Gardner DJ, Lina G, Kreiswirth BN, DeLeo FR. 2006. Is Panton-Valentine leukocidin the major virulence determinant in community-associated methicillin-resistant Staphylococcus aureus disease? J Infect Dis 194:1761–1770. doi: 10.1086/509506. [DOI] [PubMed] [Google Scholar]
- 65.Wang R, Braughton KR, Kretschmer D, Bach TH, Queck SY, Li M, Kennedy AD, Dorward DW, Klebanoff SJ, Peschel A, DeLeo FR, Otto M. 2007. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med 13:1510–1514. doi: 10.1038/nm1656. [DOI] [PubMed] [Google Scholar]
- 66.Nygaard TK, Pallister KB, DuMont AL, DeWald M, Watkins RL, Pallister EQ, Malone C, Griffith S, Horswill AR, Torres VJ, Voyich JM. 2012. Alpha-toxin induces programmed cell death of human T cells, B cells, and monocytes during USA300 infection. PLoS One 7:e36532. doi: 10.1371/journal.pone.0036532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schneewind O, Missiakas D. 2014. Genetic manipulation of Staphylococcus aureus. Curr Protoc Microbiol 32:Unit 9C.3. doi: 10.1002/9780471729259.mc09c03s32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kobayashi SD, Voyich JM, Buhl CL, Stahl RM, DeLeo FR. 2002. Global changes in gene expression by human polymorphonuclear leukocytes during receptor-mediated phagocytosis: cell fate is regulated at the level of gene expression. Proc Natl Acad Sci USA 99:6901–6906. doi: 10.1073/pnas.092148299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.DeLeo FR, Allen LA, Apicella M, Nauseef WM. 1999. NADPH oxidase activation and assembly during phagocytosis. J Immunol 163:6732–6740. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download SPECTRUM00888-21_Supp_1_seq10.pdf, PDF file, 0.6 MB (674KB, pdf)