

Abstract
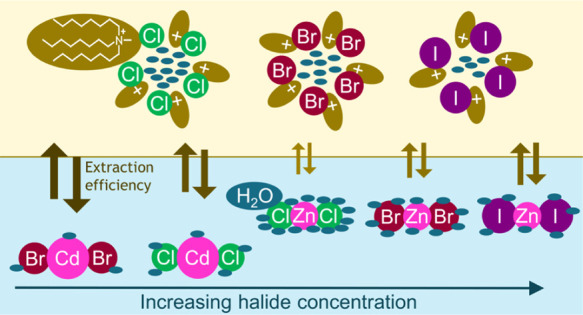
Solvent extraction is often applied to separate and purify metals on an industrial scale. Nevertheless, solvent extraction processes are challenging to develop because of the complex chemistry involved. For basic extractants, much of the chemical behavior remains poorly understood due to the conditions far from thermodynamic ideality. To elucidate the extraction mechanism, we studied the speciation and extraction of zinc(II) and cadmium(II) from chloride, bromide, and iodide media by using a basic extractant consisting of a trioctylmethylammonium cation and, respectively, a chloride, bromide, or iodide anion. These systems were specifically selected to increase the understanding of the less-studied bromide and iodide media and to focus on the effect of hard–soft interactions on solvent extraction systems. It was observed that, in general, a metal is more efficiently extracted when its hydration in the aqueous phase is lower and its stabilization in the organic phase is higher. In the investigated systems, these conditions are obtained by forming metal complexes with a lower charge density by coordinating the right number of halide anions and by selecting a halide with a lower charge density. In the organic phase, the stability of the metal complex can be increased by forming strong metal–anion bonds and by decreasing the water content. These insights might be of interest in the development and optimization of separation schemes for metals.
1. Introduction
In the metallurgical industry, mixtures of metals are often separated by a solvent extraction (or liquid–liquid extraction) process.1 Such processes work by exploiting differences in the distribution of metal ions between two immiscible liquid phases. An extractant is added to the organic phase, which selectively coordinates with some of the metal ions in the aqueous feed solution. In this way, valuable metals can be recovered from complex feed solutions or hazardous metals can be removed from waste streams.2
However, the design and optimization of solvent extraction systems are challenging because several physicochemical processes are involved. This is especially the case when employing basic extractants, which extract metal–ligand complexes from aqueous feeds with high anion concentrations.3 These anions are often halides from acids or salts introduced in the leaching step or added on purpose to increase the extraction by basic extractants. As a result of this complex chemistry, it is not completely clear how basic extractants extract metal ions. Recent investigations tackled different aspects of basic extractant chemistry, but still some questions remain unanswered.4−14
Typical basic extractants are protonated amines ([RyNH]+X–), quaternary ammonium salts ([R4N]+X–), or quaternary phosphonium salts ([R4P]+X–).1,15,16 In textbooks, these extractants are often said to be anion exchangers, which extract metals as anionic metal complexes by exchanging them for extractant anions (X–). This mechanism explains the typical observation that extraction by basic extractants is more efficient when anions coordinate more easily to the metal ion and when more anions are present in the aqueous phase.3,17,18 However, it fails to explain other observations such as the negative sequence observed for the trivalent lanthanide ions in nitrate media,19 the decrease in extraction efficiency at high acid concentration,12,18 and the extraction at high LiCl concentration of metal ions such as lanthanide(III) and nickel(II) ions that do not form negatively charged complexes in the aqueous phase.10,18,20
The issues encountered when applying the anion exchange model to all observations show that a thorough investigation of the mechanism of solvent extractions by basic extractants is necessary. In previous studies, our investigation focused on the extraction of first-row transition metals in chloride media by the basic extractant trioctylmethylammonium chloride (TOMACl).21,22 These studies focused on the influence of metal speciation and hydration in aqueous chloride media on the extraction efficiency of metals by TOMACl. To generalize the new insight, it is necessary to expand such extraction studies to other anion systems.
In this paper, we studied the speciation and extraction of zinc(II) and cadmium(II) from chloride, bromide, and iodide media by using TOMACl, trioctylmethylammonium bromide (TOMABr), and trioctylmethylammonium iodide (TOMAI), or, more generally, by TOMAX, where X = Cl, Br, or I. We also determined the amount of acid and water present in the organic phase to get a more complete picture of the composition of both phases. The choice of bromide and iodide media allows investigating other anion systems while still using anions that significantly coordinate with many metal cations. These anion systems have received less attention in the literature, while they can be relevant for separation processes.23 Zinc(II) and cadmium(II) were chosen because they coordinate well with halides24,25 and because they are chemically similar. Both metals are found in the same group of the periodic table and have the same oxidation state, making their most important chemical difference the larger ionic radius of cadmium(II). Moreover, they are not redox active, so there is no risk of oxidation of the halide ion by the metal ion. By studying zinc(II) and cadmium(II) in different halide systems, effects related to hard–soft interactions [as explained by the hard–soft acid–base (HSAB) theory] can also be investigated.
2. Results
2.1. Zinc(II) and Cadmium(II) Solvent Extractions
The extractions of 0.05 mol L–1 ZnX2 or CdX2 from aqueous HX media by 0.2 mol L–1 TOMAX (where X stands for Cl–, Br–, or I–) in toluene are depicted in Figures 1 and 2. The extraction of Cd(II) from HI media was not recorded due to practical limitations (vide infra). The anions of the acid and the extractant were always the same in a solvent extraction sample (i.e., same anion in aqueous and organic phases) to avoid exchange reactions. This also avoids so-called split-anion extractions, in which both phases deliberately have different anions to enhance extraction or selectivity. Such processes are more difficult to use for fundamental purposes.26 Nevertheless, it should still be possible to apply the considerations from this paper to split-anion extraction systems. The different extraction series will be compared in detail below following the discussion of the organic phase composition and metal speciation.
Figure 1.
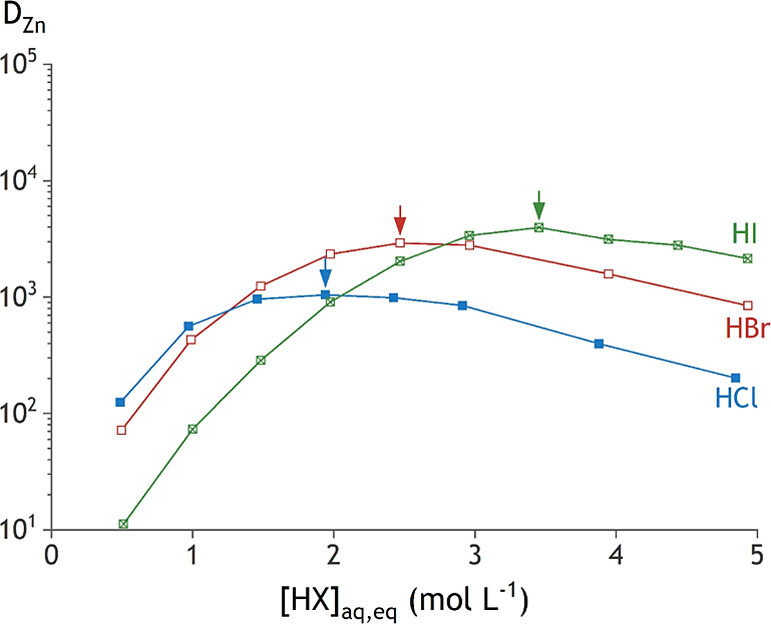
Distribution ratios for the solvent extraction of 0.05 mol L–1 ZnX2, with X being Cl– (blue box solid), Br– (brown box), or I– (green ballot box with X), by using 0.2 mol L–1 TOMAX in toluene. Arrows show the maximum extraction efficiency, which shifts to a higher anion concentration when a larger halide ion is used.
Figure 2.
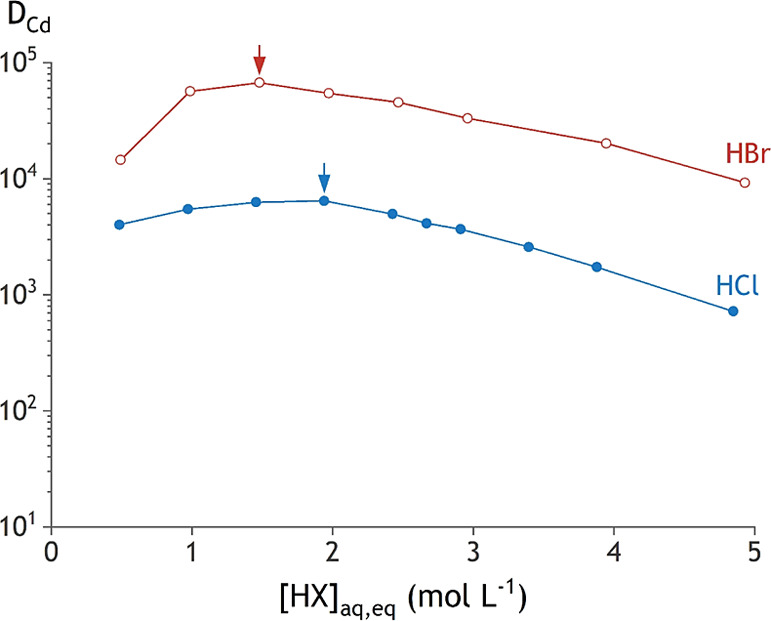
Distribution ratios for the extraction of 0.05 mol L–1 CdX2, with X being Cl– (blue circle solid) or Br– (brown circle), by using 0.2 mol L–1 TOMAX in toluene. Arrows show the maximum extraction efficiency, which shifts to a lower anion concentration when a larger halide ion is used.
A correction of the free acid and active extractant concentration was necessary for the extraction of ZnI2 from HI solutions by TOMAI. Some HI converted into I3– via oxidation due to the presence of air and via a decomposition reaction induced by light.27 This could be easily observed by a faint yellow coloration of the aqueous phase. The organic phase darkened, while the faint yellow color of the aqueous phase disappeared after shaking the HI solutions with 0.2 mol L–1 TOMAI (see the Supporting Information). This indicates the partial conversion of TOMAI into TOMAI3 by extracting I3– from the aqueous phase.28 UV–vis absorption spectroscopy was used to quantify this conversion. It showed that the conversion of TOMAI into TOMAI3 increased with the increasing HI content up to 17.5% of all TOMAI in the sample with the highest HI concentration (see the Supporting Information for more details). The triiodide ion is a weakly coordinating anion and it does not coordinate metal complexes if other ions such as iodide are present,29 but it lowers the amount of free extractant that can associate with zinc(II). This reduction in available TOMAI molecules should be considered in comparing the DZn values of all three anion systems. This is accomplished by correcting the aqueous HI concentration and by calculating the expected DZn with no TOMAI to TOMAI3 conversion, starting from the observed DZn values and the amount of converted TOMAI. The general extraction reaction appears to be as follows when (TOMA)2ZnI4 is formed in the organic phase (vide infra).30,31
| 1 |
The overbar denotes that a species resides predominantly in the organic phase and Q stands for the TOMA cation. The equation for the equilibrium constant (KEX) for this reaction is
| 2 |
This can be rewritten on a log scale with the incorporation of the observed DZn as follows
| 3 |
Now, the observed DZn can be converted into the theoretical DZn,0.2 at 0.2 mol L–1 available TOMAI by incorporating the actual concentration of TOMAI that is left based on the UV–vis absorption measurements
| 4 |
where [QI]eq is the start concentration of TOMAI (0.2 mol L–1) minus the converted TOMAI concentration.
Alternatively, a mathematical correction could be avoided by limiting the oxidization and decomposition of HI. However, this is highly impractical as light and oxygen should be avoided during all parts of the process.27 The greater experimental accuracy resulting from such manipulations does not justify the impracticalities of working in gloveboxes in the dark because an almost quantitative correction can be made mathematically.
A final remark on the extraction of ZnI2 by TOMAI should be made. The organic phase splits into two separate phases when the solution was contacted with ZnI2 in 4–5 mol L–1 HI after it was kept for more than 1 day. This third phase formation can probably be attributed to the partial conversion of TOMAI into TOMAI3 in combination with a relatively high metal loading. Thus, the instability of HI limits the applicability of solvent extraction processes with HI and basic extractants, even though they are useful for more fundamental studies. Iodination of toluene could also influence the extraction system, but such products were found (see the Supporting Information). Due to the practical limitations of HI and due to the very high DZn from HI solutions, the extraction of cadmium(II) from HI was not conducted. The even higher extraction efficiency of cadmium(II) would make it impossible to accurately determine the aqueous cadmium(II) concentration at equilibrium with inductively coupled plasma-atomic emission spectroscopy (ICP-OES), resulting in excessively large errors on DCd.
2.2. Acid and Water Distribution
Apart from the metals themselves, acids and water can also be found in the organic phase after solvent extraction. The presence of these substances in the organic phase should also be considered to understand and compare the solvent extraction of metals from different acid concentrations and in different anion systems. To quantify the amount of water and acid in the organic phase, experiments were performed without metal ions. Aqueous solutions with different concentrations of acid were contacted with 0.2 mol L–1 TOMAX in toluene, where X is Cl–, Br–, or I–. The anions of the acid and the extractant were always the same in a solvent extraction sample to avoid metathesis of the extractant.
The concentration of acid in the organic phase at equilibrium was determined using the difference in density of the initial aqueous phase and that at equilibrium (Figure 3). HCl is present to a larger extent in the TOMACl organic phase than HBr in the TOMABr phase and HI in the TOMAI phase. Based on Figure 3, one could draw the conclusion that the HBr and HI contents seem to be similar, but this is not correct as part of the HI is oxidized to I3– and extracted by TOMAI (vide supra). As a result, the actual HI content in the organic phase will be lower and an overall trend can be observed: the acids of heavier halides are distributed less favorably to the organic phase. This seems surprising since the more hydrophobic heavier halides are generally more stabilized in the organic phase.32,33 The water content in the organic phase might shed some light on this contradictory observation.
Figure 3.
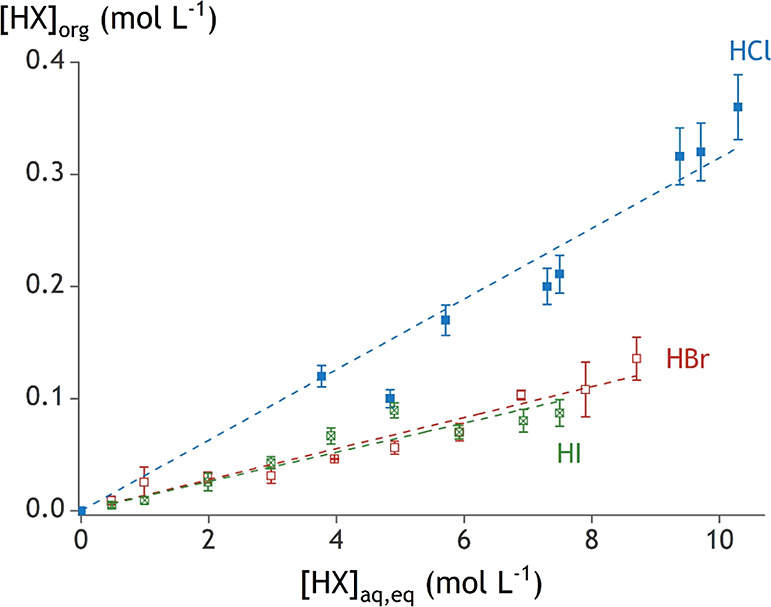
Acid content in the organic phase containing 0.2 mol L–1 TOMACl/Br/I at increasing equilibrium aqueous acid content. Error bars are included based on triplicate measurements.
The water content in the organic phase after extraction without metals was measured by Karl Fischer titration (Figure 4). A larger anion results in a more hydrophobic extractant, which explains the observed trend of decreasing water content from TOMACl > TOMABr > TOMAI. A slight additional decrease in the water content of the organic phase for the samples with HI and TOMAI can be expected with the increasing HI concentration because a part of TOMAI is converted into the even more hydrophobic TOMAI3.34
Figure 4.
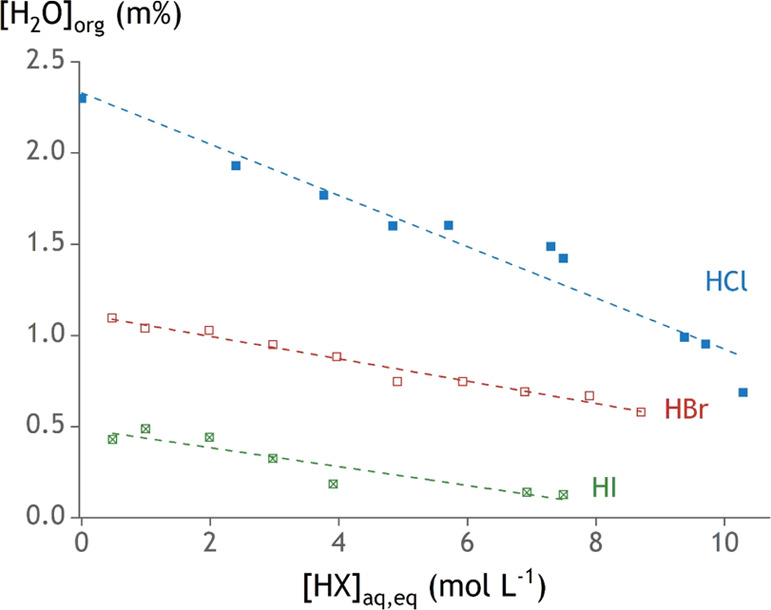
Water content in the organic phase containing 0.2 mol L–1 TOMACl/Br/I at increasing equilibrium aqueous acid content. Error bars have been omitted because the error on the measurements was less than 1%.
Extractants consisting of a quaternary ammonium cation with long alkyl chains and a counteranion typically organize into reverse micellar structures due to their surfactant-like properties.35−38 Most of the water in the organic phase is then found inside these reverse micelles. This can also explain the large amount of water that can be dissolved in organic phases with a high quaternary ammonium extractant concentration.39 It is now possible to come back to the unexpected trend in the distribution of acids to the organic phase (Figure 3). It was observed that the more hydrophilic HCl is distributed more to the organic phase than the more hydrophobic HBr. This can be explained by the presence of more water in the organic phase of those samples with HCl and TOMACl. The 0.2 mol L–1 TOMACl organic phase contains more water than the 0.2 mol L–1 TOMABr for the same acid content, suggesting the presence of more and/or larger reverse micelles. The interior of these reverse micelles has a chemical environment that is similar to that of the bulk aqueous phase and can thus accommodate a similar concentration of acid to that in the bulk aqueous phase. Of course, the micelles are microstructures for which surface effects cannot be neglected. Consequently, the content of the reverse micelles is not likely to be as homogeneous as that of the aqueous phase.
The decrease in the water content with the increasing aqueous acid concentration can thus be explained by two effects. First, an increasing acid concentration in the aqueous phase decreases the water activity. This leaves less free water in the system. Second, some water molecules might be replaced by acid in the reverse micelles when more acid is present in the system.
2.3. Raman Spectroscopy
The metal speciation can be used to interpret the origin of the differences in the extraction of zinc(II) and cadmium(II). In the literature, the speciation of zinc(II) and cadmium(II) halides has been studied by different experimental techniques, including potentiometry,24,25 vibrational40−46 and XAFS spectroscopy,47−49 and ab initio calculations.50−54 Some of these studies are helpful in determining the structure of the different species present in solution, but it is difficult to compare the speciation profiles of different metal halides with each other based solely on the preexisting literature. That is because different techniques, equipment, and analysis methods are used and not all data are available for every metal–halide combination. Most importantly, differences in ionic strength will result in different speciation profiles.
To avoid some of these issues and to fill any gaps in the speciation profiles of the metal–halide complexes, Raman spectra of zinc(II) and cadmium(II) were recorded in aqueous HCl, HBr, and HI media. The metal concentration was equal to those in the solvent extraction experiments. Zinc(II) or cadmium(II) salts with weakly coordinating anions (perchlorate and tetrafluoroborate) were added to enable analysis at zero concentration of halides and to use them as internal standards for a quantitative analysis. The raw recorded Raman spectra can be found in the Supporting Information, while the species found in the processed Raman spectra (Figure 5) are discussed below.
Figure 5.

Raman spectra of zinc(II) in aqueous HCl (A), HBr (B), and HI (C) and cadmium(II) in HCl (D) and HBr (E). The insets give an enlarged view of the peaks of the octahedral species.
The zinc(II)–chloride speciation in HCl media looks very similar to that in LiCl media, which has already been discussed extensively in our previous work.22 In water, in the absence of halides, one octahedral species is observed, with a Raman peak at 388 cm–1. This is the fully hydrated [Zn(H2O)6]2+ ion and this species is also encountered in the other zinc(II)–halide solutions. On the other end of the concentration range, one Raman peak is found at 280 cm–1. This is the tetrahedral [ZnCl4]2– complex. At intermediate HCl concentrations, only a shift in the tetrahedral peak could be observed. Most likely, this shift can be attributed to the formation of the [ZnCl3H2O]− species based on calculations by Pye et al.,53 but it cannot be entirely excluded that also some [ZnCl2(H2O)2] is formed.53
Different tetrahedral zinc(II)–bromide species can be distinguished easily with Raman spectroscopy, while only the octahedral [Zn(H2O)6]2+ is visible in the absence of added HBr.41−43 [ZnBr4]2– is found at 171 cm1, [ZnBr3H2O]− at 184 cm–1, and [ZnBr2(H2O)2] at 205 cm–1 when HBr is added.48,54 No Raman peak could be observed that could be assigned to the [ZnBr(H2O)5]+ species. At a higher temperature, such a peak was reported by Yang et al. and by Marley and Gaffney around 240 cm–1, albeit with a very low intensity.41,43
The Raman spectra of zinc(II) in HI media show a similar trend, but the tetrahedral Zn–iodide Raman peaks are even more separated. The positions of the peaks observed in this study agree well with those reported in the literature.44 Apart from the octahedral [Zn(H2O)6]2+ at 388 cm–1, three Raman peaks of tetrahedral complexes were observed: [ZnI4]2– at 173 and 121 cm–1, [ZnI3H2O]− at 138 cm–1, and [ZnI2(H2O)2] at 164 cm–1. Again, no octahedral iodidozinc(II) complex could be detected, although Wakita et al. report the presence of [ZnI(H2O)5]+.44
The cadmium(II)–halide speciation was investigated by Raman spectroscopy only in HCl and HBr media because of the stability issues with HI (vide supra) and the absence of cadmium(II) solvent extraction experiments in iodide media. The Raman peak of the fully hydrated cadmium(II) species [Cd(H2O)6]2+ is found between 356 and 360 cm–1. In both the HCl and HBr solutions, a shift in the position of the octahedral peak to lower wavenumbers was observed. This can be attributed to the formation of [CdCl(H2O)5]+ and [CdBr(H2O)5]+, respectively. After deconvolution (vide infra), peaks at 341 and 331 cm–1 are found for the monochlorido and monobromido species, respectively. These Raman peaks cannot be assigned with certainty due to a lack of literature data, but the structure of several cadmium(II) species was already obtained by various other experimental techniques.49,50 These structural data are not consistent concerning the geometry of [CdCl2(H2O)x]: Butterworth et al. found that it is octahedral, while Bazarkina et al. found that it is tetrahedral.49,50
The tetrahedral species of cadmium(II) in HCl and HBr are Raman active. In HCl solution, a change in the Raman shift of the tetrahedral peak indicates the presence of more than one tetrahedral species. [CdCl4]2– was identified at 260 cm–1, as was also done by Davies et al.45 The shift to lower wavenumbers at intermediate HCl concentrations indicates the formation of [CdCl3H2O]− and/or [CdCl2(H2O)x] if the latter is also tetrahedral. However, it is impossible to say which species cause which Raman shift without the deconvolution of all spectra. In HBr media, the Raman peak of the tetrahedral species does not shift with changing HBr concentration. This strongly suggests that only one single tetrahedral species is formed at 162 cm–1: [CdBr4]2–. This position for the Raman peak of [CdBr4]2– is also found in the literature.45,46
A change in speciation of the metal complexes in the organic phase might also significantly influence their extraction. For the zinc(II) system, the literature data show that [ZnCl4]2– is always formed, but no relevant data could be found for the cadmium(II) system.30,31 To check whether the speciation of the cadmium changes in the organic phase, Raman spectra were recorded for CdBr2 (0.05 mol L–1) in 0.2 mol L–1 TOMABr in contact with aqueous solutions with different HBr concentrations (Figure 6). The baseline of the Raman spectra is rather noisy due to the short measurement times, which were necessary to limit the high background signal of the organic phase. Nevertheless, the position of the peak of cadmium(II) in TOMABr is identical for the extractions from both 0.52 and 4.86 mol L–1 HBr. This strongly indicates a constant speciation of cadmium(II) in the organic phase over the whole aqueous acid concentration ranges employed in this study. Both peaks are found at 159 cm–1, which is close to the peak of [CdBr4]2– in the aqueous phase. This small difference between the shift in the organic and aqueous phase can be explained by the change of solvent.
Figure 6.
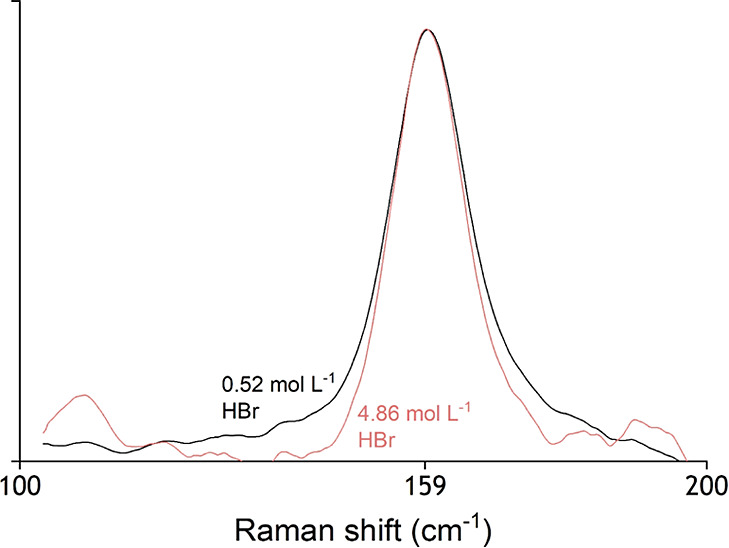
Details of the Raman spectra of 0.05 mol L–1 cadmium(II) in 0.2 mol L–1 TOMABr in toluene after extraction from 0.52 or 4.86 mol L–1 HBr.
2.4. Speciation Profiles
It is easier to compare the speciation of zinc(II) and cadmium(II) from different halide solutions using speciation profiles instead of Raman spectra. Therefore, the mole fraction (x) of each metal species (eq 5) was determined as a function of the acid concentration using the spectra in Figure 5.
| 5 |
Here, n is the number of moles and the summation in the denominator goes over every metal–halide species in solutions.
The spectra were deconvoluted with a Voigt function to obtain the best fit for liquid samples.55 An average peak center, Gaussian width, and Lorentzian width were taken for one Raman signal over the whole concentration range of an acid. This resulted in the most accurate calculation of all speciation profiles, which are shown in Figure 7.
Figure 7.

Speciation profiles of zinc(II) and cadmium(II) in aqueous HCl, HBr, and HI. Water in the first coordination sphere was omitted in the labels to increase readability.
Zinc(II) coordinates the most favorably with chloride, followed by bromide and finally iodide. This series agrees with the prediction of the HSAB concept.56 Zinc(II) is a borderline hard Lewis acid, which bonds strongly to the intermediately hard Lewis base chloride. Therefore, water molecules (hard Lewis bases according to HSAB coordinated to zinc(II) are rather easily exchanged for chloride anions. The bromide is a softer Lewis base, which explains its relatively lower bond strength with zinc(II) and more difficult exchange with water. Iodide is even softer than bromide due to its even larger ionic radius and hence larger polarizability.
The speciation profile of zinc(II) in HCl solution is shown in Figure 7, top left. A maximum mole fraction of the zinc(II) species with an intermediate number of bound chlorides is already found at 1.5 mol L–1 HCl. Only [ZnCl3H2O]− is considered in the speciation profile because the Raman spectra and the literature data indicate that only this complex is stable (vide supra).53
For zinc(II) in HBr solution (Figure 7, middle left), more fully hydrated zinc(II) is found at low HBr concentrations, and some [ZnBr(H2O)5]+ is formed. The latter was not detected in the Raman spectra, but it was incorporated to get a correct mass balance of zinc(II) in all HBr solutions. The absence of a Raman peak from [ZnBr(H2O)5]+ can be explained by a very low quantity and insensitivity of the Raman effect of that species. The intermediate species [ZnBr2(H2O)2] has a maximum mole fraction at 2.0 mol L–1 HBr. Between 2.5 and 3.0 mol L–1 HBr, the maximum of [ZnBr3H2O]− is found, and all zinc(II) is coordinated to four bromide anions above an HBr concentration between 5.0 and 8.25 mol L–1.
In HI media (Figure 7, bottom left), zinc(II) starts to coordinate with iodide only at HI concentrations higher than 0.5 mol L–1. [ZnI(H2O)5]+ is incorporated in the speciation profile, despite its absence in the Raman spectra, to fulfill the mass balance in all samples. [ZnI2(H2O)2] is maximally formed at 2.5 mol L–1, the maximum of [ZnI3H2O]− is formed between 3.0 and 4.0 mol L–1 HI, and [ZnI4]2– is formed at even higher HI concentrations. All zinc(II) ions are coordinated to four iodide ions above an HI concentration between 5.0 and 7.2 mol L–1.
Cadmium(II) and zinc(II) are in the same group in the periodic table (group 12 according to the IUPAC rules), resulting in similar chemical properties. However, cadmium(II) is found one row below zinc(II), which explains its larger ionic radius. This makes cadmium(II) a soft Lewis acid according to the HSAB concept. As a result, cadmium(II) bonds more strongly to softer Lewis bases such as bromide than to hard Lewis bases such as chloride or water. This opposite trend compared to zinc(II) is also observed in the speciation profiles (Figure 7). In general, halides coordinate at a lower halide concentration to cadmium(II) than to zinc(II) because the hard Lewis base water is more easily exchanged for the softer halides in cadmium(II) complexes.
In HCl media (Figure 7, top right), the intermediate [CdCl2(H2O)2] is maximally formed at 0.50 mol L–1 HCl, while full conversion into [CdCl4]2– is observed at 5.0 mol L–1 HCl. Note here that [CdCl2(H2O)2] and [CdCl3H2O]− could not be designated to clearly separated Raman peaks. The final mole fraction of both species was eventually obtained by taking into account the area and the Raman shift of all peaks in all spectra. In HBr media (Figure 7, bottom right), only [Cd(H2O)6]2+, [CdBr(H2O)5]+, and [CdBr4]2– are formed (vide supra). The maximum amount of [CdBr(H2O)5]+ was formed below 0.1 mol L–1 HBr, and all octahedral cadmium(II) species disappeared at 2.0 mol L–1 HBr. Above this concentration, [CdBr4]2– was the sole species present.
3. Discussion
The extent to which a metal is extracted by a basic extractant is strongly influenced by the speciation of the metal in the aqueous phase: metals are extracted more efficiently when they are less stabilized in the aqueous phase.1 Decreasing the stabilization of a metal in the aqueous phase means decreasing its effective hydration. This can be obtained by reducing the charge density of the metal cation by complexation with anions.57 Coordination of an increasing number of anions increases the size of the metal complex and decreases its charge until the positive charge of the metal cation is completely compensated by the charge of the coordinated anions, and an electrically neutral species is formed. Coordination of more anions will again increase the charge of the metal complex (with the formation of anionic complexes), which again leads to an increased charge density and hydration. This means a minimum in metal complex hydration is obtained at an intermediate acid or salt concentration where the metal complex with the lowest charge density is maximally formed.21
Note that the decrease in extraction efficiency cannot be attributed to competition with the extraction of HX2– at high HX concentrations. In an earlier publication, we reported that the association between HCl2– and TOMA is unlikely and clearly demonstrated the absence of evidence for competition between the extraction of Co(II) and HCl by TOMACl.21
The choice of acid or salt influences not only the extraction efficiency of a metal ion via complexation but also the amount of water available to hydrate the metal complexes, as the salt or acid ions are hydrated themselves.22,58 Salt cations or anions that are more strongly hydrated reduce the number of free water molecules that can stabilize the metal complexes and hence enhance the extraction of metals. This can be considered a salting-out effect. This explanation of the effects of the aqueous phase composition on the extraction by basic extractants is largely based on studies with chloride salts with different cations or HCl. A further, more encompassing investigation is obtained when also looking at different salt or acid anions.
The combined effect of these two phenomena (metal speciation and amount of free water molecules) is the determining factor to explain differences in the extraction of metals by a basic extractant at different acid or salt types and concentrations, that is, if the speciation of the metal in the organic phase is constant. Literature data support a constant speciation of zinc(II) in the organic phase.30,31 For cadmium(II), a constant speciation in the organic phase is also observed (Figure 6). The complexes of both metals coordinate with any additional halides from the extractant to form tetrahalo complexes. These complexes are then stabilized in the organic phase by electrostatic interaction with the TOMA cation in their outer coordination sphere.3
A correlation is observed between the acid concentration at which the zinc(II) species with the lowest charge density is formed, and the extraction maximum is observed. Figure 8 combines the relevant aspects from the extraction experiments (Figure 1) and the speciation profiles (Figure 7) for zinc(II). Of all tested halides, chloride forms the most stable complexes with zinc(II). The maxima of the corresponding speciation curve and DZn curve in Figure 8 are hence found at lower concentrations of HX compared to the other systems. The speciation maxima of [ZnI2(H2O)2] and the extraction maximum in iodide media are found at higher concentrations of HX because of the weaker coordination of iodide to zinc(II).
Figure 8.
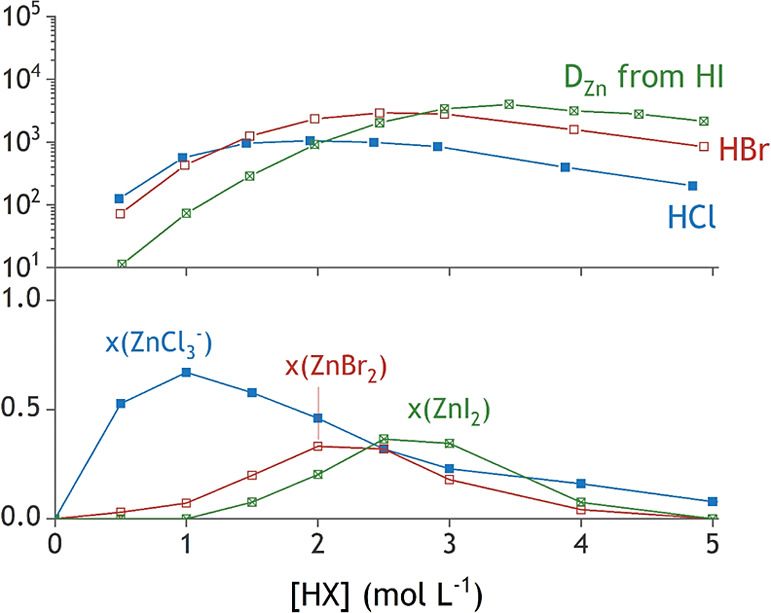
DZn (top) and the formation of the zinc(II)–halide species with the lowest charge density in the aqueous phase. The maxima coincide quite well in all tested halide systems.
In the cadmium(II) system, similar trends are observed: the halide that coordinates stronger to cadmium(II) also results in an extraction maximum at a lower halide concentration. Note here that the order of the halide series is reversed (Figure 9). The softer Lewis base bromide coordinates more strongly to the softer acid cadmium(II). This explains the maximum of DCd at 1.5 mol L–1 in bromide media compared with 2.0 mol L–1 in chloride media.
Figure 9.
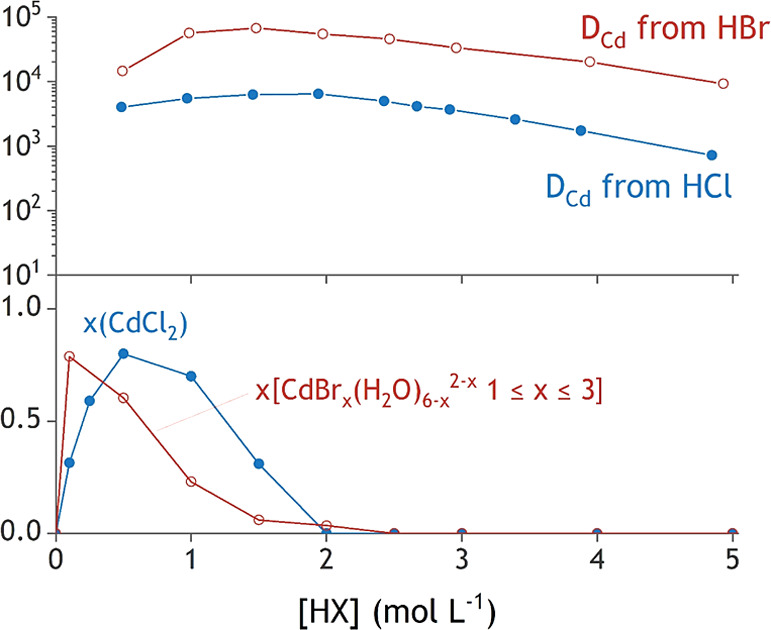
DCd (top) and the formation of the cadmium(II)–halide species with the lowest charge density in the aqueous phase.
The maximum DZn value for each halide system increases from HCl to HI media. This is in spite of the stronger zinc(II)–chloride bonds as compared to the zinc(II)–iodide bond, which could be expected to promote the formation of the tetrachloro species in the organic phase. The same trend of increasing maximum DCd value with the increasing halide ion radius is observed. For cadmium(II), this trend coincides with the increase in strength of the cadmium(II)–bromide bond as compared to that of the cadmium(II)–chloride bond. Thus, more hydrophobic anions, such as iodide, promote the extraction of metals, regardless of the differences in strength of the metal–anion bond. Two reasons can be given for this observation. First, less water is found in the organic phase containing an extractant with a more hydrophobic anion (Figure 4). Consequently, there is less water in the organic phase that can destabilize the extracted metal complexes associated with the extractant, which enhances its extraction.39 Second, the larger halides will result in larger metal–halide complexes, in which the charge of the metal complex is more delocalized. Moreover, these bonds are less polarized due to a smaller difference in electronegativity between the halide and the metal. This reduces the hydration of the metal–halide complex, which destabilizes the metal–halide complex in the aqueous phase and enhances its extraction.58
A mismatch between the halide concentration of the position of the maximum in DCd and the halide concentration at which the maximum amount of the cadmium(II) species with the lowest charge density is found. The maximum in the DCd curve is shifted to a considerably higher halide concentration compared to the maximum in the speciation curve of Figure 9. This shift is also observed for the zinc(II) systems but significantly less pronounced. This observation can be attributed to a second effect that influences the extraction, apart from the speciation of the metal in solution. All employed acids also induce salting-out of the metal complexes, resulting in an increased extraction at higher acid content. This results from hydration of the acid itself, leaving fewer free water molecules to hydrate and stabilize the metal complexes in the aqueous phase. This effect is most pronounced for the cadmium(II) extraction systems. Here, the maximum DCd is found at a very low halide concentration where a small absolute increase in acid concentration results in a larger relative increase of acid concentration than at higher absolute acid concentration. The larger relative increase in acid concentration results in a relatively larger increase in salting-out strength.
In general, cadmium(II) is more efficiently extracted than zinc(II) because of its stronger coordination with halide ions. This results not only in the formation of cadmium(II)–halide complexes in the aqueous phase at a lower acid concentration but also in a stronger tendency to form tetrahalo complexes when associating with the TOMA cation.
4. Conclusions
The differences in the solvent extraction of zinc(II) and cadmium(II) in different halide media by trioctylmethylammonium halides (TOMAX, where X = Cl, Br, or I) were investigated by looking at the aqueous and organic metal speciation, acid distribution, and water content in the organic phase. The extraction of zinc(II) and cadmium(II) by TOMAX occurs most efficiently at an anion concentration at which the zinc(II) or cadmium(II) species with the lowest charge density predominate. This observation further supports the hypothesis that the hydration of the metal complex in the aqueous phase plays a predominant role in the extraction of metals by basic extractants. The extraction of a metal is promoted by lowering its charge density via coordination of the right number of anions with a low charge density. Other positive effects on the extraction efficiency for a given extractant type and concentration include decreasing the water content of the organic phase. The use of different anions and the focus on hard–soft interaction differences allowed an in-depth investigation of the mechanism of solvent extraction by basic extractants. These new insights might be of interest in the development and optimization of separation routes for metals, especially for zinc(II) and cadmium(II) in different halide media, for which speciation profiles can now be easily compared.
5. Experimental Section
5.1. Chemicals
HNO3 (65 wt %), NaCl (99.99%), HCl (∼37 wt %), and toluene (>99.8%) were purchased from VWR (Leuven, Belgium). Zn(ClO4)2·6H2O (99.7%), Zn(BF4)2·xH2O (18.2 wt % Zn), ZnI2 (>98%), trioctylamine (TOA) (98%), and HBr (48 wt %) were purchased from Sigma-Aldrich (Overijse, Belgium). The aqueous nickel, cadmium, zinc standards (1000 mg L–1 in 2–5% HNO3), and ZnCl2 (>98%) were obtained from Chem Lab (Zedelgem, Belgium). TOMACl (98%) was purchased from J&K Scientific (Lommel, Belgium). CdCl2 (99%) and CdBr2·4H2O (98%) were purchased from Acros Organics (Geel, Belgium). HI (stabilized with 1.5% H3PO2, 57 wt %) and Cd(ClO4)2·6H2O (98%) were purchased from Alfa Aesar (Heysham, United Kingdom). ZnI2 (>98%) was purchased from Fluka Honeywell (Leuven, Belgium). Water was always of ultrapure quality, deionized to a conductivity of less than 0.055 μS cm–1 (298.15 K) with a Merck Millipore Milli-Q Reference A+ system. All chemicals were used as received, without any further purification. The bromide and iodide forms of TOMACl (TOMABr and TOMAI, respectively) were synthesized from TOMACl. Details on the synthesis of these compounds can be found in the Supporting Information. Caution! Cadmium salts are highly toxic with both acute and long-term health hazards. Contact with skin, inhalation, or ingestion should be absolutely avoided by wearing the appropriate PPE and by working in a fume cabinet while handling solids to avoid dust inhalation.
5.2. Metal-Free Solvent Extractions
Solvent extraction experiments were performed without the addition of an extractable metal ion in order to measure the water and acid uptake in the organic phase. These experiments were conducted with 5.0 mL of each phase in 20 mL glass vials. The aqueous phases containing HCl, HBr, or HI were created by adding an aliquot of a concentrated acid (HCl, HBr, or HI) to an appropriate volume of water. TOMACl, TOMABr, or TOMAI was diluted with toluene to create the organic phase with an extractant concentration of 0.2 mol L–1. The anion of the extractants was matched with the anion in the aqueous phase to allow an unambiguous investigation of the different anion systems. The vials were shaken for 30 min at 250 rpm at room temperature with a Kuhner ES-X shaker. Phase disengagement was facilitated by centrifugation for 2 min at 2000 rpm in an Eppendorf 5804 centrifuge.
The water content in the organic phases was measured using a volumetric Karl Fischer titrator (Mettler-Toledo model V30S). Water from samples was introduced to the titrator via a Stromboli oven sample changer (Mettler-Toledo). This was done to avoid the introduction of interfering solutes in the titration vessel. Acids must be excluded from samples to be measured by the Karl Fischer method. Therefore, the acid in the organic phase was neutralized with TOA prior to the Karl Fischer titration. This was done by adding an excess of TOA (0.4 mL) to 2 g of the sample. TOA was chosen as the base as its reaction products with acids are soluble in toluene. In addition, the water content of pure TOA was determined in order to calculate the correct water content in the samples. The Stromboli oven was set to 150 °C to ensure that all water in the samples evaporated while avoiding decomposition of the TOMAX and TOAHX with the release of volatile HX or CH3X (X is Cl–, Br–, or I–).59
The acid concentration in the organic phase was determined based on the density difference of the aqueous phase before and after contact with the organic phase. The density–concentration conversion for the acids was based on the measured density of the aqueous starting solutions and the volumetric preparation of the starting solutions from acid stock solutions with known concentrations. Densities of the solutions were measured with an Anton Paar DMA 4500M densitometer. This approach is justified due to the negligible solubility of TOMAX and toluene in the aqueous phase.60
5.3. Zinc(II) and Cadmium (II) Solvent Extractions
Single-element solvent extractions with zinc(II) or cadmium(II) were performed with 4.0 mL of each phase in 20 mL glass vials. The metal concentration in the aqueous phase was kept constant by adding a fixed volume of an aqueous metal stock solution [1.0 mol L–1 zinc(II) or cadmium(II) in water] to an aliquot of a concentrated acid (HCl, HBr, or HI) and water. The final concentration of zinc(II) or cadmium(II) in the aqueous samples was 0.05 mol L–1. The anion of the added metal halide was matched with the anion of the acid to avoid interference of different anions in the system. To avoid hydrolytic precipitation of zinc(II) or cadmium(II), 0.05 mol L–1 HCl, HBr, or HI was added to the zinc(II) and cadmium(II) stock solutions.
TOMACl, TOMABr, or TOMAI was diluted with toluene to create an organic phase with a concentration of 0.2 mol L–1 extractant. Again, the anion of the extractant was matched with the anion in the aqueous phase to allow an unambiguous investigation of the different anion systems. Before the actual solvent extractions, the organic phases were equilibrated with aqueous solutions with the same acid concentration. This minimizes acid concentration, water content, and volume changes during extraction. The extraction of metals was quantified by the distribution ratio of the metal M (DM)
| 6 |
where cM,aq and cM,org are the equilibrium metal concentrations in the aqueous and organic phase after extraction, respectively. DM was preferred over the percentage extraction as it represents the best underlying physical chemistry.1
All extractions were performed by shaking at 250 rpm for 30 min at room temperature, and phase disengagement was accomplished by centrifugation for 2 min at 2000 rpm. The metal concentrations in the aqueous phase before and after extraction were measured using ICP-OES with a Perkin Elmer Avio 500 spectrometer equipped with an axial/radial dual plasma view, a GemCone High Solids nebulizer, a baffled cyclonic spray chamber, and a demountable quartz torch with a 2.0 mm internal diameter alumina injector. Samples, calibration solutions, and quality control solutions were diluted with HNO3 (2 vol %), except for the extractions in HI media. These ICP-OES samples, calibration, and quality controls were diluted in HCl (2 vol %) to avoid a redox reaction between HNO3 and HI. All ICP-OES spectra were measured in triplicate. Calibration curves were made using solutions of 0.1, 1, and 10 mg L–1 of the corresponding metal, prepared from a standard solution. Quality checks were performed with 5 mg L–1 metal solutions. Ni(II) (5 mg L–1) was added to all samples, calibration solutions, and quality controls and applied as internal standards only if the quality checks failed because of matrix effects. The concentration of metal in the organic phase was calculated from the mass balance. No precipitation of third phase formation was found in the period between the experiments and the measurements. The experimental error in the metal concentration in the aqueous phase was calculated based on triplicate measurements and was less than 5%. This results only in a small error on the calculated DM, hence error bars on the graphs were omitted to increase legibility.
5.4. Raman Measurements
Raman spectra of 0.05 mol L–1 aqueous zinc(II) or cadmium(II) solutions with different HCl, HBr, or HI concentrations were recorded using a confocal Horiba Jobin Yvon LabRam HR evolution Raman microscope. The spectrometer is equipped with an 1800 grooves/mm holographic grating with a confocal attachment, a Peltier-cooled electron-multiplying CCD (SIN-EM FIVIS) for detection, and an Olympus BX41 microscope. For excitation, a green line (λ = 532.0 nm) from a solid-state laser (LAS-532-100-HREV) operating at 14 mW was employed. Raman spectra of liquid samples were collected at room temperature in quartz cuvettes (4 mL) using a ×10_VIS_LWD microscope objective to focus the laser beam at the center of the cuvette. The perchlorate salts of zinc(II) and cadmium(II) were used to avoid complexation with halides in the solutions with no or little added acid. Zn(BF4)2 replaced Zn(ClO4)2 in the samples with HI to avoid a redox reaction between perchlorate and iodide.
Blank solutions were measured, and their spectra were subtracted from the spectra of the zinc(II)-/cadmium(II)-containing samples to correct for the background signal. The blank solutions had the same composition as the samples, except for the presence of zinc(II) or cadmium(II). The spectra were processed with LabSpec6 software from Horiba and Origin 2018b software from OriginLab. The processed spectra contained a significant amount of noise due to the low intensity of the Raman signal of the measured zinc(II) and cadmium(II) complexes. Consequently, the processed spectra were smoothed using an adjacent-averaging method with a 10-point window, and any remaining background was subtracted with a baseline correction. The perchlorate peak at 462 cm–1 or the tetrafluoroborate peak at 767 cm–1 was used as the internal standard to normalize all spectra.
Acknowledgments
The authors thank the FWO Flanders (project G0B6918N and I000718N) for financial support. The research was supported by the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programme: Grant Agreement 694078—Solvometallurgy for critical metals (SOLCRIMET). The contents of this publication are the sole responsibility of the authors and do not necessarily reflect the opinion of the European Union.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c03790.
TOMACl metathesis, visual observation of TOMAI conversion into TOMAI3, raw Raman spectra of zinc(II) and cadmium(II), 13C NMR images of TOMAI organic phase, and UV–vis absorption quantification of TOMAI conversion into TOMAI3 (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Rydberg J.Solvent Extraction Principles and Practice, Revised and Expanded, 2nd ed.; Marcel Dekker: New York, 2004. [Google Scholar]
- Kentish S.; Stevens G. W. Innovations in Separations Technology for the Recycling and Re-Use of Liquid Waste Streams. Chem. Eng. J. 2001, 84, 149–159. 10.1016/S1385-8947(01)00199-1. [DOI] [Google Scholar]
- Wilson A. M.; Bailey P. J.; Tasker P. A.; Turkington J. R.; Grant R. A.; Love J. B. Solvent Extraction: The Coordination Chemistry behind Extractive Metallurgy. Chem. Soc. Rev. 2014, 43, 123–134. 10.1039/C3CS60275C. [DOI] [PubMed] [Google Scholar]
- Uchikoshi M. Determination of the Distribution of Cobalt-Chloro Complexes in Hydrochloric Acid Solutions at 298 K. J. Solution Chem. 2018, 47, 2021–2038. 10.1007/s10953-018-0831-z. [DOI] [Google Scholar]
- Lovering K.; Nayak S.; Bu W.; Uysal A. The Role of Specific Ion Effects in Ion Transport: The Case of Nitrate and Thiocyanate. J. Phys. Chem. C 2020, 124, 573–581. 10.1021/acs.jpcc.9b09288. [DOI] [Google Scholar]
- Sun P.; Huang K.; Liu H. The Nature of Salt Effect in Enhancing the Extraction of Rare Earths by Non-Functional Ionic Liquids: Synergism of Salt Anion Complexation and Hofmeister Bias. J. Colloid Interface Sci. 2019, 539, 214–222. 10.1016/j.jcis.2018.12.058. [DOI] [PubMed] [Google Scholar]
- Nayak S.; Lovering K.; Uysal A. Ion-Specific Clustering of Metal–Amphiphile Complexes in Rare Earth Separations. Nanoscale 2020, 12, 20202–20210. 10.1039/D0NR04231E. [DOI] [PubMed] [Google Scholar]
- Li Z.; Binnemans K. Hydration Counteracts the Separation of Lanthanides by Solvent Extraction. AIChE J. 2020, 66, e16545 10.1002/aic.16545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onghena B.; Papagni E.; Souza E. R.; Banerjee D.; Binnemans K.; Vander Hoogerstraete T. Speciation of Lanthanide Ions in the Organic Phase after Extraction from Nitrate Media by Basic Extractants. RSC Adv. 2018, 8, 32044–32054. 10.1039/C8RA06712K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Hoogerstraete T.; Souza E. R.; Onghena B.; Banerjee D.; Binnemans K. Mechanism for Solvent Extraction of Lanthanides from Chloride Media by Basic Extractants. J. Solution Chem. 2018, 47, 1351–1372. 10.1007/s10953-018-0782-4. [DOI] [Google Scholar]
- Deferm C.; Onghena B.; Vander Hoogerstraete T.; Banerjee D.; Luyten J.; Oosterhof H.; Fransaer J.; Binnemans K. Speciation of Indium(III) Chloro Complexes in the Solvent Extraction Process from Chloride Aqueous Solutions to Ionic Liquids. Dalton Trans. 2017, 46, 4412–4421. 10.1039/C7DT00618G. [DOI] [PubMed] [Google Scholar]
- du Preez J. G. H. Recent Advances in Amines as Separating Agents for Metal Ions. Solvent Extr. Ion Exch. 2000, 18, 679–701. 10.1080/07366290008934703. [DOI] [Google Scholar]
- Cui L.; Cheng F.; Zhou J. Behaviors and Mechanism of Iron Extraction from Chloride Solutions Using Undiluted Cyphos IL 101. Ind. Eng. Chem. Res. 2015, 54, 7534–7542. 10.1021/acs.iecr.5b01546. [DOI] [Google Scholar]
- Xiong Y.; Kuang W.; Zhao J.; Liu H. Ionic Liquid-Based Synergistic Extraction of Rare Earths Nitrates without Diluent: Typical Ion-Association Mechanism. Sep. Purif. Technol. 2017, 179, 349–356. 10.1016/j.seppur.2017.02.026. [DOI] [Google Scholar]
- Coleman C. F.; Brown K. B.; Moore J. G.; Crouse D. J. Solvent Extraction with Alkyl Amines. Ind. Eng. Chem. 1958, 50, 1756–1762. 10.1021/ie50588a032. [DOI] [Google Scholar]
- Castillo J.; Coll M. T.; Fortuny A.; Navarro Donoso P.; Sepúlveda R.; Sastre A. M. Cu(II) Extraction Using Quaternary Ammonium and Quaternary Phosphonium Based Ionic Liquid. Hydrometallurgy 2014, 141, 89–96. 10.1016/j.hydromet.2013.11.001. [DOI] [Google Scholar]
- El-Nadi Y. A. Solvent Extraction and Its Applications on Ore Processing and Recovery of Metals: Classical Approach. Sep. Purif. Rev. 2017, 46, 195–215. 10.1080/15422119.2016.1240085. [DOI] [Google Scholar]
- Seeley F. G.; Crouse D. J. Extraction of Metals from Chloride Solutions and Amines. J. Chem. Eng. Data 1966, 11, 424–429. 10.1021/je60030a043. [DOI] [Google Scholar]
- Gupta C. K.; Krishnamurthy N. Extractive Metallurgy of Rare Earths. Int. Met. Rev. 1992, 37, 197–248. 10.1179/imr.1992.37.1.197. [DOI] [Google Scholar]
- Zhang N.; Brugger J.; Etschmann B.; Ngothai Y.; Zeng D. Thermodynamic Modeling of Poorly Complexing Metals in Concentrated Electrolyte Solutions: An X-Ray Absorption and UV-Vis Spectroscopic Study of Ni(II) in the NiCl2-MgCl2-H2O System. PLoS One 2015, 10, e0119805 10.1371/journal.pone.0119805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lommelen R.; Vander Hoogerstraete T.; Onghena B.; Billard I.; Binnemans K. Model for Metal Extraction from Chloride Media with Basic Extractants: A Coordination Chemistry Approach. Inorg. Chem. 2019, 58, 12289–12301. 10.1021/acs.inorgchem.9b01782. [DOI] [PubMed] [Google Scholar]
- Lommelen R.; Onghena B.; Binnemans K. Cation Effect of Chloride Salting Agents on Transition Metal Ion Hydration and Solvent Extraction by the Basic Extractant Methyltrioctylammonium Chloride. Inorg. Chem. 2020, 59, 13442–13452. 10.1021/acs.inorgchem.0c01821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vereycken W.; Riaño S.; Gerven T. V.; Binnemans K. Extraction Behavior and Separation of Precious and Base Metals from Chloride, Bromide, and Iodide Media Using Undiluted Halide Ionic Liquids. ACS Sustainable Chem. Eng. 2020, 8, 8223–8234. 10.1021/acssuschemeng.0c01181. [DOI] [Google Scholar]
- Powell K. J.; Brown P. L.; Byrne R. H.; Gajda T.; Hefter G.; Leuz A.-K.; Sjöberg S.; Wanner H. Chemical Speciation of Environmentally Significant Metals with Inorganic Ligands. Part 5: The Zn2+ + OH–, Cl–, CO32–, SO42–, and PO43– Systems. Pure Appl. Chem. 2013, 85, 2249–2311. 10.1351/pac-rep-13-06-03. [DOI] [Google Scholar]
- Powell K. J.; Brown P. L.; Byrne R. H.; Gajda T.; Hefter G.; Leuz A.-K.; Sjöberg S.; Wanner H. Chemical Speciation of Environmentally Significant Metals with Inorganic Ligands. Part 4: The Cd2+ + OH–, Cl–, CO32–, SO42–, and PO43– Systems. Pure Appl. Chem. 2011, 83, 1163–1214. 10.1351/PAC-REP-10-08-09. [DOI] [Google Scholar]
- Larsson K.; Binnemans K. Separation of Rare Earths by Split-Anion Extraction. Hydrometallurgy 2015, 156, 206–214. 10.1016/j.hydromet.2015.04.020. [DOI] [Google Scholar]
- Husa W. J.; Shattuck P. S. The Effect of Various Iodides upon the Stability of Hydriodic Acid. J. Am. Pharm. Assoc. 1932, 21, 114–121. 10.1002/jps.3080210205. [DOI] [Google Scholar]
- Van den Bossche A.; De Witte E.; Dehaen W.; Binnemans K. Trihalide Ionic Liquids as Non-Volatile Oxidizing Solvents for Metals. Green Chem. 2018, 20, 3327–3338. 10.1039/C8GC01061G. [DOI] [Google Scholar]
- Li X.; Li Z.; Orefice M.; Binnemans K. Metal Recovery from Spent Samarium–Cobalt Magnets Using a Trichloride Ionic Liquid. ACS Sustainable Chem. Eng. 2019, 7, 2578–2584. 10.1021/acssuschemeng.8b05604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo V.; Navarro R.; Saucedo I.; Ávila M.; Guibal E. Zinc(II) Extraction from Hydrochloric Acid Solutions Using Amberlite XAD-7 Impregnated with Cyphos IL 101 (Tetradecyl(Trihexyl)Phosphonium Chloride). Sep. Sci. Technol. 2008, 43, 2434–2459. 10.1080/01496390802119002. [DOI] [Google Scholar]
- Li X.; Van den Bossche A.; Vander Hoogerstraete T.; Binnemans K. Ionic Liquids with Trichloride Anions for Oxidative Dissolution of Metals and Alloys. Chem. Commun. 2018, 54, 475–478. 10.1039/C7CC08645H. [DOI] [PubMed] [Google Scholar]
- Dupont D.; Depuydt D.; Binnemans K. Overview of the Effect of Salts on Biphasic Ionic Liquid/Water Solvent Extraction Systems: Anion Exchange, Mutual Solubility, and Thermomorphic Properties. J. Phys. Chem. B 2015, 119, 6747–6757. 10.1021/acs.jpcb.5b02980. [DOI] [PubMed] [Google Scholar]
- Kislik V. S.Chapter 2—Principles of Solvent Extraction of Organic and Mineral Acids. In Solvent Extraction; Kislik V. S., Ed.; Elsevier: Amsterdam, 2012; pp 69–111. [Google Scholar]
- Van den Bossche A.; Vereycken W.; Vander Hoogerstraete T.; Dehaen W.; Binnemans K. Recovery of Gallium, Indium, and Arsenic from Semiconductors Using Tribromide Ionic Liquids. ACS Sustainable Chem. Eng. 2019, 7, 14451–14459. 10.1021/acssuschemeng.9b01724. [DOI] [Google Scholar]
- Berkovich Y.; Garti N. Catalytic Colloidal Pd Dispersions in Water-Organic Solutions of Quaternary Ammonium Salt. Colloids Surf., A 1997, 128, 91–99. 10.1016/S0927-7757(96)03911-8. [DOI] [Google Scholar]
- Reddy T. R.; Meeravali N. N.; Reddy A. V. R. Reverse Micelle Mediated Bulk Liquid Membrane Separation of Platinum Gold and Silver From Real Samples. Sep. Sci. Technol. 2013, 48, 1859–1866. 10.1080/01496395.2013.763708. [DOI] [Google Scholar]
- Reddy T. R.; Meeravali N. N.; Reddy A. V. R. Novel Reverse Mixed Micelle Mediated Transport of Platinum and Palladium through a Bulk Liquid Membrane from Real Samples. Sep. Purif. Technol. 2013, 103, 71–77. 10.1016/j.seppur.2012.10.025. [DOI] [Google Scholar]
- Bressler E.; Braun S. Separation Mechanisms of Citric and Itaconic Acids by Water-Immiscible Amines. J. Chem. Technol. Biotechnol. 1999, 74, 891–896. . [DOI] [Google Scholar]
- Lommelen R.; Binnemans K. Thermodynamic Modeling of Salting Effects in Solvent Extraction of Cobalt(II) from Chloride Media by the Basic Extractant Methyltrioctylammonium Chloride. ACS Omega 2021, 6, 11355–11366. 10.1021/acsomega.1c00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irish D. E.; McCarroll B.; Young T. F. Raman Study of Zinc Chloride Solutions. J. Chem. Phys. 1963, 39, 3436–3444. 10.1063/1.1734212. [DOI] [Google Scholar]
- Marley N. A.; Gaffney J. S. Laser Raman Spectral Determination of Zinc Halide Complexes in Aqueous Solutions as a Function of Temperature and Pressure. Appl. Spectrosc. 1990, 44, 469–476. 10.1366/0003702904086128. [DOI] [Google Scholar]
- Morris D. F. C.; Short E. L.; Waters D. N. Zinc Chloride and Zinc Bromide Complexes—III: Structures of Species in Solution. J. Inorg. Nucl. Chem. 1963, 25, 975–983. 10.1016/0022-1902(63)80031-7. [DOI] [Google Scholar]
- Yang M. M.; Crerar D. A.; Irish D. E. Raman Spectral Studies of Aqueous Zinc Bromide Solutions to 300°C at Pressures of 9 MPa. J. Solution Chem. 1988, 17, 751–762. 10.1007/BF00647420. [DOI] [Google Scholar]
- Wakita H.; Johansson G.; Sandström M.; Goggin P. L.; Ohtaki H. Structure Determination of Zinc Iodide Complexes Formed in Aqueous Solution. J. Solution Chem. 1991, 20, 643–668. 10.1007/BF00650714. [DOI] [Google Scholar]
- Davies J. E. D.; Long D. A. The Vibrational Spectra of the Halides and Complex Halides of the Group IIB Elements. Part I. The Vibrational Spectra of Cdl3–, CdBr2, CdBr3–, CdCl2, CdCl3–, CdCl42–, and MgCl42–. J. Chem. Soc. A 1968, 2054–2058. 10.1039/J19680002054. [DOI] [Google Scholar]
- Macklin J. W.; Plane R. A. Raman Study of Stepwise Formation of Bromide Complexes of Zinc, Cadmium, and Mercury in Aqueous Solution. Inorg. Chem. 1970, 9, 821–827. 10.1021/ic50086a026. [DOI] [Google Scholar]
- D’Angelo P.; Zitolo A.; Ceccacci F.; Caminiti R.; Aquilanti G. Structural Characterization of Zinc(II) Chloride in Aqueous Solution and in the Protic Ionic Liquid Ethyl Ammonium Nitrate by x-Ray Absorption Spectroscopy. J. Chem. Phys. 2011, 135, 154509. 10.1063/1.3653939. [DOI] [PubMed] [Google Scholar]
- Liu W.; Borg S.; Etschmann B.; Mei Y.; Brugger J. An XAS Study of Speciation and Thermodynamic Properties of Aqueous Zinc Bromide Complexes at 25–150°C. Chem. Geol. 2012, 298–299, 57–69. 10.1016/j.chemgeo.2011.12.018. [DOI] [Google Scholar]
- Bazarkina E. F.; Pokrovski G. S.; Zotov A. V.; Hazemann J.-L. Structure and Stability of Cadmium Chloride Complexes in Hydrothermal Fluids. Chem. Geol. 2010, 276, 1–17. 10.1016/j.chemgeo.2010.03.006. [DOI] [Google Scholar]
- Butterworth P.; Hillier I. H.; Burton N. A.; Vaughan D. J.; Guest M. F.; Tossell J. A. Calculations of the Structures, Stabilities, Raman Spectra, and NMR Spectra of Cadmium Halide Hydroxide (CdCln(OH2)a2-n, CdBrn(OH2)a2-n), and Zinc Chloride Hydroxide (ZnCln(OH2)a2-n) Species in Aqueous Solution. J. Phys. Chem. 1992, 96, 6494–6500. 10.1021/j100194a071. [DOI] [Google Scholar]
- Mei Y.; Sherman D. M.; Liu W.; Etschmann B.; Testemale D.; Brugger J. Zinc Complexation in Chloride-Rich Hydrothermal Fluids (25–600 °C): A Thermodynamic Model Derived from Ab Initio Molecular Dynamics. Geochim. Cosmochim. Acta 2015, 150, 265–284. 10.1016/j.gca.2014.09.023. [DOI] [Google Scholar]
- Parchment O. G.; Vincent M. A.; Hillier I. H. Speciation in Aqueous Zinc Chloride. An Ab Initio Hybrid Microsolvation/Continuum Approach. J. Phys. Chem. 1996, 100, 9689–9693. 10.1021/jp960123z. [DOI] [Google Scholar]
- Pye C. C.; Corbeil C. R.; Rudolph W. W. An Ab Initio Investigation of Zinc Chloro Complexes. Phys. Chem. Chem. Phys. 2006, 8, 5428–5436. 10.1039/B610084H. [DOI] [PubMed] [Google Scholar]
- Pye C. C.; Black S. M.; Rudolph W. W. An Ab Initio Investigation of Zinc Bromo Complexes. J. Solution Chem. 2011, 40, 1932–1954. 10.1007/s10953-011-9759-2. [DOI] [Google Scholar]
- Bradley M.Curve Fitting in Raman and IR Spectroscopy: Basic Theory of Line Shapes and Applications. https://assets.thermofisher.com/TFS-Assets/CAD/Application-Notes/AN50733_E.pdf (accessed June 16, 2021).
- Pearson R. G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. 10.1021/ja00905a001. [DOI] [Google Scholar]
- Marcus Y. Thermodynamics of Solvation of Ions. Part 5.—Gibbs Free Energy of Hydration at 298.15 K. J. Chem. Soc., Faraday Trans. 1991, 87, 2995–2999. 10.1039/FT9918702995. [DOI] [Google Scholar]
- Moyer B. A.; Sun Y.. Principles of Solvent Extraction of Alkali Metal Ions: Understanding Factors Leading to Cesium Selectivity in Extraction by Solvation. Ion Exchange and Solvent Extraction; Marcel Dekker, Inc.: New York, 1997; Vol. 13, pp 295–391. [Google Scholar]
- Sato T. Thermal Decomposition of the Solvent Extracted Molybdenum (VI) Complexes from Hydrochloric Acid Solutions with Trioctylamine and Trioctylmethylammonium Chloride. J. Therm. Anal. 1991, 37, 1309–1327. 10.1007/BF01913864. [DOI] [Google Scholar]
- Xu J.; Paimin R.; Shen W.; Wang X. An Investigation of Solubility of Aliquat 336 in Different Extracted Solutions. Fibers Polym. 2003, 4, 27–31. 10.1007/BF02899326. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.