

Abstract
Hematopoietic stem cells (HSCs) and lineage committed hematopoietic progenitor cells (HPCs) undergo profound shifts in gene expression during neonatal and juvenile stages of life. Temporal changes in HSC/HPC gene expression underlie concomitant changes in self-renewal capacity, lineage biases and hematopoietic output. Moreover, they can modify disease phenotypes. For example, childhood leukemias have distinct driver mutation profiles relative to adult leukemias, and they may arise from distinct cells of origin. The putative relationship between neonatal HSC/HPC ontogeny and childhood blood disorders highlights the importance of understanding how, at a mechanistic level, HSCs transition from fetal to adult transcriptional states. In this perspective piece, we summarize recent work showing that the transition is uncoordinated and imprecisely timed. We discuss implications of these findings, including mechanisms that might enable neonatal HSCs and HPCs to acquire adult-like properties over a drawn-out period of time, in lieu of precise gene regulatory networks. The transition from fetal to adult transcriptional programs coincides with a pulse of type I interferon signaling that activates many genes associated with the adult-like state. This pulse may sensitize HSCs/HPCs to mutations that drive leukemogenesis shortly after birth. If we can understand how developmental switches modulate HSC and HPC fate after birth – both under normal circumstances and in the setting of disease-causing mutations – we can potentially reprogram these switches to treat or prevent childhood leukemias.
Introduction
Blood disorders are among the most common causes of death due to disease in children in developed countries (1, 2). They can be inherited (e.g. hemoglobinopathies, immunodeficiencies and bone marrow failure syndromes) or acquired (e.g. idiopathic aplastic anemias and leukemias/lymphomas). Key pathologies of each disease emerge against a backdrop of transcriptional and epigenetic changes, within hematopoietic stem and progenitor cells, that occur normally during childhood and potentially modify the disease phenotypes. To fully understand the mechanisms that underlie childhood blood disorders, we must also understand the mechanisms that underlie neonatal and juvenile blood ontogeny.
Properties of immature blood progenitors – including hematopoietic stem cells (HSCs), multipotent progenitors (MPPs) and lineage committed hematopoietic progenitor cells (HPCs) – change considerably between fetal and adult stages of life (3). For example, fetal HSCs reside in the liver and divide frequently without losing self-renewal capacity (4–7). In contrast, adult HSCs reside in the bone marrow, divide infrequently and exhaust their self-renewal capacity after several division cycles (3, 8–12). Fetal and adult HSCs have distinct lymphoid, myeloid and erythroid lineage biases (13–15). Most fetal HSCs regenerate myeloid and lymphoid compartments with similar efficiencies in transplantation assays (a balanced output), whereas many adult HSCs regenerate only the myeloid compartment (a myeloid-biased output) (13). Some innate immune cells (e.g. peritoneal B1 B cells and γδ T cells) arise almost exclusively from fetal HSCs (16–19). Phenotypic MPP and HPC subpopulations also have distinct lineage outputs during fetal and adult stages (20, 21). These observations suggest that hematopoietic progenitors must undergo considerable transcriptional and epigenetic reprogramming between birth and adulthood.
We are just now beginning to understand how HSCs/HPCs lose fetal identity, and acquire adult identity, as they transition through neonatal and juvenile stages of development (22). In this perspective piece, we will summarize emerging concepts that help explain how fetal progenitor cells transition to adult-like states. Furthermore, we will outline several fundamental questions, and potential answers to these questions, that require further investigation.
Fetal origins of postnatal hematopoietic cells
Sites of hemopoiesis change through the course of fetal development. The earliest blood forming cells are born in the yolk sac around 3-4 weeks gestation in humans and embryonic day (E)7.5 in mice (23–26). These primitive erythroid progenitors provide blood during early stages of embryogenesis, but they cannot engraft adult mice, and they do not contribute to lifelong hematopoiesis (27–31). The yolk sac subsequently gives rise to erythroid-myeloid progenitors (EMPs) and lymphoid restricted progenitors (23, 32–34). EMPs help sustain embryonic hematopoiesis (34), but with notable exceptions – e.g., tissue resident macrophages and microglia – their progeny do not persist beyond birth (35, 36). Definitive hematopoiesis follows next, beginning at 4-5 weeks gestation in humans and E10.5 in mice (27, 28, 37–41). During this period, HSCs with long-term multilineage repopulating activity arise from hemogenic endothelium located in the dorsal aorta, near the aorta-gonad-mesonephros, as well as from arteries within the placenta (27, 28, 37–42). Distinct, committed progenitor populations may emerge from the hemogenic endothelium, in parallel (43, 44). Definitive HSCs migrate from the aorta to the placenta and liver where their numbers expand considerably (5, 45–48). In mice, the functional HSC numbers (defined by limiting dilution transplantation assays) expand in the liver from less than 100 to ~1000 between E12.5 and E15.5 (47–49). By late gestation, HSCs, MPPs and HPCs begin to seed the bone marrow which then serves as the primary hematopoietic organ (47, 50).
Elegant lineage tracing experiments have confirmed that postnatal progenitors arise from fetal hematopoietic progenitors rather than a separate anlage. Rodewald and colleagues used a Tie2-CreER allele to label HSCs, via a Cre-inducible yellow fluorescent protein (YFP) reporter, as they emerged from the aorta-gonad-mesonephros at E10.5 (51). The authors confirmed YFP expression in HSCs, but not more committed progenitor populations, immediately after Cre activation. The YFP-expressing HSCs persisted after birth and gave rise to differentiated blood cells even in adulthood. In a follow-up paper, the same group used genetic barcoding to show that fetal HSCs give rise to adult HSCs and HSC-derived blood cells in 9- to 11-month-old mice (52). Similar results were seen by lineage tracing fetal progenitors with a Scl-CreER transgene (53). Thus, fetal HSCs can give rise to neonatal HSCs, adult HSCs and committed adult progenitors.
McKinney-Freeman and colleagues used a different strategy to quantify contributions of fetal progenitors to adult hematopoiesis. They induced Cre-mediated recombination of a Confetti reporter allele at E7-8.5, E8.5-11.5 and E11.5-14.5 (54, 55). The Confetti allele expressed mutually exclusive green, yellow, red or cyan fluorescent proteins as a consequence of stochastic rearrangement. For each age of induction, the authors were able to infer the number of fetal progenitors that gave rise to adult hematopoietic progenitors based on mouse to mouse variation in color distributions (i.e., variation increases as the number of fetal progenitors decreases). They concluded that ~600 fetal progenitors sustain lifelong hematopoiesis. Similar numbers of unique clones contributed to adult hematopoiesis irrespective of whether recombination was induced at E7-8.5, E8.5-11.5 or E11.5-14.5 (55). This surprising level of stability contrasts with transplantation data showing that fetal HSC numbers increase ~10-fold between E12.5 and E15.5 (47–49). The cause of this of the discrepancy remains unclear, but it suggests that not all fetal HSCs are fated to contribute to postnatal hematopoiesis.
Separate lineage tracing experiments have confirmed that some fetal HSCs make only limited contributions to postnatal hematopoiesis. Beaudin, Forsberg and colleagues used a Flk2-Cre allele, and a Cre-inducible Tomato to green fluorescent protein (GFP) reporter allele, to lineage trace Flk2-expressing progenitors at different stages of development (19). The authors found that all adult HSCs expressed the Tomato reporter, rather than GFP, and thus had no history of Flk2-Cre expression (19, 56). In contrast, approximately 20% of E14 fetal HSCs expressed GFP. GFP-positive HSCs could repopulate lethally irradiated mice, indicating that they were bona fide HSCs, yet they disappeared after birth. GFP-positive HSCs cycled more rapidly than GFP-negative HSCs, they displayed lymphoid bias, and they contributed disproportionately to fetal-derived B1a B-cell and γδ T cell populations (19). These observations confirmed that fetal HSC subpopulations can have distinct fates, and only a fraction of all fetal HSCs give rise to the adult HSC pool. It is not clear why Flk2-expressing HSCs disappear from the bone marrow after birth. Nevertheless, the persisting HSCs must undergo profound transcriptional and epigenetic reprogramming between birth and adulthood to account for changes in proliferation rate, hematopoietic output, lineage biases and leukemogenic potential.
Fetal and adult HSCs have distinct transcriptomes that account for differences in self-renewal and lineage differentiation
Several papers have compared transcript profiles and gene dependencies of fetal and adult HSCs (22, 57–61). For the most part, the studies focused on fetal and adult states, with little emphasis placed on the neonatal period. Nevertheless, they have provided important insights into how mechanisms of HSC self-renewal and lineage commitment change over time.
Fetal regulators.
Fetal HSCs express genes that encode transcription factors (e.g. Sox17 and Arid3a) and RNA binding proteins (e.g. Lin28b, Igf2bp1, Igf2bp2 and Igf2bp3) that promote self-renewal and fetal lymphocyte production. Lin28b and Igf2bp1-3 all enhance HSC self-renewal when ectopically expressed in adult HSCs (22, 61, 62). Furthermore, Lin28b can reprogram adult progenitors to produce fetal B and T cells, and it promotes erythroid cell production at the expense of myeloid cell production (15, 61–64). Sox17 is required for fetal HSC self-renewal (6), and ectopic SOX17 expression can induce other fetal transcripts in adult HSCs (7). Hmga2 enhances HSC self-renewal without altering fetal lineage biases (61). Arid3a promotes fetal B cell production (62, 65). Downregulation of these genes after birth likely contributes to reduced self-renewal activity and loss of fetal B cell potential in adult HSCs.
Adult regulators.
Several transcription factor-encoding genes are more highly expressed in adult HSCs than in fetal HSCs. These include Cebpa, Egr1, Esr1, Sox6, Klf9, Nfia and Bhlhe41 (22, 57, 58, 66). Cebpa and Egr1 both promote HSC quiescence (66, 67). Esr1 promotes HSC proliferation during pregnancy (68). Roles for several other transcription factors in self-renewal and HSC/HPC lineage commitment have yet to be described. Indeed, there remains a significant gap between our understanding of which transcription factors are differentially expressed during HSC/HPC ontogeny and how they account for fetal- and adult-specific fates.
Genes with temporally stable gene expression patterns but dynamic functions.
Not all genes with age-specific functions are differentially expressed through HSC ontogeny. For example, several epigenetic regulators are required to sustain HSCs after birth, but not before. These include Eed, Kat8/Mof, Kmt2a/Mll1 and Ash1l (69–72). Fetal hematopoiesis proceeds normally in the absence of these genes until late gestation (Kat8) or shortly after birth (Eed, Kmt2a, Ash1l). At that point, HSCs lose self-renewal capacity, and hematopoiesis collapses (69–72). These phenotypes are intriguing for two reasons. First, the encoded proteins place chromatin marks that one would expect necessary at all stages of life. Second, the hematopoietic collapse observed shortly after birth likely coincides with the transition from fetal to adult regulatory programs. This would push the onset of the transition earlier than previously thought.
The transition from fetal to adult transcriptional states is gradual in HSCs and HPCs, and it begins before birth
Murine HSCs have long been thought to transition from fetal- to adult-like states between 3 and 4 weeks after birth (73, 74). This conclusion has been based on observations of HSC proliferation and repopulating activity in lethally irradiated mice. In one of the earliest papers on this topic, Bowie, Eaves and colleagues evaluated repopulating activity as a function of cell cycle stage. Until 3 weeks after birth, repopulating activity was evident in G1/S/G2/M fractions of the fetal liver or bone marrow; from 4 weeks onward, it was evident primarily in the G0 fraction (73). In a follow-up paper, the same group showed that 3-week-old HSC numbers expanded after transplantation at rates similar to fetal HSCs, whereas 4-week-old HSC numbers expanded at rates similar to adult HSCs (13, 74). Finally, several groups have shown that HSCs lose B1a potential around 3-4 weeks after birth (16, 17, 64, 75). These findings all argue for an abrupt postnatal transition, but most of the assays generated categorical outputs (e.g., repopulation vs. no repopulation) that would not necessarily resolve gradual changes in HSC and HPC identity. Recent innovations – including improved HSC/HPC surface marker phenotypes, single cell RNA-seq (scRNA-seq) and low-input chromatin profiling techniques – have made it possible to revisit neonatal HSC ontogeny to better assess when the transition starts, whether it is gradual or abrupt, and how transcriptional changes are coordinated (22, 76–79).
Our lab recently performed an extensive series of scRNA-seq and epigenome profiling assays to characterize the transition from fetal to adult transcriptional states in both HSCs and HPCs (22). We found that approximately one third of the transcriptome changes as HSCs and HPCs transition from fetal to adult stages of life. These changes recapitulate differences between fetal and adult HSCs that were previously described at the population level (57, 58, 60, 61). However, the single cell resolution provided several new insights.
The transition from fetal to adult identity is gradual rather than precipitous. We used a technique called quadratic programming to assign identity scores to individual HSCs and HPCs at various stages after birth. The scores quantified how adult-like each HSC/HPC was based on its transcriptome, and they allowed us to resolve whether HSCs/HPCs convert gradually or abruptly from fetal to adult transcriptional states. We found that the transition is gradual. Individual HSCs/HPCs had similar adult identity scores at each age, and the scores increased gradually over time rather than precipitously at one specific age. Direct measurements of HSC proliferation rates also revealed a gradual transition to quiescence.
The transition reflects poorly coordinated changes in heterochronic gene expression. Neonatal HSCs and HPCs showed extensive cell to cell variation with regard to heterochronic gene expression (Figure 1). This variation was not simply due to dropout associated with the scRNA-seq technique (i.e., failure to detect expressed transcripts). Heterochronic genes (i.e., those whose expression changed over time) did not coalesce into regulatory networks; rather, the changes appeared stochastic. The probability that an HSC would express a given adult identity gene, or inactivate a given fetal identity gene, increased gradually over time. These observations suggest that heterochronic genes and enhancers function autonomously from one another rather than as part of robust networks, and changes in gene expression do not reflect a coordinated response to precisely timed stimuli.
The transition begins before birth. By birth, HSCs had begun to activate adult identity genes and inactivate fetal identity genes. These transcriptional changes were evident both in liver and bone marrow HSCs. Thus, fetal HSCs begin transitioning to adult identity much earlier than previously appreciated in mice, irrespective of where they localize. Note that we are not disavowing a role for organ- and age-specific niches in shaping HSC/HPC fate. However, the data show that activation of adult transcriptional programs during late gestation is niche-independent.
The transition coincides with a pulse of type I Interferon (IFN) signaling. A large percentage of adult identity genes are type I IFN targets. These genes were activated during late gestation, between E16 and E18, both in HSCs and HPCs (Figure 1). This was the only precisely timed switch that we identified over the course of neonatal HSC ontogeny. Its implications are discussed in greater detail below.
The transition superimposes an adult enhancer landscape onto the pre-existing fetal enhancer landscape. Consistent with prior studies, we found that HSC and HPC epigenomes undergo extensive remodeling between fetal and adult stages of life (21, 60, 80). When we evaluated neonatal HSCs and HPCs, we observed uncoordinated changes in chromatin accessibility that mirrored changes in gene expression. The enhancers bound several transcription factors that exhibited stable, rather than variable, expression across neonatal development. This raises the question of how stage-specific enhancer activity is achieved. Interestingly, enhancers near adult identity genes were commissioned through the course of development, but fetal enhancers were not decommissioned. Thus, fetal gene inactivation does not require epigenome remodeling.
Figure 1. The transition from fetal to adult transcriptional states is poorly coordinated and imprecisely timed in HSCs and HPCs.
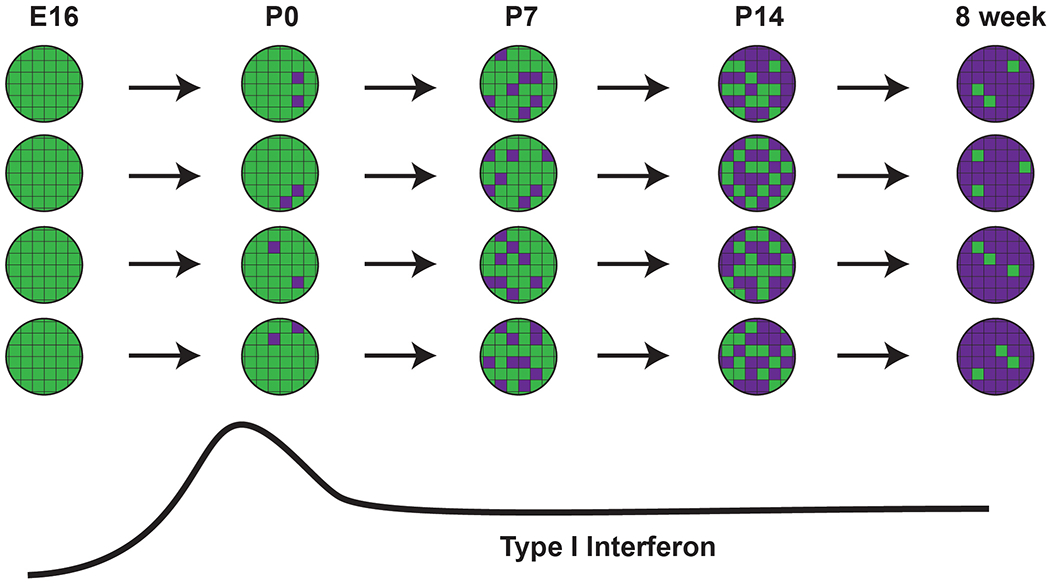
Individual cells are represented as circles, with heterochronic genes represented as boxes. Each gene converts from fetal (green) to adult (purple) states through the course of neonatal development, and each cell maintains a similar ratio of genes in fetal- and adult-like states. However, there is widespread variation among HSCs and HPCs as to which genes are active. This model predicts that all HSCs and HPCs will arrive at adult-like transcriptional states in synchrony, but the path that each cell takes to get to that point will vary.
These observations provide an important conceptual framework for understanding neonatal HSC and HPC ontogeny, but the underlying mechanisms that drive the neonatal transition remain poorly defined, particularly with regard to how the transition is initiated and timed. While more studies are needed, we offer potential explanations in the ensuing sections.
The timing problem
Several lines of evidence show that HSCs and other progenitors convert gradually from fetal- to adult-like states (22), but this raises questions as to how the gradual transition is hard-wired into cis-regulatory elements of the genome. Conventional examples of cell state changes do not offer much guidance on this point. For example, when embryonic or somatic stem cells differentiate, enhancers and super-enhancers activate lineage-specifying transcription factors, and positive feedback loops reinforce change in cell identity (81–84). Regulatory elements that might drive alternate lineage-specific programs are instead stably and heritably repressed via epigenetic alterations, including DNA methylation and repressive histone modifications (83). These changes yield robust, precipitous changes in cell identity. Neonatal HSC ontogeny does not adopt such circuitry. The transition from fetal to adult transcriptional states is uncoordinated, a majority of temporally dynamic enhancers do not qualify as super-enhancers, and the transition unfolds over weeks rather than minutes or hours (22). Fetal enhancers and promoters remain competent to drive gene expression after birth, even as transcription ceases (22). So, how might neonatal HSCs monitor and respond to the passage of time in lieu of a robust, precise gene regulatory network?
We propose that the timing problem is easily solved by the stochastic nature of the circuit. Transcription at individual loci is inherently stochastic, and it manifests as discrete bursts of mRNA synthesis that vary according to burst size (i.e., the number of transcripts generated per burst) and burst frequency (i.e., the number of bursts in a given unit of time) (85). Live imaging of individual loci, and more recently single cell RNA-seq, have shown that promoter elements generally control burst size, whereas enhancer elements control burst frequencies (86, 87). Stronger enhancers generate higher burst frequencies, and high cooperativity between strong enhancers can eliminate bursting altogether (87). One would expect that temporal changes in gene expression should follow similar rules. Thus, the noisy, uncoordinated changes in gene expression that we observed across neonatal HSC/HPC ontogeny likely reflect input from relatively weak enhancer elements.
Weak enhancers should cause adult identity gene expression to initiate over a drawn-out period of time, particularly if the enhancers act independently of one another rather than as part of a robust gene regulatory network (Figure 2). In a completely uncoordinated system, every heterochronic gene will behave as an isolated unit, and its expression will reflect a probabilistic rather than deterministic outcome. The likelihood that a given adult identity gene will be expressed at a given age, and in a given HSC, will depend on two factors: 1) the probability that its cis-regulatory elements will bind available transcription factors, and 2) the stability of the regulatory complexes. In this probabilistic system, key pioneering transcription factors are present at relatively constant levels throughout the neonatal period. Enhancers with high affinity binding sites become active earlier in life, and in a larger percentage of HSCs, than enhancers with low affinity binding sites. Thus, evolution can tune the rate at which HSCs progress from fetal to adult transcriptional states, and the order in which heterochronic genes become active across the population, by “sub-optimizing” enhancer elements (Figure 2). Of note, this model assumes that once an enhancer is pioneered, it becomes relatively stable, therein strengthening the enhancer and increasing burst frequencies. This step prevents enhancers from settling into equilibria between active and inactive states, and it keeps the arrow of developmental time moving forward. This assumption is consistent with stable changes in enhancer accessibility that take place during neonatal HSC/HPC ontogeny (22).
Figure 2. Enhancer sub-optimization can delay heterochronic gene expression after birth.
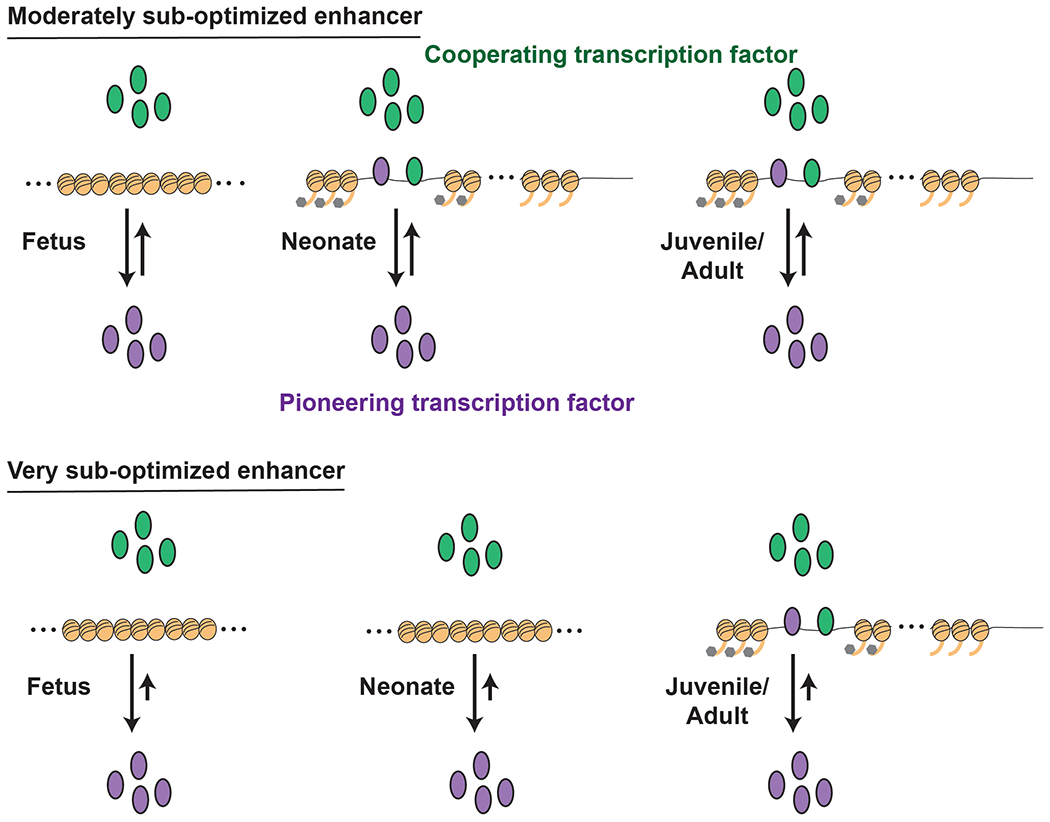
This schematic outlines a proposed explanation for the uncoordinated changes in adult identity gene expression observed in neonatal HSCs and HPCs. Pioneering transcription factors are present at similar levels throughout late fetal, neonatal and juvenile stages of development. These may include transcription factors associated with the type I IFN response. Interactions between each pioneering transcription factor and its enhancer are stochastic and shaped by the degree to which each enhancer is optimized or sub-optimized. Enhancers with moderately avid binding sites will, on average, bind the transcription factor at an earlier age than those with very low affinity sites. Once bound, the pioneering factor recruits co-factors and remodels nearby chromatin to stabilize gene expression and keep the arrow of developmental time moving forward.
Spatial regulatory programs in developing model organisms offer precedent for enhancer sub-optimization. Using Ciona intestinalis as a model, Levine and colleagues showed that an Orthodentical homeobox (Otx) enhancer requires suboptimal ETS and GATA sites to restrict Otx expression within the embryo, and binding site optimization caused ectopic Otx expression (88). In a subsequent paper, the same group showed that syntax (the order, orientation and spacing of binding sites within an enhancer) can further modulate enhancer activity (89). We propose that analogous mechanisms may control the timing of heterochronic gene expression within neonatal HSCs. In other words, evolution may optimize enhancers to drive adult genes early in neonatal development, or it may sub-optimize enhancers to delay adult gene expression.
There are caveats to the model that we propose. First, as noted above, several transcription factors exhibit fetal- or adult-specific patterns of expression (57–59, 89). These transcription factors almost certainly shape age-specific HSC and HPC fates, even if their role in timing the neonatal transition is not clear. Second, the system requires at least one chronic stimulus to initiate and sustain the transition from fetal to adult identity. Type I IFN signaling, serves as one potential stimulus. Type I IFN levels peak during late gestation and then persist at low levels after birth (far below levels observed in the context of an infection) (22). Weak, chronic IFN signaling gradually induces adult identity gene expression. Third, mechanisms must exist to inactivate fetal identity genes after birth despite sustained expression of most hematopoietic transcription factors. As noted above, we have found that fetal enhancers remain accessible and primed after birth, and they do not acquire repressive histone marks, even as gene expression declines. So, what explains the decline in fetal gene expression?
One of the most well-characterized switches in neonatal hematopoiesis, human globin switching, illustrates the key role that transcriptional repressors can play in inactivating fetal gene expression. Enhancers at the globin locus control region interact with the γ-globin promoter before birth and the β-globin promoter after birth (90, 91). These enhancers bind transcription factors, such as SCL and GATA1, that regulate erythroid gene expression at all stages of ontogeny (92). To execute the switch from γ- to β-globin expression, a transcriptional repressor, BCL11A, binds to the γ-globin promoter to displace NF-Y and recruit the NuRD repressor complex (93, 94). BCL11A translation is, in turn, inhibited by LIN28B prior to birth (95). Similar mechanisms may suppress other fetal genes during neonatal and adult stages of life, though additional studies are needed to identify candidate repressors and to understand how the interactions are timed.
Sterile inflammatory signaling and neonatal HSC/HPC identity changes
The transition from fetal to adult HSC/HPC identity begins before birth in mice, and it coincides with a pulse of type I IFN (IFNα and IFNβ) signaling that the persists at somewhat lower levels after birth (22). These cytokines induce adult gene expression programs in both HSCs and HPCs, they cause the HPC population to expand shortly before birth, and they promote Major Histocompatibility Complex-I expression in several progenitor populations (59). Type I IFN sensitizes HPCs to the FLT3 Internal Tandem Duplication (FLT3ITD) mutation as its receptor, IFNAR, is required for FLT3ITD-driven HPC expansion and target gene expression (22). This likely explains an intriguing phenomenon – FLT3ITD promotes acute myeloid leukemia initiation after birth, but not before (58). It is still not clear how type I IFN reprograms neonatal HPC fate, though the question has potential implications for neonatal immunity, lifelong blood production and childhood leukemogenesis.
The stimulus for late gestation IFNα/β production remains unclear, as well. The switch is activated even in germ free mice (22). This indicates that it reflects a normal, sterile developmental program rather than a response to infection. IFNα/β production does not localize to the liver or to other hematopoietic organs, and the cytokines do not circulate systemically in the mother (22). Instead, they are produced in the fetal skin, even under germ free conditions (22, 96). Skin undergoes an important transition between E16 and E18 in that it loses capacity for scarless healing (97, 98). This raises the intriguing possibility that normal, microbe-independent changes in the developing skin can promote adult transcriptional programs in HSCs and HPCs. Further studies are needed to more precisely identify the cells that express type I IFN (in the skin or elsewhere), and to identify the mechanisms that drive IFN expression.
The role of type I IFN during late gestation contrasts with its role in hematopoiesis at other stages of ontogeny (99). During mid-gestation, type I IFN promotes HSC emergence from the dorsal aorta (100, 101). In adulthood, type I IFN signaling promotes HSC proliferation and emergency megakaryopoiesis (102–104). IFN signaling has also drawn expanding scrutiny for its role in clonal hematopoiesis, HSC exhaustion and malignant transformation (10, 99). Thus, the late gestation/perinatal switch adds to a growing list of roles for type I IFN signaling in normal and abnormal hematopoiesis.
Neonatal HSC/HPC ontogeny and childhood leukemia initiation
The heterogeneity that we observed in neonatal HSC/HPC transcriptomes and epigenomes may help suppress rates of leukemic transformation in children. We, and others, have previously shown that pediatric leukemia driver mutations have different effects on HSC and HPC fates at different ages (58, 105–110). For example, the MLL-ENL fusion protein induces acute myeloid leukemia more efficiently during neonatal stages of development than during fetal or adult stages (106, 111). The CBFA2T3-GLIS2 fusion similarly transforms fetal HSCs/HPCs more efficiently than adult HSCs/HPCs (107). In both cases, normal developmental programs can determine whether a progenitor is competent to activate crucial effector genes that initiate leukemogenesis. The transcriptional and epigenetic heterogeneity that we observed among postnatal progenitors predicts that even at a permissive age, not every progenitor will have the requisite epigenetic landscape or transcriptional profile to support leukemia initiation (22). This may limit the number of cells that can give rise to a leukemia at any given neonatal or juvenile stage of life, and it may help explain why certain leukemogenic mutations occur far more commonly in the general population than leukemia itself (112). If we can understand which programs convey susceptibility to leukemogenic mutations, we can potentially target and re-program these vulnerabilities.
The late gestation type I IFN pulse may be one switch that potentiates leukemic transformation after birth. Childhood infection patterns have been shown to correlate with susceptibility to some subtypes of acute lymphoblastic leukemia (112), and as noted above, IFN signaling potentiates FLT3ITD target gene expression (22). These observations raise the question of whether type I IFN establishes or sustains cells of origin for a range of pediatric leukemias. Furthermore, it raises the question of whether pediatric leukemias hijack normal type I IFN-driven programs. Further studies are needed to test whether type I IFN promotes pediatric leukemia initiation, and to identify other developmental switches that may promote or suppress transformation.
Closing thoughts
We are just beginning to understand how HSCs and HPCs transition from fetal to adult transcriptional states, and a number of important questions remain. Foremost among these, we need to better understand the enhancer logic that drives the transition. What are the transcription factors? What are the enhancers? How do they interact with one another to create a gradual switch? How do they reinforce fetal, neonatal or adult lineage biases? How do they help initiate pediatric leukemias? In addition, it will be important to characterize changes in the liver and bone marrow niches that may shape neonatal HSC ontogeny, and it will be important to characterize non-transcriptional changes that may contribute to age-specific HSC/HPC fates. Finally, it will be important to characterize the fetal to adult transition as it occurs in humans. Human blood development begins much earlier in gestation than in mice, and it unfolds over a much longer period of time period. This raises the question of whether developmental switches, such as the type I interferon pulse, are conserved in humans. This question is central to ongoing efforts to model childhood leukemia initiation, and other childhood blood disorders, in relevant developmental contexts.
Highlights.
Neonatal HSCs undergo uncoordinated transcriptional reprogramming
Reprogramming can be loosely timed through enhancer sub-optimization
Type I interferon signaling promotes the transition from fetal to adult identity
Neonatal transcriptional programs can shape childhood leukemogenesis
Acknowledgements
We thank Grant Challen and Laura Schuettpelz for comments on the manuscript. We thank Samantha Morris and her lab for years of collaboration on this topic. J.A.M. is supported by grants from the NHLBI (R01HL152180 and R01HL136504), Alex’s Lemonade Stand Foundation (‘A’ Award), Hyundai Hope on Wheels, and the Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital. J.A.M. is a Leukemia & Lymphoma Society Scholar.
References
- 1.Heron M, Deaths: Leading Causes for 2018. Natl Vital Stat Rep, 2021. 70(4): p. 1–115. [PubMed] [Google Scholar]
- 2.Kyu HH, et al. Causes of death among children aged 5-14 years in the WHO European Region: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Child Adolesc Health, 2018. 2(5): p. 321–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pietras EM and Passegue E, Linking HSCs to their youth. Nat Cell Biol, 2013. 15(8): p. 885–7. [DOI] [PubMed] [Google Scholar]
- 4.Copley MR and Eaves CJ, Developmental changes in hematopoietic stem cell properties. Exp Mol Med, 2013. 45: p. e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morrison SJ, et al. The purification and characterization of fetal liver hematopoietic stem cells. Proc Natl Acad Sci U S A, 1995. 92(22): p. 10302–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim I, Saunders TL, and Morrison SJ, Sox17 dependence distinguishes the transcriptional regulation of fetal from adult hematopoietic stem cells. Cell, 2007. 130(3): p. 470–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He S, et al. Sox17 expression confers self-renewal potential and fetal stem cell characteristics upon adult hematopoietic progenitors. Genes Dev, 2011. 25(15): p. 1613–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He S, Nakada D, and Morrison SJ, Mechanisms of stem cell self-renewal. Annu Rev Cell Dev Biol, 2009. 25: p. 377–406. [DOI] [PubMed] [Google Scholar]
- 9.Bernitz JM, et al. Hematopoietic Stem Cells Count and Remember Self-Renewal Divisions. Cell, 2016. 167(5): p. 1296–1309 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walter D, et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature, 2015. [DOI] [PubMed] [Google Scholar]
- 11.Wilson A, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell, 2008. 135(6): p. 1118–29. [DOI] [PubMed] [Google Scholar]
- 12.Foudi A, et al. Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat Biotechnol, 2009. 27(1): p. 84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benz C, et al. Hematopoietic stem cell subtypes expand differentially during development and display distinct lymphopoietic programs. Cell Stem Cell, 2012. 10(3): p. 273–83. [DOI] [PubMed] [Google Scholar]
- 14.Mold JE, et al. Fetal and adult hematopoietic stem cells give rise to distinct T cell lineages in humans. Science, 2010. 330(6011): p. 1695–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rowe RG, et al. Developmental regulation of myeloerythroid progenitor function by the Lin28b-let-7-Hmga2 axis. J Exp Med, 2016. 213(8): p. 1497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosn EE, et al. Distinct B-cell lineage commitment distinguishes adult bone marrow hematopoietic stem cells. Proc Natl Acad Sci U S A, 2012. 109(14): p. 5394–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barber CL, Montecino-Rodriguez E, and Dorshkind K, Reduced production of B-1-specified common lymphoid progenitors results in diminished potential of adult marrow to generate B-1 cells. Proc Natl Acad Sci U S A, 2011. 108(33): p. 13700–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hardy RR and Hayakawa K, A developmental switch in B lymphopoiesis. Proc Natl Acad Sci U S A, 1991. 88(24): p. 11550–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beaudin AE, et al. A Transient Developmental Hematopoietic Stem Cell Gives Rise to Innate-like B and T Cells. Cell Stem Cell, 2016. 19(6): p. 768–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jassinskaja M, et al. Ontogenic shifts in cellular fate are linked to proteotype changes in lineage-biased hematopoietic progenitor cells. Cell Rep, 2021. 34(12): p. 108894. [DOI] [PubMed] [Google Scholar]
- 21.Notta F, et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science, 2016. 351(6269): p. aab2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, et al. Single-Cell Analysis of Neonatal HSC Ontogeny Reveals Gradual and Uncoordinated Transcriptional Reprogramming that Begins before Birth. Cell Stem Cell, 2020. 27(5): p. 732–747 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palis J, et al. Development of erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse. Development, 1999. 126(22): p. 5073–84. [DOI] [PubMed] [Google Scholar]
- 24.Tober J, et al. The megakaryocyte lineage originates from hemangioblast precursors and is an integral component both of primitive and of definitive hematopoiesis. Blood, 2007. 109(4): p. 1433–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Migliaccio G, et al. Human embryonic hemopoiesis. Kinetics of progenitors and precursors underlying the yolk sac----liver transition. J Clin Invest, 1986. 78(1): p. 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tavian M, et al. The human embryo, but not its yolk sac, generates lympho-myeloid stem cells: mapping multipotent hematopoietic cell fate in intraembryonic mesoderm. Immunity, 2001. 15(3): p. 487–95. [DOI] [PubMed] [Google Scholar]
- 27.Medvinsky A and Dzierzak E, Definitive hematopoiesis is autonomously initiated by the AGM region. Cell, 1996. 86(6): p. 897–906. [DOI] [PubMed] [Google Scholar]
- 28.Medvinsky AL, et al. An early pre-liver intraembryonic source of CFU-S in the developing mouse. Nature, 1993. 364(6432): p. 64–7. [DOI] [PubMed] [Google Scholar]
- 29.Sonoda T, Hayashi C, and Kitamura Y, Presence of mast cell precursors in the yolk sac of mice. Dev Biol, 1983. 97(1): p. 89–94. [DOI] [PubMed] [Google Scholar]
- 30.Yoder MC, Hiatt K, and Mukherjee P, In vivo repopulating hematopoietic stem cells are present in the murine yolk sac at day 9.0 postcoitus. Proc Natl Acad Sci U S A, 1997. 94(13): p. 6776–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cumano A, et al. Intraembryonic, but not yolk sac hematopoietic precursors, isolated before circulation, provide long-term multilineage reconstitution. Immunity, 2001. 15(3): p. 477–85. [DOI] [PubMed] [Google Scholar]
- 32.Boiers C, et al. Lymphomyeloid contribution of an immune-restricted progenitor emerging prior to definitive hematopoietic stem cells. Cell Stem Cell, 2013. 13(5): p. 535–48. [DOI] [PubMed] [Google Scholar]
- 33.McGrath KE, et al. Distinct Sources of Hematopoietic Progenitors Emerge before HSCs and Provide Functional Blood Cells in the Mammalian Embryo. Cell Rep, 2015. 11(12): p. 1892–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen MJ, et al. Erythroid/myeloid progenitors and hematopoietic stem cells originate from distinct populations of endothelial cells. Cell Stem Cell, 2011. 9(6): p. 541–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ginhoux F, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science, 2010. 330(6005): p. 841–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gomez Perdiguero E, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature, 2015. 518(7540): p. 547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muller AM, et al. Development of hematopoietic stem cell activity in the mouse embryo. Immunity, 1994. 1(4): p. 291–301. [DOI] [PubMed] [Google Scholar]
- 38.North TE, et al. Runx1 expression marks long-term repopulating hematopoietic stem cells in the midgestation mouse embryo. Immunity, 2002. 16(5): p. 661–72. [DOI] [PubMed] [Google Scholar]
- 39.de Bruijn MF, et al. Definitive hematopoietic stem cells first develop within the major arterial regions of the mouse embryo. EMBO J, 2000. 19(11): p. 2465–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ivanovs A, et al. Vast Self-Renewal Potential of Human AGM Region HSCs Dramatically Declines in the Umbilical Cord Blood. Stem Cell Reports, 2020. 15(4): p. 811–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ivanovs A, et al. Highly potent human hematopoietic stem cells first emerge in the intraembryonic aorta-gonad-mesonephros region. J Exp Med, 2011. 208(12): p. 2417–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rhodes KE, et al. The emergence of hematopoietic stem cells is initiated in the placental vasculature in the absence of circulation. Cell Stem Cell, 2008. 2(3): p. 252–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ganuza M, et al. Murine hemogenic endothelial precursors display heterogeneous hematopoietic potential ex vivo. Exp Hematol, 2017. 51: p. 25–35 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu Q, et al. Developmental trajectory of prehematopoietic stem cell formation from endothelium. Blood, 2020. 136(7): p. 845–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robin C, et al. Human placenta is a potent hematopoietic niche containing hematopoietic stem and progenitor cells throughout development. Cell Stem Cell, 2009. 5(4): p. 385–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ottersbach K and Dzierzak E, The murine placenta contains hematopoietic stem cells within the vascular labyrinth region. Dev Cell, 2005. 8(3): p. 377–87. [DOI] [PubMed] [Google Scholar]
- 47.Gekas C, et al. The placenta is a niche for hematopoietic stem cells. Dev Cell, 2005. 8(3): p. 365–75. [DOI] [PubMed] [Google Scholar]
- 48.Ema H and Nakauchi H, Expansion of hematopoietic stem cells in the developing liver of a mouse embryo. Blood, 2000. 95(7): p. 2284–8. [PubMed] [Google Scholar]
- 49.Ganuza M, et al. Clones assemble! The clonal complexity of blood during ontogeny and disease. Exp Hematol, 2020. 83: p. 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Christensen JL, et al. Circulation and chemotaxis of fetal hematopoietic stem cells. PLoS Biol, 2004. 2(3): p. E75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Busch K, et al. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature, 2015. 518(7540): p. 542–6. [DOI] [PubMed] [Google Scholar]
- 52.Pei W, et al. Polylox barcoding reveals haematopoietic stem cell fates realized in vivo. Nature, 2017. 548(7668): p. 456–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gothert JR, et al. In vivo fate-tracing studies using the Scl stem cell enhancer: embryonic hematopoietic stem cells significantly contribute to adult hematopoiesis. Blood, 2005. 105(7): p. 2724–32. [DOI] [PubMed] [Google Scholar]
- 54.Ganuza M, et al. The global clonal complexity of the murine blood system declines throughout life and after serial transplantation. Blood, 2019. 133(18): p. 1927–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ganuza M, et al. Lifelong haematopoiesis is established by hundreds of precursors throughout mammalian ontogeny. Nat Cell Biol, 2017. 19(10): p. 1153–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boyer SW, et al. All hematopoietic cells develop from hematopoietic stem cells through Flk2/Flt3-positive progenitor cells. Cell Stem Cell, 2011. 9(1): p. 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McKinney-Freeman S, et al. The transcriptional landscape of hematopoietic stem cell ontogeny. Cell Stem Cell, 2012. 11(5): p. 701–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Porter SN, et al. Fetal and neonatal hematopoietic progenitors are functionally and transcriptionally resistant to Flt3-ITD mutations. Elife, 2016. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jassinskaja M, et al. Comprehensive Proteomic Characterization of Ontogenic Changes in Hematopoietic Stem and Progenitor Cells. Cell Rep, 2017. 21(11): p. 3285–3297. [DOI] [PubMed] [Google Scholar]
- 60.Gao P, et al. Transcriptional regulatory network controlling the ontogeny of hematopoietic stem cells. Genes Dev, 2020. 34(13-14): p. 950–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Copley MR, et al. The Lin28b-let-7-Hmga2 axis determines the higher self-renewal potential of fetal haematopoietic stem cells. Nat Cell Biol, 2013. 15(8): p. 916–25. [DOI] [PubMed] [Google Scholar]
- 62.Wang S, et al. Enhancement of LIN28B-induced hematopoietic reprogramming by IGF2BP3. Genes Dev, 2019. 33(15-16): p. 1048–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yuan J, et al. Lin28b reprograms adult bone marrow hematopoietic progenitors to mediate fetal-like lymphopoiesis. Science, 2012. 335(6073): p. 1195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kristiansen TA, et al. Cellular Barcoding Links B-1a B Cell Potential to a Fetal Hematopoietic Stem Cell State at the Single-Cell Level. Immunity, 2016. 45(2): p. 346–57. [DOI] [PubMed] [Google Scholar]
- 65.Zhou Y, et al. Lin28b promotes fetal B lymphopoiesis through the transcription factor Arid3a. J Exp Med, 2015. 212(4): p. 569–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ye M, et al. C/EBPa controls acquisition and maintenance of adult haematopoietic stem cell quiescence. Nat Cell Biol, 2013. 15(4): p. 385–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Min IM, et al. The transcription factor EGR1 controls both the proliferation and localization of hematopoietic stem cells. Cell Stem Cell, 2008. 2(4): p. 380–91. [DOI] [PubMed] [Google Scholar]
- 68.Nakada D, et al. Oestrogen increases haematopoietic stem-cell self-renewal in females and during pregnancy. Nature, 2014. 505(7484): p. 555–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xie H, et al. Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell, 2014. 14(1): p. 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Valerio DG, et al. Histone acetyltransferase activity of MOF is required for adult but not early fetal hematopoiesis in mice. Blood, 2017. 129(1): p. 48–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gan T, et al. Developmentally induced Mll1 loss reveals defects in postnatal haematopoiesis. Leukemia, 2010. 24(10): p. 1732–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jones M, et al. Ash1l controls quiescence and self-renewal potential in hematopoietic stem cells. J Clin Invest, 2015. 125(5): p. 2007–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bowie MB, et al. Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific engraftment defect. J Clin Invest, 2006. 116(10): p. 2808–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bowie MB, et al. Identification of a new intrinsically timed developmental checkpoint that reprograms key hematopoietic stem cell properties. Proc Natl Acad Sci U S A, 2007. 104(14): p. 5878–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kikuchi K and Kondo M, Developmental switch of mouse hematopoietic stem cells from fetal to adult type occurs in bone marrow after birth. Proc Natl Acad Sci U S A, 2006. 103(47): p. 17852–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim I, et al. Enhanced purification of fetal liver hematopoietic stem cells using SLAM family receptors. Blood, 2006. 108(2): p. 737–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kent DG, et al. Prospective isolation and molecular characterization of hematopoietic stem cells with durable self-renewal potential. Blood, 2009. 113(25): p. 6342–50. [DOI] [PubMed] [Google Scholar]
- 78.Corces MR, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods, 2017. 14(10): p. 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schmidl C, et al. ChIPmentation: fast, robust, low-input ChIP-seq for histones and transcription factors. Nat Methods, 2015. 12(10): p. 963–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen C, et al. Spatial Genome Re-organization between Fetal and Adult Hematopoietic Stem Cells. Cell Rep, 2019. 29(12): p. 4200–4211 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Whyte WA, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell, 2013. 153(2): p. 307–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hnisz D, et al. Super-enhancers in the control of cell identity and disease. Cell, 2013. 155(4): p. 934–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brand M, et al. Polycomb/Trithorax Antagonism: Cellular Memory in Stem Cell Fate and Function. Cell Stem Cell, 2019. 24(4): p. 518–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Davidson EH, Emerging properties of animal gene regulatory networks. Nature, 2010. 468(7326): p. 911–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Raj A and van Oudenaarden A, Nature, nurture, or chance: stochastic gene expression and its consequences. Cell, 2008. 135(2): p. 216–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Larsson AJM, et al. Genomic encoding of transcriptional burst kinetics. Nature, 2019. 565(7738): p. 251–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fukaya T, Lim B, and Levine M, Enhancer Control of Transcriptional Bursting. Cell, 2016. 166(2): p. 358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Farley EK, et al. Suboptimization of developmental enhancers. Science, 2015. 350(6258): p. 325–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Farley EK, et al. Syntax compensates for poor binding sites to encode tissue specificity of developmental enhancers. Proc Natl Acad Sci U S A, 2016. 113(23): p. 6508–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sankaran VG and Orkin SH, The switch from fetal to adult hemoglobin. Cold Spring Harb Perspect Med, 2013. 3(1): p. a011643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xu J, et al. Transcriptional silencing of {gamma}-globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes Dev, 2010. 24(8): p. 783–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cantor AB and Orkin SH, Transcriptional regulation of erythropoiesis: an affair involving multiple partners. Oncogene, 2002. 21(21): p. 3368–76. [DOI] [PubMed] [Google Scholar]
- 93.Liu N, et al. Transcription factor competition at the gamma-globin promoters controls hemoglobin switching. Nat Genet, 2021. 53(4): p. 511–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xu J, et al. Corepressor-dependent silencing of fetal hemoglobin expression by BCL11A. Proc Natl Acad Sci U S A, 2013. 110(16): p. 6518–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Basak A, et al. Control of human hemoglobin switching by LIN28B-mediated regulation of BCL11A translation. Nat Genet, 2020. 52(2): p. 138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Duc-Goiran P, et al. Developmental control of IFN-alpha expression in murine embryos. Exp Cell Res, 1994. 214(2): p. 570–83. [DOI] [PubMed] [Google Scholar]
- 97.Wulff BC, et al. Mast cells contribute to scar formation during fetal wound healing. J Invest Dermatol, 2012. 132(2): p. 458–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Walmsley GG, et al. A mouse fetal skin model of scarless wound repair. J Vis Exp, 2015(95): p. 52297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Demerdash Y, et al. Yin and Yang: The dual effects of interferons on hematopoiesis. Exp Hematol, 2021. 96: p. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li Y, et al. Inflammatory signaling regulates embryonic hematopoietic stem and progenitor cell production. Genes Dev, 2014. 28(23): p. 2597–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kim PG, et al. Interferon-alpha signaling promotes embryonic HSC maturation. Blood, 2016. 128(2): p. 204–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Essers MA, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature, 2009. 458(7240): p. 904–8. [DOI] [PubMed] [Google Scholar]
- 103.Pietras EM, et al. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J Exp Med, 2014. 211(2): p. 245–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Haas S, et al. Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors. Cell Stem Cell, 2015. 17(4): p. 422–34. [DOI] [PubMed] [Google Scholar]
- 105.Magee JA, et al. Temporal changes in PTEN and mTORC2 regulation of hematopoietic stem cell self-renewal and leukemia suppression. Cell Stem Cell, 2012. 11(3): p. 415–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Okeyo-Owuor T, et al. The efficiency of murine MLL-ENL-driven leukemia initiation changes with age and peaks during neonatal development. Blood Adv, 2019. 3(15): p. 2388–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lopez CK, et al. Ontogenic Changes in Hematopoietic Hierarchy Determine Pediatric Specificity and Disease Phenotype in Fusion Oncogene-Driven Myeloid Leukemia. Cancer Discov, 2019. 9(12): p. 1736–1753. [DOI] [PubMed] [Google Scholar]
- 108.Secker KA, et al. Only Hematopoietic Stem and Progenitor Cells from Cord Blood Are Susceptible to Malignant Transformation by MLL-AF4 Translocations. Cancers (Basel), 2020. 12(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Barrett NA, et al. Mll-AF4 Confers Enhanced Self-Renewal and Lymphoid Potential during a Restricted Window in Development. Cell Rep, 2016. 16(4): p. 1039–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rowe RG, et al. The developmental stage of the hematopoietic niche regulates lineage in MLL-rearranged leukemia. J Exp Med, 2019. 216(3): p. 527–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sinha R, et al. Development of embryonic and adult leukemia mouse models driven by MLL-ENL translocation. Exp Hematol, 2020. 85: p. 13–19. [DOI] [PubMed] [Google Scholar]
- 112.Greaves M, A causal mechanism for childhood acute lymphoblastic leukaemia. Nat Rev Cancer, 2018. 18(8): p. 471–484. [DOI] [PMC free article] [PubMed] [Google Scholar]