

Abstract
The cancer-immunity cycle (CIC) is comprised of a series of events that are required for immune-mediated control of tumor growth. Interruption of one or more steps of the CIC enables tumors to evade immunosurveillance. However, attempts to restore antitumor immunity by reactivating the CIC have had thus far, limited success. Recently, numerous studies have implicated metabolic reprogramming of tumor and immune cells within the tumor microenvironment (TME) as key contributors to immune evasion. In this opinion piece, we propose that alterations in cellular metabolism during tumorigenesis promote both initiation and disruption of the CIC. We also provide a rationale for metabolically targeting the TME which may assist in improving tumor responsiveness to “chimeric antigen receptor” (CAR)-transduced T-cells or immune checkpoint blockade therapies.
Descriptive keywords: Metabolism, immunotherapy, PD-1, glycolysis, lactate, CAR-T cells, tumor immunology
The cancer-immunity cycle: a model for tumor immunogenicity and immune evasion
An optimal anticancer immune response requires a sequence of events known collectively as the “cancer-immunity cycle” (CIC) [1]. The cycle is initiated by the release of cancer-associated antigens (CAAs) from dying cancer cells. In mammals, these antigens are captured and processed by dendritic cells (DCs) and presented to naïve T-cells within tumor-draining lymph nodes. The activated, cancer antigen-specific CD8+ T-cells mobilize to and infiltrate tumors, where they recognize and eliminate cancer cells via recognition of cognate peptide antigen bound to MHC class I molecules (pMHC) present on the cancer cell surface. The subsequent release of additional CAAs initiates a new round of the CIC and amplifies the magnitude of the immune response with each subsequent round [1]. Interference with one or more events of the CIC enables tumors to evade immune-mediated destruction, which constitutes a hallmark of cancer [2].
Broadly speaking, tumors interfere with the CIC by either reducing their immunogenicity (see Glossary) or by suppressing the effector capacity of tumor-infiltrating T-cells. Therapies aimed at reversing these adaptive mechanisms, by either increasing immune recognition of tumors through the generation of synthetic “chimeric antigen receptor” (CAR)-transduced T-cells or by restoring the effector capacity of CD8+ T-cells via immune checkpoint blockade (ICB), have led to landmark clinical responses that have revolutionized the field of cancer immunotherapy in the past decade [3,4]. Yet, the overwhelming majority of patients exhibit either minimal or temporary responses to immunotherapy, suggesting that tumors interrupt the CIC at multiple points. Here, we propose that alterations in tumor metabolism are fundamental drivers of both tumor immunogenicity and immune evasion and discuss how targeting tumor metabolism can restore a functional CIC and promote durable anti-tumor immunity.
Metabolic regulation of tumor immunogenicity
To activate a host immune response, tumors must both generate and release aberrant peptides in a context that activates DC function and appropriate lymph node priming (Figure 1). Sufficient tumor immunogenicity has been proposed to require a high mutation rate, due to either i) environmental mutagens, such as in lung cancer or melanoma, or ii) impairments in endogenous DNA repair pathways, such as mismatch repair (MMR)-deficient tumors [5,6]. Alternatively, we propose that the metabolic rewiring that is essential for tumor growth fundamentally results in tumor immunogenicity. In the following sections, we describe how metabolic alterations that accompany transformation can be drivers for the elements comprising the initial stage of the CIC.
Figure 1.
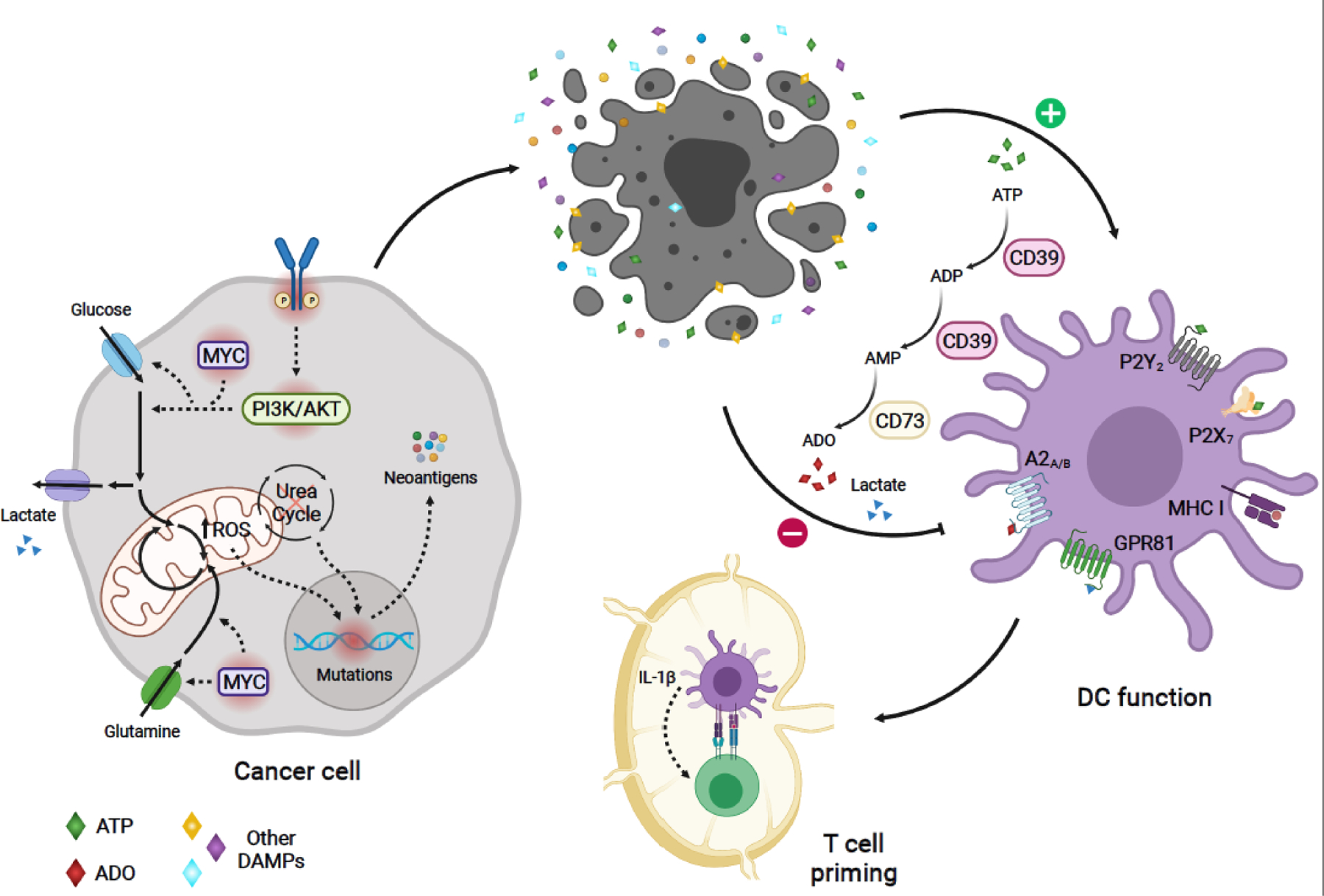
Model of metabolic regulation of tumor immunogenicity: Oncogenic-driven metabolic rewiring required for cancer cell growth can promote tumor immunogenicity. Oncogenic signaling (e.g. PI3K and MYC pathways), can boost nutrient uptake, leading to increased production of mitochondrial ROS and ROS-mediated mutagenesis [12]. Altered expression of urea cycle enzymes and transporters can result in urea cycle dysregulation, promoting diversion of nitrogen to pyrimidine synthesis, leading to increased purine to pyrimidine transversion mutations [21]. These metabolic-driven mutations generating tumor antigens can activate antitumor immune responses. Aberrant tumor growth leads to cell death and release of tumor antigens. In addition, dying cells in the TME release DAMPs, which enhance DC-mediated capture and presentation of cancer antigens to tumor antigen-specific CD8+ T-cells, as well as, priming and activation of these T-cells. One of such DAMPs, ATP, stimulates antitumor function of DCs in humans and mice [57,58]. However, sequential catabolism of ATP -- mediated by the ectonucleotidases CD39 and CD73 -- may lead to the production of ADO, which can suppress DC function through A2A and A2B receptor-mediated signaling in humans and mice [57,59–61]. Metabolic-derived lactate secreted by certain cancer cells can also hinder DC function through activation of the GPR81 receptor, as evidenced in breast cancer mouse models [73]. Solid arrows depict metabolic reactions, metabolite uptake and secretion, and modulation of cellular phenotypes. Dashed arrows indicate signaling or mutation-associated events. Oncogenic mutated molecules are surrounded by a red shadow. A2A, Adenosine A2a receptor; A2B, Adenosine A2b receptor; ADO, adenosine; DAMPs, damage-associated molecular patterns;; DC, dendritic cell; GPR81, G protein-coupled receptor 81;, bHLH transcription factor; PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; ROS, reactive oxygen species; TME, tumor microenvironment. Figure created with BioRender.com
The metabolism of oncogenically transformed cells promotes mutagenesis.
How might oncogenic activation of pathways that drive cancer cell growth promote immunogenicity? It is now well-established that growth factor-independent nutrient uptake in excess of what is needed for ATP production is a hallmark of tumorigenesis [7]. Indeed, high rates of glucose uptake are the basis for positron emission tomography (PET) imaging of tumors based on 18-fluorodeoxyglucose (FDG) uptake [7]. Increased nutrient uptake during tumorigenesis is not limited to glucose alone, as evidenced by high rates of glutamine uptake in most tumors [8,9]. Growth factor independent nutrient uptake is frequently driven by oncogenic activation of the phosphoinositol-3 kinase (PI3K) and MYC pathways, which are amongst the most frequently altered pathways in human cancers [10]. How might increased nutrient uptake lead to mutagenesis? Oncogene-driven nutrient uptake leads to the accumulation of mitochondrial reactive oxygen species (ROS), which in combination with chromatin remodeling, can lead to increased rates of mutagenesis [11–15](Figure 1). Accordingly, the inability to neutralize oncogene-dependent ROS leads to activation of cellular senescence and secretion of inflammatory factors that can initiate an immune response [16]. This is supported by multiple studies, including in vitro models in which overexpression of oncogenic MYC and RAS variants in normal human fibroblasts induce ROS-dependent cell senescence [11,16], as well as ex vivo data showing that oncogenic KRAS-induced ROS can increase the secretion of IL-1β in myeloid cells of mice and human patients with acute myeloid leukemia, chronic myelomonocytic leukemia, and juvenile myelomonocytic leukemia [17]. By contrast, stabilization of a tumor-intrinsic antioxidant program via mutations promoting inactivation of Kelch like ECH associated protein 1 (KEAP1) or stabilization of nuclear factor erythroid 2-related factor 2 (NRF2) can render tumors insensitive to ICB, as demonstrated by analysis of transcriptomic data of human patients with non-small lung cancer (NSCLC) treated with the anti-PD-L1 monoclonal antibody, atezolizumab [18,19]. Thus, elevated oncogene-driven nutrient uptake can promote mutagenesis and consequently immunogenicity by increasing intracellular ROS accumulation in certain cancers.
Furthermore, urea cycle dysregulation (UCD), which is frequently driven by oncogenic MYC activation [20], can promote diversion of nitrogen to pyrimidine synthesis by activating carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydrooratase (CAD) to support an increased rate of cell proliferation, as revealed by TCGA analysis of urea cycle enzyme expression in certain patient tumors [21]. In this analysis, augmented pyrimidine synthesis increased the pyrimidine to purine ratio, leading to pyrimidine-rich transversion mutational bias (PTMB, i.e. increased purine to pyrimidine transversion mutations) and increased numbers of hydrophobic tumor antigens relative to non-UCD cancer cells. Transcriptomic analysis of melanoma patients suggests that an increase in hydrophobic tumor antigens is associated with increased immunogenicity in the context of ICB despite not increasing global tumor mutational burden [21,22].
Collectively, these findings suggest that metabolic alterations induced by cancer cells to support growth, such as nutrient uptake-induced ROS and UCD, can elicit mutagenesis and further drive certain tumors to increase their immunogenicity (Figure 1).
Metabolic regulation of immune checkpoints
If the metabolic rewiring that characterizes oncogenic cell growth promotes immunogenicity, one might expect growth-promoting oncogenes to also activate immune evasion strategies. Analysis of the CD274 locus encoding programmed cell death ligand 1 (PD-L1), commonly referred to as PD-L1, supports this hypothesis: PD-L1 is the most well-characterized immune checkpoint expressed on tumor cells in mammals, and targeting PD-L1-driven programmed cell death 1 (PD-1) signaling has yielded significant clinical benefits in multiple malignancies, including melanoma, NSCLC, Hodgkin’s disease, and high microsatellite instability (MSI-h) cancers, among others [3]. PD-L1 expression is classically induced by local interferon gamma (IFN-γ) production during an immune response, which leads to Janus kinase (JAK)/signal transducer and activator of transcription 1 (STAT1)-dependent activation of PD-L1 gene transcription in mammalian cells [23–25]. However, PD-L1 expression can be activated by nearly every oncogenic signaling pathway that promotes tumor cell growth and proliferation [10,25,26] (pathways summarized in Figure 2). Activating mutations in growth factor signaling pathways, including the epidermal growth factor receptor (EGFR), KRAS, and mitogen-activated protein kinase (MAPK) [27–29] induce PD-L1 expression via transcriptional and post-transcriptional mechanisms. The MYC oncogene activates PD-L1 expression by directly binding the PD-L1 gene [30], whereas loss of the tumor suppressor (phosphatase and tensin homolog) PTEN leads to a PI3K-dependent increase in PD-L1 transcription [31] and translation [32]. Finally, the Hippo pathway effectors Yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ), activate TEA domain transcription factor 1 (TEAD) binding and activation of the PD-L1 enhancer [33]. Oncogenic activation of PD-L1 expression is therefore an example of how oncogene-dependent growth might require concurrent activation of immune evasion strategies to avoid accompanying tumor immunogenicity.
Figure 2.
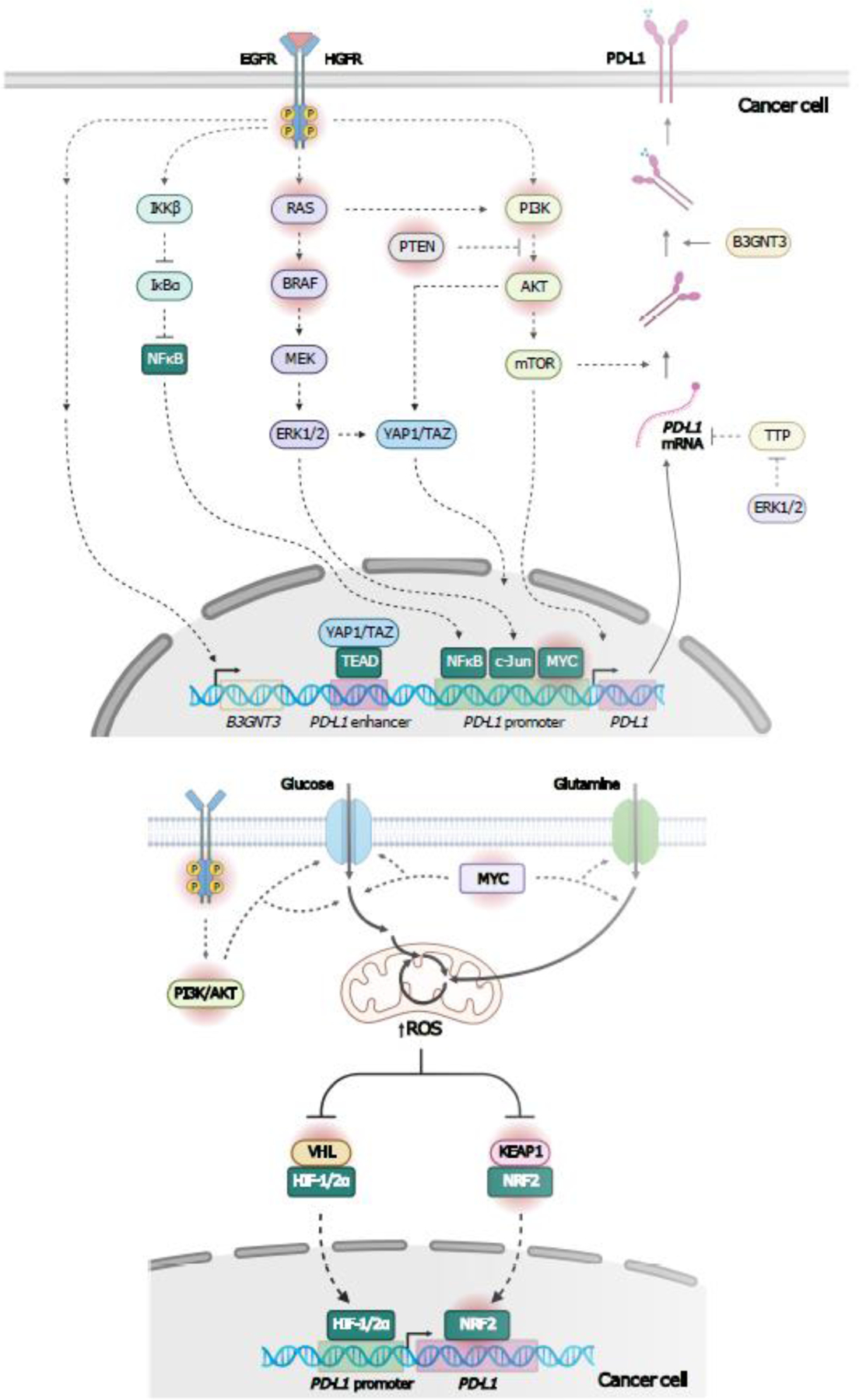
Model of Metabolic regulation of PD-L1 expression. A, Oncogenic signaling and downstream metabolic reprogramming in cancer cells can trigger immune evasion mechanisms, such as induction of PD-L1 expression [27–35]. Oncogenic signaling downstream of receptor tyrosine kinases (RTK), such as EGFR, triggers PD-L1mRNA and protein expression through multiple pathways [27,29][150]. EGFR prevents PD-L1 degradation by driving B3GNT3-mediated PD-L1 glycosylation, which stabilizes PD-L1, and PD-L1/PD-1 interactions [150]. Activation of NFκB pathway and Hippo pathway effectors, YAP/TAZ, induce PD-L1 transcription, whereas, RAS, MAPK, and PI3K/AKT pathways induce PD-L1 expression via transcriptional and post-transcriptional mechanisms [27,29]. MAPK stabilizes PD-L1 mRNA by inhibiting TTP, which promotes PD-L1 mRNA degradation by binding to AU-rich elements in the 3’UTR. MYC mediates PD-L1 transcription by directly binding the PD-L1 gene in human and mouse lung and colon cancer cells [29]. B, Enhanced metabolic activity resulting from oncogenic signaling, such as the one mediated by MYC and PI3K/AKT pathways can increase mitochondrial ROS production, and may lead to PD-L1 transcription through activation of the ROS sensing TFs, HIF-1/2α and NRF2 [36,38,40–42]. ROS inhibits the repressors of HIF-1/2α and NRF2, namely, VHL and KEAP1, respectively, triggering stabilization and nuclear translocation of theses TFs [34,35]. Mutations inhibiting VHL/HIF-1/2α and KEAP1/NRF2 interactions, also promote transcriptional activity of HIF-1/2α and NRF2 [37,41,42]. Solid arrows depict metabolic reactions, metabolite uptake, mRNA nuclear export. protein translation, and translocation. Dashed arrows indicate signaling events. Oncogenic mutated proteins are surrounded by a red shadow. AKT, AKT Serine/Threonine Kinase; B3GNT3, beta-1,3-N-acetylglucosaminyltransferase 3; EGFR, epidermal growth factor receptor; HIF-1/2α, hypoxia inducible factor 1/2 subunit alpha; KEAP1, kelch like ECH associated protein 1; MAPK, mitogen-activated protein kinase; NFκB, nuclear factor kappa B; NRF2, Nuclear Factor Erythroid 2-Related Factor 2; PD-L1, programmed cell death 1 ligand 1; PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase;; ROS, reactive oxygen species; TAZ, transcriptional co-activator with PDZ-binding motif; TPP, tristetraprolin; TFs, transcription factors, 3’UTR, 3’ untranslated region; VHL, von Hippel-Lindau tumor suppressor; YAP; Yes-associated protein. Figure created with BioRender.com
Further evidence suggests that PD-L1 expression is activated not only by oncogenic growth programs, but also by downstream consequences of oncogenic growth that might promote immunogenicity (Figure 2). For instance, as discussed above, high metabolic activity triggers ROS production, which can activate the NRF2 (also known as NFE2L2) and hypoxia inducible factor alfa (HIF-α) pathways in mouse and human cells [34,35](Figure 2). Chromatin immunoprecipitation analysis of human primary melanocytes exposed to narrow-band ultraviolet-B demonstrated that NRF2 can activate PD-L1 expression through direct binding to an enhancer in the PD-L1 regulatory region [36]. Mutations stabilizing NRF2 or inactivating its repressor, KEAP1, promote transcriptional activity of NRF2, as demonstrated by studies in NSCLC in vivo mouse models as well as in primary patient tumors [19,34,37]. Perturbation of the CUL3-KEAP1 complex (e.g. inactivating mutations in KEAP1) increases protein stability of NRF2 and NRF2-mediated transcription of PD-L1 in leukemia cell lines [38]. In mouse fibroblasts, oncogenic signaling mediated by mutant KrasG12D, BrafV619E and MycERT2 also induces transcription of Nrf2 and increases its transcriptional activity [39], suggesting that oncogenic activation of PD-L1 expression might occur via NRF2 activation.
In addition to NRF2, the HIF-1/2α pathway can promote PD-L1 expression by binding to a hypoxia-response element in the PD-L1 proximal promoter in human and mouse cell lines of various cancer types, including renal cell carcinoma [40,41]. Inactivating mutations that disrupt the function of von Hippel-Lindau tumor suppressor (VHL)-- a repressor of HIF-α subunits -- has promoted HIF-2α-mediated expression of PD-L1 in human renal cell carcinoma cell lines [41,42]. Notably, stabilization of HIFα subunits driven by oncogenic signaling, such as EML4-ALK signaling, can also induce PD-L1 mRNA and protein expression in human lung adenocarcinoma cell lines [43]. The binding of ROS-responsive transcription factors (TFs) to the PD-L1 promoter in response to oncogenic pathway activation provides further evidence in support of a role for oncogene-driven ROS promoting immunogenicity.
Importantly, PD-L1 is not the sole immunomodulatory axis that responds to oncogenic activation. The tryptophan (Trp) catabolic enzyme (TCE) indoleamine 2,3-dioxygenase 1 (IDO1) -- known to be activated by IFN-γ signaling and which drives immunosuppression through depletion of extracellular Trp or binding of Trp catabolites to the aryl hydrocarbon receptor (AHR) [44,45]-- is also subject to oncogene-driven regulation. Experiments conducted in mouse and human fibroblasts, as well as in human colon adenocarcinoma tumors and cell lines revealed that MYC activation increases Trp catabolism as well as expression and activation of AHR [46,47]. Moreover, in mouse and human breast cancer cell lines, ROS can directly promote AHR activation [48], which in turn can further promote NRF2 activation by binding to a response element in the NFE2L2 locus [49,50], thereby contributing to oncogenic and ROS-driven PD-L1 expression. Furthermore, AHR signaling can directly induce PD-L1 expression in human glioblastoma and hepatocellular carcinoma cell lines [51,52], as well as expression of the metabolic immune checkpoints IDO1, tryptophan 2,3-dioxygenase (TDO2), and interleukin 4 induced 1 (IL4I1)-- the main TCEs responsible for AHR activation-- in multiple human cancer cell lines, including glioblastoma cells [53–55].
Collectively, these findings suggest that oncogenic activation, directly or through increased production of ROS, may enhance immunogenicity and therefore require activation of multiple immunosuppressive checkpoints to limit antitumor immunity and sustain tumor growth. The extent to which oncogenic growth programs and oncogene-driven ROS cooperate to sustain robust activation of PD-L1, IDO1, and other immune checkpoints remains an area of open study.
Oncogenic metabolism promotes ‘danger signals’ that lead to DC activation.
The pathologic hallmark of tumorigenesis is growth that breaks the tissue architecture of the organ in which it is growing. This aberrant growth pattern leads invariably to growth beyond that which can be supported by an organ’s native blood supply, leading to cell death and release of cancer-associated antigens (CAA) as well as intracellular metabolites that enhance antigen presenting cell (APC) activation and antigen presentation to cancer antigen-specific T cells [56]. Many of these metabolites, known as damage-associated molecular patterns (DAMPs), activate toll-like receptors and mobilize innate immune responses (Figure 1). One such DAMP, ATP, is released by apoptotic cells in the tumor microenvironment (TME) in response to various stimuli [56]. In mouse models, ATP released by dying fibrosarcoma cells promotes the recruitment of DCs through activation of P2Y2 purinergic receptors [57]. Moreover, by using mouse genetic deficiency models (e.g. Nlrp3−/−, Casp1−/−, IL1r1−/−), ATP-mediated activation of P2X7 purinergic receptors on DCs was shown to lead to NLR family pyrin domain containing 3 (NLRP3) inflammasome activation and secretion of IL-1β, supporting the priming of antitumor CD8+ T-cells in mouse sarcoma models (Figure 1) [58].
While the aberrant growth pattern that defines tumorigenesis might promote antigen capture and APC activation through the release of DAMPs, there is evidence that tumors also respond an immune response by suppressing DAMP generation. For example, sequential catabolism of ATP -- mediated by the ectonucleotidase CD39 (also known as ectonucleoside triphosphate diphosphohydrolase 1,ENTPD1) and CD73 (also known as ecto-5’-nucleotidase, NT5E) -- expressed either by human or mouse cancer cells of multiple origins, or by stromal cells in the TME, in an IFN-γ dependent fashion, can impair DC function through direct depletion of ATP, or via the generation and signaling of adenosine (ADO) through the purinergic receptor A2B on DCs (Figure 1) [57,59–61]. A2B signaling has been associated with a tolerogenic phenotype in DCs, characterized by the expression of immunosuppressive factors, such as the metabolic enzymes arginase 2 (ARG2) and IDO1 [59]. It is noteworthy that while hypoxia is known to activate vascular endothelial growth factor (VEGF)-mediated angiogenesis in human cells via stabilization of HIF-1α, the IFN-responsive transcription factor STAT3 also activates VEGF production [62]. We therefore speculate that tumors might sense the presence of an immune response and activate angiogenesis to limit cell death and immunogenicity; however, this hypothesis requires further study.
Metabolic regulation of immune escape
Metabolic suppression of tumor antigenicity
If the metabolic rewiring that characterizes oncogenic cell growth and proliferation promotes immunogenicity, it is reasonable to ask whether further alterations in tumor cell metabolism might facilitate immune evasion similarly to the adaptations that have been shown to promote tumor growth following matrix detachment (anoikis) and under conditions of hypoxia in multiple human and mouse cell types [35,63,64]. Indeed, an increased glycolytic rate is among the most consistent metabolic features of tumors that progress following ICB and adoptive T-cell therapy (ACT), relative to either immunotherapy-naïve or -responsive tumors [65–67]. We propose multiple mechanisms by which an increase in aerobic glycolysis might suppress tumor immunogenicity. First, while oncogene-driven glucose uptake and oxidation might promote immunogenicity as discussed above, a shift from mitochondrial oxidative catabolism of glucose to aerobic glycolysis might reduce ROS-dependent DNA mutagenesis and downstream mitochondrial DNA-dependent cyclic GMP-AMP synthase (cGAS)/stimulator of interferon genes (STING) activation, as demonstrated in human non-cancer retinal and mammary epithelial cells exposed to DNA damaging stimuli [68]. Second, the increased production of lactate in glycolytic tumor cells, such as the human hepatoblastoma cell line HepG3, can directly reduce immunogenicity by suppressing retinoic acid-inducible gene I protein (RIG-I)-dependent type I IFN signaling [69]. Third, lactate production by glycolytic tumors can suppress tumor immunogenicity through modulation of antigen presenting capacity of tumor-resident APCs. Indeed, in vitro human and mouse models, as well as in vivo mouse models indicate that tumor-derived lactate can interfere with both DC function and cross-presentation of extracellular antigens to CD8+ T-cells [70–73]. Finally, an increased glycolytic rate may suppress the synthesis of tumor antigens simply by increasing the rate of cell cycle progression, which might limit the duration and extent of de novo protein synthesis, including potentially immunogenic antigens. A relationship between cell cycle progression and tumor immunogenicity is supported by pre-clinical studies of breast and colon cancer, demonstrating that selective inhibitors of cyclin-dependent kinase 4/6 (CDK4/6) can promote antitumor immunity by increasing antigen presentation in cancer cells [74,75]. Mechanistically, CDK4/6 inhibitors promote cancer cell expression of endogenous retroviral genes, leading to activation of type III IFNs and induction of antigen processing and presenting genes [74], including major histocompatibility class I (MHCI) and MHCII [74,75], in mouse and human cancer cells. Accordingly, CDK4/6 inhibitors can enhance the efficacy of ICB [74,75] and other immunotherapies, such as ACT and T cell-activating antibodies anti-OX40/anti-4-1BB in mouse models of breast cancer [76]. Taken together, these data argue that activation of aerobic glycolysis might be a potent mechanism to suppress tumor antigenicity and promote immune evasion.
Metabolic suppression of antitumor immune cell function
In addition to tumor immunogenicity, a successful antitumor immune response requires that CD4+ and particularly CD8+ T-cells have sufficient self-renewal and effector capacity within tumors. The metabolic requirements for T-cell proliferation and effector function have been discussed extensively elsewhere [77,78] and are summarized in Box 1 and Figure 3, but include the utilization of glucose as well as essential and non-essential amino acids (EAAs and NEAAs, respectively). Retention of sufficient proliferative capacity has recently been identified as a key contributor to the efficacy of ICB; this is supported by recent data from mouse models of lymphocytic choriomeningitis virus (LCMV) infection and melanoma demonstrating that ICB promotes a proliferative burst in a population of PD-1+ CD8+ tumor-infiltrating T-cells exhibiting stem-like properties [79,80], and defined as progenitor CD8+ exhausted T (Texh)-cells [81,82]. In humans and mice, this CD8+ T-cell subset expresses the TF T-cell factor 1 (TCF-1) [79,80,82]. Accordingly, higher numbers of TCF-1+ PD-1+ CD8+ Texh-cells in tumor tissues from melanoma patients have been associated with a prolonged response to ICB, compared with patients with fewer intratumoral TCF-1+ PD-1+ CD8+ Texh-cells [83].
Box 1. Metabolic requirements for T-cell proliferation and effector function.
T-cells take up nutrients from their surrounding environment to fuel bioenergetic and biosynthetic requirements. Glucose is a major nutrient required for T-cell proliferation and effector function [84,86,144]. Co-stimulation of CD28 and downstream PI3K/AKT signaling mediates activation-induced glucose uptake and metabolism in human and mouse T-cells[93,144]. In proliferating CD8+ T-cells, glucose is incorporated in the glycolytic pathway, giving rise to pyruvate and pyruvate-derived lactate or alanine [90]. The pentose phosphate pathway and 3-phosphoglycerate-derived serine contribute to nucleotide biosynthesis [90]. Furthermore, glucose catabolism contributes to the synthesis of the nucleotide sugars, UDP-glucose (UDP-Glc) and N-acetylglucosamine (UDP-GlcNAc) [90]. Glucose-derived pyruvate is also oxidized in the TCA cycle, where it enters as acetyl-CoA or oxaloacetate, contributing to the generation of TCA cycle intermediates, such as citrate, and TCA-derided amino acids, such as, aspartate and glutamine [90] -- required for lipid and nucleotide synthesis, respectively [145–148].
Amino acids also contribute to the proliferation of T-cells. The NEAA glutamine is major nutrient required in proliferating cells. Part of the activation-induced metabolic reprogramming of T cells involves increased glutamine uptake and further use in nucleotide, hexosamine (e.g. UDP-GlcNAc) and polyamine biosynthesis, as well as oxidation in the TCA cycle, all of which are implicated in cell proliferation [86,148,149]. MYC-dependent gene transcription can drive the uptake and catabolism of glutamine, as well as glucose in CD4+ and CD8+ T-cells in mice and humans [86]. Moreover, uptake and metabolism of another non-essential amino acid, serine, is essential for nucleotide biosynthesis and support of proliferation [89]. Finally, EAAs such as arginine, histidine, phenylalanine, tryptophan, valine, and methionine are also required for T-cell growth and proliferation [113,130].
Figure 3.
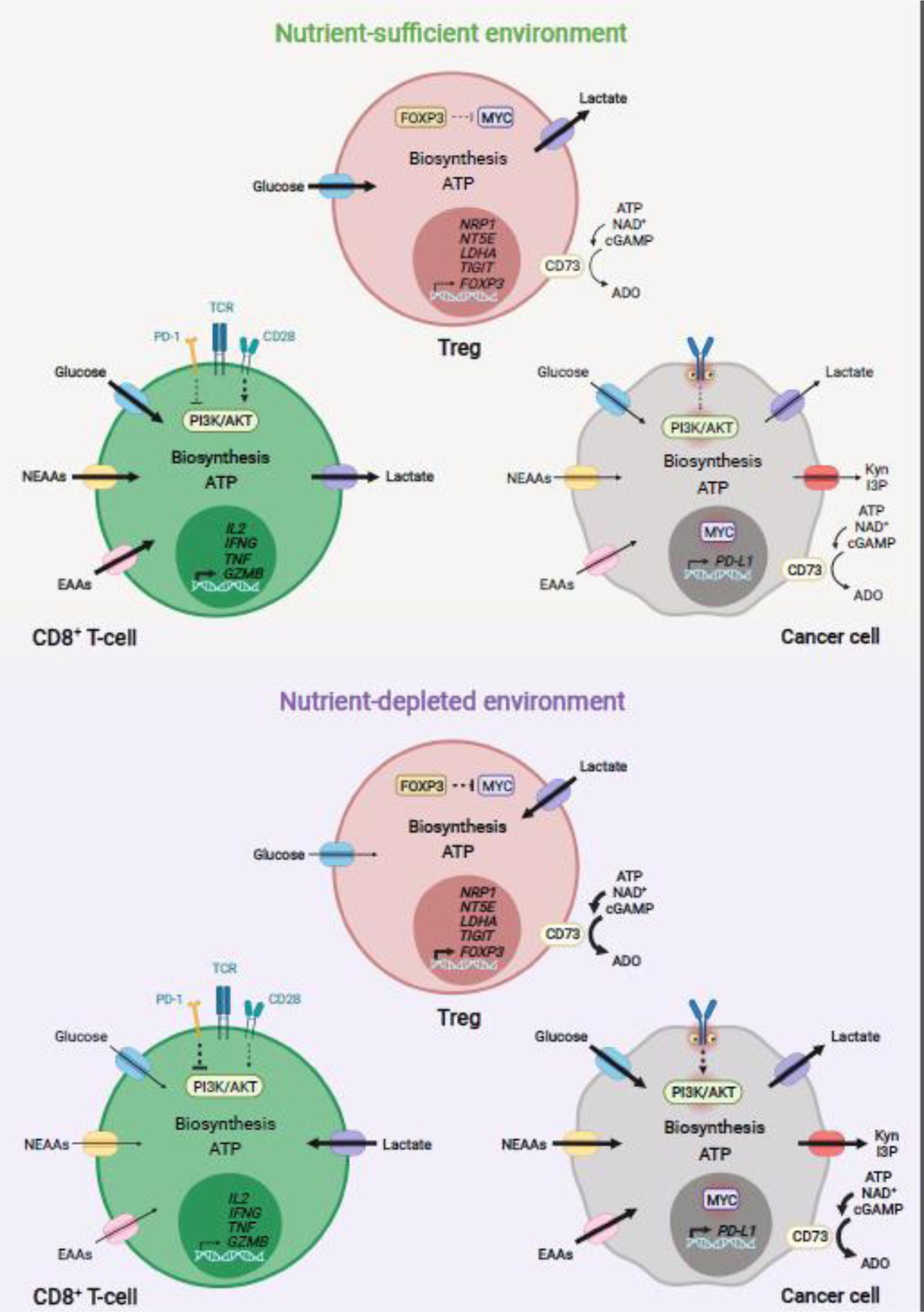
Model of metabolic modulation of antitumor immunity in the TME. In human and mouse, glucose, NEAAs and EAAs are essential for antitumor CD8+ T-cell proliferation, long-term survival, and effector function within tumors. Uptake and metabolism of these nutrients is largely modulated via ligand-receptor signaling and availability of extracellular nutrients in the TME [77,78]. Cancer cell nutrient uptake and metabolism comprise a main mechanism modulating antitumor responses in multiple human and mouse tumor entities [8,67,77,78,93]. Oncogenic signaling and immune-driven activation of metabolic enzymes are main drivers of nutrient uptake and metabolism in cancer cells [44,77,78,137]. In tumors where cancer cells have low nutrient avidity, nutrients are sufficiently taken up by antitumor CD8+ T-cells, enabling proliferation, survival, and effector function [67,93]. In this nutrient-sufficient environment, increased glucose uptake in Treg cells can impair their suppressive function [67,107]. Conversely, high nutrient avidity by cancer cells (e.g. highly glycolytic tumors), can decrease extracellular nutrient availability, leading to impairment of antitumor CD8+ T cell function [67,93,107]. In this nutrient-deprived TME, cancer cells might enhance the production and secretion of suppressors of antitumor CD8+ T-cells, including PD-L1 and lactate [93]. Expression of tryptophan catabolizing enzymes, by cancer and stromal cells, may further contribute to nutrient depletion and generation of immunosuppressive metabolites, including Kyn and I3P [44,55,137]. Ectonucleotidases, such as CD73, are expressed by multiple cancer cell types; Treg cells and other stromal cells may produce immunosuppressive ADO [60,121]. Increased uptake and oxidation of lactate can also support the proliferation and immunosuppressive function of Treg cells [106,107]. ADO, adenosine; CD73, cluster of differentiation 73; EAAs, essential amino acids; NEAAs, non-essential amino acids; I3P, indole-3-pyruvate; Kyn, kynurenine; PD-L1, programmed cell death 1 ligand 1; TME, tumor microenvironment; Treg, regulatory T cell. Figure created with BioRender.com
The mechanisms by which extracellular nutrients support T-cell proliferation and function are not limited to simply supporting macromolecular biosynthesis and proliferation. Indeed, sufficient nutrient availability, uptake, and catabolism by T-cells appears to serve as a key gatekeeper for activation of downstream functionality, including effector function and differentiation. For example, in in vitro and in vivo mouse models, activation-driven glycolysis enables effector cytokine protein synthesis in CD4+ T-cells by both promoting acetyl-coA-dependent chromatin remodeling [83] as well as preventing glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-mediated degradation of Ifng and Il2 mRNA transcripts [84]. Of note, while this acetyl-coA-dependent epigenetic effect was reported for histone H3 acetylation at the lysine 9 residue (H3K9Ac), whether other glucose-dependent acetyl-coA histone modifications such H3K18Ac, H3K14Ac, and H3K27Ac [85] are present and modulate the function of different T-cell subsets remains to be investigated. Similarly, glutamine, serine and methionine appear to support proliferation and differentiation of human and mouse CD4+ and CD8+ T-cells, both by supporting macromolecular biosynthesis as well as via genome-wide effects on chromatin [86–90]. It is important to note that the thresholds at which specific metabolite-sensitive effects on T-cell function are lost have not yet been determined, suggesting that under nutrient-limiting conditions such as those present in the TME, certain metabolite-responsive functions may be retained, while others might not.
Given the dependence of T-cells on available nutrients to sustain both proliferation and effector function [77,78], it has long been hypothesized, although not definitively proven, that environmental depletion of essential nutrients in tumors might impair antitumor immunity (Figure 3). Metabolic interference with intratumoral T-cell function can be separated into two broad categories: first, via ligand-receptor interactions that alter T-cell metabolic functions, and second, via alteration of the extracellular nutrient environment in which T-cells attempt to sustain durable immune responses. The idea that the canonical immune checkpoints cytotoxic T-lymphocyte associated protein 4 (CTLA-4) and PD-1 might suppress T-cell functions by affecting T-cell metabolism was first demonstrated by elegant in vitro work in human CD4+ T-cells showing that these checkpoints had relatively minor effects on activation-induced gene transcription but comparatively large impacts on CD28-induced glucose uptake and metabolism [91]. Subsequent studies in cell-free systems, in vitro T-cell-based systems, and in vivo human and mouse models, confirmed that PD-1 ligation predominantly impaired CD28-dependent AKT activation [92] and that blockade of CTLA-4 and PD-1 signaling promoted antitumor T-cell responses in large part by restoring glucose uptake and metabolism in CD8+ T-cells [93]. However, the inability of CTLA-4 and/or PD-1 blockade to re-invigorate antitumor immune responses in many cancer patients suggests that additional metabolic checkpoints may be present that could limit T-cell function. Indeed, numerous studies have implicated a role for mitochondrial oxidative phosphorylation in sustaining the biosynthetic requirements of activated CD4+ and CD8+ T-cells [84,90,94], including in conditions where glucose might be limiting, such as in the TME [60,67]. Moreover, mitochondrial dysfunction is a hallmark of terminal T-cell exhaustion [94–97], a cell state exhibiting, among other characteristics, reduced self-renewal capacity [98]. Mitochondrial dysfunction is driven by both PD-1 signaling as well as chronic TCR-mediated signal transduction [94,96,97]. In human and mouse CD8+ T-cells, engagement of these pathways can lead to loss of mitochondrial function by promoting mitochondrial depolarization, oxidative stress, and impairing oxidative phosphorylation, consequently reducing the proliferative capacity of antitumor T-cells [94,96,97]. In these recent studies, the severity of mitochondrial dysfunction and T-cell exhaustion correlate with antigen burden [94,96,97], which may partially explain why large solid tumors respond poorly to ICB [99,100]. Strategies preventing mitochondrial dysfunction, such as antioxidant treatments, have improved proliferation, as well as antitumor immunity in mouse Texh-cells, and demonstrate synergistic effects with ICB, in vitro and in vivo [94,96,97]. How additional immune checkpoints expressed on Texh-cells might alter T-cell metabolism remains an intriguing area of future study.
A second mechanism by which tumors can suppress T-cell function is by altering extracellular nutrient availability (Figure 3). For instance, highly glycolytic human and mouse tumors are more immunosuppressive [93,95,101] and less responsive to immunotherapy than tumors with relatively lower glycolytic activity [65–67,102]. High rates of glucose consumption within tumors need not be driven by tumor cells alone; in various human and mouse tumors, including breast and colon cancers, stromal cells including cancer associated fibroblast (CAFs) and myeloid cells exhibit high glucose consumption, and lactate excretion [8,67,93,103], thereby further compromising glucose availability [84,93,101]. Loss of mitochondrial oxidative capacity in terminally CD8+ Texh-cells can render these cells increasingly dependent on available glucose to maintain intracellular ATP pools [94]; this could increase their sensitivity to glucose depletion within the TME, potentially explaining the lack of anti-PD-1 blockade responsiveness in larger tumors, which are more likely to exhibit intratumoral glucose depletion than smaller tumors [92]. In addition to suppressing T-cell glucose uptake by activating PD-1 signaling, PD-L1 expression on tumor cell surfaces can regulate glucose uptake by tumor cells. Cell surface expression of PD-L1 has been reported to drive glucose uptake and glycolysis through AKT/mTOR signaling-dependent translation of glycolytic enzymes in mouse sarcoma cells in in vitro and in vivo models [93]. Given that PD-L1 is activated in response to immune cell-derived IFN-γ in mouse and human cells [23,24], we argue that PD-L1 upregulation on tumor cells might trigger an adaptive change in tumor metabolism that could help suppress antitumor immune responses --- a possibility that merits further attention.
Beyond reflecting the depletion of available glucose, we posit that it is likely that intratumoral accumulation of lactate directly promotes immunosuppression (Figure 3). For instance, lactate uptake by activated mouse CD8+ T-cells promotes intracellular acidification, which represses nuclear factor of activated T-cells (NFAT)-mediated induction of IFN-γ in vitro [104]. In vitro, lactate has also inhibited the proliferation of mouse CD4+ T-cells by opposing cytosolic NAD+ regeneration by lactate dehydrogenase, thereby limiting further glucose catabolism [105]. Unlike effector CD4+ and CD8+ T-cells, data from in vitro and in vivo mouse models suggests that regulatory T(Treg)-cells can readily adapt to low-glucose and lactate-rich environments through metabolic reprogramming driven by forkhead box P3 (FOXP3)-mediated suppression of MYC; this in turn limits glucose uptake [106] while increasing uptake and oxidative metabolism of lactate, supplying the requisite metabolic intermediates for proliferation and immunosuppressive function [107] (Figure 3). Finally, in vitro studies in human and mouse cells indicate that lactate may directly influence the differentiation of immune cells in the TME via lactylation of histone lysine residues, shown to induce the expression of M2-like genes such as Arg1 in macrophages during inflammatory responses [108], but whose roles in tumor-specific immune cell differentiation remain uncharacterized.
Collectively, these studies support the idea that tumors can enforce metabolic dysregulation in tumor-infiltrating T-cells, either through ligand-receptor interactions with metabolic consequences, or by remodeling the extracellular environment to interfere with T-cell metabolic homeostasis.
Metabolic enzymes that promote immunosuppression
Immune-driven activation of metabolic enzymes represents a final set of metabolic alterations to suppress antitumor immunity. The most well characterized of these families are the TCEs IDO1, TDO2, and recently, IL4I1. Across multiple human and mouse cancers, these enzymes promote immunosuppression by depleting aromatic amino acids in the TME and producing bioactive molecules which target and activate the AHR [55,109–111]. IDO1/TDO2-mediated depletion of Trp and IL4I1-mediated depletion of all aromatic amino acids can inhibit proliferation of CD8+ T-cells and induce differentiation of Treg cells through activation of the general control non-derepressible-2 (GCN2) kinase and inhibition of the mammalian target of rapamycin (mTOR) pathways in mouse models [112–114]. IDO1/TDO2-dereved kynurenines, IL4I1-derived indoles, and kynurenic acid formed by these three enzymes can also directly activate AHR [55,110], which in turn, can promote immunosuppression through several mechanisms, including upregulation of PD-1 protein expression in CD8+ T-cells [115], inhibition of CD8+ T-cell proliferation [55,110], induction of CD39 (the rate-limiting enzyme for the synthesis of the immunosuppressive metabolite, ADO, from ATP) in tumor-associated macrophages (TAMs) [51], and differentiation of Treg cells [116] and CD8+ Texh-cells [117] in mouse and/or human cells. Notably, AHR-mediated induction of CD8+ T-cell exhaustion is triggered by tryptophan hydroxylase 1 (TPH1)-derived 5-hydroxytryptophan (5-HTP) in response to continuous IL-2/STAT5 signaling in mouse in vitro models as well as human in vitro and ex vivo models [117]. IL4I1-derived H2O2 also inhibits CD4+ and CD8+ T-cell proliferation and effector function through downregulation of the TCRζ chain, leading to decreased TCR signaling [111]. Among the cytokines that can be present in the TME, IFN-γ released by activated CD8+ T-cells appears to exert a major role in inducing IDO1 and IL4I1 expression via IFN-γ receptor (IFNGR)/JAK/STAT1 pathway in multiple human and mouse cell types [118–120]. Furthermore, type I IFN produced via cGAS)/STING signaling in response to DAMPs released by stressed and dying cells present in the TME can also induce IDO1 expression in human and mouse cells [119,120]. Hence, Trp catabolism mediated by TCE appears to constitute a potent immunosuppressive mechanism in the TME.
Finally, activation of catabolic pathways leading to the extracellular formation of ADO represents a distinct mechanism by which tumors can suppress antitumor T-cell activity in the TME through the generation of immunosuppressive metabolites [60]. Specifically, ATP released by stressed or dying cancer and/or stromal cells can be sequentially metabolized to ADO by the ectonucleotidases CD39 and CD73 present on the surface of cancer and stromal cells in melanoma mouse models [60,121]. Alternatively, ADO can be formed through pathways involving sequential catabolism of NAD+ mediated by CD38, CD203 (also known as ectonucleotide pyrophosphatase/phosphodiesterase 3, ENPP3) and CD73, or sequential catabolism of cGAMP by ENPP1 and CD73 [60,122,123]. In several cancer types, including melanomas and ovarian cancer, ADO promotes immunosuppression by activating type 1 purinergic receptors, A2A and A2B, on human and mouse CD4+ and CD8+ T-cells [60,121,124]. Of note, in mouse and human melanomas, high expression of CD39 in tumor-infiltrating CD8+ T-cells defines a population exhibiting a terminally exhausted phenotype, characterized by decreased IFN-γ, tumor necrosis factor (TNF), and IL-2 production, and increased expression of immune inhibitory receptors such as PD-1, lymphocyte activating 3 (LAG-3), and T cell immunoreceptor with Ig and ITIM domains (TIGIT) [125,126]; this suggests that exhausted CD8+ T-cells can sequentially contribute to their own suppression through the extracellular synthesis of ADO. Given the constant supply of extracellular ATP released from necrotic cells in the TME during the process of rapid tumor growth, catabolic pathways giving rise to and enabling ADO signaling may represent a key immunosuppressive mechanism in certain cancers.
Targeting metabolic regulators of the cancer-immunity cycle
The discussion above supports the therapeutic potential of targeting cellular metabolism within tumors to reactivate the CIC and increase responsiveness to immunotherapies. We suggest that this can occur in large part by boosting tumor immunogenicity and/or restoring T-cell antitumor function. Indeed, several therapeutic agents targeting metabolic modulators of the CIC are currently FDA approved or under clinical development to treat various cancer types (Figure 4 and Table 1). Targeting glucose metabolism in the TME has been the most well-studied approach in this regard. In pre-clinical models, suppression of aerobic glycolysis through genetic or pharmacological inhibition of lactate dehydrogenase [65,67,70,93,107,127], which increases glucose and decreases lactate concentrations in the TME, increases tumor immunogenicity by promoting APC function and antigen presentation [70–73]. Additionally, several mouse cancer models indicate that this strategy can improve proliferation and function of antitumor T-cells by increasing available glucose and decreasing lactate-mediated immunosuppression, while simultaneously suppressing the activation of Treg cells by diminishing their lactate uptake and oxidation [65,67,93,107]. In line with this, inhibitors that target lactate secretion by cancer cells or uptake by immune cells in the TME, such as inhibitors of the lactate transporters monocarboxylate transporter 1/4 (MCT1/4) and its receptor G protein-coupled receptor 81 (GPR81), restore antitumor T-cell responses in humans and mice [73,102,107,128]. Targeting aerobic glycolysis or inhibiting lactate uptake systemically using small molecule inhibitors has shown clinical promise alone, and in the context of immunotherapy [65,67,102,107]. These findings suggest that inhibition of nutrient uptake by tumor cells might be a tractable target to enhance immunotherapy; recent studies demonstrating the therapeutic benefit of limiting glutamine catabolism constitute additional encouraging mechanisms in this vein [129]. Given the potentially suppressive role that depleting other essential and non-essential amino acids, such as methionine, serine, and cysteine, play in intratumoral T-cell suppression [89,90,130,131], targeting amino acid transporters is an area of research with prominent potential to expand.
Key Figure, Figure 4.
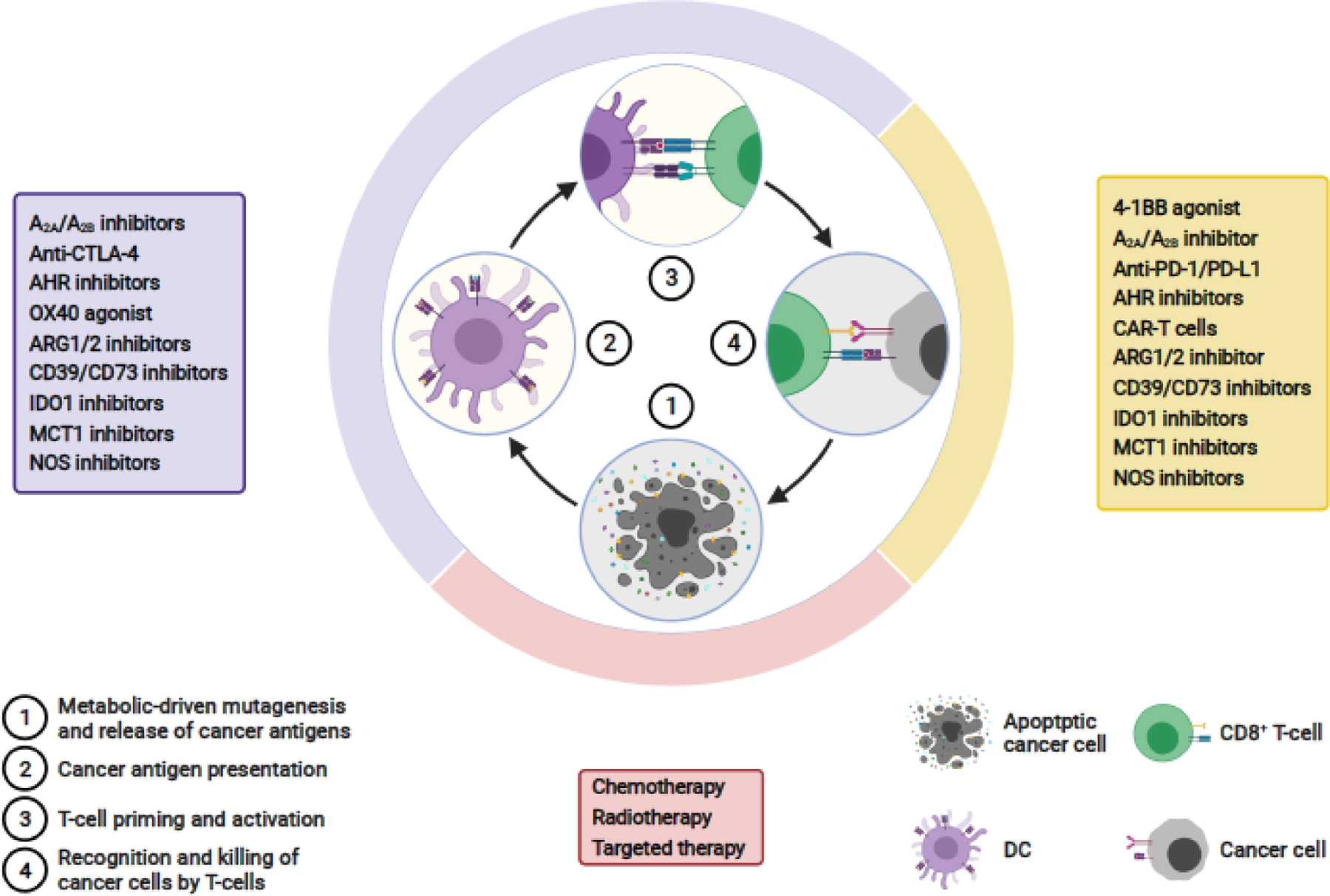
Therapies targeting metabolic modulators of the cancer-immunity cycle. Shown are the current therapeutic approaches that are either US FDA approved or in clinical development against a variety of cancers; these treatments are aimed at targeting metabolic regulators of the cancer immunity cycle. Figure is based on previously published work [1]. Figure created with BioRender.com
Table 1.
Agents in clinical trials targeting metabolic regulators of the cancer-immunity cycle
| Agenta | Combination Immunotherapy | Clinical Phase | Clinical Trial IdentifierII |
|---|---|---|---|
| A2A receptor inhibitors | |||
| AZD4635 | - | 2 | NCT03381274 |
| AZD4635 | Durvalumabc | 2 |
NCT04089553
NCT04495179 |
| CPI-444 | Atezolizumabc | 2 | NCT03337698 |
| NIR178(PBF-509) | Spartalizumabb | 2 | NCT03207867 |
| NCT02403193 | |||
| A2B receptor inhibitors | |||
| PBF-1129 | - | 1 | NCT03274479 |
| A2A/A2B receptor inhibitors | |||
| Etrumadenant (AB928) | Zimberelimabb | 2 |
NCT04660812
NCT04381832 |
| Etrumadenant (AB928) | Zimberelimabb, Domvanalimabd | 2 |
NCT04262856
NCT04791839 |
| Etrumadenant (AB928) | Atezolizumabc | 2 |
NCT03821246
NCT03555149 NCT03193190 |
| AHR inhibitors | |||
| BAY2416964 | - | 1 | NCT04069026 |
| IK-175 | Nivolumabb | 1 | NCT04200963 |
| ARG inhibitors | |||
| INCB001158 | - | 2 |
NCT03314935
NCT03837509 |
| INCB001158 | Pembrolizumabb | 2 |
NCT03361228
NCT02903914 |
| CD39 antibodies | |||
| IPH5201 | Durvalumabc | 1 | NCT04261075 |
| SRF617 | Pembrolizumabb | 1 | NCT04336098 |
| TTX-030 | Pembrolizumabb, Budigalimabb | 1 | NCT04306900 |
| Pembrolizumabb | NCT03884556 | ||
| CD73 antibodies/inhibitors | |||
| AB680 (inhibitor) | Zimberelimabb | 2 | NCT04660812 |
| BMS-986179 (antibody) | Nivolumabb | 2 | NCT02754141 |
| Oleclumab (antibody) | - | 2 | NCT03381274 |
| Oleclumab (antibody) | Durvalumabc | 2 |
NCT03616886
NCT03875573 NCT03267589 NCT04668300 |
| IDO1 inhibitors | |||
| Epacadostat | Pembrolizumabb | 3 |
NCT03361865
NCT03374488 NCT03358472 NCT03260894 |
| MCT1 inhibitors | |||
| AZD3965 | - | 1 | NCT01791595 |
| NOS inhibitors | |||
| L-NMMA | - | 2 | NCT02834403 |
| L-NMMA | Pembrolizumabb | 2 | NCT04095689 |
Agent(s) in most advanced clinical phase;
anti-PD-1;
anti-PD-L1;
anti-TIGIT.
A2A (ADORA2A), adenosine A2a receptor; A2B (ADORA2B), adenosine A2b receptor; AHR, aryl hydrocarbon receptor; ARG, arginase; CD39 (ENTPD1), cluster of differentiation 39; CD73 (NT5E), cluster of differentiation 73, IDO1, indoleamine 2,3 dioxygenase; MCT1 (SLC16A1), Monocarboxylate Transporter 1; NOS, nitric oxide synthase; TIGIT, T Cell Immunoreceptor with Ig And ITIM Domains.
These trials are listed in clinicaltrials.gov with their respective ID numbers.
A second approach to overcome immunosuppression driven by intratumoral nutrient depletion may be to enhance the metabolic fitness of effector T-cells. Restoring mitochondrial function and bioenergetics in CD8+ Texh-cells has emerged as an attractive approach to improve anticancer immunity. Both co-stimulation of 4-1BB and IL-10 can induce mitochondrial biogenesis and bolster oxidative metabolism in tumor infiltrating lymphocytes, leading to increased intratumoral T-cell survival and proliferative capacity in certain mouse or in vitro models [132–134]. Alternatively, inhibition of dysfunctional mitochondria-driven ROS through antioxidant treatment can restore oxidative phosphorylation and mitochondrial function, thereby reinvigorating the proliferative and effector function of CD8+ Texh-cells and restoring antitumor immunity, as shown in mouse melanoma models [94,97]. Of note, improving mitochondrial metabolism appears to re-program the transcriptional landscape of T-cells, restoring a ‘progenitor’ exhausted state marked by increased TCF-1 expression [94] This is intriguing because it provides evidence that it might be possible to prevent or revert the terminally exhausted phenotype of CD8+ T-cells. Given that ICB has been shown to drive Texh-cells towards a terminally exhausted phenotype [82], promoting mitochondrial metabolism might potentially enhance the durability of ICB-mediated antitumor immunity, but this remains to be rigorously tested.
Finally, inhibition of immunosuppressive enzymes and the signaling pathways triggered by their metabolic products might restore the CIC at multiple stages. DC function as well as CD8+ T-cell priming and effector function can be improved by inhibition of ADO synthesis through CD39, CD38, ENPP1 and CD73, as well as blockade of purinergic receptors, such as A2A and A2B [59,61,121–124,135]. Alternatively, enhancement of ADO degradation by adenosine deaminase (ADA) can lead to the formation of inosine [60], a metabolite that promotes antitumor T-cell activity through various mechanisms, including by serving as an alternative carbon source for CD8+ T-cell function in glucose-deprived environments [136]. As expected, targeting ADO metabolism and signaling has improved antitumor responses in combination with ICB in various models [59,122,123,135,136]. Similarly, blocking Trp catabolism through IDO1, TDO2 and IL4I1 can improve T-cell priming, proliferation, and antitumor function, as well as reduce the recruitment, differentiation and function of immunosuppressive cells such as Treg cells, and myeloid cells such as TAMs and myeloid-derived suppressor cells (MDSCs) [44,137]. The promise of blocking tryptophan catabolism to enhance ICB has been somewhat dampened by the negative results of the ECHO-301/KEYNOTE-252 randomized, placebo-controlled phase 3 trial (NCT02752074)i, in which the addition of the IDO1-selective inhibitor epacadostat to pembrolizumab did not improve progression-free survival (PFS) or overall survival (OS) for patients with unresectable stage III or IV melanoma as compared to pembrolizumab plus placebo [138]. Given that multiple other enzymes including TDO2 and IL4I1 can generate metabolites that activate AHR signaling [55], direct inhibitors of AHR might represent a more promising strategy, but this also remains to be robustly tested [44,139]. Nevertheless, the promising results attained by inhibition of immunosuppressive metabolic enzymes and their associated receptors and transporters in pre-clinical studies has prompted multiple clinical trials assessing inhibitors of these molecules as monotherapy or in combination with other therapies, including ICB (Table 1, Box 2).
Box 2. Clinician’s Corner.
The immune metabolic checkpoint, IDO1, is the most clinically well characterized metabolic repressor of antitumor CD8+ T-cell responses, as evidenced by studies of IDO1 inhibitors in combination with ICB in multiple clinical trials for the treatment of cancer patients, including patients with advance melanomas [137]. As described in the main text, the first phase 3 clinical study combining the IDO1 selective inhibitor, epacadostat, and the PD-1 inhibitor, pembrolizumab, in patients with advanced melanoma, did not accomplish its primary endpoints, i.e. PFS and OS (NCT02752074)i [138].
In a recent study, analysis of transcriptomic data of patients with advanced melanoma showed that the immunosuppressive and AHR-activating enzyme IL4I1, as well as AHR activity are induced upon treatment with the PD-1 inhibitor, nivolumab, suggesting that IL4I1 might constitute a metabolic immune checkpoint driving resistance towards ICB and/or IDO1 inhibitors [55]. Furthermore, blocking IDO1 and IL4I1 signaling by targeting the AHR in combination with ICB might also represent a promising strategy to treat multiple cancer entities [139].
Stratification of patients based on the expression of metabolic enzymes and activity of their downstream signaling pathways as in the TCE IDO1/2, TDO2, IL4I1, and AHR, respectively, might be considered to help improve responses to certain immunotherapies [44].
Targeting additional metabolic modulators of the CIC, including cancer-cell glycolysis, lactate secretion and uptake [65,67,107], and T-cell mitochondrial dysfunction [94,96,97,134], among others, might help improve responses to CAR-T and ICB therapies.
Concluding remarks
The growing understanding of how metabolic reprogramming within the TME can potentiate immunogenicity and immune evasion offer a plethora of potentially novel strategies to enhance antitumor immunity (see Outstanding questions). Here, we focused on how the metabolic rewiring of specific cell types (e.g cancer cells, DCs, CD8+ T-cells, and Treg cells) might modulate tumor immunogenicity and suppression of antitumor immunity. While we covered various metabolic pathways exerting a prominent role in the function of these cells types in the CIC, additional metabolic pathways, such as those involved in lipid uptake, synthesis, and metabolism, as well as additional stress response pathways, including the endoplasmic reticulum stress pathway, are emerging key modulators of the CIC and merit further exploration [140,141]. Moreover, how the metabolic reprogramming of additional non-malignant cells within the TME (such as CAFs, endothelial cells, TAMs, and MDSCs) modulate the CIC represents a prominent area of research that might reveal additional therapeutic targets. These may include the immunosuppressive metabolic enzymes described in this manuscript, enzymes such as ARG1/2 and nitric oxide synthase -- often expressed by stromal cells within the TME [143]. In addition, the influence of cellular metabolic reprogramming on additional events within the CIC, such as T-cell recruitment and infiltration into tumors, might provide strategies to reverse a so-called T-cell “exclusion” phenotype observed in multiple immunotherapy-refractory tumors [142]. Further characterization of cell-type- and tumor-type-specific metabolic reprogramming and how it can contribute to metabolic remodeling of the extracellular TME represents a fruitful area of investigation [8]. We posit that a deeper understanding of how specific metabolic reprogramming in tumor cells can support immune evasion will be key to preventing resistance to immune checkpoint inhibitors in the clinic. Finally, extensive analysis of metabolic networks in patients treated with CAR-T cells or ICB, can expand the identification of putative targets to be used in combination, and ideally improve patient responses to such treatments.
Outstanding questions.
In addition to oncogenic-driven ROS and UCD, which metabolic networks promote mutagenesis and subsequently enhance immunogenicity?
Do extracellular metabolites produced in response to oncogenic growth promote DC and T-cell recruitment and tumor infiltration? Alternatively, do adaptive changes in tumor metabolism promote immune exclusion?
How do oncogenic growth factors and transcription factors (TF) cooperate with ROS-sensitive TFs to regulate tumor PD-L1 expression? Do these pathways regulate additional immune checkpoints?
Are there metabolic signatures/networks that predict responsiveness to CAR-T and ICB immunotherapies? For instance, IL4I1 expression and AHR activity were recently shown to be induced in patients with advanced melanoma upon nivolumab treatment, suggesting that IL4I1 might constitute a resistant mechanism towards ICB.
What are the environmental nutrient thresholds at which specific metabolite-sensitive effects on T-cell function are lost? The TME metabolic profile in multiple cancer entities and under multiple therapeutic contexts remains to be systematically characterized.
What are the metabolic drivers of T-cell exhaustion and how can they be targeted to prevent or revert the differentiation of terminally Texh-cells? Recent studies indicate that ROS-driven mitochondrial dysfunction and impaired oxidative phosphorylation, as well as 5-HTP-mediated AHR activity, can promote T-cell exhaustion. Whether additional metabolic pathways drive T-cell exhaustion requires further investigation.
How do additional immune checkpoints known to be expressed on exhausted T-cells alter T-cell metabolism?
Highlights.
Elevated tumor glycolysis and lactate production are robust suppressors of antitumor immunity in multiple cancer subtypes.
Loss of mitochondrial function is a hallmark of CD8+ T-cell exhaustion and might be a promising metabolic target for improving patient responses to CAR-T and/or ICB therapy, pending future investigations.
IL4I1-driven Trp catabolism and aryl hydrocarbon receptor activation may constitute a resistance mechanism to immune checkpoint blockade and/or IDO1 inhibitors across cancer subtypes.
We propose that the metabolic profile of the TME promotes both initiation and disruption of the cancer-immunity cycle. Hence, targeting cellular metabolism in the TME may improve responsiveness to T-cell-based immunotherapies.
Acknowledgments
S.A.V. was supported by a Special Fellow Award from the Parker Institute for Cancer Immunotherapy, a Career Award for Medical Scientists from the Burroughs Wellcome Fund, and a Mentored Clinical Scientist Career Development Award from NCI/NIH (K08 CA237731).
Glossary
- Aerobic glycolysis
Catabolic pathway that oxidizes glucose to pyruvate and subsequently reduces pyruvate to lactate. The latter step often occurs in anaerobic or hypoxic conditions, but in rapidly proliferating cells, occurs under aerobic conditions.
- Anoikis
Programmed cell death caused by detachment of a cell from the extracellular matrix.
- CDK4/6 inhibitors
Target cyclin-dependent kinase 4 and 6; known for their ability to inhibit progression through the G1 phase of the cell cycle and consequently, inhibit cell proliferation, including cancer cells.
- Cellular senescence
phenotype adopted by proliferating cells in response to stress or damage; characterized by an arrest in the cell cycle.
- Chimeric antigen receptor (CAR) T-cell
T-cell genetically engineered to increase antigen-specific T-cell recognition of tumors. CARs contain an extracellular antigen-recognition domain and up to three intracellular signaling domains that activate T-cells. Certain CAR T-cells may be adoptively transferred to patients.
- Damage-associated molecular patterns (DAMPs)
molecules released by damage or dying cells that elicit immune responses through binding pattern recognition receptors (PRRs).
- Ectonucleotidase
enzyme that hydrolyzes nucleotide anhydride or ester bonds, producing nucleosides.
- Glycolysis
Catabolic pathway that promotes the oxidation of one molecule of glucose and produces two molecules of pyruvate.
- High mutation rate
elevated number of somatic mutations present in the genome of a cell.
- Immune checkpoint blockade (ICB)
antagonism of immune checkpoint function via therapeutic agents, mainly antibodies and inhibitors.
- Immune checkpoint
Component of an inhibitory pathway intrinsic to the immune system that regulates the duration and amplitude of immune responses.
- Immunogenicity
ability of an organism to elicit an adaptive immune response.
- Lactylation
post-translational modification of proteins consisting of the covalent addition of lactate to an amino acid residue (hitherto described for lysine) of a protein. Histone lactylation has been reported to epigenetically modulate gene expression.
- M2-like genes
subset of genes expressed by macrophages that differentiate and acquire the so-called ‘M2’ immunosuppressive/wound healing phenotype.
- Mismatch repair (MMR)-deficient tumors
tumors bearing inactivating mutations in genes encoding proteins involved in DNA mismatch repair.
- Oncogenic signaling/pathway
stimulated by mutations in one or more protooncogenes or tumor suppressor gene-encoded proteins.
- Progenitor CD8+ exhausted T (Texh)-cells
subset of exhausted CD8+T-cells characterized by TCF-1+ and PD-1+ expression. These cells possess stem-cell like properties (e.g. self-renewal capacity) and increased effector function compared to terminally exhausted CD8+ T-cells.
- Reactive oxygen species (ROS)
Oxygen-containing molecules that are or can give rise to free radicals. ROS can damage and alter function of nucleotides, proteins, and membrane lipids.
- Stem-cell like properties
resemble those of stem cells, such a self-renewal capacity and the ability to differentiate into other cells.
- T-cell exclusion phenotype
tumors exhibiting very poor infiltration of CD8+ T cells, especially in cancer-cell rich areas.
- T-cell exhaustion
T cell fate characterized by a progressive decrease of effector functions, elevated and sustained expression of immune inhibitory receptors, impaired memory and self-renewal capacity, transcriptional and epigenetic reprogramming, and altered metabolic profile.
- Tumor microenvironment (TME)
heterogenous population of cancer cells (parenchyma), non-malignant cells (stromal cells), extracellular matrix, and signaling molecules located in specific areas of tumors.
- Transversion mutation
inheritable alteration in the DNA of a cell that involves the substitution of a nucleotide containing a purine for a pyrimidine or vice versa.
- Tumor mutational burden
number of cancer cell-specific non-synonymous somatic mutations present in a tumor, per megabase of a genetic region of interest. Such mutations drive the formation of neoantigens, i.e. antigens derived from novel mutated peptides or proteins and therefore, are not present in the normal genome.
- Urea cycle dysregulation (UCD)
Metabolic phenotype caused by altered expression of enzymes and transporters associated with the urea cycle; limits the function of the urea cycle, promoting diversion of nitrogen towards pyrimidine synthesis, increasing mutagenesis, cell proliferation, and ICB responses.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Resources
This trial is listed in https://clinicaltrials.gov/ct2/show/NCT02752074
The trials in Table 1 are listed in clinicaltrials.gov with their respective ID numbers.
References
- 1.Chen DS and Mellman I (2013) Oncology meets immunology: the cancer-immunity cycle. Immunity 39, 1–10 [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 3.Ribas A and Wolchok JD (2018) Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.June CH and Sadelain M (2018) Chimeric Antigen Receptor Therapy. N. Engl. J. Med 379, 64–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Havel JJ et al. (2019) The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 19, 133–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galluzzi L et al. (2018) The hallmarks of successful anticancer immunotherapy. Sci. Transl. Med 10, [DOI] [PubMed] [Google Scholar]
- 7.Vander Heiden MG et al. (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reinfeld BI et al. (2021) Cell-programmed nutrient partitioning in the tumour microenvironment. Nature DOI: 10.1038/s41586-021-03442-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peng X et al. (2018) Molecular Characterization and Clinical Relevance of Metabolic Expression Subtypes in Human Cancers. Cell Rep. 23, 255–269.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bailey MH et al. (2018) Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 173, 371–385.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ogrunc M et al. (2014) Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. 21, 998–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wellen KE and Thompson CB (2010) Cellular metabolic stress: considering how cells respond to nutrient excess. Mol. Cell 40, 323–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang T et al. (2018) MYCN drives glutaminolysis in neuroblastoma and confers sensitivity to an ROS augmenting agent. Cell Death Dis. 9, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JV et al. (2014) Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 20, 306–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weinberg F et al. (2010) Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. U. S. A 107, 8788–8793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vafa O et al. (2002) c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol. Cell 9, 1031–1044 [DOI] [PubMed] [Google Scholar]
- 17.Hamarsheh S et al. (2020) Oncogenic Kras(G12D) causes myeloproliferation via NLRP3 inflammasome activation. Nat. Commun 11, 1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marinelli D et al. (2020) KEAP1-driven co-mutations in lung adenocarcinoma unresponsive to immunotherapy despite high tumor mutational burden. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol 31, 1746–1754 [DOI] [PubMed] [Google Scholar]
- 19.Singh A et al. (2021) NRF2 Activation Promotes Aggressive Lung Cancer and Associates with Poor Clinical Outcomes. Clin. cancer Res. an Off. J. Am. Assoc. Cancer Res 27, 877–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keshet R et al. (2018) Rewiring urea cycle metabolism in cancer to support anabolism. Nat. Rev. Cancer 18, 634–645 [DOI] [PubMed] [Google Scholar]
- 21.Lee JS et al. (2018) Urea Cycle Dysregulation Generates Clinically Relevant Genomic and Biochemical Signatures. Cell 174, 1559–1570.e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chowell D et al. (2015) TCR contact residue hydrophobicity is a hallmark of immunogenic CD8+ T cell epitopes. Proc. Natl. Acad. Sci. U. S. A 112, E1754–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Freeman GJ et al. (2000) Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med 192, 1027–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong H et al. (1999) B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med 5, 1365–1369 [DOI] [PubMed] [Google Scholar]
- 25.Cha J-H et al. (2019) Mechanisms Controlling PD-L1 Expression in Cancer. Mol. Cell 76, 359–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanchez-Vega F et al. (2018) Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 173, 321–337.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akbay EA et al. (2013) Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 3, 1355–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Azuma K et al. (2014) Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol 25, 1935–1940 [DOI] [PubMed] [Google Scholar]
- 29.Coelho MA et al. (2017) Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity 47, 1083–1099.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Casey SC et al. (2016) MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352, 227–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mittendorf EA et al. (2014) PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res 2, 361–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parsa AT et al. (2007) Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med 13, 84–88 [DOI] [PubMed] [Google Scholar]
- 33.Kim MH et al. (2018) YAP-Induced PD-L1 Expression Drives Immune Evasion in BRAFi-Resistant Melanoma. Cancer Immunol. Res 6, 255–266 [DOI] [PubMed] [Google Scholar]
- 34.Harris IS and DeNicola GM (2020) The Complex Interplay between Antioxidants and ROS in Cancer. Trends Cell Biol. 30, 440–451 [DOI] [PubMed] [Google Scholar]
- 35.Lee P et al. (2020) Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol 21, 268–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu B et al. (2018) Targeting the upstream transcriptional activator of PD-L1 as an alternative strategy in melanoma therapy. Oncogene 37, 4941–4954 [DOI] [PubMed] [Google Scholar]
- 37.Hayes JD and McMahon M (2009) NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem. Sci 34, 176–188 [DOI] [PubMed] [Google Scholar]
- 38.Papalexi E et al. (2021) Characterizing the molecular regulation of inhibitory immune checkpoints with multimodal single-cell screens. Nat. Genet 53, 322–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DeNicola GM et al. (2011) Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noman MZ et al. (2014) PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med 211, 781–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Messai Y et al. (2016) Renal Cell Carcinoma Programmed Death-ligand 1, a New Direct Target of Hypoxia-inducible Factor-2 Alpha, is Regulated by von Hippel-Lindau Gene Mutation Status. Eur. Urol 70, 623–632 [DOI] [PubMed] [Google Scholar]
- 42.Ruf M et al. (2016) PD-L1 expression is regulated by hypoxia inducible factor in clear cell renal cell carcinoma. Int. J. cancer 139, 396–403 [DOI] [PubMed] [Google Scholar]
- 43.Koh J et al. (2016) EML4-ALK enhances programmed cell death-ligand 1 expression in pulmonary adenocarcinoma via hypoxia-inducible factor (HIF)-1α and STAT3. Oncoimmunology 5, e1108514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Opitz CA et al. (2020) The therapeutic potential of targeting tryptophan catabolism in cancer. Br. J. Cancer 122, 30–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Platten M et al. (2019) Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov 18, 379–401 [DOI] [PubMed] [Google Scholar]
- 46.Venkateswaran N et al. (2019) MYC promotes tryptophan uptake and metabolism by the kynurenine pathway in colon cancer. Genes Dev. 33, 1236–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lafita-Navarro MC et al. (2018) The aryl hydrocarbon receptor regulates nucleolar activity and protein synthesis in MYC-expressing cells. Genes Dev. 32, 1303–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kubli SP et al. (2019) AhR controls redox homeostasis and shapes the tumor microenvironment in BRCA1-associated breast cancer. Proc. Natl. Acad. Sci. U. S. A 116, 3604–3613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miao W et al. (2005) Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: direct cross-talk between phase I and II drug-metabolizing enzymes. J. Biol. Chem 280, 20340–20348 [DOI] [PubMed] [Google Scholar]
- 50.Shin S et al. (2007) NRF2 modulates aryl hydrocarbon receptor signaling: influence on adipogenesis. Mol. Cell. Biol 27, 7188–7197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takenaka MC et al. (2019) Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci 22, 729–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang L-T et al. (2017) Intestine-Specific Homeobox Gene ISX Integrates IL6 Signaling, Tryptophan Catabolism, and Immune Suppression. Cancer Res. 77, 4065–4077 [DOI] [PubMed] [Google Scholar]
- 53.Litzenburger UM et al. (2014) Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget 5, 1038–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Novikov O et al. (2016) An Aryl Hydrocarbon Receptor-Mediated Amplification Loop That Enforces Cell Migration in ER-/PR-/Her2- Human Breast Cancer Cells. Mol. Pharmacol 90, 674–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sadik A et al. (2020) IL4I1 Is a Metabolic Immune Checkpoint that Activates the AHR and Promotes Tumor Progression. Cell 182, 1252–1270.e34 [DOI] [PubMed] [Google Scholar]
- 56.Galluzzi L et al. (2017) Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol 17, 97–111 [DOI] [PubMed] [Google Scholar]
- 57.Ma Y et al. (2013) Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 38, 729–741 [DOI] [PubMed] [Google Scholar]
- 58.Ghiringhelli F et al. (2009) Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med 15, 1170–1178 [DOI] [PubMed] [Google Scholar]
- 59.Perrot I et al. (2019) Blocking Antibodies Targeting the CD39/CD73 Immunosuppressive Pathway Unleash Immune Responses in Combination Cancer Therapies. Cell Rep. 27, 2411–2425.e9 [DOI] [PubMed] [Google Scholar]
- 60.Moesta AK et al. (2020) Targeting CD39 in cancer. Nat. Rev. Immunol 20, 739–755 [DOI] [PubMed] [Google Scholar]
- 61.Chen S et al. (2020) The Expression of Adenosine A2B Receptor on Antigen-Presenting Cells Suppresses CD8(+) T-cell Responses and Promotes Tumor Growth. Cancer Immunol. Res 8, 1064–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu Q et al. (2005) Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 24, 5552–5560 [DOI] [PubMed] [Google Scholar]
- 63.Kamarajugadda S et al. (2012) Glucose oxidation modulates anoikis and tumor metastasis. Mol. Cell. Biol 32, 1893–1907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schafer ZT et al. (2009) Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cascone T et al. (2018) Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. 27, 977–987.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Evangelista L et al. (2019) 18F-FDG PET/CT in non-small-cell lung cancer patients: a potential predictive biomarker of response to immunotherapy. Nucl. Med. Commun 40, 802–807 [DOI] [PubMed] [Google Scholar]
- 67.Zappasodi R et al. (2021) CTLA-4 blockade drives loss of T(reg) stability in glycolysis-low tumours. Nature 591, 652–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tigano M et al. (2021) Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nature 591, 477–481 [DOI] [PubMed] [Google Scholar]
- 69.Zhang W et al. (2019) Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell 178, 176–189.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Caronni N et al. (2018) Downregulation of Membrane Trafficking Proteins and Lactate Conditioning Determine Loss of Dendritic Cell Function in Lung Cancer. Cancer Res. 78, 1685–1699 [DOI] [PubMed] [Google Scholar]
- 71.Gottfried E et al. (2006) Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 107, 2013–2021 [DOI] [PubMed] [Google Scholar]
- 72.Nasi A et al. (2013) Dendritic cell reprogramming by endogenously produced lactic acid. J. Immunol 191, 3090–3099 [DOI] [PubMed] [Google Scholar]
- 73.Brown TP et al. (2020) The lactate receptor GPR81 promotes breast cancer growth via a paracrine mechanism involving antigen-presenting cells in the tumor microenvironment. Oncogene 39, 3292–3304 [DOI] [PubMed] [Google Scholar]
- 74.Goel S et al. (2017) CDK4/6 inhibition triggers anti-tumour immunity. Nature 548, 471–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schaer DA et al. (2018) The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep. 22, 2978–2994 [DOI] [PubMed] [Google Scholar]
- 76.Uzhachenko RV et al. (2021) Metabolic modulation by CDK4/6 inhibitor promotes chemokine-mediated recruitment of T cells into mammary tumors. Cell Rep. 35, 108944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.O’Sullivan D et al. (2019) Metabolic interventions in the immune response to cancer. Nat. Rev. Immunol 19, 324–335 [DOI] [PubMed] [Google Scholar]
- 78.Li X et al. (2019) Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat. Rev. Clin. Oncol 16, 425–441 [DOI] [PubMed] [Google Scholar]
- 79.Im SJ et al. (2016) Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Siddiqui I et al. (2019) Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 50, 195–211.e10 [DOI] [PubMed] [Google Scholar]
- 81.Beltra J-C et al. (2020) Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 52, 825–841.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Miller BC et al. (2019) Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol 20, 326–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Peng M et al. (2016) Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 354, 481–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chang C-H et al. (2013) Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhao S et al. (2016) ATP-Citrate Lyase Controls a Glucose-to-Acetate Metabolic Switch. Cell Rep. 17, 1037–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang R et al. (2011) The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Johnson MO et al. (2018) Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell 175, 1780–1795.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Roy DG et al. (2020) Methionine Metabolism Shapes T Helper Cell Responses through Regulation of Epigenetic Reprogramming. Cell Metab. 31, 250–266.e9 [DOI] [PubMed] [Google Scholar]
- 89.Ma EH et al. (2017) Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab. 25, 345–357 [DOI] [PubMed] [Google Scholar]
- 90.Ma EH et al. (2019) Metabolic Profiling Using Stable Isotope Tracing Reveals Distinct Patterns of Glucose Utilization by Physiologically Activated CD8(+) T Cells. Immunity 51, 856–870.e5 [DOI] [PubMed] [Google Scholar]
- 91.Parry RV et al. (2005) CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol 25, 9543–9553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hui E et al. (2017) T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 355, 1428–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chang C-H et al. (2015) Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 162, 1229–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vardhana SA et al. (2020) Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat. Immunol 21, 1022–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Siska PJ et al. (2017) Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI insight 2, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yu Y-R et al. (2020) Disturbed mitochondrial dynamics in CD8(+) TILs reinforce T cell exhaustion. Nat. Immunol 21, 1540–1551 [DOI] [PubMed] [Google Scholar]
- 97.Scharping NE et al. (2021) Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol 22, 205–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wherry EJ et al. (2007) Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27, 670–684 [DOI] [PubMed] [Google Scholar]
- 99.Robert C et al. (2018) Durable Complete Response After Discontinuation of Pembrolizumab in Patients With Metastatic Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 36, 1668–1674 [DOI] [PubMed] [Google Scholar]
- 100.Joseph RW et al. (2018) Baseline Tumor Size Is an Independent Prognostic Factor for Overall Survival in Patients with Melanoma Treated with Pembrolizumab. Clin. cancer Res. an Off. J. Am. Assoc. Cancer Res 24, 4960–4967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ho P-C et al. (2015) Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 162, 1217–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Renner K et al. (2019) Restricting Glycolysis Preserves T Cell Effector Functions and Augments Checkpoint Therapy. Cell Rep. 29, 135–150.e9 [DOI] [PubMed] [Google Scholar]
- 103.Becker LM et al. (2020) Epigenetic Reprogramming of Cancer-Associated Fibroblasts Deregulates Glucose Metabolism and Facilitates Progression of Breast Cancer. Cell Rep. 31, 107701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Brand A et al. (2016) LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 24, 657–671 [DOI] [PubMed] [Google Scholar]
- 105.Quinn WJ 3rd et al. (2020) Lactate Limits T Cell Proliferation via the NAD(H) Redox State. Cell Rep. 33, 108500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Angelin A et al. (2017) Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 25, 1282–1293.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Watson MJ et al. (2021) Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature 591, 645–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang D et al. (2019) Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Uyttenhove C et al. (2003) Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med 9, 1269–1274 [DOI] [PubMed] [Google Scholar]
- 110.Opitz CA et al. (2011) An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478, 197–203 [DOI] [PubMed] [Google Scholar]
- 111.Boulland M-L et al. (2007) Human IL4I1 is a secreted L-phenylalanine oxidase expressed by mature dendritic cells that inhibits T-lymphocyte proliferation. Blood 110, 220–227 [DOI] [PubMed] [Google Scholar]
- 112.Fallarino F et al. (2006) The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J. Immunol 176, 6752–6761 [DOI] [PubMed] [Google Scholar]
- 113.Cobbold SP et al. (2009) Infectious tolerance via the consumption of essential amino acids and mTOR signaling. Proc. Natl. Acad. Sci. U. S. A 106, 12055–12060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cousin C et al. (2015) The immunosuppressive enzyme IL4I1 promotes FoxP3(+) regulatory T lymphocyte differentiation. Eur. J. Immunol 45, 1772–1782 [DOI] [PubMed] [Google Scholar]
- 115.Liu Y et al. (2018) Tumor-Repopulating Cells Induce PD-1 Expression in CD8(+) T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell 33, 480–494.e7 [DOI] [PubMed] [Google Scholar]
- 116.Mezrich JD et al. (2010) An Interaction between Kynurenine and the Aryl Hydrocarbon Receptor Can Generate Regulatory T Cells. J. Immunol 185, 3190–3198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liu Y et al. (2021) IL-2 regulates tumor-reactive CD8(+) T cell exhaustion by activating the aryl hydrocarbon receptor. Nat. Immunol 22, 358–369 [DOI] [PubMed] [Google Scholar]
- 118.Chon SY et al. (1996) Cooperative role of interferon regulatory factor 1 and p91 (STAT1) response elements in interferon-gamma-inducible expression of human indoleamine 2,3-dioxygenase gene. J. Biol. Chem 271, 17247–17252 [DOI] [PubMed] [Google Scholar]
- 119.Hassanain HH et al. (1993) Differential regulation of human indoleamine 2,3-dioxygenase gene expression by interferons-gamma and -alpha. Analysis of the regulatory region of the gene and identification of an interferon-gamma-inducible DNA-binding factor. J. Biol. Chem 268, 5077–5084 [PubMed] [Google Scholar]
- 120.Munn DH and Mellor AL (2016) IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 37, 193–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Maj T et al. (2017) Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol 18, 1332–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chen L et al. (2018) CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer Discov. 8, 1156–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li J et al. (2021) Metastasis and Immune Evasion from Extracellular cGAMP Hydrolysis. Cancer Discov. 11, 1212–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mastelic-Gavillet B et al. (2019) Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8(+) T cells. J. Immunother. cancer 7, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Canale FP et al. (2018) CD39 Expression Defines Cell Exhaustion in Tumor-Infiltrating CD8(+) T Cells. Cancer Res. 78, 115–128 [DOI] [PubMed] [Google Scholar]
- 126.Simoni Y et al. (2018) Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557, 575–579 [DOI] [PubMed] [Google Scholar]
- 127.Oshima N et al. (2020) Dynamic Imaging of LDH Inhibition in Tumors Reveals Rapid In Vivo Metabolic Rewiring and Vulnerability to Combination Therapy. Cell Rep. 30, 1798–1810.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Faubert B et al. (2017) Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Leone RD et al. (2019) Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366, 1013–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bian Y et al. (2020) Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 585, 277–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang W et al. (2019) CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Menk AV et al. (2018) 4–1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J. Exp. Med 215, 1091–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kawalekar OU et al. (2016) Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 44, 380–390 [DOI] [PubMed] [Google Scholar]
- 134.Guo Y et al. (2021) Metabolic reprogramming of terminally exhausted CD8(+) T cells by IL-10 enhances anti-tumor immunity. Nat. Immunol 22, 746–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Leone RD et al. (2018) Inhibition of the adenosine A2a receptor modulates expression of T cell coinhibitory receptors and improves effector function for enhanced checkpoint blockade and ACT in murine cancer models. Cancer Immunol. Immunother 67, 1271–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang T et al. (2020) Inosine is an alternative carbon source for CD8(+)-T-cell function under glucose restriction. Nat. Metab 2, 635–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Platten M et al. (2019) Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov 18, 379–401 [DOI] [PubMed] [Google Scholar]
- 138.Long GV et al. (2019) Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet. Oncol 20, 1083–1097 [DOI] [PubMed] [Google Scholar]
- 139.Campesato LF et al. (2020) Blockade of the AHR restricts a Treg-macrophage suppressive axis induced by L-Kynurenine. Nat. Commun 11, 4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hurst KE et al. (2019) Endoplasmic Reticulum Stress Contributes to Mitochondrial Exhaustion of CD8(+) T Cells. Cancer Immunol. Res 7, 476–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Xu S et al. (2021) Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity 54, 1561–1577.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jerby-Arnon L et al. (2018) A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175, 984–997.e24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lemos H et al. (2019) Immune control by amino acid catabolism during tumorigenesis and therapy. Nat. Rev. Cancer 19, 162–175 [DOI] [PubMed] [Google Scholar]
- 144.Frauwirth KA et al. (2002) The CD28 signaling pathway regulates glucose metabolism. Immunity 16, 769–777 [DOI] [PubMed] [Google Scholar]
- 145.Bauer DE et al. (2005) ATP citrate lyase is an important component of cell growth and transformation. Oncogene 24, 6314–6322 [DOI] [PubMed] [Google Scholar]
- 146.Sullivan LB et al. (2015) Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 162, 552–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Birsoy K et al. (2015) An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 162, 540–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.DeBerardinis RJ et al. (2007) Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. U. S. A 104, 19345–19350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Swamy M et al. (2016) Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat. Immunol 17, 712–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Li C-W et al. (2018) Eradication of Triple-Negative Breast Cancer Cells by Targeting Glycosylated PD-L1. Cancer Cell 33, 187–201.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]