

SUMMARY
Duplex DNA naturally folds into a right-handed double helix in physiological conditions. Some sequences of unusual base composition may nevertheless form alternative structures, as was shown for many repeated sequences in vitro. However, evidence for the formation of noncanonical structures in living cells is difficult to gather. It mainly relies on genetic assays demonstrating their function in vivo or through genetic instability reflecting particular properties of such structures. Efforts were made to reveal their existence directly in a living cell, mainly by generating antibodies specific to secondary structures or using chemical ligands selected for their affinity to these structures. Among secondary structure-forming DNAs are G-quadruplexes, human fragile sites containing minisatellites, AT-rich regions, inverted repeats able to form cruciform structures, hairpin-forming CAG/CTG triplet repeats, and triple helices formed by homopurine-homopyrimidine GAA/TTC trinucleotide repeats. Many of these alternative structures are involved in human pathologies, such as neurological or developmental disorders, as in the case of trinucleotide repeats, or cancers triggered by translocations linked to fragile sites. This review will discuss and highlight evidence supporting the formation of alternative DNA structures in vivo and will emphasize the role of the mismatch repair machinery in binding mispaired DNA duplexes, triggering genetic instability.
KEYWORDS: DNA hairpin, G quadruplex, cruciform, fragile sites, mismatch repair, palindromes, secondary structures, trinucleotide repeats
INTRODUCTION
Canonical Right-Handed DNA Helices
Historically, fiber X-ray crystallography identified two distinct structural forms of DNA, A-DNA and B-DNA (1). A-DNA was isolated at 75% humidity, and B-DNA was isolated at higher percentages. Concomitantly, Watson and Crick identified B-DNA as a double-helix structure and proposed a model of an antiparallel double-stranded helix formed by two linear sugar-phosphate backbones that run in opposite directions (2). The two strands are connected by hydrogen bonds between the purine and pyrimidine bases, consistent with the previously enounced Chargaff’s rule stating that the purine and pyrimidine ratio should be 1:1 in all organisms (3). These bonds are called Watson-Crick bonds. Hoogsteen base pairings can be observed in alternative forms of DNA or certain protein-DNA complexes in which the purine is rotated in such a way that bonds are made between its other face and the pyrimidine (4).
A-DNA is a thicker right-handed duplex with a shorter distance between base pairs and has been described for RNA-DNA duplexes and RNA-RNA duplexes. RNA can only form A‐type double helices because of the steric restrictions of the ribose 2′ hydroxyl residue (5, 6).
Left-Handed Z-DNA Helices
Surprisingly, in the late 1970s, when single crystal X-ray diffraction was available to validate the proposed model of the double helix, the expected B-DNA structure was not the first to be observed. The crystal was obtained from the self-complementary DNA hexameric sequence d(CG)3. The alternation of guanine and cytosine residues is crucial for the formation of Z-DNA, and the crystal structure revealed a left-handed double helix (7). This unexpected result was already a hint toward the complexity of the different conformations that DNA can adopt. Since the ribose phosphate backbone followed a zig-zag, this form of DNA was called Z-DNA. It was later recognized that Z-DNA is formed due to negative supercoiling produced behind a moving RNA polymerase during transcription (8) and that its formation is favored near transcription start sites (9).
ALTERNATIVE DNA STRUCTURES
The lowest energy-level state of DNA in physiological conditions is B-DNA. However, formation of alternative structures can occur when the DNA duplex is unwound during metabolic DNA processes such as DNA replication and transcription. Alternative forms of DNA were discovered later, including G-quadruplexes, hairpins, H-DNA, cruciforms, and AT-rich DNA unwinding elements (DUE). Many repeated sequences were shown to form DNA secondary structures in vitro. However, real evidence for in vivo formation is difficult to gather. It mainly relies on genetics data underlying their function in vivo or through their instability or their propensity to trigger specific phenotypes in genetic assays. Efforts were made to isolate antibodies specifically recognizing secondary structures, aiming to reveal their existence directly in a cell. In this review, we will describe these alternative DNA forms and discuss their formation in vivo, in living cells and organisms. The role of mismatch repair (MMR), a complex machinery whose function is to detect noncanonical DNA structures, will also be highlighted, as well as its involvement in triggering non-B-DNA instability in several human neurological disorders.
G-Quadruplexes
G-quadruplexes, evidence for in vitro formation.
X-ray diffraction demonstrated very early that guanylic acids can assemble into tetrameric structures. In these tetramers, four guanine molecules form a square in which each guanine is hydrogen-bound to the two adjacent guanines by Hoogsteen bonds (10). The formation and stabilization of G-quadruplexes in solution under physiological conditions are dependent on monovalent cations, specifically K+ (11). This structure was reconstituted in a test tube using oligonucleotides encoding telomere sequences (12). A G4 consensus motif of the form G3-5N1-7 G3-5N1-7 G3-5N1-7G3-5 (where N can be any nucleotide; Fig. 1A) was adopted and used to search for G4 in eukaryotic genomes (13). In both yeast and mammals, G4 motifs are enriched in telomeric and ribosomal DNA, in transcriptional regulatory sites, and at preferred mitotic and meiotic double-strand break sites (14). In the human genome, GC-rich promoters frequently contain G-quadruplexes, some of them driving oncogene expression. This is the case of the G4 in the KRAS promoter, a known oncogene whose product is a small G-protein playing a role in cell differentiation and proliferation. The crystal structure of this G4 was very recently determined, revealing that it may adopt two stable conformations, one conformer being stabilized by a triad at the 3′ end, while the second conformer remains flexible and displays less stability (15).
FIG 1.

G quadruplexes. (A) Genetic factors stabilizing G quadruplexes. The general consensus is shown (G3 Nx G3 Nx G3 Nx), with the positions of the two lateral loops and the central loop. A stable G4 structure is formed in the presence of potassium ions if the central loop is shorter than 5 nucleotides and if lateral loops are made of one or two pyrimidine residues. If loops are longer or made of adenine residues, the G4 structure is less stable. The helicase Pif1 is known to unwind G4 in S. cerevisiae (29). (B) Stabilization of the Pu24T quadruplex by PhenDC3. Top view of the top guanine tetrad showing non-Watson-Crick bounds as orange dashed lines. Addition of PhenDC3 stabilizes the structure by stacking with the G4. (Based on data from reference 40.)
G-quadruplexes in Caenorhabditis elegans.
The deletion of a specific helicase in C. elegans led to the systematic deletion of polyglutamine tracts genome-wide. The helicase was renamed dog-1 for deletions of guanine-rich DNA. This helicase appears to be essential for the resolution of G4 motifs during replication (16). In the same organism, a reporter LacZ construct whose expression is controlled by a GC-rich promoter allowed direct visualization of cells that harbor destabilized G4 tracts within whole animals. It confirmed that only G4 DNA sites are fragile in dog-1-deficient genomes and showed that instability occurs both in early and late stages of the worm life cycle (17). C. elegans dog-1 is the homolog of human FANCJ (18), which belongs to a molecular pathway involved in Fanconia anemia, a human genetic disease characterized by many symptoms, including genomic instability and predisposition to cancer anomalies (19). Fanconia anemia proteins resolve interstrand cross-links, a dramatic type of DNA damage leading to transcription and replication arrests. A purified recombinant human FANCJ was shown to unwind G4 DNA in vitro, acting as a 5′-3′ helicase (20). All these elements point toward the in vivo formation of G4, whose structure must be resolved by specific helicases during replication to maintain genomic stability.
G-quadruplexes in Saccharomyces cerevisiae.
Minisatellites and microsatellites are DNA tandem repeats known to exhibit polymorphic length among the population. This polymorphism was historically used for forensic medicine (21), paternity tests (22), and physical mapping of genomes (23). Some minisatellites were described as being hypervariable, showing extensive and frequent length polymorphism during meiosis (24). This meiotic instability was later shown to be triggered by homologous recombination, following double-strand breaks (DSBs) occurring during meiosis, near the minisatellite (25). Similar experiments were reproduced in yeast, in which CEB1 was integrated in a chromosome, near a known meiotic hot spot. Instability of the minisatellite was shown to depend on hot spot activity and on the product of the SPO11 gene, the type VI topoisomerase responsible for meiotic double-strand breaks in yeast (26). Since it was clear that double-strand breaks destabilized CEB1 in humans as well as in yeast, the minisatellite could be used as a reporter system to detect such damage within or near the tandem repeat. Subsequent experiments by the same lab showed that mutations in RAD27 or DNA2, both genes involved in lagging strand metabolism during replication, dramatically increased CEB1 instability (27). This strongly suggested that in these mutant backgrounds, double-strand breaks occurred with a higher frequency at, or near, CEB1.
Careful examination of the minisatellite sequence showed that it was GC-rich, and computer analysis suggested that each of its repeat units could potentially form a G-quadruplex. Since it was already known that G4 can adopt different structures based on different motifs, four G4 sequences, derived from the CEB1 minisatellite, were integrated into the S. cerevisiae genome. These sequences were shown to have different conformations in vitro based on circular dichroism spectra, measuring light deviation induced by a given structure. The stability of the tract in vivo depended greatly on the predicted conformation (28).
Pif1, a replication helicase, was found to prevent genomic instability of CEB1 integrated in the Saccharomyces cerevisiae genome (29). Purified human Pif1 was shown to bind and unwind G4 structures in vitro (30). ChiP-Seq (chromatin immunoprecipitation sequencing, a technique used to sequence DNA contacting a specific protein or protein complex in vivo) revealed that Pif1 binds to G4 motifs (31). Through ChiP microarray experiments of DNA associated with DNA polymerase ε in a strain where PIF1 expression is reduced (pif1-m2 allele) and through two-dimensional (2D) gels, it was revealed that Polε accumulated at G4 sequences and that replication forks were slowed down at these loci.
Mutagenesis of another minisatellite, CEB25, and its integration in the yeast genome revealed that very short pyrimidine-containing G4 loops triggered the most instability (28). Hence, it became possible to predict the stability of a given minisatellite in vivo according to point mutations that were shown in vitro to stabilize or destabilize the G4 sequence. This work definitely proved the existence of noncanonical G quadruplexes in living yeast cells, strongly suggesting that similar structures also exist in other organisms (32). At the present time, many algorithms exist to detect and predict G4 formation according to the DNA sequence, the latest programs relying on machine learning approaches for detection (33).
G-quadruplexes in DT40 cells.
Chicken DT40 cells have been extensively used to study relationships between G4, DNA replication, and epigenetic modifications in vertebrates. The BU-1 locus, encoding a glycoprotein involved in development, contains two G-quadruplexes. One of them, forming on the leading-strand template, was shown to regulate BU-1 expression. When the G4 is present, BU-1 expression is reduced through epigenetic modification of the locus. When the G4 is reintroduced with two point mutations disrupting its formation in vitro, BU-1 expression is restored. When the wild-type G4 was reintroduced in the opposite orientation, expression of the reporter gene was not affected, proving that the G-quadruplex needs to form on the leading-strand template to downregulate BU-1 expression (34).
The chicken β-globin locus contains a G-quadruplex that is located on the leading-strand template during chromosomal replication (35). The DNA translesion synthesis polymerase REV1 was essential to replicate this quadruplex when the G4 sequence was in its wild-type orientation, but no effect was observed in the opposite orientation (G4 on lagging-strand template). REV1 encodes a multifunctional domain protein, including a PCNA-interacting domain, a ubiquitin-binding domain, and a domain of interaction with another translesion polymerase. The integrity of both ubiquitin and translesion domains, located in the C-terminal part of the protein, is required for efficient translesion synthesis (36). Loss of expression at this locus in REV1-deficient cells was associated with epigenetic changes, resulting in the incorporation of newly synthesized unmodified histones, following the uncoupling of DNA synthesis from histone recycling (37).
The following three helicases were also proven to help with replication through G-quadruplexes: FANCJ, WRN, and BLM. DT40 cells deficient for one of these helicases show expression defects similar to a rev1 mutant. The fact that FANCJ has a different polarity from the two other nucleases suggests that BLM and WRN could act in the opposite direction (3′ to 5′) of FANCJ, collaborating in unwinding the quadruplex to facilitate its replication (38).
Finally, it must be noted that use of a G4 ligand such as PhenDC3 mimics the effect of a REV1 deficiency by stabilizing the G-quadruplex and reprogramming epigenetic histone modifications, a strong argument in favor of the formation of such secondary structures in vivo (see below) (39).
Chemical ligands provide additional evidence for in vivo formation of G-quadruplexes.
Since stabilized G4 was found in the promoter of oncogenes such as c-Myc, ligands that stabilize G4 were isolated in order to serve as an anticancer therapy, by impeding the transcription of these oncogenes. Among them, the PhenDC family has a high binding affinity to G4. This property was attributed to the crescent shape and the size of the Phen-DC scaffold that fits with the structure of a G-quartet (Fig. 1B), both features being favorable for an optimal overlap between the two aromatic surfaces (40). Ligands can be used to probe the formation of G-quadruplexes in vivo, interfere with their processing, and elucidate their biological roles. As an additional proof of G4 formation in vivo, treatment of Pif1-deficient cells with the Phen-DC3 ligand further increased CEB1 instability (41).
Antibodies probing G4 formation: reality or fantasy?
Antibodies directed against the Stylonychia lemnae telomeric sequence were isolated by ribosome display (42). This ciliate is a good model to test the selected antibodies in situ since its genome contains millions of small gene-size DNA molecules, each terminated by telomeric DNA, increasing the local concentration of telomeres compared to any other organism. The affinity of these antibodies to parallel and antiparallel G4 conformations, as well as to several B-form structured DNA, was tested. Purification of DNA bound to one of the selected antibodies and analysis by circular dichroism spectroscopy revealed a parallel G4 structure, validating the specificity of this antibody. These antibodies specifically stained telomeres, suggesting the existence of telomeric G4.
Another team attempted to visualize G4 in mammalian cells using monoclonal antibodies. In particular, one antibody was raised against telomeric sequence by phage display and was called 1H6. It was used to reveal telomeres in HeLa cells and mammalian cells (43). However, this antibody was shown to cross-react with single-stranded poly(T) DNA, highlighting the difficulty of isolating antibodies specifically recognizing secondary structures and not the DNA sequence itself (44). Indeed, when developing an antibody specific to a DNA structure, all antibodies binding to double-stranded B-DNA must be counterselected. Raising monoclonal antibodies instead of polyclonal antibodies as well as a careful validation in vitro and in vivo of the antibody activity are therefore necessary.
To that end, the phage display technology was used to select a monoclonal antibody (called BG4) directed against G-quadruplexes. This antibody was used to visualize G4 structures in the nuclei of U2OS cells, as well as on metaphasic chromosomes. The number of BG4 foci increased during the transition from G1 to S phase and during S phase, proving that G4 formation is associated with DNA synthesis (45). The same antibody was used to sequence G4-containing chromatin (G4-ChIP-Seq) in 22 different breast cancer tumor xenograft models. This analysis revealed at least three G4-based cancer subtypes. G4-forming regions in 14 models out of 22 were found to be associated with more than one subtype, suggesting the existence of multiple cancer states within one single-cell model (46).
G4 detection using antibodies reveals only 1% of G-quadruplexes predicted by sequencing. Average G4-ChIP-Seq results extracted from millions of cells hide dynamic processes happening in individual cells. In order to address this problem, a G4-specific probe was recently established by fusing a far-red silicon-rhodamine fluorophore to an analogue of an established G4 ligand, pyridostatin. This probe enabled live-cell single-molecule fluorescence imaging of G-quadruplexes and revealed that secondary structure formation fluctuates between folded and unfolded states throughout the cell cycle, being maximum in S phase (47). All these recent data add strong evidence for the existence of G-quadruplexes in living cells.
Fragile Sites
Characterization of fragile sites: structure and instability.
Fragile sites cannot be detected in cells cultured in normal conditions. However, under replication stress induced by specific chemical agents, they are susceptible to breakage, on one or both chromatids, visible in metaphasic chromosomes. Fragile sites are subdivided into common fragile sites, present in all individuals, and rare fragile sites, seen in less than 5% of the individuals and segregating in a mendelian manner. There are over 100 fragile sites in the human genome, but the exact number is difficult to assess since fragile site expression depends on the replication stress applied. Common fragile sites are expressed (fragile site breakage is usually called “expression”) under partial replication stress induced by aphidicolin or camptothecin, while rare fragile sites are induced by other drugs. Folate-sensitive rare fragile sites include FRAXA and all CGG trinucleotide repeat expansions. FRA16B is induced by distamycin A, whereas FRA10B is only expressed in the presence of BrdU (48). This different pattern of expression is related to the sequence composition of these two groups of fragile sites and will be discussed below.
Common fragile sites are the largest class of fragile sites; the most studied among them are FRA3B (49) and FRA16D (50). Both were sequenced and revealed the presence of numerous repeated elements and a few AT-rich regions (Fig. 2A and B). Additionally, these sequences were found to be extensively rearranged in several cancerous cell lines, such as LS180 for FRA3B (51) and HCT116 for FRA16D (52). These fragile sites both lie within the large tumor-suppressor genes, FHIT and WWOX, respectively. Common fragile site impacts on chromosomal rearrangements have been extensively studied by mapping deletions, amplifications, and translocations occurring during early cancer development (53).
FIG 2.

Common and rare fragile sites. (A) The common FRA3B fragile site. Complete and incomplete L1 retrotransposons are shown by blue and purple arrows, respectively. Vertical dotted arrows indicate known deletion junctions, whose extents are shown by horizontal dashed lines, as heterozygous or homozygous deletions, in five cancer cell lines in which they were mapped. (Based on data from reference 51.) (B) The common FRA16D fragile site. AT-rich sequences are indicated by vertical pink arrows, with the darker shade corresponding to a higher flexibility index. Vertical dotted arrows indicate known deletion junctions, whose extents are shown by horizontal dotted lines, in two cancer cell lines in which they were mapped. (Based on data from reference 50.) (C) The rare FRA16B fragile site. It contains three AT-rich minisatellites, shown by green arrows. The most telomeric repeat is unstable, and its expansion triggers fragile site expression (58). (D) The rare FRA10B fragile site. It contains five AT-rich minisatellites, shown by green arrows. The internal repeat is unstable and prone to expansions. (Based on data from reference 59.)
The major group of rare fragile sites is the folate-sensitive group, which is associated with CGG trinucleotide repeat expansion. This group includes FRAXA, in the FMR1 gene (54, 55) (Fig. 2C), FRAXE (Xq27.3 [56]), and FRA11B (11q23.3 [57]). Other non-folate-sensitive rare fragile sites are characterized by long stretches of AT-rich tandem repeats and are induced by bromodeoxyuridine (BrdU) or distamycin A. FRA16B (58) and FRA10B (59) were cloned and sequenced, revealing that they harbor polymorphic AT-rich minisatellites. Their expression was associated with expansion of one or more of the repeats, up to several kilobases (Fig. 2D).
Common fragile site instability and replication delays.
Early on, it was shown that FRA3B was a late-replicating region (60). The FRA3B fragility mechanism was more thoroughly described through genome-wide analyses of replication timing. More precisely, DNA-combing associated with fluorescent detection of newly synthesized DNA was used in order to quantify replication fork speed. In lymphoblastoid cells, replication initiation events are excluded from a FRA3B core spanning approximately 700 kb, which forces replication forks coming from flanking regions to cover long distances in order to complete replication of the locus (61) (Fig. 3A). This delay is further exacerbated by late initiation of replication during mid-S phase. The same trend was observed for FRA16D (61). These data indicate that the fragility of some common fragile sites results from the combination of late replication completion and paucity of initiation events. It is worth noting that such a replication profile was not observed in fibroblasts, which can explain the tissue specificity observed for fragile sites.
FIG 3.

Replicating fragile sites. (A) FRA3B. In lymphoblasts, replication forks travel from large distances outside the fragile site inner core, and there is no activation of internal origins. At the end of the S phase, the whole locus is frequently underreplicated and therefore prone to breakage. In fibroblasts, the activation of several internal replication origins allows completion of replication of the locus, decreasing its fragility. (Based on data from reference 61.) (B) FRAXA. Replication proceeds from three origins within the locus. The fork traveling from ORI II is stalled by the CGG trinucleotide repeat expansion on the lagging strand template. ORI III, traveling in the other orientation, is less affected since the CGG sequence is on the leading strand template (67, 68).
Secondary structure formation in common fragile sites.
Although present in a smaller proportion, AT-rich minisatellites may also be involved in common fragile site instability. Indeed, FRA16B AT-rich repeats were also shown to form secondary structures by denaturation/renaturation experiments and migration on 4% SDS-PAGE, and by electron microscopy (62). Minisatellite-containing plasmids of various lengths were transfected into HEK293T cells, DNA was extracted and transformed into Escherichia coli, and the minisatellite repeat length was determined by restriction analysis. Frequent contractions and expansions of the repeat tract were observed, showing its natural instability in vivo. In addition, primer extension experiments on a FRA16B-containing plasmid showed discrete bands corresponding to pauses of the polymerase within AT repeats in vitro, probably due to the formation of secondary structures (62).
FlexStab is a program that calculates the local variations of the twist angle between adjacent nucleotides and identifies flexibility peaks. These peaks were shown to be composed of interrupted runs of AT-rich segments (63). Many fragile sites were shown to have such regions, including FRA3B and FRA16D. Although it was not formally proven that these common fragile sites form any kind of secondary structure in vivo, their instability may be exacerbated by the formation of such secondary structures (64). The current model does not include necessary formation of such structures, in contrast to what is proposed for CGG trinucleotide repeat-triggered rare fragile sites (65).
Rare fragile site instability and secondary structure formation.
The folate-sensitive rare fragile sites consist of expanded CGG trinucleotide repeats. These expansions, as observed for common fragile sites, also replicate very late, during the G2 phase of the cell cycle, later than unexpanded alleles at the same loci (66, 67). To account for this delay, it was proposed that CGG and AT minisatellites may form secondary structures, blocking progression of replication forks (Fig. 3B). Secondary structure formation at fragile sites was investigated in vitro. CGG repeats were shown to form tetraplexes, and DNA synthesis was blocked in vitro by 20 stretches of CGG, in a K+-dependent manner. Tetraplexes require cations small enough to fit in the cavity created by guanine tetrads and stabilize the structure, so K+ dependency is an indicator of tetraplex formation (68). Additionally, it was also shown that the stoichiometry of the structure is tetramolecular and that guanines are protected from dimethylsulfate (DMS) guanine-specific cleavage, further confirming tetraplex structure formation (69). CGG repeats could also theoretically form imperfect hairpins (70). Therefore, dynamic changes between tetraplexes and hairpins may occur in vivo in different physiological conditions (71). To better understand the impact of secondary structure formation on replication, the progression of the replication fork through CGG repeats cloned into a bacterial plasmid was followed by 2D gels. Replication fork stalling was visible in E. coli for plasmids bearing more than 30 repeats of either CGG or GCC. The stalling was abrogated when CGG repeats were interrupted by AGG motifs (72). Similar experiments in yeast showed that CGG/CCG repeats block replication fork in a length-dependent manner starting with only 10 repeats (73).
AT-rich minisatellites have the ability to form hairpins (74). A short AT-rich region from FRA16D called Flex1 was cloned in yeast and was sufficient to induce breakage at this locus, while other regions of the fragile site did not elicit fragility (75), further confirming the implication of the AT-rich minisatellite in the fragility mechanism.
In conclusion, expanded microsatellites or minisatellites form secondary structures that transiently stall replication forks. Delayed restart of a stalled fork may contribute to underreplicating a portion of the genome. During mitosis, improper chromatid disjunction of this nonreplicated region will break, leading to DSBs.
A very recent publication has shed new light on the connection between secondary structure formation and chromosome fragility. With the help of a DNA secondary structure inference program, the human genome was analyzed and predicted to contain 23,331 putative fragile sites, with sizes ranging from 1,200 to 20,000 nucleotides. Altogether, they cover 1.5% of the genome and include putative G-quadruplexes, as well as AT-rich sequences. In parallel, DSB sites were experimentally captured and sequenced. Remarkably, all predicted structured regions were enriched in DSBs, and their fragility was mediated by the TOP2 gene, suggesting that this topoisomerase preferentially targets structured regions (76).
Inverted Repeats and Cruciform Structures
Inverted repeats are sequences with an internal symmetry allowing switching between interstrand and intrastrand base pairing. As a result, these repeats can form a cruciform structure, consisting of a branch point, a stem, and a loop, where the size of the loop is dictated by the distance between inverted repeats (Fig. 4A). Inverted repeats occur nonrandomly in the genome of all organisms—phages, plasmids, mitochondria, eukaryotic viruses, and mammalian cells—and were found to be enriched at chromosomal breakpoint junctions, promoters, and replication initiation sites (77).
FIG 4.

Inverted repeats and cruciforms. (A) The unstructured palindrome is shown as complementary pink and blue arrows. In certain conditions, it may adopt a cruciform structure containing two stems and two loops of variable lengths that form a four-way junction whose structure is similar to a Holliday junction. The stability of a cruciform depends on both the stem and loop lengths. (B) Double-strand break (DSB) frequency according to the identity between direct or inverted tandem repeats in yeast. More identical repeats are more prone to form stable cruciforms, therefore inducing more frequent DSB. (Based on data from reference 96.) (C) Genetic factors involved in cruciform processing in yeast. The proteins involved in the transition between the nicked form and the capped DSB are not clearly characterized, but ligase IV (DNL4 in yeast) does not play a role in this reaction (96). (D) The number of direct or inverted Alu repeats in the human genome. The distances between repeats were classified into close (0 to 20 nucleotides [nt], left panel), medium (21 to 100 nt, middle panel), and distant (101 to 500 nt, right panel). In each panel, repeats are classified from left to right in order of decreasing identity (>90%, 81 to 90%, 71 to 80%, and 61 to 70% identity). Close inverted repeats are counterselected because they may form stable cruciforms (left panel), whereas this selection is less pronounced at longer distances. (Based on data from https://www.niehs.nih.gov/research/resources/databases/alu/index.cfm.)
Inverted repeats in Escherichia coli.
Evidence for the presence of inverted repeats in bacterial genomes was provided by genotyping bacterial strains using PCR with arbitrary primers, leading to the amplification of polymorphic inverted repeats (78) and by the use of chemical compounds and cellular stresses to modify the superhelical torsion of E. coli plasmids to extrude cruciforms (79, 80). Negative supercoiling driven by active transcription of a regulatable promoter was also shown to increase the transition of an AT-rich region into a cruciform (81). Such structures were also detected in E. coli in the early days of genetic engineering, where plasmids containing long inverted repeats were either partially deleted or would be impossible to clone in bacteria (82, 83). However, these sequences were more tolerated in sbcC (84) and sbcD mutants (85). The viability of these plasmids can be modulated by modifying the central sequence of the inverted repeat, more stable ones being less viable, arguing in favor of cruciform formation in vivo (86). Additionally, inverted repeat-containing DNA was shown to slow the replication rate of phages (87). The SbcCD operon encodes two proteins with single-stranded DNA endonuclease and double-stranded DNA exonuclease activities, which cleave and process hairpin structures in vitro (88). Using highly resolutive polyacrylamide gels, it was shown that SbcCD is a single-stranded endonuclease cleaving CTG hairpin loops and a 3′→5′ double-strand exonuclease that subsequently degrades duplex DNA to half its original length (89). Subsequent genetic analysis in recombination-deficient strains showed that DSBs were actually repaired by the RecBCD and RecA pathway of homologous recombination (90). It was therefore postulated that the complex might also process similar DNA secondary structures during replication.
Two models were proposed to explain the instability triggered by inverted repeats: (i) a hairpin structure forms, stalling replication fork progression, and cleavage at this stalled fork generates a one-ended DNA break; (ii) a hairpin structure forms after passage of the replication fork, and cleavage generates a two-ended DNA break. Pulsed-field gel electrophoresis (PFGE) followed by Southern blotting of the DNA at the inverted repeat revealed that breaks are two-ended DSBs. The two-ended nature of the break implies that the processing of the inverted repeat by SbcCD does not lead to replication fork collapse. Nevertheless, the breaks were visible only in permissive conditions for replication and were not detected when replication was inhibited at 42°C (91). Using 2D gels, inverted repeats were shown to induce replication fork stalling both in E. coli and S. cerevisiae (92). Finally, using a plasmid-based assay, a cruciform structure formed by AT-rich inverted repeats and located upstream a tightly regulated promoter was shown to form only when the promoter was active (81).
Inverted repeats in yeast.
Inverted repeats were studied in S. cerevisiae by integrating inverted human Alu repeats in the yeast genome. Such a sequence was shown to increase recombination 2,000-fold over an equivalent direct repeat in a wild-type strain (93). In this specific yeast assay, inverted repeats were more recombinogenic than direct repeats (Fig. 4B). Inverted Alu repeats that were only 86% homologous could stimulate recombination when separated by 12 bp, whereas perfectly identical repeats stimulated recombination 30-fold when separated by 100 bp. It was concluded that sequence identity and distance between repeats were synergistic in triggering homologous recombination (93).
Mre11 and Rad50 are eukaryotic homologs of SbcD and SbcC, respectively (94), and similar to what was observed for SbcD, Mre11 was shown to cleave hairpins in vitro (95). In a yeast assay, recombination frequency at inverted repeats decreased in a Δmre11 mutant, whereas no effect was observed at direct repeats. Molecular analysis and double-strand break quantification showed that DSB levels increased in Δmre11 compared to wild type, indicating that Mre11 was directly involved not in cleaving inverted repeats but, rather, in processing double-strand breaks occurring at these repeats. The same observations were made for Δrad50, Δxrs2, and Δsae2 strains, suggesting that the whole Mre11 complex was required to process DSBs at inverted Alu repeats (Fig. 4C) (96). More specifically, the endonuclease activity of the Mre11 complex is required to process double-strand breaks since similar results were obtained for strains containing the nuclease-deficient mre11-H125 or mre11-D56N alleles.
In the fission yeast Schizosaccharomyces pombe, an 80-bp inverted repeat was shown to be a meiotic recombination hot spot and was preferentially lost by gene conversion. This hot spot activity was abolished in a rad50 or rad32 (RAD32 is the S. pombe MRE11 orthologue) mutant. It was proposed that cruciform extrusion was recognized and cleaved by the Rad50-Rad32 nuclease complex (97). The same team also showed that DSBs at the inverted repeat appeared earlier than normal meiotic Rec12-dependent DSBs (S. pombe SPO11 orthologue) during premeiotic replication (98). Similarly, in budding yeast, a strong meiotic hot spot was associated with a 140-bp inverted repeat in the HIS4 gene, which was able to extrude into a cruciform structure (99). DSB formation at this site depends on genes responsible for making meiotic DSBs, including the MRE11 complex and SPO11 (100). This suggested that breaks made at this inverted repeat were induced and processed by the same machinery and within the same time frame as other meiotic breaks, underlying a subtle difference between budding and fission yeast.
A systematic genetic screen, measuring chromosomal rearrangements caused by inverted Alu repeat recombination, identified genes belonging to the DNA replication machinery, including all three replicative polymerases (Polα δ and ε), PCNA, RAD27, the MCM replicative helicase, the SGS1 helicase, and the PRI2 primase. RAD50, MRE11, and checkpoint, genes as well as telomere-protection genes, also increased chromosomal rearrangements (101). Molecular analyses showed that DSB accumulation at this locus was dependent on the Sae2 protein, showing that its function was essential to process the break. In addition, inactivation of the RAD51 recombinase suppressed chromosomal fragility observed when replication was compromised. Based on these experiments, two models were proposed to account for these chromosomal rearrangements. In replication-proficient cells, cruciform structures led to a nicked cruciform, which will be converted to a double-capped DSB, subsequently decapped by the Mre11 complex, along with Sae2, leading to a regular DSB that can be repaired by homologous recombination (Fig. 4C). In replication-compromised cells, Rad51-mediated template switching allows bypass of the replication block and leads to cruciform formation, subsequently resolved as described above (101). At the present time, it is unclear whether this model may apply to other alternative DNA secondary structures or is specific to cruciforms.
Two naturally occurring sites of chromosomal rearrangements, 20 kb apart from each other, were identified on Saccharomyces cerevisiae chromosome III. The first one (FS1) consists of two head-to-tail Ty retrotransposons, whereas the second one (FS2) was made of two head-to-head Ty elements separated by 283 bp. Strains that were engineered to produce low levels of polymerase α showed a 20-fold increase in chromosomal rearrangements involving the FS2 site. Using molecular probing, a DSB was detected at FS2, providing an explanation for the observed rearrangements and strongly suggesting that inverted Ty elements were extruded as a cruciform structure. It is, however, unfortunate that the authors did not test the effect of a mutation in RAD50 or MRE11 in their experimental system (102).
Inverted repeats in the human genome.
A large-scale survey of the human genome showed that the 1.5 million Alu sequences it contains were not randomly distributed. Direct Alu repeats are much more frequently encountered than inverted Alu sequences. This depends, at the same time, on the distance between repeats and on their sequence identity. Closely matching Alu sequences are counterselected at short distances, whereas more diverged ones are more tolerated, this homology-dependent effect decreasing with increasing distance between them. This homology and distance effect was not observed with direct Alu repeats (Fig. 4D) (93).
A well-studied inverted repeat-induced rearrangement is the t(11;22)(q23;q11.2) translocation. Balanced carriers are healthy but have infertility problems, and their offspring may have Emmanuel syndrome due to a faulty meiotic disjunction of the translocated chromosome. Their genotype contains two copies of chromosome 11, two copies of chromosome 22, and an extra translocated chromosome carrying genetic information from both chromosomes 11 and 22 (103). Breakpoint analysis of 11q23 and 22q11 revealed that these regions contain a large inverted repeat of hundreds of AT-rich base pairs named, respectively, PATRR11 and PATRR22. Small changes in the sequence of PATRR are sufficient to prevent translocation, which is in favor of a secondary structure formation that may be the cause of the translocation (104, 105). Using plasmid-based systems bearing PATRR11 and PATRR22 in HEK293 cells, it was shown that GEN1 knockdown led to a decline in hairpin-capped DSBs (106). GEN1 is an endonuclease involved in Holliday Junction resolution, a four-way DNA structure similar to cruciform DNA (Fig. 4A). However, reproducing these results in chromosome-borne construct would strengthen these observations. It is possible that, given the functional redundancy of enzymes in charge of processing three-way and four-way DNA junctions, genetic requirements are different when cruciforms are formed in a plasmid or embedded within a chromosome wrapped into chromatin. This translocation is thought to occur during spermatogenesis. Indeed, t(11;22) is detectable as a de novo translocation in sperm from normal healthy males at frequencies of 10−4 to 10−5 but not in mitotic cells (107). A model study conducted with HEK cells was carried out in order to understand how translocations arise. In this model, effective translocation between two plasmids carrying either PATRR-11 or PATRR-22 results in green fluorescent protein (GFP) expression. It was correlated with the supercoiling state of the plasmids at the time of the transfection, suggesting that secondary structure formation was sufficient to induce translocation independently of replication. Therefore, the authors suggested that excess negative supercoiling may accumulate temporarily in DNA during chromatin compaction, occurring at the latest stages of spermatogenesis, triggering the t(11;22) translocation (108). Finally, as another example, inverted gene amplification may have shaped the amplified ERBB2 locus encountered in breast cancer through breakage-fusion-bridge cycles (109).
Direct evidence for cruciform formation in vivo.
To detect cruciform structures in vivo, psoralen and UV light cross-linking were used in E. coli. A 66-bp inverted repeat integrated into a plasmid could not be detected as a cruciform by electrophoresis on agarose gels (110). Later, using the same techniques on specific inverted repeats containing AT-rich loops and GC-rich stems, the authors were able to visualize small amounts of cruciform DNA (0.01 to 1% of total DNA) (111). Finally, a PATRR11-bearing plasmid treated with psoralen and UV revealed the presence of cruciform structures under atomic force microscopy (105).
Direct visualization of cruciforms in cells was attempted with a monoclonal antibody (2D3) raised against synthetic cruciform structures. This antibody was shown to recognize such structures but not heteroduplex slipped-stranded DNA containing a hairpin on one strand only (112). Interaction of this antibody with cruciform structures was carried out in vitro using a band-shift assay and showed that 2D3 interacts with the four-way junction at the base of the cruciform (113). Later, immunoprecipitation using the same antibody revealed the presence of cruciform-containing DNA at a yeast replication origin (114). When 2D3 was tested on synthetic oligonucleotides carrying different lengths of CAG/CTG trinucleotide repeats, it was found that the antibody efficiently bound to heteroduplexes but not to homoduplexes (115). This suggests either that CAG/CTG heteroduplexes are indeed structured as four-way junctions similar to cruciforms or that 2D3 recognizes three-way junctions containing a hairpin on one strand only, a result different from what was previously reported (112). In addition, 2D3 was raised against perfect cruciforms, whereas CAG/CTG hairpins are not perfect, since A-T bases facing each other do not form Watson-Crick bonds, and the resulting structure is very different from a perfect hairpin (70, 116, 117). For these reasons, it is unclear at this time whether 2D3 specifically binds cruciforms or a panel of slipped-stranded DNA molecules.
Microsatellites and Secondary Structures
Microsatellites (also called VNTR [variable number of tandem repeats] or SSR [short sequence repeats]) are tandem repeats of short repeat unit (less than 10 bp). They were initially discovered in 1984 as a highly polymorphic sequence in the human myoglobin gene (118). Since then, they have been shown to be extremely frequent in all eukaryotes, and their length polymorphism was widely used for applications ranging from forensic medicine and paternity tests to the establishment of the first physical map of the human genome in 1996 (reviewed in reference 119). In 1991, for the first time, a human neurological disorder, the fragile X syndrome, was linked to the large expansion of a CGG trinucleotide repeat at the fragile FRAXA locus (120). Shortly after this first discovery, many other disorders were found to be strongly associated with the expansion of a trinucleotide repeat and, less frequently, with other microsatellites (GGGGCC repeats [121], CCTG repeats [122], and ATTCT repeats [123]). Interestingly, although there are 10 possible nonmonotonous trinucleotide repeats, only three of them were found to be expanded in human disorders (CAG/CTG, CGG/CCG, and GAA/TTC). Hence, it was soon proposed that expansions were probably triggered by the formation of secondary structures that would interfere with normal DNA metabolism (124). We will now be reviewing evidence proving that some of these microsatellites are able to form secondary structures in living cells.
CAG/CTG trinucleotide repeats.
CAG/CTG form imperfect hairpins in vitro; this was demonstrated using 1H nuclear magnetic resonance (NMR) (70). Experiments measuring the melting temperature (Tm) of oligonucleotides made of various numbers of CAG or CTG showed that CTG hairpins are more stable than CAG hairpins. This may be because purines occupy more space than pyrimidines and are most likely to interfere with hairpin stacking forces (125). Experiments of the denaturation/renaturation of a plasmid containing 50 CAG/CTG repeats were carried out, and their length was resolved on 4% polyacrylamide gels. Upon renaturation, 60% of the DNA was slipped-stranded. Electron microscopy comparisons with plasmids carrying 255 CAG/CTG repeats revealed compacted and bent molecules, corresponding to structured DNA fragments. The heterogeneity and complexity of these molecules increased with the number of repeats (126). Recently, FRET (Förster resonance energy transfer, in which measurement of photon transfer between two light-sensitive molecules allows determination of the distance between these two molecules) experiments revealed that parity in the number of repeats had an impact on their stability. Even numbers of CAG are stable while odd numbers induce a slipping back and forth between states (127).
Much genetics evidence argues in favor of secondary structure formation in vivo. In yeast, CAG/CTG trinucleotide repeats are more unstable when the CTG triplets are located on the lagging strand template, which is supposedly more prone to form single-stranded secondary structures, than the leading strand template. Using 2D gels to visualize replication intermediates, it was shown in E. coli that fork stalling during replication occurred for plasmids bearing CTG repeats in the lagging-strand, but not for CAG repeats. Pausing was more frequent as the length of the repeat increased (72). The same experiment was carried out in yeast and showed a mild effect on fork stalling at 80 CAG or CTG repeated sequences (73). In vivo in HeLa cells, evidence for hairpin formation was given by the use of a zinc finger nuclease (ZFN) that recognizes CTG repeats. Only one ZFN arm was found to be able to cut DNA, suggesting that a hairpin was formed and cut. This argument can, however, be debated, as hairpins formed by CTG repeats are imperfect and do not structurally mirror Watson-Crick bounds. When the same cells were serum deprived and were not cycling, no cutting was found, suggesting that hairpin formation was replication dependent. CAG/CTG repeats showed increased instability through multiple cycling division, suggesting that replication was implicated in their instability. In HeLa cells, instability of (CAG)102 and (CTG)102 was observed after 250 doublings. The instability was suppressed when the close-by replication origin was inactivated (128).
Secondary structures may form on either strand of the DNA; slippage of the DNA polymerase and the nascent strand backwards on the template strand would result in the formation of structures containing an excess of repeats on the nascent strand, resulting in expansion products. The opposite would give rise to deletion products. Long CTG repeats may also induce fork stalling and collapse. Restart subsequently involves repair and recombination machineries to pursue replication. Thus, biophysical studies showed that CAG/CTG form hairpins in vitro, and some evidence tends to confirm that they also form structures in vivo, leading to instability of the repeat tract. It is, however, unknown whether in vitro and in vivo CAG/CTG repeats display identical secondary structures or share similar properties but exhibit different structures.
Slipped-stranded DNA, recognized by 2D3 antibody (113) as the junction between B-DNA and hairpins, was found at the unstable trinucleotide repeats of the myotonic dystrophy disease locus in patient brain, heart, muscle, pancreas, and liver (129). More recently, a synthetic molecule, naphthyridine-azaquinolone (NA) was shown to specifically bind long CAG slip-outs, causing a shift on electrophoresis gel when bound to the annealing product between (CAG)50 and (CTG)30. NA was able to inhibit repeat expansions and trigger their contraction in the striatum of R6/2 mice, a model of Huntington disease (HD) harboring 150 CAG repeats. This was taken as evidence for secondary structure formation in vivo in whole animals (130). The potency of the molecule was found to be dependent on transcription, probably because secondary structures form to a greater extent during the metabolic process (131).
GAA/TTC trinucleotide repeats.
The first evidence for the formation of the non-B-DNA form was brought about by the observation that synthetic polyU-polyA ribonucleotides could hybridize in vitro in a 1:1 ratio, as predicted by the double-helix model, as well as in a 2:1 ratio, suggesting the existence of a more stable three-stranded structure (132). In triple helices, the third strand is provided by one of the strands of the same duplex DNA molecule at a mirror repeat sequence, bound by a Hoogsteen hydrogen bond. Intramolecular triplexes can be formed by T-A*T or C-G*C+ triad, where the asterisk is a Hoogsteen bond, the hyphen is a Watson-Crick bond, and C+ is a protonated cytosine. Because of the requirement of the cytosine to be protonated, this structure is called H-DNA. In contract, *H-DNA is maintained by T-A*A or C-G*G triads and is not pH dependent (133). Homopurine-homopyrimidine repeats were shown to form H-DNA, as visualized by 2D gels (Fig. 5A). Plasmids carrying different potentially forming H-DNA were subjected to 2D gels and analyzed for their propensity to undergo transition to H from under superhelical stress (134). Similarly, GAA/TTC repeats form triple helices in vitro, exhibiting specific melting curves (Fig. 5B) (135). Evidence for in vivo formation was given by antibodies targeting triple-stranded DNA (136) and single-stranded probes (137). However, none of these experiments were carried out in physiological conditions, and the transient and dynamic nature of such structures may explain why they are so difficult to detect.
FIG 5.
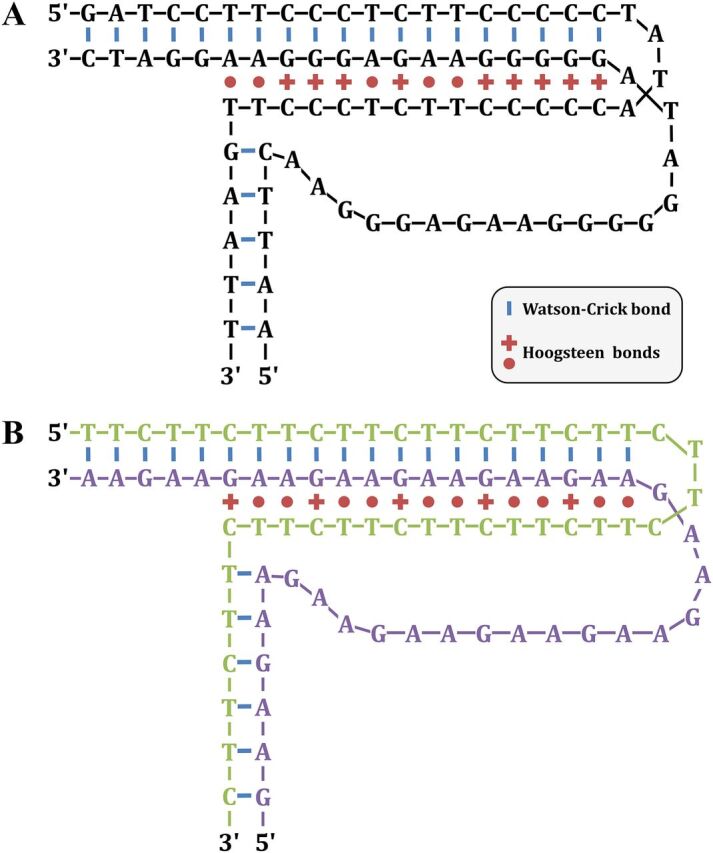
H-DNA triplex structures. (A) A polypurine-polypyrimidine H-DNA structure is shown. (Based on data from reference 134.) (B) A similar structure formed by GAA/TTC trinucleotide repeats. The GAA strand and the TTC strand are colored in green and purple, respectively, to make their visualization easier.
In S. cerevisiae, using 2D gels to monitor replication fork stalling, chromosome-borne GAA/TTC repeats transiently stalled replication forks when the GAA sequence was located on the lagging-strand template, but not in the opposite orientation (138). It was postulated that DNA polymerase stalls on the lagging strand due to GAA triplexes, while the polymerase on the leading strand continues, leading to long stretches of single-stranded DNA. The stalling region is bypassed when the stalled strand invades its sister chromatin, by template switching, and may account for large GAA repeat expansion repeats in yeast. In support of this hypothesis, RAD27 knockout in S. cerevisiae leads to a drastic increase in GAA repeat contractions (139) and expansions (140). Rad27p is a 5′ flap endonuclease, an orthologue of FEN-1 in humans, and is responsible for Okazaki fragment processing, although a possible additional role in homologous recombination was proposed (141). Mutating Rad27 residues responsible for the correct alignment of the 5′ DNA flap with the protein catalytic site increased the rate of GAA repeat expansions in yeast by a mechanism proposed to be template switching (142).
In yeast, GAA triplet repeats trigger DSB formation (143), and the GAA repeat expansion at the FXN locus in lymphoblastoid cells was linked to chromosomal breakage (144). DSBs induced by H-DNA may account for its intrinsic instability, although formal evidence is lacking to support this hypothesis, and mutants in the yeast double-strand break repair pathway do not dramatically increase GAA repeat instability (139). Finally, H-DNA colocalizes with fragile sites such as c-Myc (145) and BCL-2 loci (146).
There is some evidence that triplex structures may form in vivo within GAA/TTC repeats. In budding yeast, transcription of such repeats increases their expansion rate. Knocking out RNH1 and RNH201, encoding RNase H1 and the catalytic subunit of RNase H2, resulted in higher repeat instability when GAA repeats were transcribed (147). This suggests that RNA-DNA hybrids are linked to these expansions. When RAD52 or POL32 were knocked out, RNase H-dependent expansions returned to the wild-type level, showing that they occur through a homologous recombination mechanism involving long-range DNA synthesis. The authors hypothesized that triplex H-DNA structures, perhaps transiently stabilized by RNA-DNA hybrids, were responsible for triggering the observed expansions.
Other microsatellites.
Other microsatellites found to be unstable in vivo were shown to form secondary structures in vitro. By observing the behavior of repeat-containing oligonucleotides after enzymatic or chemical treatments, it was inferred that CAGG tetranucleotide repeats form imperfect hairpins. No structure was observed for the complementary CCTG repeat in the tested conditions, suggesting that CAGG hairpins are more stable (148). However, using the nuclear Overhauser effect, which is a more recent type of nuclear magnetic resonance, it was suggested that CCTG may form hairpins with a two-residue CT loop or a dumbbell (149). CCTG/CAGG repeats were transfected in green monkey kidney cell line COS-7. Instability was greater when CAGG was on the leading strand, and instability was length dependent (148).
DNA unpairing at ATTCT pentanucleotide repeats in supercoiled DNA was detected using 2D gels, indicating that ATTCT repeats form structures similar to DNA unwinding elements (DUE) (150). DUE were discovered in E. coli (151) and later on in S. cerevisiae, as AT-rich sequences easily unwound when located at replication origins (152). DUE are a common feature of prokaryotic and eukaryotic replication origins and act as a start point for strand separation and unwinding of the double helix. ATTCT repeats were able to trigger aberrant replication initiation in HeLa cells. Instability of the repeat may come from the refiring of replication after the fork has already passed through the repeat, leading to rereplication and massive repeat expansion (150). Very recently, NMR analysis of ATTCT repeats of different lengths showed that they adopt a very specific secondary structure in which the two first repeats form a compact minidumbbell (153). The existence of such a structure in living cells remains to be elucidated.
ATTCT repeats were linked to fragility and to expansions in a yeast reporter assay in which they were integrated in the middle of a URA3 gene. Expanded repeats directly block the expression of the URA3 gene, allowing the monitoring of expansion and contraction of the repeat tract by screening 5-fluoroorotic acid (5-FOA) resistant colonies. The expansion rate showed a 10-fold decrease in a Δrad5 mutant background, whereas contractions were unchanged. In addition, chromosomal fragility was also decreased, although to a lesser extent (154). Rad5 is involved in postreplication template switching, suggesting that this pathway triggers ATTCT expansions.
MISMATCH REPAIR ACTIVITY ON ALTERNATIVE DNA STRUCTURES
Role of the Mismatch Repair System in Microsatellite Instability: Indirect Evidence for Secondary Structure Formation In Vivo
The role of mismatch repair during replication.
The mismatch repair machinery (MMR) is a highly conserved system specialized in removing synthesis errors ignored by the editing function of DNA polymerases during genome replication. Malfunction or inactivation of this system leads to an increase in spontaneous mutations and a strong predisposition to tumor development. Tumor cells from hereditary nonpolyposis colorectal cancer patients displayed high alteration of microsatellite length (155). In yeast, the absence of a functional MMR leads to a 700-fold increase in GT repeat instability (156) and a 10- to 10,000-fold increase in polydeoxyadenine tract instability (157), suggesting that following replication, a functional MMR is required to fix small slippage errors done by the polymerase.
Mismatch repair proteins.
The MMR machinery was successfully reconstituted using bacterial (158), yeast (159), and mammalian purified proteins (160). MMR acts through a sequential mechanism starting with mismatch recognition, followed by excision of the DNA strand containing the wrong information and by subsequent resynthesis. MMR proteins were first identified in E. coli by studying mutator strains deficient for the MutS, MutL, MutH, or UvrD protein. Following mismatch recognition, MutS, through its ATP hydrolysis activity, undergoes an ADP-ATP exchange-driven conformational change into a sliding clamp and recruits the MutL heterodimer. The complex formed by MutS-MutL can translocate in either direction along DNA toward a gap between Okazaki fragments and initiate degradation of the mutated DNA strand. The resulting single-stranded gap is filled by polymerase δ. In eukaryotes, three MutS homologues (MSH genes) were shown to form heterodimers, MSH2, MSH3, and MSH6 (161). MutSα (MSH2 + MSH6) is involved in the repair of base-base mispairings and short insertion/deletions, while MutSβ (MSH2 + MSH3) acts on some base-base mispairs and both short and long insertion/deletions (162, 163).
Are DNA Secondary Structures Recognized by MMR Proteins? In Vitro Evidence
CTG or CAG hairpins are likely to be recognized by MMR proteins, which would recognize them as mismatches. Such slipped-stranded structures may be generated in vitro by annealing CAG/CTG repeat-containing oligonucleotides of either the same or different lengths, and a band-shift assay was performed to detect Msh2 binding to these mismatched heteroduplexes. Msh2 was found to bind these structures, and its affinity increased with repeat length. Furthermore, Msh2 binds more efficiently to (CAG)15 oligonucleotide and (CTG)30·(CAG)50 slipped DNA than to (CTG)15 and (CTG)50·(CAG)30 slipped DNA (164). Purified human Msh2-Msh3 protein complexes were found to lose their ATPase activity and to be stuck into a noncatalytic conformation when bound to CAG hairpins (165). However, another study, conducted using HeLa cell extracts, showed that the binding of MutSβ did not affect nucleotide binding and hydrolysis, although it confirmed the interaction with CAG hairpins (166). In spite of being contradictory regarding the effect of hairpin binding on subsequent repair steps, probably due to different experimental settings, evidence tends to confirm that MutSβ binds to CAG/CTG hairpins.
MMR Impact on Trinucleotide Repeat Instability In Vivo
Early reports pointed toward a role of MMR proteins in trinucleotide repeat instability. Loss of Msh2 in HD (Huntington disease) model R6/1 mice resulted in a strong decrease in the number of expansions of CAG repeats in somatic cells (167) and germ cells (168). Msh3 deficiency was shown to have a similar effect in R6/1 mice, while Msh6–/– mice did not exhibit any change in CAG instability (165). In mice deficient in Msh2 or Msh3, CTG repeat instability shifted from a bias toward expansions to a bias toward contractions (169, 170). Later experiments in a Msh2-mutant mice carrying a missense mutation, Msh2G674A/G674A, leading to the impairment of ATPase activity while retaining binding property to mismatches, showed a similar phenotype (171). This indicated that CTG repeat expansions require not only the binding of MutSβ to slipped-stranded DNA, but also a functional ATPase dependent catalytic activity. Similarly, Msh2 deficiency in a fragile X mouse model showed significantly reduced intergenerational instability of CGG repeats, suggesting that Msh2 is also a key factor in CGG expansions (172). Hence, hairpin binding by MutSβ is a key step toward trinucleotide repeat expansions, but the requirement for its catalytic activity strongly suggests that ATP-dependent MutL recruitment is also necessary to trigger expansions. However, the observation that Msh2 deficiency did not completely abolish expansions (168) indicates unknown roles for other DNA repair processes in promoting repeat instability and a cross talk between these DNA repair processes.
Similarly, MMR also destabilizes GAA/TTC repeats in yeast (138) and in human cells (173). In transgenic mice, the effect of the MMR was strikingly different from what was observed for CAG/CTG repeats. Msh2 or Msh3 deletion led to an increase in contractions, while the expansion rate was unchanged. In contrast, Msh6 or Pms2 inactivation led to a clear increase in expansions (174). This strongly suggests that the MMR destabilizes CAG/CTG and GAA/TTC repeats by different mechanisms.
Mechanisms underlying MMR-mediated trinucleotide repeat instability.
A reporter assay in E. coli was used to monitor the effect of CAG/CTG triplet repeats on the recombination of a tandem array of zeocin resistance genes located 6.3 kb away from the repeat tract. Recombination was eliminated in the absence of MutS, MutL, and MutH, while the hairpin endonuclease SbcCD (Mre11/Rad50) did not exhibit any effect (175). This observation suggested that MMR is critical to trigger homologous recombination near CAG/CTG repeats in E. coli, implying the formation of a recombinogenic product at, or near, the repeat tract.
In S. cerevisiae, MSH3 is required for trinucleotide repeat expansions, through successive generations, supporting an iterative model of incremental expansions, rather than a saltatory model of large expansions (176). Replication of a repeat-containing plasmid in the presence or absence of functional MMR was conducted using either cell extract from HeLa cells (MMR-proficient) or LoVo cells (MMR-deficient). The resulting replicated plasmids were then transformed in bacteria and analyzed for repeat length changes or slipped-stranded DNA formation. This revealed that heteroduplex DNA molecules are formed during lagging-strand synthesis and are eliminated via MutSβ-mediated postreplication repair (177). Using 2D gel electrophoresis, chromosome-borne CAG/CTG repeats were shown to transiently stall replication forks in S. cerevisiae. This stalling was partially alleviated in mismatch repair-deficient yeast strains. In addition, Msh2 was shown, by chromatin immunoprecipitation, to be enriched at the CAG/CTG repeat tract, suggesting that the observed reduction in replication fork stalling was not an indirect effect of MMR deficiency (178). In the same work, MSH2 overexpression led to a large increase in the number of sectored yeast colonies, the hallmark of heteroduplex DNA (Fig. 6A). Subsequent repair of stalled replication forks may then depend on the strand on which the hairpin was formed. Stalled forks may be repaired by homology-driven template switching, whereas unrepaired forks may lead to chromosome fragility (Fig. 6B). In yeast srs2Δ cells, CTG repeats undergo frequent expansions and contractions, and additional inactivation of the RAD51 recombinase or the RAD52 recombination mediator suppresses this phenotype, suggesting a role of homologous recombination in trinucleotide repeat instability in the absence of any induced DSB (179). Further 2D gel analyses of strains mutated in different domains of the Srs2 protein allowed the more precise definition of its role on CTG trinucleotide repeats: (i) Srs2 reduced chromosomal fragility through its interaction with PCNA, probably by unwinding fork-blocking CAG/CTG hairpins during replication; (ii) the helicase activity of the Srs2 protein inhibited the formation of Rad51-dependent recombination intermediates, and a mutation in this domain increases both repeat fragility and instability (Fig. 6B) (180).
FIG 6.

Role of the mismatch repair system in CAG/CTG trinucleotide repeat expansion and fragility. (A) Hairpin formed on the newly synthesized lagging strand. The damaged fork is recognized by the MMR but cannot be fixed, thus leading to a small expansion during the next S phase. Successive cycles of small expansions may occur, ultimately leading to a large expansion. Alternatively, another mechanism may directly lead to large expansions, such as those observed in some human disorders. Unrepaired heteroduplex DNA is observed in the progeny as two cell populations with different repeat tract lengths (see the text for details). (B) Hairpin formed on the lagging-strand template. The damaged fork is recognized by the mismatch repair system (MMR) and may lead to chromosomal fragility at the next S phase if checkpoints are bypassed or if the damage cannot be fixed. Template switching is a possible pathway to repair and restart the fork but may lead to trinucleotide repeat expansions and contractions by homologous recombination under the control of the Rad51 recombinase and the Srs2 helicase in yeast (see the text for details). Note that the hairpin was drawn on the lagging strand (or on its template), but the model can perfectly be reversed if its formation happens on the leading strand (or on its template).
CONCLUDING REMARKS AND FUTURE DIRECTIONS
The formation of DNA secondary structure in living cells is mainly supported by genetic evidence—instability triggered by repeated sequences, specific helicases such as Pif1 or Srs2, replication fork stalling, rare fragile sites. Although G-quadruplex formation in vivo is backed by numerous convincing experiments, many questions remain concerning the existence of other secondary structures in living cells. Even though replication forks are stalled by expanded trinucleotide repeats, is this stalling a direct effect of secondary structure formation, or is it mediated by proteins binding specifically to the expanded sequence? If secondary structures are formed within expanded microsatellites in vivo, are they similar to the structures observed in vitro? How is chromatin organized within expanded structure-forming microsatellites, and does it vary from one cell type to another, perhaps explaining differences in stability between tissues?
The development of superresolution fluorescence microscopy in living cells may open a new direction of research. In particular, the PALM technology, using genome-encoded fluorophores, may allow the observation of DNA secondary structures in vivo. The resolution attained should be sufficient to visualize large CAG/CTG hairpins, covering a hundred triplets or more. However, the fluorophore needs to bind near or at the secondary structure to be observed (181).
More direct evidence to monitor such secondary structure formation in vivo using chemical ligands or antibodies directed toward a specific structure are still elusive, since their formation is probably a transient phenomenon which does not happen in all cells and in all cell types with the same frequency. Development of an antibody is tedious and faces the challenge to prove that the antibody recognizes the structure and not the sequence. The recent evidence that a small molecule, naphthyridine-azaquinolone, induces repeat contractions in Huntington disease model cells argues in favor of the formation of CAG/CTG hairpins in vivo (130). In addition to progress in our understanding of alternative DNA conformations in vivo, better characterization of these structures could therefore hasten the development of new therapies for microsatellite expansion disorders.
ACKNOWLEDGMENTS
Work in our laboratory is generously supported by the Institut Pasteur and by the Centre National de la Recherche Scientifique (CNRS). L.P. was supported by the Fondation Blanchecape and the Association Française contre les Myopathies (AFM).
REFERENCES
- 1.Franklin RE, Gosling RG. 1953. Molecular configuration in sodium thymonucleate. Nature 171:740–741. doi: 10.1038/171740a0. [DOI] [PubMed] [Google Scholar]
- 2.Watson JD, Crick FHC. 1953. Molecular structure of nucleic acids: a structure for deoxyribose nucleic acid. Nature 171:737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 3.Chargaff E, Magasanik B, Vischer E, Green C, Doniger R, Elson D. 1950. Nucleotide composition of pentose nucleic acids from yeast and mammalian tissues. J Biol Chem 186:51–67. [PubMed] [Google Scholar]
- 4.Hoogsteen K. 1963. The crystal and molecular structure of a hydrogen-bonded complex between 1-methylthymine and 9-methyladenine. Acta Cryst 16:907–916. doi: 10.1107/S0365110X63002437. [DOI] [Google Scholar]
- 5.Arnott S, Fuller W, Hodgson A, Prutton I. 1968. Molecular conformations and structure transitions of RNA complementary helices and their possible biological significance. Nature 220:561–564. doi: 10.1038/220561a0. [DOI] [PubMed] [Google Scholar]
- 6.Xiong Y, Sundaralingam M. 2000. Crystal structure of a DNA·RNA hybrid duplex with a polypurine RNA r(gaagaagag) and a complementary polypyrimidine DNA d(CTCTTCTTC). Nucleic Acids Res 28:2171–2176. doi: 10.1093/nar/28.10.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang AH-J, Quigley GJ, Kolpak FJ, Crawford JL, van Boom JH, van der Marel G, Rich A. 1979. Molecular structure of a left-handed double helical DNA fragment at atomic resolution. Nature 282:680–686. doi: 10.1038/282680a0. [DOI] [PubMed] [Google Scholar]
- 8.Liu LF, Wang JC. 1987. Supercoiling of the DNA template during transcription. Proc Natl Acad Sci U S A 84:7024–7027. doi: 10.1073/pnas.84.20.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schroth GP, Chou PJ, Ho PS. 1992. Mapping Z-DNA in the human genome. Computer-aided mapping reveals a nonrandom distribution of potential Z-DNA-forming sequences in human genes. J Biol Chem 267:11846–11855. [PubMed] [Google Scholar]
- 10.Gellert M, Lipsett MN, Davies DR. 1962. Helix formation by guanylic acid. Proc Natl Acad Sci U S A 48:2013–2018. doi: 10.1073/pnas.48.12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sen D, Gilbert W. 1990. A sodium-potassium switch in the formation of four-stranded G4-DNA. Nature 344:410–414. doi: 10.1038/344410a0. [DOI] [PubMed] [Google Scholar]
- 12.Sundquist WI, Klug A. 1989. Telomeric DNA dimerizes by formation of guanine tetrads between hairpin loops. Nature 342:825–829. doi: 10.1038/342825a0. [DOI] [PubMed] [Google Scholar]
- 13.Huppert JL, Balasubramanian S. 2005. Prevalence of quadruplexes in the human genome. Nucleic Acids Res 33:2908–2916. doi: 10.1093/nar/gki609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huppert JL. 2010. Structure, location and interactions of G-quadruplexes. FEBS J 277:3452–3458. doi: 10.1111/j.1742-4658.2010.07758.x. [DOI] [PubMed] [Google Scholar]
- 15.Marquevielle J, Robert C, Lagrabette O, Wahid M, Bourdoncle A, Xodo LE, Mergny J-L, Salgado GF. 2020. Structure of two G-quadruplexes in equilibrium in the KRAS promoter. Nucleic Acids Res 48:9336–9345. doi: 10.1093/nar/gkaa387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheung I, Schertzer M, Rose A, Lansdorp PM. 2002. Disruption of dog-1 in Caenorhabditis elegans triggers deletions upstream of guanine-rich DNA. Nat Genet 31:405–409. doi: 10.1038/ng928. [DOI] [PubMed] [Google Scholar]
- 17.Kruisselbrink E, Guryev V, Brouwer K, Pontier DB, Cuppen E, Tijsterman M. 2008. Mutagenic capacity of endogenous G4 DNA underlies genome instability in FANCJ-defective C. elegans. Curr Biol 18:900–905. doi: 10.1016/j.cub.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 18.Youds JL, Barber LJ, Ward JD, Collis SJ, O’Neil NJ, Boulton SJ, Rose AM. 2008. DOG-1 is the Caenorhabditis elegans BRIP1/FANCJ homologue and functions in interstrand cross-link repair. Mol Cell Biol 28:1470–1479. doi: 10.1128/MCB.01641-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taniguchi T, D’Andrea AD. 2006. Molecular pathogenesis of Fanconi anemia: recent progress. Blood 107:4223–4233. doi: 10.1182/blood-2005-10-4240. [DOI] [PubMed] [Google Scholar]
- 20.Wu Y, Shin-Ya K, Brosh RM. 2008. FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol Cell Biol 28:4116–4128. doi: 10.1128/MCB.02210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hagelberg E, Gray IC, Jeffreys AJ. 1991. Identification of the skeletal remains of a murder victim by DNA analysis. Nature 352:427–429. doi: 10.1038/352427a0. [DOI] [PubMed] [Google Scholar]
- 22.Helminen P, Ehnholm C, Lokki ML, Jeffreys A, Peltonen L. 1988. Application of DNA “fingerprints” to paternity determinations. Lancet Lond Engl 331:574–576. doi: 10.1016/S0140-6736(88)91363-3. [DOI] [PubMed] [Google Scholar]
- 23.Dib C, Fauré S, Fizames C, Samson D, Drouot N, Vignal A, Millasseau P, Marc S, Hazan J, Seboun E, Lathrop M, Gyapay G, Morissette J, Weissenbach J. 1996. A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature 380:152–154. doi: 10.1038/380152a0. [DOI] [PubMed] [Google Scholar]
- 24.Buard J, Vergnaud G. 1994. Complex recombination events at the hypermutable minisatellite CEB1 (D2S90). EMBO J 13:3203–3210. doi: 10.1002/j.1460-2075.1994.tb06619.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jeffreys AJ, Murray J, Neumann R. 1998. High-resolution mapping of crossovers in human sperm defines a minisatellite-associated recombination hotspot. Mol Cell 2:267–273. doi: 10.1016/s1097-2765(00)80138-0. [DOI] [PubMed] [Google Scholar]
- 26.Debrauwère H, Buard J, Tessier J, Aubert D, Vergnaud G, Nicolas A. 1999. Meiotic instability of human minisatellite CEB1 in yeast requires DNA double-strand breaks. Nat Genet 23:367–371. doi: 10.1038/15557. [DOI] [PubMed] [Google Scholar]
- 27.Lopes J, Debrauwère H, Buard J, Nicolas A. 2002. Instability of the human minisatellite CEB1 in rad27Delta and dna2-1 replication-deficient yeast cells. EMBO J 21:3201–3211. doi: 10.1093/emboj/cdf310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piazza A, Adrian M, Samazan F, Heddi B, Hamon F, Serero A, Lopes J, Teulade-Fichou M-P, Phan AT, Nicolas A. 2015. Short loop length and high thermal stability determine genomic instability induced by G-quadruplex-forming minisatellites. EMBO J 34:1718–1734. doi: 10.15252/embj.201490702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ribeyre C, Lopes J, Boulé J-B, Piazza A, Guédin A, Zakian VA, Mergny J-L, Nicolas A. 2009. The yeast Pif1 helicase prevents genomic instability caused by G-quadruplex-forming CEB1 sequences in vivo. PLoS Genet 5:e1000475. doi: 10.1371/journal.pgen.1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanders CM. 2010. Human Pif1 helicase is a G-quadruplex DNA-binding protein with G-quadruplex DNA-unwinding activity. Biochem J 430:119–128. doi: 10.1042/BJ20100612. [DOI] [PubMed] [Google Scholar]
- 31.Paeschke K, Capra JA, Zakian VA. 2011. DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell 145:678–691. doi: 10.1016/j.cell.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piazza A, Cui X, Adrian M, Samazan F, Heddi B, Phan A-T, Nicolas AG. 2017. Non-canonical G-quadruplexes cause the hCEB1 minisatellite instability in Saccharomyces cerevisiae. Elife 6:e26884. doi: 10.7554/eLife.26884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lombardi EP, Londoño-Vallejo A. 2020. A guide to computational methods for G-quadruplex prediction. Nucleic Acids Res 48:1603. doi: 10.1093/nar/gkaa033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schiavone D, Guilbaud G, Murat P, Papadopoulou C, Sarkies P, Prioleau M-N, Balasubramanian S, Sale JE. 2014. Determinants of G quadruplex-induced epigenetic instability in REV1-deficient cells. EMBO J 33:2507–2520. doi: 10.15252/embj.201488398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prioleau M-N, Gendron M-C, Hyrien O. 2003. Replication of the chicken beta-globin locus: early-firing origins at the 5′ HS4 insulator and the rho- and betaA-globin genes show opposite epigenetic modifications. Mol Cell Biol 23:3536–3549. doi: 10.1128/mcb.23.10.3536-3549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edmunds CE, Simpson LJ, Sale JE. 2008. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol Cell 30:519–529. doi: 10.1016/j.molcel.2008.03.024. [DOI] [PubMed] [Google Scholar]
- 37.Sarkies P, Reams C, Simpson LJ, Sale JE. 2010. Epigenetic instability due to defective replication of structured DNA. Mol Cell 40:703–713. doi: 10.1016/j.molcel.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sarkies P, Murat P, Phillips LG, Patel KJ, Balasubramanian S, Sale JE. 2012. FANCJ coordinates two pathways that maintain epigenetic stability at G-quadruplex DNA. Nucleic Acids Res 40:1485–1498. doi: 10.1093/nar/gkr868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guilbaud G, Murat P, Recolin B, Campbell BC, Maiter A, Sale JE, Balasubramanian S. 2017. Local epigenetic reprogramming induced by G-quadruplex ligands. Nat Chem 9:1110–1117. doi: 10.1038/nchem.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Cian A, Delemos E, Mergny J-L, Teulade-Fichou M-P, Monchaud D. 2007. Highly efficient G-quadruplex recognition by bisquinolinium compounds. J Am Chem Soc 129:1856–1857. doi: 10.1021/ja067352b. [DOI] [PubMed] [Google Scholar]
- 41.Piazza A, Boulé J-B, Lopes J, Mingo K, Largy E, Teulade-Fichou M-P, Nicolas A. 2010. Genetic instability triggered by G-quadruplex interacting Phen-DC compounds in Saccharomyces cerevisiae. Nucleic Acids Res 38:4337–4348. doi: 10.1093/nar/gkq136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schaffitzel C, Berger I, Postberg J, Hanes J, Lipps HJ, Plückthun A. 2001. In vitro generated antibodies specific for telomeric guanine-quadruplex DNA react with Stylonychia lemnae macronuclei. Proc Natl Acad Sci U S A 98:8572–8577. doi: 10.1073/pnas.141229498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Henderson A, Wu Y, Huang YC, Chavez EA, Platt J, Johnson FB, Brosh RM, Sen D, Lansdorp PM. 2014. Detection of G-quadruplex DNA in mammalian cells. Nucleic Acids Res 42:860–869. doi: 10.1093/nar/gkt957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kazemier HG, Paeschke K, Lansdorp PM. 2017. Guanine quadruplex monoclonal antibody 1H6 cross-reacts with restrained thymidine-rich single stranded DNA. Nucleic Acids Res 45:5913–5919. doi: 10.1093/nar/gkx245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Biffi G, Tannahill D, McCafferty J, Balasubramanian S. 2013. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat Chem 5:182–186. doi: 10.1038/nchem.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hänsel-Hertsch R, Simeone A, Shea A, Hui WWI, Zyner KG, Marsico G, Rueda OM, Bruna A, Martin A, Zhang X, Adhikari S, Tannahill D, Caldas C, Balasubramanian S. 2020. Landscape of G-quadruplex DNA structural regions in breast cancer. Nat Genet 52:878–883. doi: 10.1038/s41588-020-0672-8. [DOI] [PubMed] [Google Scholar]
- 47.Di Antonio M, Ponjavic A, Radzevičius A, Ranasinghe RT, Catalano M, Zhang X, Shen J, Needham L-M, Lee SF, Klenerman D, Balasubramanian S. 2020. Single-molecule visualization of DNA G-quadruplex formation in live cells. Nat Chem 12:832–837. doi: 10.1038/s41557-020-0506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sutherland GR, Baker E, Richards RI. 1998. Fragile sites still breaking. Trends Genet 14:501–506. doi: 10.1016/s0168-9525(98)01628-x. [DOI] [PubMed] [Google Scholar]
- 49.Rassool FV, Le Beau MM, Shen ML, Neilly ME, Espinosa R, Ong ST, Boldog F, Drabkin H, McCarroll R, McKeithan TW. 1996. Direct cloning of DNA sequences from the common fragile site region at chromosome band 3p14.2. Genomics 35:109–117. doi: 10.1006/geno.1996.0329. [DOI] [PubMed] [Google Scholar]
- 50.Ried K, Finnis M, Hobson L, Mangelsdorf M, Dayan S, Nancarrow JK, Woollatt E, Kremmidiotis G, Gardner A, Venter D, Baker E, Richards RI. 2000. Common chromosomal fragile site FRA16D sequence: identification of the FOR gene spanning FRA16D and homozygous deletions and translocation breakpoints in cancer cells. Hum Mol Genet 9:1651–1663. doi: 10.1093/hmg/9.11.1651. [DOI] [PubMed] [Google Scholar]
- 51.Inoue H, Ishii H, Alder H, Snyder E, Druck T, Huebner K, Croce CM. 1997. Sequence of the FRA3B common fragile region: implications for the mechanism of FHIT deletion. Proc Natl Acad Sci U S A 94:14584–14589. doi: 10.1073/pnas.94.26.14584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alsop AE, Taylor K, Zhang J, Gabra H, Paige AJW, Edwards PAW. 2008. Homozygous deletions may be markers of nearby heterozygous mutations: the complex deletion at FRA16D in the HCT116 colon cancer cell line removes exons of WWOX. Genes Chromosomes Cancer 47:437–447. doi: 10.1002/gcc.20548. [DOI] [PubMed] [Google Scholar]
- 53.Huebner K, Croce CM. 2001. FRA3B and other common fragile sites: the weakest links. Nat Rev Cancer 1:214–221. doi: 10.1038/35106058. [DOI] [PubMed] [Google Scholar]
- 54.Kremer EJ, Pritchard M, Lynch M, Yu S, Holman K, Baker E, Warren ST, Schlessinger D, Sutherland GR, Richards RI. 1991. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science 252:1711–1714. doi: 10.1126/science.1675488. [DOI] [PubMed] [Google Scholar]
- 55.Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, Eussen BE, van Ommen G-JB, Blonden LAJ, Riggins GJ, Chastain JL, Kunst CB, Galjaard H, Caskey CT, Nelson DL, Oostra BA, Warren ST. 1991. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 56.Flynn GA, Hirst MC, Knight SJ, Macpherson JN, Barber JC, Flannery AV, Davies KE, Buckle VJ. 1993. Identification of the FRAXE fragile site in two families ascertained for X linked mental retardation. J Med Genet 30:97–100. doi: 10.1136/jmg.30.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jones C, Slijepcevic P, Marsh S, Baker E, Langdon WY, Richards RI, Tunnacliffe A. 1994. Physical linkage of the fragile site FRA11B and a Jacobsen syndrome chromosome deletion breakpoint in 11q23.3. Hum Mol Genet 3:2123–2130. doi: 10.1093/hmg/3.12.2123. [DOI] [PubMed] [Google Scholar]
- 58.Yu S, Mangelsdorf M, Hewett D, Hobson L, Baker E, Eyre HJ, Lapsys N, Paslier DL, Doggett NA, Sutherland GR, Richards RI. 1997. Human chromosomal fragile site FRA16B is an amplified AT-rich minisatellite repeat. Cell 88:367–374. doi: 10.1016/S0092-8674(00)81875-9. [DOI] [PubMed] [Google Scholar]
- 59.Hewett DR, Handt O, Hobson L, Mangelsdorf M, Eyre HJ, Baker E, Sutherland GR, Schuffenhauer S, Mao JI, Richards RI. 1998. FRA10B structure reveals common elements in repeat expansion and chromosomal fragile site genesis. Mol Cell 1:773–781. doi: 10.1016/s1097-2765(00)80077-5. [DOI] [PubMed] [Google Scholar]
- 60.Le Beau MM, Rassool FV, Neilly ME, Espinosa R, Glover TW, Smith DI, McKeithan TW. 1998. Replication of a common fragile site, FRA3B, occurs late in S phase and is delayed further upon induction: implications for the mechanism of fragile site induction. Hum Mol Genet 7:755–761. doi: 10.1093/hmg/7.4.755. [DOI] [PubMed] [Google Scholar]
- 61.Letessier A, Millot GA, Koundrioukoff S, Lachagès A-M, Vogt N, Hansen RS, Malfoy B, Brison O, Debatisse M. 2011. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 470:120–123. doi: 10.1038/nature09745. [DOI] [PubMed] [Google Scholar]
- 62.Burrow AA, Marullo A, Holder LR, Wang Y-H. 2010. Secondary structure formation and DNA instability at fragile site FRA16B. Nucleic Acids Res 38:2865–2877. doi: 10.1093/nar/gkp1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mishmar D, Rahat A, Scherer SW, Nyakatura G, Hinzmann B, Kohwi Y, Mandel-Gutfroind Y, Lee JR, Drescher B, Sas DE, Margalit H, Platzer M, Weiss A, Tsui LC, Rosenthal A, Kerem B. 1998. Molecular characterization of a common fragile site (FRA7H) on human chromosome 7 by the cloning of a simian virus 40 integration site. Proc Natl Acad Sci U S A 95:8141–8146. doi: 10.1073/pnas.95.14.8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zlotorynski E, Rahat A, Skaug J, Ben-Porat N, Ozeri E, Hershberg R, Levi A, Scherer SW, Margalit H, Kerem B. 2003. Molecular basis for expression of common and rare fragile sites. Mol Cell Biol 23:7143–7151. doi: 10.1128/mcb.23.20.7143-7151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Debatisse M, Le Tallec B, Letessier A, Dutrillaux B, Brison O. 2012. Common fragile sites: mechanisms of instability revisited. Trends Genet 28:22–32. doi: 10.1016/j.tig.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 66.Hansen RS, Canfield TK, Fjeld AD, Mumm S, Laird CD, Gartler SM. 1997. A variable domain of delayed replication in FRAXA fragile X chromosomes: X inactivation-like spread of late replication. Proc Natl Acad Sci U S A 94:4587–4592. doi: 10.1073/pnas.94.9.4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Subramanian PS, Nelson DL, Chinault AC. 1996. Large domains of apparent delayed replication timing associated with triplet repeat expansion at FRAXA and FRAXE. Am J Hum Genet 59:407–416. [PMC free article] [PubMed] [Google Scholar]
- 68.Usdin K, Woodford KJ. 1995. CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro. Nucleic Acids Res 23:4202–4209. doi: 10.1093/nar/23.20.4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fry M, Loeb LA. 1994. The fragile X syndrome d(CGG)n nucleotide repeats form a stable tetrahelical structure. Proc Natl Acad Sci U S A 91:4950–4954. doi: 10.1073/pnas.91.11.4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gacy AM, Goellner G, Juranić N, Macura S, McMurray CT. 1995. Trinucleotide repeats that expand in human disease form hairpin structures in vitro. Cell 81:533–540. doi: 10.1016/0092-8674(95)90074-8. [DOI] [PubMed] [Google Scholar]
- 71.Zumwalt M, Ludwig A, Hagerman PJ, Dieckmann T. 2007. Secondary structure and dynamics of the r(CGG) repeat in the mRNA of the fragile X mental retardation 1 (FMR1) gene. RNA Biol 4:93–100. doi: 10.4161/rna.4.2.5039. [DOI] [PubMed] [Google Scholar]
- 72.Samadashwily GM, Raca G, Mirkin SM. 1997. Trinucleotide repeats affect DNA replication in vivo. Nat Genet 17:298–304. doi: 10.1038/ng1197-298. [DOI] [PubMed] [Google Scholar]
- 73.Pelletier R, Krasilnikova MM, Samadashwily GM, Lahue R, Mirkin SM. 2003. Replication and expansion of trinucleotide repeats in yeast. Mol Cell Biol 23:1349–1357. doi: 10.1128/mcb.23.4.1349-1357.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Handt O, Sutherland GR, Richards RI. 2000. Fragile sites and minisatellite repeat instability. Mol Genet Metab 70:99–105. doi: 10.1006/mgme.2000.2996. [DOI] [PubMed] [Google Scholar]
- 75.Zhang H, Freudenreich CH. 2007. An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol Cell 27:367–379. doi: 10.1016/j.molcel.2007.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Szlachta K, Manukyan A, Raimer HM, Singh S, Salamon A, Guo W, Lobachev KS, Wang Y-H. 2020. Topoisomerase II contributes to DNA secondary structure-mediated double-stranded breaks. Nucleic Acids Res 48:6654–6671. doi: 10.1093/nar/gkaa483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pearson CE, Zorbas H, Price GB, Zannis-Hadjopoulos M. 1996. Inverted repeats, stem-loops, and cruciforms: significance for initiation of DNA replication. J Cell Biochem 63:1–22. doi:. [DOI] [PubMed] [Google Scholar]
- 78.Welsh J, McClelland M. 1990. Fingerprinting genomes using PCR with arbitrary primers. Nucleic Acids Res 18:7213–7218. doi: 10.1093/nar/18.24.7213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McClellan JA, Boublíková P, Palecek E, Lilley DM. 1990. Superhelical torsion in cellular DNA responds directly to environmental and genetic factors. Proc Natl Acad Sci U S A 87:8373–8377. doi: 10.1073/pnas.87.21.8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dayn A, Malkhosyan S, Duzhy D, Lyamichev V, Panchenko Y, Mirkin S. 1991. Formation of (dA-dT)n cruciforms in Escherichia coli cells under different environmental conditions. J Bacteriol 173:2658–2664. doi: 10.1128/jb.173.8.2658-2664.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dayn A, Malkhosyan S, Mirkin SM. 1992. Transcriptionally driven cruciform formation in vivo. Nucleic Acids Res 20:5991–5997. doi: 10.1093/nar/20.22.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Collins J. 1981. Instability of palindromic DNA in Escherichia coli. Cold Spring Harbor Symp Quant Biol 45 Pt 1:409–416. doi: 10.1101/sqb.1981.045.01.055. [DOI] [PubMed] [Google Scholar]
- 83.Hagan CE, Warren GJ. 1982. Lethality of palindromic DNA and its use in selection of recombinant plasmids. Gene 19:147–151. doi: 10.1016/0378-1119(82)90199-8. [DOI] [PubMed] [Google Scholar]
- 84.Chalker AF, Leach DR, Lloyd RG. 1988. Escherichia coli sbcC mutants permit stable propagation of DNA replicons containing a long palindrome. Gene 71:201–205. doi: 10.1016/0378-1119(88)90092-3. [DOI] [PubMed] [Google Scholar]
- 85.Gibson FP, Leach DR, Lloyd RG. 1992. Identification of sbcD mutations as cosuppressors of recBC that allow propagation of DNA palindromes in Escherichia coli K-12. J Bacteriol 174:1222–1228. doi: 10.1128/jb.174.4.1222-1228.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Davison A, Leach DRF. 1994. The effects of nucleotide sequence changes on DNA secondary structure formation in Escherichia coli are consistent with cruciform extrusion in vivo. Genetics 137:361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lindsey JC, Leach DR. 1989. Slow replication of palindrome-containing DNA. J Mol Biol 206:779–782. doi: 10.1016/0022-2836(89)90584-6. [DOI] [PubMed] [Google Scholar]
- 88.Connelly JC, Kirkham LA, Leach DRF. 1998. The SbcCD nuclease of Escherichia coli is a structural maintenance of chromosomes (SMC) family protein that cleaves hairpin DNA. Proc Natl Acad Sci U S A 95:7969–7974. doi: 10.1073/pnas.95.14.7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Connelly JC, de Leau ES, Leach DRF. 1999. DNA cleavage and degradation by the SbcCD protein complex from Escherichia coli. Nucleic Acids Res 27:1039–1046. doi: 10.1093/nar/27.4.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Leach DR, Okely EA, Pinder DJ. 1997. Repair by recombination of DNA containing a palindromic sequence. Mol Microbiol 26:597–606. doi: 10.1046/j.1365-2958.1997.6071957.x. [DOI] [PubMed] [Google Scholar]
- 91.Eykelenboom JK, Blackwood JK, Okely E, Leach DRF. 2008. SbcCD causes a double-strand break at a DNA palindrome in the Escherichia coli chromosome. Mol Cell 29:644–651. doi: 10.1016/j.molcel.2007.12.020. [DOI] [PubMed] [Google Scholar]
- 92.Voineagu I, Narayanan V, Lobachev KS, Mirkin SM. 2008. Replication stalling at unstable inverted repeats: interplay between DNA hairpins and fork stabilizing proteins. Proc Natl Acad Sci U S A 105:9936–9941. doi: 10.1073/pnas.0804510105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lobachev KS, Stenger JE, Kozyreva OG, Jurka J, Gordenin DA, Resnick MA. 2000. Inverted Alu repeats unstable in yeast are excluded from the human genome. EMBO J 19:3822–3830. doi: 10.1093/emboj/19.14.3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sharples GJ, Leach DR. 1995. Structural and functional similarities between the SbcCD proteins of Escherichia coli and the RAD50 and MRE11 (RAD32) recombination and repair proteins of yeast. Mol Microbiol 17:1215–1217. doi: 10.1111/j.1365-2958.1995.mmi_17061215_1.x. [DOI] [PubMed] [Google Scholar]
- 95.Trujillo KM, Sung P. 2001. DNA structure-specific nuclease activities in the Saccharomyces cerevisiae Rad50*Mre11 complex. J Biol Chem 276:35458–35464. doi: 10.1074/jbc.M105482200. [DOI] [PubMed] [Google Scholar]
- 96.Lobachev KS, Gordenin DA, Resnick MA. 2002. The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell 108:183–193. doi: 10.1016/s0092-8674(02)00614-1. [DOI] [PubMed] [Google Scholar]
- 97.Farah JA, Hartsuiker E, Mizuno K-I, Ohta K, Smith GR. 2002. A 160-bp palindrome is a Rad50.Rad32-dependent mitotic recombination hotspot in Schizosaccharomyces pombe. Genetics 161:461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Farah JA, Cromie G, Steiner WW, Smith GR. 2005. A novel recombination pathway initiated by the Mre11/Rad50/Nbs1 complex eliminates palindromes during meiosis in Schizosaccharomyces pombe. Genetics 169:1261–1274. doi: 10.1534/genetics.104.037515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nag DK, Kurst A. 1997. A 140-bp-long palindromic sequence induces double-strand breaks during meiosis in the yeast Saccharomyces cerevisiae. Genetics 146:835–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nasar F, Jankowski C, Nag DK. 2000. Long palindromic sequences induce double-strand breaks during meiosis in yeast. Mol Cell Biol 20:3449–3458. doi: 10.1128/mcb.20.10.3449-3458.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang Y, Saini N, Sheng Z, Lobachev KS. 2013. Genome-wide screen reveals replication pathway for quasi-palindrome fragility dependent on homologous recombination. PLoS Genet 9:e1003979. doi: 10.1371/journal.pgen.1003979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lemoine FJ, Degtyareva NP, Lobachev K, Petes TD. 2005. Chromosomal translocations in yeast induced by low levels of DNA polymerase a model for chromosome fragile sites. Cell 120:587–598. doi: 10.1016/j.cell.2004.12.039. [DOI] [PubMed] [Google Scholar]
- 103.Carter MT, St. Pierre SA, Zackai EH, Emanuel BS, Boycott KM. 2009. Phenotypic delineation of Emanuel syndrome (supernumerary derivative 22 syndrome): clinical features of 63 individuals. Am J Med Genet A 149A:1712–1721. doi: 10.1002/ajmg.a.32957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kurahashi H, Shaikh TH, Zackai EH, Celle L, Driscoll DA, Budarf ML, Emanuel BS. 2000. Tightly clustered 11q23 and 22q11 breakpoints permit PCR-based detection of the recurrent constitutional t(11;22). Am J Hum Genet 67:763–768. doi: 10.1086/303054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kurahashi H, Inagaki H, Ohye T, Kogo H, Tsutsumi M, Kato T, Tong M, Emanuel B. 2010. The constitutional t(11;22): implications for a novel mechanism responsible for gross chromosomal rearrangements. Clin Genet 78:299–309. doi: 10.1111/j.1399-0004.2010.01445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Inagaki H, Ohye T, Kogo H, Tsutsumi M, Kato T, Tong M, Emanuel BS, Kurahashi H. 2013. Two sequential cleavage reactions on cruciform DNA structures cause palindrome-mediated chromosomal translocations. Nat Commun 4:1–10. doi: 10.1038/ncomms2595. [DOI] [PubMed] [Google Scholar]
- 107.Ohye T, Inagaki H, Kogo H, Tsutsumi M, Kato T, Tong M, Macville MVE, Medne L, Zackai EH, Emanuel BS, Kurahashi H. 2010. Paternal origin of the de novo constitutional t(11;22)(q23;q11). Eur J Hum Genet 18:783–787. doi: 10.1038/ejhg.2010.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Inagaki H, Ohye T, Kogo H, Kato T, Bolor H, Taniguchi M, Shaikh TH, Emanuel BS, Kurahashi H. 2009. Chromosomal instability mediated by non-B DNA: cruciform conformation and not DNA sequence is responsible for recurrent translocation in humans. Genome Res 19:191–198. doi: 10.1101/gr.079244.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Marotta M, Onodera T, Johnson J, Budd GT, Watanabe T, Cui X, Giuliano AE, Niida A, Tanaka H. 2017. Palindromic amplification of the ERBB2 oncogene in primary HER2-positive breast tumors. Sci Rep 7:41921–41922. doi: 10.1038/srep41921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sinden RR, Broyles SS, Pettijohn DE. 1983. Perfect palindromic lac operator DNA sequence exists as a stable cruciform structure in supercoiled DNA in vitro but not in vivo. Proc Natl Acad Sci U S A 80:1797–1801. doi: 10.1073/pnas.80.7.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zheng GX, Kochel T, Hoepfner RW, Timmons SE, Sinden RR. 1991. Torsionally tuned cruciform and Z-DNA probes for measuring unrestrained supercoiling at specific sites in DNA of living cells. J Mol Biol 221:107–122. doi: 10.1016/0022-2836(91)80208-C. [DOI] [PubMed] [Google Scholar]
- 112.Frappier L, Price GB, Martin RG, Zannis-Hadjopoulos M. 1987. Monoclonal antibodies to cruciform DNA structures. J Mol Biol 193:751–758. doi: 10.1016/0022-2836(87)90356-1. [DOI] [PubMed] [Google Scholar]
- 113.Steinmetzer K, Zannis-Hadjopoulos M, Price GB. 1995. Anti-cruciform monoclonal antibody and cruciform DNA interaction. J Mol Biol 254:29–37. doi: 10.1006/jmbi.1995.0596. [DOI] [PubMed] [Google Scholar]
- 114.Callejo M, Alvarez D, Price GB, Zannis-Hadjopoulos M. 2002. The 14-3-3 protein homologues from Saccharomyces cerevisiae, Bmh1p and Bmh2p, have cruciform DNA-binding activity and associate in vivo with ARS307. J Biol Chem 277:38416–38423. doi: 10.1074/jbc.M202050200. [DOI] [PubMed] [Google Scholar]
- 115.Tam M, Erin Montgomery S, Kekis M, Stollar BD, Price GB, Pearson CE. 2003. Slipped (CTG)·(CAG) repeats of the myotonic dystrophy locus: surface probing with anti-DNA antibodies. J Mol Biol 332:585–600. doi: 10.1016/S0022-2836(03)00880-5. [DOI] [PubMed] [Google Scholar]
- 116.Mitas M, Yu A, Dill J, Kamp TJ, Chambers EJ, Haworth IS. 1995. Hairpin properties of single-stranded DNA containing a GC-rich triplet repeat: (CTG)15. Nucleic Acids Res 23:1050–1059. doi: 10.1093/nar/23.6.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yu A, Dill J, Mitas M. 1995. The purine-rich trinucleotide repeat sequences d(CAG)15 and d(GAC)15 form hairpins. Nucleic Acids Res 23:4055–4057. doi: 10.1093/nar/23.20.4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Weller P, Jeffreys A, Wilson V, Blanchetot A. 1984. Organization of the human myoglobin gene. EMBO J 3:439–446. doi: 10.1002/j.1460-2075.1984.tb01825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Richard G-F, Kerrest A, Dujon B. 2008. Comparative genomics and molecular dynamics of DNA repeats in eukaryotes. Microbiol Mol Biol Rev 72:686–727. doi: 10.1128/MMBR.00011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, Verkerk AJ, Holden JJ, Fenwick RG, Warren ST, Oostra BA, Nelson DL, Caskey CT. 1991. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 67:1047–1058. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- 121.Rowland LP, Shneider NA. 2001. Amyotrophic lateral sclerosis. N Engl J Med 344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- 122.Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LPW. 2001. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 123.Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, Khajavi M, McCall AE, Davis CF, Zu L, Achari M, Pulst SM, Alonso E, Noebels JL, Nelson DL, Zoghbi HY, Ashizawa T. 2000. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet 26:191–194. doi: 10.1038/79911. [DOI] [PubMed] [Google Scholar]
- 124.McMurray CT. 1999. DNA secondary structure: a common and causative factor for expansion in human disease. Proc Natl Acad Sci U S A 96:1823–1825. doi: 10.1073/pnas.96.5.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Petruska J, Arnheim N, Goodman MF. 1996. Stability of intrastrand hairpin structures formed by the CAG/CTG class of DNA triplet repeats associated with neurological diseases. Nucleic Acids Res 24:1992–1998. doi: 10.1093/nar/24.11.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pearson CE, Wang YH, Griffith JD, Sinden RR. 1998. Structural analysis of slipped-strand DNA (S-DNA) formed in (CTG)n. (CAG)n repeats from the myotonic dystrophy locus. Nucleic Acids Res 26:816–823. doi: 10.1093/nar/26.3.816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xu P, Pan F, Roland C, Sagui C, Weninger K. 2020. Dynamics of strand slippage in DNA hairpins formed by CAG repeats: roles of sequence parity and trinucleotide interrupts. Nucleic Acids Res 48:2232–2245. doi: 10.1093/nar/gkaa036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liu G, Chen X, Bissler JJ, Sinden RR, Leffak M. 2010. Replication-dependent instability at (CTG)•(CAG) repeat hairpins in human cells. Nat Chem Biol 6:652–659. doi: 10.1038/nchembio.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Axford MM, Wang Y-H, Nakamori M, Zannis-Hadjopoulos M, Thornton CA, Pearson CE. 2013. Detection of slipped-DNAs at the trinucleotide repeats of the myotonic dystrophy type I disease locus in patient tissues. PLoS Genet 9:e1003866. doi: 10.1371/journal.pgen.1003866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nakamori M, Panigrahi GB, Lanni S, Gall-Duncan T, Hayakawa H, Tanaka H, Luo J, Otabe T, Li J, Sakata A, Caron M-C, Joshi N, Prasolava T, Chiang K, Masson J-Y, Wold MS, Wang X, Lee MYWT, Huddleston J, Munson KM, Davidson S, Layeghifard M, Edward L-M, Gallon R, Santibanez-Koref M, Murata A, Takahashi MP, Eichler EE, Shlien A, Nakatani K, Mochizuki H, Pearson CE. 2020. A slipped-CAG DNA-binding small molecule induces trinucleotide-repeat contractions in vivo. Nat Genet 52:146–159. doi: 10.1038/s41588-019-0575-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nakamori M, Sobczak K, Puwanant A, Welle S, Eichinger K, Pandya S, Dekdebrun J, Heatwole CR, McDermott MP, Chen T, Cline M, Tawil R, Osborne RJ, Wheeler TM, Swanson MS, Moxley RT, Thornton CA. 2013. Splicing biomarkers of disease severity in myotonic dystrophy. Ann Neurol 74:862–872. doi: 10.1002/ana.23992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Felsenfeld G, Rich A. 1957. Studies on the formation of two- and three-stranded polyribonucleotides. Biochim Biophys Acta 26:457–468. doi: 10.1016/0006-3002(57)90091-4. [DOI] [PubMed] [Google Scholar]
- 133.Malkov VA, Voloshin ON, Veselkov AG, Rostapshov VM, Jansen I, Soyfer VN, Frank-Kamenetskii MD. 1993. Protonated pyrimidine-purine-purine triplex. Nucleic Acids Res 21:105–111. doi: 10.1093/nar/21.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mirkin SM, Lyamichev VI, Drushlyak KN, Dobrynin VN, Filippov SA, Frank-Kamenetskii MD. 1987. DNA H form requires a homopurine-homopyrimidine mirror repeat. Nature 330:495–497. doi: 10.1038/330495a0. [DOI] [PubMed] [Google Scholar]
- 135.Gacy AM, Goellner GM, Spiro C, Chen X, Gupta G, Bradbury EM, Dyer RB, Mikesell MJ, Yao JZ, Johnson AJ, Richter A, Melancon SB, McMurray CT. 1998. GAA instability in Friedreich’s ataxia shares a common, DNA-directed and intraallelic mechanism with other trinucleotide diseases. Mol Cell 1:583–593. doi: 10.1016/S1097-2765(00)80058-1. [DOI] [PubMed] [Google Scholar]
- 136.Lee JS, Burkholder GD, Latimer LJ, Haug BL, Braun RP. 1987. A monoclonal antibody to triplex DNA binds to eucaryotic chromosomes. Nucleic Acids Res 15:1047–1061. doi: 10.1093/nar/15.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ohno M, Fukagawa T, Lee JS, Ikemura T. 2002. Triplex-forming DNAs in the human interphase nucleus visualized in situ by polypurine/polypyrimidine DNA probes and antitriplex antibodies. Chromosoma 111:201–213. doi: 10.1007/s00412-002-0198-0. [DOI] [PubMed] [Google Scholar]
- 138.Shishkin AA, Voineagu I, Matera R, Cherng N, Chernet BT, Krasilnikova MM, Narayanan V, Lobachev KS, Mirkin SM. 2009. Large-scale expansions of Friedreich’s ataxia GAA REPEATS in yeast. Mol Cell 35:82–92. doi: 10.1016/j.molcel.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Khristich AN, Armenia JF, Matera RM, Kolchinski AA, Mirkin SM. 2020. Large-scale contractions of Friedreich’s ataxia GAA repeats in yeast occur during DNA replication due to their triplex-forming ability. Proc Natl Acad Sci U S A 117:1628–1637. doi: 10.1073/pnas.1913416117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhang Y, Shishkin AA, Nishida Y, Marcinkowski-Desmond D, Saini N, Volkov KV, Mirkin SM, Lobachev KS. 2012. Genome-wide screen identifies pathways that govern GAA/TTC repeat fragility and expansions in dividing and nondividing yeast cells. Mol Cell 48:254–265. doi: 10.1016/j.molcel.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Harrington JJ, Lieber MR. 1994. The characterization of a mammalian DNA structure-specific endonuclease. EMBO J 13:1235–1246. doi: 10.1002/j.1460-2075.1994.tb06373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tsutakawa SE, Thompson MJ, Arvai AS, Neil AJ, Shaw SJ, Algasaier SI, Kim JC, Finger LD, Jardine E, Gotham VJB, Sarker AH, Her MZ, Rashid F, Hamdan SM, Mirkin SM, Grasby JA, Tainer JA. 2017. Phosphate steering by Flap Endonuclease 1 promotes 5′-flap specificity and incision to prevent genome instability. 1. Nat Commun 8:15855. doi: 10.1038/ncomms15855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kim H-M, Narayanan V, Mieczkowski PA, Petes TD, Krasilnikova MM, Mirkin SM, Lobachev KS. 2008. Chromosome fragility at GAA tracts in yeast depends on repeat orientation and requires mismatch repair. EMBO J 27:2896–2906. doi: 10.1038/emboj.2008.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kumari D, Hayward B, Nakamura AJ, Bonner WM, Usdin K. 2015. Evidence for chromosome fragility at the frataxin locus in Friedreich ataxia. Mutat Res 781:14–21. doi: 10.1016/j.mrfmmm.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kinniburgh AJ. 1989. A cis-acting transcription element of the c-myc gene can assume an H-DNA conformation. Nucleic Acids Res 17:7771–7778. doi: 10.1093/nar/17.19.7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Raghavan SC, Chastain P, Lee JS, Hegde BG, Houston S, Langen R, Hsieh C-L, Haworth IS, Lieber MR. 2005. Evidence for a triplex DNA conformation at the bcl-2 major breakpoint region of the t(14;18) translocation. J Biol Chem 280:22749–22760. doi: 10.1074/jbc.M502952200. [DOI] [PubMed] [Google Scholar]
- 147.Neil AJ, Liang MU, Khristich AN, Shah KA, Mirkin SM. 2018. RNA-DNA hybrids promote the expansion of Friedreich’s ataxia (GAA)n repeats via break-induced replication. Nucleic Acids Res 46:3487–3497. doi: 10.1093/nar/gky099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Dere R, Napierala M, Ranum LPW, Wells RD. 2004. Hairpin structure-forming propensity of the (CCTG.CAGG) tetranucleotide repeats contributes to the genetic instability associated with myotonic dystrophy type 2. J Biol Chem 279:41715–41726. doi: 10.1074/jbc.M406415200. [DOI] [PubMed] [Google Scholar]
- 149.Lam SL, Wu F, Yang H, Chi LM. 2011. The origin of genetic instability in CCTG repeats. Nucleic Acids Res 39:6260–6268. doi: 10.1093/nar/gkr185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Potaman VN, Bissler JJ, Hashem VI, Oussatcheva EA, Lu L, Shlyakhtenko LS, Lyubchenko YL, Matsuura T, Ashizawa T, Leffak M, Benham CJ, Sinden RR. 2003. Unpaired structures in SCA10 (ATTCT)n·(AGAAT)n repeats. J Mol Biol 326:1095–1111. doi: 10.1016/s0022-2836(03)00037-8. [DOI] [PubMed] [Google Scholar]
- 151.Kowalski D, Eddy MJ. 1989. The DNA unwinding element: a novel, cis-acting component that facilitates opening of the Escherichia coli replication origin. EMBO J 8:4335–4344. doi: 10.1002/j.1460-2075.1989.tb08620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Umek RM, Kowalski D. 1990. The DNA unwinding element in a yeast replication origin functions independently of easily unwound sequences present elsewhere on a plasmid. Nucleic Acids Res 18:6601–6605. doi: 10.1093/nar/18.22.6601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Guo P, Lam SL. 2020. Minidumbbell structures formed by ATTCT pentanucleotide repeats in spinocerebellar ataxia type 10. Nucleic Acids Res 48:7557–7568. doi: 10.1093/nar/gkaa495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Cherng N, Shishkin AA, Schlager LI, Tuck RH, Sloan L, Matera R, Sarkar PS, Ashizawa T, Freudenreich CH, Mirkin SM. 2011. Expansions, contractions, and fragility of the spinocerebellar ataxia type 10 pentanucleotide repeat in yeast. Proc Natl Acad Sci U S A 108:2843–2848. doi: 10.1073/pnas.1009409108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Parsons R, Li GM, Longley MJ, Fang WH, Papadopoulos N, Jen J, de la Chapelle A, Kinzler KW, Vogelstein B, Modrich P. 1993. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell 75:1227–1236. doi: 10.1016/0092-8674(93)90331-j. [DOI] [PubMed] [Google Scholar]
- 156.Strand M, Prolla TA, Liskay RM, Petes TD. 1993. Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair. Nature 365:274–276. doi: 10.1038/365274a0. [DOI] [PubMed] [Google Scholar]
- 157.Tran HT, Keen JD, Kricker M, Resnick MA, Gordenin DA. 1997. Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol Cell Biol 17:2859–2865. doi: 10.1128/mcb.17.5.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lahue RS, Au KG, Modrich P. 1989. DNA mismatch correction in a defined system. Science 245:160–164. doi: 10.1126/science.2665076. [DOI] [PubMed] [Google Scholar]
- 159.Bowen N, Smith CE, Srivatsan A, Willcox S, Griffith JD, Kolodner RD. 2013. Reconstitution of long and short patch mismatch repair reactions using Saccharomyces cerevisiae proteins. Proc Natl Acad Sci U S A 110:18472–18477. doi: 10.1073/pnas.1318971110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Constantin N, Dzantiev L, Kadyrov FA, Modrich P. 2005. Human mismatch repair: reconstitution of a nick-directed bidirectional reaction. J Biol Chem 280:39752–39761. doi: 10.1074/jbc.M509701200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Jiricny J. 2006. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol 7:335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 162.Warren JJ, Pohlhaus TJ, Changela A, Iyer RR, Modrich PL, Beese LS. 2007. Structure of the human MutSα DNA lesion recognition complex. Mol Cell 26:579–592. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 163.Gupta S, Gellert M, Yang W. 2011. Mechanism of mismatch recognition revealed by human MutSβ bound to unpaired DNA loops. Nat Struct Mol Biol 19:72–78. doi: 10.1038/nsmb.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pearson CE, Ewel A, Acharya S, Fishel RA, Sinden RR. 1997. Human MSH2 binds to trinucleotide repeat DNA structures associated with neurodegenerative diseases. Hum Mol Genet 6:1117–1123. doi: 10.1093/hmg/6.7.1117. [DOI] [PubMed] [Google Scholar]
- 165.Owen BAL, Yang Z, Lai M, Gajec M, Gajek M, Badger JD, Hayes JJ, Edelmann W, Kucherlapati R, Wilson TM, McMurray CT. 2005. (CAG)(n)-hairpin DNA binds to Msh2-Msh3 and changes properties of mismatch recognition. Nat Struct Mol Biol 12:663–670. doi: 10.1038/nsmb965. [DOI] [PubMed] [Google Scholar]
- 166.Tian L, Hou C, Tian K, Holcomb NC, Gu L, Li G-M. 2009. Mismatch recognition protein MutSbeta does not hijack (CAG)n hairpin repair in vitro. J Biol Chem 284:20452–20456. doi: 10.1074/jbc.C109.014977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Manley K, Shirley TL, Flaherty L, Messer A. 1999. Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat Genet 23:471–473. doi: 10.1038/70598. [DOI] [PubMed] [Google Scholar]
- 168.Kovtun IV, McMurray CT. 2001. Trinucleotide expansion in haploid germ cells by gap repair. Nat Genet 27:407–411. doi: 10.1038/86906. [DOI] [PubMed] [Google Scholar]
- 169.Foiry L, Dong L, Savouret C, Hubert L, Te Riele H, Junien C, Gourdon G. 2006. Msh3 is a limiting factor in the formation of intergenerational CTG expansions in DM1 transgenic mice. Hum Genet 119:520–526. doi: 10.1007/s00439-006-0164-7. [DOI] [PubMed] [Google Scholar]
- 170.Savouret C, Brisson E, Essers J, Kanaar R, Pastink A, Te Riele H, Junien C, Gourdon G. 2003. CTG repeat instability and size variation timing in DNA repair-deficient mice. EMBO J 22:2264–2273. doi: 10.1093/emboj/cdg202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Tomé S, Holt I, Edelmann W, Morris GE, Munnich A, Pearson CE, Gourdon G. 2009. MSH2 ATPase domain mutation affects CTG•CAG repeat instability in transgenic mice. PLoS Genet 5:e1000482. doi: 10.1371/journal.pgen.1000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lokanga RA, Zhao X-N, Usdin K. 2014. The mismatch repair protein MSH2 is rate limiting for repeat expansion in a fragile X premutation mouse model. Hum Mutat 35:129–136. doi: 10.1002/humu.22464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Halabi A, Fuselier KTB, Grabczyk E. 2018. GAA•TTC repeat expansion in human cells is mediated by mismatch repair complex MutLγ and depends upon the endonuclease domain in MLH3 isoform one. Nucleic Acids Res 46:4022–4032. doi: 10.1093/nar/gky143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ezzatizadeh V, Pinto RM, Sandi C, Sandi M, Al-Mahdawi S, Te Riele H, Pook MA. 2012. The mismatch repair system protects against intergenerational GAA repeat instability in a Friedreich ataxia mouse model. Neurobiol Dis 46:165–171. doi: 10.1016/j.nbd.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Blackwood JK, Okely EA, Zahra R, Eykelenboom JK, Leach DRF. 2010. DNA tandem repeat instability in the Escherichia coli chromosome is stimulated by mismatch repair at an adjacent CAG·CTG trinucleotide repeat. Proc Natl Acad Sci U S A 107:22582–22586. doi: 10.1073/pnas.1012906108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Williams GM, Surtees JA. 2015. MSH3 promotes dynamic behavior of trinucleotide repeat tracts in vivo. Genetics 200:737–754. doi: 10.1534/genetics.115.177303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Slean MM, Panigrahi GB, Castel AL, Pearson AB, Tomkinson AE, Pearson CE. 2016. Absence of MutSβ leads to the formation of slipped-DNA for CTG/CAG contractions at primate replication forks. DNA Repair (Amst) 42:107–118. doi: 10.1016/j.dnarep.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Viterbo D, Michoud G, Mosbach V, Dujon B, Richard G-F. 2016. Replication stalling and heteroduplex formation within CAG/CTG trinucleotide repeats by mismatch repair. DNA Repair (Amst) 42:94–106. doi: 10.1016/j.dnarep.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 179.Kerrest A, Anand RP, Sundararajan R, Bermejo R, Liberi G, Dujon B, Freudenreich CH, Richard G-F. 2009. SRS2 and SGS1 prevent chromosomal breaks and stabilize triplet repeats by restraining recombination. Nat Struct Mol Biol 16:159–167. doi: 10.1038/nsmb.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Nguyen JHG, Viterbo D, Anand RP, Verra L, Sloan L, Richard G-F, Freudenreich CH. 2017. Differential requirement of Srs2 helicase and Rad51 displacement activities in replication of hairpin-forming CAG/CTG repeats. Nucleic Acids Res 45:4519–4531. doi: 10.1093/nar/gkx088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Henriques R, Griffiths C, Rego EH, Mhlanga MM. 2011. PALM and STORM: unlocking live-cell super-resolution. Biopolymers 95:322–331. doi: 10.1002/bip.21586. [DOI] [PubMed] [Google Scholar]