

Abstract
Diabetic patients suffer augmented severity of myocardial infarction. Excessive activation of the mammalian target of rapamycin (mTOR) and decreased activation of STAT3 are implicated in diabetic complications. Considering the potent cardioprotective effect of mTOR inhibitor, rapamycin, we hypothesized that reperfusion therapy with rapamycin would reduce infarct size in the diabetic hearts through STAT3 signaling. Hearts from adult male db/db or wild type (WT) C57 mice were isolated and subjected to 30 min of normothermic global ischemia and 60 min of reperfusion in Langendorff mode. Rapamycin (100 nM) was infused at the onset of reperfusion. Myocardial infarct size (IS) was significantly reduced in rapamycin-treated mice (13.3 ± 2.4 %) compared to DMSO vehicle control (35.9 ± 0.9 %) or WT mice (27.7 ± 1.1 %). Rapamycin treatment restored phosphorylation of STAT3 and enhanced AKT phosphorylation (target of mTORC2), but significantly reduced ribosomal protein S6 phosphorylation (target of mTORC1) in the diabetic heart. To determine the cause and effect relationship of STAT3 in cardioprotection, inducible cardiac-specific STAT3-deficient (MCM TG:STAT3flox/flox) and WT mice (MCM TG:STAT3flox/flox) were made diabetic by feeding high fat diet (HFD). Rapamycin given at reperfusion reduced IS in WT mice but not in STAT3-deficient mice following I/R. Moreover, cardiomyocytes isolated from HFD-fed WT mice showed resistance against necrosis (trypan blue staining) and apoptosis (TUNEL assay) when treated with rapamycin during reoxygenation following simulated ischemia. Such protection was absent in cardiomyocytes from HFD-fed STAT3-deficient mice. STAT3 signaling plays critical role in reducing IS and attenuates cardiomyocyte death following reperfusion therapy with rapamycin in diabetic heart.
Keywords: Diabetes, Ischemia/reperfusion, mTOR, Rapamycin, STAT3
Introduction
Diabetes mellitus (diabetes) is an important prognostic factor in pathogenesis of cardiovascular diseases including coronary artery disease, acute myocardial infarction (MI) and congestive heart failure [20]. The most prevalent form of diabetes mellitus is type 2 diabetes (T2D). The underlying metabolic causes of T2D are the combination of impairment in insulin-mediated glucose disposal (insulin resistance) and defective secretion of insulin by pancreatic β-cells. Several clinical and epidemiological studies indicate that patients with diabetes had an increased mortality rate after myocardial infarction (MI) compared with non-diabetic patients [26, 32, 33].
The important role of mammalian target of rapamycin (mTOR) in maintaining intracellular energy metabolism, nutrient availability and cellular growth is well documented [35]. Persistent hyperactivation of mTOR has been implicated in diverse disorders, including cancer and obesity-related metabolic pathologies [42, 44, 46]. Chronic increase in mTOR activity in T2D causes insulin resistance which contributes to hyperinsulinemia and hyperglycemia [14, 28, 39, 44]. Therefore, signaling pathways of mTOR may offer exciting prospects for the development of novel therapies in treating diabetes-related complications. In fact, a recent study from our laboratory reported beneficial effect of rapamycin (Sirolimus®) in improving metabolic status and preventing cardiac dysfunction in T2D mice through attenuation of oxidative stress as well as alteration of contractile and glucose metabolic proteins [8]. In addition, previous studies have shown that diabetic hearts have decreased phosphorylation of signal transducers and activator of transcription 3 (STAT3), which was associated with insulin resistance and dilated cardiomyopathy [15, 24]. More importantly, STAT3 signaling plays an essential role in rapamycin-induced protection against myocardial I/R injury [9]. In addition, post-conditioning and other pharmacological cardioprotective modalities including insulin therapy and IL-11 have been shown to protect against I/R injury through STAT3 activation [3, 17, 37, 43]. Thus considering such an important role of STAT3 in protection, we hypothesized that mTOR inhibition with rapamycin at the time of reperfusion after ischemia may trigger similar cardioprotective signaling in the diabetic heart. We further contemplated that αMHC-MerCreMer (MCM) diabetic mouse in which STAT3 is deleted only in the heart following tamoxifen treatment would fail to respond to reperfusion therapy with rapamycin.
Methods
Animals
Adult male (16–18 weeks) leptin receptor null, homozygous db/db mice (strain: BKS.Cg-Dock7m+/+Leprdb/J) and their corresponding wild type mice (C57/BL/6J) were purchased from the Jackson Laboratories (Bar Harbor, ME). Inducible, cardiac-specific STAT3-deficient mice (MCM TG:STAT3flox/flox) and wild type (WT) non-transgenic control mice (MCM NTG:STAT3flox/flox) were created by interbreeding STAT3flox/flox mice with tamoxifen-inducible αMHC-MCM TG (MCM) mice [6]. STAT3-deficient and WT male mice (8–10 weeks, n = 24/group) were fed high fat diet (HFD) (containing 60 % fat calories, Bio-Serv Company, NJ) for 16 weeks to induce obesity and diabetes (glucose >250 mg/dL), after which they received tamoxifen (20 mg/kg/day i.p.) for 10 days. The animal experimental protocols were approved by the Institutional Animal Care and Use Committee of the Virginia Commonwealth University. The animal and experiments were conducted the Guide for the care and Use of Laboratory Animals for Biomedical Research published by the National Institutes of Health (No. 85–23, revised 1996).
Experimental groups
For global I/R protocol, three groups were used: (1) C57 (n = 4), (2) db/db (n = 4)-DMSO and db/db-Rapamycin (100 nM) (LC Laboratories, MA, USA) (n = 6). Isolated hearts were infused with DMSO (solvent for rapamycin) and rapamycin. The dose of rapamycin was chosen based on our previous studies on rapamycin-induced cardioprotection against I/R injury [9, 29]. To investigate STAT3 signaling, four groups were used (n = 5): (1) WT-DMSO, (2) WT-Rapamycin, (3) STAT3-deficient-DMSO and (4) STAT3-deficient-Rapamycin. Also, we isolated primary adult cardiomyocytes from these groups of mice (n = 4). Protein was analyzed from the hearts following global I/R (n = 3). Experimental protocol is documented in Fig. 1.
Fig. 1.
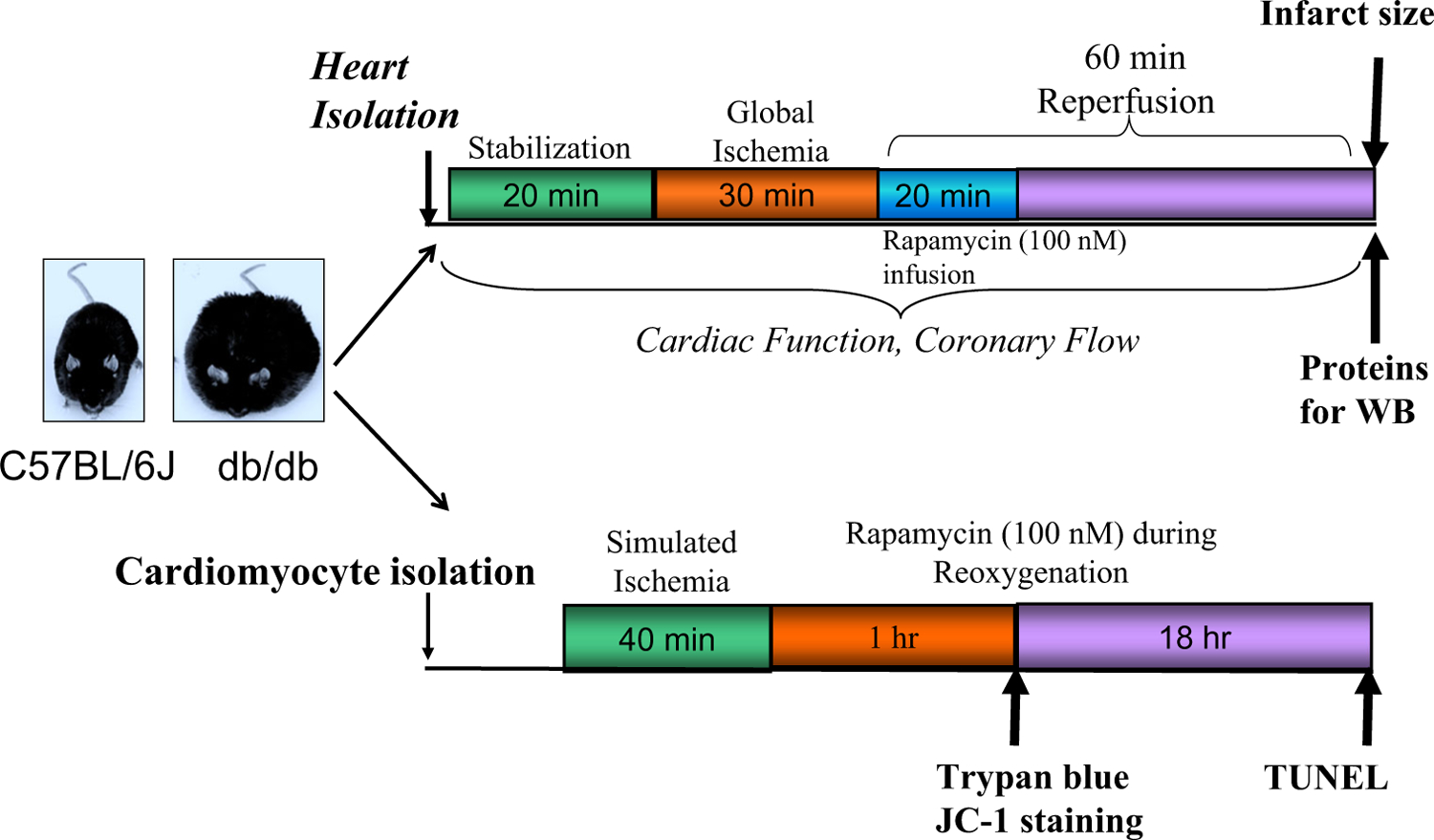
Experimental design: isolated hearts from C57 and diabetic (db/db) mice were subjected to 30 min no-flow global ischemia (I) and 60 min reperfusion (R) in Langendorff perfusion system with Krebs-Henseleit (K-H) buffer. Rapamycin (RAPA, 100 nM) or volume-matched DMSO (solvent of rapamycin) was infused (intra-coronary) during the first 20 min of reperfusion. Myocardial infarct size was measured and proteins were isolated following I/R. Isolated adult mouse cardiomyocytes from C57 and db/db mice were subjected to 40 min of simulated ischemia (SI) and 1 or 18 h of reoxygenation (RO). During RO, cardiomyocytes were treated with rapamycin (100 nM). Cell necrosis and mitochondrial membrane potential were assessed after 1 h of RO. Apoptosis was examined following 18 h of RO
Langendorff isolated perfused heart and ischemia/reperfusion protocol
The methodology of isolated perfused heart has been described previously in detail [9, 10]. In brief, mice were anesthetized with sodium pentobarbital (Nembutal® sodium solution; 100 mg/kg i.p.). The heart was removed and aorta was cannulated on a 20-gauge blunt needle connected to Langendorff perfusion system. The heart was perfused at a constant pressure of 55 mmHg with modified Krebs-Henseleit solution (in mM): NaCl 118, NaHCO3 24, CaCl2 2.5, KCl 4.7, KH2PO4 1.2, MgSO4 1.2, Glucose 11, and EDTA 0.5, continuously gassed with 95 % O2 + 5 % CO2 (pH 7.34–7.49). Buffer and heart temperature was maintained at 37 °C. A force–displacement transducer (Grass, Model FT03) was attached to the apex through a No. 5 surgical thread and a rigid metal hook. The resting tension of the isolated heart was adjusted to ~0.30 g. Ventricular contractile force was recorded by Powerlab 8SP computerized data acquisition system connected to the force transducer. After 30 min of stabilization, the hearts were subjected to 30 min of no-flow global ischemia followed by 60 min of reperfusion. Hearts were infused with rapamycin (100 nM) or DMSO (vehicle control) at the onset of reperfusion for 20 min. Coronary flow rate was calculated by timed collection of the outflow perfusate. After the end of reperfusion, the heart was weighed, frozen and cut into 6–8 transverse slices from apex to base of equal thickness (~1 mm). The slices were stained with 10 % tetrazolium chloride (TTC) for 30 min and infarct size was determined by computer morphometry using image J software.
Isolation of cardiomyocytes and simulated ischemia/reoxygenation protocol
The ventricular cardiomyocytes from C57, db/db, STAT3-deficient and WT mice were isolated using enzymatic technique as previously reported [11]. The cardiomyocytes were subjected to simulated ischemia (SI) for 40 min by incubating in ischemic buffer under hypoxic conditions (1 % O2 and 5 % CO2) followed by reoxygenation (RO) for 1 or 18 h under normoxic conditions [11]. During reoxygenation, cardiomyocytes were treated with rapamycin (100 nM) or an equivalent volume of vehicle i.e., DMSO.
Measurement of necrosis, apoptosis and mitochondrial membrane potential
Cardiomyocyte necrosis was assessed by trypan blue exclusion assay following 40 min of SI and 1 h of reoxygenation. Apoptosis was determined after prolonged reoxygenation for 18 h by TUNEL staining (BD Biosciences, CA) which detected nuclear DNA fragmentation via a fluorescence assay [11]. Mitochondrial membrane potential was assessed using cationic lipophilic probe JC-1 as described previously [11].
Western blot analysis
Total soluble protein was extracted from the whole heart tissue after completion of experimental protocol as described above. The extraction buffer consisted of 20 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.2 mM PMSF, 1 μg/mL pepstatin, 05 μg/mL leupeptin, 2 mM NaF, 0.2 mM Na3VO4, 5 mM β-mecaptoethanol. The homogenate was centrifuged at 14,000g for 15 min at 4 °C and the supernatant was recovered. Protein (50 μg) from each sample was separated by SDS-PAGE and transferred onto nitrocellulose membrane [10–12]. The membrane was incubated overnight with primary antibody at a dilution 1:1000 for each of the respective proteins (i.e., phosphotyrosine705-STAT3, STAT3, phospho-Ser235/236-S6, S6, phosphor Ser473-AKT and AKT from Cell Signaling, MA; and GAPDH from Santa Cruz Biotechnology, TX). The membrane was washed and incubated with horseradish peroxidase conjugated secondary antibody (1:2000 dilutions) for 1 h. The blots were developed using a chemiluminescent system (ECL Plus; Amersham Biosciences).
Data analysis and statistics
Statistical analysis was performed with GraphPad Prism 4.0 (Graphpad Software Inc.). Data are presented as mean ± SE. Statistical comparisons between 2 groups were performed with the unpaired Student t test. The differences between more than 2 groups were analyzed with one-way analysis of variance followed by Student–New-man–Keuls post hoc test for pair-wise comparison; p < 0.05 was considered to be statistically significant.
Results
Effect of rapamycin on ischemia/reperfusion injury
The average infarct size in the hearts from db/db mice following 30 min of global ischemia and 60 min of reperfusion was higher as compared to the corresponding hearts from C57 background mice (n = 4, *p < 0.05 vs C57, Fig. 2a). Rapamycin treatment at reperfusion reduced infarct size as compared to DMSO-treated diabetic hearts or C57 hearts (n = 6, αp < 0.001 vs db/db and C57). Cardiac function (rate-force product, measured as product of heart rate and ventricular developed force) and coronary flow rate improved in rapamycin-treated db/db hearts following I/R as compared to DMSO-treated hearts (Fig. 2b, c).
Fig. 2.
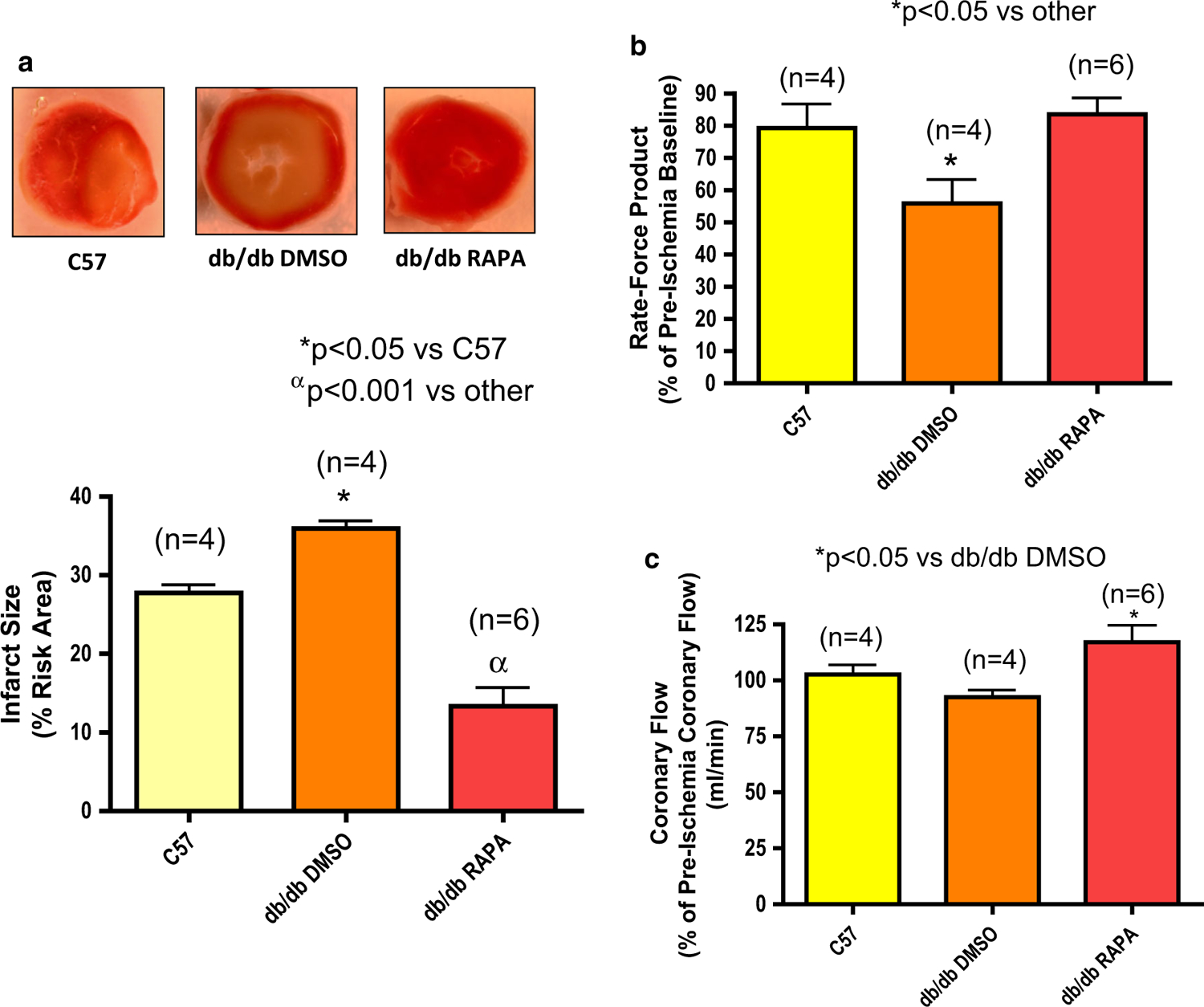
Reperfusion therapy with rapamycin (RAPA) reduces infarct size of diabetic mouse (db/db) heart following ischemia/reperfusion (I/R). a Myocardial infarct size in C57 and db/db mice following I/R with the treatment of rapamycin (RAPA, 100 nM) or DMSO (solvent of rapamycin) at the time of reperfusion. b Product of heart rate and ventricular developed force (% of pre-ischemic baseline). c Coronary flow (% of pre-ischemic coronary flow)
STAT3 Signaling in rapamycin-induced protection
The phosphorylation of STAT3 (Tyr-705) and total STAT3 were significantly reduced in the diabetic hearts as compared to C57 hearts (Fig. 3). Rapamycin treatment significantly increased phosphorylated as well as total STAT3 when compared to vehicle-treated db/db hearts (Fig. 3). The ratios of p-STAT3 to STAT3 and GAPDH were significantly higher with rapamycin treatment in the hearts of db/db mice. The phosphorylation of AKT was enhanced with concomitant reduction of total AKT after I/R in diabetic hearts as compared to C57 hearts which was further increased following rapamycin treatment. The ratios of p-AKT to AKT and GAPDH were significantly higher in rapamycin-treated hearts of db/db mice. As phosphorylated STAT3 and Akt have survival and anti-apoptotic effects, the results suggest that these signaling molecules may be involved in the beneficial effects of rapamycin during reperfusion injury. The phosphorylation of ribosomal protein S6 (by mTORC1) was significantly enhanced in the hearts of db/db mice as compared with C57 mice. Rapamycin treatment significantly reduced phosphorylation of S6 in both diabetic and non-diabetic hearts (Fig. 3).
Fig. 3.
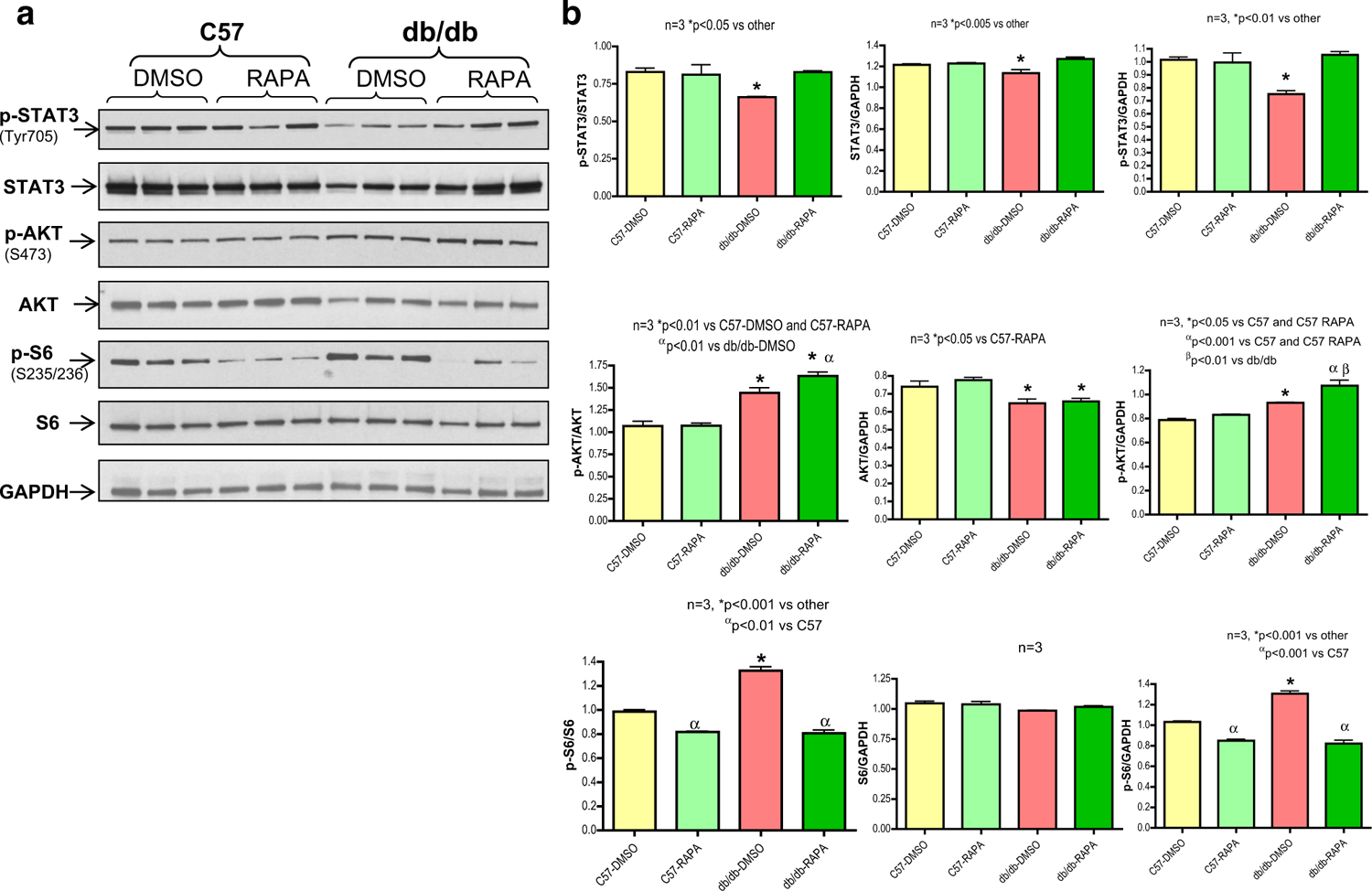
Rapamycin restores phosphorylation of STAT3 in diabetic heart, but inhibits mTORC1. a Representative immunoblots of phospho-STAT3, STAT3, phospho-AKT, AKT, phospho-S6, S6 and GAPDH in hearts of C57 and db/db mice following ischemia/reperfusion (I/R) as well as infusion of rapamycin (RAPA, 100 nM) or DMSO (solvent of rapamycin) at reperfusion. b Densitometric analysis of the ratios of phosphorylated (p) to total protein, total protein to GAPDH and phosphorylated proteins to GAPDH
The cause and effect of STAT3 in rapamycin-induced protection was determined using αMHC-MerCreMer-STAT3-deficient diabetic mice in which STAT3 is deleted only in the heart following tamoxifen treatment. To induce obesity and diabetes, STAT3-deficient (MCM TG:STAT3flox/flox) and corresponding WT mice (MCM TG:STAT3flox/flox) were fed HFD for 16 weeks. Body weight and glucose levels were not different between WT and STAT3-deficient mice (24–25 weeks old-age match) after feeding with regular chow (WT: BW-30.56 ± 0.95 gm, Glucose: 133 ± 10.3 mg/dL; and STAT3-deficient: BW-30.15 ± 1.4 gm, Glucose: 117 ± 5.54 mg/dL; n = 8). Following 16 weeks of feeding with HFD, body weight and glucose levels were increased in both WT and STAT3-deficient mice, but there was no significant difference between the two strains of mice (Table 1). The expression of STAT3 was decreased by 67 % in cardiac-specific STAT3-deficient mice (MCM TG:STAT3flox/flox) following 10 days of tamoxifen treatment as compared to WT littermates (MCM NTG:STAT3flox/flox) (Fig. 4a). Rapamycin treatment reduced infarct size in HFD-fed WT (MCM NTG:STAT3flox/flox) mice following I/R as compared to HFD-fed vehicle-treated WT (MCM NTG:STAT3flox/flox) mice (n = 5, p < 0.001 vs DMSO, Fig. 4b). The infarct-limiting effect of rapamycin was blunted in HFD-fed STAT3-deficient mice (MCM TG:STAT3flox/flox) (Fig. 4b). Cardiac function (rate-force function) was also improved in rapamycin-treated HFD-fed WT (MCM NTG:STAT3flox/flox) mice (n = 5, p < 0.01 vs vehicle-treated WT mice (Fig. 4c). A severe ventricular dysfunction was noticed in STAT3-deficient mice following rapamycin treatment at reperfusion. The post-ischemic coronary flow rate did not change between groups (Fig. 4d).
Table 1.
Body weight, blood glucose level and heart weight in WT (MCM NTG:STATflox/flox) and STAT3-deficient (MCM TG:STATflox/flox) mice following fed with high fat diet for 16 weeks N = 10 per group
| Mouse strain | MCM NTG:STATflox/flox | MCM TG:STATflox/flox |
|---|---|---|
| Body weight (gm) | 43.96 ± 1.21 | 46.92 ± 0.91 |
| Glucose (mg/dL) | 255.4 ± 3.49 | 265 ± 5.61 |
| Heart weight (mg) | 252.4 ± 7.15 | 283.2 ± 16.86 |
Fig. 4.
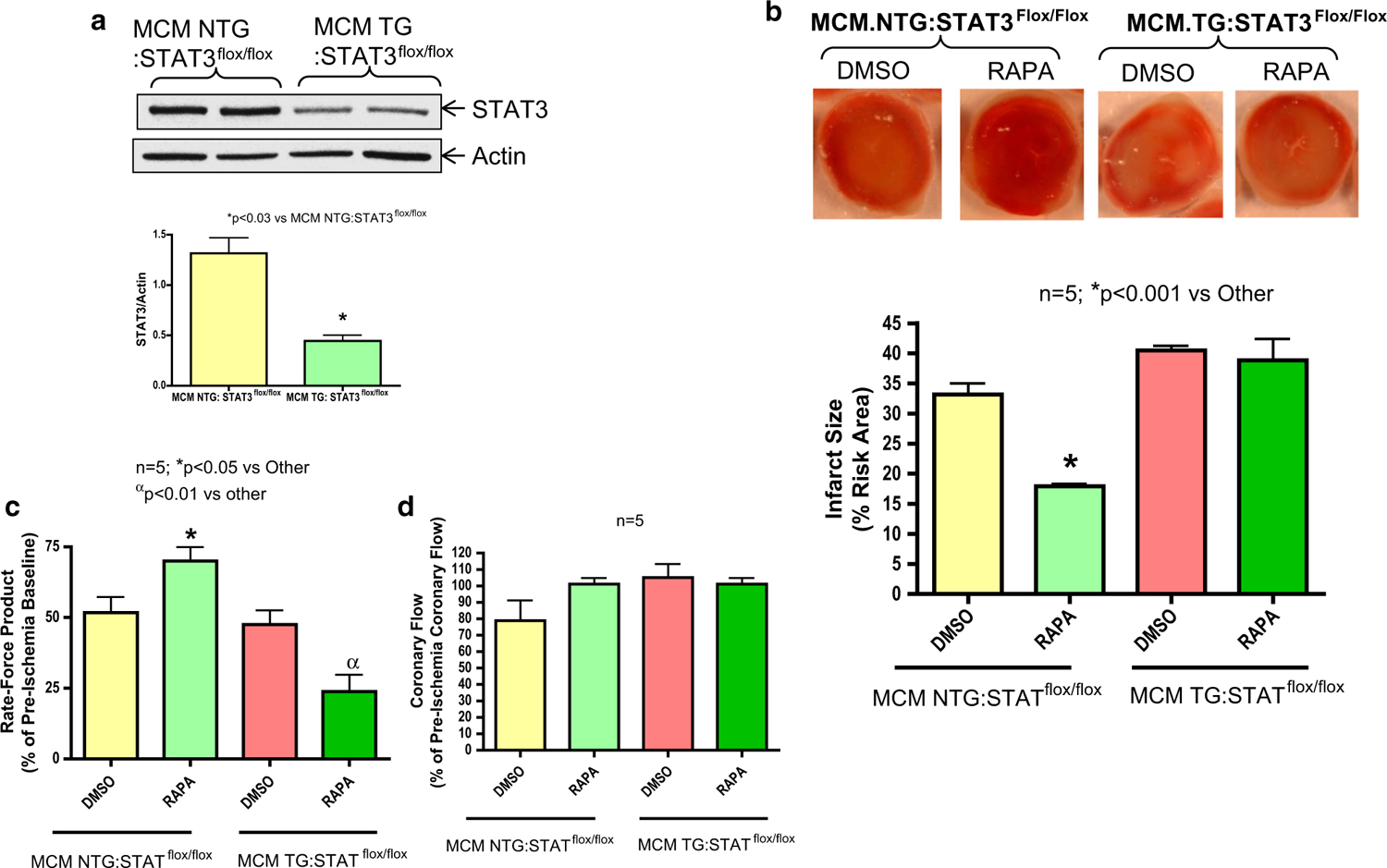
Cardiac-specific STAT3 deficiency abolishes infarct-limiting effect of rapamycin (RAPA) against ischemia/reperfusion (I/R) injury in diabetic mice. a STAT3-deficient (MCM TG:STAT3flox/flox) and WT (MCM NTG:STAT3flox/flox) male mice (8–10 weeks) were fed high fat diet (HFD) for 16 weeks, after which they received tamoxifen (20 mg/kg/day i.p.) for 10 days. Representative immunoblots of STAT3 and GAPDH in hearts of HFD-fed and tamoxifen treated STAT3-deficient and WT mice. Densitometric analysis representing fold change in STAT3/GAPDH ratio. b Myocardial infarct sizes of WT and STAT3-deficient mice following global I/R as well as infusion of rapamycin (RAPA, 100 nM) or DMSO (solvent of rapamycin) at reperfusion. c Product of heart rate and ventricular developed force (% of pre-ischemic baseline). d Coronary flow rate (% of pre-ischemic coronary flow)
Role of STAT3 signaling in protection of cardiomyocytes with rapamycin
Necrosis and apoptosis following SI-RO were significantly higher in primary cardiomyocytes isolated from diabetic mice as compared to C57 mice (Figs. 5, 6). Mitochondrial membrane potential assessed by JC-1 aggregate/monomer ratio was also reduced significantly in cardiomyocytes from db/db mice as compared to C57 mice (Fig. 7). Rapamycin treatment at reoxygenation caused significant reduction in necrosis, apoptosis and improvement in mitochondrial membrane potential in cardiomyocytes isolated from db/db mice. Interestingly, the magnitude of improvement in the mitochondrial membrane potential following rapamycin treatment was significantly higher in cardiomyocytes from db/db mice as compared to C57 mice (Fig. 7). Necrosis and apoptosis were also reduced significantly with rapamycin treatment in cardiomyocytes from HFD-fed WT mice following SI. Rapamycin-induced protection in cardiomyocytes was lost in HFD-fed STAT3-deficient mice (Fig. 8).
Fig. 5.
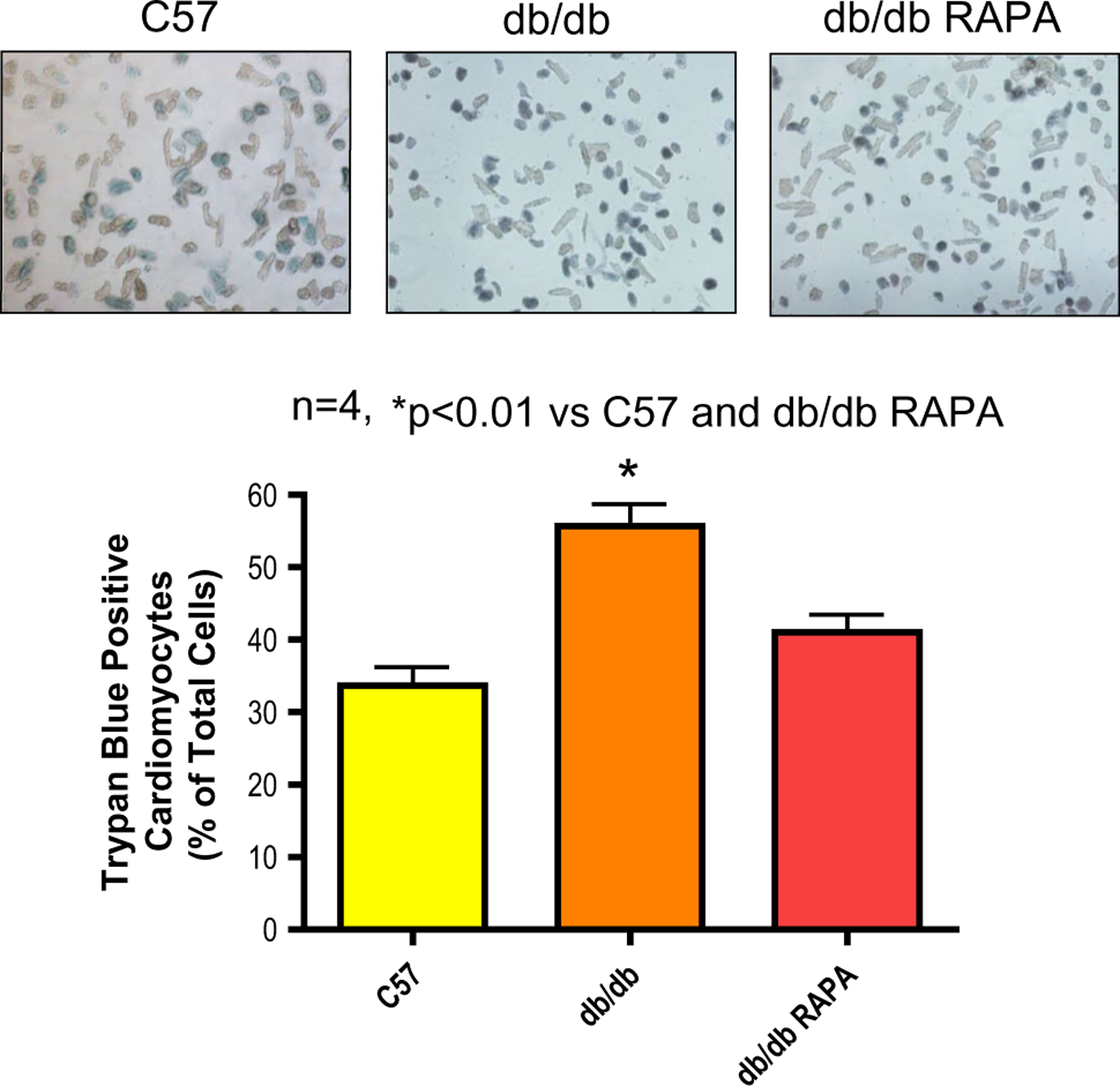
Rapamycin protects cardiomyocytes of diabetic mice (db/db) against necrosis following simulated ischemia and reoxygenation (SI/RO). Cardiomyocytes (isolated from C57 and db/db mice) were subjected to SI/RO. During RO (1 h), cardiomyocytes were treated with rapamycin (RAPA, 100 nM) or DMSO (solvent of RAPA). Cardiomyocyte necrosis was determined by trypan blue staining following 40 min SI and 1 h RO
Fig. 6.
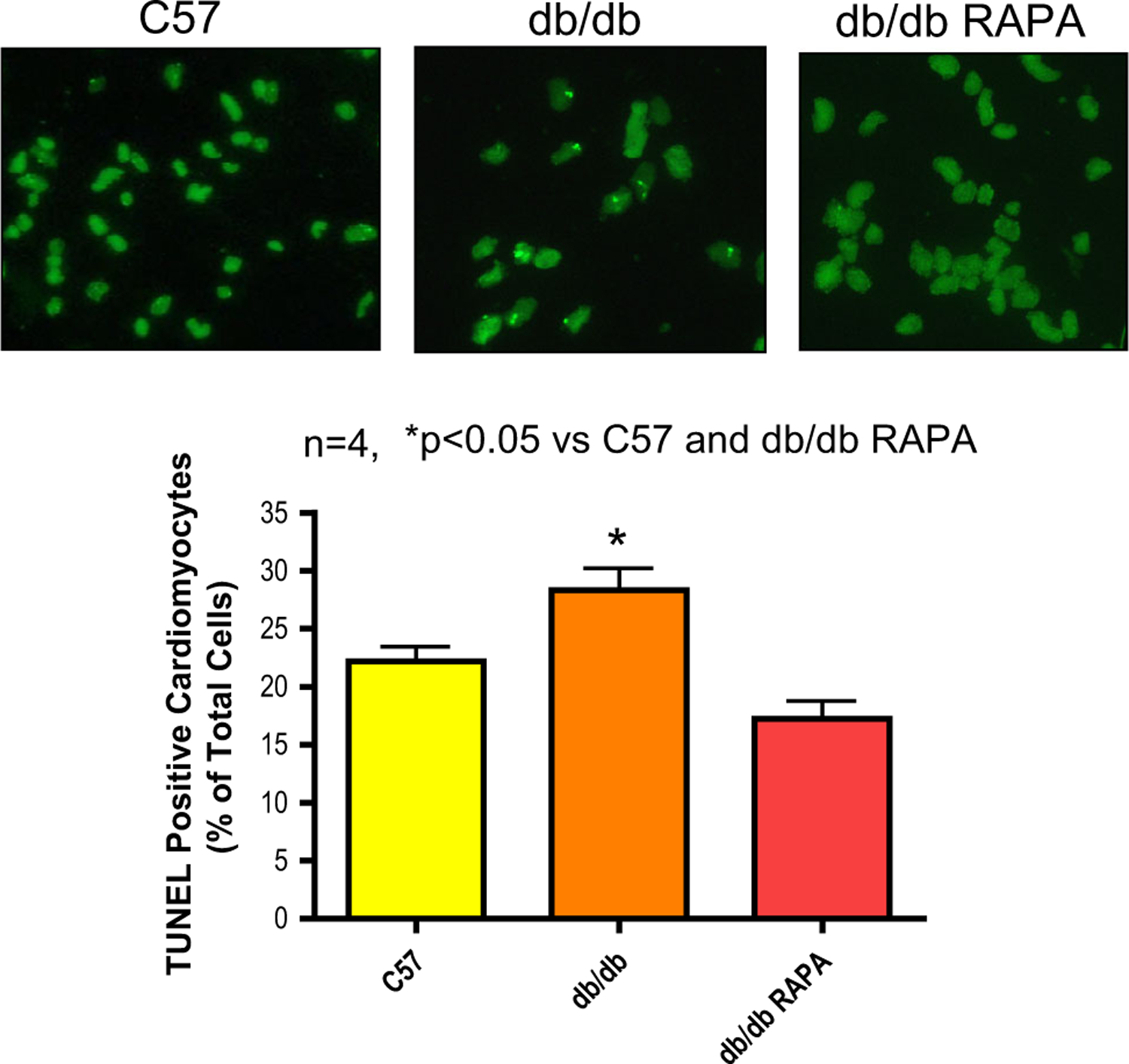
Reduction of apoptosis in cardiomyocytes of diabetic mice (db/db) with rapamycin treatment during reoxygenation (SI/RO). Cardiomyocytes (isolated from C57 and db/db mice) were subjected to SI/RO. During RO (18 h), cardiomyocytes were treated with rapamycin (RAPA, 100 nM) or DMSO (solvent of RAPA). Cardiomyocyte apoptosis was determined by TUNEL assay following 40 min SI and 18 h RO
Fig. 7.
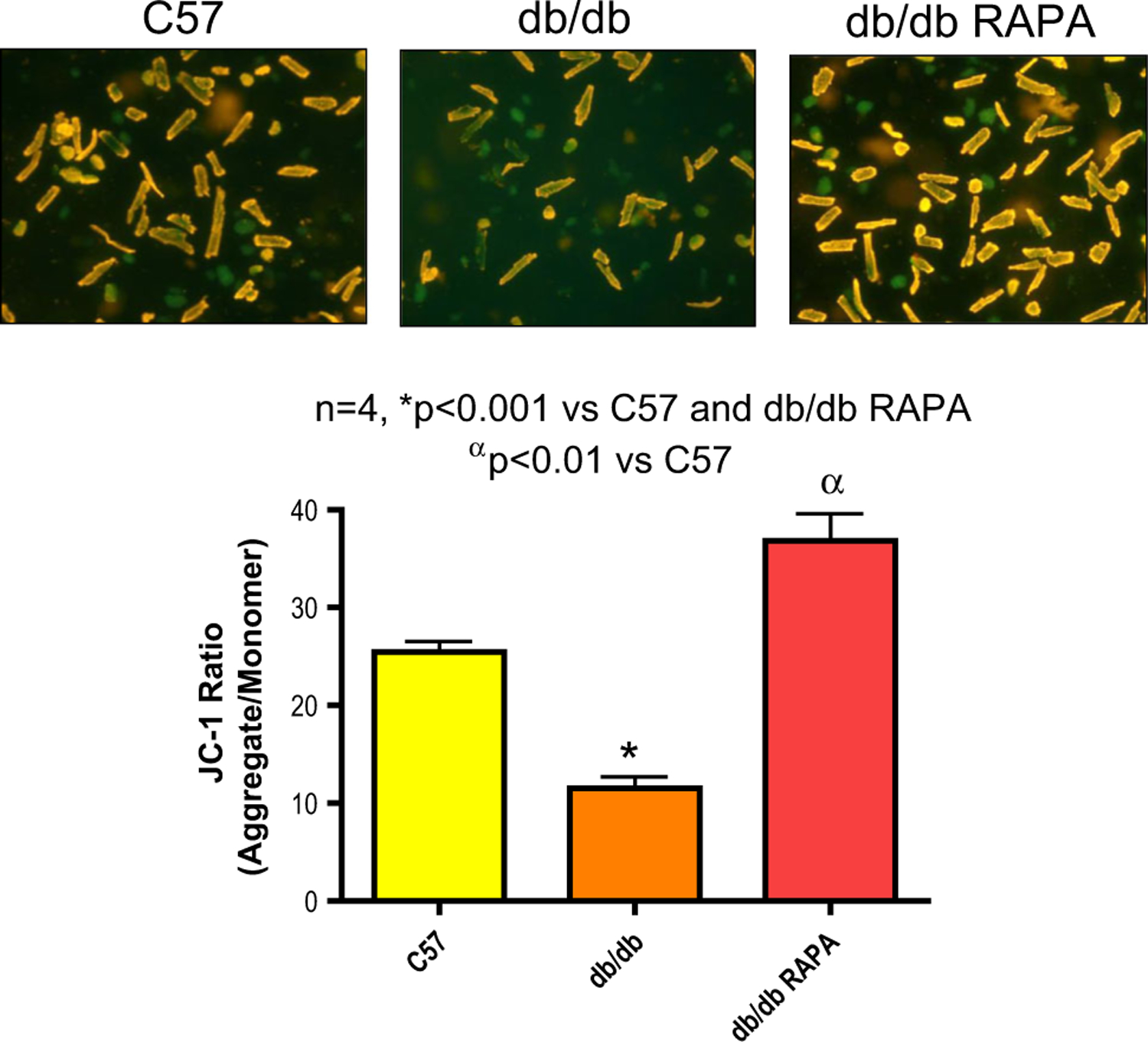
Rapamycin preserves mitochondrial membrane potential in cardiomyocytes of diabetic mice (db/db) following SI/RO. Isolated murine cardiomyocytes from C57 and db/db mice were subjected to SI/RO. During RO (1 h), cardiomyocytes were treated with rapamycin (RAPA, 100 nM) or DMSO (solvent of RAPA). Cardiomyocyte membrane potential was examined by using cationic lipophilic probe JC-1. Quantification of JC-1 aggregate (red/orange color)/monomer (in green color) ratio indicates the preservation of mitochondrial membrane potential
Fig. 8.
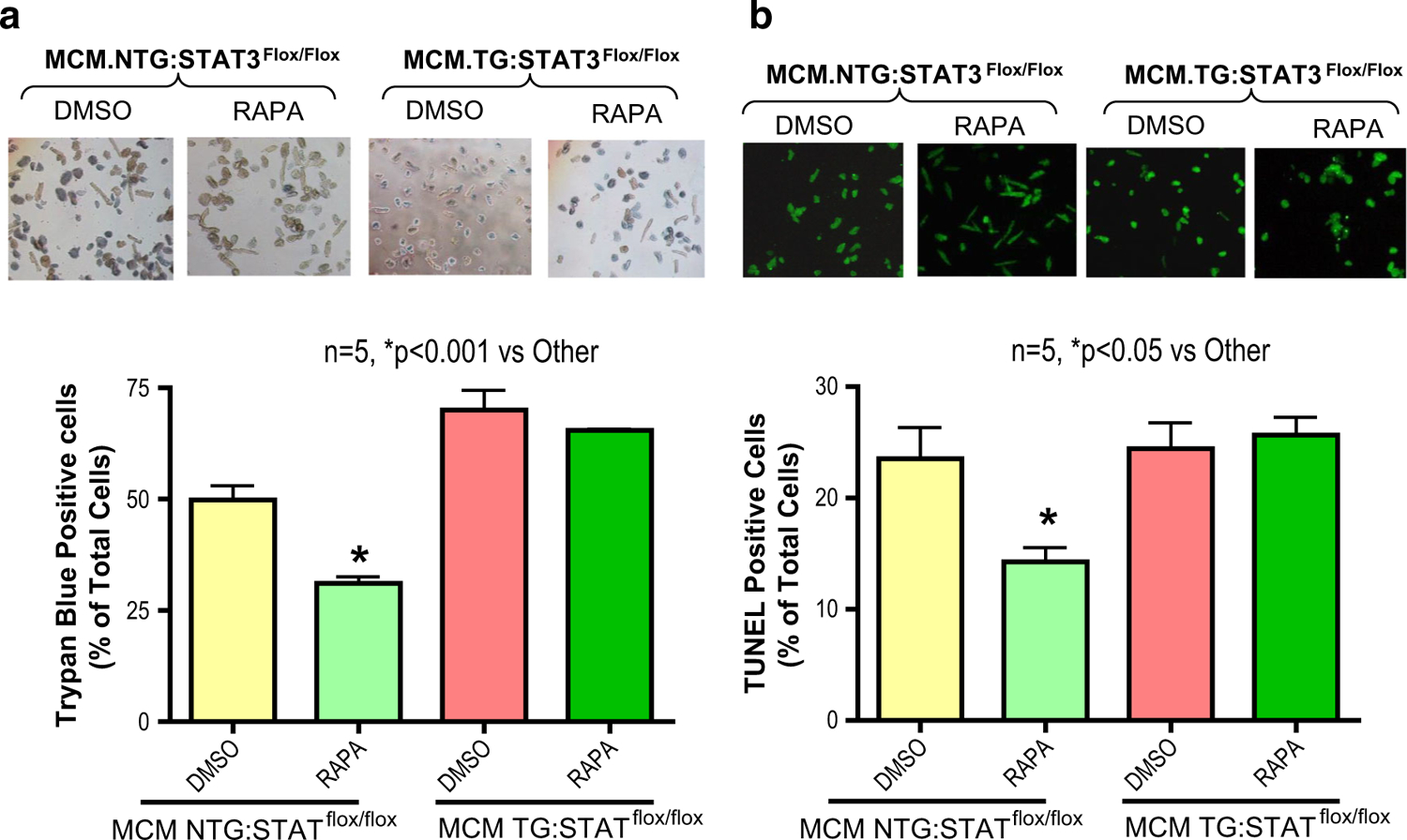
STAT3-deficiency abrogates rapamycin-induced protection against necrosis/apoptosis in cardiomyocytes of diabetic mice following simulated ischemia and reoxygenation (SI/RO). Cardiomyocytes (isolated from high fat diet fed and tamoxifen treated STAT3-deficient, MCM TG:STAT3flox/flox and WT, MCM NTG:STAT3flox/flox male mice) were subjected to SI/RO. During RO, cardiomyocytes were treated with rapamycin (RAPA, 100 nM) or DMSO (solvent of RAPA). a Cardiomyocyte necrosis was determined by trypan blue staining following 40 min SI and 1 h RO. b Cardiomyocyte apoptosis was determined by TUNEL assay following 40 min SI and 18 h RO
Discussion
Ischemic heart disease continues to be the underlying cause of most acute myocardial infarctions, congestive heart failure, arrhythmias, and sudden cardiac death. Protection of the heart against injury caused by infarction remains a challenge for the cardiologists and emergency room physicians. Although, reperfusion is mandatory to salvage ischemic myocardium from infarction, reperfusion itself contributes to irreversible ischemic myocardial injury by excess formation of reactive oxygen species, intracellular calcium overload, mitochondrial dysfunction, activation of intracellular proteolysis, and uncoordinated excess contractile activity [21, 22, 48]. Currently there are no therapies approved by the FDA that can directly protect the heart against the deleterious effects of reperfusion injury. The present study shows for the first time that mTOR inhibitor, rapamycin administered at the onset of reperfusion following global ischemia significantly reduced myocardial infarct size in diabetic hearts (Fig. 2). Moreover, rapamycin treatment improved cardiac function as measured by rate-force product and coronary flow rates in diabetic heart following I/R injury as compared to vehicle-treated diabetic hearts. At the cellular level, rapamycin treatment during reoxygenation following simulated ischemia reduced necrosis, apoptosis, and preserved mitochondrial membrane potential in adult primary cardiomyocytes isolated from diabetic mice (Figs. 5, 6, 7). In summary, these results provide novel information on the protective effect of rapamycin against reperfusion injury in diabetic heart and cardiomyocytes.
mTOR exists in two functionally distinct complexes referred to as mTOR complex 1 (mTORC1) and mTORC2, which were originally defined as being rapamycin-sensitive and -insensitive, respectively [40]. mTOR is critical for the induction of cardiac hypertrophy after stress, and inhibiting this signaling complex improves cardiac function in pathological settings [7]. In mice and rats, mTOR inhibition with rapamycin regressed remodeling by pressure overload and blunted the development of cardiac dysfunction [34, 41, 49]. In contrast, mTOR deletion caused lethal, dilated cardiomyopathy that was sufficient to induce the development of heart failure [1]. The involvement of mTORC1 activation in the pathogenesis of insulin resistance in T2D has been considered [25, 46], although its role following acute MI has never been studied. Rapamycin preferentially inhibits mTORC1, although recent studies suggest that prolonged treatment with the drug also inhibits mTORC2 thereby leading to insulin resistance [30, 40]. Therefore, a more selective approach toward mTORC1 inhibition (without disrupting mTORC2) may be important for treatment of diabetes-induced cardiovascular diseases. In this regard, we recently showed that chronic rapamycin treatment with relatively low dose i.e. 0.25 mg/kg prevented cardiac dysfunction in T2D mice with inhibition of mTORC1, without interfering with mTORC2 [8].
In the present study, rapamycin infusion inhibited enhanced phosphorylation of S6 ribosomal protein (target of mRORC1) following I/R in the diabetic heart (Fig. 3). Moreover, rapamycin inhibited S6 phosphorylation in hearts of normoglycemic mice without interfering with AKT phosphorylation on Ser473, a target of mTORC2. The AKT phosphorylation was significantly increased following I/R in diabetic hearts as compared to C57 hearts. Interestingly, AKT phosphorylation was further enhanced with rapamycin suggesting that it specifically inhibited mTORC1 activity in normoglycemic and diabetic hearts, whereas activated mTORC2 in the diabetic heart. Thus activation of mTORC2 may provide beneficial effect of cardioprotection by rapamycin treatment at reperfusion in diabetic heart following MI. It has been reported that the diabetic state interferes with the intrinsic protective-adaptive mechanism of myocardial ischemic injury [15, 27], which may contribute to enhanced AKT phosphorylation with expanded infarct size.
Our results also showed that phosphorylation of STAT3 (Tyr-705) and total STAT3 were significantly reduced in the diabetic as compared to normoglycemic hearts (Fig. 3). Rapamycin significantly increased both the total and phosphorylated STAT3 in relation to vehicle-treated db/db hearts (Fig. 3). These results were also duplicated in inducible, cardiac-specific STAT3-deficient mice rendered diabetic by feeding HFD. Rapamycin treatment at reperfusion exerted robust infarct-sparing effect in HFD-fed WT littermates which was associated with improved functional recovery after global I/R injury (Fig. 4). Conversely, the infarct-limiting effect of rapamycin was abolished in HFD-fed STAT3-deficient mice. Furthermore, rapamycin treatment at reoxygenation in cardiomyocytes isolated from HFD-fed STAT3-deficient mice failed to protect against SI/RO, thereby corroborating the essential role of STAT3 in mediating this protection (Fig. 8).
A number of studies have suggested that STAT3 pathway is a fundamental cardioprotective component of the intrinsic survival signaling, which prevents apoptosis, hypertrophy and inflammation [19, 36, 47]. STAT3 plays a critical role in ischemic as well as pharmacological postconditioning [16, 17, 22, 23, 37, 43]. Although STAT3 is a well-known transcription factor which is involved in several physiological and pathological processes in the heart, its mitochondrial location and function was identified only recently. STAT3 is located in the matrix of subsarcolemmal and interfibrillar mitochondria [4, 22]. Mitochondria from mice with cardiomyocyte-specific deletion of STAT3 displayed reduced activities of complexes I and II of the electron transport chain and decreased oxygen consumption [4, 45]. STAT3 contributed to cardioprotection against I/R injury by stimulation of mitochondrial respiration and inhibition of mitochondrial permeability transition pore (MPTP) [4]. ADP-stimulated respiration was reduced in mitochondria of cardiomyocyte-specific STAT3-KO mice as compared to wild type mice [4]. Inhibition of mitochondrial STAT3 by Stattic impaired cardiomyocyte mitochondrial function through formation of reactive oxygen species [5]. In the aged mouse heart, the reduced level of STAT3 protein contributed to the loss of cardioprotection by ischemic post-conditioning [3]. Interestingly, the unresponsiveness of diabetic myocardium to ischemic pre- or post-conditioning has been linked to a defect in insulin receptor substrate-1-associated PI3K/AKT activity and tyrosine phosphorylation of STAT3 [15, 38]. The impairment of protective signaling pathway related to the activation of STAT3 and AKT are the major mechanisms responsible for increased post-ischemic myocardial injury in diabetes [15, 31]. STAT3 signaling also plays an essential role in rapamycin-induced cardioprotection against I/R injury in non-diabetic mice [9]. Prior studies reveal a cross-talk between STAT3 and PI3K/AKT signaling in the cardiac myocytes [15, 18]. STAT3 activation is required for initiation of PI3K/AKT signaling, which is known to enhance post-conditioning mediated reduction in myocardial infarct size [15, 17, 18]. Using cardiomyocytes-specific STAT3 knock-out mice, Goodman et al. [18] confirmed that JAK/STAT signaling may provide upstream initiation of RISK pathway signaling via PI3K/AKT activation which leads to cardioprotection following post-conditioning. In a previous study, we reported that pretreatment with rapamycin (1 h) induced phosphorylation of STAT3 in the hearts of CD-1 mice [9]. However, in the present study, hearts were infused with 100 nM rapamycin at the onset of reperfusion for 20 min following 30 min of no-flow global ischemia in Langendorff mode. Based on these differences in the experimental protocol and strain of mice, we believe that rapamycin treatment during reperfusion does not activate STAT3 in WT C57 mice. However, pretreatment with rapamycin-induced phosphorylation of STAT3 in the hearts of CD-1 (WT) mice. Thus it appears that that reperfusion therapy with rapamycin in diabetic heart provides protection through STAT3-AKT signaling pathway.
In summary, we have provided compelling evidence that mTOR inhibition by infusion of clinically-approved anti-rejection drug, rapamycin protects against myocardial reperfusion injury. A definite role of STAT3 in protection is provided by failure of rapamycin to protect against I/R injury in cardiac-specific STAT3-deficient mice. Considering that sirolimus-eluting stents are highly effective in reducing the risk for major cardiac events and are safe in diabetic patients with coronary artery disease [2, 13], it is tempting to propose that rapamycin may be developed as a promising clinical approach to combat myocardial infarction and improve prognosis in diabetic patients. However, one limitation of this study is that we used isolated perfused hearts that were subjected to global I/R injury as opposed to the in vivo model of myocardial infarction. Unfortunately, the left coronary artery occlusion in the db/db or HFD fed diabetic mice is challenging because of high mortality as compared to non-diabetic wild type mice. Despite this limitation, our results using two independent models i.e., isolated heart and adult cardiomyocytes provide convincing evidence on the efficacy of rapamycin in reducing reperfusion injury in diabetic mice.
Acknowledgments
This work was supported by grants from the National Institutes of Health R37 HL051045, R01 HL079424, R01 HL093685, and R01 HL118808 (RCK), the American Heart Association 10SDG3770011 and 14GRNT20010003 (FNS), and A. D. Williams’ Fund of Virginia Commonwealth University Grant UL1RR031990 (AD).
Footnotes
Conflict of interest On behalf of all authors, the corresponding author states that there is no conflict of interest.
References
- 1.Aoyagi T, Kusakari Y, Xiao CY, Inouye BT, Takahashi M, Scherrer-Crosbie M, Rosenzweig A, Hara K, Matsui T (2012) Cardiac mTOR protects the heart against ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 303:H75–H85. doi: 10.1152/ajpheart.00241.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baumgart D, Klauss V, Baer F, Hartmann F, Drexler H, Motz W, Klues H, Hofmann S, Volker W, Pfannebecker T, Stoll HP, Nickenig G (2007) One-year results of the SCORPIUS study: a German multicenter investigation on the effectiveness of sirolimus-eluting stents in diabetic patients. J Am Coll Cardiol 50:1627–1634. doi: 10.1016/j.jacc.2007.07.035 [DOI] [PubMed] [Google Scholar]
- 3.Boengler K, Buechert A, Heinen Y, Roeskes C, Hilfiker-Kleiner D, Heusch G, Schulz R (2008) Cardioprotection by ischemic postconditioning is lost in aged and STAT3-deficient mice. Circ Res 102:131–135. doi: 10.1016/j.yjmcc.2011.01.002 [DOI] [PubMed] [Google Scholar]
- 4.Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R (2010) Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res Cardiol 105:771–785. doi: 10.1007/s00395-010-0124-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boengler K, Ungefug E, Heusch G, Schulz R (2013) The STAT3 inhibitor stattic impairs cardiomyocyte mitochondrial function through increased reactive oxygen species formation. Curr Pharm Des 19:6890–6895. doi: 10.2174/138161281939131127115940 [DOI] [PubMed] [Google Scholar]
- 6.Bolli R, Stein AB, Guo YR, Wang OL, Rokosh G, Dawn B, Molkentin JD, Sanganalmath SK, Zhu YQ, Xuan YT (2011) A murine model of inducible, cardiac-specific deletion of STAT3: its use to determine the role of STAT3 in the upregulation of cardioprotective proteins by ischemic preconditioning. J Mol Cell Cardiol 50:589–597. doi: 10.1016/j.yjmcc.2011.01.002 [DOI] [PubMed] [Google Scholar]
- 7.Boluyt MO, Zheng JS, Younes A, Long X, O’Neill L, Silverman H, Lakatta EG, Crow MT (1997) Rapamycin inhibits alpha 1-adrenergic receptor-stimulated cardiac myocyte hypertrophy but not activation of hypertrophy-associated genes. Evidence for involvement of p70 S6 kinase. Circ Res 81:176–186. doi: 10.1161/01.RES.81.2.176 [DOI] [PubMed] [Google Scholar]
- 8.Das A, Durrant D, Koka S, Salloum FN, Xi L, Kukreja RC (2014) Mammalian target of rapamycin (mTOR) inhibition with rapamycin improves cardiac function in type 2 diabetic mice: potential role of attenuated oxidative stress and altered contractile protein expression. J Biol Chem 289:4145–4160. doi: 10.1074/jbc.M113.521062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Das A, Salloum FN, Durrant D, Ockaili R, Kukreja RC (2012) Rapamycin protects against myocardial ischemia-reperfusion injury through JAK2-STAT3 signaling pathway. J Mol Cell Cardiol 53:858–869. doi: 10.1016/j.yjmcc.2012.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Das A, Salloum FN, Xi L, Rao YJ, Kukreja RC (2009) ERK phosphorylation mediates sildenafil-induced myocardial protection against ischemia-reperfusion injury in mice. Am J Physiol Heart Circ Physiol 296:H1236–H1243. doi: 10.1152/ajpheart.00100.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das A, Xi L, Kukreja RC (2005) Phosphodiesterase-5 inhibitor sildenafil preconditions adult cardiac myocytes against necrosis and apoptosis. Essential role of nitric oxide signaling. J Biol Chem 280:12944–12955. doi: 10.1074/jbc.M404706200 [DOI] [PubMed] [Google Scholar]
- 12.Das A, Xi L, Kukreja RC (2008) Protein kinase G-dependent cardioprotective mechanism of phosphodiesterase-5 inhibition involves phosphorylation of ERK and GSK3beta. J Biol Chem 283:29572–29585. doi: 10.1074/jbc.M801547200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Waha A, Dibra A, Kufner S, Baumgart D, Sabate M, Maresta A, Schomig A, Kastrati A (2011) Long-term outcome after sirolimus-eluting stents versus bare metal stents in patients with diabetes mellitus: a patient-level meta-analysis of randomized trials. Clin Res Cardiol 100:561–570. doi: 10.1007/s00392-010-0278-8 [DOI] [PubMed] [Google Scholar]
- 14.Drake JC, Alway SE, Hollander JM, Williamson DL (2010) AICAR treatment for 14 days normalizes obesity-induced dys-regulation of TORC1 signaling and translational capacity in fasted skeletal muscle. Am J Physiol Regul Integr Comp Physiol 299:R1546–R1554. doi: 10.1152/ajpregu.00337.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drenger B, Ostrovsky IA, Barak M, Nechemia-Arbely Y, Ziv E, Axelrod JH (2011) Diabetes blockade of sevoflurane postconditioning is not restored by insulin in the rat heart: phosphorylated signal transducer and activator of transcription 3- and phosphatidylinositol 3-kinase-mediated inhibition. Anesthesiology 114:1364–1372. doi: 10.1097/ALN.0b013e31820efafd [DOI] [PubMed] [Google Scholar]
- 16.Duan W, Yang Y, Yan J, Yu S, Liu J, Zhou J, Zhang J, Jin Z, Yi D (2012) The effects of curcumin post-treatment against myocardial ischemia and reperfusion by activation of the JAK2/STAT3 signaling pathway. Basic Res Cardiol 107:263. doi: 10.1007/s00395-012-0263-7 [DOI] [PubMed] [Google Scholar]
- 17.Fuglesteg BN, Suleman N, Tiron C, Kanhema T, Lacerda L, Andreasen TV, Sack MN, Jonassen AK, Mjos OD, Opie LH, Lecour S (2008) Signal transducer and activator of transcription 3 is involved in the cardioprotective signalling pathway activated by insulin therapy at reperfusion. Basic Res Cardiol 103:444–453. doi: 10.1007/s00395-008-0728-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodman MD, Koch SE, Fuller-Bicer GA, Butler KL (2008) Regulating RISK: a role for JAK-STAT signaling in postconditioning? Am J Physiol Heart Circ Physiol 295:H1649–H1656. doi: 10.1152/ajpheart.00692.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gross ER, Hsu AK, Gross GJ (2006) The JAK/STAT pathway is essential for opioid-induced cardioprotection: JAK2 as a mediator of STAT3, Akt, and GSK-3 beta. Am J Physiol Heart Circ Physiol 291:H827–H834. doi: 10.1152/ajpheart.00003.2006 [DOI] [PubMed] [Google Scholar]
- 20.Grundy SM, Benjamin IJ, Burke GL, Chait A, Eckel RH, Howard BV, Mitch W, Smith SC Jr, Sowers JR (1999) Diabetes and cardiovascular disease: a statement for healthcare professionals from the American Heart Association. Circulation 100:1134–1146. doi: 10.1161/01.CIR.100.10.1134 [DOI] [PubMed] [Google Scholar]
- 21.Heusch G (2013) Cardioprotection: chances and challenges of its translation to the clinic. Lancet 381:166–175. doi: 10.1016/S0140-6736(12)60916-7 [DOI] [PubMed] [Google Scholar]
- 22.Heusch G (2015) Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res 116:674–699. doi: 10.1161/CIRCRESAHA.116.305348 [DOI] [PubMed] [Google Scholar]
- 23.Heusch G, Musiolik J, Gedik N, Skyschally A (2011) Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circ Res 109:1302–1308. doi: 10.1161/CIRCRESAHA.111.255604 [DOI] [PubMed] [Google Scholar]
- 24.Hilfiker-Kleiner D, Hilfiker A, Fuchs M, Kaminski K, Schaefer A, Schieffer B, Hillmer A, Schmiedl A, Ding Z, Podewski E, Podewski E, Poli V, Schneider MD, Schulz R, Park JK, Wollert KC, Drexler H (2004) Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ Res 95:187–195. doi: 10.1161/01.RES.0000134921.50377.61 [DOI] [PubMed] [Google Scholar]
- 25.Howell JJ, Manning BD (2011) mTOR couples cellular nutrient sensing to organismal metabolic homeostasis. Trends Endocrinol Metab 22:94–102. doi: 10.1016/j.tem.2010.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacoby JJ, Kalinowski A, Liu MG, Zhang SS, Gao Q, Chai GX, Ji L, Iwamoto Y, Li E, Schneider M, Russell KS, Fu XY (2003) Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc Natl Acad Sci USA 100:12929–12934. doi: 10.1073/pnas.2134694100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kersten JR, Montgomery MW, Ghassemi T, Gross ER, Toller WG, Pagel PS, Warltier DC (2001) Diabetes and hyperglycemia impair activation of mitochondrial K(ATP) channels. Am J Physiol Heart Circ Physiol 280:H1744–H1750 [DOI] [PubMed] [Google Scholar]
- 28.Khamzina L, Veilleux A, Bergeron S, Marette A (2005) Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology 146:1473–1481. doi: 10.1210/en.2004-0921 [DOI] [PubMed] [Google Scholar]
- 29.Khan S, Salloum F, Das A, Xi L, Vetrovec GW, Kukreja RC (2006) Rapamycin confers preconditioning-like protection against ischemia-reperfusion injury in isolated mouse heart and cardiomyocytes. J Mol Cell Cardiol 41:256–264. doi: 10.1016/j.yjmcc.2006.04.014 [DOI] [PubMed] [Google Scholar]
- 30.Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, Davis JG, Salmon AB, Richardson A, Ahima RS, Guertin DA, Sabatini DM, Baur JA (2012) Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 335:1638–1643. doi: 10.1126/science.1215135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, Liu Z, Wang J, Wong GT, Cheung CW, Zhang L, Chen C, Xia Z, Irwin MG (2013) Susceptibility to myocardial ischemia reperfusion injury at early stage of type 1 diabetes in rats. Cardiovasc Diabetol 12:133. doi: 10.1186/1475-2840-12-133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo M, Guan X, Luczak ED, Lang D, Kutschke W, Gao Z, Yang J, Glynn P, Sossalla S, Swaminathan PD, Weiss RM, Yang B, Rokita AG, Maier LS, Efimov IR, Hund TJ, Anderson ME (2013) Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J Clin Invest 123:1262–1274. doi: 10.1172/JCI65268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mak KH, Moliterno DJ, Granger CB, Miller DP, White HD, Wilcox RG, Califf RM, Topol EJ (1997) Influence of diabetes mellitus on clinical outcome in the thrombolytic era of acute myocardial infarction. GUSTO-I Investigators. Global utilization of streptokinase and tissue plasminogen activator for occluded coronary arteries. J Am Coll Cardiol 30:171–179. doi: 10.1016/S0735-1097(97)00118-6 [DOI] [PubMed] [Google Scholar]
- 34.McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T, Izumo S (2004) Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation 109:3050–3055. doi: 10.1161/01.CIR.0000130641.08705.45 [DOI] [PubMed] [Google Scholar]
- 35.Meric-Bernstam F, Gonzalez-Angulo AM (2009) Targeting the mTOR signaling network for cancer therapy. J Clin Oncol 27:2278–2287. doi: 10.1200/JCO.2008.20.0766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Negoro S, Kunisada K, Tone E, Funamoto M, Oh H, Kishimoto T, Yamauchi-Takihara K (2000) Activation of JAK/STAT pathway transduces cytoprotective signal in rat acute myocardial infarction. Cardiovasc Res 47:797–805. doi: 10.1016/S0008-6363(00)00138-3 [DOI] [PubMed] [Google Scholar]
- 37.Obana M, Miyamoto K, Murasawa S, Iwakura T, Hayama A, Yamashita T, Shiragaki M, Kumagai S, Miyawaki A, Takewaki K, Matsumiya G, Maeda M, Yoshiyama M, Nakayama H, Fujio Y (2012) Therapeutic administration of IL-11 exhibits the postconditioning effects against ischemia-reperfusion injury via STAT3 in the heart. Am J Physiol Heart Circ Physiol 303:H569–H577. doi: 10.1152/ajpheart.00060.2012 [DOI] [PubMed] [Google Scholar]
- 38.Park SY, Cho YR, Finck BN, Kim HJ, Higashimori T, Hong EG, Lee MK, Danton C, Deshmukh S, Cline GW, Wu JJ, Bennett AM, Rothermel B, Kalinowski A, Russell KS, Kim YB, Kelly DP, Kim JK (2005) Cardiac-specific overexpression of peroxisome proliferator-activated receptor-alpha causes insulin resistance in heart and liver. Diabetes 54:2514–2524. doi: 10.2337/diabetes.54.9.2514 [DOI] [PubMed] [Google Scholar]
- 39.Ranieri SC, Fusco S, Panieri E, Labate V, Mele M, Tesori V, Ferrara AM, Maulucci G, De SM, Martorana GE, Galeotti T, Pani G (2010) Mammalian life-span determinant p66shcA mediates obesity-induced insulin resistance. Proc Natl Acad Sci USA 107:13420–13425. doi: 10.1073/pnas.1008647107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM (2006) Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22:159–168. doi: 10.1016/j.molcel.2006.03.029 [DOI] [PubMed] [Google Scholar]
- 41.Shioi T, McMullen JR, Tarnavski O, Converso K, Sherwood MC, Manning WJ, Izumo S (2003) Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation 107:1664–1670. doi: 10.1161/01.CIR.0000057979.36322.88 [DOI] [PubMed] [Google Scholar]
- 42.Sung MM, Koonen DP, Soltys CL, Jacobs RL, Febbraio M, Dyck JR (2011) Increased CD36 expression in middle-aged mice contributes to obesity-related cardiac hypertrophy in the absence of cardiac dysfunction. J Mol Med (Berl) 89:459–469. doi: 10.1007/s00109-010-0720-4 [DOI] [PubMed] [Google Scholar]
- 43.Tian Y, Zhang W, Xia D, Modi P, Liang D, Wei M (2011) Postconditioning inhibits myocardial apoptosis during prolonged reperfusion via a JAK2-STAT3-Bcl-2 pathway. J Biomed Sci 18:53. doi: 10.1186/1423-0127-18-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turdi S, Kandadi MR, Zhao J, Huff AF, Du M, Ren J (2011) Deficiency in AMP-activated protein kinase exaggerates high fat diet-induced cardiac hypertrophy and contractile dysfunction. J Mol Cell Cardiol 50:712–722. doi: 10.1016/j.yjmcc.2010.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, Moh A, Moghaddas S, Chen Q, Bobbili S, Cichy J, Dulak J, Baker DP, Wolfman A, Stuehr D, Hassan MO, Fu XY, Avadhani N, Drake JI, Fawcett P, Lesnefsky EJ, Larner AC (2009) Function of mitochondrial Stat3 in cellular respiration. Science 323:793–797. doi: 10.1126/science.1164551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wullschleger S, Loewith R, Hall MN (2006) TOR signaling in growth and metabolism. Cell 124:471–484. doi: 10.1016/j.cell.2006.01.016 [DOI] [PubMed] [Google Scholar]
- 47.Xuan YT, Guo Y, Han H, Zhu Y, Bolli R (2001) An essential role of the JAK-STAT pathway in ischemic preconditioning. Proc Natl Acad Sci USA 98:9050–9055. doi: 10.1073/pnas.161283798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yellon DM, Hausenloy DJ (2007) Myocardial reperfusion injury. N Engl J Med 357:1121–1135. doi: 10.1056/NEJMra071667 [DOI] [PubMed] [Google Scholar]
- 49.Zhang D, Contu R, Latronico MV, Zhang J, Rizzi R, Catalucci D, Miyamoto S, Huang K, Ceci M, Gu Y, Dalton ND, Peterson KL, Guan KL, Brown JH, Chen J, Sonenberg N, Condorelli G (2010) MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J Clin Invest 120:2805–2816. doi: 10.1172/JCI43008 [DOI] [PMC free article] [PubMed] [Google Scholar]