

Abstract
Several non-redundant features of the tumour microenvironment facilitate immunosuppression and limit anticancer immune responses. These include physical barriers to immune infiltration, the recruitment of suppressive immune cells and the upregulation of ligands on tumour cells that bind to inhibitory receptors on immune cells. Recent insights into the importance of the metabolic restrictions imposed by the tumour microenvironment on antitumour T cells have begun to inform immunotherapeutic anticancer strategies. Therapeutics that target metabolic restrictions, such as low glucose levels, a low pH, hypoxia and the generation of suppressive metabolites, have shown promise as combination therapies for different types of cancer. In this Review, we discuss the metabolic aspects of the antitumour T cell response in the context of immune checkpoint blockade, adoptive cell therapy and treatment with oncolytic viruses, and discuss emerging combination strategies.
Since the FDA approval of the first checkpoint inhibitor in 2011, the field of cancer immunotherapy has grown exponentially: checkpoint blockade therapies with monoclonal antibodies targeting CTLA4 or PD1/PDL1, as monotherapy or in combination, are now approved for at least 13 cancer indications. The FDA has issued its first approval for an oncolytic virus (OV) (talimogene laherparepvec (T-VEC)), and adoptive transfer of chimeric antigen receptor (CAR) T cells has become the standard of care for select haematological malignancies1. However, many patients with cancer will not respond to immunotherapy, and, in the case of CAR T cells, their application for the treatment of solid tumours has largely been unsuccessful. Although many soluble, cellular and cell-intrinsic factors can contribute to resistance to immunotherapy, the role of metabolic pathways in determining T cell fate and function in antitumour immunity has recently started to attract considerable interest.
Anticancer immunotherapeutic strategies based on checkpoint inhibition, adoptive cell therapy (ACT) and OVs all require sustained effector T cell responses to induce durable clinical responses. These T cells, whether endogenous or adoptively transferred, must enter the tumour microenvironment (TME) and thrive within it. Tumour hypoxia, a low pH, suppressive metabolites and low nutrient availability can all prevent T cells from acquiring optimal effector functions. Each of these immunotherapies affects the T cell response at a different stage: blocking CTLA4 releases restrictions on T cell priming and activation; blocking PD1 releases restrictions on activation of the PD1low progenitor T cell population in the TME (see BOX 1); and OV therapy induces the priming and activation of new antiviral and anti-tumour responses (see BOX 2). ACT (see BOX 3) bypasses restrictions on activation and expansion; however, cells may experience metabolic stress owing to these expansion conditions and experience metabolic restrictions upon entry into the tumour (FIG. 1). At present, the metabolic restraints experienced by effector T cells in the tumour are not addressed in the context of these therapeutic interventions. Although many patients respond to these therapies without additional targeting of metabolic barriers, addressing the metabolic challenges of immune responses in the TME in combination with current therapeutic interventions may increase response rates.
Box 1 |. Checkpoint blockade.
Checkpoint blockade describes a therapeutic approach in which certain inhibitory receptors on immune cells (‘checkpoint molecules’) or their ligands are inhibited, usually with monoclonal antibodies. The first monoclonal antibodies to be approved for checkpoint blockade targeted the checkpoint molecules CTLA4 and PD1. Ipilimumab, a fully humanized anti-CTLA4 antibody, was approved in 2011 for its ability to induce long-term survival in patients with metastatic melanoma 144. This was followed in 2014 by monoclonal antibodies that block PD1, first pembrolizumab and shortly after nivolumab, also for the treatment of patients with melanoma145. These checkpoint blockers, alone or in combination, have since been approved for a number of different types of cancer. However, depending on their target, they act at different stages of T cell differentiation.
CTLA4.
T cells require three types of signal for proper activation: TCR signalling, co-stimulatory signalling and cytokine signalling. Without co-stimulatory signalling through the surface receptor CD28, T cells proceed to an anergic state146. CTLA4 binds CD80 and CD86, the same ligands bound by CD28 (REFS147–150). This may sequester these ligands and prevent successful T cell activation151. Regulatory T cells (Treg cells) constitutively express CTLA4 (REF.152), and mice lacking CTLA4 (REFS153,154), or mice with a Treg cell-specific knockout of Ctla4 (REF.155), have rampant and widespread autoimmunity. Blocking CTLA4 in a mouse model of melanoma led to the regression of established tumours and generated memory responses that were sufficient to protect against rechallenge156.
CTLA4 blockade acts on T cells during the priming stage. This occurs primarily in secondary lymphoid organs, such as the tumour-draining lymph nodes, where naive T cells are activated. It is thought that the blocking of CLTA4 allows CD28–ligand binding and the subsequent complete activation of the cells, which would otherwise be restrained by CTLA4-mediated suppression. Although mouse models showed that CTLA4-targeted treatment can selectively deplete Treg cells through antibody-dependent cellular cytotoxicity, there is currently much discussion around whether this contributes to responses in humans157.
PD1.
PD1 is expressed by immature T cells in the thymus during T cell receptor selection158–160, as well as by mature T cells after activation161. The binding of PD1 to its ligands, PDL1 or PDL2, which are expressed on cells in inflamed peripheral tissues, results in the inhibition of certain T cell signalling pathways that are important for T cell activation, proliferation, survival, cytokine production and metabolic switching162,163, which was reviewed extensively in REF.164.
Unlike CTLA4, PD1 is upregulated only after cells are activated, and it is involved in preventing autoimmunity in the periphery165. Inflammatory cytokines such as interferon-γ can induce an upregulation of PDL1 in tumour cells, leading to a dampened immune response166. PD1 expression is highly upregulated in T cells classified as ‘exhausted’, along with other inhibitory (checkpoint) receptors such as LAG3, TIM3 and TIGIT167,168. T cell exhaustion, characterized as an intrinsic failure to respond robustly to antigens and perform multiple effector functions, was first observed in chronic infection with lymphocytic choriomeningitis virus169,170 and eventually in cancer171. Although PD1 blockade can restore T cell activity in models of infection and cancer172,173, it is now generally appreciated that blocking PD1 or its ligands does not restore the activity of the terminally differentiated, exhausted PD1hi T cells, but rather acts on less differentiated, stem-like T cells, which are characterized by the expression of the transcription factor TCF7 (REFS174–176). The blockade of PD1 is thought to induce the differentiation of these progenitor cells into cytotoxic T cells that can mediate tumour killing.
Box 2 |. Adoptive cell therapy.
Adoptive cell therapy (ACT) uses tumour-infiltrating lymphocytes (TILs) or peripheral blood mononuclear cells from patients or healthy donors as ‘living drugs’. The cells are isolated from the donor or patient, expanded in vitro and readministered to the patient. T cells for ACT are expanded in vitro under culturing conditions that allow rapid proliferation and expansion. Patients who respond well to ACT generally show long-term maintenance of the transferred T cells, as well as strong expression of a tumour antigen that is targeted by the product. Two chimeric antigen receptor (CAR) T cell products, Kymriah and Yescarta, which both target CD19, received approval by the FDA in 2017 (REF.177).
TIL therapy.
T cells can be isolated from tumour tissues and cultured in vitro for extended periods in the presence of high doses of IL-2. Thus, one way to potentially treat inaccessible and/or undetectable metastases is to expand T cells from an accessible lesion and administer the expanded TILs to the patient. Isolating TILs directly from the tumour, combined with expanding them outside the suppressive tumour microenvironment, allows the activation and expansion of tumour-specific T cells that can be reimplanted in high numbers. This method has been shown to induce complete and durable responses in patients with metastatic melanoma178, and FDA approval for a TIL therapy is expected to be imminent. However, it has been challenging to isolate T cells with specificity for tumour antigens; to circumvent this limitation, studies are ongoing to identify neoantigen-specific T cells as well as to engineer tumour-specific T cell receptors into patient lymphocytes. However, the production of personalized T cell receptor-engineered lymphocytes is expensive and time-consuming, and cancer cells can escape by reducing their expression of the targeted antigen. Although cells used for adoptive TIL therapy are expanded in excess nutrient and oxygen conditions, once they are in the tumour, they experience the same nutrient restrictions as endogenous cells, which may affect effector function such as cytokine production. Also, these transferred T cells must infiltrate the tumour, which is another ‘metabolically expensive’ process.
CAR T cells.
CAR T cells are lymphocytes derived from the patient that are engineered to express hybrid receptors that combine two parts: an extracellular domain to recognize the antigen of interest, which is generally composed of a single-chain variable fragment (scFv) of an antibody, and an intracellular signalling domain that is engineered to provide activation signals, which is typically derived from the signalling molecule CD28 or 4–1BB179. CAR T cells are expanded in vitro in culturing conditions that contain excess nutrients and oxygen. The most successful example of CAR T cell therapy is the use of CD19-targeted CARs to treat patients with B cell leukaemia and lymphoma, where complete remission rates of 83% for patients with acute lymphoblastic leukaemia and 43% for patients with diffuse B cell lymphoma have been reported180–187. Cellular targets for the treatment of other haematological malignancies are currently under investigation179. The main cause of relapse in patients is dysfunction and poor persistence of the CAR T cells, and CAR T cells generated from more memory-like T cells are more likely to yield complete responses than those generated from effector T cells188.
Despite the success of CAR T cells in haematological cancers, there has been little success with using this approach for the treatment of solid tumours. As mentioned above, antigenic targets that are selectively expressed by tumour cells can be difficult to identify. Furthermore, CAR T cells must first infiltrate the tumour, and, once inside, they encounter a number of metabolic barriers. Enhancing the metabolism of CAR T cells to overcome these barriers may benefit patients with solid tumours.
Box 3 |. Oncolytic viruses.
Oncolytic viruses (OVs) are viruses that selectively infect tumour cells. In addition, to increase safety, many OVs are engineered to enhance their selectivity for tumour tissue over healthy tissue. Lysis occurs by immunogenic cell death mechanisms and leads to the infiltration of the tumour by cytotoxic effector T cells that clear the tumour103. There are currently a number of OVs in preclinical investigation and in clinical trials, with both intratumoural and intravenous routes of administration. Viruses currently under study for their utility as OVs include herpesvirus, vaccinia virus, vesicular stomatitis virus, Newcastle disease virus, reovirus, Coxsackie virus and measles virus103. Only one OV, the herpesvirus talimogene laherparepvec (T-VEC), has so far received approval by the FDA (2014)113. Currently, Pexa-Vec, a genetically engineered vaccinia virus, is the only OV in phase III clinical trials, while several others, including a poliovirus (PVS-RIP) and an adenovirus (DNX-240), are currently in phase II trials189.
Although it was originally thought that the immune system is detrimental to OV therapy, as it was feared that it could clear the virus and thereby impair its ability to induce the lysis of tumour cells, it is now generally accepted that the immune response to the OV is crucial to tumour clearance190–192. When tumour-bearing mice are treated with CD8+ T cell depleting antibodies, the efficacy of OV therapy is lost, illustrating the importance of the T cell response193–197. Both tumour- and virus-targeted T cells can be detected in response to OV therapy, and the relative importance (and crosstalk) between these subsets remains incompletely understood198. Mouse models show an abscopal effect in non-injected lesions, memory responses, and a reduction in suppressive immune cell populations199–201. However, the de novo T cells generated in response to OV therapy experience many of the same metabolic barriers that restrict trafficking, proliferation and effector functions within the tumour as those observed for other T cell-based immunotherapies.
Fig. 1 |. Metabolic barriers acting on different phases of immunotherapeutic response.
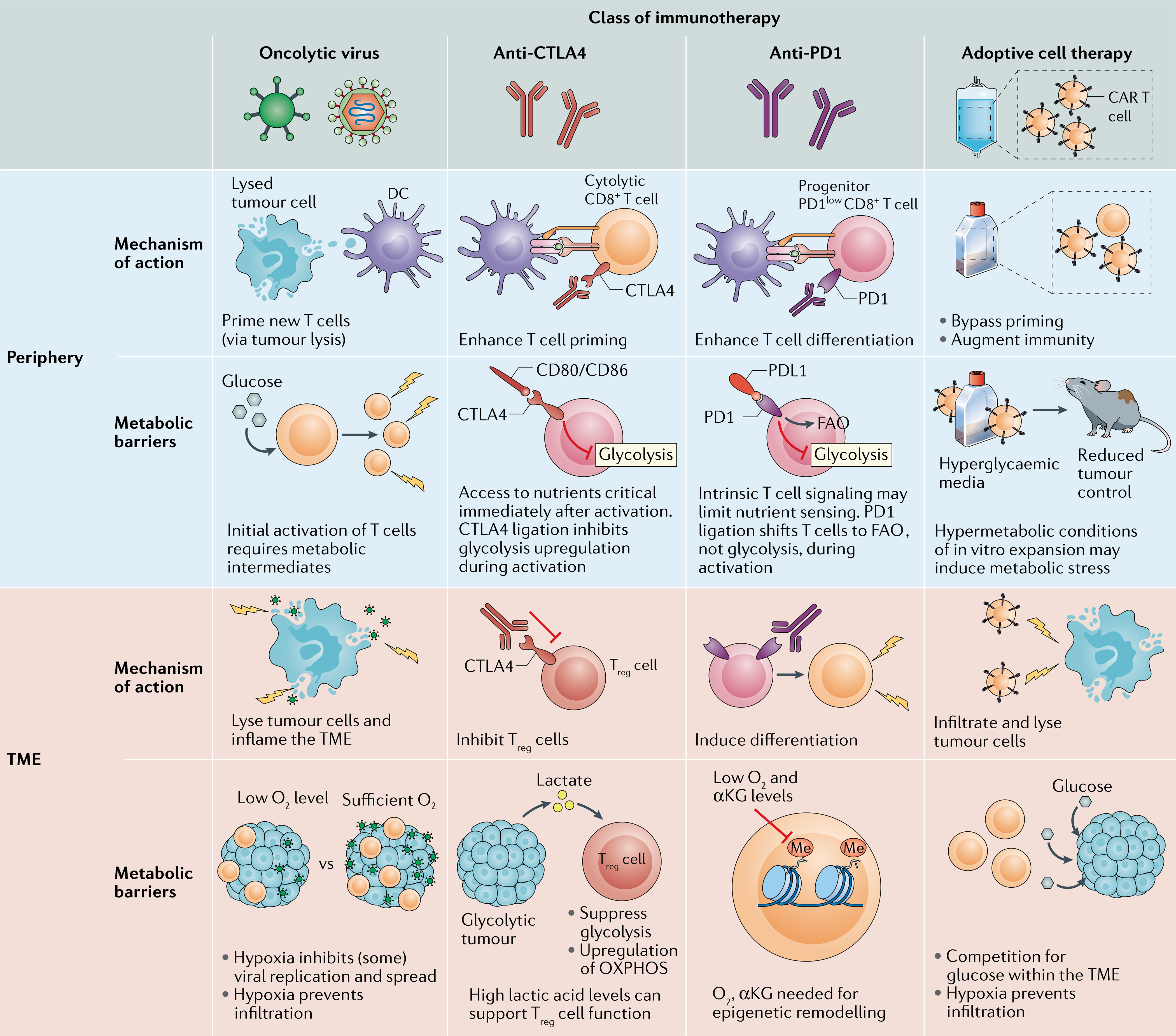
Different classes of immunotherapy act on the immune response at different stages of cell differentiation and activation. This is dependent on the mechanism of action of the drug as well as the location of the immune cells. The metabolic requirements and barriers experienced by the adaptive immune response to each treatment differ by location and treatment intervention. This figure highlights the mechanism of action and metabolic barriers to treatment, both in the periphery and in the tumour microenvironment (TME), for each type of immunotherapy covered in this Review. αKG, α-ketoglutarate; CAR, chimeric antigen receptor; DC, dendritic cell; FAO, fatty acid oxidation; OXPHOS, oxidative phosphorylation; Treg cell, regulatory T cell.
Here, we discuss the roles of different metabolic pathways in antitumour immune cell function, with a specific focus on the three approved classes of immunotherapy: checkpoint blockade, adoptive cell therapy and OVs. Furthermore, we discuss how targeting metabolic barriers may improve therapeutic responses.
Metabolic relationships in the TME
A hallmark of cancer is continual growth and cellular proliferation while avoiding cell death, which has a considerable energetic cost1. Although it has long been known that the intrinsic metabolism of tumour cells can shape the TME1, until recently the effects of such a metabolically altered tissue on non-transformed cells such as infiltrating immune cells was not appreciated. However, the interaction between nutrients, metabolites and immune cells is likely to play a major role in immune editing and in tumoural escape (FIG. 2). The ability of tumour cells to utilize glycolysis for energy (ATP) generation, despite the presence of sufficient levels of oxygen for oxidative phosphorylation (OXPHOS), which results in a more efficient pathway for ATP generation, was discovered by Otto Warburg in the early twentieth century, and is termed the ‘Warburg effect’2,3. The utilization of glucose for glycolysis and the rapid cellular proliferation leaves the TME depleted of nutrients for infiltrating immune cells such as effector T cells, and simultaneously enriches the TME in immunosuppressive metabolites4,5. However, nutrient and oxygen availability are not homogeneous within the TME, with pockets of nutrient depletion and hypoxia resulting from differential ‘fuel’ uptake by heterogeneous tumour cell populations. Such variation can depend on the tumour type and tumour location6–10. Residence within a nutrient-restricted tumour site may exert metabolic stress on T cells, which can affect their differentiation and function. However, some nutrients can also be overabundant within the TME compared with plasma as shown in a mouse model of pancreatic ductal adenocarcinoma10. High glucose utilization by the tumour also results in the generation of large amounts of lactate as a by-product, which lowers the pH of the TME. This can impair the cytolytic activity and cytokine production of T cells11. Moreover, lactate, irrespective of its pH-lowering property, is a metabolite with diverse effects on immune cell populations, and has been shown to polarize macrophages towards the tolerogenic M2 type and shift the metabolism of regulatory T cells (Treg cells) to maintain their activity in low-glucose environments12–14. Tumour cells that undergo rapid proliferation also require an increased blood flow to transport nutrients and oxygen into the TME and often produce angiogenic factors. This can result in the formation of a disorganized vasculature and the formation of pockets of extreme hypoxia in the tumour, which restricts immune cell infiltration and leads to the build-up of immunosuppressive metabolic by-products8,9. Intriguingly, many of these metabolic changes, although detrimental to effector T cell function, can support — or at least not negatively affect — suppressive immune populations such as Treg cells or suppressive myeloid populations13,14.
Fig. 2 |. Metabolic suppression in the tumour microenvironment.
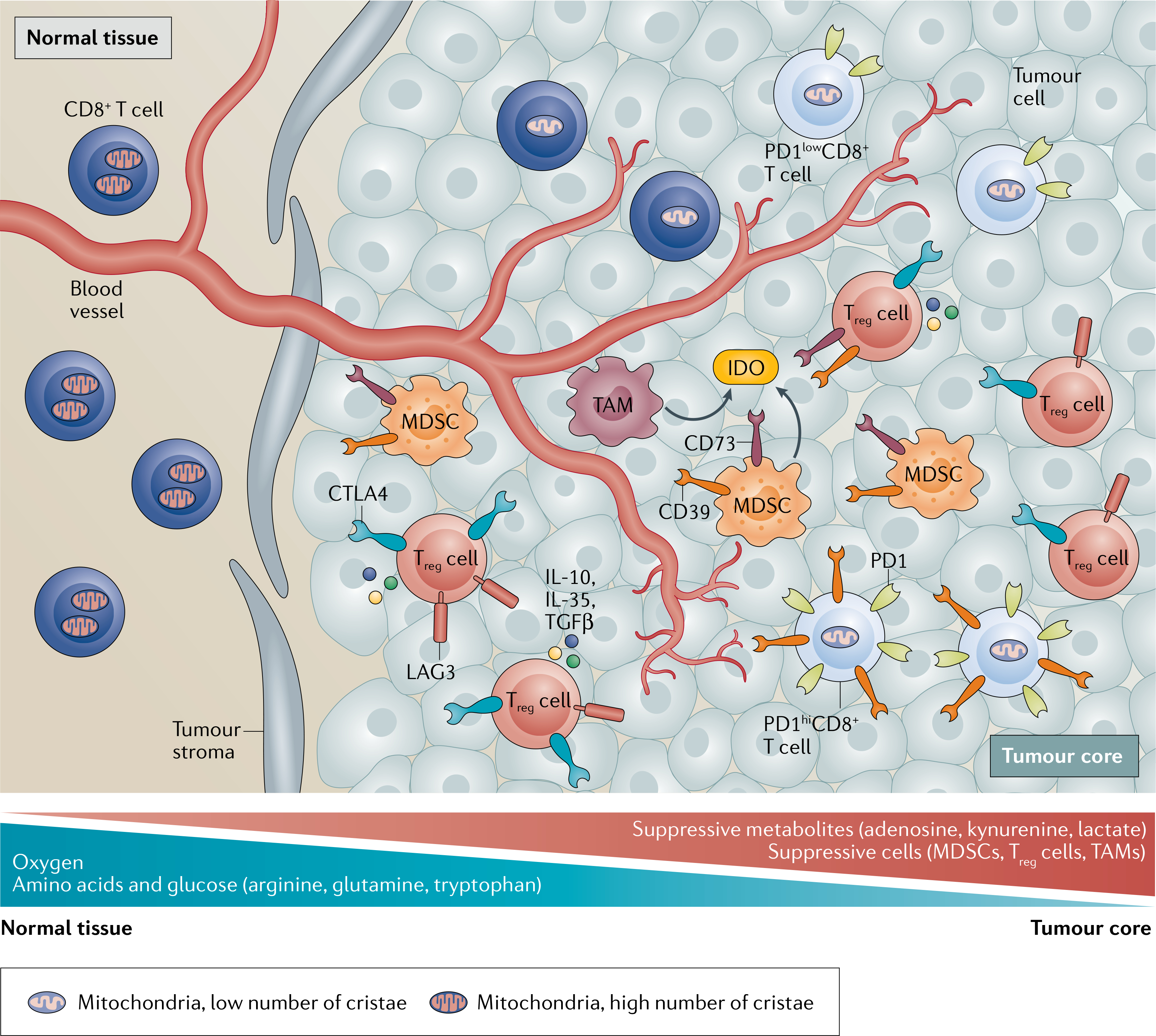
The tumour microenvironment imparts metabolic stress on effector CD4+ T cells and CD8+ T cells through a variety of mechanisms. The rapid proliferation of tumour cells is supported by high rates of glycolysis, which leads to the local depletion of oxygen and glucose levels, resulting in hypoxic regions that contain high levels of lactate. Dysregulated vasculature, a result of increased tumour proliferation, results in damaged and leaky vessels, which reduce blood flow. This reduces oxygen and nutrient levels in the tumour core and prevents metabolites that suppress CD8+ T cells from being transported out of the tumour. Suppressive myeloid cells such as myeloid-derived suppressor cells (MDSCs) as well as regulatory T cells (Treg cells) are abundant within the tumour, and their distinct metabolic profiles allow them to thrive on tumour-derived metabolites. Furthermore, MDSCs, tumour-associated macrophages (TAMs) and Treg cells produce suppressive metabolites such as adenosine and kynurenine. Adenosine is produced from ATP by the ectoenzymes CD73 and CD39, which are expressed on the surface of these suppressive cells. MDSCs and TAMs produce arginase 1, which utilizes arginine, and indoleamine 2,3-dioxygenase (IDO), which metabolizes tryptophan into suppressive kynurenine, and reduce the availability of these amino acids within the tumour. These mechanisms starve the T cells in the tumour microenvironment of amino acids. Treg cells exert their immunosuppressive effects through the expression of inhibitory molecules such as CTLA4 and LAG3, as well as by secreting immunosuppressive cytokines such as IL-10, TGFβ and IL-35. CD8+ T cells enter the tumour as PD1− or PD1low cells with intact mitochondria. Continuous T cell receptor stimulation and hypoxia induce exhaustion in tumour-specific CD8+ T cells, characterized by the co-expression of multiple inhibitory receptors (TIM3, LAG3, PD1, TIGIT and so on) and by the loss of functional mitochondria. As a result of the suppressive tumour microenvironment, the most terminally exhausted T cells (PD1hi T cells) can no longer control tumour growth. Although PD1low progenitor T cells can be reinvigorated by checkpoint blockade, terminally exhausted PD1hi T cells cannot, highlighting the need to understand how these cells may be additionally modulated to enhance response to PD1 blockade.
Metabolic reprogramming plays a central role in the differentiation and activation of T cells as the switch from quiescence to activation, proliferation, differentiation and infiltration carries a heavy energetic burden. Therefore, metabolic pathways have an immunoregulatory role in T cells, which was reviewed in REFS15–18. The role of metabolism in altering the epigenome and the influence this has on cell fate was recently reviewed19.
Different subsets of differentiated T cells utilize distinct metabolic programmes20–22. Conventional naive T cells exist in a resting state, potentially for a lifetime, with minimal metabolic activity. However, newly activated T cells (effector T cells) rapidly upregulate glucose uptake and associated aerobic glycolysis, which enables nucleotide synthesis for rapid proliferation and ATP production, and relieves translational inhibition to promote cytokine production5,23. It also frees up intermediates from the tricarboxylic acid (TCA) cycle to generate metabolic by-products that are involved in cellular signalling, biosynthetic reactions such as the production of amino acids and epigenetic modifications15. For example, in parallel to upregulating glucose metabolism, T cells also engage metabolic programmes to rapidly take up and metabolize glutamine, which enters the TCA cycle as α-ketoglutarate24. Glutamine can support OXPHOS through the TCA cycle, but α-ketoglutarate can also directly interact with the demethylase machinery to promote epigenetic modulation15. Furthermore, both resting and activated T cells take up exogenous lipids and can oxidize intracellular stores of lipids25,26. β-Oxidation is a very efficient method to generate ATP in the presence of oxygen. Thus, a newly activated conventional T cell can fuel its proliferation and differentiation through multiple nutrient sources, creating a clonal population with diverse effector functions.
However, not all T cells uniformly engage in these metabolic transitions after activation. Treg cells have low rates of glycolysis and predominantly utilize fatty acid oxidation for their metabolism27. It is unclear whether this is the default pathway for energy generation in the absence of glycolysis or whether this is the intrinsic metabolic pathway for Treg cells. Treg cells do upregulate the transcription of fatty acid transporters; however, they also can take up alternative metabolites, such as butyrate and lactate28. Regardless, this pathway of energy generation may allow Treg cells to function optimally within the TME, whereas the activity of conventional CD4+ T cells and CD8+ T cells is impaired. Notably, Treg cell metabolism is an understudied field, and many aspects of the metabolic requirements of T cells, especially in the context of specific tissues and disease states, have yet to be elucidated.
Given that chemotaxis and motility impose heavy energetic demands on cells, the metabolism of T cells influences their trafficking in and out of secondary lymphoid organs (SLOs) and tissues. During activation of naive T cells, the PI3K–AKT–mTOR axis downregulates the expression of the SLO homing receptors CD62L, CCR7 and S1P1, while at the same time increasing the expression of the chemokine receptors CXCR3 and CCR5 to allow egress from the SLO. By contrast, during the differentiation of activated effector T cells into long-lived memory cells, low mTOR activity leads to increased CD62L and CCR7 expression, which allows increased trafficking and surveillance29. During this differentiation step there is also a metabolic transition to an increase in mitochondrial oxidative metabolism, which is promoted by IL-15, an important cytokine that supports memory T cell differentiation30. Indeed, in memory T cells, the sensing of nutrients such as glucose and amino acids and relative ATP levels via signalling by the mTOR complex stimulates mitochondrial biogenesis, which creates an increase in mitochondrial respiratory capacity and allows for efficient usage of metabolic pathways under stress situations such as nutrient deprivation30,31. Most long-lived cells rely on mitochondrial metabolism for energy generation, and thus energy generation through OXPHOS, which occurs in the mitochondrial membrane, supports the long-term persistence of these cells. In summary, T cells require various fuel sources to effectively activate, proliferate, differentiate, home to tissue sites, search for antigens and perform their effector functions. As the metabolic pathways utilized during each of these steps can be distinct, the differential access to nutrients at various tissue sites can alter the path of T cell differentiation and function. This is critically important in anticancer immunity, in which the ‘normal’ rules of activation, tissue homing, antigen recognition and cytotoxicity of T cells are skewed by the suppressive mechanisms of the TME.
Checkpoint blockade
Immune checkpoint molecules, which are generally co-inhibitory receptors expressed by immune cells such as T cells, are critical for the prevention of autoimmunity and immunopathology; however, in the context of cancer, they can dampen antitumour immune responses. Checkpoint molecules or their ligands are expressed by a variety of cell types within the TME. Here, we focus on the checkpoint molecules most widely targeted in the clinic, CTLA4 and PD1 (BOX 1).
Immunotherapy via checkpoint blockade depends on a pre-existing antitumour response, a smouldering immune cell infiltrate that is capable of reinvigoration. However, patients may either have very low numbers of tumour-specific T cells or lack an antitumour response entirely.
Although a number of different mechanisms of resistance to immune responses may be at play within the TME32, a low pH and low oxygen availability, the presence of suppressive metabolites, and reduced glucose and nutrient availability impose metabolic barriers on T cells33. Moreover, T cells also generate their own intrinsic metabolic barriers to activation, which can be promoted by checkpoint molecules. For example, signalling through CTLA4 and PD1 interferes with glycolysis by reducing AKT activation, although this is achieved through different mechanisms34,35. Whereas PD1 binding reduces PI3K signalling through the inhibitory domains of its cytoplasmic tail, CTLA4 binding does not interfere with PI3K signalling and instead may reduce AKT activity through protein phosphatase 2A (PP2A)34. Additionally, PD1 ligation may also induce a metabolic shift towards increased fatty acid oxidation36–38; however, such a shift has not been observed in response to CTLA4 activation36. CTLA4 binds CD28 on T cells in SLOs during T cell priming and may prevent activation-induced glycolysis34, thereby reducing the propensity of activated T cells to become fully differentiated effector cells. By contrast, the expression of PD1 by T cells is elevated late, and most prominently by continuous activation; in the context of cancer, PD1 ligation occurs when the T cell encounters PDL1-expressing cells in the TME. Thus, PD1 ligation may limit the increase in glycolysis that occurs upon T cell activation and thereby limit T cell proliferation and effector functions within the TME. As both CTLA4 and PD1 prevent the upregulation of T cell glycolysis during activation, augmenting CLTA4-targeted and PD1-targeted checkpoint blockade with therapeutic approaches that increase glycolysis may enhance patient responses.
Mitochondrial metabolism.
Mitochondria are crucially important for cellular function; beyond production of ATP, they also generate critical precursors for lipids, proteins and nucleic acids and aid in adaptation to extracellular and intracellular stresses such as DNA damage, nutrient and oxygen deprivation, and endoplasmic reticulum and oxidative stress39. As a result, changes to mitochondrial function have a profound impact on cell survival and function. For instance, we found that T cells isolated from the TME of mouse B16 melanomas show reduced mitochondrial function and a poor ultrastructure (the ultrastructure is necessary for electron transport chain activity), as well as reduced mitochondrial mass40. This was found to be a consequence of the repression of transcriptional programmes associated with mitochondrial biogenesis, most notably the transcriptional co-activator PGC1α. These changes were concomitant with the development of the exhausted phenotype, such that terminally exhausted T cells bore the most severe mitochondrial deficiencies40. Importantly, these mitochondrial deficiencies were also observed in exhausted T cells in chronic viral infections41. Treatment with PD1 blockade does not reverse the mitochondrial deficiencies of tumour-infiltrating T cells40. However, experiments in mice have shown that an increase of mitochondrial biogenesis in T cells, by genetic or pharmacologic means, not only restores their function in the tumour but also shows synergistic effects with PD1 blockade40,42,43. Enforcing mitochondrial biogenesis and increasing fatty acid oxidation with the PPARα agonist bezafibrate, in combination with PDL1-targeted therapy, reduced CD8+ T cell reliance on glycolysis and improved T cell tumour infiltration compared with PDL1-targeted therapy alone in mice44. Co-stimulatory therapy, for example by using agonistic antibodies to the co-stimulatory molecule 4–1BB, can also promote mitochondrial biogenesis in human and mouse T cells and synergize with PD1-targeted therapy in mouse models45,46. More recently it was shown that chronic antigen stimulation of T cells in the TME can directly impact mitochondrial function and lead to the development of the exhausted phenotype. Chronic antigen stimulation, hypoxic conditions and PD1 signalling all contribute to the generation of mitochondrial reactive oxygen species, which further promote the exhausted T cell phenotype by inducing epigenetic alterations47–49. Targeting this oxidant imbalance can synergize with checkpoint blockade and improve antitumour functionality47–49. These data suggest that mitochondria play a critical role in counteracting the ‘exhausted’ phenotype, such that T cells that are metabolically fitter, with a superior mitochondrial phenotype and ability to compete for nutrients, are more likely to respond to PD1 blockade.
Hypoxia.
Mitochondria require oxygen for optimal function, which can be limiting within the TME. Tumours can differ with regards to hypoxia by both the tissue of origin and the region within the tumour50. Hypoxia is typically increased in the tumour core owing to factors such as an irregular vasculature that is poorly perfused by oxygen, an increased distance from the vasculature to the tumour core and the propensity of the tumour itself to consume oxygen51. Mouse tumours with an increased oxidative metabolism, and therefore severer hypoxia, were found to be infiltrated by more terminally exhausted T cells and showed decreased immune activity. These tumours did not respond to PD1-targeted therapy. These findings correlate with patient data, where patients with tumours that show higher utilization of oxidative metabolism were more likely to fail to respond to PD1 blockade52.
As oxygen is a key determinant for both mitochondrial function and the consequent energetic and epigenetic changes needed for the function and differentiation of immune cells, and in particular T cells, alleviating hypoxia can facilitate antitumour immunity and boost anticancer immunotherapy responses. For example, it was shown that supplemental oxygen can decrease the levels of immunosuppressive adenosine in mice with lung tumours, increase the levels of CD8+ T cell infiltration and inflammatory cytokine production, and decrease Treg cell-mediated immunosuppression, resulting in increased survival53. When tumour-bearing mice were treated with metformin, which can act on tumour cells to reduce their mitochondrial activity, tumour hypoxia was decreased and the T cell response to anti-PD1 was improved54. Similar results have been shown with use of a hypoxia-activated prodrug, evofosfamide, which reduced tumour hypoxia in a mouse model of prostate cancer, and anti-VEGF antibodies that normalize the vasculature in CT26, a mouse model of colorectal cancer55,56.
Taken together, these data suggest that insufficient oxygen levels either prevent successful T cell activation or support T cell exhaustion, which may contribute to resistance to checkpoint blockade. Determining the metabolic profile of the tumour of a patient may therefore identify responders and inform potential combination therapies.
Glycolysis and glutaminolysis.
Tumour cells outcompete effector T cells for glucose in the TME2,57. CD4+ T cells and CD8+ T cells also require intermediates generated during glycolysis for proliferation and cytokine production5,23,58. For example, 2-phosphoenolpyruvate (PEP), an intermediate upstream of glycolytic metabolism, is crucial for Ca2+ signalling and nuclear translocation of the transcription factor NFAT in response to T cell receptor stimulation4. Likewise, glycolysis itself can control T cell effector functions by inhibiting the RNA-binding ‘moonlighting’ functions of glycolytic enzymes such as GAPDH and LDH5,23. These metabolic enzymes have been shown to bind mRNA during periods of metabolic glycolysis, preventing effective translation. In this way, the shift from glycolysis to oxidative metabolism may aid in sustaining effector functions despite low levels of glucose in the TME. Modulating this metabolic switch in effector T cells within the tumour has shown promise in preclinical studies43,45,46.
As tumour cells have high rates of glycolysis, they are also likely to produce large amounts of the by-product lactate2; indeed, serum lactate levels have been shown to correlate with higher tumour burden in patients with a variety of cancer types59. Lactic acid has a suppressive effect on CD4+ T cells and CD8+ T cells by reducing proliferation and cytokine production59. Lactate can impair effector T cells by depleting intracellular NAD+ levels because LDH utilizes the lactate to generate NADH. Intriguingly, this does not occur in suppressive Treg cells; NAD+ generated through mitochondrial metabolism allows Treg cells to continue to function in high-lactate environments, where conventional T cells are suppressed13. We recently showed that deletion of the lactate transporter MCT1 on Treg cells selectively inhibits immunosuppression in lactate-rich TMEs60. It also reduces the motility of CD4+ T cells and CD8 T+ cells, which may reduce their infiltration of the TME and their movement within the TME12. Blocking the lactate transporters MCT1 and MCT4 with a pharmacological inhibitor reduced tumour acidification by preventing lactate release. In a mouse model of breast cancer, blockade of MCT1/MCT4 in combination with checkpoint blockade enhanced responses compared with checkpoint blockade alone61.
Glutamine is a critical amino acid for cellular function and has an equally important role in the generation of energy and metabolic intermediates. Upon entry into the cell, glutamine is converted by the enzyme glutaminase into glutamate, which is further converted into α-ketoglutarate, which enters the TCA cycle and leads to the production of metabolic intermediates that can be used to produce lipids, nucleic acids and proteins. Many cancer cells lack the enzymes to synthesize glutamine and, instead, are dependent on exogenous glutamine sources62. However, this depends on the tumour type and oncogenic driver, and indeed some tumour types, such as non-small-cell lung carcinoma, actually show increased glutamine synthesis62. Glutamine metabolism facilitates T cell activation and proliferation, and, as a result, a lack of glutamine in the TME due to high consumption by the tumour may inhibit T cell activity24,63. It was shown that treatment of MC38-tumour bearing mice (a mouse model of colon cancer) with a prodrug of the glutamine antagonist 6-diazo-5-oxo-l-norleucine, which binds the active site of a number of glutamine-utilizing enzymes, decreased both glucose and glutamine metabolism within the tumour and increased the availability of both nutrients within the TME64. Co-administration of PD1-targeted checkpoint blockade induced complete responses as well as a memory response upon tumour rechallenge. These responses were due to the metabolic plasticity in CD8+ tumour-infiltrating T cells, which, in contrast to tumour cells, can increase acetate metabolism, thereby allowing OXPHOS and acetyl-CoA formation64. These more oxidative CD8+ T cells are more active and longer lived, as previously described. However, the effectiveness of this combination therapy may be lower for tumours that do not utilize exogenous glutamine.
Other metabolic pathways.
Methionine, an essential amino acid, is important for T cell differentiation. Reduced intracellular methionine levels can lead to a reduction in the levels of the epigenetic methyl donor S-adenosyl-l-methionine65,66. For example, it was shown that CD8+ T cells cultured in low-methionine conditions or isolated from mouse or human tumours had reduced levels of S-adenosyl-l-methionine as well as reduced dimethylation at lysine 79 of histone H3 (H3K79me2), resulting in decreased expression of STAT5 (REF.66). STAT5 signalling is critical for T cell responses to IL-7 and IL-15, which are critical cytokines for T cell memory development67. Human colorectal tumour cells have higher expression of the methionine transporter SLC43A2 than T cells, which may allow them to outcompete T cells for methionine in the TME. Pharmacological inhibition of this transporter in mouse ovarian cancer (ID8) and melanoma (B16F10) models increased methionine availability for T cells, and a combination of this drug with checkpoint blockade improved tumour control and increased T cell cytokine production66. These data suggest that altered metabolite availability may cause the altered epigenetic profile observed in exhausted tumour-infiltrating T cells. Targeting methylation in T cells in combination with checkpoint blockade may enhance T cell activity68.
A lack of extracellular arginine has also been shown to reduce T cell proliferation69. Arginase 1 (ARG1) is expressed by myeloid-derived suppressor cells in the TME and can deplete arginine levels within tumours70. Some success was observed when ARG1 inhibitors were used in combination with checkpoint blockade70. However, as low levels of arginine impact mainly on T cell proliferation, and the major site of T cell proliferation is in the draining lymph node, it is unclear how critical the targeting of this pathway is as arginine levels in the draining lymph node have not been measured70.
Adoptive cell therapy
Therapeutic T cells for ACT are activated and expanded in vitro in a medium containing excess nutrients, relieving these cells of the metabolic barriers that may exist during priming, expansion and initial differentiation within the tumour site. Once transferred, these cells must enter the tumour, which is a metabolically taxing effort, as well as perform effector functions in the nutrient-restricted TME. ACT provides a unique opportunity to counteract these metabolic barriers during the pretransfer expansion. This can be achieved by manipulating the cells before transfer to improve their metabolic fitness, or by supporting the metabolism of the transferred T cells though systemic therapy.
Metabolic modulation to increase the generation of memory cells.
The persistence of the transferred cells and their differentiation into memory cells are critical aspects for ACT. For example, it was shown that CAR T cell persistence correlates with better tumour clearance and increased survival71. Deriving cells for adoptive transfer from central memory T cells yields a more persistent memory phenotype in vivo72,73. A large body of work has demonstrated that the differentiation of T cells towards memory cells is largely determined by the prevalence of particular metabolic pathways. IL-15 promotes mitochondrial biogenesis and the expression of carnitine palmitoyltransferase 1A (CPT1A), an enzyme involved in fatty acid oxidation. In memory T cells, CPT1A supports a shift from glycolysis to oxidative metabolism30. This metabolic shift can be enforced through culturing conditions or through the ex vivo genetic manipulation of T cells engineered for ACT.
Both inhibiting glycolysis with use of the glycolysis inhibitor 2-deoxy-d-glucose74 and culturing cells in conditions that reduce the generation of mitochondrial reactive oxygen species75 were shown to support ‘stemness’ (self-renewing capacity) and improve antitumour activity of T cells used for ACT. Although CD19-targeted CAR T cell products used in the clinic are generated from effector T cells and rely on glycolysis for energy generation, CAR T cells generated from memory T cells mainly utilize OXPHOS76. Tweaking ex vivo expansion conditions, such as supplementing the media with IL-15 (REF.77), a combination of IL-7 and IL-15 (REF.78) or l-arginine to enhance OXPHOS79, preserves the memory phenotype during the expansion phase. Treatment with rapamycin has also been shown to promote the differentiation of memory T cells, in part by inhibiting mTOR, and by promoting oxidative metabolism and bolstering mitochondrial mass and function31. This appears to improve their persistence and their expansion and cytokine production after rechallenge80. This memory phenotype can also be enforced by engineering CAR T cells with enhanced mitochondrial metabolism. This can be done by substituting the CD28-derived signalling portion of the chimeric receptor with a construct containing a signalling domain derived from the co-stimulatory molecule 4–1BB, resulting in cells with increased mitochondrial biogenesis, increased spare respiratory capacity and fatty acid oxidation, and better persistence after ACT compared with CD28-based CAR T cells81.
However, the usual culturing conditions for ACT products may not support the development of efficacious long-lived memory T cells. The typical culture medium is hyperglycaemic compared with human plasma (25 mM versus 4.5–5.5 mM), along with many other differences in amino acid and metabolite concentrations82. Blocking glycolysis with 2-deoxy-d-glucose during the ex vivo culturing of the cells to be used for ACT improved tumour clearance after cells were injected, demonstrating the potentially damaging effect of high glucose concentrations during expansion74. Also, CAR T cells cultured for a shorter period appear to perform better than those cultured for longer periods83. This was attributed to a lower degree of differentiation of product cells at the time of transfer, but may also be a result of less time spent in hyperglycaemic conditions.
Thus, altering T cell metabolism, through the design of the CAR or by treating cells with metabolic modulators during expansion, may help stabilize a memory-like phenotype, thereby increasing the persistence and efficacy of T cells. Importantly, these strategies are used before the CAR T cell product reaches the patient and are relatively easy to implement.
Nutrient availability.
Highly glycolytic tumours are more likely to show resistance to ACT treatment as these conditions impair T cell trafficking and T cell cytotoxicity, indicating that transferred cells require glucose84. Cells can be engineered to overcome the lack of glucose in the TME to improve antitumour activity. As low levels of glucose in the tumour decrease PEP levels and T cell receptor signalling after stimulation, counteracting this by increasing PEP levels can improve ACT. Enforced expression of the enzyme phosphoenolpyruvate carboxy-kinase 1 (PCK1), which converts oxaloacetate into PEP, in transferred T cells can increase intracellular PEP levels, support the survival of the T cells and allow for better control of tumour growth4. By culturing of CAR T cells in conditions that mimic the TME, the cells can be ‘acclimatized’ to low nutrient availability. For example, it was shown that T cells that were expanded in media containing low levels of glutamine had effective antitumour functions after adoptive transfer compared with cells cultured in traditional media. These low-nutrient-cultured cells were skewed towards a memory-like phenotype and expressed fewer inhibitory receptors after transfer85. As observed with checkpoint blockade, reducing the reliance of T cells on glycolysis improves their antitumour function. However, in contrast to checkpoint blockade, ACT allows interventions before the cells are transferred back into the patient, whereas interventions targeting T cell metabolism in the context of checkpoint blockade must be administered as combination therapies.
Hypoxia.
As discussed above, low oxygen levels present a crucial barrier to T cell function in the TME. Cellular responses to hypoxia are primarily controlled by the activation of the transcription factor hypoxia-inducible factor 1α (HIF1α). This induces changes in proliferation, metabolism and other important cellular mechanisms to protect the cell from damage and death86. This is especially important in the context of solid tumours, where oxygen levels can fall as low as 0.3%50. Clinically approved CD19-targeted CAR T cells were shown to have reduced effector functions and lower proliferation rates under hypoxic conditions87. Developing CAR T cell-based therapies for solid tumours has been particularly challenging, which may, in part, be due to their poor performance in a low-oxygen environment. Adapting therapeutic T cells to hypoxia by expanding them in media containing low oxygen levels was shown to increase their antitumour activity after transfer88. Deletion of HIF1α in therapeutic T cells before transfer into mouse lung (LLC) or melanoma (B16F10) models led to a decrease in tumour infiltration and an increased tumour burden, demonstrating that the ability to adapt to low oxygen levels is crucial for T cell responses89. It has been shown that it is more efficacious to use effector T cells for ACT than naive or memory T cells90. There is, however, overwhelming evidence that CAR T cells generated from memory T cells are vastly superior in terms of their persistence and durability of responses compared with those generated from effector cells. Instead of deriving cells for ACT from a population that may fare better in hypoxia, combining the use of memory T cells with hypoxia relief may be a better strategy. Together, these data show the importance of the ability of T cells to adapt to hypoxia for antitumour activity.
TME-specific metabolites.
Certain metabolites present in the TME can have suppressive effects on the antitumour immune response. The ectoenzymes CD39 and CD73, which metabolize ATP into adenosine, are expressed by Treg cells, B cells, tumour-associated macrophages, tumour cells and endothelial cells of the tumour vasculature. Although ATP is immunostimulatory, adenosine suppresses the effector functions of immune cells91–93. For example, it has been shown that antagonism of adenosine A2A receptor in mice improves the effector function of CD8+ T cells94. Targeting the adenosine A2A receptor in combination with CAR T cell therapy in mouse models of breast cancer increases treatment efficacy95.
Moreover, ionic imbalance of Ca2+, K+ and Cl− in the TME, as a result of a low pH96, the high usage of ion-gated channels by the tumour97,98 and the release of ions by necrotic tumours can affect T cell function. This is because ion-gated Ca2+ signalling is important for T cell survival and function99–101. It has also been shown that overexpressing a K+ efflux pump in T cells to improve the shuttling of excess K+ out of the cell increased interferon-γ production, tumour clearance and survival after T cell transfer in a mouse model99. This demonstrates that the imbalance of ions in the TME may disrupt T cell signalling and contribute to a loss of T cell function. Engineering cells to be better equipped to function under such conditions may improve ACT therapy for solid tumours.
As previously described, low arginine levels can also affect T cell proliferation. In paediatric patients with acute myeloid leukaemia, it was found that arginine levels in peripheral blood were lower than in healthy controls; this may impair the proliferation and maintenance of CAR T cells after transfer102. CAR T cells engineered to express the arginine-synthesizing enzymes OTC and ASS showed better proliferation in arginine-depleted media and increased survival in mice compared with CAR T cells without these enzymes102.
Oncolytic viruses
OVs selectively infect and lyse tumour cells (see BOX 2). Cell death occurs in an immunogenic fashion, generating a primarily CD8+ T cell-driven adaptive immune response capable of memory responses. Both primary infected lesions as well as metastatic sites may be targeted by newly generated CD8+ T cells103. Thus, similarly to ACT, OVs are an attractive modality for patients with little to no existing immune infiltrate, as they generate a de novo immune response against tumour and viral antigens released during tumour lysis. For T cell-mediated antiviral and antitumour responses elicited by the OV, naive T cells must be appropriately activated, must mature and must travel to the tumour. These T cells then encounter the same metabolic challenges as discussed for T cells activated by checkpoint blockade or transferred by ACT.
Enhancing viral replication and lytic capacity.
A large body of work in the OV field has centred on increasing viral replication and lytic ability. Recently, metabolic modulators have been investigated for their potential to increase OV replication and OV capacity for cellular lysis. However, the mechanisms of action of OV therapy are incompletely understood, and it is currently unclear to what extent viral replication and lytic capability are required for efficacy104.
The skewing of tumour cell metabolism to favour OXPHOS can render tumour cells more permissible to viral replication and lysis. Treatment of tumour-bearing mice with the OV Newcastle disease virus (NDV) and dichloroacetate (DCA), a pyruvate dehydrogenase kinase inhibitor that induces a shift to OXPHOS, allowed increased NDV replication. The release of lactate by tumour cells and their uptake of glucose were decreased, and survival in mice was increased in comparison to treatment with NDV alone105. Although it was not directly studied, one might expect that the reduction in lactate release and glucose uptake by the tumours of the DCA-treated animals in this study may have improved cytotoxic T cell responses, in addition to possible effects on the OV. Treating tumour cells with DCA and oncolytic reovirus or oncolytic measles virus also increased tumour cell lysis and survival in preclinical models106,107. Similar results were observed for oncolytic adenoviruses when glycolysis was blocked with the glycolysis inhibitor 2-deoxy-d-glucose108. However, these studies did not examine the effects of an increase in oxidative metabolism in tumour cells on the immune response. Tumours with an enhanced oxidative metabolism tend to be more hypoxic, which can impede the T cell response. Therefore, it may be useful to identify tumours with oxidative metabolism as a selection criterion for OV treatment, instead of skewing tumour metabolism towards OXPHOS as part of a treatment strategy.
Modulation of other metabolic pathways and transporters can uncover previously unknown metabolites required for viral replication and tumour death. For example, vaccinia virus requires glutamine and the TCA cycle, but not glucose and glycolysis, for sufficient replication109,110. Another study found that tumour cells resistant to infection and lysis by the oncolytic M1 virus were rendered sensitive when treated in combination with an inhibitor of the mevalonate pathway111. In a CRISPR screen of solute carriers that altered vesicular stomatitis virus replication, a tumour cell line with deletion of the zinc exporter SLC30A1 showed reduced viral replication and tumour death112. Studies such as these identify viral requirements for replication and lysis, which could help to identify patients with OV-responsive tumours.
Combination therapy with OVs.
OVs, especially if based on DNA viruses, can be engineered to deliver genes encoding immunologic or metabolic modulators to tumour cells. For example, the herpesvirus-based OV T-VEC, which is approved for the treatment of melanoma, carries a gene encoding the immune stimulatory cytokine GM-CSF, which increases activation and antigen presentation by dendritic cells113. There are also efforts to engineer OVs to encode other cytokines or even PD1-targeted blocking antibodies114. This strategy can also be extended to metabolic modulators. By engineering an oncolytic strain of vaccinia virus to enforce tumour cell production of the immunometabolic adipokine leptin, we demonstrated remodelling of the TME and improved antitumour immunity115. Tumour-infiltrating T cells from animals treated with this OV showed higher interferon-γ and TNF production, an increased mitochondrial mass, increased respiratory capacity and increased proliferation, and the leptin-encoding OV allowed longer survival than the same OV without the leptin gene in a mouse melanoma model115. Importantly, the secretion of leptin into the TME enhanced the development of memory precursor T cells, which conferred a survival benefit during secondary tumour challenge.
Many OVs, such as vaccinia virus, NDV and vesicular stomatitis virus, have been shown to induce vascular collapse in the tumour. This was initially thought to be beneficial as it maintains the virus within the tumour and increases tumour cell death; however, it also impairs infiltration by immune cells and may increase local hypoxia encountered by the immune infiltrate. Pretreatment of tumour-bearing mice with 3TSR, a recombinant thrombospondin 1 protein that binds to CD36 and inhibits angiogenesis (thereby promoting vascular normalization), followed by NDV reduced hypoxia, increased tumour regression and increased CD8+ T cell infiltration compared with monotherapy with either 3TSR or NDV116. This demonstrates the importance of oxygen perfusion for tumour control and immune cell infiltration during treatment with OVs.
The study of metabolism, OVs and the immune response is a growing field. The insights gained from these studies will help the identification of patients more likely to respond to OV therapy. The OV T-VEC is currently being investigated in combination with PD1-targeted checkpoint blockade117,118. OVs are being used in preclinical models in combination with CAR T cells to improve responses in solid tumours119. The virus can be engineered to produce cytokines such as RANTES and IL-15 (REF.120), or TNF and IL-2 (REF.121) in the tumour to improve trafficking and increase persistence of the CAR T cell.
Outlook
Interest in the field of ‘immunometabolism’ has increased significantly in recent years, revealing not only fundamental basic insights into immunoregulation from a metabolic perspective but also a plethora of potential therapeutic applications. Cancer cells, which are highly dependent on metabolic pathways that can facilitate their high rates of proliferation, are an attractive target for such interventions.
First, as these therapies move towards application in the clinic, it is important to appreciate the heterogeneity of the patient population. With regard to their reliance on glycolysis, there is variation between different types of tumour, between patients with the same types of tumour, between different tumours in the same patient and even across regions of the same tumour122. How can such information be integrated and used to design immunotherapeutic strategies that take advantage of the tumour-specific metabolism of a patient? Using flux analysis, we have shown that patients with tumours showing enhanced oxidative metabolism are less likely to respond to PD1 blockade52; however, such tumours appear to be more sensitive to oncolytic virotherapy105–108. Likewise, high rates of tumour cell glycolysis have been shown to pose a barrier for adoptive cell therapies. Metabolic imaging (PET scans), hypoxia staining and the analysis of biopsy samples may help clinicians determine the best combinatorial treatment strategies for a patient.
Second, heterogeneity in tumour cell metabolism must also be appreciated within the larger context of a patient’s own systemic metabolism. Obesity is a growing problem in the developed world, and diet-driven and body mass-driven changes in systemic metabolism may affect the initial oncogenic transformation of cancer cells and their progress, as well as their impact on anticancer immune responses123,124. Indeed, although obesity and metabolic syndrome are risk factors for most types of cancer125, surprisingly it was found that patients with obesity may respond more readily to immunotherapy126–128. Furthermore, as obesity drives changes in the levels of hormones such as leptin, adiponectin and ghrelin, which all have immunomodulatory function, it remains critically important to determine how these metabolic, hormonal and immunologic functions may intersect, starting first in appropriately designed mouse models and then expanding to the patient population129–133.
Third, a number of critical metabolites are derived not from the patient’s own cells but from the patient’s microbiota134, so individuals harbour distinct consortia of microorganisms that may each have unique metabolic outputs. The immunomodulatory effect of microorganism-derived metabolites on the immune system is just starting to be appreciated: for instance, short-chain fatty acids such as butyrate can modulate Treg cell function135–140. However, how a diverse set of microorganisms ultimately shapes the quality of a patient’s immune response remains unclear. As obesity and diet are also known to affect the microbiota141,142 and changes to diet can rapidly change the microbiota and its production of short-chain fatty acids in as little as 5 days143, dietary or microbiota alterations may be beneficial in combinatorial therapies.
Finally, as more diverse types of immunotherapy start to enter the clinic, it remains crucial to continue to evaluate their basic mechanistic underpinnings to determine how they may modulate immune metabolism. The checkpoint molecules LAG3, TIM3 and TIGIT, as well as adenosine, represent a new wave of targets for immunomodulation, and the immunometabolic effects of targeting these molecules are just beginning to come to light. A more comprehensive understanding of the immunometabolic impact of these therapies may allow physicians, armed with the knowledge of their patient’s own TME, to design combinatorial strategies that allow the metabolic barriers to immunotherapy to be overcome.
Immune editing
The process by which tumour cells mutate and evolve to evade detection and elimination by the immune system.
Tumoural escape
The ability of tumour cells to escape from elimination by immune cells, for example through immune editing.
Glycolysis
The process by which cytosolic glucose is converted into pyruvate, which is then shuttled into the mitochondria, where it feeds into the tricarboxylic acid cycle. However, under conditions of low oxygen, pyruvate is usually fermented into lactic acid, which regenerates oxidized nicotinamide adenine dinucleotide (NAD+) and accelerates upstream glycolysis. Otto Warburg discovered that in tumour cells this fermentation can also occur under oxygen-sufficient conditions. This phenomenon is termed ‘aerobic glycolysis’ or the ‘Warburg effect’.
Oxidative phosphorylation
(OXPHOS). The process by which metabolic intermediates from the tricarboxylic acid cycle are oxidized, generating a proton gradient that powers ATP synthase to produce ATP.
Tricarboxylic acid (TCA) cycle
A series of biochemical reactions in which acetyl-CoA is metabolized to generate reducing intermediates for the electron transport chain (generating ATP through oxidative phosphorylation), as well as numerous metabolic intermediates for various synthetic pathways. Acetyl-CoA can be fed into the TCA cycle by the decarboxylation of pyruvate, β-oxidation of fatty acids or α-ketoglutarate through the production of amino acids.
Fatty acid oxidation
The process by which fatty acid chains are broken down by β-oxidation and subsequently combined with CoA to form acetyl-CoA and enter the tricarboxylic acid cycle.
PI3K–AKT–mTOR axis
A signalling pathway that is activated through extracellular or intracellular stimulation of phosphoinositide-3 kinase (PI3K), which leads to the activation of AKT (also known as protein kinase B) and mechanistic target of rapamycin (mTOR). The manner in which PI3K and AKT are activated can alter the downstream effects of mTOR activation and the activity of its various ternary complexes. This axis controls proliferation, translation, the cell cycle, glucose metabolism, autophagy and cell survival.
Flux analysis
The analysis of the oxygen consumption rate and extracellular acidification rate of a cell population to determine its rate of glycolysis or oxidative phosphorylation.
Acknowledgements
The authors thank members of the Delgoffe laboratory for helpful discussions during the course of writing this Review. K.D. was supported by T32CA082084 and 1F31CA247129. G.M.D. is supported by an NIH Director’s New Innovator Award (DP2AI136598), the Mark Foundation for Cancer Research Emerging Leader Award, the Cancer Research Institute Lloyd J. Old STAR Award and the Sy Holzer Endowed Immunotherapy Fund.
Footnotes
Competing interests
G.M.D. declares competing financial interests and has submitted patents covering the use of PGC1α in cell therapies, metabolic manipulation of culture conditions for cell therapies, and the use of leptin and other adipokines in oncolytic viruses that are licensed or pending and is entitled to a share in net income generated from the licencing of these patent rights for commercial development. G.M.D. consults for and/or is on the scientific advisory boards of BlueSphere Bio, Century Therapeutics, Novasenta, Pieris Pharmaceuticals and Kalivir, has grants from bluebird bio, Novasenta, Pfizer, Pieris Pharmaceuticals, TCR2 and Western Oncolytics/Kalivir, and owns stock in Novasenta and BlueSphere Bio. K.D. declares no competing interests.
Peer review information
Nature Reviews Immunology thanks Jeffrey Rathmell and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
References
- 1.Muir A & Vander Heiden MG The nutrient environment affects therapy. Science 360, 962–963 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Warburg O, Wind F & Negelein E The metabolism of tumors in the body. J. Gen. Physiol. 8, 519–530 (1927). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeBerardinis RJ & Chandel NS We need to talk about the Warburg effect. Nat. Metab. 2, 127–129 (2020). [DOI] [PubMed] [Google Scholar]
- 4. Ho P-C et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 162, 1217–1228 (2015). This study shows the mechanism by which glucose deprivation in the tumour can restrict antitumour T cell activity.
- 5.Chang C-H et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fukumura D, Duda DG, Munn LL & Jain RK Tumor microvasculature and microenvironment: novel insights through intravital imaging in pre-clinical models. Microcirculation 17, 206–225 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hensley CT et al. Metabolic heterogeneity in human lung tumors. Cell 164, 681–694 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeBerardinis RJ & Chandel NS Fundamentals of cancer metabolism. Sci. Adv. 2, e1600200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Viallard C & Larrivée B Tumor angiogenesis and vascular normalization: alternative therapeutic targets. Angiogenesis 20, 409–426 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Sullivan MR et al. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife 8, e44235 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erra Díaz F, Dantas E & Geffner J Unravelling the interplay between extracellular acidosis and immune cells. Mediators Inflamm. 2018, 1218297 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haas R et al. Lactate regulates metabolic and pro-inflammatory circuits in control of T cell migration and effector functions. PLoS Biol. 13, e1002202 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Angelin A et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. 25, 1282–1293.e7 (2017). This study shows that FOXP3 can reprogramme T cells to downregulate glycolysis and upregulate OXPHOS to increase survival in low-glucose environments.
- 14.Colegio OR et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sugiura A & Rathmell JC Metabolic barriers to T cell function in tumors. J. Immunol. 200, 400–407 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andrejeva G & Rathmell JC Similarities and distinctions of cancer and immune metabolism in inflammation and tumors. Cell Metab. 26, 49–70 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shyer JA, Flavell RA & Bailis W Metabolic signaling in T cells. Cell Res. 30, 649–659 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang L & Romero P Metabolic control of CD8+ T cell fate decisions and antitumor immunity. Trends Mol. Med. 24, 30–48 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Dai Z, Ramesh V & Locasale JW The evolving metabolic landscape of chromatin biology and epigenetics. Nat. Rev. Genet. 21, 737–753 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wahl DR, Byersdorfer CA, Ferrara JLM, Opipari AW & Glick GD Distinct metabolic programs in activated T cells: opportunities for selective immunomodulation. Immunol. Rev. 249, 104–115 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kishton RJ, Sukumar M & Restifo NP Metabolic regulation of T cell longevity and function in tumor immunotherapy. Cell Metab. 26, 94–109 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Makowski L, Chaib M & Rathmell JC Immunometabolism: from basic mechanisms to translation. Immunol. Rev. 295, 5–14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Menk AV et al. Early TCR signaling induces rapid aerobic glycolysis enabling distinct acute T cell effector functions. Cell Rep. 22, 1509–1521 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang R et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882 (2011). This article shows that MYC signalling is required for the induction of glycolysis and glutaminolysis that occurs with T cell activation.
- 25.MacIver NJ, Michalek RD & Rathmell JC Metabolic regulation of T lymphocytes. Annu. Rev. Immunol. 31, 259–283 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Howie D, Ten Bokum A, Necula AS, Cobbold SP & Waldmann H The role of lipid metabolism in T lymphocyte differentiation and survival. Front. Immunol. 8, 1949 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michalek RD et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 186, 3299–3303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kempkes RWM, Joosten I, Koenen HJPM & He X Metabolic pathways involved in regulatory T cell functionality. Front. Immunol. 10, 2839 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marelli-Berg FM & Jangani M Metabolic regulation of leukocyte motility and migration. J. Leukoc. Biol. 104, 285–293 (2018). [DOI] [PubMed] [Google Scholar]
- 30. van der Windt GJW et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 36, 68–78 (2012). This article shows that IL-15 signalling promotes mitochondrial biogenesis and induction of CPT1A to increase fatty acid oxidation and spare respiratory capacity to enable long-lived memory cells.
- 31.Araki K et al. mTOR regulates memory CD8 T-cell differentiation. Nature 460, 108–112 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalbasi A & Ribas A Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 20, 25–39 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pavlova NN & Thompson CB The emerging hallmarks of cancer metabolism. Cell Metab. 23, 27–47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parry RV et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 25, 9543–9553 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riley JL PD-1 signaling in primary T cells. Immunol. Rev. 229, 114–125 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patsoukis N et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 6, 6692 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ogando J et al. PD-1 signaling affects cristae morphology and leads to mitochondrial dysfunction in human CD8+ T lymphocytes. J. Immunother. Cancer 7, 151 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tkachev V et al. Programmed death-1 controls T cell survival by regulating oxidative metabolism. J. Immunol. 194, 5789–5800 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spinelli JB & Haigis MC The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 20, 745–754 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Scharping NE et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity 45, 374–388 (2016). This article shows that tumour-infiltrating T cells lose their capacity to generate mitochondria through the loss of PGC1α, which controls mitochondrial biogenesis, and that restoration of PGC1α restores T cell function.
- 41.Bengsch B et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8+ T cell exhaustion. Immunity 45, 358–373 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chamoto K et al. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc. Natl Acad. Sci. USA 114, E761–E770 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siska PJ et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight 2, e93411 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chowdhury PS, Chamoto K, Kumar A & Honjo T PPAR-induced fatty acid oxidation in T cells increases the number of tumor-reactive CD8+ T cells and facilitates anti-PD-1 therapy. Cancer Immunol. Res. 6, 1375–1387 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Menk AV et al. 4–1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J. Exp. Med. 215, 1091–1100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Teijeira A et al. Mitochondrial morphological and functional reprogramming following CD137 (4–1BB) costimulation. cancer. Immunol. Res. 6, 798–811 (2018). [DOI] [PubMed] [Google Scholar]
- 47. Scharping NE et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 22, 205–215 (2021). Together with Yu et al. (2020) and Vardhana et al. (2020) this article shows that T cell mitochondrial reactive oxygen species, triggered by chronic antigen stimulation, drive epigenetic alterations and T cell exhaustion.
- 48.Yu Y-R et al. Disturbed mitochondrial dynamics in CD8+ TILs reinforce T cell exhaustion. Nat. Immunol. 21, 1540–1551 (2020). [DOI] [PubMed] [Google Scholar]
- 49.Vardhana SA et al. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat. Immunol. 21, 1022–1033 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McKeown SR Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br. J. Radiol. 87, 20130676 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bertout JA, Patel SA & Simon MC The impact of O2 availability on human cancer. Nat. Rev. Cancer 8, 967–975 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Najjar YG et al. Tumor cell oxidative metabolism as a barrier to PD-1 blockade immunotherapy in melanoma. JCI Insight 4, e124989 (2019). This article shows in patients with melanoma that oxidative tumours, which are more hypoxic than glycolytic tumours, are resistant to PD1 blockade through a more exhausted T cell phenotype.
- 53.Hatfield SM et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl Med. 7, 277ra30 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scharping NE, Menk AV, Whetstone RD, Zeng X & Delgoffe GM Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol. Res. 5, 9–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jayaprakash P et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J. Clin. Invest. 128, 5137–5149 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Voron T et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 212, 139–148 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ward PS & Thompson CB Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 21, 297–308 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gerriets VA et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Invest. 125, 194–207 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fischer K et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 109, 3812–3819 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Watson MJ et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature 10.1038/s41586-020-03045-2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Renner K et al. Restricting glycolysis preserves T cell effector functions and augments checkpoint therapy. Cell Rep. 29, 135–150.e9 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Cluntun AA, Lukey MJ, Cerione RA & Locasale JW Glutamine metabolism in cancer: understanding the heterogeneity. Trends Cancer 3, 169–180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakaya M et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 40, 692–705 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leone RD et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366, 1013–1021 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roy DG et al. Methionine metabolism shapes T helper cell responses through regulation of epigenetic reprogramming. Cell Metab. 31, 250–266.e9 (2020). [DOI] [PubMed] [Google Scholar]
- 66.Bian Y et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 585, 277–282 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tripathi P et al. STAT5 is critical to maintain effector CD8+ T cell responses. J. Immunol. 185, 2116–2124 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Emran AA et al. Targeting DNA methylation and EZH2 activity to overcome melanoma resistance to immunotherapy. Trends Immunol. 40, 328–344 (2019). [DOI] [PubMed] [Google Scholar]
- 69.Rodriguez PC et al. L-arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J. Immunol. 171, 1232–1239 (2003). [DOI] [PubMed] [Google Scholar]
- 70.Grzywa TM et al. Myeloid cell-derived arginase in cancer immune response. Front. Immunol. 11, 938 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Porter DL et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl Med. 7, 303ra139 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lugli E et al. Superior T memory stem cell persistence supports long-lived T cell memory. J. Clin. Invest. 123, 594–599 (2013). This article demonstrates that a pool of stem cell memory T cells are the precursors to central memory cells.
- 73.Berger C et al. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J. Clin. Invest. 118, 294–305 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sukumar M et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Invest. 123, 4479–4488 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pilipow K et al. Antioxidant metabolism regulates CD8+ T memory stem cell formation and antitumor immunity. JCI Insight 3, e122299 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sabatino M et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood 128, 519–528 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alizadeh D et al. IL15 enhances CAR-T cell antitumor activity by reducing mTORC1 activity and preserving their stem cell memory phenotype. Cancer Immunol. Res. 7, 759–772 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xu Y et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood 123, 3750–3759 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Geiger R et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell 167, 829–842.e13 (2016). This articles shows that elevating L-arginine levels can shift T cell metabolism from glycolysis to OXPHOS and support the antitumour function of T cells.
- 80.Rao RR, Li Q, Odunsi K & Shrikant PA The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and eomesodermin. Immunity 32, 67–78 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kawalekar OU et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity 44, 380–390 (2016). [DOI] [PubMed] [Google Scholar]
- 82.Ackermann T & Tardito S Cell culture medium formulation and its implications in cancer metabolism. Trends Cancer 5, 329–332 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ghassemi S et al. Reducing ex vivo culture improves the antileukemic activity of chimeric antigen receptor (CAR) T cells. Cancer Immunol. Res. 6, 1100–1109 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cascone T et al. Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab. 27, 977–987.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nabe S et al. Reinforce the antitumor activity of CD8+ T cells via glutamine restriction. Cancer Sci. 109, 3737–3750 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Majmundar AJ, Wong WJ & Simon MC Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 40, 294–309 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Berahovich R et al. Hypoxia selectively impairs CAR-T cells in vitro. Cancers 11, 602 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gropper Y et al. Culturing CTLs under hypoxic conditions enhances their cytolysis and improves their anti-tumor function. Cell Rep. 20, 2547–2555 (2017). [DOI] [PubMed] [Google Scholar]
- 89.Palazon A et al. An HIF-1α/VEGF-A axis in cytotoxic t cells regulates tumor progression. Cancer Cell 32, 669–683.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu Y et al. Glycolysis determines dichotomous regulation of T cell subsets in hypoxia. J. Clin. Invest. 126, 2678–2688 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ohta A et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl Acad. Sci. USA 103, 13132–13137 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huang S, Apasov S, Koshiba M & Sitkovsky M Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood 90, 1600–1610 (1997). [PubMed] [Google Scholar]
- 93.Maj T et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 18, 1332–1341 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hatfield SM & Sitkovsky M A2A adenosine receptor antagonists to weaken the hypoxia-HIF-1α driven immunosuppression and improve immunotherapies of cancer. Curr. Opin. Pharmacol. 29, 90–96 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Beavis PA et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J. Clin. Invest. 127, 929–941 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kato Y et al. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 13, 89 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kunzelmann K Ion channels and cancer. J. Membr. Biol. 205, 159–173 (2005). [DOI] [PubMed] [Google Scholar]
- 98.Kunzelmann K Ion channels in regulated cell death. Cell Mol. Life Sci. 73, 2387–2403 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Eil R et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature 537, 539–543 (2016). This article shows that necrosis within the tumour can release K+ ions into the microenvironment, which interferes with T cell receptor signalling.
- 100.Omilusik K et al. The CaV1.4 calcium channel is a critical regulator of T cell receptor signaling and naive T cell homeostasis. Immunity 35, 349–360 (2011). [DOI] [PubMed] [Google Scholar]
- 101.Trebak M & Kinet J-P Calcium signalling in T cells. Nat. Rev. Immunol. 19, 154–169 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fultang L et al. Metabolic engineering against the arginine microenvironment enhances CAR-T cell proliferation and therapeutic activity. Blood 136, 1155–1160 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bommareddy PK, Shettigar M & Kaufman HL Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 18, 498–513 (2018). [DOI] [PubMed] [Google Scholar]
- 104.Davola ME & Mossman KL Oncolytic viruses: how “lytic” must they be for therapeutic efficacy? Oncoimmunology 8, e1581528 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Meng G et al. Targeting aerobic glycolysis by dichloroacetate improves Newcastle disease virus-mediated viro-immunotherapy in hepatocellular carcinoma. Br. J. Cancer 122, 111–120 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li C et al. Dichloroacetate blocks aerobic glycolytic adaptation to attenuated measles virus and promotes viral replication leading to enhanced oncolysis in glioblastoma. Oncotarget 6, 1544–1555 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kennedy BE et al. Inhibition of pyruvate dehydrogenase kinase enhances the antitumor efficacy of oncolytic reovirus. Cancer Res. 79, 3824–3836 (2019). [DOI] [PubMed] [Google Scholar]
- 108.Dyer A et al. Antagonism of glycolysis and reductive carboxylation of glutamine potentiates activity of oncolytic adenoviruses in cancer cells. Cancer Res. 79, 331–345 (2019). [DOI] [PubMed] [Google Scholar]
- 109.Pant A, Cao S & Yang Z Asparagine is a critical limiting metabolite for vaccinia virus protein synthesis during glutamine deprivation. J. Virol. 10.1128/JVI.01834-18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Greseth MD & Traktman P De novo fatty acid biosynthesis contributes significantly to establishment of a bioenergetically favorable environment for vaccinia virus infection. PLoS Pathog. 10, e1004021 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liang J et al. Inhibition of the mevalonate pathway enhances cancer cell oncolysis mediated by M1 virus. Nat. Commun. 9, 1524 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moskovskich A et al. The transporters SLC35A1 and SLC30A1 play opposite roles in cell survival upon VSV virus infection. Sci. Rep. 9, 10471 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Andtbacka RHI et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 33, 2780–2788 (2015). [DOI] [PubMed] [Google Scholar]
- 114.Fountzilas C, Patel S & Mahalingam D Review: oncolytic virotherapy, updates and future directions. Oncotarget 8, 102617–102639 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rivadeneira DB et al. Oncolytic viruses engineered to enforce leptin expression reprogram tumor-infiltrating T cell metabolism and promote tumor clearance. Immunity 51, 548–560.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Matuszewska K et al. Combining vascular normalization with an oncolytic virus enhances immunotherapy in a preclinical model of advanced-stage ovarian cancer. Clin. Cancer Res. 25, 1624–1638 (2019). [DOI] [PubMed] [Google Scholar]
- 117.Twumasi-Boateng K, Pettigrew JL, Kwok YYE, Bell JC & Nelson BH Oncolytic viruses as engineering platforms for combination immunotherapy. Nat. Rev. Cancer 18, 419–432 (2018). [DOI] [PubMed] [Google Scholar]
- 118.Guo ZS et al. Oncolytic immunotherapy: conceptual evolution, current strategies, and future perspectives. Front. Immunol. 8, 555 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Guedan S & Alemany R CAR-T cells and oncolytic viruses: joining forces to overcome the solid tumor challenge. Front. Immunol. 9, 2460 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nishio N et al. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 74, 5195–5205 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Watanabe K et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight 3, e99573 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Warmoes MO & Locasale JW Heterogeneity of glycolysis in cancers and therapeutic opportunities. Biochem. Pharmacol. 92, 12–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cordain L et al. Origins and evolution of the Western diet: health implications for the 21st century. Am. J. Clin. Nutr. 81, 341–354 (2005). [DOI] [PubMed] [Google Scholar]
- 124.Deng T, Lyon CJ, Bergin S, Caligiuri MA & Hsueh WA Obesity, inflammation, and cancer. Annu. Rev. Pathol. 11, 421–449 (2016). [DOI] [PubMed] [Google Scholar]
- 125.Moore LL, Chadid S, Singer MR, Kreger BE & Denis GV Metabolic health reduces risk of obesity-related cancer in Framingham study adults. Cancer Epidemiol. Biomarkers Prev. 23, 2057–2065 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. McQuade JL et al. Association of body-mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: a retrospective, multicohort analysis. Lancet Oncol. 19, 310–322 (2018). This article, along with Cortellini et al. (2019) and Wang et al. (2019), shows that patients with obesity based on body mass index respond better to immunotherapy than patients with normal body mass index.
- 127.Cortellini A et al. A multicenter study of body mass index in cancer patients treated with anti-PD-1/PD-L1 immune checkpoint inhibitors: when overweight becomes favorable. J. Immunother. Cancer 7, 57 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wang Z et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat. Med. 25, 141–151 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Matarese G & La Cava A The intricate interface between immune system and metabolism. Trends Immunol. 25, 193–200 (2004). [DOI] [PubMed] [Google Scholar]
- 130.Cao H Adipocytokines in obesity and metabolic disease. J. Endocrinol. 220, T47–T59 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jung UJ & Choi M-S Obesity and its metabolic complications: the role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int. J. Mol. Sci. 15, 6184–6223 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Weidinger C, Ziegler JF, Letizia M, Schmidt F & Siegmund B Adipokines and their role in intestinal inflammation. Front. Immunol. 9, 1974 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Francisco V et al. Adipokines and inflammation: is it a question of weight? Br. J. Pharmacol. 175, 1569–1579 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wikoff WR et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl Acad. Sci. USA 106, 3698–3703 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chang PV, Hao L, Offermanns S & Medzhitov R The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl Acad. Sci. USA 111, 2247–2252 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Furusawa Y et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450 (2013). This article shows that butyrate produced in the gut by commensal microorganisms can induce Treg cell differentiation and maintain gut homeostasis.
- 137.Smith PM et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Luu M et al. Regulation of the effector function of CD8+ T cells by gut microbiota-derived metabolite butyrate. Sci. Rep. 8, 14430 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Bachem A et al. Microbiota-derived short-chain fatty acids promote the memory potential of antigen-activated CD8+ T cells. Immunity 51, 285–297.e5 (2019). This article shows that germ-free mice have impaired memory T cell responses and that microbially derived butyrate promotes CD8+ T cell memory formation by shifting the metabolism of CD8+ T cells to OXPHOS.
- 140.Sun M et al. Microbiota-derived short-chain fatty acids promote Th1 cell IL-10 production to maintain intestinal homeostasis. Nat. Commun. 9, 3555 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zmora N, Suez J & Elinav E You are what you eat: diet, health and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 16, 35–56 (2019). [DOI] [PubMed] [Google Scholar]
- 142.Castaner O et al. The gut microbiome profile in obesity: a systematic review. Int. J. Endocrinol. 2018, 4095789 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.David LA et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Hodi FS et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010). The clinical trial results in this article resulted in the approval of ipilimumab for treatment of patients with melanoma.
- 145.Topalian SL et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Harding FA, McArthur JG, Gross JA, Raulet DH & Allison JP CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 356, 607–609 (1992). [DOI] [PubMed] [Google Scholar]
- 147.Brunet JF et al. A new member of the immunoglobulin superfamily–CTLA-4. Nature 328, 267–270 (1987). [DOI] [PubMed] [Google Scholar]
- 148.Linsley PS et al. Human B7–1 (CD80) and B7–2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1, 793–801 (1994). [DOI] [PubMed] [Google Scholar]
- 149.Linsley PS et al. CTLA-4 is a second receptor for the B cell activation antigen B7. J. Exp. Med. 174, 561–569 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lenschow DJ et al. Long-term survival of xenogeneic pancreatic islet grafts induced by CTLA4lg. Science 257, 789–792 (1992). [DOI] [PubMed] [Google Scholar]
- 151.Qureshi OS et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332, 600–603 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Takahashi T et al. Immunologic self-tolerance maintained by CD25+CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 192, 303–310 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tivol EA et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3, 541–547 (1995). [DOI] [PubMed] [Google Scholar]
- 154.Waterhouse P et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 270, 985–988 (1995). [DOI] [PubMed] [Google Scholar]
- 155.Wing K et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275 (2008). [DOI] [PubMed] [Google Scholar]
- 156.Leach DR, Krummel MF & Allison JP Enhancement of antitumor immunity by CTLA-4 blockade. Science 271, 1734–1736 (1996). [DOI] [PubMed] [Google Scholar]
- 157.Wei SC, Duffy CR & Allison JP Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 8, 1069–1086 (2018). [DOI] [PubMed] [Google Scholar]
- 158.Ishida Y, Agata Y, Shibahara K & Honjo T Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 11, 3887–3895 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nishimura H, Honjo T & Minato N Facilitation of beta selection and modification of positive selection in the thymus of PD-1-deficient mice. J. Exp. Med. 191, 891–898 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Nishimura H et al. Developmentally regulated expression of the PD-1 protein on the surface of double-negative (CD4-CD8-) thymocytes. Int. Immunol. 8, 773–780 (1996). [DOI] [PubMed] [Google Scholar]
- 161.Agata Y et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 8, 765–772 (1996). [DOI] [PubMed] [Google Scholar]
- 162.Freeman GJ et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192, 1027–1034 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Latchman Y et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2, 261–268 (2001). [DOI] [PubMed] [Google Scholar]
- 164.Sharpe AH & Pauken KE The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 18, 153–167 (2018). [DOI] [PubMed] [Google Scholar]
- 165.Pardoll DM The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ribas A Adaptive immune resistance: how cancer protects from immune attack. Cancer Discov. 5, 915–919 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wherry EJ & Kurachi M Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 15, 486–499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.McLane LM, Abdel-Hakeem MS & Wherry EJ CD8 T cell exhaustion during chronic viral infection and cancer. Annu. Rev. Immunol. 37, 457–495 (2019). [DOI] [PubMed] [Google Scholar]
- 169.Zajac AJ et al. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 188, 2205–2213 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Gallimore A et al. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J. Exp. Med. 187, 1383–1393 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lee PP et al. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat. Med. 5, 677–685 (1999). [DOI] [PubMed] [Google Scholar]
- 172.Barber DL et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687 (2006). [DOI] [PubMed] [Google Scholar]
- 173.Iwai Y et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl Acad. Sci. USA 99, 12293–12297 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Miller BC et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 20, 326–336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Sade-Feldman M et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175, 998–1013.e20 (2018). This article shows the correlation of TCF7+ T cells and response to immunotherapy.
- 176. Chen Z et al. TCF-1-centered transcriptional network drives an effector versus exhausted CD8 T cell-fate decision. Immunity 51, 840–855.e5 (2019). This article, along with Miller et al. (2019) and Sade-Feldman (2018), demonstrates the importance of a TCF7+ progenitor CD8+ T cell population in response to anti-PD1 therapy (TCF7 is also known as TCF1).
- 177.Sadelain M CD19 CAR T cells. Cell 171, 1471 (2017). [DOI] [PubMed] [Google Scholar]
- 178.Rosenberg SA et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 17, 4550–4557 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Guedan S, Ruella M & June CH Emerging cellular therapies for cancer. Annu. Rev. Immunol. 37, 145–171 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Schuster SJ et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med. 377, 2545–2554 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kalos M et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl Med. 3, 95ra73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Brentjens RJ et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl Med. 5, 177ra38 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Maude SL et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 371, 1507–1517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Park JH et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic Leukemia. N. Engl. J. Med. 378, 449–459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Turtle CJ et al. CD19 CAR-T cells of defined CD4+:CD8+composition in adult B cell ALL patients. J. Clin. Invest. 126, 2123–2138 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lee DW et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385, 517–528 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Maude SL et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 378, 439–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Fraietta JA et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 24, 563–571 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hamid O, Ismail R & Puzanov I Intratumoral immunotherapy — update 2019. Oncologist 25, e423–e438 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Filley AC & Dey M Immune system, friend or foe of oncolytic virotherapy? Front. Oncol. 7, 106 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lemos de Matos A, Franco LS & McFadden G Oncolytic viruses and the immune system: the dynamic duo. Mol. Ther. Methods Clin. Dev. 17, 349–358 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Gujar S, Pol JG, Kim Y, Lee PW & Kroemer G Antitumor benefits of antiviral immunity: an underappreciated aspect of oncolytic virotherapies. Trends Immunol. 39, 209–221 (2018). [DOI] [PubMed] [Google Scholar]
- 193.Rojas JJ, Sampath P, Hou W & Thorne SH Defining effective combinations of immune checkpoint blockade and oncolytic virotherapy. Clin. Cancer Res. 21, 5543–5551 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Woller N et al. Viral infection of tumors overcomes resistance to PD-1-immunotherapy by broadening neoantigenome-directed T-cell responses. Mol. Ther. 23, 1630–1640 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ricca JM et al. Pre-existing immunity to oncolytic virus potentiates its immunotherapeutic efficacy. Mol. Ther. 26, 1008–1019 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Liu Z, Ravindranathan R, Kalinski P, Guo ZS & Bartlett DL Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat. Commun. 8, 14754 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Nakao S et al. Intratumoral expression of IL-7 and IL-12 using an oncolytic virus increases systemic sensitivity to immune checkpoint blockade. Sci. Transl Med. 12, eaax7992 (2020). [DOI] [PubMed] [Google Scholar]
- 198.Zamarin D et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl Med. 6, 226ra32 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Oh E, Choi I-K, Hong J & Yun C-O Oncolytic adenovirus coexpressing interleukin-12 and decorin overcomes Treg-mediated immunosuppression inducing potent antitumor effects in a weakly immunogenic tumor model. Oncotarget 8, 4730–4746 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Fend L et al. Oncolytic virotherapy with an armed vaccinia virus in an orthotopic model of renal carcinoma is associated with modification of the tumor microenvironment. Oncoimmunology 5, e1080414 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Patel MR et al. Vesicular stomatitis virus expressing interferon-β is oncolytic and promotes antitumor immune responses in a syngeneic murine model of non-small cell lung cancer. Oncotarget 6, 33165–33177 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]