

Abstract
Due to the limitations of existing medications, there is a critical need for new drugs to treat visceral leishmaniasis. Since arylimidamides and antifungal azoles both show oral activity in murine visceral leishmaniasis models, a molecular hybridization approach was employed where arylimidamide and azole groups were separated by phenoxyalkyl linkers in an attempt to capitalize on the favorable antileishmanial properties of both series. Among the target compounds synthesized, greater antileishmanial potency against intracellular Leishmania donovani was observed as the linker length increased from two to eight carbons and when an imidazole ring was employed as the terminal group compared to a 1,2,4-triazole group. Compound 24c (N-(4-((8-(1H-imidazol-1-yl)octyl)oxy)-2-isopropoxyphenyl) picolinimidamide) displayed activity against L. donovani intracellular amastigotes with an IC50 value of 0.53 μM. When tested in a murine visceral leishmaniasis model, compound 24c at a dose of 75 mg/kg/day p.o. for five consecutive days resulted in a modest 33% decrease in liver parasitemia compared to the control group, indicating that further optimization of these molecules is needed. While potent hybrid compounds bearing an imidazole terminal group were also strong inhibitors of recombinant CYP51 from L. donovani as assessed by a fluorescence-based assay, additional targets are likely to play an important role in the antileishmanial action of these compounds.
Keywords: Leishmaniasis, azole antifungal, arylimidamide, CYP51, molecular hybridization
Leishmaniasis continues to be an important public health problem. According to recent World Health Organization estimates, approximately 30,000 and over one million new cases of visceral leishmaniasis and cutaneous leishmaniasis occur each year, respectively, and over 1 billion people live in areas where leishmaniasis occurs.1 The visceral form of leishmaniasis is the most serious; without drug treatment, symptomatic visceral leishmaniasis is usually fatal. Treatment of visceral leishmaniasis generally involves the use of pentavalent antimonials, amphotericin B, paromomycin, miltefosine, or combinations of two of these drugs. Pentavalent antimonials suffer from cardiotoxicity and the loss of efficacy on the Indian subcontinent.2 Amphotericin B is expensive when given as a liposomal formulation and nephrotoxic when administered as the more economical deoxycholate formulation; this drug is more effective for treating visceral leishmaniasis in India than in East Africa,3 although higher doses of amphotericin B are effective against East African visceral leishmaniasis.4 Like the antimonials and amphotericin B, paromomycin is given by injection (for three weeks in the case of this aminoglycoside). As with amphotericin B, paromomycin is more effective in treating visceral leishmaniasis on the Indian subcontinent than in East Africa.5 Miltefosine has the advantage of being the only oral drug of the four listed above, but it is relatively expensive, is teratogenic, and its efficacy has decreased for the treatment of visceral disease in recent years.2 Given the weaknesses of the current antileishmanial drugs, the need for new, effective oral treatments is clear.
The potency of arylimidamides (AIAs) against Leishmania has been known for some time. Initially termed reversed amidines, AIAs displayed potent activity against Leishmania donovani6, 7 and Leishmania species responsible for cutaneous disease.7, 8 Such compounds contain pyridyl imidamide groups at both ends of the molecule and are thus referred to as bis-AIAs. Mono-AIAs (which contain only one pyridyl imidamide group) were less potent than bis-AIAs, but several mono-AIAs nevertheless showed submicromolar antileishmanial activity in vitro.9 The most promising AIA identified to date, the bis-AIA 1 (DB766), also displayed good in vivo activity in rodent models of visceral leishmaniasis when given orally, but >90% inhibition of liver parasitemia was not observed at achievable doses.10, 11 Orally available azole-containing antifungal drugs have also shown antileishmanial activity12, 13 and have occasionally been used in the clinic against leishmaniasis, with varying success.14–16 Although the target of the AIAs has not been determined, azole antifungal drugs are thought to act on sterol-14α-demethylase (CYP51), a critical enzyme in the sterol biosynthesis pathway in both fungi and kinetoplastid parasites such as Leishmania.17 Inhibition of this target is believed to occur through coordination of the active site heme group of the enzyme by an imidazole or triazole nitrogen lone pair, while the remainder of the molecule makes important contacts within the substrate binding channel of CYP51.17
We demonstrated that oral combinations of 1 with posaconazole displayed moderately synergistic activity in a murine model of visceral leishmaniasis as defined by the combination index method, while combinations of 1 with ketoconazole were less effective in this model.11 It was striking that posaconazole and ketoconazole, which were over two orders of magnitude less potent in our hands against intracellular L. donovani in vitro compared to 1, displayed comparable, if not superior, efficacy to 1 in the murine visceral leishmaniasis model.11 In addition, L. donovani axenic amastigotes which became resistant to compound 1 due to prolonged pressure with 1 in culture exhibited hypersensitivity to posaconazole and ketoconazole.18 Based on these observations above, as well as the desire to take advantage of polypharmacology,19 the synthesis of AIA-azole hybrid compounds displaying features of mono-AIAs together with features of azole-containing antifungal drugs was undertaken. Molecular hybridization is a strategy in which two or more structural components possessing distinct pharmacologic activities are incorporated in one molecule to attack different targets.20 This strategy has been used widely in infectious disease drug discovery in an attempt to address resistance issues,21–24 to improve pharmacokinetic properties,24 and even to combat different infections using the same molecule.25 Structures of the frontrunner bis-AIA 1, the mono-AIA 2 (DB2002), the azole antifungals posaconazole (3) and ketoconazole (4), and the scaffold of AIA-azole hybrid compounds are shown in Figure 1. The synthesis and evaluation of an initial series of such hybrid compounds containing a phenoxyalkyl linker against L. donovani is described here.
Figure 1.
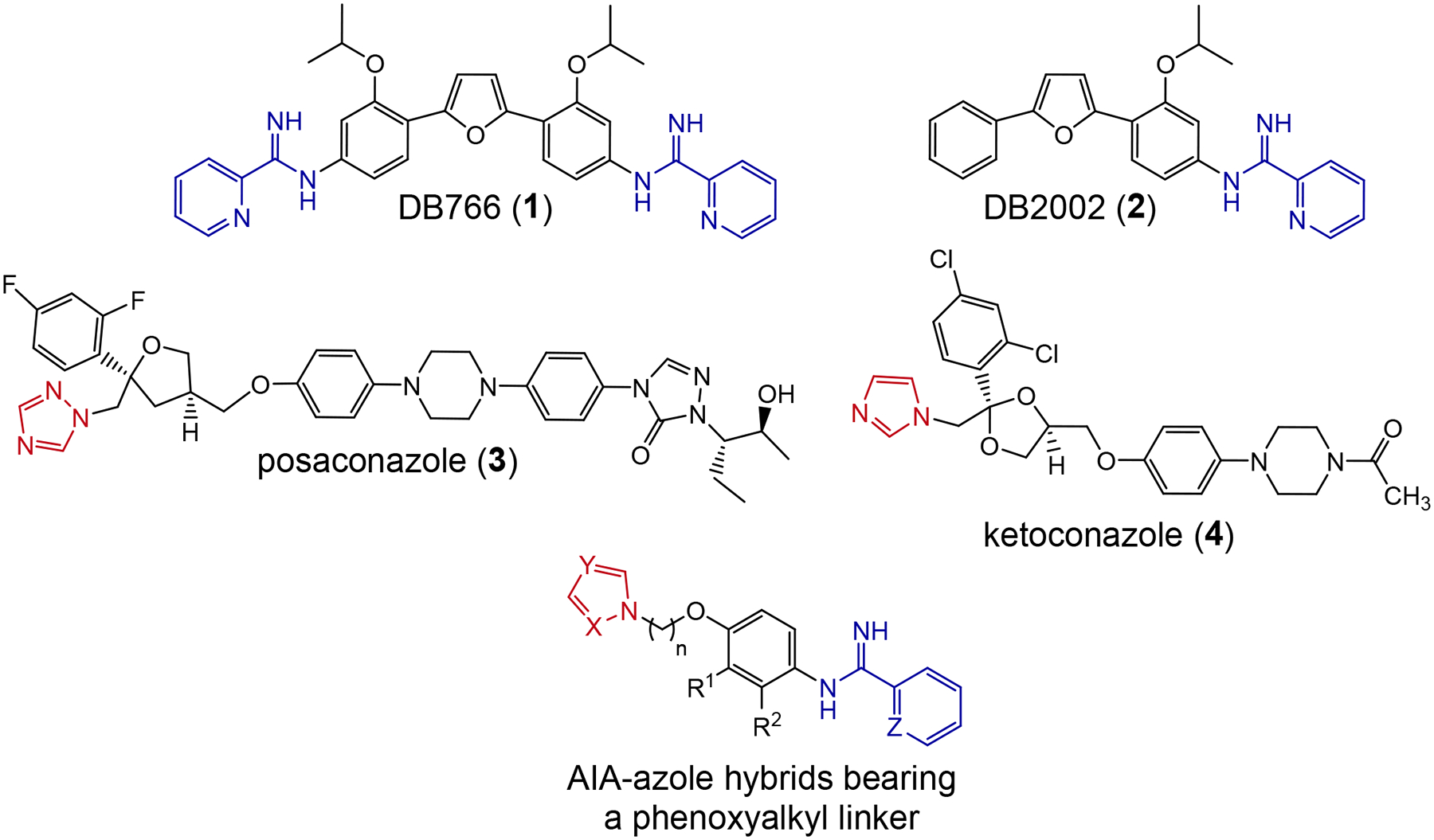
Structures of 1-4 and the AIA-azole scaffold explored here.
RESULTS
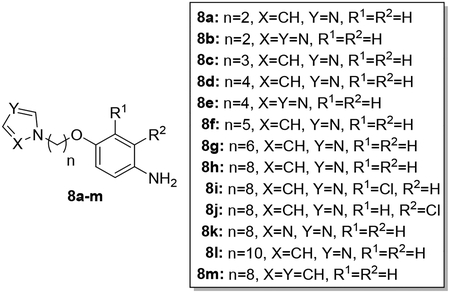
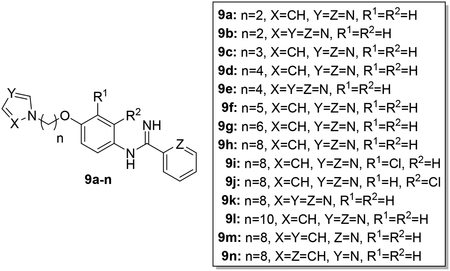
The synthesis of AIA azole hybrids 9a-l is shown in Scheme 1. According to a previously published procedure,26 reaction of dibromo alkanes of different chain lengths with 5a-c in dry acetonitrile under basic conditions provided 6a-i. Similarly, reaction of 6a-i with imidazole or 1,2,4-triazole under the same basic conditions afforded compounds 7a-l. Tin chloride-mediated reduction of the nitro group yielded 8a-l. Compounds 8a-l rapidly changed within less than an hour to dark gummy materials, possibly due to auto-oxidation upon exposure to air. This is reminiscent of our earlier observation regarding the auto-oxidation of 1-benzyl-2,2,4-trimethyl-1,2-dihydroquinolin-6-ols,27 which contain a similar structural motif. Arylamines 8a-l were thus used without further purification to yield target compounds 9a-l through reaction with the pyridyl thioimidate salt under the general conditions reported previously.7 Since the resultant AIA azole hybrids were stable as free bases, permitting satisfactory elemental analysis and testing in biological assays, conversion to the salt form as mentioned in Reid et al.7 was not necessary.
Scheme 1.
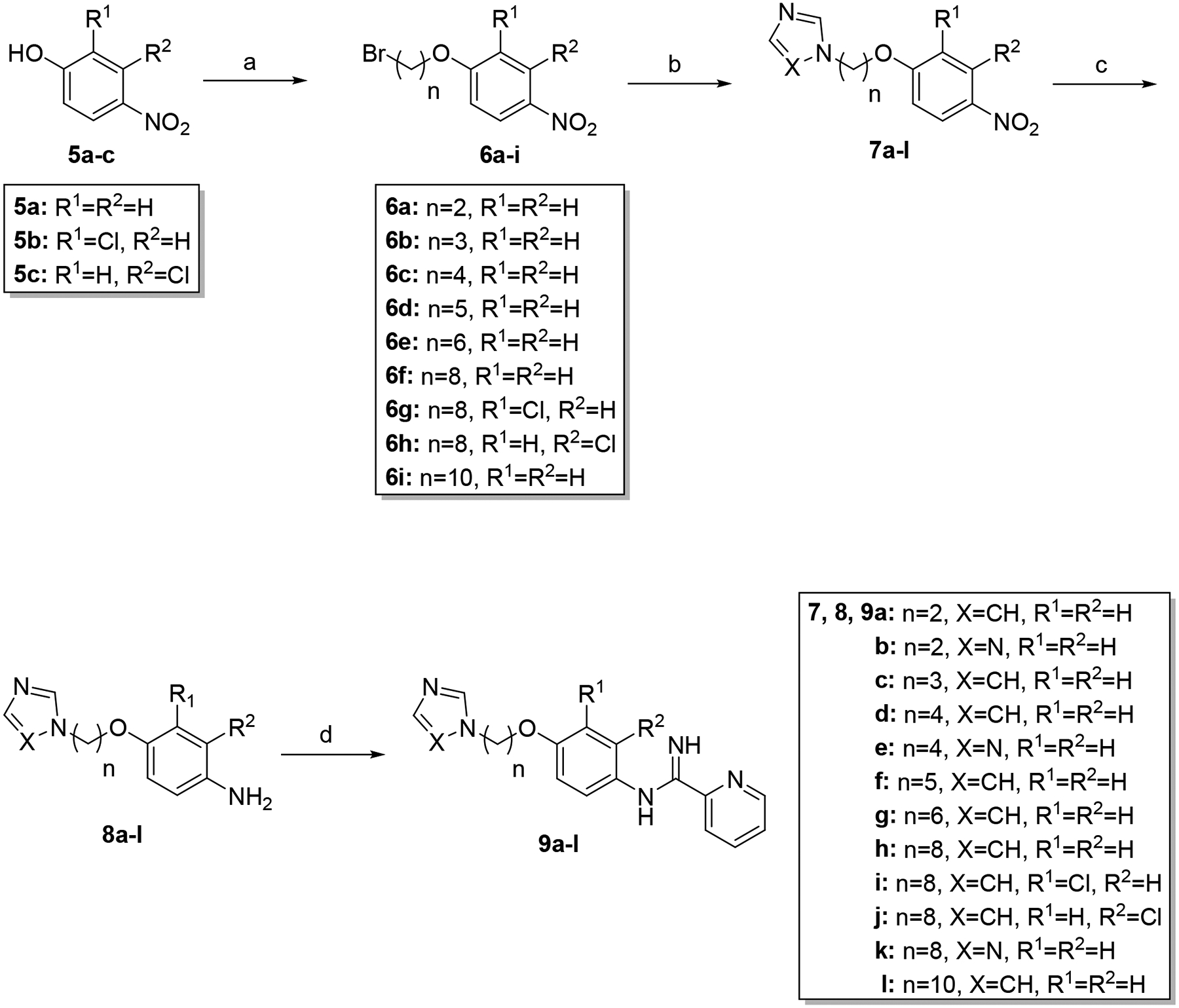
Synthesis of 9a-l. Reagents and conditions: a) dibromoalkane, K2CO3, CH3CN, reflux (62–93%); b) imidazole or 1,2,4-triazole, K2CO3, CH3CN (49–86%); c) SnCl2·2H2O, EtOAc (81–99%); d) naphthalen-2-ylmethyl pyridine-2-carbimidothioate·HBr, CH3CN/EtOH (1:3), rt (33–70%).
The method for preparing AIA-azole hybrids possessing additional terminal groups is shown in Scheme 2. Compound 9m contains a pyrrole group rather than an imidazole or triazole substitution at the azole terminus while a phenyl group is present in compound 9n instead of a pyridine at the imidamide end. The route used to synthesize these molecules is similar to the one shown in Scheme 1 except that the installation of the pyrrole ring in target compound 9m required the use of a stronger base (KOH) along with the more polar DMF as a solvent to facilitate the reaction.28 In addition, the reduction of nitroaromatic intermediate 7m to the corresponding aniline 8m could be accomplished with either tin(II)chloride dehydrate or Zn metal/ammonium chloride. Target compound 9n was obtained using the phenyl thioimidate salt rather than the pyridyl thioimidate salt.
Scheme 2.
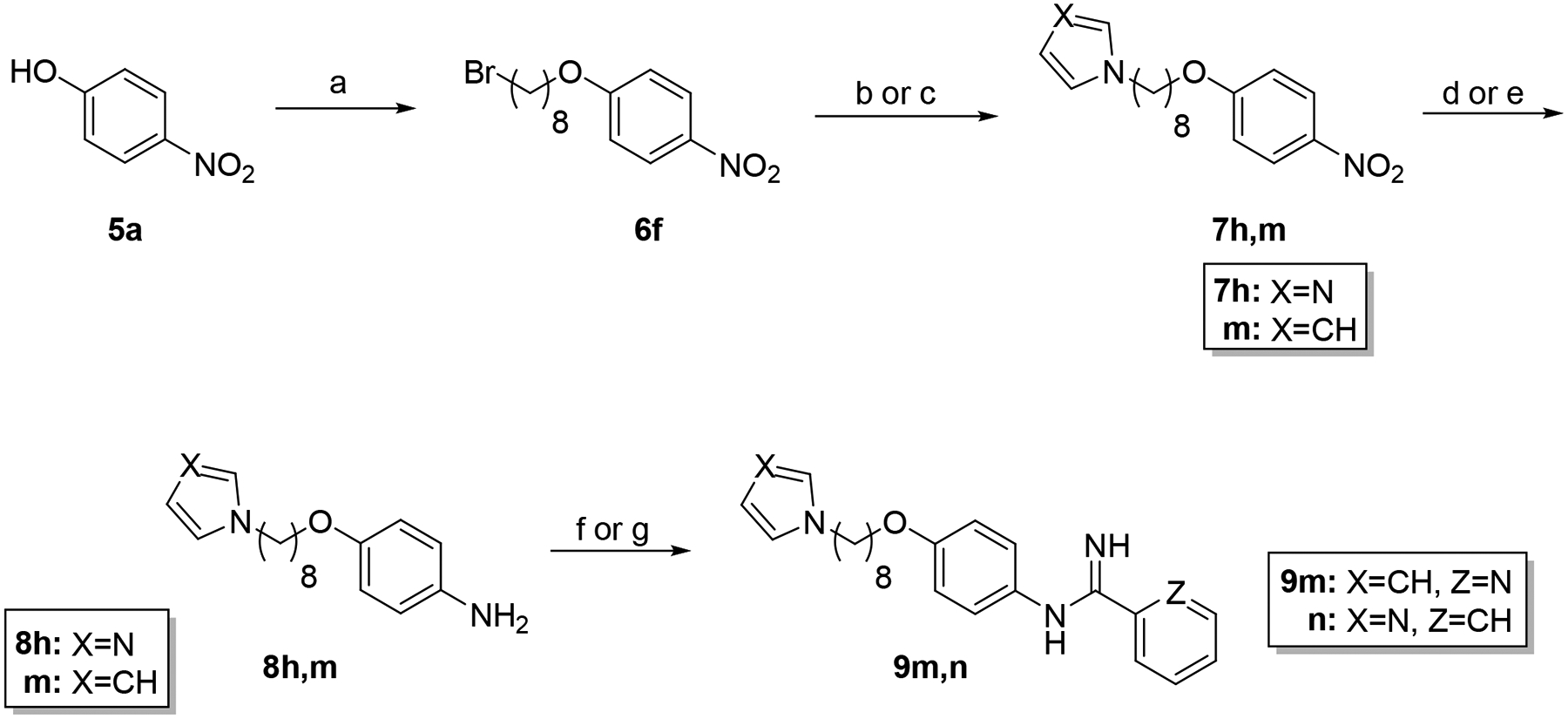
Synthesis of 9m,n. Reagents and conditions: a) 1,8-dibromooctane, K2CO3, CH3CN, reflux (92%); b) imidazole, K2CO3, CH3CN, reflux (86% 7h); c) pyrrole, KOH, DMF, reflux (35% 7m); d) SnCl2·2H2O, EtOAc (85% 8h); e) Zn, NH4Cl, MeOH (85% 8m); f) naphthalen-2-ylmethyl benzimidothioate·HBr, CH3CN/EtOH (1:3), rt (22% 9n); g) naphthalen-2-ylmethyl pyridine-2-carbimidothioate·HBr, CH3CN/EtOH (1:3), rt (17% 9m).
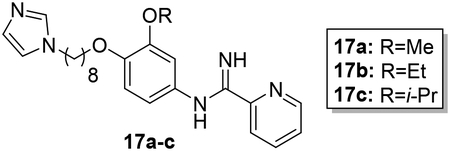
Synthesis of hybrid target compounds 17a-c containing alkoxy substitutions meta to the imidamide group is shown in Scheme 3. MOMCl protection of 4-nitrocatechol (10) was carried out according to a previous method29 followed by reaction of methyl, ethyl, or isopropyl iodide with the protected nitrophenol 11 under basic conditions, providing compounds 12a-c. Deprotection under acidic conditions followed by reaction of the resulting phenols 13a-c with dibromooctane under basic conditions in dry acetonitrile provided the intermediate monobromo derivatives 14a-c, which were transformed to 15a-c by treatment with imidazole in the presence of potassium carbonate. Nitro reduction followed by reaction of the resulting arylamines 16a-c with the pyridyl thioimidate salt as described above yielded target compounds 17a-c.
Scheme 3.
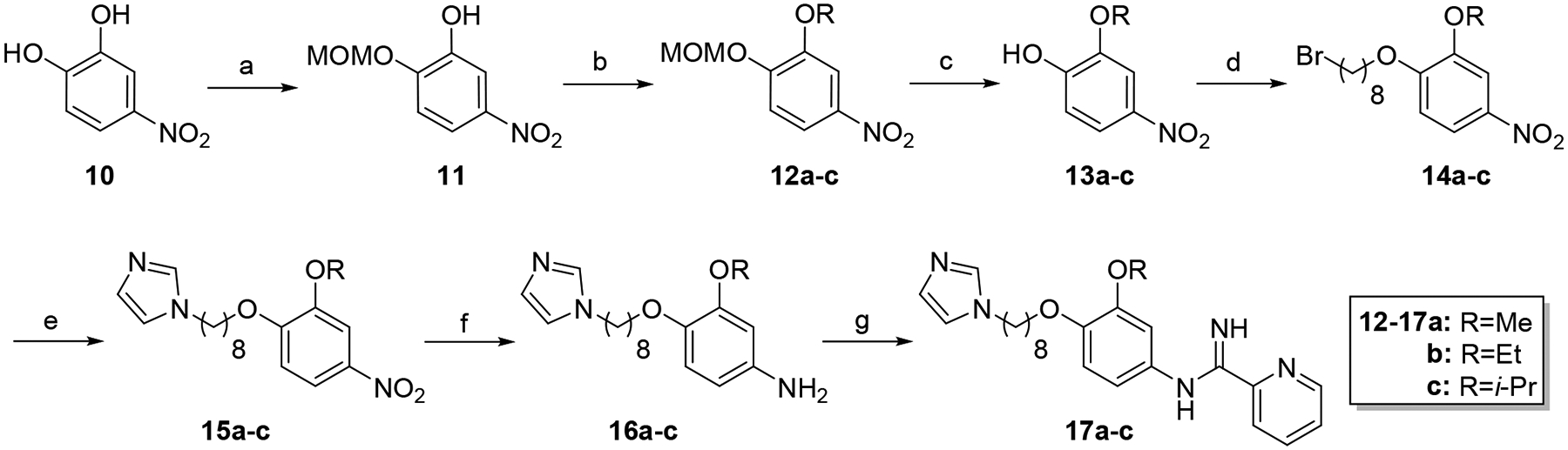
Synthesis of 17a-c. Reagents and conditions: a) MOMCl, K2CO3, DMF, 30°C (48%); b) RI, K2CO3, CH3CN, sealed tube, 80°C (91–97%); c) HCl, MeOH/CH2Cl2, rt (81–90%); d) 1,8-dibromooctane, K2CO3, CH3CN, reflux (84–92%); e) imidazole, K2CO3, CH3CN, reflux (73–90%); f) SnCl2·2H2O, EtOAc, reflux (98–99%); g) naphthalen-2-ylmethyl pyridine-2-carbimidothioate·HBr, CH3CN/EtOH (1:3), rt (31–45%).
Scheme 4 shows the preparation of AIA azole target compounds 24a-c bearing alkoxy groups ortho to the imidamide moiety. Installation of the alkyl groups through reaction of alkyl iodides with 5-fluoro-2-nitrophenol (18) followed by replacement of fluorine via nucleophilic aromatic substitution using sodium hydroxide as reported by Lebold and Kerr30 yielded nitrophenols 20a-c. Conversion of 20a-c to the desired target compounds 24a-c followed the general synthetic strategy described above.
Scheme 4.
Synthesis of 24a-c. Reagents and conditions: a) RI, K2CO3, sealed tube, DMF, 80°C (64–82%); b) NaOH, DMSO/H2O, reflux (74–85%); c) 1,8-dibromooctane, K2CO3, CH3CN, reflux (76–94%); d) imidazole, K2CO3, CH3CN, reflux (79–81%); e) SnCl2·2H2O, EtOAc, reflux (95–100%); f) naphthalen-2-ylmethyl pyridine-2-carbimidothioate·HBr, CH3CN/EtOH (1:3), rt (34–45%).
Synthesis of the AIA azole hybrid lacking a phenoxy linker (28) is illustrated in Scheme 5. Compound 26 was prepared through reaction of potassium phthalimide (25) with 1,8-dibromooctane. Reaction of 26 with imidazole under basic conditions followed by subjecting the intermediate to Gabriel synthesis conditions provided the primary amine 27 according to a previously published procedure.31 Reaction of 27 with the pyridyl thioimidate salt under the conditions described earlier yielded target compound 28 in low yield.
Scheme 5.
Synthesis of 28. Reagents and conditions: a) 1,8-dibromooctane, DMF, 90°C (60%); b) imidazole, K2CO3, DMF, 80°C (62%); c) NH2NH2.H2O, EtOH, reflux (67%); d) naphthalen-2-ylmethyl pyridine-2-carbimidothioate·HBr, CH3CN/EtOH (1:3), rt (28%).
Structure-activity and structure-toxicity relationships.
In designing the hybrid target compounds, the effect of three factors was examined: 1) chain length, 2) identity of the terminal groups, and 3) substitution of the phenoxy linker. Data from the evaluation of the potency of the hybrid compounds against intracellular L. donovani and their effect against two mammalian cell lines is shown in Table 1. Increasing the length of the alkyl chain improved the antileishmanial potency but also increased activity against both J774 macrophages and HepG2 cells. The best balance of potency and toxicity in this series was achieved with an octyl chain; compound 9h possessed an IC50 value of 1.0 μM against L. donovani intracellular amastigotes and IC50 values of 11 μM and 12 μM against J774 macrophages and HepG2 cells, respectively. Increasing the chain length to ten carbons (compound 9l) did not improve the antileishmanial activity but decreased the selectivity against both the macrophage and hepatocarcinoma cell lines. Compounds containing imidazole as the azole terminal group showed greater antileishmanial potency than the corresponding triazoles. For the compounds bearing four carbon alkyl linkers, imidazole 9d was at least five-fold more potent against intracellular L. donovani than triazole 9e. In the eight carbon linker series, replacement of imidazole with triazole led to a >10-fold decrease in potency when 9h is compared with 9k (a precise IC50 value could not be calculated for compound 9k in the intracellular L. donovani assay because of toxicity to the host cells at higher concentrations, but ~50% inhibition of intracellular parasitemia was observed for 9k at a concentration of 12.5 μM as indicated in Table 1). Little effect on antileishmanial potency and mammalian cell toxicity was observed when the imidazole ring was replaced with pyrrole (compare 9h to 9m). Regarding the arylimidamide moiety, replacement of the 2-pyridyl group with a phenyl moiety (compound 9n) resulted in a 22-fold increase in potency against J774 cells, a 2.4-fold increase in HepG2 activity, and no increase in antileishmanial potency compared to 9h. In terms of substitutions on the phenoxy portion of the linker, placement of chlorine atoms either meta (9i) or ortho (9j) to the imidamide end did not increase antileishmanial potency but may have decreased antileishmanial selectivity compared to 9h. Minor effects on antileishmanial potency and selectivity were observed upon substitution of the phenoxy ring with alkoxy groups. The most favorable substitutions of those tested appear to be ethoxy and isopropoxy groups ortho to the imidamide (compounds 24b and 24c, respectively). These changes resulted in slight increases in antileishmanial potency, similar selectivity for intracellular L. donovani vs. J774 macrophages compared to 9h, and slight improvements in selectivity for intracellular parasites vs. HepG2 cells compared to 9h. For example, compound 24c displayed sub-micromolar potency against L. donovani with selectivity indexes (IC50 vs. mammalian cell line/IC50 vs. L. donovani) of 13 and 23 for intracellular parasites compared to J774 macrophages and HepG2 cells, respectively. Notably, the removal of the phenoxy linker (compound 28) resulted in a total loss of activity against L. donovani and HepG2 cells. The higher hydrophilicity of compound 28 and the presumed higher pKa value of its imidamide moiety may contribute to its lack of antileishmanial activity.
Table 1.
In Vitro Antileishmanial Activity, Mammalian Cell Toxicity, and Inhibition of L. donovani CYP51 Activity for AIA-Azole Hybrids
| Compound | X | Y | Z | R1 | R2 | n | IC50 against L. donovani (μM)a | IC50 against J774 macrophages (μM)a | IC50 against HepG2 (μM)a | IC50 against recombinant L. donovani CYP51 (μM)a |
|---|---|---|---|---|---|---|---|---|---|---|
| 9a | CH | N | N | H | H | 2 | >50 | >50 | >50 | 21 ± 1 |
| 9b | N | N | N | H | H | 2 | >100 | >50 | >100 | 35 ± 2 |
| 9c | CH | N | N | H | H | 3 | >25 | >50 | >50 | 22 ± 0 |
| 9d | CH | N | N | H | H | 4 | 18 ± 3 | >50 | >20 | 6.4 ± 0.1 |
| 9e | N | N | N | H | H | 4 | >100 | >50 | >100 | 89 ± 4 |
| 9f | CH | N | N | H | H | 5 | 13 ± 5 | >50 | 42 ± 4 | 4.8 ± 0.1 |
| 9g | CH | N | N | H | H | 6 | 8.8 ± 0.6 | >40 | 20 ± 3 | 1.3 ± 0.1 |
| 9h | CH | N | N | H | H | 8 | 1.0 ± 0.0 | 11 ± 1 | 12 ± 3 | 0.36 ± 0.03 |
| 9i | CH | N | N | Cl | H | 8 | 1.3 ± 0.2 | 6.8 ± 0.7 | 7.7 ± 1.5 | 0.18 ± 0.01 |
| 9j | CH | N | N | H | Cl | 8 | 2.1 ± 0.2 | 8.6 ± 1.2 | 8.6 ± 1.2 | 0.15 ± 0.01 |
| 9k | N | N | N | H | H | 8 | 51 ± 5%b | 36 ± 1 | 32 ± 5 | 1.9 ± 0.1 |
| 9l | CH | N | N | H | H | 10 | 1.4 ± 0.2 | 4.9 ± 0.9 | 6.9 ± 1.0 | 0.22 ± 0.01 |
| 9m | CH | CH | N | H | H | 8 | 1.7 ± 0.3 | 11 ± 1 | 13 ± 2 | 14 ± 1 |
| 9n | CH | N | CH | H | H | 8 | 1.2 ± 0.1c | 0.48 ± 0.02 | 4.9 ± 1.0 | 0.34 ± 0.01 |
| 17a | CH | N | N | OMe | H | 8 | 2.2 ± 0.3 | 16 ± 1 | 23 ± 2 | 0.57 ± 0.01 |
| 17b | CH | N | N | OEt | H | 8 | 1.9 ± 0.3 | 9.0 ± 1.3 | 13 ± 0 | 0.53 ± 0.02 |
| 17c | CH | N | N | OiPr | H | 8 | 0.97 ± 0.07 | 7.8 ± 0.3 | 18 ± 1 | 0.29 ± 0.01 |
| 24a | CH | N | N | H | OMe | 8 | 1.5 ± 0.4 | 9.3 ± 0.5 | 16 ± 2 | 0.36 ± 0.01 |
| 24b | CH | N | N | H | OEt | 8 | 0.73 ± 0.15 | 6.8 ± 0.1 | 12 ± 2 | 0.34 ± 0.01 |
| 24c | CH | N | N | H | OiPr | 8 | 0.53 ± 0.05 | 6.8 ± 0.2 | 12 ± 2 | 0.33 ± 0.01 |
| 28 | - | - | - | - | - | - | >100 | Not tested | >100c | Not tested |
| Amphotericin B | 0.045 ± 0.003 | Not tested | Not tested | Not tested | ||||||
| Podophyllotoxin | Not tested | 0.025 ± 0.001 | Not tested | Not tested | ||||||
| Doxorubicin | Not tested | Not tested | 0.20 ± 0.02 | Not tested | ||||||
| Quinacrine | Not tested | Not tested | 5.5 ± 0.7 | Not tested | ||||||
| Ketoconazole | 50 ± 7%d | Not tested | Not tested | 0.048 ± 0.004 | ||||||
| Posaconazole | 47 ± 2%e | Not tested | Not tested | 0.059 ± 0.001 | ||||||
| Fluconazole | 16 ± 3%d | Not tested | Not tested | 0.96 ± 0.03 |
Mean ± standard error of the mean (S.E.M., n=3);
Inhibition of parasitemia at 12.5 μM (mean ± S.E.M. (n=3));
Mean ± range (n=2);
Inhibition of parasitemia at 50 μM (mean ± S.E.M. (n=3));
Inhibition of parasitemia at 7.5 μM (mean ± S.E.M. (n=3)).
Binding to L. donovani CYP51.
Purified CYP51 showed good purity with a single major band in both SDS-PAGE and Western blot analyses (Figure 2A) and exhibited a characteristic absorbance at 450 nm (the active P450 form) upon dithionite reduction and carbon monoxide (CO)-binding (Figure 2B). The P450 form of the protein was stabilized by lanosterol as evidenced by a reduction in the inactive P420 form (Figure 2B),32 likely due to its binding at the active site. When titrated with 24c or 9h, CYP51 exhibited typical Type II binding spectra (Figure 2C and 2D) with an absorbance maximum at 431 nm and a minimum at 412–413 nm, indicating direct binding to the active site of CYP51. A Type II binding with CYP protein shows an absorption maximum above the 415–418 nm Soret band range (red shifted Soret band for low spin Fe3+), and is indicative of inhibitory ligand binding as the ligand replaces the water molecule to form the sixth coordination with the heme iron, stabilizes low spin Fe3+, and prevents substrate binding.33
Figure 2.
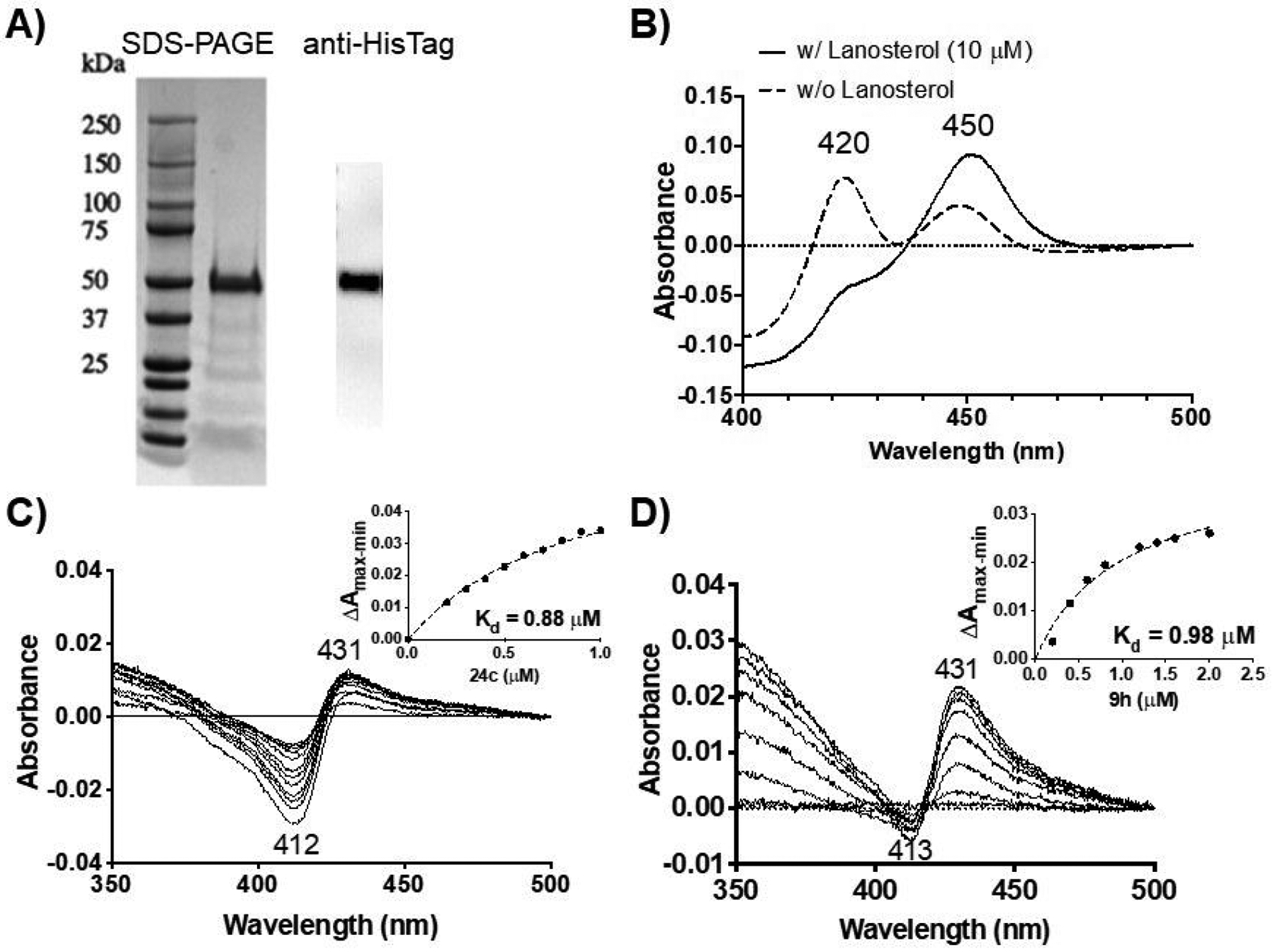
Gel electrophoresis (A), reduced CO-difference spectra (B), and ligand binding spectra (C and D) of the purified L. donovani CYP51. The expected molecular weight of the CYP51-32c construct is 53 kDa. The P450 content was determined to be 7.1 nmol/mg for the batch of purified CYP51 used in this experiment. Various concentrations of 24c (C) and 9h (D) were titrated into CYP51 before recording binding spectra and determining dissociation constants (Kd).
Inhibition of L. donovani CYP51.
A fluorescence-based inhibition assay was developed for the L. donovani CYP51 using 7-benzyloxy-4-trifluoromethylcoumarin (BFC) as substrate, which was readily converted via oxidative O-debenzylation to its fluorescent metabolite 7-hydroxy-4-trifluoromethylcoumarin (HFC, excitation 410 nm, emission 538 nm) by the purified CYP51 in the presence of cumene hydroperoxide (CuOOH) as a cofactor (Figure 3A). CuOOH has previously been shown to enable CYP-catalyzed reactions in the absence of CYP natural cofactors (NADPH:P450 oxidoreductase and NADPH).34
Figure 3.
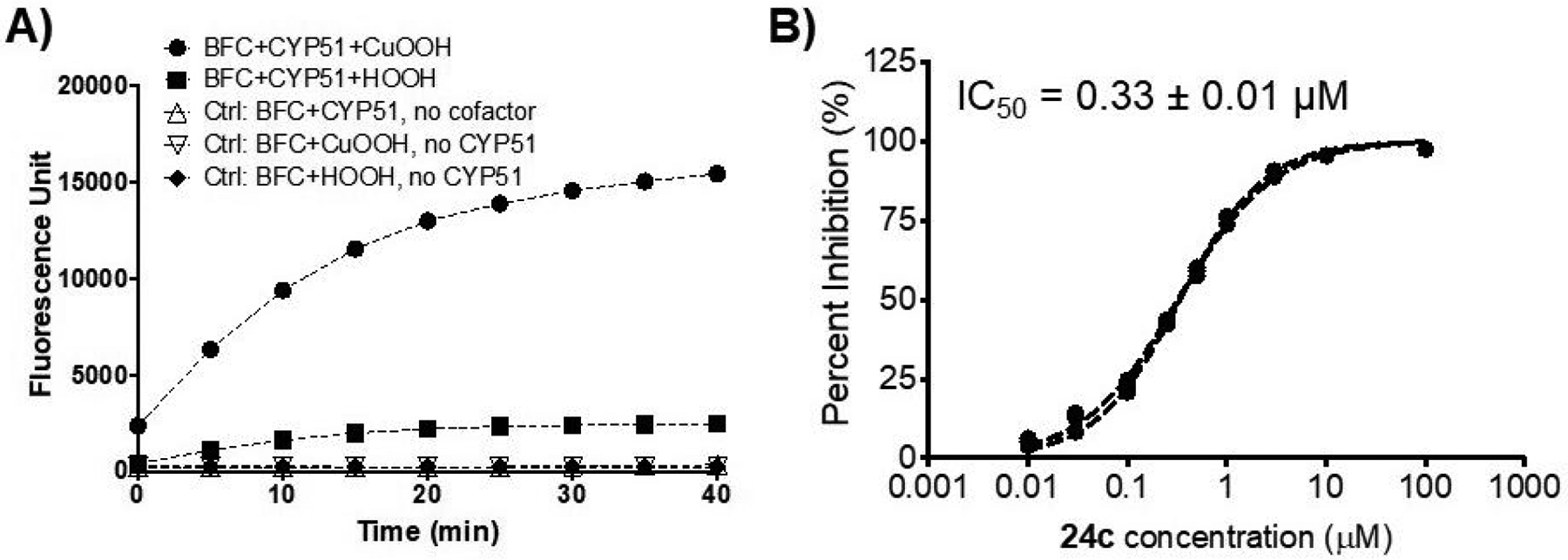
Fluorescence-based inhibition assay for L. donovani CYP51 (A) and inhibition by 24c (B). The fluorogenic substrate BFC (50 μM) was incubated with purified CYP51-32c (60 nM) in the presence of cofactor CuOOH (0.1 mM) or hydrogen peroxide (HOOH; 25 mM) at 37°C. Control incubations did not contain either the cofactor (but with CYP) or the CYP protein (but with CuOOH or HOOH). IC50 value (mean ± SE of triplicate determinations) is shown for 24c.
The azole antifungal drugs posaconazole, ketoconazole and fluconazole exhibited strong-to-moderate inhibition of the L. donovani CYP51 with IC50 values (mean ± S.E.M. of triplicate determinations) of 0.059 ± 0.001, 0.048 ± 0.004, and 0.96 ± 0.03 μM, respectively. Table 1 also shows the inhibition of CYP51-mediated conversion of BFC to HFC by the hybrid compounds. A representative inhibition curve is shown for 24c (Figure 3B). Generally consistent with observations regarding antiparasitic potency, compounds possessing longer linkers display greater inhibition of CYP51. In the imidazole series, inhibitory activity increases as chain length increases from 3 carbons (9c) through 10 carbons (9l). Hybrids bearing an imidazole ring at the “azole end” of the molecule have higher potency than those containing the triazole ring. This is clearly illustrated by comparing four carbon linker containing imidazole 9d and triazole 9e (CYP51 IC50 values of 6.4 μM and 89 μM, respectively) and eight carbon linker containing imidazole 9h and triazole 9k (CYP51 IC50 values of 0.36 μM and 1.9 μM, respectively). The importance of the imidazole ring in conveying CYP51 inhibition is further demonstrated through comparison of the pyrrole terminal compound 9m and the corresponding imidazole terminal compound 9h. Lacking nitrogen at position 3 of the heterocycle, compound 9m displays an IC50 value of 14 μM against CYP51 as opposed to an IC50 value of 0.36 μM for corresponding imidazole compound 9h as mentioned earlier. This observation for 9m represents a departure from the antiparasitic SAR described above. Within the series of imidazole-containing phenoxyalkyl hybrids bearing eight carbon linkers, the substitution on the phenoxyalkyl ring and the opposite terminal group have relatively little influence on CYP51 inhibition, as phenoxyalkyl substituted compounds 17a-c and 24a-c along with phenyl terminal compound 9n show CYP51 IC50 values similar to that of 9h. It should be noted, however, that Cl substituted compounds 9i and 9j are about 2-fold more potent as inhibitors of L. donovani CYP51 compared to 9h.
Inhibition of human CYP3A4.
CYP3A enzymes are critical in drug metabolism, being expressed in the liver, intestine, and other tissues.35 Since several azole drugs are potent inhibitors of CYP3A4, the inhibition of recombinant human CYP3A4 by hybrid compounds 9h and 24c was evaluated using BFC as a substrate.36 Compounds 9h and 24c were found to be strong inhibitors of recombinant human CYP3A4, displaying IC50 values of 0.047 ± 0.003 μM and 0.080 ± 0.009 μM, respectively. For comparison, azole antifungal drugs ketoconazole, posaconazole, and fluconazole exhibited IC50 values of 0.0070 ± 0.0003 μM, 0.15 ± 0.01 μM, and 18 ± 2 μM, respectively. Fluconazole is known to be a relatively weak inhibitor of CYP3A4.37
Microsomal stability.
Selected hybrid compounds were also evaluated for their in vitro microsomal stability (Table 2). These compounds were incubated with either human or mouse liver microsomes at a substrate concentration of 3 μM; disappearance of the compound of interest was measured over time by LC-MS or LC-UV detection. All compounds exhibited t1/2 > 50 min. Compounds 9l (bearing a ten carbon linker) and 24c (containing an eight carbon linker and an isopropoxy substitution ortho to the imidamide) were less stable in the presence of both human and mouse liver microsomes, possessing t1/2 of 52–68 min under these conditions. Compound 9h (containing an eight carbon linker and an unsubstituted phenoxy ring) displayed t1/2 = 70 min in the presence of mouse liver microsomes but was not degraded by human liver microsomes over a period of one hour. For the rest of the hybrid compounds tested, 80% or more remained unchanged in the presence of either human or mouse liver microsomes after a 60 min incubation. Metabolic stability data for AIAs 1 and 2 are provided for comparison.
Table 2.
In vitro metabolic stability of selected hybrid compounds.
| Compound | Human liver microsome t1/2 or percent remaininga | Mouse liver microsome t1/2 or percent remaininga |
|---|---|---|
| 9h | 100% after 60 min | 70 min |
| 9l | 57 min | 68 min |
| 17a | 100% after 60 min | 100% after 60 min |
| 17c | >80% after 60 min | 100% after 60 min |
| 24a | >80% after 60 min | >80% after 60 min |
| 24b | >80% after 60 min | 100% after 60 min |
| 24c | 52 minb | 67 minb |
| 1 | >50% after 120 minc | >50% after 120 minc |
| 2 | 100%, 111% after 60 mind | 44%, 61% after 60 mind |
Substrate concentration 3 μM, measured by LC-MS detection unless otherwise noted
Measured by UV detection
Measured at substrate concentrations of 1 μM and 10 μM, from Wang et al.10
Measured by UV detection at substrate concentrations of 1 μM and 10 μM, respectively.
In vivo evaluation of compound 24c.
Since 24c displayed a t1/2 of >60 minutes in the presence of mouse liver microsomes and was the most potent hybrid compound tested in the phenoxyalkyl hybrid series, it was evaluated for antileishmanial efficacy in vivo. When 24c was given by oral gavage at a dose of 100 mg/kg/day for five days to a pair of female 6–8 week old BALB/c mice, no weight loss, signs of overt toxicity, or obvious effects on the GI tract, liver, spleen, or kidneys were observed upon necropsy performed one day after administration of the final dose. Oral doses of 24c at 75 mg/kg/day × 5 and 37.5 mg/kg/day × 5 were then administered to L. donovani-infected female BALB/c mice starting at one week post infection (these doses were chosen based on compound availability). Liver parasitemia was then determined three days after the final dose (Figure 4). In animals treated with 75 mg/kg 24c, 33 ± 7% (mean ± S.E.M.) reduction in liver parasitemia was observed, while the liver parasite burden was reduced by 17 ± 5% in those animals given 24c at 37.5 mg/kg. Consistent with previous studies in this murine visceral leihsmaniasis model, oral treatment with miltefosine at 10 mg/kg/day × 5 led to a 95 ± 2% reduction of liver infection.7, 11
Figure 4.
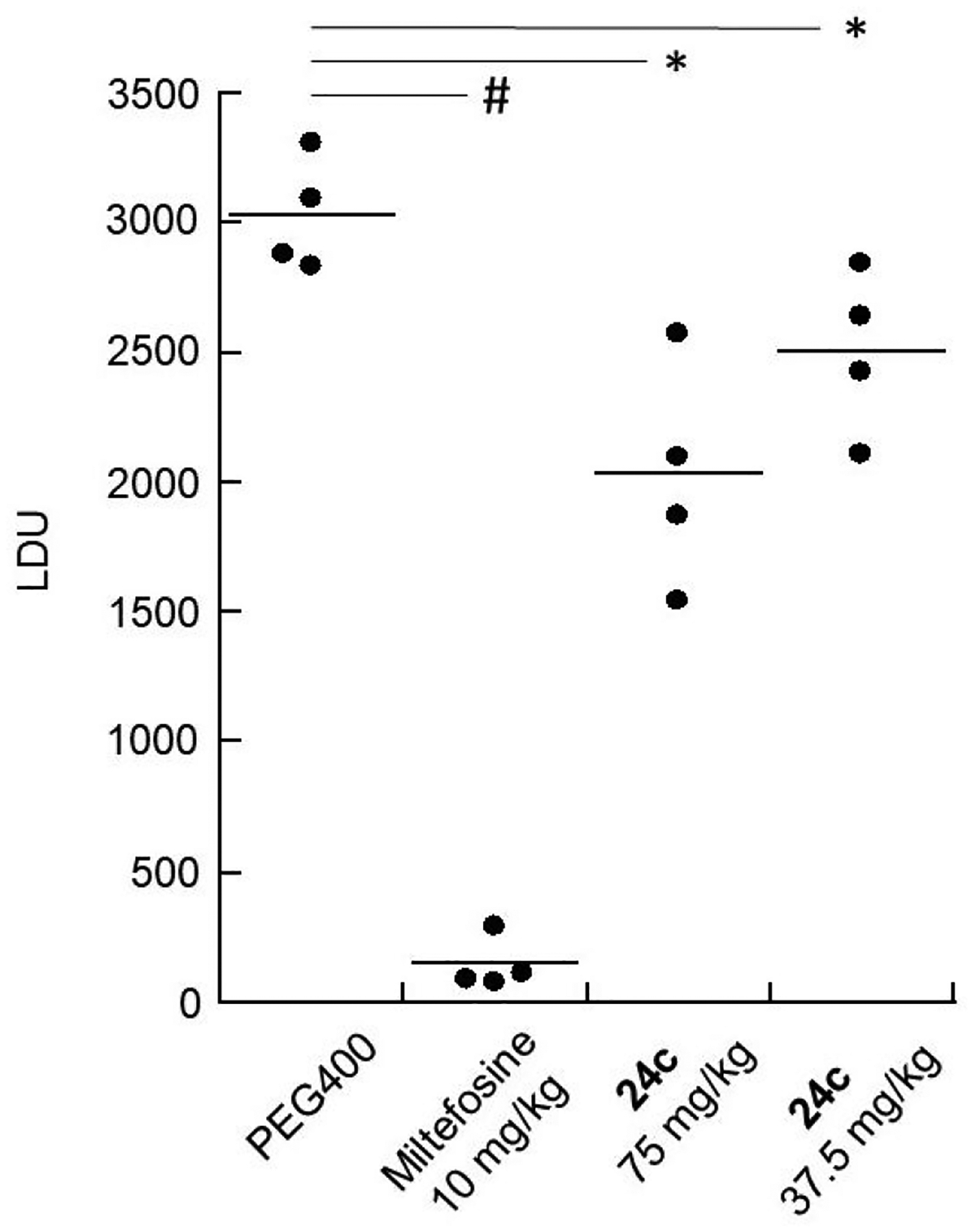
Efficacy of compound 24c in the murine model of visceral leishmaniasis. Compound 24c, miltefosine, and vehicle were administered by oral gavage for five consecutive days to groups of four L. donovani-infected female BALB/c mice. Liver parasite burdens are expressed as Leishman-Donovan units (LDU), with horizontal bars indicating the mean for each group. #, p < 0.001 compared to PEG400 control group; *, p < 0.05 compared to PEG400 control group as assessed by two-tailed Student’s t test.
DISCUSSION
Molecular hybridization is a powerful approach that has been widely used in the search for promising antileishmanial and antitrypanosomal candidates.38–41 The design and synthesis of hybrid compounds displaying structural features of both antifungal azoles and AIAs may be a promising strategy toward the discovery of new antileishmanial leads because members of both classes of compounds display in vivo antileishmanial activity when given orally. Although they are not FDA approved for antileishmanial treatment, antifungal azoles are considered for off-label use against leishmaniasis,42 while AIAs have shown oral activity in rodent models of visceral leishmaniasis.9, 10, 43 It is widely accepted that antifungal azoles exert their activity against fungi by inhibiting CYP51, the 14α-demethylase enzyme crucial for sterol biosynthesis; this enzyme is also inhibited by azoles displaying antileishmanial activity.44 However, it has been speculated that the development of new antileishmanial therapies targeting sterol biosynthesis may require the discovery of more powerful inhibitors, drug combinations, or molecular hybridization approaches.17, 45 While the precise mechanism(s) of action of AIAs remain(s) unknown, L. donovani half knockout parasites for CYP5122A1, a protein involved in Leishmania ergosterol biosynthesis,46 were less susceptible to 1 than wild type parasites but were more susceptible to ketoconazole.18 Work with these hybrid compounds may uncover additional insights regarding Leishmania sterol metabolism that could aid antileishmanial drug design.
In terms of their potency against intracellular L. donovani, the hybrid compounds reported here are comparable to the mono-AIAs.9 The most potent compounds reported thus far from each group exhibit high nanomolar IC50 values against these intracellular parasites and show toxicity to J774 macrophages at concentrations near 10 μM. There are caveats to this comparison; the mono-AIAs appear to be slightly more selective for Leishmania and the intracellular L. donovani assays reported in the two studies were performed by different methods. Nonetheless, considering their comparable in vitro biological activities given the available data, it is appropriate to ask whether the hybrid target compounds reported here are simply a subset of the mono-AIAs. Although the target(s) of the AIAs remain unknown, the interactions of active compounds from both series with L. donovani CYP51 are discussed below.
Azole antifungals disrupt sterol biosynthesis in Leishmania.47 Fluconazole inhibits CYP51, the 14α-demethylase involved in sterol biosynthesis, from Leishmania as shown by Hargrove et al.48 and as demonstrated in the present work. Imidazole-containing compounds VNI, VFV, and their analogs also inhibit Leishmania CYP5144, 49 and display in vitro and in vivo antileishmanial effects.44 Molecules consisting of an imidazole group and an aromatic substituent connected by a hydrocarbon chain were identified by high throughput screening as strong binders to T. cruzi CYP51 and also displayed submicromolar potency against intracellular T. cruzi amastigotes.50 Most of the hybrid compounds reported here that display single-digit micromolar or greater potency against intracellular L. donovani also inhibit Leishmania CYP51 (Table 1); imidazole containing hybrid compounds are more active against both Leishmania CYP51 and against intracellular parasites than the corresponding triazole compounds. While these data show the ability of selected hybrid compounds to bind to this parasite enzyme involved in sterol biosynthesis, they do not clearly demonstrate that CYP51 is the target for these compounds. Given that the bis-AIA 1 and the related mono-AIA 2 are weak inhibitors of L. donovani CYP51 at concentrations 5–10 times greater than their IC50 against intracellular L. donovani (M.Z. Wang, unpublished observations), CYP51 inhibition does not appear to be required in order for molecules possessing an arylimidamide group to display potent antileishmanial activity. In addition, the pyrrole containing compound 9m is only slightly less potent against intracellular L. donovani than the corresponding imidazole 9h, yet it is 39-fold less active as a CYP51 inhibitor. The protein CYP5122A1 also has a role in ergosterol synthesis in Leishmania donovani, although its precise function is currently unknown.46 Given the range of inhibitors that have been identified for T. cruzi CYP51, it may also be possible to target CYPs in Leishmania with a variety of inhibitors. In an attempt to test the hypothesis mentioned above that improved inhibitors of Leishmania sterol biosynthesis may result in the development of new therapeutics, the identification of new CYP450 inhibitors in Leishmania and the characterization of their effects on this parasite is an area of active investigation in our laboratories.
Like ketoconazole and posaconazole, two hybrid compounds (9h and 24c) are shown to be potent inhibitors of human CYP3A4, which poses a risk for drug-drug interactions with molecules that are metabolized by human CYP3A4. It was reported that human CYP51 is resistant to inhibition by azole antifungals, including posaconazole, ketoconazole and fluconazole.51 As such, in this first report on AIA-azole hybrid compounds, these agents were not evaluated for inhibition of human CYP51. Future studies should consider the inhibition of a wider panel of human CYPs by the AIA-azole hybrid compounds, especially those CYPs commonly involved in drug metabolism, to prioritize hits for further optimization and development.
The AIA-azole hybrid agents described here hold promise as antileishmanials based on their strong activity against intracellular parasites (Table 1) and the opportunity they present to explore a wide range of chemical space in the design of new derivatives to improve potency and drug-like properties. The data shown in Table 2 also indicate that, when compared to AIAs 1 and 2, the inclusion of the azole group in the hybrid molecules does not have a clear negative effect on metabolic stability. Lead compound 24c did not display signs of toxicity when administered orally to BALB/c mice at doses up to 100 mg/kg, although the in vivo efficacy of this compound in a murine model of visceral leishmaniasis was modest (Figure 4). Work is in progress to improve the antileishmanial properties of the AIA-azole hybrids, to describe their interactions with potential target molecules, and to examine their influence on sterol metabolism in Leishmania.
METHODS
General Chemistry Methods.
Reactions were carried out and monitored and compounds were purified according to the techniques described earlier52 unless noted otherwise. Commercial solvents and reagents were purchased from Sigma-Aldrich, VWR, Fisher Scientific, or Combi-Blocks and were used without further purification. NMR spectra were recorded on Bruker AV300 or DRX400 or Avance III HD Ascend 700 MHz spectrometers at 300 °K unless noted and were calibrated using the residual undeuterated solvent peak (CDCl3: δ 7.26 ppm for 1H NMR, 77.16 ppm for 13C NMR; DMSO-d6: δ 2.50 ppm for 1H NMR, 39.52 ppm for 13C NMR).53 Proton (1H) and carbon (13C) NMR data are reported as outlined previously.52 High resolution mass spectra (HRMS) and melting points were obtained as described earlier.52
Synthesis of 4-nitrophenoxy alkyl bromides (6a-i).
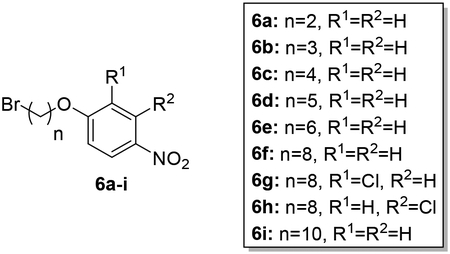
To a solution of 4-nitrophenols 5a-c (2.9–14.4 mmol) in dry acetonitrile (20 mL) was added 2 equivalents of K2CO3 (5.8–28.8 mmol) and 5 equivalents of dibromoalkane (14.4–71.8 mmol). The mixture was stirred at 80 °C overnight according to a previously published procedure.26, 54 After completion of the reaction, the suspension was filtered and the filtrate was concentrated under reduced pressure. The crude product was subjected to silica gel chromatography employing ethyl acetate/hexanes 1:10 as the eluent to afford the product in 62–93% yield.
1-(2-Bromoethoxy)-4-nitrobenzene (6a).54
White powder, 2.2 g, yield 62% starting from 2.0 g 4-nitrophenol (5a, 14.4 mmol).
1-(3-Bromopropoxy)-4-nitrobenzene (6b).54
White powder, 3.0 g, yield 80% starting from 2.0 g 4-nitrophenol (5a, 14.4 mmol).
1-(4-Bromobutoxy)-4-nitrobenzene (6c).54
White powder, 2.5 g, yield 63% starting from 2.0 g 4-nitrophenol (5a, 14.4 mmol).
1-(5-Bromopentoxy)-4-nitrobenzene (6d).54
White powder, 3.2 g, yield 77% starting from 2.0 g 4-nitrophenol (5a, 14.4 mmol).
1-(6-Bromohexoxy)-4-nitrobenzene (6e).54
Yellow oil, 3.30 g, yield 76% starting from 2.0 g 4-nitrophenol (5a, 14.4 mmol).
1-((8-Bromooctyl)oxy)-4-nitrobenzene (6f).54
White powder, 1.10 g, yield 92% starting from 0.5 g 4-nitrophenol (5a, 3.60 mmol).
1-((8-Bromooctyl)oxy)-2-chloro-4-nitrobenzene (6g).
Yellow oil, 0.98 g, yield 93% starting from 0.5 g 2-chloro-4-nitrophenol (5b, 2.88 mmol). 1H NMR (300 MHz, CDCl3) δ 1.31–1.56 (m, 8H), 1.80–1.94 (m, 4H), 3.40 (t, J = 6.8 Hz, 2H), 4.13 (t, J = 6.4 Hz, 2H), 6.96 (d, J = 9.1 Hz, 1H), 8.13 (dd, J = 9.1, 2.7 Hz, 1H), 8.28 (d, J = 2.7 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 25.9, 28.2, 28.7, 28.9, 29.2, 32.9, 34.0, 70.0, 111.8, 123.6, 124.1, 126.2, 141.1, 159.9.
4-((8-Bromooctyl)oxy)-2-chloro-1-nitrobenzene (6h).
Yellow oil, 0.97 g, yield 92% starting from 0.5 g 3-chloro-4-nitrophenol (5c, 2.88 mmol). 1H NMR (300 MHz, CDCl3) δ 1.30–1.52 (m, 8H), 1.75–1.92 (m, 4H), 3.41 (t, J = 6.7 Hz, 2H), 4.02 (t, J = 6.4 Hz, 2H), 6.85 (dd, J = 9.1, 2.6 Hz, 1H), 6.99 (d, J = 2.6 Hz, 1H), 7.99 (d, J = 9.1 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 25.9, 28.2, 28.7, 29.0, 29.2, 32.9, 34.0, 69.3, 113.5, 117.4, 128.2, 129.8, 140.6, 162.7.
1-(10-Bromodecyoxy)-4-nitrobenzene (6i).54
White powder, 1.0 g, yield 78%, starting from 0.5 g 4-nitrophenol (5a, 3.60 mmol).
Synthesis of 4-nitrophenoxy alkyl azoles (7a-l) and pyrrole 7m.
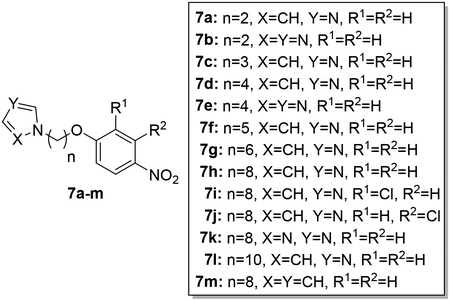
Synthesis of 7a-l.
To a solution of 6a-i (1.40–5.5 mmol) in dry acetonitrile (20 mL) was added 2 equivalents K2CO3 (2.80–11.00 mmol) and 2 equivalents of imidazole or triazole (2.80–11.00 mmol) and the mixture was stirred at 80 °C overnight. After the reaction was completed, the suspension was filtered and the filtrate was concentrated under reduced pressure. The crude product was subjected to silica gel chromatography employing DCM/MeOH 20:1 as the eluent to afford the product in 51–86% yield.
1-(2-(4-Nitrophenoxy)ethyl)-1H-imidazole (7a).
White powder, 0.72 g, yield 76% starting from 1.00 g 6a (4.06 mmol). 1H NMR (300 MHz, CDCl3) δ 4.28–4.33 (m, 2H), 4.37–4.43 (m, 2H), 6.93 (d, J = 9.3 Hz, 2H), 7.03 (t, 1.2 Hz, 1H), 7.08 (s, 1H), 7.60 (s, 1H), 8.20 (d, J = 9.3 Hz, 2H).
1-(2-(4-Nitrophenoxy)ethyl)-1H-1,2,4-triazole (7b).
White powder, 0.86 g, yield 75% starting from 1.20 g 6a (4.87 mmol). 1H NMR (300 MHz, CDCl3) δ 4.44 (t, J = 4.9 Hz, 2H), 4.62 (t, J = 5.0 Hz, 2H), 6.91 (d, J = 9.2 Hz, 2H), 7.96 (s, 1H), 8.14–8.23 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 49.0, 66.4, 114.6, 126.1, 142.3, 144.1, 152.6, 162.8.
1-(3-(4-Nitrophenoxy)propyl)-1H-imidazole (7c).55
White powder, 1.0 g, yield 78% starting from 1.35 g 6b (5.19 mmol).
1-(4-(4-Nitrophenoxy)butyl)-1H-imidazole (7d).55
White powder, 0.73 g, yield 51% starting from 1.5 g 6c (5.47 mmol).
1-(4-(4-Nitrophenoxy)butyl)-1H-1,2,4-triazole (7e).55
White powder, 1.10 g, yield 76% starting from 1.5 g 6c (5.47 mmol).
1-(5-(4-Nitrophenoxy)pentyl)-1H-imidazole (7f).
White powder, 0.96 g, yield 67% starting from 1.5 g 6d (5.20 mmol); 1H NMR (300 MHz, CDCl3) δ 1.42–1.54 (m, 2H), 1.78–1.92 (m, 4H), 3.97 (t, J = 7.0 Hz, 2H), 4.02 (t, J = 6.1 Hz, 2H), 6.88–6.95 (m, 3H), 7.05 (s, 1H), 7.46 (s, 1H), 8.17 (d, J = 9.2 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 23.3, 28.6, 30.9, 46.9, 68.4, 114.5, 118.8, 126.0, 129.7, 137.2, 141.6, 164.0.
1-(6-(4-Nitrophenoxy)hexyl)-1H-imidazole (7g).
White powder, 1.10 g, yield 72% starting from 1.6 g 6e (5.29 mmol); 1H NMR (300 MHz, CDCl3) δ 1.30–1.42 (m, 2H), 1.44–1.55 (m, 2H), 1.75–1.86 (m, 4H), 3.94 (t, J = 7.0 Hz, 2H), 4.01 (t, J = 6.3 Hz, 2H), 6.88–6.94 (m, 3H), 7.04 (s, 1H), 7.45 (s, 1H), 8.18 (d, J = 9.2 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 25.6, 26.4, 28.9, 31.1, 47.0, 68.6, 114.5, 118.8, 126.0, 129.6, 137.2, 141.6, 164.2.
1-(8-(4-Nitrophenoxy)octyl)-1H-imidazole (7h).
Off-white powder, 0.41 g, yield 86% starting from 0.5 g 6f (1.51 mmol); 1H NMR (300 MHz, CDCl3) δ 1.25–1.50 (m, 8H), 1.72–1.85 (m, 4H), 3.93 (t, J = 7.1 Hz, 2H), 4.03 (t, J = 6.4 Hz, 2H), 6.88–6.96 (m, 3H), 7.05 (s, 1H), 7.45 (s, 1H), 8.19 (d, J = 9.3 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 25.9, 26.6, 29.0, 29.1, 29.2, 31.2, 47.1, 68.9, 114.5, 118.9, 126.0, 129.6, 137.2, 141.5, 164.3.
1-(8-(2-Chloro-4-nitrophenoxy)octyl)-1H-imidazole (7i).
Yellow oil, 0.41 g, yield 60%, starting from 0.71 g 6f (1.94 mmol); 1H NMR (400 MHz, CDCl3) δ 1.25–1.41 (m, 6H), 1.43–1.53 (m, 2H), 1.72–1.81 (m, 2H), 1.81–1.90 (m, 2H), 3.92 (t, J = 7.1 Hz, 2H), 4.11 (t, J = 6.4 Hz, 2H), 6.89 (s, 1H), 6.95 (d, J = 9.1 Hz, 1H), 7.04 (s, 1H), 7.44 (s, 1H), 8.12 (dd, J = 9.1, 2.7 Hz, 1H), 8.27 (d, J = 2.7 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 25.9, 26.6, 28.8, 29.0, 29.1, 31.1, 47.1, 69.9, 111.8, 118.8, 123.5, 124.1, 126.1, 129.5, 137.2, 141.1, 159.8.
1-(8-(3-Chloro-4-nitrophenoxy)octyl)-1H-imidazole (7j).
Synthesis of 7j followed the general synthesis procedure except for the use of 1.2 equivalents of imidazole (115 mg, 1.69 mmol) and 1.2 equivalents of potassium carbonate (231 mg, 1.66 mmol) with gentle reflux overnight. Yellow oil, 0.24 g, yield 49% starting from 0.51 g 6f (1.39 mmol). 1H NMR (400 MHz, CDCl3) δ 1.22–1.48 (m, 8H), 1.71–1.83 (m, 4H), 3.92 (t, J = 7.1 Hz, 2H), 4.00 (t, J = 6.4 Hz, 2H), 6.83 (dd, J = 9.2, 2.6 Hz, 1H), 6.88 (s, 1H), 6.98 (d, J = 2.6 Hz, 1H), 7.03 (s, 1H), 7.44 (s, 1H), 7.97 (d, J = 9.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 25.8, 26.6, 28.9, 29.1, 29.2, 31.1, 47.1, 69.2, 113.5, 117.3, 118.8, 128.1, 129.5, 129.8, 137.2, 140.6, 162.6.
1-(8-(4-Nitrophenoxy)octyl)-1H-1,2,4-triazole (7k).
White powder, 0.41 g, yield 70% starting from 0.6 g 6f (1.81 mmol); 1H NMR (400 MHz, CDCl3) δ 1.24–1.49 (m, 8H), 1.75–1.84 (m, 2H), 1.85–1.94 (m, 2H), 4.03 (t, J = 6.4 Hz, 2H), 4.16 (t, J = 7.1 Hz, 2H), 6.92 (d, J = 9.2 Hz, 2H), 7.93 (s, 1H), 8.04 (s, 1H), 8.18 (d, J = 9.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 25.9, 26.5, 29.0, (overlapped 2Cs), 29.2, 29.9, 49.8, 68.9, 114.5, 126.0, 141.5, 142.9, 152.1, 164.3.
1-(10-(4-Nitrophenoxy)decyl)-1H-imidazole (7l).
Off-white powder, 0.32 g, yield 65%, starting from 0.5 g 6i (1.40 mmol); 1H NMR (300 MHz, CDCl3) δ 1.22–1.50 (m, 12H), 1.71–1.86 (m, 4H), 3.91 (t, J = 7.1 Hz, 2H), 4.03 (t, J = 6.5 Hz, 2H), 6.88–6.95 (m, 3H), 7.04 (s, 1H), 7.45 (s, 1H), 8.18 (d, J = 9.2 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 26.0, 26.6, 29.1, 29.2, 29.35, 29.45, 29.49, 31.2, 47.1, 69.0, 114.5, 118.9, 126.0, 129.5, 137.2, 141.5, 164.3.
Synthesis of 1-(8-(4-nitrophenoxy)octyl)-1H-pyrrole (7m).
Compound 7m was synthesized according to a previously published procedure.28 To a solution of pyrrole (0.21 g, 3.13 mmol) in DMF (5 mL) was added 1.14 equivalent of potassium hydroxide (0.20 g, 3.56 mmol) and the suspension was stirred at rt for 15 min. 1.16 equivalent of 6f (1.2 g, 3.56 mmol) was added and the mixture was stirred at 80 °C for 16h. After the reaction was complete, the mixture was quenched with water, extracted with ethyl acetate, and dried over sodium sulfate. The organic layer was evaporated under reduced pressure and purified by column chromatography using hexanes/DCM (5:1) as the eluent to yield the product as a yellow oil, 0.35 g, 35%. 1H NMR (300 MHz, CDCl3) δ 1.25–1.52 (m, 8H), 1.72–1.87 (m, 4H), 3.87 (t, J = 7.1 Hz, 2H), 4.04 (t, J = 6.5 Hz, 2H), 6.14 (t, J = 2.1 Hz, 2H), 6.65 (t, J = 2.1 Hz, 2H), 6.94 (d, J = 9.3 Hz, 2H), 8.19 (d, J = 9.3 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 26.0, 26.8, 29.0, 29.2, 29.3, 31.6, 49.7, 68.9, 107.9, 114.5, 120.6, 126.0, 141.5, 164.3.
Synthesis of 4-aminophenoxy alkyl azoles (8a-l) and pyrrole 8m.
To a solution of 7a-m (0.60–1.70 mmol) in ethyl acetate (20 mL) was added 5 equivalents of tin(II) chloride dihydrate (3.0–8.5 mmol) and the mixture was refluxed for 8–12 h. After the reaction was complete, the suspension was made basic with 1N NaOH solution (pH = 11) and extracted with ethyl acetate (30 mL × 3). The organic layer was dried over sodium sulfate and filtered. The filtrate was evaporated under reduced pressure yielding the product in 81–99% yield which was taken directly to the next step without further purification. For this reason, only 1H NMR spectra were obtained for 8a-l. Since the compounds were crude, 1H NMR spectra for the anilines reported in this manuscript often contain solvent peaks such as DCM and ethyl acetate along with traces of starting material in some cases (occasionally resulting in slightly higher than expected integration for the alkyl protons, particularly the most upfield alkyl multiplet).
4-(2-(1H-imidazol-1-yl)ethoxy)aniline (8a).
Yellow powder, 0.10 g, yield 82% starting from 0.14 g 7a (0.60 mmol). 1H NMR (300 MHz, DMSO-d6) δ 4.08 (t, J = 5.2 Hz, 2H), 4.27 (t, J = 5.2 Hz, 2H), 4.62 (brs, ArNH2), 6.49 (d, J = 8.8 Hz, 2H), 6.63 (d, J = 8.8 Hz, 2H), 6.88 (s, 1H), 7.21 (s, 1H), 7.65 (s, 1H).
4-(2-(1H-1,2,4-triazol-1-yl)ethoxy)aniline (8b).
Yellow powder, 0.28 g, yield 81% starting from 0.40 g 7b (1.70 mmol). 1H NMR (300 MHz, CDCl3) δ 3.26–3.65 (brs, ArNH2), 4.24 (t, J = 5.0 Hz, 2H), 4.50 (t, J = 5.0 Hz, 2H), 6.60 (d, J = 9.0 Hz, 2H), 6.67 (d, J = 9.3 Hz, 2H), 7.94 (s, 1H), 8.20 (s, 1H).
4-(3-(1H-imidazol-1-yl)propoxy)aniline (8c).
Brown oil, 0.26 g, yield 99% starting from 0.30 g 7c (1.21 mmol). 1H NMR (300 MHz, CDCl3) δ 2.12–2.23 (m, 2H), 2.86–3.93 (brs, ArNH2), 3.83 (t, J = 5.7 Hz, 2H), 4.17 (t, J = 6.8 Hz, 2H), 6.63 (d, J = 8.8 Hz, 2H), 6.72 (d, J = 8.9 Hz, 2H), 6.91 (s, 1H), 7.05 (s, 1H), 7.48 (s, 1H).
4-(4-(1H-imidazol-1-yl)butoxy)aniline (8d).
Buff powder, 0.25 g, yield 95% starting from 0.3 g 7d (1.14 mmol); 1H NMR (300 MHz, DMSO-d6) δ 1.67–1.78 (m, 2H), 1.91–2.02 (m, 2H), 3.09–3.46 (brs, ArNH2), 3.88 (t, J = 6.0 Hz, 2H), 4.00 (t, J = 7.1 Hz, 2H), 6.62 (d, J = 8.9 Hz, 2H), 6.71 (d, J = 8.9 Hz, 2H), 6.92 (s, 1H), 7.05 (s, 1H), 7.47 (s, 1H).
4-(4-(1H-1,2,4-triazol-1-yl)butoxy)aniline (8e).
Buff powder, 0.32 g, yield 91% starting from 0.40 g 7e (1.52 mmol); 1H NMR (300 MHz, CDCl3) δ 1.68–1.79 (m, 2H), 2.02–2.13 (m, 2H), 3.16–3.69 (brs, ArNH2), 3.91 (t, J = 6.0 Hz, 2H), 4.24 (t, J = 7.1 Hz, 2H), 6.62 (d, J = 8.9 Hz, 2H), 6.71 (d, J = 8.9 Hz, 2H), 7.93 (s, 1H), 8.06 (s, 1H).
4-((5-(1H-imidazole-1-yl)pentyl)oxy)aniline (8f).
Brown oil, 0.29 g, yield 99% starting from 0.33 g 7f (1.19 mmol); 1H NMR (300 MHz, CDCl3) δ 1.40–1.52 (m, 2H), 1.70–1.89 (m, 4H), 3.08–3.41 (brs, ArNH2), 3.86 (t, J = 6.2 Hz, 2H), 3.94 (t, J = 7.1 Hz, 2H), 6.62 (d, J = 8.9 Hz, 2H), 6.71 (d, J = 8.9 Hz, 2H), 6.90 (s, 1H), 7.05 (s, 1H), 7.47 (s, 1H).
4-((6-(1H-imidazole-1-yl)hexyl)oxy)aniline (8g).
Brown oil, 0.30 g, yield 92% starting from 0.35 g 7g (1.20 mmol); 1H NMR (300 MHz, CDCl3) δ 1.29–1.41 (m, 2H), 1.42–1.54 (m, 2H), 1.67–1.86 (m, 4H), 2.80–3.35 (brs, ArNH2), 3.86 (t, J = 6.3 Hz, 2H), 3.93 (t, J = 7.1 Hz, 2H), 6.63 (d, J = 8.9 Hz, 2H), 6.72 (d, J = 8.9 Hz, 2H), 6.89 (s, 1H), 7.05 (s, 1H), 7.46 (s, 1H).
4-((8-(1H-imidazole-1-yl)octyl)oxy)aniline (8h).
Orange powder, 0.23 g, yield 85%, starting from 0.30 g 7h (0.95 mmol); 1H NMR (400 MHz, CDCl3) δ 1.23–1.36 (m, 6H), 1.37–1.46 (m, 2H), 1.67–1.80 (m, 4H), 3.18–3.58 (brs, ArNH2), 3.86 (t, J = 6.5 Hz, 2H), 3.90 (t, J = 7.1 Hz, 2H), 6.62 (brd, J = 8.7 Hz m, 2H), 6.72 (brd, J = 8.7 Hz, 2H), 6.89 (s, 1H), 7.04 (s, 1H), 7.44 (s, 1H).
4-((8-(1H-imidazole-1-yl)octyl)oxy)-3-chloroaniline (8i).
Yellow oil, 0.17 g, yield 93%, starting from 0.20 g 7i (0.57 mmol); 1H NMR (300 MHz, CDCl3) δ 1.24–1.39 (m, 6H), 1.40–1.51 (m, 2H), 1.70–1.82 (m, 4H), 2.95–3.70 (brs, ArNH2), 3.91 (t, J = 6.7 Hz, 4H), 6.52 (dd, J = 8.6, 2.8 Hz, 1H), 6.73 (d, J = 2.8 Hz, 1H), 6.75 (d, J = 8.6 Hz, 1H), 6.89 (s, 1H), 7.04 (s, 1H), 7.45 (s, 1H).
4-((8-(1H-imidazole-1-yl)octyl)oxy)-2-chloroaniline (8j).
Yellow oil, 0.23 g, yield 97%, starting from 0.26 g 7j (0.74 mmol); 1H NMR (400 MHz, CDCl3) δ 1.23–1.45 (m, 8H), 1.66–1.81 (m, 4H), 3.61–3.80 (brs, ArNH2, partially under other peaks), 3.84 (t, J = 6.5 Hz, 2H), 3.91 (t, J = 7.1 Hz, 2H), 6.62–6.71 (m, 2H), 6.83 (d, J = 2.4 Hz, 1H), 6.89 (s, 1H), 7.04 (s, 1H), 7.46 (s, 1H).
4-((8-(1H-1,2,4-triazol-1-yl)octyl)oxy)aniline (8k).
Buff powder, 0.21 g, yield 93%, starting from 0.25 g 7k (0.78 mmol); 1H NMR (400 MHz, CDCl3) δ 1.23–1.46 (m, 8H), 1.66–1.75 (m, 2H), 1.83–1.91 (m, 2H), 3.26–3.53 (brs, ArNH2), 3.85 (t, J = 6.5 Hz, 2H), 4.14 (t, J = 7.1 Hz, 2H), 6.62 (brd, J = 8.8 Hz, 2H), 6.72 (brd, J = 8.8 Hz, 2H), 7.92 (s, 1H), 8.03 (s, 1H).
4-((10-(1H-imidazole-1-yl)decyl)oxy)aniline (8l).
Buff powder, 0.20 g, yield 88%, starting from 0.25 g 7l (0.72 mmol); 1H NMR (300 MHz, CDCl3) δ 1.23–1.47 (m, 12H), 1.67–1.81 (m, 4H), 3.07–3.64 (brs, ArNH2), 3.83–3.95 (m, 4H), 6.63 (d, J = 9.0 Hz, 2H), 6.73 (d, J = 8.9 Hz, 2H), 6.89 (t, J = 1.2 Hz, 1H), 7.05 (s, 1H), 7.45 (s, 1H).
4-((8-(1H-pyrrol-1-yl)octyl)oxy)aniline (8m).
The general procedure listed above was used to prepare 8m, but this material was also prepared via a Zn-mediated reduction which is described here. To a solution of 7m (170 mg, 0.54 mmol) in MeOH (10 mL) was added ammonium chloride (285 mg, 5.30 mmol) and Zn (350 mg, 5.40 mmol) and the mixture was stirred at room temperature for 3 h. After the reaction was complete, the reaction mixture was filtered through celite. The celite was washed with MeOH (10 mL), then the combined MeOH filtrate was evaporated under reduced pressure to obtain a crude solid. To the crude solid was added water (20 mL), followed by extraction with DCM (2×20 mL). The organic layers were collected, dried over anhydrous sodium sulfate and evaporated under reduced pressure to obtain the title product as yellow oil (130 mg, 85%). 1H NMR (300 MHz, CDCl3) δ 1.23–1.47 (m, 8H), 1.68–1.80 (m, 4H), 3.83–3.89 (m, 4H), 6.13 (t, J = 2.1 Hz, 2H), 6.64 (t, J = 2.1 Hz, 2H), 6.67 (d, J = 8.8 Hz, 2H), 6.74 (d, J = 8.8 Hz, 2H).
Synthesis of hybrid target compounds 9a-n.
Synthesis of AIA azole hybrid target compounds followed previously published procedures for the synthesis of arylimidamides.6, 7, 56 Two equivalents of S-(2-naphthylmethyl)-2-pyridyl thioimidate hydrobromide (0.76–1.38 mmol) or S-(2-naphthylmethyl) benzimidothioate hydrobromide (1.04 mmol) were added to one equivalent of a cooled solution of the arylamines 8a-m (0.38–0.69 mmol) in dry acetonitrile:ethanol (3:10 mL) in an ice bath for 15 minutes, then this solution was warmed to room temperature. The reaction mixture was stirred at room temperature for 24–48 h. After the disappearance of the starting material, the organic solvent was evaporated under reduced pressure to yield a crude oil product. Dry ether (100 mL) was added to the crude material and the mixture was stirred at room temperature overnight. The precipitate was filtered and washed with dry ether. The solid was dissolved in ethanol (2 mL), then the solution was cooled to 0 °C in an ice bath and 10% NaOH was added until the pH reached approximately 11. The free base was extracted with ethyl acetate (3 × 30 mL). The organic layer was washed with distilled water, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure yielding a waxy or solid product which was further purified by column chromatography on triethylamine neutralized silica using DCM:MeOH as an eluent (100:0.5 to 100:3). The product was further crystalized using hexanes/ethyl acetate to yield the final product in 33–70% yield.
N-(4-(2-(1H-imidazol-1-yl)ethoxy)phenyl) picolinimidamide (9a).
Yellow powder; 63 mg; yield 42% (starting from 0.10 g of 8a, 0.49 mmol); mp 141–144 °C; 1H NMR (400 MHz, DMSO-d6) δ 4.21 (t, J = 5.2 Hz, 2H), 4.35 (t, J = 5.1 Hz, 2H), 6.26–6.59 (brs, imidamide NHs), 6.82–6.93 (m, 5H), 7.25 (t, J = 1.2 Hz, 1H), 7.53 (ddd, J = 7.5, 4.8, 1.2 Hz, 1H), 7.68 (s, 1H), 7.93 (td, J = 7.7, 1.8 Hz, 1H), 8.29 (d, J = 7.9 Hz, 1H), 8.61 (ddd, J = 4.8, 1.7, 0.9 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ 45.7, 67.4, 115.3, 119.7, 121.1, 122.4, 125.3, 128.3, 137.0, 137.6, 143.6, 148.0, 151.6, 151.8, 153.4; HRMS (ESI) m/z (M +H)+ calcd for C17H18N5O, 308.15059; found, 308.15057; Anal. Calcd for C17H17N5O: C, 66.43; H, 5.58; N, 22.79. Found: C, 66.15; H, 5.71; N, 22.49.
N-(4-(2-(1H-1,2,4-triazol-1-yl)ethoxy)phenyl) picolinimidamide (9b).
Buff powder; 107 mg; yield 59% (starting from 0.12 g of 8b, 0.58 mmol); mp 115–118 °C; 1H NMR (400 MHz, CDCl3) δ 4.33 (t, J = 5.0 Hz, 2H), 4.56 (t, J = 5.0 Hz, 2H), 5.40–6.21 (brs, imidamide NHs), 6.86 (brd, J = 8.8 Hz, 2H), 6.93 (brd, J = 8.6 Hz, 2H), 7.38 (ddd, J = 7.4, 4.9, 1.1 Hz, 1H), 7.80 (td, J = 7.8, 1.7 Hz, 1H), 7.96 (s, 1H), 8.23 (s, 1H), 8.38 (d, J = 8.0 Hz, 1H), 8.56 (br d, J = 4.4 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 49.6, 66.2, 115.9, 121.6, 122.8, 125.3, 136.9, 144.2, 148.0, 151.7, 152.2, 153.1, 154.0; HRMS (ESI) m/z (M +H)+ calcd for C16H17N6O, 309.14584; found, 309.14569; Anal. Calcd for C16H16N6O: C, 62.32; H, 5.23; N, 27.26. Found: C, 62.39; H, 5.28; N, 27.18.
N-(4-(3-(1H-imidazol-1-yl)propoxy)phenyl) picolinimidamide (9c).
Yellow powder; 85 mg; yield 38% (starting from 0.15 g of 8c, 0.69 mmol); mp 115–118 °C; 1H NMR (700 MHz, CDCl3) δ 2.20–2.25 (m, 2H), 3.91 (t, J = 5.7 Hz, 2H), 4.21 (t, J = 6.8 Hz, 2H), 5.35–6.49 (brs, imidamide NHs), 6.90 (d, J = 8.8 Hz, 2H), 6.94 (t, J = 1.2 Hz, 1H), 6.97 (brd, J = 8.6 Hz, 2H), 7.07 (s, 1H), 7.39 (ddd, J = 7.5, 4.8, 1.1 Hz, 1H), 7.49 (s, 1H), 7.81 (td, J = 7.7, 1.7 Hz, 1H), 8.41 (d, J = 7.9 Hz, 1H), 8.57 (ddd, J = 4.8, 1.7, 0.9 Hz, 1H); 13C NMR (176 MHz, CDCl3) δ 31.0, 43.6, 64.1, 115.7, 119.1, 121.6, 122.8, 125.3, 129.8, 136.9, 137.6, 143.5, 148.0, 151.7, 153.2, 154.7; HRMS (ESI) m/z (M +H)+ calcd for C18H20N5O, 322.16624; found, 322.16592; Anal. Calcd for C18H19N5O: C, 67.27; H, 5.96; N, 21.79. Found: C, 67.42; H, 5.98; N, 21.78.
N-(4-(4-(1H-imidazol-1-yl)butoxy)phenyl) picolinimidamide (9d).
Buff powder; 132 mg; yield 65% (starting from 0.14 g of 8d, 0.60 mmol); mp 103–105 °C; 1H NMR (400 MHz, CDCl3) δ 1.74–1.82 (m, 2H), 1.96–2.05 (m, 2H), 3.97 (t, J = 6.0 Hz, 2H), 4.03 (t, J = 7.0 Hz, 2H), 5.43–6.30 (brs, imidamide NHs), 6.89 (d, J = 8.9 Hz, 2H), 6.92–6.98 (m, 3H), 7.07 (t, J = 0.9 Hz, 1H), 7.38 (ddd, J = 7.5, 4.8, 1.2 Hz, 1H), 7.49 (s, 1H), 7.80 (td, J = 7.7, 1.7 Hz, 1H), 8.40 (d, J = 7.9 Hz, 1H), 8.56 (ddd, J = 4.8, 1.7, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 26.5, 28.2, 46.9, 67.5, 115.7, 118.9, 121.6, 122.7, 125.2, 129.7, 136.9, 137.2, 143.3, 148.0, 151.8, 153.1, 154.9; HRMS (ESI) m/z (M +H)+ calcd for C19H22N5O, 336.18189; found, 336.18155; Anal. Calcd for C19H21N5O: C, 68.04; H, 6.31; N, 20.88. Found: C, 68.20; H, 6.35; N, 20.77.
N-(4-(4-(1H-1,2,4-triazol-1-yl)butoxy)phenyl) picolinimidamide (9e).
Buff powder; 141 mg; yield 70% (starting from 0.14 g of 8e, 0.60 mmol); mp 109–111 °C; 1H NMR (400 MHz, CDCl3) δ 1.75–1.83 (m, 2H), 2.08–2.16 (m, 2H), 3.99 (t, J = 6.0 Hz, 2H), 4.27 (t, J = 7.0 Hz, 2H), 5.32–6.31 (brs, imidamide NHs), 6.89 (d, J = 8.9 Hz, 2H), 6.96 (brd, J = 8.7 Hz, 2H), 7.38 (ddd, J = 7.5, 4.8, 1.2 Hz, 1H), 7.80 (td, J = 7.8, 1.8 Hz, 1H), 7.95 (s, 1H), 8.08 (s, 1H), 8.40 (d, J = 7.9 Hz, 1H), 8.56 (ddd, J = 4.8, 1.7, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 26.3, 27.1, 49.5, 67.5, 115.6, 121.62, 122.7, 125.2, 136.9, 143.1, 143.3, 148.0, 151.8, 152.1, 153.1, 154.9; HRMS (ESI) m/z (M +H)+ calcd for C18H21N6O, 337.17714; found, 337.17712; Anal. Calcd for C18H20N6O: C, 64.27; H, 5.99; N, 24.98. Found: C, 64.43; H, 6.00; N, 24.93.
N-(4-((5-(1H-imidazol-1-yl)pentyl)oxy)phenyl) picolinimidamide (9f).
Light brown powder; 70 mg; yield 33% (starting from 0.15 g of 8f, 0.61 mmol); mp 128–131 °C; 1H NMR (700 MHz, CDCl3) δ 1.47–1.53 (m, 2H), 1.78–1.83 (m, 2H), 1.84–1.89 (m, 2H), 3.94 (t, J = 6.2 Hz, 2H), 3.97 (t, J = 7.2 Hz, 2H), 5.37–6.48 (brs, imidamide NHs), 6.89 (d, J = 8.8 Hz, 2H), 6.91 (t, J = 1.1 Hz, 1H) 6.95 (brd, J = 7.5 Hz, 2H), 7.06 (s, 1H), 7.38 (ddd, J = 7.5, 4.8, 1.1 Hz, 1H), 7.47 (s, 1H), 7.80 (td, J = 7.7, 1.7 Hz, 1H), 8.40 (d, J = 8.0 Hz, 1H), 8.57 (ddd, J = 4.8, 1.6, 0.8 Hz, 1H); 13C NMR (176 MHz, CDCl3) δ 23.4, 28.9, 31.0, 47.0, 67.8, 115.6, 118.9, 121.6, 122.6, 125.2, 129.6, 136.9, 137.2, 143.2, 148.0, 151.8, 153.1, 155.1; HRMS (ESI) m/z (M +H)+ calcd for C20H24N5O, 350.19754; found, 350.19757; Anal. Calcd for C20H23N5O: C, 68.74; H, 6.63; N, 20.04. Found: C, 68.45; H, 6.60; N, 19.90.
N-(4-((6-(1H-imidazol-1-yl)hexyl)oxy)phenyl) picolinimidamide (9g).
Light brown powder; 70 mg; yield 33% (starting from 0.15 g of 8g, 0.57 mmol); mp 127–129 °C; 1H NMR (400 MHz, CDCl3) δ 1.33–1.43 (m, 2H), 1.48–1.56 (m, 2H), 1.73–1.87 (m, 4H), 3.92–3.97 (m, 4H), 5.61–6.12 (brs, imidamide NHs), 6.88–6.98 (m, 5H), 7.06 (s, 1H), 7.39 (ddd, J = 7.4, 4.9, 1.0 Hz, 1H), 7.46 (s, 1H), 7.81 (td, J = 7.8, 1.7 Hz, 1H), 8.41 (d, J = 8.0 Hz, 1H), 8.57 (d, J = 4.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 25.8, 26.5, 29.3, 31.2, 47.1, 68.0, 115.7, 118.9, 121.6, 122.6, 125.2, 129.6, 136.9, 137.2, 143.1, 148.0, 151.9, 153.1, 155.2; HRMS (ESI) m/z (M +H)+ calcd for C21H26N5O, 364.21319; found, 364.21298; Anal. Calcd for C21H25N5O: C, 69.40; H, 6.93; N, 19.27. Found: C, 69.43; H, 6.81; N, 19.13.
N-(4-((8-(1H-imidazol-1-yl)octyl)oxy)phenyl) picolinimidamide (9h).
Light brown powder; 85 mg; yield 44% (starting from 0.14 g of 8h, 0.49 mmol); mp 89–92 °C; 1H NMR (400 MHz, CDCl3) δ 1.28–1.40 (m, 6H), 1.41–1.50 (m, 2H), 1.73–1.82 (m, 4H), 3.90–3.97 (m, 4H), 5.47–6.30 (brs, imidamide NHs), 6.89–6.93 (m, 3H), 6.96 (d, J = 8.9 Hz, 2H), 7.05 (t, J = 1.0 Hz, 1H), 7.39 (ddd, J = 7.5, 4.8, 1.2 Hz, 1H), 7.45 (s, 1H), 7.81 (td, J = 7.8, 1.8 Hz, 1H), 8.42 (d, J = 8.0 Hz, 1H), 8.57 (ddd, J = 4.9, 1.7, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 26.1, 26.6, 29.1, 29.3, 29.4, 31.2, 47.2, 68.3, 115.7, 118.9, 121.7, 122.7, 125.2, 129.6, 136.9, 137.2, 142.7, 148.0, 151.8, 153.2, 155.4; HRMS (ESI) m/z (M +H)+ calcd for C23H30N5O, 392.24449; found, 392.24451; Anal. Calcd for C23H29N5O: C, 70.56; H, 7.47; N, 17.89. Found: C, 70.27; H, 7.29; N, 17.70.
N-(4-((8-(1H-imidazol-1-yl)octyl)oxy)-3-chlorophenyl) picolinimidamide (9i).
Yellow powder; 99 mg; yield 54% (starting from 0.14 g of 8i, 0.43 mmol); mp 102–104 °C; 1H NMR (400 MHz, CDCl3) δ 1.25–1.41 (m, 6H), 1.45–1.55 (m, 2H), 1.74–1.86 (m, 4H), 3.92 (t, J = 7.1 Hz, 2H), 4.01 (t, J = 6.4 Hz, 2H), 5.42–6.39 (brs, imidamide NHs), 6.86 (dd, J = 8.6, 2.4 Hz, 1H), 6.90 (t, J = 1.2 Hz, 1H), 6.93 (d, J = 8.6 Hz, 1H), 7.05 (s, 1H), 7.08 (d, J = 2.4 Hz, 1H), 7.39 (ddd, J = 7.5, 4.8, 1.2 Hz, 1H), 7.45 (s, 1H), 7.81 (td, J = 7.7, 1.7 Hz, 1H), 8.37 (d, J = 8.0 Hz, 1H), 8.57 (ddd, J = 4.8, 1.7, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 26.0, 26.6, 29.1, 29.2, 29.3, 31.2, 47.2, 69.7, 115.0, 118.9, 120.8, 121.7, 123.6, 123.9, 125.4, 129.6, 137.0, 137.2, 143.6, 148.0, 150.7, 151.5, 153.3; HRMS (ESI) m/z (M +H)+ and ((M+2) +H)+ calcd for C23H29N5ClO, 426.20551 and 428.20261; found, 426.20490 and 428.20236; Anal. Calcd for C23H28N5ClO: C, 64.85; H, 6.63; N, 16.44. Found: C, 64.81; H, 6.67; N, 16.32.
N-(4-((8-(1H-imidazol-1-yl)octyl)oxy)-2-chlorophenyl) picolinimidamide (9j).
White powder; 71 mg; yield 39% (starting from 0.14 g of 8j, 0.43 mmol); mp 74–76 °C; 1H NMR (400 MHz, CDCl3) δ 1.24–1.40 (m, 6H), 1.40–1.50 (m, 2H), 1.72–1.83 (m, 4H), 3.89–3.95 (m, 4H), 5.40–6.38 (brs, imidamide NHs), 6.82 (dd, J = 8.7, 2.7 Hz, 1H), 6.90 (s, 1H), 6.95 (d, J = 8.7 Hz, 1H), 7.00 (d, J = 2.7 Hz, 1H), 7.05 (s, 1H), 7.40 (ddd, J = 7.4, 4.8, 1.0 Hz, 1H), 7.46 (s, 1H), 7.82 (td, J = 7.7, 1.6 Hz, 1H), 8.46 (d, J = 8.0 Hz, 1H), 8.58 (d, J = 4.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 26.0, 26.6, 29.1, 29.27, 29.30, 31.2, 47.2, 68.6, 114.7, 116.2, 118.9, 122.0, 123.4, 125.4, 126.7, 129.6, 137.0, 137.2, 139.7, 148.0, 151.3, 153.4, 155.6; HRMS (ESI) m/z (M +H)+ and ((M+2) +H)+ calcd for C23H29N5ClO, 426.20551 and 428.20261; found, 426.20663 and 428.20416; Anal. Calcd for C23H28N5ClO: C, 64.85; H, 6.63; N, 16.44. Found: C, 64.68; H, 6.58; N, 16.38.
N-(4-((8-(1H-1,2,4-triazol-1-yl)octyl)oxy)phenyl) picolinimidamide (9k).
Light brown powder; 85 mg; yield 39% (starting from 0.16 g of 8k, 0.55 mmol); mp 98–100 °C; 1H NMR (700 MHz, CDCl3) δ 1.28–1.39 (m, 6H), 1.42–1.48 (m, 2H), 1.74–1.79 (m, 2H), 1.87–1.92 (m, 2H), 3.94 (t, J = 6.4 Hz, 2H), 4.16 (t, J = 7.1 Hz, 2H), 5.29–6.44 (brs, imidamide NHs), 6.91 (d, J = 8.8 Hz, 2H), 6.96 (brd, J = 8.6 Hz, 2H), 7.38 (ddd, J = 7.4, 4.9, 1.0 Hz, 1H), 7.80 (td, J = 7.7, 1.7 Hz, 1H), 7.93 (s, 1H), 8.04 (s, 1H), 8.41 (d, J = 7.9 Hz, 1H), 8.56 (d, J = 4.7 Hz, 1H); 13C NMR (176 MHz, CDCl3) δ 26.1, 26.5, 29.1, 29.3, 29.4, 29.9, 49.8, 68.3, 115.7, 121.7, 122.7, 125.2, 136.9, 142.9, 148.0, 151.7, 152.0, 153.2, 155.4; HRMS (ESI) m/z (M +H)+ calcd for C22H29N6O, 393.23974; found, 393.23932; Anal. Calcd for C22H28N6O: C, 67.32; H, 7.19; N, 21.41. Found: C, 67.18; H, 7.16; N, 21.47.
N-(4-((10-(1H-imidazol-1-yl)decyl)oxy)phenyl) picolinimidamide (9l).
Light brown powder; 61 mg; yield 38% (starting from 0.12 g of 8l, 0.38 mmol); mp 104–106 °C; 1H NMR (400 MHz, CDCl3) δ 1.24–1.39 (m, 10H), 1.41–1.50 (m, 2H), 1.73–1.81 (m, 4H), 3.89–3.97 (m, 4H total, overlapped), 3.91 (t, J = 7.1 Hz, overlapped), 3.94 (t, J = 6.5 Hz, overlapped), 5.38–6.29 (brs, imidamide NHs), 6.89–6.98 (m, 5H), 7.05 (s, 1H), 7.38 (ddd, J = 7.4, 4.8, 1.1 Hz, 1H), 7.45 (s, 1H), 7.80 (td, J = 7.7, 1.7 Hz, 1H), 8.41 (d, J = 8.0 Hz, 1H), 8.56 (ddd, J = 4.8, 1.7, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 26.2, 26.7, 29.2, 29.46, 29.47, 29.50, 29.55, 31.2, 47.2, 68.4, 115.7, 118.9, 121.7, 122.7, 125.2, 129.5, 136.9, 137.2, 142.5, 148.0, 151.8, 153.2, 155.4; HRMS (ESI) m/z (M +H)+ calcd for C25H34N5O, 420.27579; found, 420.27637; Anal. Calcd for C25H33N5O: C, 71.57; H, 7.93; N, 16.69. Found: C, 71.27; H, 7.66; N, 16.47.
N-(4-((8-(1H-pyrrol-1-yl)octyl)oxy)phenyl) picolinimidamide (9m).
The synthesis of 9m follows the general synthesis of 9a-l with the modification of using 1.5 equivalents of S-(2-naphthylmethyl)-2-pyridyl thioimidate hydrobromide and, after completion of the reaction, no diethyl ether treatment was performed. The compound was purified by column chromatography using hexanes/DCM (1:1.5) on triethylamine neutralized silica. After column chromatography purification step, the powder was triturated with diethyl ether/hexanes (3 × 10 mL) followed by filtration. The filtrate was further crystalized using hexanes/ethyl acetate to yield the final product Light brown powder, 58 mg, yield 17% (starting from 0.25 g of 8m, 0.87 mmol); mp 93–95 °C; 1H NMR (400 MHz, CDCl3) δ 1.26–1.40 (m, 6H), 1.42–1.51 (m, 2H), 1.73–1.82 (m, 4H), 3.87 (t, J = 7.2 Hz, 2H), 3.94 (t, J = 6.5 Hz, 2H), 5.52–6.09 (brs, imidamide NHs), 6.14 (t, J = 2.1 Hz, 2H), 6.65 (t, J = 2.1 Hz, 2H), 6.89–6.99 (m, 4H), 7.39 (ddd, J = 7.5, 4.8, 1.1 Hz, 1H), 7.81 (td, J = 7.8, 1.7 Hz, 1H), 8.41 (d, J = 8.0 Hz, 1H), 8.57 (d, J = 4.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 26.2, 26.9, 29.3, 29.4, 29.5, 31.7, 49.8, 68.4, 107.9, 115.7, 120.6, 121.6, 122.6, 125.2, 136.9, 143.0, 148.0, 151.9, 153.0, 155.4; HRMS (ESI) m/z (M +H)+ calcd for C24H31N4O, 391.24924; found, 391.24988; Anal. Calcd for C24H30N4O: C, 73.81; H, 7.74; N, 14.35. Found: C, 74.03; H, 7.89; N, 14.25.
N-(4-((8-(1H-imidazol-1-yl)octyl)oxy)phenyl)benzimidamide (9n).
Buff powder; 45 mg; yield 22% (starting from 0.15 g of 8h, 0.52 mmol); mp 107–109 °C; 1H NMR (700 MHz, CDCl3) δ 1.28–1.38 (m, 6H), 1.42–1.48 (m, 2H), 1.74–1.80 (m, 4H), 3.90–3.95 (m, 4H), 4.68–5.18 (brs, imidamide NHs), 6.87–6.94 (m, 5H), 7.04 (s, 1H), 7.41–7.48 (m, 4H), 7.85 (brs, 2H); 13C NMR (176 MHz, CDCl3) δ 26.1, 26.6, 29.1, 29.3, 29.4, 31.2, 47.1, 68.3, 115.6, 118.9, 122.7, 126.9, 128.7, 129.5, 130.6, 136.1, 137.2, 142.6, 155.3; HRMS (ESI) m/z (M +H)+ calcd for C24H31N4O, 391.24924; found, 391.24905; Anal. Calcd for C24H30N4O: C, 73.81; H, 7.74; N, 14.35. Found: C, 73.53; H, 7.80; N, 14.26.
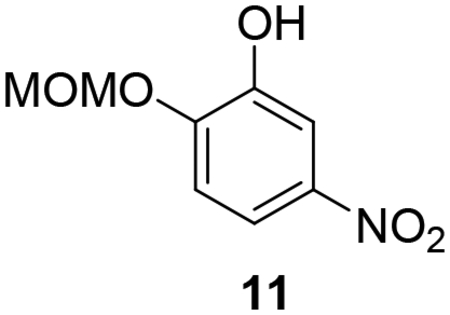
2-(Methoxymethoxy)-5-nitrophenol (11).
Compound 11 was synthesized following a previously published procedure.29 To a solution of 4-nitrocatechol (10, 1.0 g, 6.44 mmol) in dry DMF (10 mL) was added 1.2 equivalent of potassium carbonate (1.06 g, 7.67 mmol) and 1.1 equivalent of MOMCl (0.57 g, 7.08 mmol). The reaction mixture was heated to 40 °C for 2 hours. After completion of the reaction, the solvent was removed under reduced pressure, water was added and the product was extracted with ethyl acetate (3 × 30 mL). The combined organic layer was washed with brine, dried over sodium sulfate, and evaporated under reduced pressure. The crude product was purified by column chromatography using hexanes/ethyl acetate (4:1) as eluent yielding the product as a buff powder, 0.61 g, 48%.
Synthesis of 2-alkoxy-1-(methoxymethoxy)-4-nitrobenzenes (12a-c).
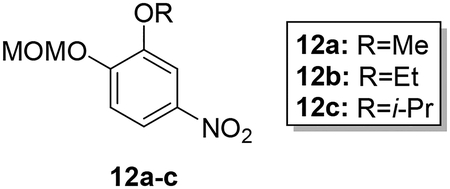
To a solution of 11 (1.30–1.50 mmol) in dry acetonitrile (5 mL) was added 2 equivalents of potassium carbonate (2.60–3.00 mmol) and 3 equivalents of alkyl iodide (3.90–4.50 mmol). The mixture was heated in a sealed tube at 80 °C for 4–5 hours. After the reaction was complete, the solution was cooled and the solvent was removed under reduced pressure. Ice was added to precipitate the pure product, which was filtered as a white powder in 91–97% yield.
2-Methoxy-1-(methoxymethoxy)-4-nitrobenzene (12a).
0.29 g, yield 97%, starting from 0.28 g 11 (1.40 mmol). 1H NMR (300 MHz, CDCl3) δ 3.51 (s, 3H), 3.96 (s, 3H), 5.32 (s, 2H), 7.21 (d, J = 8.9 Hz, 1H), 7.77 (d, J = 2.6 Hz, 1H), 7.86 (dd, J = 8.9, 2.6 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 56.5, 56.8, 95.3, 107.1, 114.4, 117.6, 142.5, 149.6, 152.2.
2-Ethoxy-1-(methoxymethoxy)-4-nitrobenzene (12b).
0.28 g, yield 95%, starting from 0.26 g 11 (1.30 mmol). 1H NMR (300 MHz, CDCl3) δ 1.49 (t, J = 7.0 Hz, 3H), 3.51 (s, 3H), 4.18 (q, J = 7.0 Hz, 2H), 5.31 (s, 2H), 7.20 (d, J = 9.0 Hz, 1H), 7.75 (d, J = 2.6 Hz, 1H), 7.84 (dd, J = 9.0, 2.6 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 14.7, 56.7, 65.1, 95.4, 108.2, 115.0, 117.4, 142.6, 149.0, 152.4.
2-Isopropoxy-1-(methoxymethoxy)-4-nitrobenzene (12c).
0.33 g, yield 91%, starting from 0.30 g 11 (1.50 mmol). 1H NMR (300 MHz, CDCl3) δ 1.40 (d, J = 6.1 Hz, 6H), 3.51 (s, 3H), 4.62 (sep, J = 6.1 Hz, 1H), 5.28 (s, 2H), 7.18 (d, J = 9.0 Hz, 1H), 7.77 (d, J = 2.6 Hz, 1H), 7.83 (dd, J = 9.0, 2.6 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 22.0, 56.7, 72.4, 95.3, 110.7, 115.5, 117.5, 142.6, 148.1, 153.4.
Synthesis of 2-alkoxy-4-nitrobenzenes (13a-c).
To a cooled solution of 12a-c (1.11–1.31 mmol) in methanol/DCM (6:3) at 0 °C was added 4N HCl (1 mL) and the mixture was stirred for 30 min. The mixture was then warmed to rt and allowed to stir for another 2 hours. After the reaction was complete, ethyl acetate (30 mL) was added and the product was extracted (2 × 30 mL) with ethyl acetate. The combined organic layer was washed with water and brine, dried over sodium sulfate, and evaporated under reduced pressure. The crude product was purified by column chromatography affording the product in 82–89% yield.
2-Methoxy-4-nitrophenol (13a).57
Chromatography solvent: hexanes/DCM 1:1, yellow powder, 0.18 g, yield 81% starting from 0.28 g 12a (1.31 mmol).
2-Ethoxy-4-nitrophenol (13b).58
Chromatography solvent: hexanes/DCM 2:1 to 2:3, yellow powder, 0.18 g, yield 89% starting from 0.25 g 12b (1.11 mmol).
2-Isopropoxy-4-nitrophenol (13c).
Chromatography solvent: hexanes/DCM 2:1, yellow waxy oil, 0.23 g, yield 90% starting from 0.31 g 12c (1.29 mmol); 1H NMR (300 MHz, CDCl3) δ 1.42 (d, J = 6.1 Hz, 6H), 4.71 (sep, J = 6.1 Hz, 1H), 6.31 (brs, 1H), 6.97 (d, J = 8.8 Hz, 1H), 7.75 (d, J = 2.5 Hz, 1H), 7.84 (dd, J = 8.8, 2.5 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 22.0, 72.7, 108.4, 114.1, 118.4, 141.2, 144.3, 152.6.
Synthesis of 1-((8-bromooctyl)oxy)-2-alkoxy-4-nitrobenzenes (14a-c).
Compounds 14a-c were prepared from 13a-c (0.92–1.11 mmol) following the general synthesis of compounds 6a-i.
1-((8-Bromooctyl)oxy)-2-methoxy-4-nitrobenzene (14a).
Yellowish white powder, 0.33 g, yield 91% starting from 0.17 g 13a (1.00 mmol); 1H NMR (300 MHz, CDCl3) δ 1.30–1.54 (m, 8H), 1.80–1.93 (m, 4H), 3.40 (t, J = 6.8 Hz, 2H), 3.94 (s, 3H), 4.10 (t, J = 6.7 Hz, 2H), 6.88 (d, J = 9.0 Hz, 1H), 7.73 (d, J = 2.6 Hz, 1H), 7.88 (dd, J = 8.9, 2.6 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 25.9, 28.2, 28.7, 28.9, 29.2, 32.8, 34.0, 56.4, 69.6, 106.8, 110.9, 117.9, 141.4, 149.2, 154.3.
1-((8-Bromooctyl)oxy)-2-ethoxy-4-nitrobenzene (14b).
Yellowish white powder, 0.32 g, yield 92% starting from 0.17 g 13b (0.92 mmol); 1H NMR (300 MHz, CDCl3) δ 1.31–1.52 (m, 11H total, overlapped), 1.48 (t, J = 7.0 Hz, overlapped) 1.80–1.92 (m, 4H), 3.40 (t, J = 6.8 Hz, 2H), 4.09 (t, J = 6.7 Hz, 2H, overlapped), 4.15 (q, J = 7.0 Hz, 2H, overlapped), 6.88 (d, J = 8.9 Hz, 1H), 7.73 (d, J = 2.6 Hz, 1H), 7.87 (dd, J = 8.9, 2.6 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 14.7, 25.9, 28.2, 28.8, 29.0, 29.2, 32.9, 34.0, 65.1, 69.6, 108.2, 111.2, 117.8, 141.4, 148.6, 154.7.
1-((8-Bromooctyl)oxy)-2-isopropoxy-4-nitrobenzene (14c).
White powder, 0.36 g, yield 84% starting from 0.22 g 13c (1.11 mmol); 1H NMR (300 MHz, CDCl3) δ 1.31–1.54 (m, 14H total, overlapped), 1.38 (d, J = 6.1 Hz, overlapped), 1.80–1.92 (m, 4H), 3.41 (t, J = 6.8 Hz, 2H), 4.07 (t, J = 6.5 Hz, 2H), 4.57 (sep, J = 6.0 Hz, 1H), 6.88 (d, J = 9.0 Hz, 1H), 7.76 (d, J = 2.6 Hz, 1H), 7.87 (dd, J = 9.0, 2.6 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 22.1, 25.9, 28.2, 28.8, 29.0, 29.2, 32.9, 34.0, 69.5, 72.8, 111.7, 111.7, 118.3, 141.3, 147.5, 156.0.
Synthesis of 1-(8(2-alkoxy-4-nitrophenoxy)octyl)-1H-imidazoles (15a-c).
Compounds 15a-c were synthesized from 14a-c (0.83–0.98 mmol) following the general synthesis of compounds 7a-l.
1-(8(2-Methoxy-4-nitrophenoxy)octyl)-1H-imidazole (15a).
Yellowish white powder, 0.23 g, yield 74% starting from 0.32 g 14a (0.89 mmol); 1H NMR (300 MHz, CDCl3) δ 1.23–1.50 (m, 8H), 1.71–1.90 (m, 4H), 3.88–3.94 (m, 5H total, overlapped), 3.94 (s, overlapped), 4.08 (t, J = 6.7 Hz, 2H), 6.85–6.89 (m, 2H), 7.04 (s, 1H), 7.44 (s, 1H), 7.72 (d, J = 2.6 Hz, 1H), 7.87 (dd, J = 8.9, 2.6 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 25.9, 26.6, 28.9, 29.1, 29.2, 31.1, 47.1, 56.4, 69.5, 106.8, 110.9, 117.9, 118.8, 129.5, 137.2, 141.4, 149.2, 154.3.
1-(8(2-Ethoxy-4-nitrophenoxy)octyl)-1H-imidazole (15b).
Yellowish white powder, 0.22 g, yield 73% starting from 0.31 g 14b (0.83 mmol); 1H NMR (300 MHz, CDCl3) δ 1.23–1.51 (m, 11H total, overlapped), 1.47 (t, J = 7.0 Hz, overlapped), 1.72–1.90 (m, 4H), 3.92 (t, J = 7.1 Hz, 2H), 4.07 (t, J = 6.7 Hz, 2H), 4.14 (q, J = 7.0 Hz, 2H), 6.84–6.90 (m, 2H), 7.04 (s, 1H), 7.45 (s, 1H), 7.72 (d, J = 2.6 Hz, 1H), 7.86 (dd, J = 8.9, 2.6 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 14.7, 25.9, 26.6, 28.9, 29.1, 29.2, 31.2, 47.1, 65.1, 69.5, 108.2, 111.2, 117.8, 118.9, 129.5, 137.2, 141.4, 148.6, 154.6.
1-(8(2-Isopropoxy-4-nitrophenoxy)octyl)-1H-imidazole (15c).
Yellow oil, 0.33 g, yield 90% starting from 0.38 g 14c (0.98 mmol); 1H NMR (300 MHz, CDCl3) δ 1.24–1.50 (m, 14H total, overlapped), 1.36 (d, J = 6.1 Hz, overlapped) 1.71–1.88 (m, 4H), 3.91 (t, J = 7.1 Hz, 2H), 4.05 (t, J = 6.6 Hz, 2H), 4.55 (sep, J = 6.1 Hz, 1H), 6.84–6.89 (m, 2H), 7.03 (s, 1H), 7.43 (s, 1H), 7.74 (d, J = 2.7 Hz, 1H), 7.85 (dd, J = 9.0, 2.7 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 22.1, 25.9, 26.6, 28.9, 29.1, 29.2, 31.1, 47.1, 69.4, 72.7, 111.6, 111.7, 118.2, 118.8, 129.5, 137.1, 141.3, 147.5, 155.9.
Synthesis of 4-((8-(1H-imidazol-1-yl)octyl)oxy)-3-alkoxyanilines (16a-c).
Compounds 16a-c were synthesized from 15a-c (0.49–0.86 mmol) following the general synthesis of compounds 8a-m.
4-((8-(1H-imidazol-1-yl)octyl)oxy)-3-methoxyaniline (16a).
Orange oil, 0.27 g, yield 99% starting from 0.30 g 15a (0.86 mmol); 1H NMR (300 MHz, CDCl3) δ 1.23–1.46 (m, 8H), 1.70–1.81 (m, 4H), 3.00–3.39 (brs, ArNH2), 3.79 (s, 3H), 3.86–3.93 (m, 4H), 6.19 (dd, J = 8.4, 2.6 Hz, 1H), 6.29 (d, J = 2.6 Hz, 1H), 6.70 (d, J = 8.4 Hz, 1H), 6.88 (t, J = 1.1 Hz, 1H), 7.04 (s, 1H), 7.44 (s, 1H).
4-((8-(1H-imidazol-1-yl)octyl)oxy)-3-ethoxyaniline (16b).
Brown oil, 0.16 g, yield 98% starting from 0.18 g 15b (0.49 mmol); 1H NMR (300 MHz, CDCl3) δ 1.24–1.48 (m, 11H total, overlapped), 1.40 (t, J = 7.0 Hz, overlapped), 1.68–1.82 (m, 4H), 1.92–2.43 (brs, ArNH2), 3.86–3.95 (m, 4H), 4.01 (q, J = 7.0 Hz, 2H), 6.20 (dd, J = 8.4, 2.6 Hz, 1H), 6.30 (d, J = 2.7 Hz, 1H), 6.72 (d, J = 8.4 Hz, 1H), 6.89 (brs, 1H), 7.04 (s, 1H), 7.45 (s, 1H).
1-((8-(1H-imidazol-1-yl)octyl)oxy)-3-isopropoxyaniline (16c).
Brown oil, 0.27 g, yield 98% starting from 0.30 g 15c (0.79 mmol); 1H NMR (300 MHz, CDCl3) δ 1.22–1.47 (m, 14H total, overlapped), 1.30 (d, J = 6.1 Hz, overlapped), 1.66–1.80 (m, 4H), 3.08–3.43 (brs, ArNH2), 3.84–3.93 (m, 4H), 4.42 (sep, J = 6.1 Hz, 1H), 6.22 (dd, J = 8.4, 2.7 Hz, 1H), 6.31 (d, J = 2.7 Hz, 1H), 6.71 (d, J = 8.4 Hz, 1H), 6.88 (t, J = 1.1 Hz, 1H), 7.03 (s, 1H), 7.44 (s, 1H).
Synthesis of N-(4-((8-(1H-imidazol-1-yl)octyl)oxy)3-alkoxyphenyl) picolinimidamides (17a-c).
Compounds 17a-c were synthesized from 16a-c (0.45–0.69 mmol) following the general synthesis of compounds 9a-m.
N-(4-((8-(1H-imidazol-1-yl)octyl)oxy)3-methoxyphenyl) picolinimidamide (17a).
Yellowish white powder, 130 mg, yield 45% starting from 220 mg 16a (0.69 mmol); mp 117–119 °C; 1H NMR (400 MHz, CDCl3) δ 1.28–1.39 (m, 6H), 1.41–1.51 (m, 2H), 1.74–1.87 (m, 4H), 3.84 (s, 3H), 3.92 (t, J = 7.2 Hz, 2H), 3.99 (t, J = 6.8 Hz, 2H), 5.50–6.34 (brs, imidamide NHs), 6.55 (dd, J = 8.4, 2.3 Hz, 1H), 6.62 (d, J = 2.3 Hz, 1H), 6.87–6.91 (m, 2H), 7.05 (s, 1H), 7.39 (ddd, J = 7.5, 4.8, 1.2 Hz, 1H), 7.45 (s, 1H), 7.81 (td, J = 7.7, 1.7 Hz, 1H), 8.41 (d, J = 8.0 Hz, 1H), 8.57 (ddd, J = 4.8, 1.7, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 26.0, 26.6, 29.1, 29.3, 29.4, 31.2, 47.2, 56.0, 69.6, 106.5, 112.8, 114.6, 118.9, 121.6, 125.3, 129.6, 136.9, 137.2, 143.5, 144.6, 148.0, 150.6, 151.7, 153.2; HRMS (ESI) m/z (M +H)+ calcd for C24H32N5O2, 422.25505; found, 422.25616; Anal. Calcd for C24H31N5O2: C, 68.38; H, 7.41; N, 16.61. Found: C, 68.18; H, 7.46; N, 16.38.
N-(4-((8-(1H-imidazol-1-yl)octyl)oxy)3-ethoxyphenyl) picolinimidamide (17b).
Yellowish white powder, 65 mg, yield 33% starting from 150 mg 16b (0.45 mmol); mp 100–102 °C; 1H NMR (400 MHz, CDCl3) δ 1.25–1.51 (m, 11H total, overlapped), 1.42 (t, J = 7.0 Hz, overlapped), 1.74–1.84 (m, 4H), 3.92 (t, J = 7.1 Hz, 2H), 3.98 (t, J = 6.7 Hz, 2H), 4.06 (q, J = 7.0 Hz, 2H), 5.49–6.23 (brs, imidamide NHs), 6.54 (d, J = 8.3 Hz, 1H), 6.61 (s, 1H), 6.87–6.91 (m, 2H), 7.05 (s, 1H), 7.38 (ddd, J = 7.5, 4.8, 1.1 Hz, 1H), 7.45 (s, 1H), 7.80 (td, J = 7.8, 1.7 Hz, 1H), 8.40 (d, J = 7.9 Hz, 1H), 8.56 (d, J = 4.9 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 15.0, 26.1, 26.6, 29.1, 29.3, 29.5, 31.2, 47.2, 64.5, 70.0, 108.0, 113.0, 115.7, 118.9, 121.6, 125.2, 129.6, 136.9, 137.2, 143.9, 145.0, 148.0, 150.1, 151.8, 153.1; HRMS (ESI) m/z (M +H)+ calcd for C25H34N5O2, 436.27070; found, 436.27029; Anal. Calcd for C25H33N5O2: C, 68.94; H, 7.64; N, 16.08. Found: C, 69.09; H, 7.54; N, 16.03.
N-(4-((8-(1H-imidazol-1-yl)octyl)oxy)3-isopropoxyphenyl) picolinimidamide (17c).
Yellowish white powder, 90 mg, yield 31% starting from 220 mg 16c (0.63 mmol); mp 88–91 °C; 1H NMR (400 MHz, CDCl3) δ 1.28–1.39 (m, 12H), 1.42–1.52 (m, 2H), 1.74–1.82 (m, 4H), 3.92 (t, J = 7.2 Hz, 2H), 3.97 (t, J = 6.6 Hz, 2H), 4.48 (sep, J = 6.1 Hz, 1H), 5.50–6.30 (brs, imidamide NHs), 6.57 (d, J = 8.4 Hz, 1H), 6.64 (s, 1H), 6.88–6.91 (m, 2H), 7.05 (s, 1H), 7.38 (ddd, J = 7.5, 4.9, 1.2 Hz, 1H), 7.45 (s, 1H), 7.80 (td, J = 7.8, 1.7 Hz, 1H), 8.40 (d, J = 8.0 Hz, 1H), 8.57 (ddd, J = 4.8, 1.6, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 22.4, 26.1, 26.7, 29.2, 29.3, 29.6, 31.2, 47.2, 70.0, 72.1, 111.7, 114.1, 116.3, 118.9, 121.6, 125.2, 129.6, 136.9, 137.2, 143.9, 146.4, 148.0, 149.1, 151.8, 153.1; HRMS (ESI) m/z (M +H)+ calcd for C26H36N5O2, 450.28635; found, 450.28671; Anal. Calcd for C26H35N5O2: C, 69.46; H, 7.85; N, 15.58. Found: C, 69.19; H, 7.77; N, 15.28.
Synthesis of 4-fluoro-2-alkoxy-1-nitrobenzenes (19a-c).
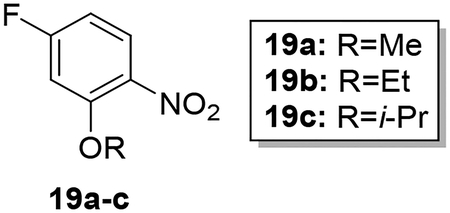
To a solution of 18 (0.50–0.55 g, 3.18–3.50 mmol) in dry DMF (5 mL) was added 1.5 equivalent of potassium carbonate (4.77–5.25 mmol) and 3 equivalent of alkyl iodide (9.54–10.50 mmol) and the mixture was heated in a sealed tube at 80 °C for 4–5 hours. After the reaction was complete, the solution was cooled down and the solvent was removed under reduced pressure. The crude product was purified by column chromatography to afford the products 24a-c in 64–82% yield.
4-Fluoro-2-methoxy-1-nitrobenzene (19a).59
Chromatography solvent: hexanes/DCM 3:1 yellow powder, 0.43 g, yield 79% starting from 0.50 g 18 (3.18 mmol).
4-Fluoro-2-ethoxy-1-nitrobenzene (19b).59
Chromatography solvent: hexanes/DCM 4:1 yellow powder, 0.53 g, yield 82% starting from 0.55 g 18 (3.50 mmol).
4-Fluoro-2-isopropoxy-1-nitrobenzene (19c).59
Chromatography solvent: hexanes/DCM 4:1 yellow oil, 0.42 g, yield 64% starting from 0.52 g 18 (3.30 mmol).
Synthesis of 3-alkoxy-4-nitrophenols (20a-c).
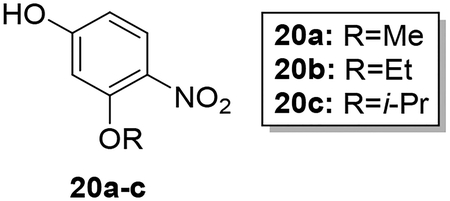
Compounds 20a-c were prepared according to a previously published procedure.30 To a solution of 19a-c (1.30–1.80 mmol) in DMSO (5 mL) was added NaOH (0.5 g) dissolved in distilled water (5 mL) and the mixture was stirred vigorously at 80 °C for 20 hours. After completion of the reaction, the solution was cooled and the pH was rendered acidic with 6N HCl. The product was extracted with ethyl acetate (3 × 30 mL), washed with water and brine and dried over sodium sulfate. The combined organic layer was evaporated under reduced pressure then purified by column chromatography using hexanes/ethyl acetate 2:1 as eluent to afford the pure product in 74–85% yield.
3-Methoxy-4-nitrophenol (20a).60
Yellow powder, 0.32 g, yield 83%, starting from 0.39 g 19a (2.27 mmol). 1H NMR (300 MHz, DMSO-d6) δ 3.86 (s, 3H), 6.47 (dd, J = 9.1, 2.4 Hz, 1H), 6.60 (d, J = 2.4 Hz, 1H), 7.88 (dd, J = 9.1 Hz, 1H), 10.87 (brs); 13C NMR (75 MHz, DMSO-d6) δ 56.4, 100.4, 107.5, 128.3, 130.9, 155.7, 164.0.
3-Ethoxy-4-nitrophenol (20b).
Yellow powder, 0.36 g, yield 74%, starting from 0.49 g 19b (2.64 mmol). 1H NMR (300 MHz, DMSO-d6) δ 1.35 (t, J = 6.9 Hz, 3H), 4.13 (q, J = 6.9 Hz, 2H), 6.46 (dd, J = 9.0, 2.3 Hz, 1H), 6.58 (d, J = 2.3 Hz, 1H), 7.86 (d, J = 9.0 Hz, 1H), 10.83 (brs); 13C NMR (75 MHz, DMSO-d6) δ 14.3, 64.8, 101.0, 107.5, 128.1, 131.1, 154.8, 163.8.
3-Isopropoxy-4-nitrophenol (20c).61
Yellow powder, 0.30 g, yield 85%, starting from 0.36 g 19c (1.80 mmol).
Synthesis of 4-((8-bromooctyl)oxy)-2-alkoxy-1-nitrobenzenes (21a-c).
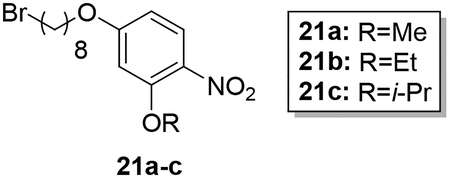
Compounds 21a-c were synthesized from 20a-c (1.32–2.06 mmol) following the general synthesis of compounds 6a-i.
4-((8-Bromooctyl)oxy)-2-methoxy-1-nitrobenzene (21a).
Yellow powder, 0.67 g, yield 90% starting from 0.35 g 20a (2.06 mmol); 1H NMR (300 MHz, CDCl3) δ 1.33–1.53 (m, 8H), 1.75–1.91 (m, 4H), 3.41 (t, J = 6.8 Hz, 2H), 3.94 (s, 3H), 4.02 (t, J = 6.5 Hz, 2H), 6.48 (dd, J = 9.0, 2.5 Hz, 1H), 6.52 (d, J = 2.4 Hz, 1H), 7.98 (d, J = 9.0 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 26.0, 28.2, 28.8, 29.1, 29.2, 32.9, 34.0, 56.6, 68.9, 100.2, 105.3, 128.6, 132.9, 155.9, 164.5.
4-((8-Bromooctyl)oxy)-2-ethoxy-1-nitrobenzene (21b).
Yellow powder, 0.51 g, yield 76% starting from 0.33 g 20b (1.80 mmol); 1H NMR (300 MHz, CDCl3) δ 1.32–1.52 (m, 11H total, overlapped), 1.48 (t, J = 7.0 Hz, overlapped) 1.75–1.91 (m, 4H), 3.41 (t, J = 6.8 Hz, 2H), 4.00 (t, J = 6.5 Hz, 2H), 4.15 (q, J = 7.0 Hz, 2H), 6.46 (dd, J = 9.0, 2.4 Hz, 1H), 6.50 (d, J = 2.3 Hz, 1H), 7.95 (d, J = 9.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 14.7, 26.0, 28.2, 28.8, 29.1, 29.2, 32.9, 34.1, 65.5, 68.8, 101.0, 105.3, 128.4, 133.2, 155.2, 164.3.
4-((8-Bromooctyl)oxy)-2-isopropoxy-1-nitrobenzene (21c).
White powder, 0.48 g, yield 94% starting from 0.26 g 20c (1.32 mmol); 1H NMR (400 MHz, CDCl3) δ 1.30–1.50 (m, 14 H total, overlapped), 1.39 (d, J = 6.1 Hz, overlapped) 1.74–1.89 (m, 4H), 3.40 (t, J = 6.8 Hz, 2H), 3.99 (t, J = 6.5 Hz, 2H), 4.62 (sep, J = 6.1 Hz, 1H), 6.45 (dd, J = 9.1, 2.5 Hz, 1H), 6.50 (d, J = 2.5 Hz, 1H), 7.89 (d, J = 9.1 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 22.0, 25.9, 28.1, 28.7, 29.1, 29.2, 32.8, 34.0, 68.7, 72.8, 102.5, 105.4, 128.2, 134.2, 154.1, 164.0.
Synthesis of 1-(8(3-alkoxy-4-nitrophenoxy)octyl)-1H-imidazoles (22a-c).
Compounds 22a-c were synthesized from 21a-c (0.85–1.38 mmol) following the general synthesis of compounds 7a-l.
1-(8-(3-Methoxy-4-nitrophenoxy)octyl)-1H-imidazole (22a).
Yellow powder, 0.28 g, yield 81% starting from 0.36 g 21a (1.00 mmol); 1H NMR (300 MHz, CDCl3) δ 1.24–1.50 (m, 8H), 1.72–1.84 (m, 4H), 3.89–3.95 (m, 5H total, overlapped), 3.93 (s, overlapped), 4.00 (t, J = 6.5 Hz, 2H), 6.47 (dd, J = 9.1, 2.5 Hz, 1H), 6.51 (d, J = 2.4 Hz, 1H), 6.89 (s, 1H), 7.05 (s, 1H), 7.45 (s, 1H), 7.98 (d, J = 9.0 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 26.0, 26.6, 29.09, 29.11, 29.3, 31.2, 47.1, 56.6, 68.8, 100.2, 105.3, 118.9, 128.6, 129.6, 132.9, 137.2, 155.9, 164.5.
1-(8-(3-Ethoxy-4-nitrophenoxy)octyl)-1H-imidazole (22b).
Yellow powder, 0.25 g, yield 81% starting from 0.32 g 21b (0.85 mmol); 1H NMR (300 MHz, CDCl3) δ 1.25–1.50 (m, 11H total, overlapped), 1.47 (t, J = 7.0 Hz, overlapped), 1.72–1.83 (m, 4H), 3.92 (t, J = 7.1 Hz, 2H), 3.99 (t, J = 6.5 Hz, 2H), 4.14 (q, J = 7.0 Hz, 2H), 6.45 (dd, J = 9.0, 2.5 Hz, 1H), 6.49 (d, J = 2.4 Hz, 1H), 6.89 (s, 1H), 7.04 (s, 1H), 7.45 (s, 1H), 7.94 (d, J = 9.0 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 14.6, 25.9, 26.6, 29.08, 29.10, 29.2, 31.1, 47.1, 65.5, 68.7, 101.0, 105.3, 118.9, 128.4, 129.6, 133.3, 137.2, 155.1, 164.3.
1-(8-(3-Isopropoxy-4-nitrophenoxy)octyl)-1H-imidazole (22c).
Yellow oil, 0.35 g, yield 79% starting from 0.46 g 21c (1.18 mmol); 1H NMR (300 MHz, CDCl3) δ 1.25–1.48 (m, 14H total, overlapped), 1.40 (d, J = 6.1 Hz, overlapped), 1.73–1.81 (m, 4H), 3.92 (t, J = 7.1 Hz, 2H), 3.98 (t, J = 6.5 Hz, 2H), 4.61 (sep, J = 6.1 Hz, 1H), 6.45 (dd, J = 9.1, 2.5 Hz, 1H), 6.49 (d, J = 2.4 Hz, 1H), 6.89 (s, 1H), 7.04 (s, 1H), 7.45 (s, 1H), 7.90 (d, J = 9.1 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 22.0, 26.0, 26.6, 29.10, 29.11, 29.3, 31.2, 47.1, 68.7, 72.8, 102.5, 105.4, 118.9, 128.2, 129.6, 134.3, 137.2, 154.1, 164.0.
Synthesis of 4-((8-(1H-imidazol-1-yl)octyl)oxy)-3-alkoxyanilines (23a-c).
Compounds 23a-c were synthesized from 22a-c (0.66–0.86 mmol) following the general synthesis of compounds 8a-m.
4-((8-(1H-imidazol-1-yl)octyl)oxy)-2-methoxyaniline (23a).
Yellow oil, 0.26 g, yield 95% starting from 0.30 g 22a (0.86 mmol); 1H NMR (300 MHz, CDCl3) δ 1.23–1.48 (m, 8H), 1.67–1.82 (m, 4H), 2.88–3.30 (brs, ArNH2), 3.83 (s, 3H), 3.87 (t, J = 6.4 Hz, 2H), 3.91 (t, J = 7.1 Hz, 2H), 6.33 (dd, J = 8.4, 2.6 Hz, 1H), 6.44 (d, J = 2.6 Hz, 1H), 6.62 (d, J = 8.4 Hz, 1H), 6.89 (t, J = 1.1 Hz, 1H), 7.05 (s, 1H), 7.45 (s, 1H).
4-((8-(1H-imidazol-1-yl)octyl)oxy)-2-ethoxyaniline (23b).
Brown oil, 0.21 g, yield 95% starting from 0.24 g 22b (0.66 mmol); 1H NMR (300 MHz, CDCl3) δ 1.26–1.47 (m, overlapped, 11H total), 1.42 (t, J = 7.0 Hz),1.67–1.83 (m, 4H), 2.34–3.02 (brs, ArNH2), 3.86 (t, J = 6.6 Hz, 2H), 3.92 (t, J = 7.1 Hz, 2H), 4.03 (q, J = 7.0 Hz, 2H), 6.32 (dd, J = 8.4, 2.6 Hz, 1H), 6.43 (d, J = 2.6 Hz, 1H), 6.63 (d, J = 8.4 Hz, 1H), 6.89 (s, 1H), 7.05 (s, 1H), 7.46 (s, 1H).
4-((8-(1H-imidazol-1-yl)octyl)oxy)-2-isopropoxyaniline (23c).
Brown oil, 0.24 g, yield 100% starting from 0.26 g 22c (0.69 mmol); 1H NMR (300 MHz, CDCl3) δ 1.25–1.47 (m, overlapped, 14H total), 1.34 (d, J = 6.0 Hz), 1.67–1.82 (m, 4H), 3.02–3.36 (brs, ArNH2), 3.85 (t, J = 6.5 Hz, 2H), 3.91 (t, J = 7.1 Hz, 2H), 4.48 (sep, J = 6.1 Hz, 1H), 6.32 (dd, J = 8.5, 2.6 Hz, 1H), 6.44 (d, J = 2.6 Hz, 1H), 6.62 (d, J = 8.5 Hz, 1H), 6.89 (s, 1H), 7.04 (s, 1H), 7.45 (s, 1H).
Synthesis of N-(4-((8-(1H-imidazol-1-yl)octyl)oxy)2-alkoxyphenyl) picolinimidamides (24a-c).
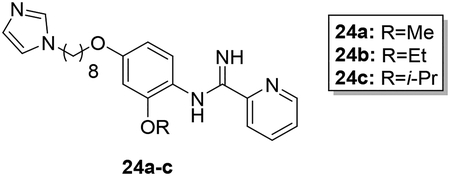
Compounds 24a-c were synthesized from 23a-c (0.54–0.63 mmol) following the general synthesis of compounds 9a-m.
N-(4-((8-(1H-imidazol-1-yl)octyl)oxy)-2-methoxyphenyl) picolinimidamide (24a).
Yellowish white powder, 120 mg, yield 45% starting from 200 mg 23a (0.63 mmol); mp 109–112°C; 1H NMR (400 MHz, CDCl3) δ 1.26–1.40 (m, 6H), 1.41–1.51 (m, 2H), 1.73–1.83 (m, 4H), 3.80 (s, 3H), 3.90–3.97 (m, 4H), 5.53–6.18 (brs, imidamide NHs), 6.50 (dd, J = 8.5, 2.6 Hz, 1H), 6.56 (d, J = 2.6 Hz, 1H), 6.88–6.99 (brs, 2H total, overlapped), 6.90 (t, J = 1.2 Hz), 7.05 (t, J = 1.0 Hz, 1H), 7.37 (ddd, J = 1.2, 4.8, 7.5 Hz, 1H), 7.46 (s, 1H), 7.80 (td, J = 7.7, 1.7 Hz, 1H), 8.47 (d, J = 8.0 Hz, 1H). 8.56 (ddd, J = 0.9, 1.7, 4.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 26.1, 26.6, 29.2, 29.4, 29.5, 31.2, 47.2, 56.0, 68.3, 100.7, 105.6, 118.9, 122.0, 123.0, 125.2, 129.6, 136.9, 137.2, 147.9, 151.7, 152.0, 153.6, 156.5; HRMS (ESI) m/z (M +H)+ calcd for C24H32N5O2, 422.25505; found, 422.25452; Anal. Calcd for C24H31N5O2: C, 68.38; H, 7.41; N, 16.61. Found: C, 68.47; H, 7.54; N, 16.61.
N-(4-((8-(1H-imidazol-1-yl)octyl)oxy)-2-ethoxyphenyl) picolinimidamide (24b).
Buff powder, 100 mg, yield 42% starting from 180 mg 23b (0.54 mmol); mp 86–89°C; 1H NMR (700 MHz, CDCl3) δ 1.28–1.38 (m, 9H total, overlapped), 1.34 (t, J = 7.0 Hz), 1.43–1.48 (m, 2H), 1.74–1.81 (m, 4H), 3.91–3.95 (m, 4H), 4.03 (q, J = 7.0 Hz, 2H), 5.51–6.41 (brs, imidamide NHs), 6.51 (dd, J = 8.5, 2.5 Hz, 1H), 6.56 (d, J = 2.5 Hz, 1H), 6.90 (s, 1H), 6.98 (brs, 1H), 7.05 (s, 1H), 7.39 (ddd, J = 0.9, 4.8, 7.4 Hz, 1H), 7.45 (s, 1H), 7.81 (td, J = 7.8, 1.6 Hz, 1H), 8.47 (d, J = 7.8 Hz, 1H), 8.57 (d, J = 4.6 Hz, 1H);13C NMR (176 MHz, CDCl3) δ 15.0, 26.1, 26.6, 29.1, 29.3, 29.5, 31.2, 47.1, 64.5, 68.3, 102.2, 106.1, 118.9, 122.0, 123.5, 125.2, 129.6, 137.0, 137.2, 148.0, 151.2, 151.5, 153.5, 156.5; HRMS (ESI) m/z (M +H)+ calcd for C25H34N5O2, 436.27070; found, 436.27139; Anal. Calcd for C25H33N5O2: C, 68.94; H, 7.64; N, 16.08. Found: C, 68.66; H, 7.65; N, 15.86.
N-(4-((8-(1H-imidazol-1-yl)octyl)oxy)-2-isopropoxyphenyl) picolinimidamide (24c).
Yellow powder, 92 mg, yield 34% starting from 210 mg 23c (0.60 mmol); mp 82–85 °C; 1H NMR (700 MHz, CDCl3) δ 1.25 (brd, J = 5.9 Hz, 6H), 1.28–1.38 (m, 6H), 1.42–1.48 (m, 2H), 1.74–1.80 (m, 4H), 3.92 (t (apparent), J = 6.9 Hz, 4H), 4.44 (sep, J = 5.9 Hz, 1H), 5.38–6.16 (brs, imidamide NHs), 6.55 (dd, J = 8.5, 2.5 Hz, 1H), 6.57 (d, J = 2.5 Hz, 1H), 6.80–6.98 (brs, 2H total, overlapped), 6.90 (s), 7.05 (s, 1H), 7.36–7.39 (m, 1H), 7.46 (s, 1H), 7.78–7.82 (m, 1H), 8.43 (brs, 1H), 8.57 (d, J = 3.9 Hz, 1H); 13C NMR (176 MHz, CDCl3) δ 22.4, 26.1, 26.6, 29.2, 29.3, 29.5, 31.2, 47.2, 68.3, 72.1, 105.9, 107.7, 118.9, 121.8, 123.7, 125.0, 129.6, 133.8, 136.8, 137.2, 147.9, 149.8, 152.0, 152.6, 156.1; HRMS (ESI) m/z (M +H)+ calcd for C26H36N5O2, 450.28635; found, 450.28778; Anal. Calcd for C26H35N5O2: C, 69.46; H, 7.85; N, 15.58. Found: C, 69.40; H, 7.90; N, 15.37.
2-(8-bromooctyl) isoindoline-1, 3-dione (26).
To a solution of 1,8-dibromooctane (8.8 g, 32.4 mmol) in dry DMF (30 mL) was added phthalimide potassium salt (2.0 g, 10.8 mmol) and the mixture was stirred at 90 °C for 24h. The reaction mixture was extracted with CH2Cl2 (2 × 30 mL) and washed with 0.1N NaOH (50 mL). The combined organic layer was dried over anhydrous Na2SO4, filtered and evaporated under reduced pressure. The crude product was purified by column chromatography using hexanes/ethyl acetate 15:1 as eluent to afford the pure product as a white powder, 2.2 g, yield 60%; 1H NMR (400 MHz, CDCl3) δ 1.25–1.46 (m, 8H), 1.62–1.72 (m, 2H), 1.79–1.87 (m, 2H), 3.38 (t, J = 6.9 Hz, 2H), 3.67 (t, J = 7.3 Hz, 2H), 7.67–7.73 (m, 2H), 7.81–7.86 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 26.8, 28.2, 28.6, 28.7, 29.1, 32.9, 34.1, 38.1, 123.3, 132.3, 134.0, 168.6.
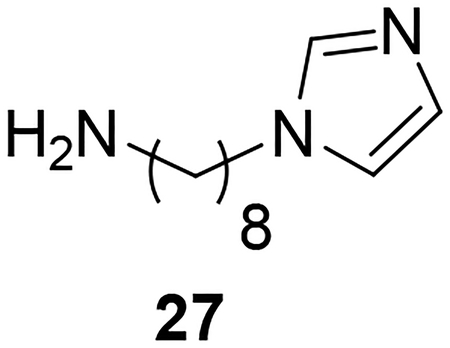
8-(1H-imidazol-1-yl)octan-1-amine (27).
Compound 27 was prepared over two steps with slight modification to a previously published procedure.31 To a solution of 26 (2.0 g, 5.91 mmol) in dry DMF (20 mL) was added 1.5 equivalents K2CO3 (1.22 g, 8.86 mmol) and 2 equivalents of imidazole (0.80g, 11.82 mmol) and the mixture was stirred at 80 °C for 12h. After the reaction was completed, the suspension was filtered and the filtrate was concentrated under reduced pressure. The crude product was subjected to silica gel chromatography using DCM/MeOH 10:0.3 as the eluent to afford the pure product as a white powder, 1.20 g, yield 62%. The 2-(8-(1H-imidazol-1-yl)octyl)isoindoline-1,3-dione product (1.1. g, 3.38 mmol) was heated to reflux with 6 equivalents of 80% NH2NH2·H2O (1.2 mL, 20.28 mmol) in absolute ethanol (40 mL) under a N2 atmosphere for 8h. The mixture was cooled and treated with 4N HCl (12 mL) and heated to reflux for a further 6 h. The suspension was filtered and the filtrate was concentrated under reduced pressure. The solution was rendered alkaline with 2N NaOH then the product was extracted with DCM (100 mL). The organic layer was dried over anhydrous sodium sulfate and evaporated under reduced pressure to afford the product as yellow oil which was taken to the next step without further purification, 0.44 g, yield 67%; 1H NMR (300 MHz, CDCl3) δ 1.19–1.34 (m, 8H), 1.35–1.48 (m, 2H), 1.67–1.87 (m, 4H), 2.66 (t, J = 7.0 Hz, 2H), 3.89 (t, J = 7.1 Hz, 2H), 6.87 (s, 1H), 7.02 (s, 1H), 7.44 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 26.6, 26.8, 29.1, 29.3, 31.1, 33.6, 42.2, 47.1, 118.8, 129.4, 137.2
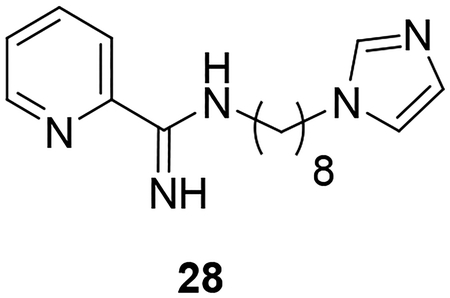
N-(8-(1H-imidazol-1-yl)octyl)picolinimidamide (28).
The synthesis of 28 follows the general synthesis and workup procedure for 9a-l with the modification of using 2.2 equivalents of S-(2-naphthylmethyl)-2-pyridyl thioimidate hydrobromide. The compound was further purified by column chromatography using DCM/methanol/triethylamine (10:1.5:0.5) followed by trituration from hexanes/ether (3 × 10 mL). White powder, 65 mg, yield 28% (starting from 0.15 g of 26, 0.77 mmol); mp 65–67 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.16–1.40 (m, 8H), 1.53–1.62 (m, 2H), 1.64–1.73 (m, 2H), 3.15 (t, J = 7.0 Hz, 2H), 3.93 (t, J = 7.1 Hz, 2H), 6.86 (s, 1H), 7.14 (t, J = 1.1 Hz, 1H), 7.45 (ddd, J = 7.4, 4.8, 1.1 Hz, 1H), 7.59 (s, 1H), 7.86 (td, J = 7.7, 1.7 Hz, 1H), 8.12 (d, J = 8.0 Hz, 1H), 8.55 (ddd, J = 4.8, 1.6, 0.9 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ 25.9, 27.0, 28.5, 28.8, 30.0, 30.6, 45.4, 45.9, 119.2, 120.5, 124.7, 128.3, 136.8, 137.2, 147.8, 151.9, 154.1; HRMS (ESI) m/z (M +H)+ calcd for C17H26N5, 300.21827; found, 300.21811; Anal. Calcd for C17H25N5: C, 68.19; H, 8.42; N, 23.39. Found: C, 68.30; H, 8.33; N, 23.35.
Biological Assays
IC50 determinations (general).
For in vitro cell-based susceptibility assays, validity criteria concerning Z’ scores and R2 values for dose-response curves were as defined in Abdelhameed et al.52 For colorimetric assays employing J774 macrophages and HepG2 cells, an additional validity criterion was that the mean absorbance of the positive control wells was ≥ 1.0. Absolute IC50 values were determined for all assays as described earlier.11
L. donovani-infected macrophage assay.
The species identity of the LV82 strain L. donovani parasites used in this work was verified by HaeIII-mediated restriction fragment length polymorphism (RFLP) analysis of the ribosomal internal transcribed spacer region obtained by PCR from promastigote genomic DNA.62 Evaluation of the activity of hybrid compounds against intracellular L. donovani was performed as outlined by Abdelhameed et al.52
J774 macrophage toxicity assay.
J774 murine macrophages were confirmed to be of mouse origin by species-specific PCR evaluation and to be free of Mycoplasma contamination by IDEXX BioResearch (Columbia, MO). The assay to determine the toxicity of hybrid compounds on J774 murine macrophages was conducted as described by Zhu et al.63
HepG2 toxicity assay.
HepG2 cells were authenticated as an exact match to ATCC HB-8065 (HepG2) by the American Type Culture Collection (ATCC, Manassas, VA) using Short Tandem Repeat analysis.64 The HepG2 assay was performed as described previously65 with minor modifications. HepG2 cells were maintained in a complete MEM Alpha medium (MEM Alpha (Life Technologies) containing 10% heat-inactivated FBS [Sigma-Aldrich], 50 U/mL penicillin, and 50 μg/mL streptomycin (Life Technologies)). Cells were trypsinized with TrypLE Express (Life Technologies) and were plated at a density of 5 × 103 cells/well in 100 μL volume in 96-well plates. Cells were allowed to adhere overnight at 37 °C in a 5% CO2 atmosphere. The medium was then replaced with medium containing either compound or control drug and test plates were allowed to incubate for approximately 72 hours at 37 °C in a 5% CO2 atmosphere. Following this incubation, medium was removed and replaced with 100 μL/well of fresh medium and 25 μL/well of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, 5 mg/mL in sterile PBS). After an additional 2–3 hour incubation, 100 μL of SDS lysis buffer (100 mg/mL SDS in 50% aqueous DMF) was added to lyse the cells. After an additional 3–5 hour incubation, the absorbance in each well was read at 570 nm with the aid of a SpectraMax M5 microplate reader (Molecular Devices, Sunnyvale, CA).
L. donovani CYP51 binding spectra and dissociation constant determination.
To examine the binding of hybrid compounds to L. donovani CYP51 (XP_003859085.1), a plasmid containing an N-terminal truncated construct (CYP51-32c; removal of 31 N-terminal amino acids to increase solubility) and a C-terminal histidine tag (6xHis; to facilitate affinity purification) was selected for heterologous expression in Escherichia coli and protein purification as described previously for L. infantum CYP5148 with modifications. Emulgen 911, instead of Triton X-100, was used as detergent to solubilize CYP51 from E. coli cell homogenate. Purified CYP51 was analyzed by SDS-PAGE and Western blot (anti-His tag) analysis for purity and by carbon monoxide (CO)-difference spectra upon reduction by sodium dithionite as previously described.32 To determine dissociation constants (Kd) of CYP-ligand complexes, titration of L. donovani CYP51 with selected AA hybrid compounds was performed as described previously66 with modifications. Titration of CYP51 (0.5 μM) was carried out in 30 mM potassium phosphate buffer, pH7.4, containing 0.1 mM EDTA and 20% glycerol. The difference spectra were obtained by recording the absorbance in the sample cuvette versus the absorbance in the reference cuvette. For compounds without absorbance interference in the 350 to 500 nm range, both reference and sample cuvettes contained the same amount of the protein. Compounds were added to the sample cuvette from stock solutions in DMSO and the corresponding volume of DMSO was added to the reference cuvette. For compounds with absorbance interference in 350 to 500 nm range, only sample cuvette contained protein solution. Compounds were then added to both sample and reference cuvettes from a stock solution in DMSO. The dissociation constants were calculated by fitting the equation to the Δ(Amax − Amin) versus substrate concentration curves.
L. donovani CYP51 inhibition assay.
A fluorescence-based inhibition assay was developed for the L. donovani CYP51 using 7-benzyloxy-4-trifluoromethylcoumarin (BFC; Corning Inc.) as substrate and its fluorescent metabolite 7-hydroxy-4-trifluoromethylcoumarin (HFC, excitation 410 nm, emission 538 nm). Cumene hydroperoxide (CuOOH) was used as a cofactor to enable the CYP-catalyzed reactions in place of CYP natural cofactors (NADPH:P450 oxidoreductase and NADPH).34 Inhibition assays were performed in 96-well plates in a 200 μL volume. A stock solution of fluorogenic substrate BFC (5 mM) was prepared in 1:1 [v/v] DMSO:DMA (dimethylacetamide). The hybrid compounds (1–10 mM) were dissolved in DMSO. The reaction mixture contained CYP51 (60 nM P450), BFC (50 μM), and various concentrations (0.01–100 μM) of hybrid compounds in a phosphate buffer (100 mM, pH 7.4) containing MgCl2 (3.3 mM). DMSO was used as negative control. The reaction was initiated with the addition of 100 μM CuOOH (Alfa Aesar) and monitored at 37°C for emission at 538 nm under excitation at 410 nm on a Tecan Infinite® M200 Pro microplate reader. For each compound, the percent inhibition at different concentrations was calculated as (1 – RFUcompound/RFUnegative control) × 100, where RFU is the relative fluorescence unit. The following two-parameter logistic equation was fitted to the percent inhibition versus the logarithm of the compound concentration curves to obtain the IC50 values.
Microsomal stability determinations.
Metabolic stability of selected hybrid compounds (3 μM) was evaluated using human and mouse liver microsomes (0.5 mg/mL) supplemented with NADPH (1 mM). Microsomal incubations were carried out as described previously10, 11 with modifications. Reactions were allowed to proceed for up to 60 min at 37°C, then aliquots (10 μL each) were removed and quenched with ice-cold acetonitrile (200 uL) containing an internal standard (2 nM). After centrifugation (2,000 × g), the supernatants (100 μL) were dried down with nitrogen gas, then reconstituted with 15% methanol (150 μL) and analyzed by UPLC-MS/MS to quantify the amount of hybrid compound remaining. For LC-UV detection, reaction aliquots (200 μL) were removed and quenched with ice-cold acetonitrile (100 ul) containing an internal standard (5 μM). After centrifugation (2,000 × g), the supernatants (250 μL) were dried down with nitrogen gas, then reconstituted with 15% methanol (100 μL) and quantified. Control incubations were run as described above in the absence of NADPH. Microsomal half-life (t1/2) values were obtained by fitting the one-phase exponential decay equation (C = C0 × e−kt; t1/2 = 0.693/k) to percent substrate remaining versus time curves.
In vivo experiments.
Experiments assessing the in vivo toxicity and antileishmanial efficacy of compound 24c were conducted according to procedures approved by the Institutional Animal Care and Use Committee at The Ohio State University. In vivo toxicity. The toxicity of compound 24c to 6–8 week old female BALB/c mice was assessed as outlined by Joice et al.11 Compound 24c was dissolved in 50% PEG400/50% water and was administered by oral gavage. No particles or cloudiness were noted when the required solution of 24c (10 mg/mL) was prepared. In vivo efficacy. The efficacy of compound 24c and miltefosine in a murine visceral leishmaniasis model were determined according to the methods described by Joice et al.11 In the present study, the uninfected vehicle control group received PEG-400, compound 24c was dissolved in 50% PEG400/50% water, and miltefosine was dissolved in water. Administration of the test agents and vehicle was by oral gavage.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the Office of the Assistant Secretary of Defense for Health Affairs under the Peer Reviewed Medical Research Program through award no. W81XWH-14-2-0017 (to K.A.W.), by the National Institutes of Health through R01 AI139198 (to M.Z.W.), and by the Neglected Disease Drug Discovery Fund of The Ohio State University College of Pharmacy. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense or the U.S. Army. We thank Dr. Chunhua Yuan for assistance in collecting NMR spectra. We also thank Dr. Pankaj Sharma for assistance with chemical characterization and Dr. Liva Rakotondraibe for assistance in interpreting NMR spectra.
ABBREVIATIONS
- AIA
arylimidamide
- BFC
7-benzyloxy-4-trifluoromethylcoumarin
- CuOOH
cumene hydroperoxide
- HFC
7-hydroxy-4-(trifluoromethyl)coumarin
- LDU
Leishman-Donovan units
- S.E.M.
standard error of the mean
Footnotes
SUPPORTING INFORMATION
This information is available free of charge on the ACS Publications website: 1H and 13C NMR spectra for all target compounds.
REFERENCES
- 1.World Health Organization, Leishmaniasis. www.who.int/health-topics/leishmaniasis#tab=tab_1, accessed October 28, 2020.
- 2.Sundar S; Chakravarty J, An update on pharmacotherapy for leishmaniasis. Expert Opin. Pharmacother 2015, 16, 237–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rodrigo C; Weeratunga P; Fernando S; Rajapakse S, Amphotericin B for treatment of visceral leishmaniasis: systematic review and meta-analysis of prospective comparative clinical studies including dose-ranging studies. Clin. Microbiol. Infect 2018, 24, 591–598. [DOI] [PubMed] [Google Scholar]
- 4.Tamiru A; Tigabu B; Yifru S; Diro E; Hailu A, Safety and efficacy of liposomal amphotericin B for treatment of complicated visceral leishmaniasis in patients without HIV, North-West Ethiopia. BMC Infect. Dis 2016, 16, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hailu A; Musa A; Wasunna M; Balasegaram M; Yifru S; Mengistu G; Hurissa Z; Hailu W; Weldegebreal T; Tesfaye S; Makonnen E; Khalil E; Ahmed O; Fadlalla A; El-Hassan A; Raheem M; Mueller M; Koummuki Y; Rashid J; Mbui J; Mucee G; Njoroge S; Manduku V; Musibi A; Mutuma G; Kirui F; Lodenyo H; Mutea D; Kirigi G; Edwards T; Smith P; Muthami L; Royce C; Ellis S; Alobo M; Omollo R; Kesusu J; Owiti R; Kinuthia J, Geographical variation in the response of visceral leishmaniasis to paromomycin in East Africa: a multicentre, open-label, randomized trial. PLoS Negl. Trop. Dis 2010, 4, e709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stephens C; Brun R; Salem M; Werbovetz K; Tanious F; Wilson W; Boykin D, The activity of diguanidino and “reversed” diamidino 2,5-diarylfurans versus Trypanosoma cruzi and Leishmania donovani. Bioorg. Med. Chem. Lett 2003, 13, 2065–2069. [DOI] [PubMed] [Google Scholar]
- 7.Reid C; Farahat A; Zhu X; Pandharkar T; Boykin D; Werbovetz K, Antileishmanial bis-arylimidamides: DB766 analogs modified in the linker region and bis-arylimidamide structure-activity relationships. Bioorg. Med. Chem. Lett 2012, 22, 6806–6810. [DOI] [PubMed] [Google Scholar]
- 8.Collar C; Zhu X; Werbovetz K; Boykin D; Wilson W, Molecular factors governing inhibition of arylimidamides against Leishmania: conservative computational modeling to improve chemotherapies. Bioorg. Med. Chem 2011, 19, 4552–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu X; Farahat A; Mattamana M; Joice A; Pandharkar T; Holt E; Banerjee M; Gragg J; Hu L; Kumar A; Yang S; Wang M; Boykin D; Werbovetz K, Synthesis and pharmacological evaluation of mono-arylimidamides as antileishmanial agents. Bioorg. Med. Chem. Lett 2016, 26, 2551–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang M; Zhu X; Srivastava A; Liu Q; Sweat J; Pandharkar T; Stephens C; Riccio E; Parman T; Munde M; Mandal S; Madhubala R; Tidwell R; Wilson W; Boykin D; Hall J; Kyle D; Werbovetz K, Novel arylimidamides for the treatment of visceral leishmaniasis. Antimicrob. Agents Chemother 2010, 54, 2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joice A; Yang S; Farahat A; Meeds H; Feng M; Li J; Boykin D; Wang M; Werbovetz K, Antileishmanial efficacy and pharmacokinetics of DB766-azole combinations. Antimicrob. Agents Chemother 2018, 62, e01129–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Macedo-Silva S; Urbina J; de Souza W; Rodrigues J, In vitro activity of the antifungal azoles itraconazole and posaconazole against Leishmania amazonensis. PLoS One 2013, 8, e83247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kulkarni M; Reddy N; Gude T; McGwire B, Voriconazole suppresses the growth of Leishmania species in vitro. Parasitol. Res 2013, 112, 2095–2099. [DOI] [PubMed] [Google Scholar]
- 14.Rashid J; Wasunna K; Gachihi G; Nyakundi P; Mbugua J; Kirigi G, The efficacy and safety of ketoconazole in visceral leishmaniasis. East Afr. Med. J 1994, 71, 392–395. [PubMed] [Google Scholar]
- 15.Sundar S; Singh V; Agrawal N; Gibbs D; Murray H, Treatment of kala-azar with oral fluconazole. Lancet 1996, 348, 614. [DOI] [PubMed] [Google Scholar]
- 16.Paniz Mondolfi A; Stavropoulos C; Gelanew T; Loucas E; Perez Alvarez A; Benaim G; Polsky B; Schoenian G; Sordillo E, Successful treatment of Old World cutaneous leishmaniasis caused by Leishmania infantum with posaconazole. Antimicrob. Agents Chemother 2011, 55, 1774–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lepesheva G; Friggeri L; Waterman M, CYP51 as drug targets for fungi and protozoan parasites: past, present and future. Parasitology 2018, 145, 1820–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pandharkar T; Zhu X; Mathur R; Jiang J; Schmittgen T; Shaha C; Werbovetz K, Antileishmanial mechanism of action of the arylimidamide DB766: azole interactions and role of CYP5122A1. Antimicrob. Agents Chemother 2014, 58, 4682–4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anighoro A; Bajorath J; Rastelli G, Polypharmacology: challenges and opportunities in drug discovery. J. Med. Chem 2014, 57, 7874–7887. [DOI] [PubMed] [Google Scholar]
- 20.Ivasiv V; Albertini C; Gonçalves A; Rossi M; Bolognesi M, Molecular hybridization as a tool for designing multitarget drug candidates for complex diseases. Curr. Top. Med. Chem 2019, 19, 1694–1711. [DOI] [PubMed] [Google Scholar]
- 21.Burgess S; Kelly J; Shomloo S; Wittlin S; Brun R; Liebmann K; Peyton D, Synthesis, structure-activity relationship, and mode-of-action studies of antimalarial reversed chloroquine compounds. J. Med. Chem 2010, 53, 6477–6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gunsaru B; Burgess S; Morrill W; Kelly J; Shomloo S; Smilkstein M; Liebman K; Peyton D, Simplified reversed chloroquines to overcome malaria resistance to quinoline-based drugs. Antimicrob. Agents Chemother 2017, 61, e01913–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Musonda C; Whitlock G; Witty MJ; Brun R; Kaiser M, Chloroquine-astemizole hybrids with potent in vitro and in vivo antiplasmodial activity. Bioorg. Med. Chem. Lett 2009, 19, 481–484. [DOI] [PubMed] [Google Scholar]
- 24.Han S; Sang Y; Wu Y; Tao Y; Pannecouque C; De Clercq E; Zhuang C; Chen FE, Molecular hybridization-inspired optimization of diarylbenzopyrimidines as HIV-1 nonnucleoside reverse transcriptase inhibitors with improved activity against K103N and E138K mutants and pharmacokinetic profiles. ACS Infect. Dis 2020, 6, 787–801. [DOI] [PubMed] [Google Scholar]
- 25.Aminake M; Mahajan A; Kumar V; Hans R; Wiesner L; Taylor D; de Kock C; Grobler A; Smith PJ; Kirschner M; Rethwilm A; Pradel G; Chibale K, Synthesis and evaluation of hybrid drugs for a potential HIV/AIDS-malaria combination therapy. Bioorg. Med. Chem 2012, 20, 5277–5289. [DOI] [PubMed] [Google Scholar]
- 26.Song H; Lee Y; Roh E; Seo J; Oh K; Lee B; Han H; Shin K, Discovery of potent and selective rhodanine type IKKβ inhibitors by hit-to-lead strategy. Bioorg. Med. Chem. Lett 2012, 22, 5668–5674. [DOI] [PubMed] [Google Scholar]
- 27.Fotie J; Kaiser M; Delfin D; Manley J; Reid C; Paris J; Wenzler T; Maes L; Mahasenan K; Li C; Werbovetz K, Antitrypanosomal activity of 1,2-dihydroquinolin-6-ols and their ester derivatives. J. Med. Chem 2010, 53, 966–982. [DOI] [PubMed] [Google Scholar]
- 28.Venning A; Bohan P; Alexanian E, Palladium-catalyzed, ring-forming aromatic C-H alkylations with unactivated alkyl halides. J. Am. Chem. Soc 2015, 137, 3731–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tavares F; Al-Barazanji K; Bigham E; Bishop M; Britt C; Carlton D; Feldman P; Goetz A; Grizzle M; Guo Y; Handlon A; Hertzog D; Ignar D; Lang D; Ott R; Peat A; Zhou H, Potent, selective, and orally efficacious antagonists of melanin-concentrating hormone receptor 1. J. Med. Chem 2006, 49, 7095–7107. [DOI] [PubMed] [Google Scholar]
- 30.Lebold T; Kerr M, Total syntheses of clausamines A-C and clausevatine D. Org. Lett 2008, 10, 997–1000. [DOI] [PubMed] [Google Scholar]
- 31.Wright W Jr.; Press J; Chan P; Marsico J; Haug M; Lucas J; Tauber J; Tomcufcik A, Thromboxane synthetase inhibitors and antihypertensive agents. 1. N-[(1H-imidazol-1-yl)alkyl]aryl amides and N-[(1H-1,2,4-triazol-1-yl)alkyl]aryl amides. J. Med. Chem 1986, 29, 523–530. [DOI] [PubMed] [Google Scholar]
- 32.Guengerich F; Martin M, Purification of Cytochromes P450. In Cytochrome P450 Protocols, Phillips I; Shephard E, Eds. Humana Press: Totowa, NJ, 2006. [Google Scholar]
- 33.Sevrioukova I; Poulos T, Current approaches for investigating and predicting cytochrome P450 3A4-ligand interactions. Adv. Exp. Med. Biol 2015, 851, 83–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chefson A; Zhao J; Auclair K, Replacement of natural cofactors by selected hydrogen peroxide donors or organic peroxides results in improved activity for CYP3A4 and CYP2D6. Chembiochem 2006, 7, 916–919. [DOI] [PubMed] [Google Scholar]
- 35.Liu YT; Hao HP; Liu CX; Wang GJ; Xie HG, Drugs as CYP3A probes, inducers, and inhibitors. Drug Metab. Rev 2007, 39, 699–721. [DOI] [PubMed] [Google Scholar]
- 36.Donato M; Jiménez N; Castell J; Gómez-Lechón M, Fluorescence-based assays for screening nine cytochrome P450 (P450) activities in intact cells expressing individual human P450 enzymes. Drug Metab. Dispos 2004, 32, 699–706. [DOI] [PubMed] [Google Scholar]
- 37.Niwa T; Shiraga T; Takagi A, Effect of antifungal drugs on cytochrome P450 (CYP) 2C9, CYP2C19, and CYP3A4 activities in human liver microsomes. Biol. Pharm. Bull 2005, 28, 1805–1808. [DOI] [PubMed] [Google Scholar]
- 38.Qiao Z; Wang Q; Zhang F; Wang Z; Bowling T; Nare B; Jacobs R; Zhang J; Ding D; Liu Y; Zhou H, Chalcone-benzoxaborole hybrid molecules as potent antitrypanosomal agents. J. Med. Chem 2012, 55, 3553–3557. [DOI] [PubMed] [Google Scholar]
- 39.Tyagi V; Khan S; Shivahare R; Srivastava K; Gupta S; Kidwai S; Srivastava K; Puri S; Chauhan P, A natural product inspired hybrid approach towards the synthesis of novel pentamidine based scaffolds as potential anti-parasitic agents. Bioorg. Med. Chem. Lett 2013, 23, 291–296. [DOI] [PubMed] [Google Scholar]
- 40.Harrison J; Brand S; Smith V; Robinson D; Thompson S; Smith A; Davies K; Mok N; Torrie LS; Collie I; Hallyburton I; Norval S; Simeons F; Stojanovski L; Frearson J; Brenk R; Wyatt P; Gilbert I; Read K, A molecular hybridization approach for the design of potent, highly selective, and brain-penetrant N-myristoyltransferase inhibitors. J. Med. Chem 2018, 61, 8374–8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Upadhyay A; Chandrakar P; Gupta S; Parmar N; Singh SK; Rashid M; Kushwaha P; Wahajuddin M; Sashidhara K; Kar S, Synthesis, biological evaluation, structure-activity relationship, and mechanism of action studies of quinoline-metronidazole derivatives against experimental visceral leishmaniasis. J. Med. Chem 2019, 62, 5655–5671. [DOI] [PubMed] [Google Scholar]
- 42.Centers for Disease Control, Parasites - Leishmaniasis: Resources for health professionals. https://www.cdc.gov/parasites/leishmaniasis/health_professionals/index.html#tx, accessed January 21, 2021.
- 43.Zhu X; Liu Q; Yang S; Parman T; Green C; Mirsalis J; Soeiro M; de Souza E; da Silva C; Batista D; Stephens C; Banerjee M; Farahat A; Munde M; Wilson W; Boykin D; Wang M; Werbovetz K, Evaluation of arylimidamides DB1955 and DB1960 as candidates against visceral leishmaniasis and Chagas disease – in vivo efficacy, acute toxicity, pharmacokinetics and toxicology studies. Antimicrob. Agents Chemother 2012, 56, 3690–3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lepesheva G; Hargrove T; Rachakonda G; Wawrzak Z; Pomel S; Cojean S; Nde P; Nes W; Locuson C; Calcutt M; Waterman M; Daniels J; Loiseau P; Villalta F, VFV as a new effective CYP51 structure-derived drug candidate for Chagas disease and visceral leishmaniasis. J. Infect. Dis 2015, 212, 1439–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Emami S; Tavangar P; Keighobadi M, An overview of azoles targeting sterol 14α-demethylase for antileishmanial therapy. Eur. J. Med. Chem 2017, 135, 241–259. [DOI] [PubMed] [Google Scholar]
- 46.Verma S; Mehta A; Shaha C, CYP5122A1, a novel cytochrome P450 is essential for survival of Leishmania donovani. PLoS One 2011, 6, e25273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beach D; Goad L; Holz G Jr., Effects of antimycotic azoles on growth and sterol biosynthesis of Leishmania promastigotes. Mol. Biochem. Parasitol 1988, 31, 149–162. [DOI] [PubMed] [Google Scholar]
- 48.Hargrove T; Wawrzak Z; Liu J; Nes W; Waterman M; Lepesheva G, Substrate preferences and catalytic parameters determined by structural characteristics of sterol 14α-demethylase (CYP51) from Leishmania infantum. J. Biol. Chem 2011, 286, 26838–26848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Friggeri L; Hargrove T; Rachakonda G; Blobaum A; Fisher P; de Oliveira G; da Silva C; Soeiro M; Nes W; Lindsley C; Villalta F; Guengerich F; Lepesheva G, Sterol 14α-demethylase structure-based optimization of drug candidates for human infections with the protozoan trypanosomatidae. J. Med. Chem 2018, 61, 10910–10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gunatilleke S; Calvet C; Johnston J; Chen C; Erenburg G; Gut J; Engel J; Ang K; Mulvaney J; Chen S; Arkin M; McKerrow J; Podust L, Diverse inhibitor chemotypes targeting Trypanosoma cruzi CYP51. PLoS Negl. Trop. Dis 2012, 6, e1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hargrove T; Friggeri L; Wawrzak Z; Sivakumaran S; Yazlovitskaya E; Hiebert S; Guengerich F; Waterman M; Lepesheva G, Human sterol 14α-demethylase as a target for anticancer chemotherapy: towards structure-aided drug design. J. Lipid Res 2016, 57, 1552–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abdelhameed A; Liao X; McElroy C; Joice A; Rakotondraibe L; Li J; Slebodnick C; Guo P; Wilson W; Werbovetz K, Synthesis and antileishmanial evaluation of thiazole orange analogs. Bioorg. Med. Chem. Lett 2020, 30, 126725. [DOI] [PubMed] [Google Scholar]
- 53.Gottlieb H; Kotlyar V; Nudelman A, NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem 1997, 62, 7512–7515. [DOI] [PubMed] [Google Scholar]
- 54.Narlawar R; Lane J; Doddareddy M; Lin J; Brussee J; Ijzerman A, Hybrid ortho/allosteric ligands for the adenosine A1 receptor. J. Med. Chem 2010, 53, 3028–3037. [DOI] [PubMed] [Google Scholar]
- 55.Salerno L; Pittalà V; Romeo G; Modica M; Siracusa M; Di Giacomo C; Acquaviva R; Barbagallo I; Tibullo D; Sorrenti V, Evaluation of novel aryloxyalkyl derivatives of imidazole and 1,2,4-triazole as heme oxygenase-1 (HO-1) inhibitors and their antitumor properties. Bioorg. Med. Chem 2013, 21, 5145–5153. [DOI] [PubMed] [Google Scholar]
- 56.Stephens C; Tanious F; Kim S; Wilson W; Schell W; Perfect J; Franzblau S; Boykin D, Diguanidino and “reversed” diamidino 2,5-diarylfurans as antimicrobial agents. J. Med. Chem 2001, 44, 1741–1748. [DOI] [PubMed] [Google Scholar]
- 57.Imoto M; Matsui Y; Takeda M; Tamaki A; Taniguchi H; Mizuno K; Ikeda H, A probable hydrogen-bonded Meisenheimer complex: an unusually high SNAr reactivity of nitroaniline derivatives with hydroxide ion in aqueous media. J. Org. Chem 2011, 76, 6356–6361. [DOI] [PubMed] [Google Scholar]
- 58.Shi M; Cui SC; Yin WP, Highly efficient catalytic nitration of phenolic compounds by nitric acid with a recoverable and reusable Zr or Hf oxychloride complex and KSF. Eur. J. Org. Chem 2005, 2379–2384. [Google Scholar]
- 59.Sythana S; Naramreddy S; Kavitake S; Vinod Kumar C; Bhagat P, Nonpolar solvent a key for highly regioselective SNAr reaction in the case of 2,4-difluoronitrobenzene. Org. Process Res. Dev 2014, 18, 912–918. [Google Scholar]
- 60.Ménard D; Niculescu-Duvaz I; Dijkstra H; Niculescu-Duvaz D; Suijkerbuijk B; Zambon A; Nourry A; Roman E; Davies L; Manne H; Friedlos F; Kirk R; Whittaker S; Gill A; Taylor R; Marais R; Springer C, Novel potent BRAF inhibitors: toward 1 nM compounds through optimization of the central phenyl ring. J. Med. Chem 2009, 52, 3881–3891. [DOI] [PubMed] [Google Scholar]
- 61.Yasuma T; Takakura N Preparation of fused ring compounds as glucokinase activators. WO 2009125873, October 15, 2009. [Google Scholar]
- 62.Schönian G; Nasereddin A; Dinse N; Schweynoch C; Schallig H; Presber W; Jaffe C, PCR diagnosis and characterization of Leishmania in local and imported clinical samples. Diagn. Microbiol. Infect. Dis 2003, 47, 349–358. [DOI] [PubMed] [Google Scholar]
- 63.Zhu X; Van Horn K; Barber M; Yang S; Wang MZ; Manetsch R; Werbovetz K, SAR refinement of antileishmanial N2,N4-disubstituted quinazoline-2,4-diamines. Bioorg. Med. Chem 2015, 23, 5182–5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Capes-Davis A; Reid Y; Kline M; Storts D; Strauss E; Dirks W; Drexler H; MacLeod R; Sykes G; Kohara A; Nakamura Y; Elmore E; Nims R; Alston-Roberts C; Barallon R; Los G; Nardone R; Price P; Steuer A; Thomson J; Masters J; Kerrigan L, Match criteria for human cell line authentication: where do we draw the line? Int. J. Cancer 2013, 132, 2510–2519. [DOI] [PubMed] [Google Scholar]
- 65.Werbovetz K; Riccio E; Furimsky A; Richard J; He S; Iyer L; Mirsalis J, Evaluation of antitrypanosomal dihydroquinolines for hepatotoxicity, mutagenicity, and methemoglobin formation In vitro. Int. J. Toxicol 2014, 33, 282–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hargrove T; Wawrzak Z; Alexander P; Chaplin J; Keenan M; Charman S; Perez C; Waterman M; Chatelain E; Lepesheva G, Complexes of Trypanosoma cruzi sterol 14α-demethylase (CYP51) with two pyridine-based drug candidates for Chagas disease: structural basis for pathogen selectivity. J. Biol. Chem 2013, 288, 31602–31615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.