

Abstract
Melanomas can have multiple co-existing cell states, including proliferative (PRO) versus invasive (INV) subpopulations that represent a “go or grow” tradeoff; however, how these populations interact is poorly understood. Using a combination of zebrafish modeling and analysis of patient samples we show that INV and PRO cells form spatially structured heterotypic clusters and cooperate in the seeding of metastasis, maintaining cell state heterogeneity. INV cells adhere tightly to each other, and form clusters with a rim of PRO cells. Intravital imaging demonstrated cooperation in which INV cells facilitate dissemination of less metastatic PRO cells. We identified the TFAP2 neural crest transcription factor as a master regulator of clustering and PRO/INV states. Isolation of clusters from patients with metastatic melanoma revealed a subset with heterotypic PRO-INV clusters. Our data suggest a framework for the co-existence of these two divergent cell populations, in which heterotypic clusters promote metastasis via cell-cell cooperation.
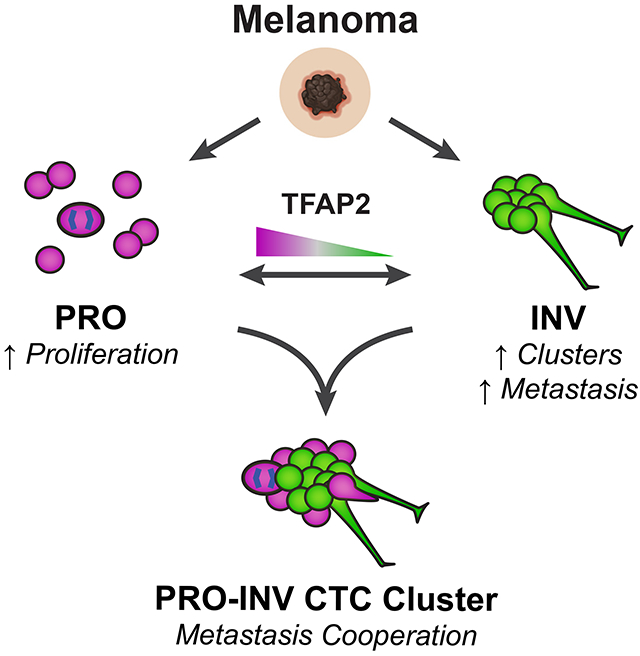
Graphical abstract
eTOC
Proliferative (PRO) and invasive (INV) cell states coexist in melanoma. Campbell et al. demonstrate that PRO and INV cells cooperate in metastasis via heterotypic circulating tumor cell (CTC) clusters and identify TFAP2 as a key mediator. Their work highlights a role for collective dissemination in melanoma metastasis.
INTRODUCTION
Tumors contain subpopulations of cells differing both through genetic and non-genetic mechanisms (Hinohara and Polyak, 2019). One form of heterogeneity is transcriptional, often referred to as cell state (Hinohara and Polyak, 2019; Hoek and Goding, 2010), that can drive a range of phenotypes including pigmentation and invasiveness (Houghton et al., 1987). Increasing transcriptomic evidence points to at least four distinct cell states in melanoma (Rambow et al., 2018; Tsoi et al., 2018; Wouters et al., 2020), with the most consistently identified states being proliferative (PRO) versus invasive (INV). The PRO vs. INV state is in part controlled by the melanocyte master transcription factor MITF (Carreira et al., 2006; Eichhoff et al., 2010), with the PRO cells being MITFHI and INV cells being MITFLO, although many other genes have been linked to these states (Tirosh et al., 2016; Verfaillie et al., 2015). Individual cells tend to be PRO or INV, but not both (Hoek et al., 2008; Hoek et al., 2006; Rambow et al., 2015; Tirosh et al., 2016; Verfaillie et al., 2015; Widmer et al., 2012)—a tradeoff reminiscent of the “grow or go” hypothesis (Hatzikirou et al., 2010; Matus et al., 2015). However, some data posit the existence of biphenotypic cells, with individual cells having characteristics of both PRO and INV cells (Tuncer et al., 2019). Despite evidence that these and other (Baron et al., 2020; Rambow et al., 2019; Rambow et al., 2018; Tsoi et al., 2018) subpopulations exist in tumors, little is known about how these states co-exist within the tumor, or whether they cooperate to promote tumorigenic phenotypes such as metastasis.
While certain cell states exhibit resistance to MAPK inhibitor therapy (Rambow et al., 2018; Tsoi et al., 2018), the role of the PRO/INV populations is best studied in the context of metastasis. Analogous to epithelial-to-mesenchymal transition (EMT) in epithelial cancers, it was hypothesized that during metastasis PRO cells could phenotype switch to an INV state and back in response to microenvironmental cues such as Wnt5A, EDN3, hypoxia, inflammation or nutrient deprivation (Carreira et al., 2006; Eichhoff et al., 2010; Falletta et al., 2017; Hoek et al., 2008; Hoek et al., 2006; Kim et al., 2017; Pinner et al., 2009; Weeraratna et al., 2002; Widmer et al., 2012). The extent to which cells are stable versus switching between these states has implications in whether new therapies should target specific states versus plasticity itself. While this switching model likely explains metastases in some patients, it does not fully explain why these cell state subpopulations co-exist in nearly all tumors examined.
Cooperation—a social behavior where one individual increases the fitness of another—is widely studied in ecology and evolution (Ågren et al., 2019; Archetti and Pienta, 2019; Foster, 2011; Hauser et al., 2009; Korolev et al., 2014). Cancer cells interact with each other, competing for resources but also possibly cooperating with each other to potentiate malignancy (Axelrod et al., 2006). The role of cooperation between cancer cells remains understudied, especially in vivo. In breast cancer there are reports of specific subpopulations that facilitate the overall growth of the tumor, demonstrating how cooperation can provide a selective pressure for the maintenance of heterogeneity within tumors (Cleary et al., 2014; Marusyk et al., 2014). In melanoma, mixing PRO and INV cells from different patients leads to polyclonal metastatic seeding (Chapman et al., 2014; Rowling et al., 2020); however, whether PRO and INV cells from the same tumor physically interact and cooperate, and the mechanistic links between cell state and cooperation, remain unknown.
Using a combination of zebrafish modeling and analysis of patient samples we show that coexisting PRO and INV cell states form heterotypic clusters that cooperate in metastasis. Circulating tumor cell clusters are a potent mechanism for metastasis and are associated with worse outcome (Aceto et al., 2014; Fidler, 1973; Gkountela et al., 2019; Glaves, 1983; Liotta et al., 1976; Long et al., 2016; Luo et al., 2014; Mayhew and Glaves, 1984; Szczerba et al., 2019; Watanabe, 1954). We find that PRO and INV states spontaneously form spatially ordered clusters, with a rim of PRO cells surrounding a dense core of INV cells. Unexpectedly, we find that INV cells express higher levels of adhesion molecules. We find that clustering is regulated by the developmental neural crest transcription factor TFAP2, which also mediates the PRO vs. INV state and metastatic capacity. While phenotype switching is a likely mechanism of metastasis in some patients, our data provide an alternative mechanism by which relatively fixed cell states physically cooperate via clustering to promote metastasis.
RESULTS
Characterization of PRO/INV cell states
To address the question of how PRO and INV populations interact, we utilized a zebrafish model of melanoma that allows for longitudinal single cell analysis of these heterogeneous subpopulations in metastasis formation (Cagan et al., 2019; Heilmann et al., 2015). From a transgenic melanoma in a BRAFV600E;p53−/− animal (Ceol et al., 2011; Kaufman et al., 2016; Patton et al., 2005; White et al., 2011) we generated a low-passage zebrafish melanoma cell line, ZMEL1 (Heilmann et al., 2015), and phenotyped multiple cultures to identify populations enriched for either proliferative (ZMEL1-PRO) or invasive (ZMEL1-INV) phenotypes (Figure 1a). Consistent with the previous characterization of PRO and INV states (Verfaillie et al., 2015; Widmer et al., 2012), we observed a small but consistent difference in net proliferation, and a more substantial motility difference, between the ZMEL1-PRO and -INV states (Figure 1b-c, Figure S1a-b). To confirm that this recapitulates human PRO and INV states, we performed RNA-sequencing analysis (RNA-seq) on these two ZMEL1 populations and found a strong association between ZMEL1-INV and -PRO states and published human INV and PRO gene signatures (Hoek et al., 2006; Tirosh et al., 2016; Verfaillie et al., 2015; Widmer et al., 2012), respectively, with the INV signature from Hoek et al. (Hoek et al., 2006) the top gene set (Figure 1d-e, Figure S1c, Tables S1, S5).
Figure 1. PRO and INV coexist in zebrafish melanoma, with INV cells metastasizing more frequently due to increased extravasation.

a. Proliferative (PRO) and invasive (INV) subpopulations were identified from the ZMEL1 zebrafish melanoma cell line, which was originally isolated from a transgenic zebrafish and can be transplanted into transparent casper zebrafish. b-c. Tracking of individual cells by time-lapse microscopy (both p<0.001 by linear regression, N=4 independent experiments). b. Growth curves (mean ± SE of mean, smoothed with moving window average of 5 time points) and doubling time (mean [95% CI]: 27.0 h [26.9, 27.1] vs. 29.7 h [29.6, 29.9] for ZMEL1-PRO and -INV, respectively). c. (left) Representative displacements of 500 tracks, and (right) model estimates ± 95% CI for alpha, the slope of the log-log plot of mean squared displacement vs. lag time (tau) for each ZMEL1-PRO and -INV. Larger alpha indicates more persistent motion, with α=1 for diffusive and α=2 for projectile motion. d. (left) The INV signature (Hoek et al., 2006) was the top gene set by Gene Set Association Analysis (GSAA) of ZMEL1-INV vs. -PRO RNA-seq. (Right) Dual waterfall plot of GSAA ranked by false discovery rate (FDR). Literature PRO/INV gene sets are indicated with an asterisk and colored according to FDR. e. Heatmap of genes in Hoek INV signature that are differentially expressed between ZMEL1-PRO and -INV (log2 fold change cutoff ± 1.5, padj < 0.05). Human ortholog gene names are used for clarity (see Figure S1e for zebrafish gene names). f. Segmentation of representative images of ZMEL1-PRO and -INV tumors and distant metastases (e.g. to caudal region [box]) at 3 days post-transplant (3dpt). Original images shown in Figure S1e. g. Quantification of caudal metastases seeded by ZMEL1 populations at 3 dpt (OR [95% CI]: 11.62 [1.43, 94.53]; p=0.022 by logistic regression; N=3 independent experiments with PRO/INV 10/10, 31/33, and 13/13 fish per group, respectively; n=110 fish total; plot shows mean ± SD). h. Representative composite images at 1 dpt from time lapse confocal microscopy of ZMEL1 cells transplanted intravenously in larval zebrafish. Arrowhead indicates group of cells invading from the notochord (NC) and caudal hematopoietic tissue (CHT) into the tail fin mesenchyme (TF). Images are representative of n=13 fish per cell type. See Video S1 for full time lapse. i. Quantification of caudal tissue invasion by imaging at 4-6dpt (N=3 independent experiments with PRO/INV 23/23, 21/21, and 19/23 fish per group, respectively; OR [95% CI]: 13.58 [5.56, 33.18]; p<0.001 by logistic regression, plot shows mean ± SD). j. Relative number of ZMEL1-PRO and -INV cells isolated and quantified by flow cytometry from primary tumors and metastases of fish transplanted with a 1:1 mixture of ZMEL1-PRO and -INV (primary tumors from n=6 fish; metastases from n=4 fish; p= 0.51 and p=0.031, respectively, by one-sample two-sided t-test with Bonferroni correction). See also Figure S1, Tables S1,S5,S6, and Video S1.
To compare the metastatic potential of ZMEL1-PRO and ZMEL1-INV—a far more stringent assay than in vitro migration—we transplanted each population orthotopically into the subcutaneous tissue of transparent casper zebrafish and followed their growth and metastasis by whole-fish fluorescence microscopy (Heilmann et al., 2015) (Figure 1f-g, Figure S1d-g). Fish harboring ZMEL1-INV tumors were significantly more likely to have distant metastases three days post-transplant (3 dpt), particularly in the caudal region of the fish, an anatomical region relatively resistant to metastasis (Heilmann et al., 2015). ZMEL1-PRO formed more round, exophytic tumors compared to the more invasive tumors of ZMEL1-INV, but their engraftment efficiency was similar (Figure 1j), suggesting an increased metastatic potential of the ZMEL1-INV cells. To investigate this difference in detail, we transplanted each population intravenously in larval casper zebrafish where we followed the seeding of metastases by confocal time-lapse microscopy. ZMEL1-INV cells extravasated more effectively than ZMEL1-PRO cells within the first dpt (Figure 1h, Video S1). To quantify this difference, we tracked metastatic progression in similarly transplanted larval fish by daily whole-fish imaging; ZMEL1-INV cells invaded into the caudal tissue in a significantly higher proportion of fish at the experiment endpoint (4-6 dpt, Figure 1i). Since the cells were injected intravenously, these findings indicate that extravasation—a key step of metastatic spread—is more effective in INV cells than PRO cells. To characterize the growth dynamics of these tumors and metastases, we co-transplanted PRO and INV cells in a 1:1 ratio and then isolated cells from resultant primary tumors and metastases. Primary tumors showed similar engraftment efficiency and maintained the initial 1:1 ratio of PRO and INV cells. Metastases, initially seeded more efficiently by INV cells, became dominated by PRO cells over time, highlighting the increased in vivo proliferative potential of this population (Figure 1j). Given the agreement, both phenotypic and transcriptomic, between these ZMEL1 populations and established PRO and INV cell states, we utilized ZMEL1-PRO and ZMEL1-INV to further characterize the relationship of these cell states.
PRO/INV cells form heterotypic clusters
To identify functional processes differentiating PRO and INV populations, we performed Gene Ontology (GO) association analysis on our RNA-seq data. This analysis unexpectedly revealed a strong association between the INV state and signatures of enhanced cell-cell adhesion, with many adhesion genes upregulated (Figure 2a-b, Figure S2a-c, Table S5). This association was surprising, as the classical model of cell invasion involves the loss of cell adhesion and the gain of individual motility, the opposite of what we observed (Gupta et al., 2005; Li et al., 2015; Padmanaban et al., 2019). To test this paradoxical finding, we utilized a three-dimensional (3D) cluster formation assay in low-attachment plates, which allows cells to spontaneously aggregate over 1-3 days with minimal contribution from cell proliferation. Under these conditions, while the ZMEL1-PRO cells tended to stay as individual cells or small clusters, the ZMEL1-INV population formed strikingly large, spherical clusters (Figure 2c-d, Figure S2d-e, Video S2) in agreement with increased adhesive properties. To test whether the association between invasiveness and cell clustering is a general feature of melanoma, we compared the INV signature defined by Hoek et al. (Hoek et al., 2006) with that of the cell-cell adhesion genes most associated with ZMEL1-INV in a panel of 56 melanoma cell lines available in the Cancer Cell Line Encyclopedia (CCLE) (Ghandi et al., 2019) and 472 clinical melanoma samples from The Cancer Genome Atlas (TCGA) (Cancer Genome Atlas Network, 2015). In both cohorts, the expression of cell-cell adhesion genes correlated strongly with the INV cell state (Figure S2f-g). To validate this finding functionally, we assayed the cluster-forming ability of a panel of nine human melanoma cell lines. We observed a strong correlation between cluster formation and the INV state, consistent with our zebrafish and transcriptomic findings (Figure 2e). Taken together, these results indicate that melanomas that are invasive and metastatic tend to form cluster aggregates.
Figure 2. Cluster formation by INV state drives spatial patterning of melanoma clusters.

a. (left) Plot of top Gene Ontology (GO) gene set by GSAA for ZMEL1-INV vs. -PRO RNA-seq. (right) Dual waterfall plot of top/bottom 250 gene sets from GO analysis (for full plot see Figure S2a). Adhesion GO gene sets are indicated with an asterisk and colored according to false discovery rate (FDR) b. Heatmap of genes in adhesion GO gene sets (FDR < 0.05, n=3) that are differentially expressed between ZMEL1-PRO and -INV (log2 fold change cutoff ± 1.5, padj < 0.05). Human ortholog gene names are used for clarity (see Figure S2b-c for absolute expression and zebrafish gene names). c. (top) Schematic of assay and (bottom) quantification of cluster formation in low-bind plates after 2 days (N=6 independent experiments, p<0.001 by two-sided t-test, plot shows mean ± SD). d. Representative images of clusters formed after 3 days. e. Human melanoma cell lines ranked by cluster forming ability (left to right: low to high) with PRO/INV gene expression scores (Hoek et al., 2006) (Spearman correlation and Bonferroni-corrected p-values shown, scale bar 100μm). f. Co-clusters of 1:1 mixture of ZMEL1-PRO and -INV (inset scale 50 μm). g. (left) 3D opacity rendering and (right) central slice (slice 54 of 115) of confocal imaging of co-cluster of ZMEL1-PRO and ZMEL1-INV. h. Co-clusters of 1:1 mixture of ZMEL1-INV and ZMEL1-INV with either control (sg_scr) or cdh1 (sg_cdh1) sgRNA (inset scale 50 μm). f-h are composite images. See also Figure S2, Tables S1,S5,S6, and Videos S2,S3.
Individual primary patient melanomas comprise both PRO and INV subpopulations, and disseminated metastases preserve that diversity (Tirosh et al., 2016), raising the question of whether these subpopulations interact. Circulating tumor cell (CTC) clusters—comprised either of tumor cells or tumor and microenvironmental cells—are increasingly recognized for their role in promoting metastatic spread, facilitating diversity at metastatic sites (Aceto et al., 2014; Cheung et al., 2016; Gkountela et al., 2019; Maddipati and Stanger, 2015; Szczerba et al., 2019). Because the ZMEL1-PRO and -INV populations were isolated from a single primary tumor, we sought to establish whether the two could interact in clusters. Differential labeling of the PRO vs. INV cells revealed that the two cell states consistently generated co-clusters with a coherent spatial structure, with ZMEL1-INV cells at the core and ZMEL1-PRO cells at the rim, reminiscent of developmental cadherin sorting (Foty and Steinberg, 2005) (Figure 2f-g, Figure S2h-i, Video S3). Indeed, CRISPR/Cas9 induced deletion of cdh1 in ZMEL1-INV partially phenocopied ZMEL1-PRO, both decreasing the cluster size relative to INV clusters and causing spatial sorting of mixed clusters (Figure 2h, Figure S2j-m). Deletion of cdh1 alone was insufficient, however, to induce changes in the metastatic rate of ZMEL1-INV (Figure S2n-o), suggesting that a broader set of adhesion and invasion genes, and not only cdh1, underlies the observed phenotypes. This stereotyped spatial organization of ZMEL1-PRO and -INV clusters in an assay that models the behavior of CTC clusters (Cheung et al., 2016; Gkountela et al., 2019) motivated us to investigate whether interaction between these two populations would play a role in vivo.
PRO-INV heterotypic clusters cooperate in metastasis
To assay PRO-INV interactions in vivo during metastatic dissemination, we transplanted a 1:1 mixture of the ZMEL1-PRO and -INV populations intravenously as single cells in zebrafish larvae and followed them by confocal time-lapse microscopy (Figure 3a, Video S4). We observed that these transplanted single cell populations—as confirmed by microscopic inspection and in vitro cluster formation assays (Figure S2d)—spontaneously formed intravascular tumor cell clusters comprised of cells from one or both cell states, consistent both with previous intravital imaging (Liu et al., 2018) and with the detection of CTC clusters heterogenous for the melanoma marker S100 in the blood of patients (Khoja et al., 2014). More notably, we observed that nearly half (11 out of 24) of ZMEL1-PRO extravasation events were co-extravasations of heterotypic tumor cell clusters with ZMEL1-INV (Figure S3a). While imaging of actively circulating CTC clusters was beyond the resolution of our assay, we detected a pattern of collective motility suggesting that cells from the same heterotypic cluster rearranged in vivo to extravasate collectively, with ZMEL1-INV cells behaving as leader cells and ZMEL1-PRO as followers. These detailed observations suggest that the PRO and INV states known to coexist in primary tumors can form heterotypic clusters and interact in the seeding of metastases.
Figure 3. PRO and INV cooperate in metastasis via co-extravasation.

a. In the first 24 hours after intravenous transplant of ZMEL1-PRO and -INV, a PRO-INV mixed cluster (left, arrowhead) extravasated from the caudal hematopoietic tissue (CHT) into the tail fin mesenchyme (TF)—with ZMEL1-INV leading (middle, arrow) and ZMEL1-PRO following (right, arrow). Composite images shown. b. Segmented representative image of adult zebrafish with orthotopic transplant of 1:1 mixture of ZMEL1-PRO and -INV. Arrowheads indicate polyclonal metastases, including to the kidney and caudal regions (left and right boxes, respectively). Original image shown in Figure S3b. c. Number of fish co-transplanted with a 1:1 mixture of ZMEL1-PRO and -INV that have no caudal metastases (None), caudal metastases comprised of exclusively PRO or INV, or caudal metastases formed by PRO-INV co-metastasis (Co-Met) (N=5 independent experiments with 17, 15, 16, 15, and 17 fish each; 80 fish total; p<0.001 by Mantel-Haenszel’s test for null hypothesis of no interaction). d. Percentage of fish with ZMEL1-PRO caudal metastasis 3 dpt following orthotopic transplant of ZMEL1-PRO or a 1:1 mixture of ZMEL1 PRO:INV (OR [95% CI]: 3.31 [1.10, 9.96]; p=0.033 by logistic regression). e. Proportion of fish with ZMEL1-INV caudal metastasis 3 dpt following orthotopic transplant of ZMEL1-INV or a 1:1 mixture of ZMEL1 PRO:INV (OR [95% CI]: 1.32 [0.61, 2.88]; p=0.49 by logistic regression). For c-e: N=5 independent experiments with PRO/MIX/INV 18/17/18, 13/15/14, 15/16/15, 12/15/15, and 15/17/16 fish per group, respectively; 231 fish total; plots show mean ± SD. f. Number of ZMEL1-PRO cells in Boyden Chamber assay migrating per 20X field alone or mixed with ZMEL1-INV (p=0.042 by linear regression). g. Number of ZMEL1-INV cells in Boyden Chamber assay migrating per 20X field alone or mixed with ZMEL1-PRO (p=0.91 by linear regression). For f-g: N=3 independent experiments for each EGFP and tdTomato labeling; plots show mean ± SD. See also Figure S3 and Video S4.
To test the consequences of PRO-INV interaction in a more physiological setting, we next assessed their interaction after orthotopic transplantation in adult zebrafish. We transplanted primary tumors of each population alone and as a 1:1 mixture and followed their growth and metastasis by whole-fish fluorescence microscopy. In the group with mixed primary tumors, we observed a significantly higher number of fish with polyclonal metastasis than would be expected based on the metastatic rate of each subpopulation alone if they did not interact (Figure 3b-c, Figure S3b-c). Strikingly, we also observed that the less metastatic ZMEL1-PRO population had an increased rate of caudal metastases in mixed tumors compared to when it was transplanted alone (Figure 3d, Figure S3d-h), showing that this population benefited from cell-cell interaction with the INV cells. Moreover, the more metastatic ZMEL1-INV population did not become less metastatic (Figure 3e), meaning that they did not pay a significant cost for giving this benefit to ZMEL1-PRO. This type of interaction, where one individual (INV) increases the fitness of another (PRO), is formally defined as cooperation (Foster, 2011) (see Figure S3n for a schematic representation of social interactions including cooperation). To further characterize the benefit to the ZMEL1-PRO population, we performed transplants at various PRO:INV mixing ratios (1:4, 4:1, and 9:1) consisting of tdTomato-expressing ZMEL1-PRO cells mixed with EGFP-expressing ZMEL1 cells (either PRO or INV) and then quantified the metastases (Figure S3i-j). This confirmed that when ZMEL1-INV cells comprise at least half of the primary tumor, the PRO subpopulation has an increased rate of metastasis, providing context to which patients may exhibit such metastatic interaction. We observed a similar cooperative interaction in vitro in dual-color Boyden Chamber migration assays (Figure 3f-g), confirming that ZMEL1-PRO invades better when mixed with -INV cells independently of the microenvironment. Experiments with conditioned media further suggested this interaction is mediated by direct cell-cell contact (Figure S3k-l) and not via soluble factors, and there was no evidence for ZMEL1-INV clusters protecting ZMEL1-PRO from apoptosis (Figure S3m). The in vivo cooperative benefit was only evident early in metastatic dissemination (3 dpt vs 7 dpt, Figure S3g-h), indicating that this cooperation is particularly beneficial when both primary tumors and the number of disseminating tumor cells are small. Taken together, these data show that the formation of heterotypic clusters enables the collective extravasation of PRO and INV, facilitating cooperation that preserves cell state diversity in early metastatic lesions (Figure S3n) (Foster, 2011; Hauser et al., 2009).
TFAP2 mediates the PRO/INV state and clustering
Although several molecular mechanisms have been shown to regulate the PRO and INV state in melanoma (including MITF, AXL, WNT5A and BRN2 and their up- and downstream regulatory networks (Cheng et al., 2015; Falletta et al., 2017; Fane et al., 2019; Hoek et al., 2008; Hoek et al., 2006; Pinner et al., 2009; Rambow et al., 2015; Rambow et al., 2019; Rambow et al., 2018; Shakhova et al., 2015; Tirosh et al., 2016; Verfaillie et al., 2015; Weeraratna et al., 2002; Widmer et al., 2012)), there is no known connection between these programs and the formation of tumor cell clusters. To identify the mechanism regulating clustering in the INV population, we performed motif analysis on 1 kilobase regions associated with genes differentially expressed between ZMEL1-PRO and -INV cells (Figure 4a, Table S3). The top motif whose target genes were enriched in the PRO cells putatively binds the NFIC and TFAP2A transcription factors. One of the TFAP2 family members itself, tfap2e, was also one of the most differentially expressed genes between the PRO and INV cells, with its expression being over 100-fold higher in the PRO versus INV cells (Figure S4a, Table S1). The TFAP2 family of transcription factors plays essential roles in neural crest and melanocyte cell fate during development (de Croze et al., 2011; Hoffman et al., 2007; Kaufman et al., 2016; Li and Cornell, 2007; Luo et al., 2002; Seberg et al., 2017a; Seberg et al., 2017b; Van Otterloo et al., 2010) and has been implicated as part of a regulatory network promoting the PRO state (Hoek et al., 2006; Rambow et al., 2015; Tirosh et al., 2016; Verfaillie et al., 2015). This raised the hypothesis that TFAP2 was acting as a master regulator of the clustering phenotype observed in the INV population. To test this, we performed RNA-seq of ZMEL1 cells in 3D (clustered) culture, and asked which genes were differentially expressed in 3D compared to 2D (non-clustered) conditions (Figure 4b, Figure S4b, Tables S2,S3). In both PRO and INV, we again found enrichment of a motif that binds TFAP2—specifically, TFAP2E—when looking at up- and downregulated genes together. This is consistent with the known redundancy of tfap2a and tfap2e in zebrafish (Van Otterloo et al., 2010), and highly suggestive of a role for TFAP2 in mediating clustering.
Figure 4. TFAP2 distinguishes PRO vs. INV state and modulates clustering and metastasis.

a. HOMER de-novo motif analysis on genes upregulated in ZMEL1-PRO vs. -INV (log2 fold change cutoff ± 1.5, padj < 0.05, ±500bp of transcription start site [TSS]). b. HOMER de-novo motif analysis of genes differentially expressed between ZMEL1-INV in 3D (clusters) vs. 2D (no clusters) (log2 fold change cutoff ± 1.5, padj < 0.05, ±500bp of TSS). c. Cluster size after 2 days in ZMEL1-PRO with CRISPR-Cas9 inactivation of tfap2a and tfap2e alone or in combination (sg_tfap2a/e) versus control (sg_scr) (p-values by linear regression; N=3 independent experiments). d. Representative images of clusters formed after 2 days from ZMEL1-PRO with sg_scr vs. sg_tfap2a/e. e. Growth of ZMEL1-PRO orthotopic primary tumors with sg_scr vs. sg_tfap2a/e (p=0.011 by linear regression; N=3 independent experiments with sg_scr/sg_tfap2a/e 24/22, 22/22, 24/24 fish per group, respectively; n=138 fish total). f. Representative composite image of extravasated (arrows) and partially extravasated (arrow-head) ZMEL1-PRO cells with sg_tfap2a/e following intravenous transplant in casper fish with kdrl-RFP transgene labeling the vasculature. g. Proportion of larval fish intravenously transplanted with ZMEL1-PRO with sg_scr or sg_tfap2a/e with extravasated cells at 1 dpt, as quantified from confocal time lapse microscopy (OR [95% CI]: 2.20 [1.05, 4.61]; p=0.038 by logistic regression; N=3 independent experiments with sg_scr/sg_tfap2a/e 20/20, 22/23, and 22/22 fish per group, respectively; n=129 fish total). See also Figure S4 and Tables S2,S3.
We next sought to test whether TFAP2 plays a functional role in melanoma cluster formation and metastasis. We performed CRISPR/Cas9 deletion of tfap2a and tfap2e in ZMEL1-PRO, which typically forms poor clusters, and found a significant increase in clustering only in the context of tfap2a/e double knockout (Figure 4c-d, Figure S4c-e). We also found that the tfap2a/e knockout compared with a non-targeting control had a small but reproducible decrease in cell proliferation, along with an increase in the persistence of migration (Figure S4f-i), consistent with the phenotype differences between the INV and PRO populations. We next wanted to determine whether this phenotypic switch mediated by TFAP2 translated to an in vivo effect on metastasis. We orthotopically transplanted control or tfap2a/e knockout cells into adult casper fish and measured both primary tumor growth and metastatic dissemination. The tfap2a/e knockout cells formed primary tumors that grew significantly slower than controls (Figure 4e), which was expected from their slower in vitro proliferation. Despite this decrease in primary tumor growth, we found similar rates of overall and caudal metastasis, suggesting that loss of tfap2a/e induces a higher proportion of cells to metastasize (Figure S4j-k). To test this idea more directly, we assessed the effect of tfap2a/e on metastasis in a proliferation-independent assay by intravenous transplant. Time lapse confocal microscopy revealed that loss of tfap2a/e led to metastatic extravasation in a significantly higher proportion of fish (Figure 4f-g), consistent with a report that TFAP2A overexpression in human cells slows metastatic spread (Huang et al., 1998). Taken together, these data suggest that TFAP2 is not only a major regulator of the PRO vs. INV cell state, but that it also controls tumor cell clustering and regulates metastasis via an effect on extravasation.
TFAP2 correlates with clustering in human melanoma
We next wanted to determine whether the effects of TFAP2 we observed in the zebrafish were conserved in human melanoma. TFAP2A is a member of several gene expression profiles describing the proliferative state (Rambow et al., 2015; Tirosh et al., 2016; Verfaillie et al., 2015), including the Hoek et al. set (Hoek et al., 2006), and is critical for melanoma cell proliferation (Figure S5a). Consistent with this, increased expression in primary tumors of either TFAP2A or genes associated with the PRO state is associated with worse clinical outcomes in two large independent clinical cohorts (Figure S5b-g), likely reflecting the known prognostic effects of mitotic rate and primary tumor size in melanoma TNM staging (Gershenwald et al., 2017; Thompson et al., 2011). We next examined TFAP2A expression in a panel of 56 melanoma cell lines (CCLE) (Ghandi et al., 2019) and 472 clinical melanoma samples (TCGA) (Cancer Genome Atlas Network, 2015), and asked how this correlated with their PRO/INV signatures defined by Hoek et al. (Hoek et al., 2006). In both cohorts, we confirmed that the PRO and INV states were strongly anti-correlated. Samples with higher TFAP2A expression exhibited a more PRO gene signature, and conversely, samples with lower TFAP2A expression exhibited a more INV signature (Figure 5a, Figure S5h). As expected based on these results, TFAP2A expression was correlated with MITF expression in these cohorts (Figure S5i-j). Further, we asked whether the association between TFAP2A expression and the PRO/INV signatures was maintained at the level of single cells. We analyzed available single cell RNA-seq data across a panel of 23 human melanoma patients (Jerby-Arnon et al., 2018), and found a similar relationship: individual cells with high TFAP2A tend to have a higher PRO score, whereas cells with low TFAP2A tend to have a higher INV score (Figure S5k). Within the TCGA dataset, tumor samples collected from primary sites had higher levels of TFAP2A compared to metastatic lesions despite similar expression of pan-melanoma markers (Figure 5b, Figure S5l-m), in agreement with a prior report (Tellez et al., 2007). Further, in the two patients for which paired primary and metastatic samples were available, TFAP2A expression was lower in the metastatic lesion. A direct measurement of the relative ratio of TFAP2HI to TFAP2LO cells in the tumors, and its correlation with patient prognosis will await future longitudinal prospective single cell analysis. Next, we examined TFAP2A expression in the panel of human melanoma cell lines used in Figure 2e and found that cluster-forming lines had lower TFAP2A expression than non-clustering lines (Figure S5n-o). To test this association across cells that better preserve the heterogeneity observed clinically in melanoma, we examined a panel of four short-term human melanoma cultures (Raaijmakers et al., 2015). Cluster formation correlated strongly with lower levels of TFAP2A (Figure 5c), consistent with our observation that TFAP2 loss drives melanoma clustering. Collectively, our data confirm that the association we had discovered in zebrafish—between TFAP2, the PRO/INV state and tumor cell clustering—also occurs in human melanoma.
Figure 5. TFAP2 correlates with clustering in human melanoma and regulates genes associated with metastasis and cell-cell adhesion.

a. Human melanoma cell lines in the Cancer Cell Line Encyclopedia (CCLE, n=56) plotted as PRO and INV scores (Hoek et al., 2006) and colored according to TFAP2A mRNA expression. Pearson correlation coefficients between TFAP2A and PRO/INV scores are shown on axes. b. TFAP2A mRNA expression in The Cancer Genome Atlas (TCGA) melanoma (SKCM) cohort comparing primary tumors and metastases (p<0.001 by Wilcoxon rank sum test with Bonferroni correction). c. Low-passage human melanoma cell lines ranked by increased cluster forming ability (left to right) with TFAP2A expression quantified by immunofluorescence (plot and top; Spearman correlation shown; scale bar 20 μm) and clustering (bottom, scale bar 500 μm). d. GSAA was run using gene sets and GO gene sets with FDR < 0.05 from INV vs. PRO RNA-seq (n=39 gene sets; cyan points in Figures 1d and 2a). Bars show Normalized Association Score (NAS) for CRISPR (ZMEL1-PRO sg_tfap2a/e vs. sg_scr) and INV vs. PRO for each gene set, with black outline representing FDR<0.05 for CRISPR experiment. e. Plot of INV signature (Hoek et al., 2006) by GSAA for ZMEL1-PRO sg_tfap2a/e vs. sg_scr RNA-seq. f. Heatmap of top genes in Hoek INV and GO Adhesion gene sets that are differentially expressed between ZMEL1-PRO sg_tfap2a/e and sg_scr (log2 fold change cutoff ± 0.5, padj < 0.05). Asterisks (*) indicate genes with associated TFAP2A CUT&RUN peaks. Human ortholog gene names are used for clarity (see Figure S5p for zebrafish gene names). See also Figure S5 and Tables S4,S5,S6.
TFAP2 regulates genes associated with metastasis and cell-cell adhesion
To gain further insight into the mechanism by which TFAP2 regulates melanoma phenotypes, we performed RNA-seq of the tfap2a/e knockout cells versus controls. We first validated that the tfap2a/e knockout recapitulated the observed differences between ZMEL1-PRO and -INV by performing gene set association analysis (GSAA) using the gene sets that had passed false discovery cutoff (FDR < 0.05) in our ZMEL1-INV vs. -PRO RNA-seq analysis. We observed a high concordance in the top dysregulated pathways—including multiple INV and GO adhesion gene sets associated with TFAP2 loss—confirming that TFAP2 regulates pathways distinguishing ZMEL1-PRO and -INV (Figure 5d-e, Tables S4,S5). Specific genes upregulated upon TFAP2 loss and associated with either the INV state or adhesion include several with known functions in melanoma metastasis (Figure 5f, Figure S5p; e.g. TGFBI (Lauden et al., 2014), VEGFC (Streit and Detmar, 2003), CTGF (Finger et al., 2014), and CDH2 (Mrozik et al., 2018)). In order to understand the mechanism by which TFAP2 regulates PRO/INV state and cell-cell adhesion, we performed TFAP2A CUT&RUN (Cleavage Under Targets and Release Using Nuclease) in SKMEL28 cells, the human melanoma cell line with the highest expression of PRO-state genes out of those we characterized. This allowed us to understand the genes bound by TFAP2A in melanoma (Rambow et al., 2015; Seberg et al., 2017b). Consistent with the known roles of TFAP2 as both a transcriptional activator and repressor (Ren and Liao, 2001; Seberg et al., 2017b), we observed significant enrichment for TFAP2A peaks in genes that are upregulated upon tfap2a/e knockout in ZMEL1-PRO cells (Figure 5f asterisks, Figure S5q-s; e.g. TGFBI, CDH2), suggesting it acts as a repressor of those loci. We did not observe evidence of a stress response or changes in mitfa expression resulting from tfap2a/e knockout (Table S5), lending further support to a model of direct regulation by TFAP2. Taken together, these data highlight the direct and pleiotropic effects of TFAP2 loss on metastatic spread, further confirming a role for TFAP2 in cell state and suggesting downstream mediators.
PRO-INV heterotypic CTC clusters exist in the blood of melanoma patients
To further elucidate the translational relevance of our findings and to better characterize the role of PRO-INV heterotypic CTC clusters in metastatic dissemination, we isolated CTC clusters from the blood of patients with metastatic melanoma (Figure 6a). We sampled the peripheral blood of nine patients with advanced metastatic melanoma and performed microfluidics-based capture of CTCs through a procedure that allows isolation of CTC clusters (Gkountela et al., 2019; Szczerba et al., 2019; Xu et al., 2015). We were able to identify CTC clusters from four of these nine patients (44%), from which we isolated a total of 32 CTC clusters, consistent with prior rates of cluster detection (Khoja et al.; Long et al.; Luo et al.; Ruiz et al.; Sarioglu et al., 2015). We characterized the cell states present in these clusters by staining with antibodies against TFAP2A for PRO cells, SOX9 for INV cells, and CD45 to exclude immune cells (Figure 6b, Figure S6). Overall, we found that 19% of these clusters were heterotypic, composed of a mixture of PRO and INV cells (Figure 6c). As expected, this result varied across patients, with some patients having no clusters and the others having a mix of homotypic and heterotypic clusters (Figure 6d, Table S7). These results provide important validation that PRO-INV heterotypic CTC clusters occur clinically in a subset of patients with metastatic melanoma.
Figure 6. PRO-INV heterotypic CTC clusters exist in the blood of melanoma patients.

a. Schematic of experimental design. b. (left) IF staining for TFAP2A and SOX9 (composite image) and (right) nuclear quantification in a PRO-INV heterotypic melanoma CTC cluster. Scale bar is 5μm. c-d. Classification of human melanoma CTC clusters based on quantification of nuclear TFAP2A and SOX9 staining as (c) homotypic (PRO-only or INV-only) vs. PRO-INV heterotypic, and (d) PRO, PRO-INV, and INV. For c-d n=32 clusters from 4 patients. 5 additional patients were analyzed with no CTC clusters identified. For d each stacked box represents one CTC cluster. See also Figure S6 and Table S7.
Longitudinal single-cell RNA-seq reveals stability of PRO but not INV state
The above data suggest a model in which PRO and INV cell clusters, regulated by TFAP2, form the unit of initial metastatic seeding in certain patients. However, once seeding has occurred, it is still possible that either of these cell states can undergo phenotype switching and contribute to metastatic outgrowth. This possibility was suggested by our finding that metastases tend to become dominated by PRO cells over time (Figure 1j). To test this more formally, we conducted a large-scale longitudinal analysis of cell state at the single cell level, interrogating the effects of cell-cell interaction, tumor formation, and metastasis. We performed single-cell RNA-seq of over 40,000 ZMEL1 cells from both the PRO and INV cell states across four different conditions: (1) in vitro individual culture; (2) in vitro co-culture; (3) in vivo primary tumors; and (4) in vivo metastatic lesions (Figure 7a). Strikingly, ZMEL1-PRO and -INV subpopulations were highly pure in vitro and—despite substantial gene expression changes associated with the dramatic microenvironmental pressures in vivo—remained largely discrete throughout all conditions (Figure 7b). In order to quantify the stability of the two populations, we calculated PRO and INV scores for each cell based on gene sets derived from ZMEL1 bulk RNA-seq, and trained a classifier based on in vitro individual culture samples (Figure 7c-d). Consistent with our results from conditioned media experiments (Figure S3k-l), we observed very little effect of co-culture upon the transcriptomes of ZMEL1-PRO and -INV cells, with both populations remaining more than 99% pure. Strikingly, a fraction of INV cells in tumors, especially from the metastases, upregulated PRO-state genes, increasingly occupying a PRO/INV double-positive state. This is in contrast to PRO cells, which remained stable in the PRO state, and is consistent with prior in vivo evidence that an INV-to-PRO switch is favored (Pinner et al., 2009). Further validating a role for TFAP2 as a master regulator of melanoma cell state, we found that ZMEL1-INV cells that gained a PRO-like gene expression program also reactivated tfap2e (Figure 7e, Figure S7a). Overall, these data support a model of cooperation whereby clusters comprised of distinct PRO and INV subpopulations promote co-metastatic seeding, and metastatic outgrowth is increasingly dominated by PRO-like cells.
Figure 7. Longitudinal single-cell RNA-seq reveals stability of PRO but not INV state.

a. Schematic of experiment. Prior to flow cytometry and 10X single-cell RNA-seq, ZMEL1-PRO and -INV cells were either grown in vitro (individual or co-culture) or isolated from zebrafish orthotopically transplanted with a 1:1 mixture of PRO and INV (primary tumors or metastases). b. Uniform Manifold Approximation and Projection (UMAP) (McInnes et al., 2018) dimensionality reduction of 40,293 ZMEL1 cells sequenced as in (a). Individual culture (IND); co-culture (CO); primary tumors (PRI); metastases (MET). c. PRO and INV scores based on ZMEL1 bulk RNA-seq plotted for all cells (gray) with ZMEL1-PRO (purple) and ZMEL1-INV (green) for the indicated condition overlaid. Diagonal line represents the classifier used in (d). d. Confusion matrices comparing initial cell identity with observed cell classification based on a linear classifier trained on in vitro individual culture samples. e. ZMEL1-INV cells plotted as in (c) colored according to tfap2e mRNA expression reveal re-activation of tfap2e upon metastatic dissemination. See also Figure S7.
To interrogate signaling pathways that could be important for PRO-INV interaction in vivo, we analyzed our single-cell data using CellPhoneDB. The analysis identified ligand-receptor pairs that were significantly enriched between PRO and INV cells (Efremova et al., 2020) (Figure S7b-c). Among the most enriched candidate receptor-ligand pairings between the PRO/INV cells in vivo were IGF2-IGF2R and VEGFA-EFNB2. The IGF pathway, in particular, is interesting: IGF signaling has previously been shown by us and others to be an important regulator of melanoma cell growth (Lissanu Deribe, 2016; Weiss et al., 2020; Zhang et al., 2020). While it has traditionally been assumed that IGF ligands come solely from the stroma, these data suggest that the melanoma cells themselves may be one source of such ligands. In addition, a longstanding observation in the melanoma literature has been the phenomenon of vasculogenic mimicry, in which tumor cells take on characteristics of blood vessels, and this has been shown to be mediated in part by the VEGF axis (Frank et al., 2011; Maniotis et al., 1999; Schnegg et al., 2015; Xu et al., 2019). Future experiments aimed at disrupting these pathways will be important to functionally confirm their effect on metastatic cooperation.
DISCUSSION
Both individual and collective mechanisms of metastasis can occur in melanoma (Long et al., 2016) and other cancers (Pearson, 2019; Reichert et al., 2018). Phenotype switching between PRO and INV states has long been postulated to be a mechanism for individual seeding of metastasis in melanoma (Hoek et al., 2008; Kim et al., 2017; Pinner et al., 2009; Vandamme and Berx, 2014). Separately, circulating tumor cell (CTC) clusters, a mode of collective metastasis, have been shown to have increased metastatic potential (Aceto et al., 2014; Cheung et al., 2016), and patients with detected CTC clusters have worse clinical outcomes (Giuliano et al., 2018; Long et al., 2016). Cooperation has previously been reported both in epithelial cancers (Celià-Terrassa et al., 2012; Neelakantan et al., 2017; Tsuji et al., 2009) and between melanoma PRO and INV states in the context of primary tumor collective cell invasion (Chapman et al., 2014) and metastatic tropism (Rowling et al., 2020), but the mechanisms that explain the relationship between the PRO/INV states and cooperative metastasis have remained unknown. We provide a mechanism that explains how these two subpopulations, which coexist in the primary tumor, cooperate in metastasis formation. We find that PRO and INV cells form heterotypic clusters, which are controlled by the neural crest transcription factor TFAP2. We provide direct evidence of the presence of heterotypic PRO-INV CTC clusters in a subset of melanoma patients, providing an important translational link to our mechanistic work in the zebrafish. Our data on heterotypic CTC clusters in patients are consistent with the hypothesis that in individual patients, either individual or collective migration may predominate. An alternative explanation for the observed cooperation and polyclonal metastasis is that they result from single-cell phenomena such as the formation of aggregates at the site of extravasation or through sequential seeding. We believe this alternative is less likely because—as we showed—PRO-INV heterotypic CTC clusters do exist in melanoma patients. This is further supported by reported melanoma CTC cluster detection rates between 2 and 55 percent (Khoja et al.; Long et al.; Luo et al.; Ruiz et al.; Sarioglu et al., 2015), and the observation that certain melanoma patients exhibit polyclonal metastatic seeding (Rabbie et al., 2020; Sanborn et al., 2015). While the phenotype switching model predicts dynamic switching of individual cells between PRO and INV states (analogous to an epithelial-to-mesenchymal transition) as a necessary feature of individual metastasis (Hoek et al., 2008; Kim et al., 2017; Pinner et al., 2009; Vandamme and Berx, 2014), our finding that PRO and INV can cooperate while remaining as distinct phenotypic populations suggests that tumors can preserve diversity during initial metastatic seeding without the need for large-scale cell state switching on a rapid time scale.
Our data do not exclude the possibility that phenotype switching, or plasticity, is an operative mechanism that promotes metastasis. Our single cell analysis of metastatic outgrowth demonstrates that INV cells, once they arrive, can still switch to a double-positive PRO/INV state, indicating that phenotype switching in the INV to PRO direction may be important after initial seeding. Instead, our data indicate that dynamic switching, at least on a short time scale, is not an absolute requirement. It is likely that in patients—where tumors are extraordinarily heterogeneous at both genetic and epigenetic levels—both mechanisms (fixed cell state and plastic cell states) can co-exist and may both be important. Recent data suggest that within tumors, there may be “high plasticity” cell states which are more prone to switching compared to other cells (Marjanovic et al., 2020). This must be balanced with the observation that populations enriched for metastatic phenotypes can be isolated and passaged in vitro, demonstrating at least transient stability of such traits (Bos et al., 2009; Laughney et al., 2020; Malladi et al., 2016; Minn et al., 2005; Pencheva et al., 2012). It is likely that the relative balance between fixed versus plastic cell states is governed by the epigenetic state of the cell, since chromatin-related proteins such as JARID1 are known to be involved in determination of cell state (Harmeyer et al., 2017; Roesch et al., 2010). A major unanswered question, however, is what might be the molecular driver of such plasticity. Going from the PRO to INV state is linked to molecules such as WNT5A (Dissanayake et al., 2007; Weeraratna et al., 2002), which drives subsequent metastatic ability, and could act in both paracrine and autocrine ways. Conversely, fewer molecules driving the INV to PRO direction are known. We previously demonstrated a role for EDN3 in this process (Kim et al., 2017), and given the known role of endothelin signaling in melanocyte and melanoma proliferation, this is likely one such factor. One important consideration is whether one direction (i.e. PRO-to-INV versus INV-to-PRO) is favored over the other, which would be enabled by monitoring of cell states in real time as they traverse the bloodstream. While obtaining such samples from patients would be ideal, animal models might provide insights, although even in those models such experiments are still technically challenging. Previous work along this line has suggested that INV cells marked by BRN2 may be biased towards switching more readily to the PRO state, which would be consistent with our single-cell data (Pinner et al., 2009). Defining the range of mechanisms that mediate plasticity, and whether some cells are more easily switched than others, remains an important area for future exploration.
Our data demonstrate that cell cluster formation driven by TFAP2 loss is a pro-metastatic feature of INV cells, with pleiotropic increases in cell-cell adhesion and cell clustering enabling cooperation with PRO cells. Further, we demonstrate the functional role of TFAP2 in regulating cell state and clustering. TFAP2A is known to have overlapping downstream targets with MITF, the best characterized driver of the PRO state, and promoters of these targets are frequently bound simultaneously by TFAP2A and MITF (Seberg et al., 2017b). Despite this, they exert at least partially independent functions, as evidenced by only partial rescue of tfap2a/e knockout with overexpression of mitfa (Van Otterloo et al., 2010) and our own data showing that CRISPR knockout of tfap2a/e did not alter expression of mitfa. How TFAP2 itself is regulated in this context, however, remains an open question. DNA methylation has been linked to expression of PRO/INV genes (Verfaillie et al., 2015) and to TFAP2A expression (Hallberg et al., 2014; Zeng et al., 2013); however, further work is required to fully elucidate these relationships. This mechanism is consistent with the recent report that breast cancer epigenetic state and CTC cluster formation are tightly linked (Gkountela et al., 2019), and suggests that clusters may act to potentiate an already more metastatic cell population. Given that the INV state in melanoma is also associated with increased resistance to targeted therapy (Konieczkowski et al., 2014; Muller et al., 2014; Verfaillie et al., 2015), pharmacologic disruption of CTC clusters could be an attractive target to slow metastasis and decrease the distant spread of drug-resistant cells.
Limitations of the study
The current study relies on a zebrafish model, which was used primarily to quantify and dissect the mechanisms of clustering and extravasation and to identify TFAP2. However, it has limitations compared to the various ways in which cells can extravasate in human patients. The zebrafish is also limited in that prospective identification of PRO and INV cells from tumors is not currently feasible. Additionally, visualization of actively circulating clusters—and thus monitoring cell states in real time as they traverse the bloodstream—is beyond the resolution of this study. Further defining the timescale of PRO-INV plasticity, and its relation to CTC clusters, remains an important area for future investigation.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Richard M. White (whiter@mskcc.org).
Materials Availability
Cell lines generated in this work are available upon request.
Data and code availability
All RNA-seq data generated in this study have been deposited at NCBI GEO (GSE151679), with bulk RNA-seq counts and differential expression tables in the Supplemental Information. TFAP2A CUT&RUN data are available via the NCBI GEO repository (GSE153020). This paper analyzes existing, publicly available data with accession numbers listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-hs_CDH1 | BD | Cat # 610181 |
| Goat anti-dr_Tfap2a | LifeSpan Biosciences | Cat # LS-C87212 |
| Rabbit anti-dr_Tfap2e | Fisher | Cat # PA5-72631 |
| Rabbit anti-hs_TFAP2A | Cell Signaling | Cat # 3215 |
| Rabbit anti-hs_cyclophilin B | Fisher | Cat # PA1-027A |
| Anti-mouse Alexa Fluor® 488 | Cell Signaling | Cat # 4408S |
| Anti-rabbit Alexa Fluor® 594 | Cell Signaling | Cat # 8889S |
| Mouse anti-hs_H3 | Cell Signaling | Cat # 14269 |
| Rabbit anti-TFAP2A | Abcam | Cat # ab108311 |
| Rabbit anti-Sox9 | Sigma | Cat # HPA001758 |
| Mouse anti-TFAP2A | Santa Cruz Biotechnology | Cat # sc-12726 |
| Rat anti-CD45 | ThermoFisher | Cat # MA5-17687 |
| Donkey anti-mouse AF488 | ThermoFisher | Cat # A21202 |
| Donkey anti-rabbit AF647 | ThermoFisher | Cat # A31573 |
| Donkey anti-rat DyLight 555 | ThermoFisher | Cat # SA5-10027 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Human metastatic melanoma patient blood samples | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Tricaine-S | Syndel | Cat # MS-222 |
| Hoechst 33342 | Fisher | Cat # H1399 |
| poly-D-lysine | Sigma | Cat # P-0899 |
| Rabbit IgG | Millipore | Cat # 12-370 |
| Concanavalin A-coated magnetic beads | Bang Laboratories, Inc | Cat # BP531 |
| pA-MNase | Dr. Steve Henikoff | N/A |
| Liberase TL | Sigma | Cat # 5401020001 |
| Alt-R® S.p. Cas9 Nuclease V3 | Integrated DNA Technologies | Cat # 1081059 |
| Alt-R® CRISPR-Cas9 tracrRNA, ATTO™ 550 | Integrated DNA Technologies | Cat # 1075928 |
| Cas9 Electroporation Enhancer | Integrated DNA Technologies | Cat # 1075916 |
| Alt-R® CRISPR-Cas9 crRNA (see Oligonucleotides below for sequences) | Integrated DNA Technologies | N/A |
| Neon Transfection System 100 μL Kit | Fisher | Cat # MPK10025 |
| Critical commercial assays | ||
| Neon Transfection System | Fisher | Cat # MPK5000 |
| Ultra-Low Attachment Surface 96-well plate, flat well | Corning | Cat # 3474 |
| Ultra-Low Attachment Surface 96-well plate, round well | Corning | Cat # 7007 |
| Transwell assay: 3.0μm pore insert / 24-well plate | Corning | Cat # 353492 / 353504 |
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat # G7572 |
| Caspase-Glo® 3/7 Assay System | Promega | Cat # G8091 |
| RNeasy Plus Mini kit | Qiagen | Cat # 74034 |
| QiaShredder | Qiagen | Cat # 79656 |
| Parsortix Cell Separation Cassette | ANGLE (Xu et al., 2015) | Cat # GEN3D6.5 |
| Chromium Single Cell 3’ Library & Gel Bead Kit v3.1 | 10X Genomics | Cat # 1000128 |
| Chromium Single Cell 3’ Chip G | 10X Genomics | Cat # 1000127 |
| Deposited data | ||
| Bulk and single-cell RNA-seq data | This paper | GEO # GSE151679 |
| TFAP2A CUT&RUN data | This paper | GEO # GSE153020 |
| Cancer Cell Line Encyclopedia (CCLE) RNA-seq | Broad CCLE (https://portals.broadinstitute.org/ccle) | RSEM genes TPM, version 20180929 |
| The Cancer Genome Atlas (TCGA) Skin Cutaneous Melanoma (SKCM) mRNA expression | Broad GDAC Firehose (http://firebrowse.org/) | v2 RSEM genes normalized, version 2016_01_28 |
| Human melanoma single-cell RNA-seq | (Jerby-Arnon et al., 2018) | GEO # GSE115978 |
| Broad Institute Dependency Map (DepMap) | Broad Institute (https://depmap.org/portal/) | CRISPR (Avana) Public 19Q3 dataset |
| AVAST-M melanoma cohort | (Corrie et al., 2018; Garg et al., 2021) | EGA # EGAD00001006401 |
| Leeds Melanoma Cohort | (Nsengimana et al., 2018) | EGA # EGAS00001002922 |
| Experimental models: Cell lines | ||
| ZMEL1-PRO (mitfa: EGFP) | This paper | N/A |
| ZMEL1-PRO (mitfa: tdTomato) | This paper | N/A |
| ZMEL1-PRO (mitfa: EGFP & Ub: nls-mCherry) | This paper | N/A |
| ZMEL1-INV (mitfa: EGFP) | This paper | N/A |
| ZMEL1-INV (mitfa: tdTomato) | This paper | N/A |
| ZMEL1-INV (mitfa: EGFP & Ub: nls-mCherry) | This paper | N/A |
| MEWO | ATCC | Cat # HTB-65 |
| SH4 | ATCC | Cat # CRL-7724 |
| SKMEL28 | ATCC | Cat # HTB-72 |
| A375 | ATCC | Cat # CRL-1619 |
| HS294T | ATCC | Cat # HTB-140 |
| A2058 | ATCC | Cat # CRL-11147 |
| RPMI7951 | ATCC | Cat # HTB-66 |
| HMCB | ATCC | Cat # CRL-9607 |
| HS895T | ATCC | Cat # CRL-7637 |
| M000921 | (Raaijmakers et al., 2015) | N/A |
| M111031 | (Raaijmakers et al., 2015) | N/A |
| M990922 | (Raaijmakers et al., 2015) | N/A |
| MM121224 | (Raaijmakers et al., 2015) | N/A |
| Experimental models: Organisms/strains | ||
| Casper zebrafish | White Lab | N/A |
| Casper;kdrl-RFP zebrafish | This paper | N/A |
| Oligonucleotides | ||
| sg_scr Alt-R crRNA | (Wang, T., et al., Science, 2015) | AACCTACGGGCTACGATACG |
| sg_cdh1_1 Alt-R crRNA | This paper | AGTGGCAAAAGACTAGGCAA |
| sg_cdh1_2 Alt-R crRNA | This paper | TCATGCAAACGGAGTGGACG |
| sg_tfap2a_1 Alt-R crRNA | This paper | TTGTGGACGTGAATTCCCCA |
| sg_tfap2a_2 Alt-R crRNA | This paper | CATGTATTCAGCTATCGCCT |
| sg_tfap2a_3 Alt-R crRNA | This paper | ATAGTTGATGCGGTAACCCG |
| sg_tfap2a_4 Alt-R crRNA | This paper | CTCAACCACAACACCCGGGA |
| sg_tfap2e_1 Alt-R crRNA | This paper | GGCAGGGGTCCCGAATCCGT |
| sg_tfap2e_2 Alt-R crRNA | This paper | TACTCCCAGAGCCAGGACGG |
| sg_tfap2e_3 Alt-R crRNA | This paper | TGAGACTGAGTTCCCTGCGC |
| sg_tfap2e_4 Alt-R crRNA | This paper | TGCACCGCGTGCATCCCCAG |
| Recombinant DNA | ||
| Ub: nls-mCherry | This paper | N/A |
| Mitfa: tdTomato | This paper | N/A |
| Software and algorithms | ||
| Zebrafish image analysis pipeline | (Heilmann et al., 2015) | https://github.com/SiljaHeilmann/Matlab-fish-image-analysis |
| Volocity v6.3 | PerkinElmer | https://www.perkinelmer.com |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| IDL tracking methods | John Crocker, David Grier, and Eric Weeks | http://site.physics.georgetown.edu/matlab/ |
| FASTQC | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| TRIMMOMATIC | (Bolger et al., 2014) | http://www.usadellab.org/cms/?page=trimmomatic |
| STAR | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| SeQC | (DeLuca et al., 2012) | https://bioconductor.org/packages/release/data/experiment/html/seqc.html |
| DESeq2 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| GSAA | (Xiong et al., 2012; Xiong et al., 2014) | http://gsaa.unc.edu/ |
| DIOPT | (Hu et al., 2011) | https://www.flyrnai.org/diopt |
| HOMER | (Heinz et al., 2010) | http://homer.ucsd.edu/homer |
| JASPAR | (Khan et al., 2017) | http://jaspar.genereg.net |
| Trim Galore version 0.6.3 | Developed by Felix Krueger at the Babraham Institute | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore |
| Bowtie2 version 2.1.0 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| ChIPseeker version 1.18.0 | (Yu et al., 2015) | https://bioconductor.org/packages/release/bioc/html/ChIPseeker.html |
| CellRanger version 3.1.0 | 10X Genomics | https://www.10xgenomics.com/ |
| Seurat version 3.1.4 | (Stuart et al., 2019) | https://satijalab.org/seurat/index.html |
| CellPhoneDB | (Efremova et al., 2020) | N/A |
| MATLAB | Mathworks | https://www.mathworks.com/products/matlab.html |
| R | R Foundation for Statistical Computing | https://www.r-project.org/ |
| Zen | Zeiss | https://www.zeiss.com |
| STATA v14 | StataCorp | https://www.stata.com/stata14/ |
| Other | ||
A MATLAB-based image analysis pipeline for quantifying melanoma in zebrafish was previously published (Heilmann et al., 2015).
All other data reported in this paper will be shared by the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
The establishment of the ZMEL1 zebrafish melanoma cell line from a tumor in a mitfa-BRAFV600E/p53−/− zebrafish was described previously (Heilmann et al., 2015; Kim et al., 2017). ZMEL1 was grown at 28°C in a humidified incubator in DMEM (Gibco #11965) supplemented with 10% FBS (Seradigm), 1X penicillin/streptomycin/glutamine (Gibco #10378016), and 1X GlutaMAX (Gibco #35050061). The ZMEL1-PRO and -INV populations were identified based on phenotyping of multiple concurrent ZMEL1 cultures. ZMEL1 populations were validated by RNA-seq confirming expression of expected transgenes. Human melanoma cell lines were maintained in DMEM (Gibco #11965) supplemented with 10% FBS (Seradigm), 1X penicillin/streptomycin/glutamine (Gibco #10378016), with the exception of HMCB, which was maintained in MEM (Gibco #11095080) supplemented with 10% FBS (Seradigm), 1% Sodium Pyruvate (Gibco #11360070), 1% MEM-Non-Essential Amino Acids (Gibco #11140050), 10mM HEPES (Gibco #15630080), and 1X penicillin/streptomycin (Gibco #15140122). Low-passage human melanoma cell lines were established and cultured as previously described (Raaijmakers et al., 2015). Cells were routinely confirmed to be free from mycoplasma (Lonza Mycoalert). Human cell lines were either purchased directly from ATCC or verified by STR profiling.
Zebrafish husbandry
Zebrafish were housed in a dedicated facility maintained at 28.5°C with a light/dark cycle (14 hours on, 10 hours off). Casper zebrafish used in this study were obtained by crossing casper zebrafish (White et al., 2008) with Ekkwil (https://zfin.org/action/genotype/view/ZDB-GENO-990520-2) and subsequently incrossing. Zebrafish constitutively expressing RFP under the kdrl promoter to label vasculature (https://zfin.org/ZDB-TGCONSTRCT-070117-163) were generously provided by Dr. Leonard Zon (Boston Children’s Hospital and Dana-Farber Cancer Institute), and were crossed with casper zebrafish to obtain casper;kdrl-RFP zebrafish. All anesthesia was performed using Tricaine-S (MS-222, Syndel USA, Ferndale, WA) with a 4g/L, pH 7.0 stock. Previous work demonstrated no difference in melanoma growth and metastasis between male and female fish (Heilmann et al., 2015); thus animals of both sexes were used for all studies. All procedures adhered to Memorial Sloan Kettering Cancer Center IACUC protocol number 12-05-008.
Human blood samples
Peripheral blood samples were obtained from consenting and de-identified stage IV melanoma patients in accordance with relevant institutional and national policies (Kantonal Ethics Commission of Zürich, BASEC-Nr.PB.2017-00494).
METHOD DETAILS
Cloning
Zebrafish-specific expression plasmids were generated by Gateway Cloning (Fisher) into the pDestTol2pA2 backbone (Tol2kit, plasmid #394) (Kwan et al., 2007). nls-mCherry (Tol2kit, plasmid #233) was cloned under the ubiquitin promoter (Baron et al., 2020) (Ub: nls-mCherry). tdTomato (Zhang et al., 2018) was cloned under the zebrafish mitfa promoter (Heilmann et al., 2015) (mitfa: tdTomato).
Fluorescently labeled cell lines
The ZMEL1 cell line constitutively expresses EGFP under the mitfa promoter (Heilmann et al., 2015). ZMEL1 lines additionally expressing nls-mCherry under the ubiquitin promoter for cell tracking experiments were generated with the Neon Transfection System (Fisher) followed by FACS sorting. ZMEL1 lines expressing tdTomato under the mitfa promoter were generated through CRISPR/Cas9 mutation of the constitutive EGFP (see CRISPR/Cas9 below) followed by Neon Transfection and FACS sorting.
CRISPR/Cas9
The Alt-R CRISPR-Cas9 System (Integrated DNA Technologies) was used for CRISPR-Cas9 experiments following the manufacturer’s protocols for use with the Neon Transfection System (Fisher) and adherent cells. Successful nucleofection was confirmed by visualizing ATTO 550 labeled tracrRNA one day post-nucleofection. Successful loss of full-length protein expression was verified by visualizing loss of EGFP expression (EGFP) or by Western blot (cdh1, tfap2a, tfap2e). Control scramble sgRNA (sg_scr) sequence was used from Wang et al. (Wang et al., 2015). sgRNA sequences were designed using CHOPCHOP (Labun et al., 2019) and GuideScan (Perez et al., 2017) and are listed in the key resources table.
Cluster formation assay
ZMEL1 cells were trypsinized, centrifuged at 300g for 3 minutes, and resuspended in standard culture media. A Corning Ultra-Low Attachment Surface 96-well plate (#3474) was seeded with 5x104 cells/well in a final volume of 100uL. Clusters were allowed to form over the course of 2-3 days in a humidified 28°C incubator, or were used for time-lapse microscopy on a Zeiss AxioObserver Z1 equipped with an incubation chamber using a 5x/0.16NA objective. For human melanoma cell lines, a round bottom Corning Ultra-Low Attachment Surface 96-well plate (#7007) was seeded with 5x103 cells/well in a final volume of 100uL. Clusters were allowed to form over the course of 24-48h in a humidified 37°C incubator. Cell lines were ranked according to their relative abilities to form dense three-dimensional clusters.
Cluster quantification
The average cluster size of each image was quantified using a MATLAB implementation of the characteristic length scale equation from Smeets et al (Smeets et al., 2016). To quantify cluster mixing, background corrected images for EGFP and tdTomato were each segmented using a low and high threshold to improve detection of small and large clusters, respectively. Segmentation from each channel was merged, and the coefficient of variation (CV) of cluster area was calculated for clusters filtered to have a size corresponding to 1 cell or larger. The composition of each cluster (filtered to have a size corresponding to a size of approximately 4 cells or larger) was classified as red-only, green-only, or red-green mix. Cluster spatial sorting was calculated for individual large (equivalent diameter greater than 45μm) mixed red-green clusters by calculating the weighted average of radial intensity profiles for each channel. The difference between the weighted averages for each channel was calculated and normalized by the radius of the cluster. With this dimensionless metric, small values correspond to well-mixed clusters, whereas larger values correspond to a high degree of spatial segregation.
Cluster confocal microscopy
ZMEL1 clusters were fixed at room temperature for 45 minutes by adding an equal volume of 4% PFA in PBS to 2-day cultures of clusters (2% final PFA concentration). Fixed clusters were washed once with PBS and counterstained with Hoechst 33342 (Fisher H1399) and transferred to a 96-well glass-bottom plate (Mattek, PBK96G-1.5-5-F) before imaging. Individual clusters were imaged on a Leica TCS SP5-II inverted point-scanning confocal microscope with a or 40x/1.10NA objective. 3D reconstruction was performed using Volocity (PerkinElmer, v6.3). Individual slices were visualized with ImageJ.
Cell tracking
Time-lapse microscopy was performed on a Zeiss AxioObserver Z1 equipped with an incubation chamber. A 96-well plate (Corning 353072) was seeded with 1.2x104 ZMEL1 cells (PRO or INV) admixed such that 1/6 of the population stably expressed nls-mCherry under the Ubi promoter, and allowed to adhere overnight. Cells were imaged every 5 minutes for 24 hours. Centroids of nuclei were identified, and tracks generated using a MATLAB implementation of the IDL tracking methods developed by John Crocker, David Grier, and Eric Weeks (physics.georgetown.edu/matlab/). For each imaging location, growth was calculated based on the number of nuclei present at each time point, assuming equal numbers of cells move in and out of the field of view over time. For each track, mean squared displacement (MSD) was calculated as previously described (Gorelik and Gautreau, 2014) over a range of lag times (5≤τ<100 min). The log-log plot of MSD vs. τ provides information about both the diffusion coefficient (intercept) and persistence (slope, α) of cells. For a cell moving randomly, α=1; and for a cell moving along a straight line, α=2 (Gorelik and Gautreau, 2014). N=4 independent replicates were performed, each consisting of 6 technical replicates per cell type. Growth rates were quantified with a linear mixed-effects model using the fitlme function in MATLAB with the model, ‘log(cell_number) ~ time + cell_type:time + (1. replicate)’. Motility (α) was quantified as the slope of the linear model, ‘log(MSD) ~ 1 + cell_type*log(τ)’. Growth plots represent the smoothed (moving window average of 5 time points [= 25 min]) average cell number ± SE normalized to the cell number at time zero.
Boyden chamber migration
Cell migration of PRO/PRO, INV/INV, and PRO/INV mixtures was quantified using a Boyden chamber (transwell) assay. A 1:1 mixture of ZMEL1 cells labeled with EGFP and tdTomato (5x104 cells total per well) were added to each transwell insert (Corning, 353492, 3.0μm pore) in a 24-well plate (Corning, 353504) in 500uL of complete media. The lower chamber was filled with 500μL of complete media and cells were allowed to migrate for 2 days. Cells were fixed for 15 minutes at room temperature with 4% PFA in PBS, washed once with PBS, and non-migrated cells removed with cotton-tipped swabs. Migrated cells were stained with Hoechst 33342 (Fisher H1399) and ≥9 fields/well imaged on a Zeiss AxioObserver Z1 with a 10x/0.45NA objective. Nuclei were segmented using intensity thresholding of background-corrected Hoechst staining followed by an intensity-based watershed step to separate adjacent objects. Cell identity was established based on fluorophore expression within the mask defining each nucleus.
Conditioned media
Conditioned media was collected from confluent 10cm dishes of ZMEL1-PRO and -INV cultures following 2-3 days of growth and filtered through a 0.45μm syringe filter (Fisher #09-720-005) to remove viable cells. Filtered conditioned media or fresh complete media was mixed 1:1 with fresh complete media and used for subsequent assays. For proliferation assays, ZMEL1-PRO or -INV cells (1.4x104 cells/well) were plated in white wall 96-well plates (Corning #3610) in a 1:1 mixture of fresh complete media with either ZMEL1-PRO or -INV conditioned media or fresh complete media. Relative proliferation was measured by CellTiter-Glo® Luminescent Cell Viability Assay (Promega) according to manufacturer’s protocol two days after plating. For Boyden Chamber assays, ZMEL1-PRO or -INV cells (5x104 cells total per well) were added to each transwell insert (Corning, 353492, 3.0um pore) in a 24-well plate (Corning, 353504) in 500uL of fresh complete media. The lower chamber was filled with either ZMEL1-PRO or -INV conditioned media or fresh complete media and cells were allowed to migrate for 2 days. Non-migrated cells were removed with cotton-tipped swabs. Migrated cells were stained with Hoechst 33342 (Fisher H1399) and 26 fields/well imaged on a Zeiss AxioObserver Z1 with a 10x/0.45NA objective. Centroids of nuclei were identified and counted using a MATLAB implementation of the IDL tracking methods developed by John Crocker, David Grier, and Eric Weeks (physics.georgetown.edu/matlab/).
Cluster caspase-3/7 assay
ZMEL1 cells were plated into cluster formation assay as described above using multiple ratios of PRO:INV cells (1:0, 4:1, 1:1, 1:4, and 0:1) with two identical plates per replicate. After 2 days, one plate was used for quantification of caspase-3/7 activity using Caspase-Glo® 3/7 Assay System (Promega) and the other for quantification of cell number using CellTiter-Glo® Luminescent Cell Viability Assay (Promega) each according to manufacturer’s protocol. For each replicate caspase-3/7 activity was normalized to cell number. The effect of various PRO:INV mixing ratios was quantified by linear regression with the fraction of INV cells as a continuous variable.
Larval transplantation
Transplantation of ZMEL1 cells into 2dpf casper zebrafish larvae was performed as previously described (Heilmann et al., 2015; Kim et al., 2017). Briefly, ZMEL1 cells were prepared by trypsinization, centrifuged at 300g for 3 minutes, and resuspended at a concentration of either 2.5x107 or 5.0x107 cells/mL in 9:1 DPBS:H2O (Gibco 14190-144). Cells were injected into the Duct of Cuvier of 2dpf casper or casper;kdrl-RFP (labeling the vasculature with RFP) fish using a Picoliter Microinjector (Warner Instruments, PLI-100A) with a glass capillary needle (Sutter, Q100-50-10) made on a laser-based needle puller (Sutter, P-2000). For mixing studies, ZMEL1-PRO and ZMEL1-INV differentially labeled with EGFP or tdTomato were mixed at a 1:1 ratio prior to injection. Fish with successful transplants based on the presence of circulating cells and/or cells arrested in the caudal vasculature were either used for time-lapse confocal microscopy (see “Zebrafish confocal time-lapse imaging”) or individually housed and followed by daily imaging on a Zeiss AxioZoom V16 fluorescence microscope.
Adult transplantation
Transplantation of ZMEL1 cells into adult casper zebrafish was performed as previously described (Heilmann et al., 2015; Kim et al., 2017). Briefly, adult zebrafish were irradiated on two sequential days with 15 Gy on a cesium irradiator (Shepherd) and were transplanted 3-4 days later. On the day of transplant, ZMEL1 cells were prepared by trypsinization, washed once with DPBS, and resuspended at a concentration of 1.67x108 cells/mL in DPBS (Gibco 14190-144). Cells were injected subcutaneously, caudal to the cloaca, on the ventral side of zebrafish anesthetized with Tricaine-S. Fish were imaged on days 1, 3, and 7 post-transplant on a Zeiss AxioZoom V16 fluorescence microscope. For 1:1 mixing studies, fish were injected with an equivalent final concentration of either ZMEL1-PRO, ZMEL1-INV, or a 1:1 mixture of the two populations differentially labeled with EGFP or tdTomato. A total of N=5 independent mixing experiments were performed, N=3 for INV-EGFP/PRO-tdTomato and N=2 for INV-tdTomato/PRO-EGFP, each with at least 12 fish per group (n=231 fish total). For variable ratio mixing studies, fish were injected with an equivalent final concentration of tdTomato-labeled ZMEL1-PRO mixed at a 1:4, 4:1, or 9:1 ratio with EGFP-labeled ZMEL1-PRO or ZMEL1-INV. A total of N=3 independent variable ratio mixing experiments were performed, each with at least 16 fish per group (n=334 fish total).
Zebrafish imaging and image quantification
Whole-fish larval imaging
Larval zebrafish transplanted as described above were anesthetized with Tricaine-S and imaged on a bed of 2% agarose (KSE Scientific, BMK-A1705) in E3. Images were manually scored for cells that had invaded the tail fin parenchyma at experiment endpoint.
Adult imaging
Adult zebrafish were imaged as previously described (Heilmann et al., 2015). Briefly, on days 1, 3, and 7 post-transplant fish were anesthetized with Tricaine-S and imaged on a bed of 2% agarose using a monocolor camera for fluorescence and brightfield, and color camera for observing pigmentation. Fluorescence images were manually scored for the following pre-specified binary outcomes:
Distant metastases: tumor not adjacent or contiguous with primary tumor
Caudal metastases: distant metastasis caudal to anal fin
Dorsal metastases: distant metastasis on dorsum of fish, near insertion of dorsal fin
For each binary outcome, the population composition was also scored (PRO, INV, or both). Primary tumor growth was also quantified over time for each fish using a previously described custom MATLAB pipeline (Heilmann et al., 2015). This pipeline was utilized with adaptive threshold segmentation to allow easier visualization of representative images.
Zebrafish larval confocal time-lapse imaging
Larval zebrafish were transplanted as described above. Fish with successful intravenous transplants were anesthetized with Tricaine-S and embedded in 1% low-melt agarose (Sigma A9045) in E3 containing 0.28ug/mL Tricaine-S in a glass-bottom square-well 96-well plate (Arrayit 96-well Microplate SuperClean, Cat M96FC, Lot 150901). Wells were filled with E3 containing 0.28ug/mL Tricaine-S and the plate sealed with a PCR microseal (BioRad Microseal 'B' Film, #MSB1001). Up to 45 larval zebrafish per experiment were imaged in parallel on a GE IN Cell Analyzer 6000 every 15-20 minutes for 24-30h. A single z-stack was acquired for each fish using a 10X/0.45NA objective and 8-10um z-steps. Because the ventral edge of the caudal vein exists as a single plane at this developmental stage (2-3dpf) (Isogai et al., 2001), maximum intensity projections were generated for each fish and manually scored for the presence of ZMEL1 cells that extravasate ventrally from the caudal vein and invade into the tail fin.
Western blot
Cell lysates were collected by sonication in RIPA buffer (Thermo #89901) with 1X Halt Protease and Phosphatase Inhibitor Cocktail (Thermo #78441) followed by centrifugation (14,000rpm for 10min at 4°C) and collection of the supernatant. Protein concentration was quantified by Bradford (Sigma B6916-500mL) according to manufacturer’s protocol. Samples were mixed with 6X reducing loading buffer (Boston BioProducts #BP-111R) and denatured at 95°C for 10 minutes. Samples were run on a Mini PROTEAN TGX gel (BioRad) and transferred using Turbo Mini Nitrocellulose Transfer Pack (Bio-Rad, catalog #1704158). Membranes were blocked with 5% nonfat dry milk in TBST (1X TBS + 0.1% Tween 20) for 1 hour before incubation with primary antibody in PBS overnight at 4°C. Membranes were washed with TBST and incubated with secondary antibody in 5% nonfat dry milk for 1 hour at room temperature. Membranes were washed with TBST and developed with ECL (Amersham, RPN2109) using an Amersham Imager 600 (GE) or chemiluminescence film. Antibodies: mouse anti-hs_CDH1 (BD #610181, lot 8082613), goat anti-dr_Tfap2a (LifeSpan Biosciences, #LS-C87212, lot 113877), rabbit anti-dr_Tfap2e (Fisher, #PA5-72631, lot UA2709682A), rabbit anti-hs_TFAP2A (Cell Signaling, #3215, clone C83E10, lot 2), rabbit anti-hs_cyclophilin B (Fisher #PA1-027A, lot SD248938).
Immunofluorescence
Cells were allowed to adhere for 2 days to glass chamber slides (PEZGS0816) coated with poly-D-lysine (Sigma #P-0899). Slides were fixed for 15 minutes with 4% PFA in PBS, washed 3 times with PBS, and permeabilized with 0.1% Triton X-100 (Fisher #BP151-100) in PBS for 15 min. Slides were blocked with 5% donkey serum (Sigma #S30-M), 1% BSA (Fisher #BP1600-100), and 0.1% Triton X-100 in PBS for 1 hour, followed by incubation in primary antibody overnight at 4°C. Slides were washed 3 times with PBS and incubated in secondary antibody (anti-mouse Alexa Fluor® 488 [Cell Signaling, #4408S]; anti-rabbit Alexa Fluor® 594 [Cell Signaling, #8889S]) for 1.5 hours, followed by 3 washes with PBS and counterstain with (1:10,000) Hoechst 33342 in PBS for 45 minutes. Slides were washed with PBS and mounted in ProLong Glass Antifade Mountant (Fisher #P36984). All incubations were carried out at room temperature unless otherwise noted. Antibodies: rabbit anti-hs_TFAP2A (Cell Signaling, #3215, clone C83E10, lot 2), mouse anti-hs_H3 (Cell Signaling, #14269, clone 1B1B2, lot 6). Slides were imaged in a minimum of 9 fields on a Zeiss AxioObserver Z1 using a 20x/0.80NA objective. Nuclei were segmented using intensity thresholding of background-corrected Hoechst staining followed by an intensity-based watershed step to separate adjacent objects. TFAP2A expression was quantified for each nucleus as the ratio of TFAP2A to Histone H3 staining intensity.
RNA-seq
Samples
For 2D culture, three replicate cultures at 70-80% confluence for each ZMEL1-PRO and -INV were utilized. For 3D culture, three replicate cultures for each ZMEL1-PRO and -INV grown in Corning Ultra-Low Attachment Surface 6-well plates (Corning #3471) for 48 hours were utilized. One individual replicate of RNA-seq of ZMEL1-PRO grown in 3D culture was excluded due to low RIN score, poor SeQC metrics and poor clustering with other replicate samples. For tfap2a/e CRISPR, three independent batches of ZMEL1-PRO cells nucleofected with either sg_scr or sg_tfap2a/e and grown in 2D conditions were utilized at 8-16 days post nucleofection.
Sequencing and analysis
Total RNA was extracted with the RNeasy Plus Mini kit with QiaShredder (Qiagen). Purified RNA was delivered to GENEWIZ (South Plainfield, NJ) for mRNA preparation with the TruSeq RNA V2 kit (Illumina) and 100bp (2D) or 150bp (3D and CRISPR) paired-end sequencing on the Illumina HiSeq2500. After quality control with FASTQC (Babraham Bioinformatics) and trimming with TRIMMOMATIC (Bolger et al., 2014) when necessary, reads were aligned to GRCz10 (Ensembl version 81) using STAR (Dobin et al., 2013), with quality control via SeQC (DeLuca et al., 2012). Differential expression was calculated with DESeq2 (Love et al., 2014) using the output of the --quantMode GeneCounts feature of STAR. The rlog function was used to generate log2 transformed normalized counts. Pathway and Gene Ontology (GO) analysis were performed with GSAA using the following parameters: gsametric Weighted_KS, demetric Signal2Noise, permute gene_set, rnd_type no_balance, scoring_scheme weighted, norm MeanDiv (Xiong et al., 2012; Xiong et al., 2014). A full list of gene sets used for GSAA can be found in Table S5. Ortholog mapping between zebrafish and human was performed with DIOPT (Hu et al., 2011) (Table S6). Only orthologs with a DIOPT score greater than 6 were used for GSAA and heatmap generation. In cases of more than one zebrafish ortholog of a given human gene, the zebrafish gene with the highest average expression was selected. De-novo motif analysis was performed with the HOMER (Heinz et al., 2010) function findMotifs.pl, using the zebrafish genome (GRCz10) and searching for motifs of lengths 8, 10, and 16 within ± 500bp of the TSS of differentially expressed genes. Motifs were annotated using JASPAR (Khan et al., 2017). A Cell-Cell Adhesion gene set was defined from the Core Enrichment genes from comparing ZMEL1-INV vs. -PRO to the gene set, GO Cell Cell Adhesion Via Plasma Membrane Adhesion Molecules.
TFAP2A CUT&RUN
Sample preparation
Anti-TFAP2A Cleavage Under Targets and Release Using Nuclease (CUT&RUN) sequencing was performed in wild-type and TFAP2A;TFAP2C double-mutant SKMEL28 cell lines as described (Skene and Henikoff, 2017) with minor modifications. Cells in log-phase culture (approximately 80% confluent) were harvested by cell scraping (Corning), centrifuged at 600g (Eppendorf, centrifuge 5424) and washed twice in calcium-free wash-buffer (20 mM HEPES, pH7.5, 150 mM NaCl, 0.5 mM spermidine and protease inhibitor cocktail, cOmplete Mini, EDTA-free Roche). Pre-activated Concanavalin A-coated magnetic beads (Bangs Laboratories, Inc) were added to cell suspensions (2x105 cells) and incubated at 4°C for 15 mins. Antibody buffer (wash-buffer with 2mM EDTA and 0.03% digitonin) containing rabbit anti-TFAP2A (abcam, ab108311) or rabbit IgG (Millipore, 12-370) was added and cells were incubated overnight at 4°C. The next day, cells were washed in dig-wash buffer (wash buffer containing 0.025% digitonin) and pA-MNase was added at a concentration of 500 μg/ mL (pA-MNase generously received from Dr. Steve Henikoff). The pA-MNase reactions were quenched with 2X Stop buffer (340mM NaCl, 20mM EDTA, 4mM EGTA, 0.05% Digitonin, 100 μg/ mL RNAse A, 50 μg/ mL Glycogen and 2 pg/ mL sonicated yeast spike-in control). Released DNA fragments were Phosphatase K (1μL/mL, Thermo Fisher Scientific) treated for 1 hr at 50°C and purified by phenol/chloroform-extracted and ethanol-precipitated. Fragment sizes analysed using an 2100 Bioanalyzer (Agilent). All CUT&RUN experiments were performed in duplicate.
Library preparation and data analysis
CUT&RUN libraries were prepared using the KAPA Hyper Prep Kit (Roche). Quality control post-library amplification was conducted using the 2100 Bioanalyzer (Agilent) for fragment analysis. Libraries were pooled to equimolar concentrations and sequenced with paired-end 100 bp reads on an Illumina HiSeq X platform. Paired-end FastQ files were processed through FastQC (Babraham Bioinformatics) for quality control. Reads were trimmed using Trim Galore Version 0.6.3 (Developed by Felix Krueger at the Babraham Institute) and Bowtie2 version 2.1.0 (Langmead and Salzberg, 2012) was used to map the reads against the hg19 genome assembly. The mapping parameters and peak calling analysis was performed as previously described (Meers et al., 2019). Called peaks were annotated with ChIPseeker v1.18.0 (Yu et al., 2015), with Distal Intergenic peaks excluded from downstream analysis. P-values for overlap with differential expression gene sets were calculated by comparing against overlap with randomly selected gene sets (n=10,000 iterations).
Isolation of human CTC clusters
Peripheral blood samples were collected from 9 consenting and de-identified patients with stage IV metastatic melanoma within 6 months of death (Kantonal Ethics Commission of Zürich, BASEC-Nr.PB.2017-00494) and enriched for CTCs on a Parsortix Cell Separation Cassette (GEN3D6.5, ANGLE) (Xu et al., 2015). In-cassette staining was performed following fixation with 4% paraformaldehyde and permeabilization with 0.2% Triton-100X and 4% donkey serum (DS) in PBS. Samples were incubated for 1 hour with primary antibodies—rabbit anti-Sox9 (HPA001758, Sigma, 1:50), mouse anti-TFAP2A (sc-12726, Santa Cruz Biotechnology, 1:50) and rat anti-CD45 (MA5-17687, ThermoFisher, 1:500)-followed by 1 hour incubation with secondary antibodies-donkey anti-mouse AF488 (A21202, ThermoFisher, 1:800), donkey anti-rabbit AF647 (A31573, ThermoFisher, 1:800), and donkey anti-rat DyLight 555 (SA5-10027, ThermoFisher, 1:1000)—and subsequent DAPI staining. Images of CTC clusters were acquired with Leica DMI 4000/6000 using 500ms exposure for AF488 and AF647, and 300ms exposure for DyLight 555 acquisitions. Presumptive CTC clusters were manually identified as two or more adjacent TFAP2A-positive cells. Nuclei were segmented using intensity thresholding of background-corrected DAPI staining followed by a distance-based watershed step and manual curation to separate adjacent objects, with subsequent exclusion of CD45-positive cells. Nuclear TFAP2A and SOX9 intensity were quantified for each cell from background-corrected images and each cell classified as PRO vs. INV based on the relative expression of each marker.
Single-cell RNA-seq (scRNA-seq)
Sample preparation
In vitro samples were cultured under standard conditions with either ZMEL1-PRO (tdTomato) and -INV (EGFP) separately (individual culture) or mixed together in a 1:1 ratio for 11 days (co-culture). Cells were trypsinized, resuspended in DMEM supplemented with 2% FBS, and flow sorted using a SY3200 (Sony) for DAPI-negative pure ZMEL1-PRO and -INV populations prior to droplet-based scRNA-seq. For in vivo samples, adult casper zebrafish were transplanted with a 1:1 mixture of ZMEL1-PRO (tdTomato) and -INV (EGFP) cells as described above (Adult transplantation). Tumors were allowed to grow for 6 days (primary tumors) or 13 days (metastases). At experimental timepoint, tumors were surgically excised and minced with a fresh scalpel (primary tumors from n=6 fish; metastases from n=4 fish). Each sample was placed in a 15mL tube containing 3mL of 0.9X DPBS with 0.16 mg/mL of Liberase TL (Sigma #5401020001), incubated at room temperature for 15 minutes followed by trituration using a wide-bore P1000 (Fisher #2069G), and incubated for an additional 15 minutes. 500μL FBS was added and each sample was triturated again and passed through a 70μm cell strainer (Corning #352350). Samples were centrifuged 500g for 5 minutes, resuspended in DMEM supplemented with 2% FBS, and flow sorted using a SY3200 (Sony) for DAPI-negative pure ZMEL1-PRO and -INV populations prior to droplet-based scRNA-seq.
Droplet-based scRNA-seq
For droplet-based scRNA-seq, experiments were performed using the 10X Genomics Chromium platform, with the Chromium Single Cell 3′ Library & Gel Bead Kit v3.1 (1000128) and Chromium Single Cell 3′ Chip G (1000127). ~8,000 cells per condition were centrifuged 300g for 5 minutes and resuspended in DMEM supplemented with 10% FBS and loaded to each channel for GEM generation and barcoded single cell libraries were generated according to manufacturer’s instructions. Libraries were diluted to 2nM and 75bp paired end sequencing was performed using the Illumina NextSeq 500. Between 150-200 million paired reads were generated for each library.
scRNA-seq processing and analysis
Raw sequencing data were processed using the CellRanger 3.1.0 pipeline developed by 10X Genomics. First, a custom zebrafish genome was generated based on GRCz10 with the addition of exogenous transgenes EGFP, tdTomato, and human BRAF-V600E using the command cellranger mkref. Next, the command cellranger count was utilized to perform alignment, filtering, barcode counting and UMI counting of all the samples. The Seurat R package (Version 3.1.4) (Stuart et al., 2019) was used for quality control, normalization and dimensionality reduction. Low quality cells with a) features less than 200 or greater than 5000, b) total counts less than 5000, or c) mitochondrial content greater than 15%, were discarded from the analysis. After filtering, normalization was performed using Seurat’s SCTransform procedure with default parameters to perform a regularized negative binomial regression based on the 3,000 most variable genes. Uniform Manifold Approximation and Projection (UMAP) (McInnes et al., 2018) dimensionality reduction was performed using default parameters. The log transformed normalized count data were extracted from Seurat and used for downstream analysis in MATLAB. Plots of PRO vs. INV scores were generated as described in Tirosh et al. (Tirosh et al., 2016) using PRO and INV gene sets defined from the top 250 most differentially expressed genes in each population by ZMEL1 bulk RNA-seq. Briefly, log-transformed data were mean-centered. For each gene set a control gene set was defined to control for variations in sequencing depth and library complexity by randomly selecting 100 genes from the same expression bin (n=25 bins), such that a 50 gene set would have a control gene set of 5,000 genes. The score for each sample was defined as the mean expression of the gene set minus the mean expression of the respective control gene set. A binary classifier for PRO versus INV state was defined by logistic regression on in vitro individual culture samples and used to classify cells from all conditions. Candidate ligand-receptor pairings for in vivo samples were identified using CellPhoneDB (Efremova et al., 2020).
Analysis of publicly available RNA-seq data
Cancer Cell Line Encyclopedia (CCLE)
RNA-seq expression data (RSEM genes TPM, version 20180929) were downloaded from the Broad CCLE (https://portals.broadinstitute.org/ccle). Plots of PRO vs. INV scores and their correlations with TFAP2A mRNA expression were generated as for ZMEL1 scRNA-seq data above using log-transformed [log2(TPM+1)] and mean-centered data with PRO and INV gene sets from Hoek et al. (Hoek et al., 2006).
The Cancer Genome Atlas (TCGA)
Skin Cutaneous Melanoma (SKCM) mRNA expression (v2 RSEM genes normalized, version 2016_01_28, 472 samples from 469 patients) was downloaded from the Broad GDAC Firehose (http://firebrowse.org/). PRO and INV gene expression scores and their correlations with TFAP2A mRNA expression were calculated and plotted as described for CCLE data above. Log-transformed normalized expression of TFAP2A and the pan-melanoma markers (Gaynor et al., 1981; Xiong et al., 2019) S100A1 and S100B were compared between samples from primary tumors and metastatic sites (Wilcoxon rank sum test with Bonferroni correction).
Human melanoma single-cell RNA-seq
Processed data for human melanoma single-cell RNA-seq (Jerby-Arnon et al., 2018) were downloaded from NCBI GEO (GSE115978). PRO and INV gene expression scores and their correlations with TFAP2A mRNA expression were calculated and plotted as described for CCLE data above.
DepMap
TFAP2A was queried through the Broad Institute Dependency Map (DepMap) portal (https://depmap.org/portal/) using the CRISPR (Avana) Public 19Q3 dataset.
External validation using the AVAST-M melanoma cohort
Bulk RNA-seq data from 194 primary melanoma patients were extracted from the phase III adjuvant AVAST-M melanoma cohort (accession number EGAD00001006401) (Corrie et al., 2018; Garg et al., 2021). Variance stabilizing transformation (VST) was applied to the raw counts using the varianceStabilizingTransformation function from the package DESeq2 (Love et al., 2014) (v1.22.2).
TFAP2A survival analysis
VST normalized expression data were used as a continuous variable in a multivariate Cox regression model, using the coxph function of the survival package (Therneau, 2020; Therneau and Grambsch, 2000) (v2.42-3) in R (v3.5.1). Progression-free survival was calculated as the time from diagnosis to the last follow-up or death/progression to metastatic disease, whichever occurred first. The following clinical covariates were considered in the multivariate Cox regression model; age at diagnosis, gender, stage (AJCC 7th edition), ECOG (Eastern Cooperative Oncology Group Performance Status), NClass (regional lymph involvement) and treatment (bevacizumab or placebo) (Garg et al., 2021).
PRO/INV survival analysis
VST expression data corresponding to the genes listed in the PRO and INV gene sets were extracted (Hoek et al., 2006; Tirosh et al., 2016; Verfaillie et al., 2015; Widmer et al., 2012). For each sample, the expression values of these genes were standardized to have zero mean and unit standard deviation. The mean of these standardized expression values was computed to obtain a vector score corresponding to the PRO and INV expression score vectors. These PRO and INV expression score vectors were divided into “high” and “low” expression groups using the median cut-off. Cox regression models were then fitted by means of the coxph function of the survival package (Therneau, 2020; Therneau and Grambsch, 2000) (v2.42-3) in R (v3.5.1). The hazard ratio (HR) (95% CI) and p-values corresponding to the “high” expression score group were reported in both univariate and multivariate analyses.
External validation using the Leeds Melanoma Cohort
Primary tumor expression of TFAP2A as well as PRO and INV signatures were tested for association with melanoma specific survival and relapse-free survival by Cox proportional hazards in a large population-based cohort (n=703, accession number EGAS00001002922) (Nsengimana et al., 2018). Signature scores were created by averaging z-transformed gene expressions and dichotomized by median split.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis and figure generation were performed in MATLAB (Mathworks, R2016a). RNA-seq analysis was performed in R (R Foundation for Statistical Computing, 3.4.0). Image processing and analysis was performed using MATLAB, Zen (Zeiss), ImageJ (NIH), and Volocity (PerkinElmer, v6.3). The Leeds Melanoma Cohort was analyzed in STATA v14 (StataCorp, Texas, USA). Unless otherwise noted, bar plots represent mean ± standard deviation (SD) of independent experiments, and dots represent means of independent experiments. Unless otherwise indicated, statistical details of experiments are included in figure legends. Abbreviations for p-values are as follows: * p<0.05, ** p<0.01, *** p<0.001.
Supplementary Material
Table S1 (Related to Figures 1-2)
ZMEL1-INV vs. -PRO RNA-seq results. Filtered differential expression (DE; absolute log2 fold change ≥ 1.5, padj < 0.05); all DE, log normalized counts, and raw counts.
Table S2 (Related to Figure 4)
ZMEL1 3D vs. 2D culture RNA-seq results for ZMEL1-PRO and ZMEL1-INV. Filtered differential expression (DE; absolute log2 fold change ≥ 1.5, padj < 0.05); all DE, log normalized counts, and raw counts.
Table S3 (Related to Figure 4)
Annotation of top HOMER motif for 1000bp region (transcription start site ± 500bp) associated with genes upregulated in ZMEL1-PRO and with genes differentially expressed in 3D vs. 2D culture for each ZMEL1-PRO and ZMEL1-INV. Annotations presented for genes differentially expressed in ZMEL1-PRO and -INV and for top motif for each ZMEL1-PRO and ZMEL1-INV.
Table S4 (Related to Figure 5)
ZMEL1-PRO sg_tfap2a/e vs. sg_scr RNA-seq results. Filtered differential expression (DE; absolute log2 fold change ≥ 0.5, padj < 0.05); all DE, log normalized counts, and raw counts.
Table S5 (Related to Figures 1, 2, and 5)
List of all gene sets used for GSAA, including sources and full gene lists. Results from GSAA analyses. Results table and details for gene sets with false discovery rate (FDR) below 0.05 (up to 6 per analysis).
Table S6 (Related to Figures 1, 2, and 5)
Full zebrafish to human ortholog mapping table. Orthologs with DIOPT Score < 2 were excluded. A DIOPT Score greater than 6 was considered sufficient for use in GSAA.
Video S1 (Related to Figure 1)
Time lapse confocal microscopy of ZMEL1 cells transplanted intravenously in larval zebrafish. Arrowhead indicates group of cells invading from the notochord (NC) and caudal hematopoietic tissue (CHT) into the tail fin mesenchyme (TF).
Video S2 (Related to Figure 2)
Time lapse microscopy of ZMEL1 cluster formation in low-bind plates over 3 days.
Video S3 (Related to Figure 2)
Time lapse microscopy of cluster formation of 1:1 mixture of ZMEL1-PRO and -INV in low-bind plates over 3 days.
Video S4 (Related to Figure 3)
Time lapse confocal microscopy following intravenous transplant of ZMEL1-PRO and -INV. A mixed cluster of both ZMEL1-PRO and -INV populations (11h:00m, arrowhead) extravasated from the caudal hematopoietic tissue (CHT) into the tail fin mesenchyme (TF)—with ZMEL1-INV leading (19h:00m, arrow) and ZMEL1-PRO following (23h:40m, arrow).
HIGHLIGHTS.
Cluster formation is a pro-metastatic phenotype associated with the INV state
PRO and INV cooperate in metastasis via heterotypic circulating tumor cell clusters
TFAP2 modulates PRO vs. INV state, clustering and metastasis
ACKNOWLEDGEMENTS
This work was supported by the NIH Research Program Grant R01CA229215 to J.B.X. and R.M.W. and the NIH Director’s New Innovator Award DP2CA186572, the Melanoma Research Alliance, The Pershing Square Sohn Cancer Research Alliance, The Mark Foundation for Cancer Research, The Alan and Sandra Gerry Metastasis Research Initiative at the Memorial Sloan Kettering Cancer Center, The Harry J. Lloyd Foundation, and the Starr Cancer Consortium (all to R.M.W.). N.R.C. was supported by the NIH Kirschstein-NRSA Predoctoral Fellowship F30CA220954, a Medical Student Research Grant from the Melanoma Research Foundation, and the NIH Medical Scientist Training Program Grant T32GM007739. C.K. was supported by a postdoctoral fellowship from the American Association for Anatomy. This work was also supported by the NIH Research Program Grant R01AR062547 to R.A.C. The Leeds Melanoma Cohort was funded by Cancer Research UK grants C588/A19167, C8216/A6129, and C588/A10721 and NIH grant R01CA83115. D.J.A. was supported by the Medical Research Council Dermatlas Project (MR/V000292/1). The AVAST-M melanoma trial was funded by Cancer Research UK grants C7535/A6408 and C2195/A8466. We thank the University Research Priority Program (URPP) of the University of Zurich for access to the melanoma biobank and early passage cultures. This work was also supported by funding awarded to the Aceto lab from the European Research Council (101001652), the Swiss National Science Foundation (PP00P3_190077), the Swiss Cancer League (KLS-4834-08-2019) and the ETH Zurich. M.K.S. is a H2020 Marie Skłodowska-Curie Actions (847012) and EMBO Long Term Fellow. E.M. was supported by the NIH Individual Predoctoral to Postdoctoral Fellow Transition Award K00CA223016. The results published here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. Portions of figures created with BioRender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
M.R.M. receives research funding from GRAIL. D.J.A. is a paid consultant for Microbiotica and receives researching funding from AstraZeneca and OpenTargets. M.P.L. receives research funding from Roche and Novartis. N.A. is a paid consultant for companies with an interest in liquid biopsy. R.M.W. is a paid consultant to N-of-One Therapeutics, a subsidiary of Qiagen. R.M.W. is on the Scientific Advisory Board of Consano, but receives no income for this. R.M.W. receives royalty payments for the use of the casper line from Carolina Biologicals.
REFERENCES
- Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H, et al. (2014). Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158, 1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ågren JA, Davies NG, and Foster KR (2019). Enforcement is central to the evolution of cooperation. Nature Ecology & Evolution 3, 1018–1029. [DOI] [PubMed] [Google Scholar]
- Archetti M, and Pienta KJ (2019). Cooperation among cancer cells: applying game theory to cancer. Nat Rev Cancer 19, 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod R, Axelrod DE, and Pienta KJ (2006). Evolution of cooperation among tumor cells. Proc Natl Acad Sci U S A 103, 13474–13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron M, Tagore M, Hunter MV, Kim IS, Moncada R, Yan Y, Campbell NR, White RM, and Yanai I (2020). The Stress-Like Cancer Cell State Is a Consistent Component of Tumorigenesis. Cell Systems 11, 536–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, Minn AJ, van de Vijver MJ, Gerald WL, Foekens JA, et al. (2009). Genes that mediate breast cancer metastasis to the brain. Nature 459, 1005–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagan RL, Zon LI, and White RM (2019). Modeling Cancer with Flies and Fish. Dev Cell 49, 317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network (2015). Genomic Classification of Cutaneous Melanoma. Cell 161, 1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira S, Goodall J, Denat L, Rodriguez M, Nuciforo P, Hoek KS, Testori A, Larue L, and Goding CR (2006). Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev 20, 3426–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celià-Terrassa T, Meca-Cortés Ó, Mateo F, Martínez de Paz A, Rubio N, Arnal-Estapé A, Ell BJ, Bermudo R, Díaz A, Guerra-Rebollo M, et al. (2012). Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. The Journal of Clinical Investigation 122, 1849–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceol CJ, Houvras Y, Jane-Valbuena J, Bilodeau S, Orlando DA, Battisti V, Fritsch L, Lin WM, Hollmann TJ, Ferré F, et al. (2011). The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature 471, 513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman A, Fernandez del Ama L, Ferguson J, Kamarashev J, Wellbrock C, and Hurlstone A (2014). Heterogeneous tumor subpopulations cooperate to drive invasion. Cell Rep 8, 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng PF, Shakhova O, Widmer DS, Eichhoff OM, Zingg D, Frommel SC, Belloni B, Raaijmakers MIG, Goldinger SM, Santoro R, et al. (2015). Methylation-dependent SOX9 expression mediates invasion in human melanoma cells and is a negative prognostic factor in advanced melanoma. Genome Biol 16, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KJ, Padmanaban V, Silvestri V, Schipper K, Cohen JD, Fairchild AN, Gorin MA, Verdone JE, Pienta KJ, Bader JS, et al. (2016). Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc Natl Acad Sci U S A 113, E854–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary AS, Leonard TL, Gestl SA, and Gunther EJ (2014). Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature 508, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrie PG, Marshall A, Nathan PD, Lorigan P, Gore M, Tahir S, Faust G, Kelly CG, Marples M, Danson SJ, et al. (2018). Adjuvant bevacizumab for melanoma patients at high risk of recurrence: survival analysis of the AVAST-M trial. Ann Oncol 29, 1843–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Croze N, Maczkowiak F, and Monsoro-Burq AH (2011). Reiterative AP2a activity controls sequential steps in the neural crest gene regulatory network. Proc Natl Acad Sci U S A 108, 155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca DS, Levin JZ, Sivachenko A, Fennell T, Nazaire MD, Williams C, Reich M, Winckler W, and Getz G (2012). RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics 28, 1530–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dissanayake SK, Wade M, Johnson CE, O'Connell MP, Leotlela PD, French AD, Shah KV, Hewitt KJ, Rosenthal DT, Indig FE, et al. (2007). The Wnt5A/protein kinase C pathway mediates motility in melanoma cells via the inhibition of metastasis suppressors and initiation of an epithelial to mesenchymal transition. J Biol Chem 282, 17259–17271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremova M, Vento-Tormo M, Teichmann SA, and Vento-Tormo R (2020). CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat Protoc 15, 1484–1506. [DOI] [PubMed] [Google Scholar]
- Eichhoff OM, Zipser MC, Xu M, Weeraratna AT, Mihic D, Dummer R, and Hoek KS (2010). The immunohistochemistry of invasive and proliferative phenotype switching in melanoma: a case report. Melanoma Res 20, 349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falletta P, Sanchez-Del-Campo L, Chauhan J, Effern M, Kenyon A, Kershaw CJ, Siddaway R, Lisle R, Freter R, Daniels MJ, et al. (2017). Translation reprogramming is an evolutionarily conserved driver of phenotypic plasticity and therapeutic resistance in melanoma. Genes Dev 31, 18–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fane ME, Chhabra Y, Smith AG, and Sturm RA (2019). BRN2, a POUerful driver of melanoma phenotype switching and metastasis. Pigment Cell Melanoma Res 32, 9–24. [DOI] [PubMed] [Google Scholar]
- Fidler IJ (1973). The relationship of embolic homogeneity, number, size and viability to the incidence of experimental metastasis. Eur J Cancer 9, 223–227. [DOI] [PubMed] [Google Scholar]
- Finger EC, Cheng CF, Williams TR, Rankin EB, Bedogni B, Tachiki L, Spong S, Giaccia AJ, and Powell MB (2014). CTGF is a therapeutic target for metastatic melanoma. Oncogene 33, 1093–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster KR (2011). The sociobiology of molecular systems. Nature Reviews Genetics 12, 193–203. [DOI] [PubMed] [Google Scholar]
- Foty RA, and Steinberg MS (2005). The differential adhesion hypothesis: a direct evaluation. Dev Biol 278, 255–263. [DOI] [PubMed] [Google Scholar]
- Frank NY, Schatton T, Kim S, Zhan Q, Wilson BJ, Ma J, Saab KR, Osherov V, Widlund HR, Gasser M, et al. (2011). VEGFR-1 expressed by malignant melanoma-initiating cells is required for tumor growth. Cancer Res 71, 1474–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg M, Couturier D-L, Nsengimana J, Fonseca NA, Wongchenko M, Yan Y, Lauss M, Jönsson GB, Newton-Bishop J, Parkinson C, et al. (2021). Tumour gene expression signature in primary melanoma predicts long-term outcomes. Nat Commun 12, 1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaynor R, Herschman HR, Irie R, Jones P, Morton D, and Cochran A (1981). S100 protein: a marker for human malignant melanomas? Lancet 1, 869–871. [DOI] [PubMed] [Google Scholar]
- Gershenwald J, Scolyer R, Hess K, and et al. (2017). Melanoma of the Skin. In AJCC Cancer Staging Manual: 8th Edition, Amin MB, ed. (Chicago: American Joint Committee on Cancer; ), p. 563. [Google Scholar]
- Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER, Barretina J, Gelfand ET, Bielski CM, Li H, et al. (2019). Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuliano M, Shaikh A, Lo HC, Arpino G, De Placido S, Zhang XH, Cristofanilli M, Schiff R, and Trivedi MV (2018). Perspective on Circulating Tumor Cell Clusters: Why It Takes a Village to Metastasize. Cancer Res 78, 845–852. [DOI] [PubMed] [Google Scholar]
- Gkountela S, Castro-Giner F, Szczerba BM, Vetter M, Landin J, Scherrer R, Krol I, Scheidmann MC, Beisel C, Stirnimann CU, et al. (2019). Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 176, 98–112 e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaves D (1983). Correlation between circulating cancer cells and incidence of metastases. Br J Cancer 48, 665–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelik R, and Gautreau A (2014). Quantitative and unbiased analysis of directional persistence in cell migration. Nat Protocols 9, 1931–1943. [DOI] [PubMed] [Google Scholar]
- Gupta PB, Kuperwasser C, Brunet JP, Ramaswamy S, Kuo WL, Gray JW, Naber SP, and Weinberg RA (2005). The melanocyte differentiation program predisposes to metastasis after neoplastic transformation. Nat Genet 37, 1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallberg AR, Vorrink SU, Hudachek DR, Cramer-Morales K, Milhem MM, Cornell RA, and Domann FE (2014). Aberrant CpG methylation of the TFAP2A gene constitutes a mechanism for loss of TFAP2A expression in human metastatic melanoma. Epigenetics 9, 1641–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmeyer KM, Facompre ND, Herlyn M, and Basu D (2017). JARID1 Histone Demethylases: Emerging Targets in Cancer. Trends Cancer 3, 713–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzikirou H, Basanta D, Simon M, Schaller K, and Deutsch A (2010). ‘Go or Grow’: the key to the emergence of invasion in tumour progression? Mathematical Medicine and Biology: A Journal of the IMA 29, 49–65. [DOI] [PubMed] [Google Scholar]
- Hauser M, McAuliffe K, and Blake PR (2009). Evolving the ingredients for reciprocity and spite. Philos Trans R Soc Lond B Biol Sci 364, 3255–3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilmann S, Ratnakumar K, Langdon E, Kansler E, Kim I, Campbell NR, Perry E, McMahon A, Kaufman C, van Rooijen E, et al. (2015). A quantitative system for studying metastasis using transparent zebrafish. Cancer Res 75, 4272–4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinohara K, and Polyak K (2019). Intratumoral Heterogeneity: More Than Just Mutations. Trends Cell Biol 29, 569–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek KS, Eichhoff OM, Schlegel NC, Dobbeling U, Kobert N, Schaerer L, Hemmi S, and Dummer R (2008). In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res 68, 650–656. [DOI] [PubMed] [Google Scholar]
- Hoek KS, and Goding CR (2010). Cancer stem cells versus phenotype-switching in melanoma. Pigment Cell Melanoma Res 23, 746–759. [DOI] [PubMed] [Google Scholar]
- Hoek KS, Schlegel NC, Brafford P, Sucker A, Ugurel S, Kumar R, Weber BL, Nathanson KL, Phillips DJ, Herlyn M, et al. (2006). Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res 19, 290–302. [DOI] [PubMed] [Google Scholar]
- Hoffman TL, Javier AL, Campeau SA, Knight RD, and Schilling TF (2007). Tfap2 transcription factors in zebrafish neural crest development and ectodermal evolution. J Exp Zool B Mol Dev Evol 308, 679–691. [DOI] [PubMed] [Google Scholar]
- Houghton AN, Real FX, Davis LJ, Cordon-Cardo C, and Old LJ (1987). Phenotypic heterogeneity of melanoma. Relation to the differentiation program of melanoma cells. J Exp Med 165, 812–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Flockhart I, Vinayagam A, Bergwitz C, Berger B, Perrimon N, and Mohr SE (2011). An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics 12, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Jean D, Luca M, Tainsky MA, and Bar-Eli M (1998). Loss of AP-2 results in downregulation of c-KIT and enhancement of melanoma tumorigenicity and metastasis. EMBO J 17, 4358–4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogai S, Horiguchi M, and Weinstein BM (2001). The vascular anatomy of the developing zebrafish: an atlas of embryonic and early larval development. Dev Biol 230, 278–301. [DOI] [PubMed] [Google Scholar]
- Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, Leeson R, Kanodia A, Mei S, Lin JR, et al. (2018). A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175, 984–997 e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman CK, Mosimann C, Fan ZP, Yang S, Thomas AJ, Ablain J, Tan JL, Fogley RD, van Rooijen E, Hagedorn EJ, et al. (2016). A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science 351, aad2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, van der Lee R, Bessy A, Chèneby J, Kulkarni SR, Tan G, et al. (2017). JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res 46, D260–D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoja L, Shenjere P, Hodgson C, Hodgetts J, Clack G, Hughes A, Lorigan P, and Dive C (2014). Prevalence and heterogeneity of circulating tumour cells in metastatic cutaneous melanoma. Melanoma Res 24, 40–46. [DOI] [PubMed] [Google Scholar]
- Kim IS, Heilmann S, Kansler ER, Zhang Y, Zimmer M, Ratnakumar K, Bowman RL, Simon-Vermot T, Fennell M, Garippa R, et al. (2017). Microenvironment-derived factors driving metastatic plasticity in melanoma. Nat Commun 8, 14343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A, Frederick DT, Barzily-Rokni M, Straussman R, Haq R, et al. (2014). A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov 4, 816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolev KS, Xavier JB, and Gore J (2014). Turning ecology and evolution against cancer. Nat Rev Cancer 14, 371–380. [DOI] [PubMed] [Google Scholar]
- Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, Parant JM, Yost HJ, Kanki JP, and Chien CB (2007). The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn 236, 3088–3099. [DOI] [PubMed] [Google Scholar]
- Labun K, Montague TG, Krause M, Torres Cleuren YN, Tjeldnes H, and Valen E (2019). CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res 47, W171–W174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauden L, Siewiera J, Boukouaci W, Ramgolam K, Mourah S, Lebbe C, Charron D, Aoudjit F, Jabrane-Ferrat N, and Al-Daccak R (2014). TGF-beta-induced (TGFBI) protein in melanoma: a signature of high metastatic potential. J Invest Dermatol 134, 1675–1685. [DOI] [PubMed] [Google Scholar]
- Laughney AM, Hu J, Campbell NR, Bakhoum SF, Setty M, Lavallée V-P, Xie Y, Masilonis I, Carr AJ, Allaj V, et al. (2020). Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat Med 26, 259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FZ, Dhillon AS, Anderson RL, McArthur G, and Ferrao PT (2015). Phenotype switching in melanoma: implications for progression and therapy. Front Oncol 5, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, and Cornell RA (2007). Redundant activities of Tfap2a and Tfap2c are required for neural crest induction and development of other non-neural ectoderm derivatives in zebrafish embryos. Dev Biol 304, 338–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liotta LA, Saidel MG, and Kleinerman J (1976). The significance of hematogenous tumor cell clumps in the metastatic process. Cancer Res 36, 889–894. [PubMed] [Google Scholar]
- Lissanu Deribe Y (2016). Interplay between PREX2 mutations and the PI3K pathway and its effect on epigenetic regulation of gene expression in NRAS-mutant melanoma. Small GTPases 7, 178–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Taftaf R, Kawaguchi M, Chang YF, Chen W, Entenberg D, Zhang Y, Gerratana L, Huang S, Patel DB, et al. (2018). Homophilic CD44 interactions mediate tumor cell aggregation and polyclonal metastasis in patient-derived breast cancer models. Cancer Discov 9, 96–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long E, Ilie M, Bence C, Butori C, Selva E, Lalvee S, Bonnetaud C, Poissonnet G, Lacour JP, Bahadoran P, et al. (2016). High expression of TRF2, SOX10, and CD10 in circulating tumor microemboli detected in metastatic melanoma patients. A potential impact for the assessment of disease aggressiveness. Cancer Med 5, 1022–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo T, Matsuo-Takasaki M, Thomas ML, Weeks DL, and Sargent TD (2002). Transcription factor AP-2 is an essential and direct regulator of epidermal development in Xenopus. Dev Biol 245, 136–144. [DOI] [PubMed] [Google Scholar]
- Luo X, Mitra D, Sullivan RJ, Wittner BS, Kimura AM, Pan S, Hoang MP, Brannigan BW, Lawrence DP, Flaherty KT, et al. (2014). Isolation and molecular characterization of circulating melanoma cells. Cell Rep 7, 645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddipati R, and Stanger BZ (2015). Pancreatic Cancer Metastases Harbor Evidence of Polyclonality. Cancer Discov 5, 1086–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malladi S, Macalinao, Danilo G, Jin X, He L, Basnet H, Zou Y, de Stanchina E, and Massagué J (2016). Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 165, 45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe'er J, Trent JM, Meltzer PS, and Hendrix MJ (1999). Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol 155, 739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marjanovic ND, Hofree M, Chan JE, Canner D, Wu K, Trakala M, Hartmann GG, Smith OC, Kim JY, Evans KV, et al. (2020). Emergence of a High-Plasticity Cell State during Lung Cancer Evolution. Cancer Cell 38, 229–246.e213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marusyk A, Tabassum DP, Altrock PM, Almendro V, Michor F, and Polyak K (2014). Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature 514, 54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matus DQ, Lohmer LL, Kelley LC, Schindler AJ, Kohrman AQ, Barkoulas M, Zhang W, Chi Q, and Sherwood DR (2015). Invasive Cell Fate Requires G1 Cell-Cycle Arrest and Histone Deacetylase-Mediated Changes in Gene Expression. Dev Cell 35, 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayhew E, and Glaves D (1984). Quantitation of tumorigenic disseminating and arrested cancer cells. Br J Cancer 50, 159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes L, Healy J, and Melville J (2018). UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv, eprint 1802.03426. [Google Scholar]
- Meers MP, Tenenbaum D, and Henikoff S (2019). Peak calling by Sparse Enrichment Analysis for CUT&RUN chromatin profiling. Epigenetics Chromatin 12, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, and Massague J (2005). Genes that mediate breast cancer metastasis to lung. Nature 436, 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrozik KM, Blaschuk OW, Cheong CM, Zannettino ACW, and Vandyke K (2018). N-cadherin in cancer metastasis, its emerging role in haematological malignancies and potential as a therapeutic target in cancer. BMC Cancer 18, 939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller J, Krijgsman O, Tsoi J, Robert L, Hugo W, Song C, Kong X, Possik PA, Cornelissen-Steijger PD, Geukes Foppen MH, et al. (2014). Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun 5, 5712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelakantan D, Zhou H, Oliphant MUJ, Zhang X, Simon LM, Henke DM, Shaw CA, Wu MF, Hilsenbeck SG, White LD, et al. (2017). EMT cells increase breast cancer metastasis via paracrine GLI activation in neighbouring tumour cells. Nat Commun 8, 15773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nsengimana J, Laye J, Filia A, O'Shea S, Muralidhar S, Pozniak J, Droop A, Chan M, Walker C, Parkinson L, et al. (2018). beta-Catenin-mediated immune evasion pathway frequently operates in primary cutaneous melanomas. J Clin Invest 128, 2048–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanaban V, Krol I, Suhail Y, Szczerba BM, Aceto N, Bader JS, and Ewald AJ (2019). E-cadherin is required for metastasis in multiple models of breast cancer. Nature 573, 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton EE, Widlund HR, Kutok JL, Kopani KR, Amatruda JF, Murphey RD, Berghmans S, Mayhall EA, Traver D, Fletcher CD, et al. (2005). BRAF mutations are sufficient to promote nevi formation and cooperate with p53 in the genesis of melanoma. Current biology : CB 15, 249–254. [DOI] [PubMed] [Google Scholar]
- Pearson GW (2019). Control of Invasion by Epithelial-to-Mesenchymal Transition Programs during Metastasis. J Clin Med 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pencheva N, Tran H, Buss C, Huh D, Drobnjak M, Busam K, and Tavazoie SF (2012). Convergent multi-miRNA targeting of ApoE drives LRP1/LRP8-dependent melanoma metastasis and angiogenesis. Cell 151, 1068–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez AR, Pritykin Y, Vidigal JA, Chhangawala S, Zamparo L, Leslie CS, and Ventura A (2017). GuideScan software for improved single and paired CRISPR guide RNA design. Nat Biotechnol 35, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinner S, Jordan P, Sharrock K, Bazley L, Collinson L, Marais R, Bonvin E, Goding C, and Sahai E (2009). Intravital imaging reveals transient changes in pigment production and Brn2 expression during metastatic melanoma dissemination. Cancer Res 69, 7969–7977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raaijmakers MIG, Widmer DS, Maudrich M, Koch T, Langer A, Flace A, Schnyder C, Dummer R, and Levesque MP (2015). A new live-cell biobank workflow efficiently recovers heterogeneous melanoma cells from native biopsies. Exp Dermatol 24, 377–380. [DOI] [PubMed] [Google Scholar]
- Rabbie R, Ansari-Pour N, Cast O, Lau D, Scott F, Welsh SJ, Parkinson C, Khoja L, Moore L, Tullett M, et al. (2020). Multi-site clonality analysis uncovers pervasive heterogeneity across melanoma metastases. Nat Commun 11, 4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambow F, Job B, Petit V, Gesbert F, Delmas V, Seberg H, Meurice G, Van Otterloo E, Dessen P, Robert C, et al. (2015). New Functional Signatures for Understanding Melanoma Biology from Tumor Cell Lineage-Specific Analysis. Cell Reports 13, 840–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambow F, Marine JC, and Goding CR (2019). Melanoma plasticity and phenotypic diversity: therapeutic barriers and opportunities. Genes Dev 33, 1295–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambow F, Rogiers A, Marin-Bejar O, Aibar S, Femel J, Dewaele M, Karras P, Brown D, Chang YH, Debiec-Rychter M, et al. (2018). Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 174, 843–855 e819. [DOI] [PubMed] [Google Scholar]
- Reichert M, Bakir B, Moreira L, Pitarresi JR, Feldmann K, Simon L, Suzuki K, Maddipati R, Rhim AD, Schlitter AM, et al. (2018). Regulation of Epithelial Plasticity Determines Metastatic Organotropism in Pancreatic Cancer. Dev Cell 45, 696–711 e698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, and Liao WS (2001). Transcription factor AP-2 functions as a repressor that contributes to the liver-specific expression of serum amyloid A1 gene. J Biol Chem 276, 17770–17778. [DOI] [PubMed] [Google Scholar]
- Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, Basu D, Gimotty P, Vogt T, and Herlyn M (2010). A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 141, 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowling EJ, Miskolczi Z, Nagaraju R, Wilcock DJ, Wang P, Telfer B, Li Y, Lasheras-Otero I, Redondo-Munoz M, Sharrocks AD, et al. (2020). Cooperative behaviour and phenotype plasticity evolve during melanoma progression. Pigment Cell Melanoma Res 33, 695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz C, Li J, Luttgen MS, Kolatkar A, Kendall JT, Flores E, Topp Z, Samlowski WE, McClay E, Bethel K, et al. (2015). Limited genomic heterogeneity of circulating melanoma cells in advanced stage patients. Phys Biol 12, 016008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanborn JZ, Chung J, Purdom E, Wang NJ, Kakavand H, Wilmott JS, Butler T, Thompson JF, Mann GJ, Haydu LE, et al. (2015). Phylogenetic analyses of melanoma reveal complex patterns of metastatic dissemination. Proc Natl Acad Sci U S A 112, 10995–11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarioglu AF, Aceto N, Kojic N, Donaldson MC, Zeinali M, Hamza B, Engstrom A, Zhu H, Sundaresan TK, Miyamoto DT, et al. (2015). A microfluidic device for label-free, physical capture of circulating tumor cell clusters. Nat Methods 12, 685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnegg CI, Yang MH, Ghosh SK, and Hsu MY (2015). Induction of Vasculogenic Mimicry Overrides VEGF-A Silencing and Enriches Stem-like Cancer Cells in Melanoma. Cancer Res 75, 1682–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seberg HE, Van Otterloo E, and Cornell RA (2017a). Beyond MITF: Multiple transcription factors directly regulate the cellular phenotype in melanocytes and melanoma. Pigment Cell Melanoma Res 30, 454–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seberg HE, Van Otterloo E, Loftus SK, Liu H, Bonde G, Sompallae R, Gildea DE, Santana JF, Manak JR, Pavan WJ, et al. (2017b). TFAP2 paralogs regulate melanocyte differentiation in parallel with MITF. PLoS Genet 13, e1006636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakhova O, Cheng P, Mishra PJ, Zingg D, Schaefer SM, Debbache J, Häusel J, Matter C, Guo T, Davis S, et al. (2015). Antagonistic Cross-Regulation between Sox9 and Sox10 Controls an Anti-tumorigenic Program in Melanoma. PLoS Genet 11, e1004877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, and Henikoff S (2017). An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeets B, Alert R, Pešek J, Pagonabarraga I, Ramon H, and Vincent R (2016). Emergent structures and dynamics of cell colonies by contact inhibition of locomotion. Proceedings of the National Academy of Sciences 113, 14621–14626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit M, and Detmar M (2003). Angiogenesis, lymphangiogenesis, and melanoma metastasis. Oncogene 22, 3172–3179. [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM III, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczerba BM, Castro-Giner F, Vetter M, Krol I, Gkountela S, Landin J, Scheidmann MC, Donato C, Scherrer R, Singer J, et al. (2019). Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 566, 553–557. [DOI] [PubMed] [Google Scholar]
- Tellez CS, Davis DW, Prieto VG, Gershenwald JE, Johnson MM, McCarty MF, and Bar-Eli M (2007). Quantitative analysis of melanocytic tissue array reveals inverse correlation between activator protein-2alpha and protease-activated receptor-1 expression during melanoma progression. J Invest Dermatol 127, 387–393. [DOI] [PubMed] [Google Scholar]
- Therneau TM (2020). A Package for Survival Analysis in R.
- Therneau TM, and Grambsch PM (2000). Modeling Survival Data: Extending the Cox Model (New York: Springer; ). [Google Scholar]
- Thompson JF, Soong SJ, Balch CM, Gershenwald JE, Ding S, Coit DG, Flaherty KT, Gimotty PA, Johnson T, Johnson MM, et al. (2011). Prognostic significance of mitotic rate in localized primary cutaneous melanoma: an analysis of patients in the multi-institutional American Joint Committee on Cancer melanoma staging database. J Clin Oncol 29, 2199–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, Wong DJL, Atefi M, Shirazi R, Wang X, et al. (2018). Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 33, 890–904 e895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji T, Ibaragi S, and Hu GF (2009). Epithelial-mesenchymal transition and cell cooperativity in metastasis. Cancer Res 69, 7135–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuncer E, Calçada RR, Zingg D, Varum S, Cheng P, Freiberger SN, Deng C-X, Kleiter I, Levesque MP, Dummer R, et al. (2019). SMAD signaling promotes melanoma metastasis independently of phenotype switching. The Journal of Clinical Investigation 129, 2702–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Otterloo E, Li W, Bonde G, Day KM, Hsu MY, and Cornell RA (2010). Differentiation of zebrafish melanophores depends on transcription factors AP2 alpha and AP2 epsilon. PLoS Genet 6, e1001122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandamme N, and Berx G (2014). Melanoma cells revive an embryonic transcriptional network to dictate phenotypic heterogeneity. Front Oncol 4, 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verfaillie A, Imrichova H, Atak ZK, Dewaele M, Rambow F, Hulselmans G, Christiaens V, Svetlichnyy D, Luciani F, Van den Mooter L, et al. (2015). Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat Commun 6, 6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, and Sabatini DM (2015). Identification and characterization of essential genes in the human genome. Science 350, 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S (1954). The metastasizability of tumor cells. Cancer 7, 215–223. [DOI] [PubMed] [Google Scholar]
- Weeraratna AT, Jiang Y, Hostetter G, Rosenblatt K, Duray P, Bittner M, and Trent JM (2002). Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell 1, 279–288. [DOI] [PubMed] [Google Scholar]
- Weiss JM, Hunter MV, Tagore M, Ma Y, Misale S, Simon-Vermot T, Campbell NR, Newell F, Wilmott JS, Johansson PA, et al. (2020). Anatomic position determines oncogenic specificity in melanoma. bioRxiv, 2020.2011.2014.383083. [Google Scholar]
- White RM, Cech J, Ratanasirintrawoot S, Lin CY, Rahl PB, Burke CJ, Langdon E, Tomlinson ML, Mosher J, Kaufman C, et al. (2011). DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature 471, 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RM, Sessa A, Burke C, Bowman T, LeBlanc J, Ceol C, Bourque C, Dovey M, Goessling W, Burns CE, et al. (2008). Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell 2, 183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmer DS, Cheng PF, Eichhoff OM, Belloni BC, Zipser MC, Schlegel NC, Javelaud D, Mauviel A, Dummer R, and Hoek KS (2012). Systematic classification of melanoma cells by phenotype-specific gene expression mapping. Pigment Cell Melanoma Res 25, 343–353. [DOI] [PubMed] [Google Scholar]
- Wouters J, Kalender-Atak Z, Minnoye L, Spanier KI, De Waegeneer M, Bravo González-Blas C, Mauduit D, Davie K, Hulselmans G, Najem A, et al. (2020). Robust gene expression programs underlie recurrent cell states and phenotype switching in melanoma. Nat Cell Biol 22, 986–998. [DOI] [PubMed] [Google Scholar]
- Xiong Q, Ancona N, Hauser ER, Mukherjee S, and Furey TS (2012). Integrating genetic and gene expression evidence into genome-wide association analysis of gene sets. Genome Res 22, 386–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Q, Mukherjee S, and Furey TS (2014). GSAASeqSP: A Toolset for Gene Set Association Analysis of RNA-Seq Data. Scientific Reports 4, 6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong T.-f., Pan F.-q., and Li D (2019). Expression and clinical significance of S100 family genes in patients with melanoma. Melanoma Res 29, 23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Mao X, Imrali A, Syed F, Mutsvangwa K, Berney D, Cathcart P, Hines J, Shamash J, and Lu YJ (2015). Optimization and Evaluation of a Novel Size Based Circulating Tumor Cell Isolation System. PLoS ONE 10, e0138032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Zong Y, Gao Y, Sun X, Zhao H, Luo W, and Jia S (2019). VEGF Induce Vasculogenic Mimicry of Choroidal Melanoma through the PI3k Signal Pathway. Biomed Res Int 2019, 3909102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang L-G, and He Q-Y (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Zeng L, Jarrett C, Brown K, Gillespie KM, Holly JM, and Perks CM (2013). Insulin-like growth factor binding protein-3 (IGFBP-3) plays a role in the anti-tumorigenic effects of 5-Aza-2'-deoxycytidine (AZA) in breast cancer cells. Exp Cell Res 319, 2282–2295. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen F, Fonseca NA, He Y, Fujita M, Nakagawa H, Zhang Z, Brazma A, Group PTW, Group PSVW, et al. (2020). High-coverage whole-genome analysis of 1220 cancers reveals hundreds of genes deregulated by rearrangement-mediated cis-regulatory alterations. Nat Commun 11, 736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YM, Zimmer MA, Guardia T, Callahan SJ, Mondal C, Di Martino J, Takagi T, Fennell M, Garippa R, Campbell NR, et al. (2018). Distant Insulin Signaling Regulates Vertebrate Pigmentation through the Sheddase Bace2. Dev Cell 45, 580–594.e587. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 (Related to Figures 1-2)
ZMEL1-INV vs. -PRO RNA-seq results. Filtered differential expression (DE; absolute log2 fold change ≥ 1.5, padj < 0.05); all DE, log normalized counts, and raw counts.
Table S2 (Related to Figure 4)
ZMEL1 3D vs. 2D culture RNA-seq results for ZMEL1-PRO and ZMEL1-INV. Filtered differential expression (DE; absolute log2 fold change ≥ 1.5, padj < 0.05); all DE, log normalized counts, and raw counts.
Table S3 (Related to Figure 4)
Annotation of top HOMER motif for 1000bp region (transcription start site ± 500bp) associated with genes upregulated in ZMEL1-PRO and with genes differentially expressed in 3D vs. 2D culture for each ZMEL1-PRO and ZMEL1-INV. Annotations presented for genes differentially expressed in ZMEL1-PRO and -INV and for top motif for each ZMEL1-PRO and ZMEL1-INV.
Table S4 (Related to Figure 5)
ZMEL1-PRO sg_tfap2a/e vs. sg_scr RNA-seq results. Filtered differential expression (DE; absolute log2 fold change ≥ 0.5, padj < 0.05); all DE, log normalized counts, and raw counts.
Table S5 (Related to Figures 1, 2, and 5)
List of all gene sets used for GSAA, including sources and full gene lists. Results from GSAA analyses. Results table and details for gene sets with false discovery rate (FDR) below 0.05 (up to 6 per analysis).
Table S6 (Related to Figures 1, 2, and 5)
Full zebrafish to human ortholog mapping table. Orthologs with DIOPT Score < 2 were excluded. A DIOPT Score greater than 6 was considered sufficient for use in GSAA.
Video S1 (Related to Figure 1)
Time lapse confocal microscopy of ZMEL1 cells transplanted intravenously in larval zebrafish. Arrowhead indicates group of cells invading from the notochord (NC) and caudal hematopoietic tissue (CHT) into the tail fin mesenchyme (TF).
Video S2 (Related to Figure 2)
Time lapse microscopy of ZMEL1 cluster formation in low-bind plates over 3 days.
Video S3 (Related to Figure 2)
Time lapse microscopy of cluster formation of 1:1 mixture of ZMEL1-PRO and -INV in low-bind plates over 3 days.
Video S4 (Related to Figure 3)
Time lapse confocal microscopy following intravenous transplant of ZMEL1-PRO and -INV. A mixed cluster of both ZMEL1-PRO and -INV populations (11h:00m, arrowhead) extravasated from the caudal hematopoietic tissue (CHT) into the tail fin mesenchyme (TF)—with ZMEL1-INV leading (19h:00m, arrow) and ZMEL1-PRO following (23h:40m, arrow).
Data Availability Statement
All RNA-seq data generated in this study have been deposited at NCBI GEO (GSE151679), with bulk RNA-seq counts and differential expression tables in the Supplemental Information. TFAP2A CUT&RUN data are available via the NCBI GEO repository (GSE153020). This paper analyzes existing, publicly available data with accession numbers listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-hs_CDH1 | BD | Cat # 610181 |
| Goat anti-dr_Tfap2a | LifeSpan Biosciences | Cat # LS-C87212 |
| Rabbit anti-dr_Tfap2e | Fisher | Cat # PA5-72631 |
| Rabbit anti-hs_TFAP2A | Cell Signaling | Cat # 3215 |
| Rabbit anti-hs_cyclophilin B | Fisher | Cat # PA1-027A |
| Anti-mouse Alexa Fluor® 488 | Cell Signaling | Cat # 4408S |
| Anti-rabbit Alexa Fluor® 594 | Cell Signaling | Cat # 8889S |
| Mouse anti-hs_H3 | Cell Signaling | Cat # 14269 |
| Rabbit anti-TFAP2A | Abcam | Cat # ab108311 |
| Rabbit anti-Sox9 | Sigma | Cat # HPA001758 |
| Mouse anti-TFAP2A | Santa Cruz Biotechnology | Cat # sc-12726 |
| Rat anti-CD45 | ThermoFisher | Cat # MA5-17687 |
| Donkey anti-mouse AF488 | ThermoFisher | Cat # A21202 |
| Donkey anti-rabbit AF647 | ThermoFisher | Cat # A31573 |
| Donkey anti-rat DyLight 555 | ThermoFisher | Cat # SA5-10027 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Human metastatic melanoma patient blood samples | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Tricaine-S | Syndel | Cat # MS-222 |
| Hoechst 33342 | Fisher | Cat # H1399 |
| poly-D-lysine | Sigma | Cat # P-0899 |
| Rabbit IgG | Millipore | Cat # 12-370 |
| Concanavalin A-coated magnetic beads | Bang Laboratories, Inc | Cat # BP531 |
| pA-MNase | Dr. Steve Henikoff | N/A |
| Liberase TL | Sigma | Cat # 5401020001 |
| Alt-R® S.p. Cas9 Nuclease V3 | Integrated DNA Technologies | Cat # 1081059 |
| Alt-R® CRISPR-Cas9 tracrRNA, ATTO™ 550 | Integrated DNA Technologies | Cat # 1075928 |
| Cas9 Electroporation Enhancer | Integrated DNA Technologies | Cat # 1075916 |
| Alt-R® CRISPR-Cas9 crRNA (see Oligonucleotides below for sequences) | Integrated DNA Technologies | N/A |
| Neon Transfection System 100 μL Kit | Fisher | Cat # MPK10025 |
| Critical commercial assays | ||
| Neon Transfection System | Fisher | Cat # MPK5000 |
| Ultra-Low Attachment Surface 96-well plate, flat well | Corning | Cat # 3474 |
| Ultra-Low Attachment Surface 96-well plate, round well | Corning | Cat # 7007 |
| Transwell assay: 3.0μm pore insert / 24-well plate | Corning | Cat # 353492 / 353504 |
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat # G7572 |
| Caspase-Glo® 3/7 Assay System | Promega | Cat # G8091 |
| RNeasy Plus Mini kit | Qiagen | Cat # 74034 |
| QiaShredder | Qiagen | Cat # 79656 |
| Parsortix Cell Separation Cassette | ANGLE (Xu et al., 2015) | Cat # GEN3D6.5 |
| Chromium Single Cell 3’ Library & Gel Bead Kit v3.1 | 10X Genomics | Cat # 1000128 |
| Chromium Single Cell 3’ Chip G | 10X Genomics | Cat # 1000127 |
| Deposited data | ||
| Bulk and single-cell RNA-seq data | This paper | GEO # GSE151679 |
| TFAP2A CUT&RUN data | This paper | GEO # GSE153020 |
| Cancer Cell Line Encyclopedia (CCLE) RNA-seq | Broad CCLE (https://portals.broadinstitute.org/ccle) | RSEM genes TPM, version 20180929 |
| The Cancer Genome Atlas (TCGA) Skin Cutaneous Melanoma (SKCM) mRNA expression | Broad GDAC Firehose (http://firebrowse.org/) | v2 RSEM genes normalized, version 2016_01_28 |
| Human melanoma single-cell RNA-seq | (Jerby-Arnon et al., 2018) | GEO # GSE115978 |
| Broad Institute Dependency Map (DepMap) | Broad Institute (https://depmap.org/portal/) | CRISPR (Avana) Public 19Q3 dataset |
| AVAST-M melanoma cohort | (Corrie et al., 2018; Garg et al., 2021) | EGA # EGAD00001006401 |
| Leeds Melanoma Cohort | (Nsengimana et al., 2018) | EGA # EGAS00001002922 |
| Experimental models: Cell lines | ||
| ZMEL1-PRO (mitfa: EGFP) | This paper | N/A |
| ZMEL1-PRO (mitfa: tdTomato) | This paper | N/A |
| ZMEL1-PRO (mitfa: EGFP & Ub: nls-mCherry) | This paper | N/A |
| ZMEL1-INV (mitfa: EGFP) | This paper | N/A |
| ZMEL1-INV (mitfa: tdTomato) | This paper | N/A |
| ZMEL1-INV (mitfa: EGFP & Ub: nls-mCherry) | This paper | N/A |
| MEWO | ATCC | Cat # HTB-65 |
| SH4 | ATCC | Cat # CRL-7724 |
| SKMEL28 | ATCC | Cat # HTB-72 |
| A375 | ATCC | Cat # CRL-1619 |
| HS294T | ATCC | Cat # HTB-140 |
| A2058 | ATCC | Cat # CRL-11147 |
| RPMI7951 | ATCC | Cat # HTB-66 |
| HMCB | ATCC | Cat # CRL-9607 |
| HS895T | ATCC | Cat # CRL-7637 |
| M000921 | (Raaijmakers et al., 2015) | N/A |
| M111031 | (Raaijmakers et al., 2015) | N/A |
| M990922 | (Raaijmakers et al., 2015) | N/A |
| MM121224 | (Raaijmakers et al., 2015) | N/A |
| Experimental models: Organisms/strains | ||
| Casper zebrafish | White Lab | N/A |
| Casper;kdrl-RFP zebrafish | This paper | N/A |
| Oligonucleotides | ||
| sg_scr Alt-R crRNA | (Wang, T., et al., Science, 2015) | AACCTACGGGCTACGATACG |
| sg_cdh1_1 Alt-R crRNA | This paper | AGTGGCAAAAGACTAGGCAA |
| sg_cdh1_2 Alt-R crRNA | This paper | TCATGCAAACGGAGTGGACG |
| sg_tfap2a_1 Alt-R crRNA | This paper | TTGTGGACGTGAATTCCCCA |
| sg_tfap2a_2 Alt-R crRNA | This paper | CATGTATTCAGCTATCGCCT |
| sg_tfap2a_3 Alt-R crRNA | This paper | ATAGTTGATGCGGTAACCCG |
| sg_tfap2a_4 Alt-R crRNA | This paper | CTCAACCACAACACCCGGGA |
| sg_tfap2e_1 Alt-R crRNA | This paper | GGCAGGGGTCCCGAATCCGT |
| sg_tfap2e_2 Alt-R crRNA | This paper | TACTCCCAGAGCCAGGACGG |
| sg_tfap2e_3 Alt-R crRNA | This paper | TGAGACTGAGTTCCCTGCGC |
| sg_tfap2e_4 Alt-R crRNA | This paper | TGCACCGCGTGCATCCCCAG |
| Recombinant DNA | ||
| Ub: nls-mCherry | This paper | N/A |
| Mitfa: tdTomato | This paper | N/A |
| Software and algorithms | ||
| Zebrafish image analysis pipeline | (Heilmann et al., 2015) | https://github.com/SiljaHeilmann/Matlab-fish-image-analysis |
| Volocity v6.3 | PerkinElmer | https://www.perkinelmer.com |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| IDL tracking methods | John Crocker, David Grier, and Eric Weeks | http://site.physics.georgetown.edu/matlab/ |
| FASTQC | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| TRIMMOMATIC | (Bolger et al., 2014) | http://www.usadellab.org/cms/?page=trimmomatic |
| STAR | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| SeQC | (DeLuca et al., 2012) | https://bioconductor.org/packages/release/data/experiment/html/seqc.html |
| DESeq2 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| GSAA | (Xiong et al., 2012; Xiong et al., 2014) | http://gsaa.unc.edu/ |
| DIOPT | (Hu et al., 2011) | https://www.flyrnai.org/diopt |
| HOMER | (Heinz et al., 2010) | http://homer.ucsd.edu/homer |
| JASPAR | (Khan et al., 2017) | http://jaspar.genereg.net |
| Trim Galore version 0.6.3 | Developed by Felix Krueger at the Babraham Institute | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore |
| Bowtie2 version 2.1.0 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| ChIPseeker version 1.18.0 | (Yu et al., 2015) | https://bioconductor.org/packages/release/bioc/html/ChIPseeker.html |
| CellRanger version 3.1.0 | 10X Genomics | https://www.10xgenomics.com/ |
| Seurat version 3.1.4 | (Stuart et al., 2019) | https://satijalab.org/seurat/index.html |
| CellPhoneDB | (Efremova et al., 2020) | N/A |
| MATLAB | Mathworks | https://www.mathworks.com/products/matlab.html |
| R | R Foundation for Statistical Computing | https://www.r-project.org/ |
| Zen | Zeiss | https://www.zeiss.com |
| STATA v14 | StataCorp | https://www.stata.com/stata14/ |
| Other | ||
A MATLAB-based image analysis pipeline for quantifying melanoma in zebrafish was previously published (Heilmann et al., 2015).
All other data reported in this paper will be shared by the lead contact upon request.