

Abstract
Defense against genome invaders universally relies on RNA-guided immunity. Prokaryotic CRISPR-Cas and eukaryotic RNA interference pathways recognize targets by complementary base-pairing, which places the sequences of their guide RNAs at the center of self/nonself discrimination. Here, we explore the sequence space of PIWI-interacting RNAs (piRNAs), the genome defense of animals, and establish functional priority among individual sequences. Our results reveal that only the topmost abundant piRNAs are commonly present in every cell, whereas rare sequences generate cell-to-cell diversity in flies and mice. We identify a skewed distribution of sequence abundance as a hallmark of piRNA populations and show that quantitative differences of more than a 1000-fold are established by conserved mechanisms of biogenesis. Finally, our genomics analyses and direct reporter assays reveal that abundance determines function in piRNA-guided genome defense. Taken together, we identify an effective sequence space and untangle two classes of piRNAs that differ in complexity and function. The first class represents the topmost abundant sequences and drives silencing of genomic parasites. The second class sparsely covers an enormous sequence space. These rare piRNAs cannot function in every cell, every individual, or every generation but create diversity with potential for adaptation in the ongoing arms race with genome invaders.
Retroviruses and other foreign nucleic acids pose a threat to genome integrity (Slotkin and Martienssen 2007; Kazazian and Moran 2017). In the ongoing arms race with invading nucleic acids, host genomes accumulated scars, eliminated deleterious mutations, and selected for the rare advantageous insertions, but above all, they devised defense pathways (Cosby et al. 2019). RNA-guided mechanisms, CRISPR pathways and RNA interference (RNAi), protect the integrity of genomes from bacteria to humans (Williams et al. 2015; Koonin 2019). Animal germ cells employ a specialized RNAi pathway, PIWI proteins and their PIWI-interacting small RNAs (piRNAs), to establish lasting epigenetic restriction of mobile genetic elements (Ozata et al. 2019; Ophinni et al. 2019). Loss of key piRNA pathway genes universally results in sterility of the animal and threatens the survival of the species (Iwasaki et al. 2015; Czech et al. 2018; Ozata et al. 2019).
Specificity of genome defense is imperative, because failing to silence a single parasite or wrongly restricting a single essential host gene is deleterious. Target specificity is determined by complementary base-pairing and places the sequences of piRNAs at the center of self/nonself discrimination (Brennecke et al. 2008; Paul 2010; Wasik et al. 2015; Janssen et al. 2018). Mature piRNAs are ∼30 nucleotides (nt) in length and generated from hundreds of precursors that can be more than a thousand times their size (Supplemental Fig. S1A). A single precursor can give rise to hundreds or thousands of different piRNAs and is consumed in the process (Iwasaki et al. 2015; Czech et al. 2018; Ozata et al. 2019). Although core mechanisms of piRNA silencing are conserved from flies to mice, the sequences of piRNAs and thus their target repertoire are variable and poorly understood (Parhad and Theurkauf 2019; Özata et al. 2020; Zhang et al. 2020).
In Drosophila, a genomic region of more than 100 kilobases (kb), flamenco (lncRNA:flam), has long been known as a major transposon control region (Supplemental Fig. S1B; Lin and Spradling 1997; Sarot et al. 2004; Wu et al. 2020). This essential piRNA cluster looks like a transposon graveyard, with densely packed fragments of endogenous retroviruses (Brennecke et al. 2007). It is suggested to produce a single transcript that captures most transposon fragments in antisense orientation, so that the resulting piRNAs identify these very elements by sequence complementarity. Insertion of a novel sequence into a piRNA cluster promotes silencing of complementary targets, and a single fortunate insertion of a genomic invader could provide immunity against the parasite (Muerdter et al. 2012; Yu et al. 2019; Zhang et al. 2020). Upon association with PIWI-proteins, piRNAs become sequence-specific guides that trigger transcriptional and post-transcriptional restriction (Siomi et al. 2011).
piRNAs are generated from their long precursors by the conserved endonuclease Zucchini (Zuc)/Pld6 or by the piRNA-guided nuclease activity of PIWI proteins themselves (Brennecke et al. 2007; Gunawardane et al. 2007; Ipsaro et al. 2012; Nishimasu et al. 2012; Czech et al. 2018). The Zuc-processor complex has a characteristic bias to generate piRNAs with a uridine at the 5′-most position (1U-bias) (Stein et al. 2019; Izumi et al. 2020). Additional sequence motifs, RNA structures, RNA-binding proteins, and piRNA-guided cleavage itself have been implicated to instruct patterns of piRNA biogenesis and shape the piRNA sequence space (Han et al. 2015; Ishizu et al. 2015; Mohn et al. 2015; Pandey et al. 2017; Rogers et al. 2017; Izumi et al. 2020). However, a universal signature remains elusive. Here, we identify conserved rules that govern the production of abundant piRNAs and establish functional priority among individual piRNA sequences.
Results
A single cell contains only a fraction but never the entire set of piRNA sequences
piRNAs comprise millions of unique sequences in flies and mice (Fig. 1A). To better understand the sequence diversity of piRNAs, we estimated the total expected number of piRNAs as a function of our sampling efforts. Using an optimized application of species accumulation curves for large-scale sequencing data (Deng et al. 2015), we predict millions of unique sequences in a saturated population of about half a billion Piwi-piRNAs and hundreds of millions Miwi- or Mili-piRNAs (Fig. 1B). The large number of piRNAs presents a biological dilemma because there is a physical constraint on how many piRNAs a cell can accommodate. We calculated the average number of piRNA molecules in a single Drosophila ovarian somatic sheath cell (OSC), a cell culture model for Piwi-piRNA biology (Saito et al. 2009). To estimate the number of piRNA molecules in a single cell, we combined a known number of synthetic reference oligonucleotides with a defined number of counted cells and prepared small RNAs (20–30 nt) for sequencing (Supplemental Figs. S1C, S2). The resulting data enable a relative quantification of endogenous small RNAs to the synthetic oligonucleotides, which provide a reference for the original cell count (Bissels et al. 2009; Farazi et al. 2011). To account for experimental differences in ligation efficacy of 3′ 2′-O-methylated piRNAs compared to microRNAs (miRNAs), we calculated a correction factor based on publicly available data sets from the Zamore lab (Supplemental Table S6; Gainetdinov et al. 2018). We used unique molecular identifiers (UMIs) to accurately represent the original number of small RNA molecules and varying adapter-terminal nucleotides to minimize ligation bias (Supplemental Figs. S2, S3; Hafner et al. 2011; Kivioja et al. 2012; Fu et al. 2018; Anastasakis et al. 2021). Combining calculations from eight biological replicates, we estimated that the total number of piRNAs in a single cell ranges from ∼500,000 to ∼800,000 and does not exceed one million (Fig. 1C; Supplemental Table S7). Our estimates bolster previous observations in mice that place piRNAs among the most abundant molecules in a cell, with numbers potentially close to ribosome (Gainetdinov et al. 2018). However, despite the large number of piRNAs within a cell, there are more unique sequences than the total number of molecules that a single cell can contain. Our data imply that each cell contains only a fraction but never the entire set of piRNA sequences. With the essential role of piRNAs in germ cells, the heterogeneous complement of single cells could be a key contributor to reproductive polymorphisms and epigenetic variability.
Figure 1.

The sequence diversity of piRNAs exceeds the capacity of an individual cell and generates cell-to-cell variability. (A) Sequence diversity (sequences/reads) of piRNAs associated with different PIWI proteins in flies and mice. Piwi-piRNAs in ovarian somatic sheath cells (OSCs) (mean ± SD, n = 3; this study). Piwi-, Aubergine- (Aub), and Argonaute 3- (AGO3) piRNAs in Drosophila ovaries (GEO: GSE83698) (n = 1). Miwi/Piwil1- and Mili/Piwil2-piRNAs in primary spermatocytes (BioProject: PRJNA421205) (n = 2, range indicated) (Supplemental Table S1). For comparison: microRNAs (miRNAs) according to miRBase annotation from total small RNA data sets (GEO: GSE83698 and SRA: SRR3715418). (B) Prediction of piRNA populations according to species accumulation curves based on experimental sampling. Piwi-piRNAs from OSCs (this study) (n = 3) and Mili- and Miwi- piRNAs from primary spermatocytes (SPI) (n = 2) (BioProject: PRJNA421205). The number of sampled reads (x-axis) and sequences (y-axis) is indicated by dotted lines. (C) The average number of Piwi-piRNAs and miRNAs in a single cell. Numbers based on calibrated sequencing of total small RNAs from OSCs (median, 25th–75th percentile, data points for the eight biological replicates are indicated [n = 8]) (Supplemental Fig. S1C). Mouse data from primary spermatocytes (SPI), secondary spermatocytes (SPII), and round spermatids (RS) from Gainetdinov et al. (2018) are shown for comparison.
A skewed distribution of sequence abundance results in a few common and many rare piRNAs
Next, we aimed to identify the group of piRNAs that is common to all cells. Based on our estimate that a single ovarian somatic sheath cell cannot contain more than one million Piwi-piRNAs, we posit that a piRNA needs to occur more than once in a million to be potentially present in every cell. To track the abundance of individual piRNA sequences, we calculated their concentration (in parts per million, ppm) and ranked them by this measure of sequence abundance (Fig. 2A). Our analysis revealed that the abundance of individual piRNA sequences is highly skewed and varies by more than 1000-fold. Less than five percent of the sequences can be present in every cell (Fig. 2A, red dotted line). Most of the sequences are seen less than once in a million and about half are only represented by a single read in an average data set (Fig. 2A, bottom). The skewed distribution of sequence abundance is a conserved feature of piRNAs in flies and mice, based on publicly available data (Hayashi et al. 2016; Gainetdinov et al. 2018), and identifies a small group of abundant sequences that dominate piRNA populations (Fig. 2B,C). We observed a variable sequence abundance for PRG1-associated worm piRNAs (21U-RNAs) using publicly available data (Supplemental Fig. S4; Gu et al. 2012). Their distribution was less skewed, perhaps due to the more precise molecular definition of these 21U-RNAs that originate from individual mini-genes rather than long precursors and are processed by mechanisms that are not conserved in flies or mice (Batista et al. 2008; Weick and Miska 2014; Iwasaki et al. 2015; Ketting and Cochella 2021).
Figure 2.

A skewed distribution of sequence abundance results in a few common and many rare piRNAs. The abundance of individual piRNA sequences (Abundance) varies by ∼1000-fold—ranging from ∼0.1 to more than 1000 reads per million (ppm)—in flies (A,B) and mice (C) (n = 1). (A) Individual Piwi-piRNA sequences from OSCs (S, orange) were ranked by their abundance in reads per million (Supplemental Table S2) (n = 1). Cumulative distribution of corresponding reads (R, gray). The fraction of sequences that is only represented by a single molecule in a representative data set is indicated below. (B) Sequence abundance and cumulative read distribution as in A for piRNAs that were associated with Piwi, Aubergine (Aub), and Argonaute 3 (AGO3) in Drosophila ovaries (GEO: GSE83698), and (C) Mili/Piwil2 and Miwi/Piwil1 in murine primary spermatocytes (SPI) (BioProject: PRJNA421205). (D) Only 13% of the most abundant sequences can be commonly found in three independent data sets but make up 88% of all sampled piRNAs. Violin plots depict the abundance of individual sequences (S) and cumulative reads (R) for Piwi-piRNAs in three biological data sets. Common piRNA sequences are found in all three replicates (purple). Rare piRNAs are only observed in one of the three samples (teal). Schematic Venn diagrams indicate the intersecting sets (n = 3) (Supplemental Table S2). (E) Common and rare piRNAs (analysis as in D) from the intersection of two biological replicates (n = 2) for Mili- and Miwi-piRNAs from murine primary spermatocytes (BioProject: PRJNA421205).
In agreement with the small fraction of highly abundant sequences in flies and mice, <20% of the topmost abundant Piwi-piRNAs and about 30% of mouse piRNAs can be commonly found in independent biological data sets (Fig. 2D,E). Our results reveal differences in sequence abundance as a characteristic of piRNA populations and raise two main questions: Why are some piRNAs so much more abundant than others, and how much does abundance matter for function?
Abundant and rare piRNAs originate from the same precursors
With the goal to identify mechanisms that determine the abundance of individual piRNA sequences, we hypothesized that abundant and rare piRNAs either originate from different long precursors or are generated by different processing mechanisms. We observe that about half of all common Piwi-piRNAs originate from piRNA clusters and, in particular, from the flamenco region (Fig. 3A, inset). Notably, almost all rare Piwi-piRNAs originate from piRNA clusters, too. However, they were not enriched for flamenco-derived sequences. When we systematically compared piRNAs across 450 clusters, we observed that the mean sequence abundance of common piRNAs varies about 100-fold between different clusters and suggests a ranking of these piRNA-generating regions (Fig. 3A). Only the top-ranked clusters produced sequences with a mean abundance greater than one in a million and thus the potential to be present in all cells (red dotted line). Our results show that the abundance of individual piRNAs is linked to their genomic origin and that only a few top-ranked clusters produce highly abundant piRNAs.
Figure 3.

Precursor and processing preferences determine piRNA sequence abundance. (A) A small number of top-ranked piRNA clusters (Supplemental Table S3), led by flamenco (lncRNA:flam), produces common sequences (purple) with an average abundance of more than one read per million (red dotted line). Clusters were ranked by the mean abundance of common sequences (Supplemental Table S3). The sequence abundance of common and rare Piwi-piRNAs for each cluster is shown (mean, median, 25th–75th percentile; n = 3). Inset: The fraction of common and rare Piwi-piRNAs that originate from flamenco (lncRNA:flam) and other piRNA clusters. (B) Common and rare Piwi-piRNAs are generated by the Zuc-processor. Metagene analyses reveal the phased preference of the Zuc-processor for uridine (U) in the first position of the observed piRNAs (colored box) as well as in the first position of proceeding and preceding piRNAs. Inset: Length distribution of common and rare piRNAs in nucleotides (nt). (C) Common piRNAs have a stronger 1U-bias than rare piRNAs from the same cluster. The bias for uridine (1U-bias) in the first position—observed over expected 1U frequencies (obs/exp)—of Piwi-piRNAs (clusters ranked as in A and D). (D) Uridine in the first position (1U) is indicative of higher sequence abundance. The mean abundance of piRNAs that start with a uridine (1U) was compared to that of piRNAs that start with either adenosine (1A), guanosine (1G) or cytidine (1C). The mean abundance of 1U-, 1A-, 1G-, and 1C-sequences relative to the mean abundance of all sequences from the same cluster (relative sequence abundance) is shown (clusters ranked as in A and C). (E,F), Most of the 1000 topmost abundant Piwi-piRNAs exhibit a 1U, originate from flamenco (lncRNA:flam) (flam) and show antisense-complementarity (as) to gypsy endogenous retroviruses. Annotated fractions (E) and genomic position within flamenco (lncRNA:flam) (F). Multimapping sequences are represented with all possible coordinates within flamenco (lncRNA:flam) (F).
However, this simple relationship of precursor and product cannot explain the groups of rare piRNAs that originate from all piRNA clusters (Fig. 3A). To test if abundant and rare piRNAs are produced by different processing mechanisms, we probed for the preference of the Zuc-processor to generate piRNAs with uridine in the first position (1U) (Han et al. 2015; Mohn et al. 2015; Stein et al. 2019). We observe the characteristic phased 1U-signature for common and rare piRNAs, though the preference for 1U is less pronounced in the rare group (Fig. 3B). When we normalized the observed to the expected 1U frequencies (1U-bias) for each cluster, we observed that rare piRNAs consistently exhibit less bias for uridine in the first position than abundant piRNAs from the same piRNA precursor (Fig. 3C). The differences in the 1U-bias and in sequence abundance were particularly pronounced for piRNAs that originate from the top-ranked piRNA clusters.
Sequence preferences modulate abundance
Indeed, when we grouped piRNA sequences solely by the identity of their first nucleotide, we observed that the presence of a 1U alone was indicative of higher sequence abundance, with the biggest differences for the top-ranked piRNA clusters (Fig. 3D). Among non-1U-piRNAs, sequences with adenosine (A), cytidine (C), or guanosine (G) in the first position were equally lower in abundance. Taken together, our results suggest that the topmost abundant piRNAs originate from a few top-ranked precursors and harbor a uridine in the first position. To test this hypothesis, we characterized the top 1000 most abundant Piwi-piRNAs (Fig. 3E,F). Ninety-eight percent of these top 1000 sequences start with uridine, and 88% can be generated by a single piRNA cluster, flamenco (lncRNA:flam), the only known essential piRNA cluster (Sarot et al. 2004). More than 80% of these 1000 topmost abundant sequences show antisense complementarity to endogenous retroviruses of the gypsy family in accordance with the known function of flamenco (lncRNA:flam) in controlling these elements (Sarot et al. 2004; Brennecke et al. 2007). Our results reveal that the abundance of individual piRNAs is determined by their genomic origin and by the identity of their first nucleotide and suggest that the abundance of individual piRNAs is regulated during piRNA biogenesis.
A conserved mechanism discriminates abundant from rare piRNAs in flies and mice
We observe similar signatures for mouse Mili- and Miwi-piRNAs from primary spermatocytes mining publicly available data from the Zamore group (Fig. 4A; Gainetdinov et al. 2018). Both common and rare piRNAs originate from the same pachytene piRNA precursors but exhibit marked differences in mean sequence abundance. Both piRNA groups exhibit a 1U-signature, with reduced 1U-preference in rare piRNAs. Indeed, the 1U-bias of rare piRNAs is generally lower than that of common piRNAs (Fig. 4B). Both groups, abundant and rare piRNAs, exhibit the characteristic 3′-end processing signatures of murine piRNAs, including PNLDC1-dependent trimming and the +1U-signature of the preprocessing event, likely generated by the murine (m)Zuc-processor (Supplemental Fig. S5A,B; Gainetdinov et al. 2018). These results suggest that abundant and rare piRNAs are generated by the same processing mechanisms.
Figure 4.

Precursor and processing preferences determine piRNA sequence abundance in mice. (A) piRNA precursors can be ranked by the mean sequence abundance of common piRNAs. All precursors, even top-ranked ones, also produced rare piRNAs. The sequence abundance of common and rare piRNAs associated with Mili or Miwi in primary spermatocytes (SPI) is shown for individual pachytene piRNA precursors (mean, median, 25th–75th percentile; n = 2). Publicly available source data: BioProject: PRJNA421205. (B) Common piRNAs have a stronger 1U-bias than rare piRNAs from the same precursor. The bias for uridine (1U-bias) in the first position—observed over expected 1U frequencies (obs/exp)—of Miwi- and Mili-piRNAs is shown for common and rare sequences from individual pachytene piRNA precursors (precursors are ranked as in A). (C) Uridine in the first position (1U) is indicative of higher sequence abundance. The mean abundance of piRNAs that start with a uridine (1U) was compared to that of piRNAs that start with either adenosine (1A), guanosine (1G), or cytidine (1C). The mean abundance of 1U-, 1A-, 1G-, and 1C-sequences relative to the mean abundance of all sequences from the same cluster (relative sequence abundance) is shown (pachytene piRNA precursors are ranked as in A and B).
Finally, we asked, if the 1U-bias determines differences in piRNA sequence abundance also in mice. We grouped all piRNAs from each precursor by their first nucleotide into 1U-, 1C-, 1A-, and 1G-piRNAs and calculated their relative abundance. Like for Drosophila Piwi-piRNAs, the presence of a 1U alone correlates with increased sequence abundance for Mili- and Miwi-piRNAs. Overall, our comprehensive cross-species analyses identified a conserved signature that discriminates abundant from rare piRNAs. Our data show that piRNA abundance depends on processing preferences and suggest that the position of uridines across precursors shapes the composition of mature piRNA populations. This simple conserved mechanism could enable up-regulation of essential piRNAs and suppression of auto-aggressive sequences during purifying selection.
Abundance determines function
Although it is obvious that piRNAs that cannot be present in every cell cannot act in every cell, we wanted to know if more subtle changes in piRNA abundance affect piRNA function. To directly measure how much piRNA abundance impacts piRNA-guided silencing, we developed a reporter assay. Our design aimed at providing a quantitative readout for piRNA-mediated silencing at the single-cell level. We placed target sites with complementarity to endogenous piRNA-generating regions in the 3′ UTR of a green fluorescent protein (GFP) and expressed a red fluorophore (mCherry) from the same plasmid as normalization control (Fig. 5A; Post et al. 2014). Both fluorophores were driven by the same minimal promoter and separated by an insulator element to avoid spreading of silencing. We transfected our sensor into ovarian somatic sheath cells and evaluated the expression of both fluorophores after 48 h. In the absence of piRNA-targeting, both fluorophores were expressed, and most cells appeared yellow (Fig. 5A). We anticipated that repression of GFP by endogenous piRNAs would result in “red-only,” GFP-silenced, cells. To quantify the silencing effect at single-cell resolution, we analyzed cells by flow cytometry. In the absence of a piRNA-target-sequence, 76% of the cells expressed both green and red fluorophores (Fig. 5B). As expected, a 460-nt-long target with antisense (as) complementarity to flamenco (flam[as]-460) resulted in decreased green fluorescence, and 71% of the cells appeared fully silenced (“red-only”) (Fig. 4C; Supplemental Fig. S4A). When this flamenco (lncRNA:flam) target-sequence was split in half (flam[as]-230-A and B), either part resulted in about 50% fully silenced cells (Supplemental Figs. S6A–C, S7). Shortening the target region to 100 nt (flam[as]-100) further reduced the occurrence of red-only cells to 2% (Supplemental Fig. S6D). Sensors with 230-nt complementarity to the piRNA-producing regions of l(3)80Fj (CG17514), traffic jam (tj), pathetic (path), and Cyclin B (CycB) resulted in up to 33% fully silenced cells (Supplemental Fig. S6E–I). Overall, we observed a range of piRNA-guided restriction for different sensors, from a barely measurable effect to almost complete silencing.
Figure 5.

Silencing directly correlates with piRNA abundance. (A) A reporter assay for Piwi-piRNA silencing. GFP (green) reports silencing by endogenous piRNAs. mCherry (red) serves as a control. GFP and mCherry are expressed from the same plasmid by individual promoters and separated by an insulator sequence. Different target sites with antisense complementarity to piRNA-generating regions were inserted into the 3′ UTR of GFP (piRNA target) (Supplemental Table S4). Sensors were expressed in ovarian somatic sheath cells. Expression of the dual color reporter was visualized (A) and measured by flow cytometry (B,C) 48 h after transfection. (B) Without any target site (nontarget), the sensor expressed both fluorophores in 76% of the transfected cells. (C) A flamenco (lncRNA:flam)-target sensor (flam[as]-460) showed complete silencing of GFP in 71% of the transfected cells (“red only” cells). (D) piRNA abundance correlates with silencing. Correlation of “red-only” (GFP-silenced) cells and the total abundance of complementary piRNAs (in parts per million). Pearson correlation coefficient (r2). piRNA-sensors with different target sequences complementary to flamenco (lncRNA:flam) (varying target length: 100, 230, 460 nt) or complementarity to other piRNA-producing regions: traffic jam (tj), pathetic (path), l(3)80Fj (CG17514), and two different intervals of Cyclin B (*) (constant length 230 nt) (Supplemental Table S4). The dispersion of data points might be partly caused by quantifying fully complementary piRNAs as a surrogate for targeting piRNAs (y-axis), and by only considering completely silenced (“red-only”) cells (x-axis). Future improvements in our understanding of piRNA:target engagement will enable refinement of these scores.
We next asked, whether the abundance of complementary endogenous piRNAs influenced the fraction of fully silenced, “red only,” cells (Fig. 5D). We observed a positive correlation between the total number of fully complementary piRNAs, as a proxy for targeting piRNAs, and the number of fully silenced cells (Pearson correlation coefficient, r2 = 0.75; two-tailed P-value = 0.002717). Our results suggest that the combined abundance of complementary piRNAs determines the efficacy of target restriction.
Discussion
Overall, a model emerges that attributes the significance of individual piRNA sequences to their abundance and abundance to mechanisms of piRNA biogenesis (Fig. 6). The topmost abundant sequences originate from a few piRNA-generating regions and are characterized by preferences of the Zuc-processor, resulting in a strong 1U-bias. This class of piRNAs represents only a small fraction of the observed sequence space but dominates the functional piRNA molecules in every cell. This outstanding group of silencers has the potential to define nonself for future generations.
Figure 6.
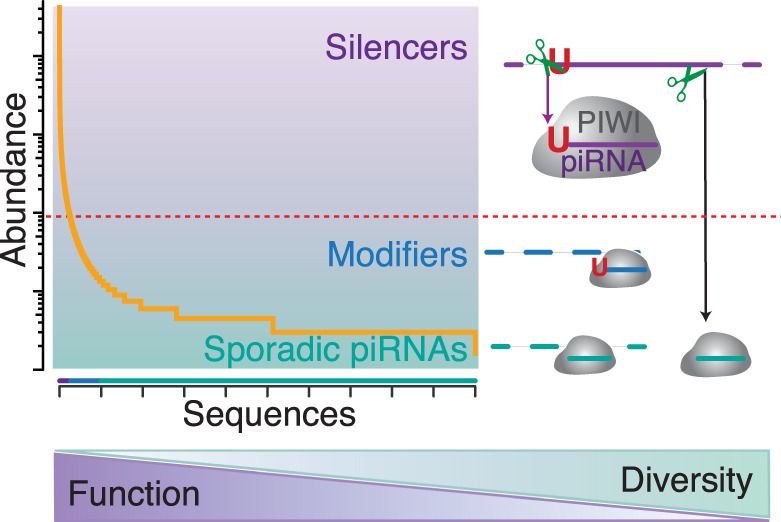
Two classes of piRNAs separate silencing from diversity. Based on the limited number of piRNAs in a single cell (Fig. 1) and the skewed distribution of their sequence abundance (Fig. 2), only the topmost abundant piRNA sequences can be present in every cell. These commonly detected sequences originate from a few top-ranked piRNA-generating regions and exhibit a strong preference for uridine at their 5′-most position (1U) (Figs. 3, 4). The 1U-preference of the Zuc-processor complex modulates the abundance of individual piRNAs (Fig. 3). Abundance correlates with function (Figs. 3E,F, 5). Based on their sequence abundance, piRNAs can be divided into functional classes. A few topmost abundant sequences dominate piRNA silencing (silencers). A biological threshold (red dotted line) separates these piRNAs from the bulk of low abundant sequences that cannot be present in every cell. Rare piRNA establishes cell-to-cell diversity. On convergent targets, these piRNAs could act as Modifiers and promote reproductive polymorphism. In isolation, extremely low abundant piRNAs might never act. However, over time, these highly diverse sporadic sequences could serve as a resource for evolutionary tinkering and bolster adaptation to novel genomic invaders. The functional sequence space of piRNAs is concise and can be regulated to ensure careful self/nonself discrimination.
An intermediate group of modifiers contains sequences that are not present in every cell but are commonly found in different animals. These piRNAs could collaborate and modulate the efficacy of piRNA-guided restriction on converging targets. These modifiers establish functional cell-to-cell diversity and could contribute to previously observed polymorphisms in piRNA-guided restriction (Ryazansky et al. 2017).
Finally, innumerous piRNA sequences are extremely rare. They seem to be functionally insignificant at first glance. However, these sporadic piRNAs could provide substrate for evolutionary tinkering (Palazzo and Koonin 2020). The sequences themselves or the mechanisms that generate them present an opportunity for purifying selection to revise the arsenal of piRNA ammunition in response to novel genome invaders (Yu et al. 2019).
Self/nonself discrimination is at the heart of every self-defense, and regulatory mechanisms are required to avoid auto-aggression. In the ongoing arms race with genomic parasites, purifying selection could act on entire piRNA-generating regions and within individual precursors to shape the functional piRNA sequence space and successfully control a new invader.
Methods
Generation of Piwi-piRNA data sets
piRNAs are defined by their association with PIWI proteins as PIWI-interacting RNAs. Therefore, we focused all analyses in this manuscript on bona fide piRNAs that were extracted from immunopurified PIWI-piRNA complexes (original and publicly available data sets).
Piwi-piRNAs were extracted from ovarian somatic sheath cells by immunoprecipitation of endogenous or endogenously FLAG-tagged Piwi (eF-Piwi): The anti-Piwi antibody (Stein et al. 2019) and Surebeads Protein A magnetic beads (Bio-Rad 1614013) were used to immunoprecipitate endogenous Piwi-piRNAs from wild-type OSCs. Anti-FLAG M2 magnetic beads (Sigma-Aldrich M8823) were used to immunopurify eF-Piwi from OSCeF-Piwi (Marlin Andrews et al. 2020). Three biological replicates were prepared for each sample type. Cells and plasmids are available through the Drosophila Genomics Resource Center. To optimize the purification of Piwi-piRNA complexes from ovarian somatic sheath cells, we generated an endogenously FLAG-HA-tagged piwi-allele in OSCs (OSC:eFH-piwi) that produced an N-terminally tagged Piwi protein that functionally emulates wild-type Piwi (Marlin Andrews et al. 2020). OSC:eFH-piwi cells and associated reagents are being deposited at the Drosophila Genomics Resource Center (DGRC). We characterized eF-Piwi-piRNAs and established that they matched Piwi-piRNAs with respect to their length profile, genomic origin, and targeting potential (Marlin Andrews et al. 2020). The presence of the high-affinity tag on Piwi protein allowed for increasing stringency of the purification using a high-salt wash (0.5 M NaCl) to improve removal of contaminating RNA fragments. Finally, we integrated 10 unique molecular identifiers during the preparation of small RNAs for Illumina sequencing, which allowed us to remove PCR duplicates and precisely quantitate individual piRNA sequences.
Generating endogenously tagged Piwi in ovarian somatic sheath cells
Ovarian somatic sheath cells were purchased from the Drosophila Genomics Resource Center (DGRC cell line 288) and were grown in Shields and Sang M3 insect medium supplemented with 10% heat-inactivated FBS, 10% fly extract (DGRC), 0.6 mg/mL reduced L-glutathione (Sigma-Aldrich G6013), and 5 μg/mL insulin (Sigma-Aldrich I9278) at 25°C. Endogenous piwi was tagged with 3xFLAG-3xHA using the CRISPR-Cas9 system. The tag was inserted immediately upstream of the piwi gene in frame with the ATG to produce N-terminally tagged Piwi protein. First, sgRNA sequence 5′-GCGAGTGCCAAAAAGTAACAA-3′ was cloned into the pU6-BbsI-chiRNA plasmid (Addgene plasmid 45946). Next, we generated a donor plasmid that contained homology regions for recombination into piwi, a 3xFLAG-3xHA tag. A puromycin resistance gene driven by an independent promoter was placed in an intron. The donor plasmid, the sgRNA plasmid, and a modified pAc-sgRNA-Cas9 (Addgene plasmid 49330) were cotransfected in OSCs using Xfect Transfection Reagent (Takara 631318). Immediately after transfection, the cells were treated with SCR7 at a final concentration of 5 μM to block nonhomologous DNA end joining. After 48 h, pooled selection of edited cells was started by 2 μg/mL puromycin treatment (Marlin Andrews et al. 2020).
piRNA preparation for Illumina sequencing
Piwi-piRNAs were extracted from ovarian somatic sheath cells by immunoprecipitation of endogenous or endogenously FLAG-tagged Piwi (eF-Piwi): First, the anti-Piwi antibody and Surebeads Protein A magnetic beads (Bio-Rad 1614013) were used to immunoprecipitate endogenous Piwi-piRNAs from wild-type OSCs. Second, anti-FLAG M2 magnetic beads (Sigma-Aldrich M8823) were used to immunopurify eF-Piwi from OSCeF-Piwi. Three biological replicates were prepared for each sample type.
Cell extracts were prepared in cold lysis buffer (20 mM Tris HCl pH 7.4, 250 mM NaCl, 2 mM MgCl2, 1% NP-40) supplemented with 1× Halt Protease & Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific 1861281), and cleared from insoluble material by centrifugation at 13000×g for 15 min. The soluble lysates were used for immunoprecipitation (input). Immunoprecipitation was performed at 4°C overnight. Next, the beads were washed with a high-salt buffer (20 mM Tris HCl pH 7.4, 500 mM NaCl, 2 mM MgCl2, 1% NP-40), followed by three washes with lysis buffer. One percent of the immunoprecipitate was evaluated for the precipitation of Piwi and eF-Piwi protein by Western blotting. The copurifying RNA was recovered with TRIzol using the Direct-zol RNA MiniPrep kit (Zymo Research R2051).
The libraries were prepared using the general protocol described by Hafner et al. (2012) with the following modifications. Small RNAs were ligated to 3′ indexed adaptors with 2 UMIs (Supplemental Table S5), and ligation products were recovered from a 12% urea PAGE gel by extracting 48- to 58-nt-long fragments (corresponding to 19- to 29-nt-long input small RNAs). Next, 3′-ligated RNA was ligated to the 5′ DNA-RNA hybrid adaptor, containing eight UMIs, and ligation products (82- to 92-nt-long fragments) were recovered from a 10% urea PAGE gel. The adapter ligated RNAs were converted to DNA and amplified as described above for the calibrated small RNA samples. The final small RNA samples were sequenced using an Illumina HiSeq 3000 for 50 cycles (single-end, SE-50).
Sequence diversity: calculating sequences and reads ratio (Fig. 1A)
The sequences to reads ratio was calculated for: OSC samples (FH-Piwi biological replicates, n = 3, this study), Drosophila ovary samples (NCBI Gene Expression Omnibus [GEO; https://www.ncbi.nlm.nih.gov/geo/] accession number GSE83698; fly-Piwi-IP = NCBI Sequence Read Archive [SRA; https://www.ncbi.nlm.nih.gov/sra] accession number SRR3715419; fly-Aub-IP = SRR3715420; fly-AGO3-IP = SRR3715421; fly-tsRNA = SRR3715418; n = 1), and mouse testis samples (NCBI BioProject [https://www.ncbi.nlm.nih.gov/bioproject] accession number PRJNA421205; MILI(SPI) = SRR7760331, SRR7760329; MIWI(SPI) = SRR7760307, SRR7760307; mouse-tsRNA = SRR7760319, SRR7760317; n = 2). For OSC and mouse samples with multiple replicates, the mean ratio and standard deviation were calculated. To calculate sequence to read ratio for miRNAs, miRNA annotation files (GFF3; miRBase; Release 22.1) were used to extract all sequences overlapping with the genomic intervals from total small RNA (tsRNA) samples.
Predictions of piRNA populations based on species accumulation curves (Fig. 1B)
Species accumulation curves provide an estimate for the number of unique species (here, sequences) as a function of sampling effort (here, reads). Dr. Andrew Smith's group has adapted this method to predict the complexity of high-throughput sequencing data and to estimate the sequencing depth (number of reads) required to observe a saturated population (Daley and Smith 2013; Deng et al. 2015). We used their most recent algorithm to estimate the size of piRNA populations based on sampling efforts (Deng et al. 2015). First, unique sequences with identical abundance were grouped to generate multiplicity tables for each piRNA data set. In multiplicity tables, each row contains the number of sequences that appeared exactly n times in the data set. The species accumulation curve was then used to predict the total number of piRNA sequences and reads in a saturated population. Prediction trajectories were constructed using the preseqR package (Deng et al. 2015, 2018). The program uses the observed frequencies to estimate the number of species (here, piRNAs) under deeper sequencing conditions. This prediction relies on the assumption that the total number of species is finite.
Calibrated total small RNA sample preparation for Illumina sequencing (Fig. 1C)
To quantify the average number of piRNAs in a single cell, we used an established reference approach (Farazi et al. 2011; Gainetdinov et al. 2018). Small RNA samples were prepared according to Hafner et al. (2012) and Anastasakis et al. (2021). In brief, total small RNAs from one million OSCs were extracted using a PureLink miRNA Isolation kit (Thermo Fisher Scientific K157001) and were spiked with a calibrator mix (see below for preparation, calibrators 1–4) (Supplemental Table S5; Supplemental Fig. S2). The calibrated small RNA samples were ligated to an adenylated and fluorescently labeled 3′ adaptor using T4 RNA ligase 2 truncated KQ (NEB M0373S) and were subsequently separated on a 12% polyacrylamide urea gel. Adaptor-ligated RNAs 48 to 58 nt long (corresponding to 19- to 29-nt-long input RNAs) were extracted and ligated to the 5′ adaptor using T4 RNA ligase 1 (NEB M0204). A total of 10 variable nucleotides (unique molecular identifiers) were included in the adaptor sequences (eight in the 5′ adaptor and two in the 3′ adaptor) (Supplemental Table S5). The 10 UMIs were used for removal of PCR duplicates during analysis. After 5′ ligation, RNA was recovered using an Oligo Clean and Concentrator kit (Zymo Research D4060) and used as template for cDNA synthesis using a SuperScript IV First-Strand Synthesis System kit (Invitrogen 18091050). The generated cDNA was amplified by PCR and the libraries were purified on a Pippin Gel Cassette (Sage Science, 3% agarose w/ethidium bromide cassette CSD3010) using the Pippin Prep system. Quality of the samples was assessed on a DNA Screen Tape (Agilent D1000) using a TapeStation (Agilent). The calibrated small RNA samples were sequenced using an Illumina NextSeq 550, and 50-nt-long single-end reads were obtained.
Reference oligonucleotides used for calibrated small RNA sample preparation
Four synthetic 26-nt-long RNA sequences were used as calibrators (Supplemental Table S5) to enable an absolute quantification of 19- to 29-nt-long small RNAs. All calibrators had a 5′ phosphate and a 3′ hydroxyl group. Each calibrator was designed with a distinct 5′ and 3′ end to reduce potential ligation bias. Calibrator sequences were designed to be absent from the D. melanogaster genome (dm6). A defined number of reference molecules was added to total small RNAs from 1 million ovarian somatic sheath cells (calibrator1 = 0.5 fmol, calibrator2 = 5 fmol, calibrator3 = 5 fmol, and calibrator4 = 0.5 fmol). To minimize surface absorption during preparation of calibrator dilutions at the nanomolar range, we added an 11-nt-long DNA carrier (TCG AAG TAT TC) at a final concentration of 500 nM, as suggested by Max et al. (2018).
Estimating the small RNA content of a single cell based on calibrated small RNA data sets
The calibrator sequences for all reference oligonucleotides were extracted using grep and cutadapt (v2.8) (Martin 2021). Mature miRNAs were identified using miRBase annotation (dm6; Release 22.1). The 24- to 29-nt-long reads, corresponding to Piwi-piRNAs, were aligned to the Drosophila melanogaster genome (dm6) using Bowtie (v1.2.3) (Langmead 2010).
Estimation of kcorr for 3′-2′-O-methylated RNAs: piRNAs are 2′ O-methylated at their 3′ end, which results in a lower ligation efficiency during sample preparation and a potential to underestimate their abundance relative to nonmodified small RNAs like miRNAs. To correct for this experimental bias, we calculated a correction factor (kcorr) based on publicly available data that compared nonmodified reference oligonucleotides to 3′ 2′-O methylated oligonucleotides (BioProject: PRJNA421205) (Gainetdinov et al. 2018). We extracted counts for nonmodified and modified reference oligonucleotides from seven data sets (SRA: SRR7760317, SRR7760319, SRR7760321, SRR7760326, SRR7760323, SRR7760377, SRR7760373) and calculated a median correction factor of 3.4 for the most reproducible concentration of reference oligonucleotides (1 fmol) (Supplemental Table S6). We applied this correction factor (kcorr = 3.4) to estimate a median abundance of 6 × 105 piRNAs per ovarian somatic sheath cell (Supplemental Table S7). This estimate is presented in Figure 1C. To avoid an underestimation of piRNA abundance, we calculated a correction factor for the 25th percentile of reference oligonucleotides (kcorr 25%tile = 4) that results in an estimated 7 × 105 piRNAs per OSC.
Sequence abundance, cumulative read distribution, and reproducibility (violin) plots (Fig. 2)
We generated multiplicity tables for each data set, where each row contains the count of unique sequences (SEQ) that appear exactly n times in the data set (multiplicity, MULT). To calculate read abundance (READ), SEQ was multiplied by MULT. To normalize the abundance to library size, we calculated parts per million. To depict the sequence abundance and cumulative reads distributions (Fig. 2A), counts were converted into a cumulative library fraction and plotted relative to normalized abundance using geom_step(). Violin plots were generated using normalized sample abundances and the geom_violin() function, setting the adjust value to 1, and scaling the width of violins to the total number of reads in the library. The “total” violin plot corresponds to all reads from three biological replicates; “common” violin to reads that are present in each of the three samples, and “rare” to reads only present in single biological sample.
Metagene analysis of nucleotide frequencies (Fig. 3)
Only uniquely mapping sequences (NH = 1) were considered for this analysis. Reads were aligned to either the 5′ end (5′ metagene: position 1 showing piRNA start 1) or the 3′ end (3′ metagene: position +1 indicates the first nucleotide after the 3′ end of the observed piRNA). The original genomic interval was extended 50 nt upstream and downstream to represent a 100-nt interval. Nucleotide frequencies were plotted as lines using ggplot() (Wickham 2016). The location of the actual piRNA was highlighted by the colored box. Size distribution of piRNAs was calculated by counting piRNAs at different sizes and plotting their normalized frequencies as a bar plot.
Analysis of Piwi-piRNA clusters and pachytene piRNA precursors (Figs. 3 and 4)
Piwi-piRNA clusters were defined using Piwi-piRNA sequencing data (this study) according to the original definition (genomic intervals with ≥1 uniquely mapping read per kilobase and ≥5 kb total length). Coordinates for mouse pachytene precursors were obtained from Li et al. (2013). To include multimapping reads, piRNAs were mapped to the cluster/precursor file using Rsubread (Liao et al. 2019). Only clusters/precursors with at least 100 uniquely mapping (NH = 1) sequences were considered for further analyses (Piwi-piRNA clusters) (Supplemental Table S3). Read abundance was normalized to library size by calculating parts per million. To rank the clusters, we used the mean sequence abundance per cluster. Mean values were then plotted alongside a box plot (showing median abundance value and 25th to 75th percentile) for each group (Fig. 3A).
To investigate the contribution of first nucleotide identity to sequence abundance, all reads were split into 1U- and non-1U (starting with 1A, 1C, or 1G) groups or divided into 1U-, 1A-, 1G-, 1C-groups. Abundance ratio of 1U/non-1U was plotted as a line of best fit. Enrichment of first nucleotide abundances over cluster mean was plotted as a line of best fit. To plot the 1U-bias, the observed frequency for each nucleotide was normalized to the expected frequency based on the genomic sequence of each piRNA cluster (obs/exp). Trends were highlighted using a line of best fit with “lm” setting. Sequence logos were generated using the ggseqlogo package for R (Wagih 2017). Genomic read coverage was visualized in R using Gviz (Hahne and Ivanek 2016) and ggbio (Yin et al. 2012) packages.
Calculating coverage and sequence distribution across flamenco (lncRNA:flam) (Fig. 3F)
Only perfectly mapping sequences (NM = 0) were considered for the analysis. The 1000 most abundant sequences (top 1000) were selected from a single biological replicate ranked by read abundance. To determine coverage of the flamenco region, we selected only the mapping positions overlapping with flamenco coordinates from among all possibilities. The sequences were divided into unique mapping (NH = 1) and multimapping (NH > 1), duplicated to account for read abundance, and coverage data was plotted using the Gviz package as polygons with window value set at 250. We then selected only sequences that mapped within the flamenco region uniquely and grouped them depending on the first nucleotide into 1-U or non-1U groups. The position within the flamenco region and abundance (ppm) for sequences in each group was plotted.
Modeling mappability across flamenco (lncRNA:flam) (Fig. 3F, heat map)
To generate a model of mappability across flamenco (Chr X: 21,631,891–21,632,731, +strand), we first extracted the genomic sequence. This sequence was split into all possible fragments in an 18- to 32-nt size range similar to our libraries’ distribution. Fragments were then mapped to the fly genome (dm6), and multimapping sequences were identified. Reads mapping to the flamenco region were selected, and for each read size (18–32), we calculated a per nucleotide multimapping score. This score was normalized by the contribution factor determined from the library's size distribution. The normalized scores for each read size were summed up to yield a final mappability score. The final score ranges from 0 (all unique coverage) to 1 (all multimapping coverage). The data were plotted using the Gviz package as a heat map with a window value set at 20,000.
Dual color reporter assay (Fig. 5 and Supplemental Figs. S6, S7)
Plasmid design and construction
To create the dual-color reporter plasmid, the sequence of mCherry-T2A-(puromycin-resistance) was retrieved from pCDH-CMV-mCherry-T2A-Puro (Addgene plasmid 72264) and was cloned into pUC19 at the HindIII cleavage site using HiFi assembly (NEB E2621S). The physical DNA sequence of gypsy insulator was a gift from Dr. Brian Oliver's group (NIDDK) (sequence as in pCFD6; Addgene 73915) and was inserted downstream of the mCherry-T2A-puromycin resistance stretch using restriction cloning (AscI-XhoI). EGFP was synthesized as a g-block by Integrated DNA Technologies (IDT) and was inserted downstream of the gypsy insulator using HiFi assembly at the XhoI cleavage site. Both the EGFP and the mCherry are driven by a 595-nt sequence upstream of the Piwi gene that contains the core Piwi promoter. The sensor sequences corresponding to complementary sequences of piRNA-generating regions were synthesized as g-blocks by IDT and ligated into the 3′ UTR of EGFP at the SrfI restriction site (Fig. 5; Supplemental Fig. S3; Supplemental Table S4).
Transfection of ovarian somatic sheath cells
Each plasmid containing different piRNA sensors was transfected into wild-type OSCs using the Xfect Transfection Reagent (Takara 631318). In short, OSCs (∼50% confluency) were transfected with plasmid DNA (30 μg DNA per 10-cm dish) in Shields and Sang M3 insect medium. The cells were incubated at 25°C for 3 h, after which the Shields and Sang M3 insect medium containing the transfection mixture was removed and replaced with complete medium (see cell culture section). For Piwi-piRNA reporter assays, cells were harvested 48 h after transfection.
Microscopy
Transfected OSCs were cultured on glass bottom culture chambers (Ibidi 80427) coated with 1 mg/mL concanavalin A (Sigma-Aldrich L7647). For imaging, full medium was replaced with Shields and Sang M3 insect medium. The cells were examined using a Nikon Eclipse Ti2 inverted microscope, and images were taken using an Andor iXon Ultra 888 EMCCD camera. Data were collected using the Nikon Elements software.
Flow cytometry (FACS) analysis of OSCs expressing piRNA-reporter constructs
For quantitative flow cytometry (FACS) analysis, cells were harvested using trypsinization and washed and resuspended in PBS 48 h after transfection (Fig. 5; Supplemental Fig. S3; Supplemental Table S4). The cell suspension was then centrifuged, and the cell pellets were resuspended in Shields and Sang M3 insect medium and filtered through a 35-μm cell strainer (Stellar Scientific FSC-9005-IW) to generate a uniform single cell suspension and eliminate cell aggregates. We used forward and side scatter to eliminate dead cells from the analysis. FACS analysis was performed on a BD FACS Symphony A5 flow cytometer equipped with 355-, 405-, 445-, 488-, 561-, 640-, and 786-nm laser lines using DIVA 8.0.1 software (BD). The intensities of mCherry (red) and GFP (green) were measured for individual cells. Nontransfected cells were used to determine background signals, and thresholds are indicated in the flow diagrams. As a measure of silencing efficacy, we determined the fraction of cells that expressed mCherry (sensor) above background and GFP (control) comparable to our nontransfected background control. The fraction of these red-only cells or fully-silenced cells was determined for each sensor construct and is plotted on the x-axis in Figure 5D. All data were analyzed with FlowJo software version 9.4.6 (Treestar).
Correlation of piRNA-guided silencing and piRNA abundance (Fig. 5D)
Piwi-piRNAs were mapped to each individual sensor and all the perfectly mapping antisense reads were counted. piRNA counts were normalized to library size by calculating parts per million. These normalized counts (in ppm) were plotted against the percentage of red-only cells (as a percentage of all transfected cells according to the FACS analyses). The line of the best fit and R2 (Pearson) value were calculated to highlight the relationship between data.
Basic bioinformatic processing of sequencing files
Converting raw FASTQ files into size optimized FASTA files
The 5′ and 3′ adapter constant regions were removed using cutadapt (v2.3). Sequences were collapsed using 10 UMIs to eliminate PCR duplicates. UMIs were then removed and sequences ≥19 nt were exported into FASTA files. To optimize file size, reads were collapsed by unique sequences, and the abundance of each sequence was recorded in the FASTA header (SAMPLE_NAME-S[id#]M[abundance#]) where “id” corresponds to unique sequence identifier and “abundance” (M or MULT) represents number of times this sequence was repeated. The collapsed unique read names and sequences were then exported in FASTA format. Our raw and processed files (*UNIQSEQS.FASTA) files are available online (GEO: GSE156058). Publicly available data sets (GEO: GSE83698; BioProject: PRJNA421205) were processed according to the provided instructions.
Elimination of fragments from abundant cellular RNAs and genome mapping
The FASTA files were first mapped against structural RNAs (tRNA, rRNA, snRNA, snoRNA; from UCSC annotations for dm6 and mm10) using STAR aligner (v2.5.2b) (Dobin et al. 2013). The unmapped sequences were then mapped to the genome using STAR aligner (v2.5.2b). Multimapping (NH) of up to 100 positions was allowed. BAM files were loaded and filtered in R (R Core Team 2020) using custom scripts. For most analysis, we used primary alignments (flag = 0 and 16) of perfectly mapping reads (NM = 0). We used 18- to 32-nt-long reads from fly and 18- to 50-nt-long reads from mouse piRNA libraries, respectively. For all data sets, we loaded NH mapping tags and extracted sequence abundance from FASTA header (M or MULT).
Merging and intersecting sequencing files
Piwi-piRNA samples were sequenced with increasing read depth to assess appropriate sampling of the piRNA population during a pilot study. In brief, three biological samples were sequenced on three independent Illumina HiSeq 3000 lanes. Each technical replicate was prepared and mapped independently. Three technical replicates for each sample were merged and intersected in R before analysis using custom scripts.
Software availability
All computational tools used in this study are available at GitHub (https://github.com/HaaseLab/piRNA_Diversity) and as Supplemental Code.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE156058. Key count tables are provided as Supplemental Tables S1–S4.
Supplementary Material
Acknowledgments
We thank V.G. Cheung, S. Gottesman, and T.S. Macfarlan for critical comments on the manuscript; M. Hafner, N.R. Guydosh, and all members of the Haase, Hafner, and Guydosh labs for discussions; D. Anastasakis, K. Bettridge, B. Hayward, and K. McJunkin for experimental advice, and S. Mitra for technical assistance; the NHLBI flow cytometry and genomics cores, especially A. Saxena, the National Institutes of Health (NIH) high-performance computing group, and NIH Medical Arts, especially E. He for help with the model figure. This work was supported by the intramural research program of the National Institute of Diabetes and Digestive and Kidney Diseases (ZIA DK075111-07).
Author contributions: P.G., P.K., and D.S. performed experiments and analyzed data with help from A.M., C.M.A., A.R.E., and C.S.; A.D.H. conceived the project and wrote the manuscript with help from P.G., P.K., and D.S.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.275478.121.
Competing interest statement
The authors declare no competing interests.
References
- Anastasakis DG, Jacob A, Konstantinidou P, Meguro K, Claypool D, Cekan P, Haase AD, Hafner M. 2021. A non-radioactive, improved PAR-CLIP and small RNA cDNA library preparation protocol. Nucleic Acids Res 49: e45. 10.1093/nar/gkab011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Ruby JG, Claycomb JM, Chiang R, Fahlgren N, Kasschau KD, Chaves DA, Gu W, Vasale JJ, Duan S, et al. 2008. PRG-1 and 21U-RNAs interact to form the piRNA complex required for fertility in C. elegans. Mol Cell 31: 67–78. 10.1016/j.molcel.2008.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissels U, Wild S, Tomiuk S, Holste A, Hafner M, Tuschl T, Bosio A. 2009. Absolute quantification of microRNAs by using a universal reference. RNA 15: 2375–2384. 10.1261/rna.1754109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ. 2007. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128: 1089–1103. 10.1016/j.cell.2007.01.043 [DOI] [PubMed] [Google Scholar]
- Brennecke J, Malone CD, Aravin AA, Sachidanandam R, Stark A, Hannon GJ. 2008. An epigenetic role for maternally inherited piRNAs in transposon silencing. Science 322: 1387–1392. 10.1126/science.1165171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosby RL, Chang N-C, Feschotte C. 2019. Host-transposon interactions: conflict, cooperation, and cooption. Genes Dev 33: 1098–1116. 10.1101/gad.327312.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech B, Munafò M, Ciabrelli F, Eastwood EL, Fabry MH, Kneuss E, Hannon GJ. 2018. piRNA-guided genome defense: from biogenesis to silencing. Annu Rev Genet 52: 131–157. 10.1146/annurev-genet-120417-031441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley T, Smith AD. 2013. Predicting the molecular complexity of sequencing libraries. Nat Methods 10: 325–327. 10.1038/nmeth.2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C, Daley T, Smith AD. 2015. Applications of species accumulation curves in large-scale biological data analysis. Quant Biol 3: 135–144. 10.1007/s40484-015-0049-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C, Daley T, Calabrese P, Ren J, Smith AD. 2018. Estimating the number of species to attain sufficient representation in a random sample. arXiv:1607.02804v3 [stat.ME].
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farazi TA, Horlings HM, Hoeve ten JJ, Mihailovic A, Halfwerk H, Morozov P, Brown M, Hafner M, Reyal F, van Kouwenhove M, et al. 2011. MicroRNA sequence and expression analysis in breast tumors by deep sequencing. Cancer Res 71: 4443–4453. 10.1158/0008-5472.CAN-11-0608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Wu P-H, Beane T, Zamore PD, Weng Z. 2018. Elimination of PCR duplicates in RNA-seq and small RNA-seq using unique molecular identifiers. BMC Genomics 19: 531. 10.1186/s12864-018-4933-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov I, Colpan C, Arif A, Cecchini K, Zamore PD. 2018. A single mechanism of biogenesis, initiated and directed by PIWI proteins, explains piRNA production in most animals. Mol Cell 71: 775–790.e5. 10.1016/j.molcel.2018.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Lee H-C, Chaves D, Youngman EM, Pazour GJ, Conte D Jr, Mello CC. 2012. Capseq and CIP-TAP identify Pol II start sites and reveal capped small RNAs as C. elegans piRNA precursors. Cell 151: 1488–1500. 10.1016/j.cell.2012.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardane LS, Saito K, Nishida KM, Miyoshi K, Kawamura Y, Nagami T, Siomi H, Siomi MC. 2007. A Slicer-mediated mechanism for repeat-associated siRNA 5′ end formation in Drosophila. Science 315: 1587–1590. 10.1126/science.1140494 [DOI] [PubMed] [Google Scholar]
- Hafner M, Renwick N, Brown M, Mihailović A, Holoch D, Lin C, Pena JTG, Nusbaum JD, Morozov P, Ludwig J, et al. 2011. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA 17: 1697–1712. 10.1261/rna.2799511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Renwick N, Farazi TA, Mihailović A, Pena JTG, Tuschl T. 2012. Barcoded cDNA library preparation for small RNA profiling by next-generation sequencing. Methods 58: 164–170. 10.1016/j.ymeth.2012.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne F, Ivanek R. 2016. Statistical genomics: methods and protocols. In Visualizing genomic data using Gviz and Bioconductor (ed. Mathé E, Davis S), pp. 335–351. Springer, New York. 10.1007/978-1-4939-3578-9_16 [DOI] [PubMed] [Google Scholar]
- Han BW, Wang W, Li C, Weng Z, Zamore PD. 2015. piRNA-guided transposon cleavage initiates Zucchini-dependent, phased piRNA production. Science 348: 817–821. 10.1126/science.aaa1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi R, Schnabl J, Handler D, Mohn F, Ameres SL, Brennecke J. 2016. Genetic and mechanistic diversity of piRNA 3′-end formation. Nature 539: 588–592. 10.1038/nature20162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ipsaro JJ, Haase AD, Knott SR, Joshua-Tor L, Hannon GJ. 2012. The structural biochemistry of Zucchini implicates it as a nuclease in piRNA biogenesis. Nature 491: 279–283. 10.1038/nature11502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizu H, Iwasaki YW, Hirakata S, Ozaki H, Iwasaki W, Siomi H, Siomi MC. 2015. Somatic primary piRNA biogenesis driven by cis-acting RNA elements and trans-acting Yb. Cell Rep 12: 429–440. 10.1016/j.celrep.2015.06.035 [DOI] [PubMed] [Google Scholar]
- Iwasaki YW, Siomi MC, Siomi H. 2015. PIWI-Interacting RNA: its biogenesis and functions. Annu Rev Biochem 84: 405–433. 10.1146/annurev-biochem-060614-034258 [DOI] [PubMed] [Google Scholar]
- Izumi N, Shoji K, Suzuki Y, Katsuma S, Tomari Y. 2020. Zucchini consensus motifs determine the mechanism of pre-piRNA production. Nature 578: 311–316. 10.1038/s41586-020-1966-9 [DOI] [PubMed] [Google Scholar]
- Janssen A, Colmenares SU, Karpen GH. 2018. Heterochromatin: guardian of the genome. Annu Rev Cell Dev Biol 34: 265–288. 10.1146/annurev-cellbio-100617-062653 [DOI] [PubMed] [Google Scholar]
- Kazazian HH Jr, Moran JV. 2017. Mobile DNA in health and disease. N Engl J Med 377: 361–370. 10.1056/NEJMra1510092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketting RF, Cochella L. 2021. Concepts and functions of small RNA pathways in C. elegans. Curr Top Dev Biol 144: 45–89. 10.1016/bs.ctdb.2020.08.002 [DOI] [PubMed] [Google Scholar]
- Kivioja T, Vähärautio A, Karlsson K, Bonke M, Enge M, Linnarsson S, Taipale J. 2012. Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods 9: 72–74. 10.1038/nmeth.1778 [DOI] [PubMed] [Google Scholar]
- Koonin EV. 2019. CRISPR: a new principle of genome engineering linked to conceptual shifts in evolutionary biology. Biol Philos 34: 9. 10.1007/s10539-018-9654-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B. 2010. Aligning short sequencing reads with Bowtie. Curr Protoc Bioinformatics Chapter 11: Unit 11.7. 10.1002/0471250953.bi1107s32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XZ, Roy CK, Dong X, Bolcun-Filas E, Wang J, Han BW, Xu J, Moore MJ, Schimenti JC, Weng Z, et al. 2013. An ancient transcription factor initiates the burst of piRNA production during early meiosis in mouse testes. Mol Cell 50: 67–81. 10.1016/j.molcel.2013.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. 2019. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res 47: e47. 10.1093/nar/gkz114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Spradling AC. 1997. A novel group of pumilio mutations affects the asymmetric division of germline stem cells in the Drosophila ovary. Development 124: 2463–2476. 10.1242/dev.124.12.2463 [DOI] [PubMed] [Google Scholar]
- Marlin Andrews C, Konstantinidou P, Genzor P, Stoyko D, Elchert AR, Benner L, Sobti S, Katz EY, Meng Q, Haase AD. 2020. Functional tagging of endogenous proteins and rapid selection of cell pools (rapid generation of endogenously tagged piwi in ovarian somatic sheath cells). bioRxiv 10.1101/2020.12.18.423517 [DOI] [PMC free article] [PubMed]
- Martin M. 2021. Cutadapt removed adapter sequences from high-throughput sequencing reads. EMBnet Journal 17: 10–12. 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- Max KEA, Bertram K, Akat KM, Bogardus KA, Li J, Morozov P, Ben-Dov IZ, Li X, Weiss ZR, Azizian A, et al. 2018. Human plasma and serum extracellular small RNA reference profiles and their clinical utility. Proc Natl Acad Sci 115: E5334–E5343. 10.1073/pnas.1714397115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn F, Handler D, Brennecke J. 2015. piRNA-guided slicing specifies transcripts for Zucchini-dependent, phased piRNA biogenesis. Science 348: 812–817. 10.1126/science.aaa1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muerdter F, Olovnikov I, Molaro A, Rozhkov NV, Czech B, Gordon A, Hannon GJ, Aravin AA. 2012. Production of artificial piRNAs in flies and mice. RNA 18: 42–52. 10.1261/rna.029769.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimasu H, Ishizu H, Saito K, Fukuhara S, Kamatani MK, Bonnefond L, Matsumoto N, Nishizawa T, Nakanaga K, Aoki J, et al. 2012. Structure and function of Zucchini endoribonuclease in piRNA biogenesis. Nature 491: 284–287. 10.1038/nature11509 [DOI] [PubMed] [Google Scholar]
- Ophinni Y, Palatini U, Hayashi Y, Parrish NF. 2019. piRNA-guided CRISPR-like immunity in eukaryotes. Trends Immunol 40: 998–1010. 10.1016/j.it.2019.09.003 [DOI] [PubMed] [Google Scholar]
- Ozata DM, Gainetdinov I, Zoch A, O'Carroll D, Zamore PD. 2019. PIWI-interacting RNAs: small RNAs with big functions. Nat Rev Genet 20: 89–108. 10.1038/s41576-018-0073-3 [DOI] [PubMed] [Google Scholar]
- Özata DM, Yu T, Mou H, Gainetdinov I, Colpan C, Cecchini K, Kaymaz Y, Wu P-H, Fan K, Kucukural A, et al. 2020. Evolutionarily conserved pachytene piRNA loci are highly divergent among modern humans. Nat Ecol Evol 4: 156–168. 10.1038/s41559-019-1065-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo AF, Koonin EV. 2020. Functional long non-coding RNAs evolve from junk transcripts. Cell 183: 1151–1161. 10.1016/j.cell.2020.09.047 [DOI] [PubMed] [Google Scholar]
- Pandey RR, Homolka D, Chen K-M, Sachidanandam R, Fauvarque M-O, Pillai RS. 2017. Recruitment of Armitage and Yb to a transcript triggers its phased processing into primary piRNAs in Drosophila ovaries. PLoS Genet 13: e1006956. 10.1371/journal.pgen.1006956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parhad SS, Theurkauf WE. 2019. Rapid evolution and conserved function of the piRNA pathway. Open Biol 9: 180181. 10.1098/rsob.180181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul WE. 2010. Self/Nonself—Immune Recognition and Signaling: a new journal tackles a problem at the center of immunological science. Self Nonself 1: 2–3. 10.4161/self.1.1.10682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post C, Clark JP, Sytnikova YA, Chirn G-W, Lau NC. 2014. The capacity of target silencing by Drosophila PIWI and piRNAs. RNA 20: 1977–1986. 10.1261/rna.046300.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2020. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.R-project.org/. [Google Scholar]
- Rogers AK, Situ K, Perkins EM, Toth KF. 2017. Zucchini-dependent piRNA processing is triggered by recruitment to the cytoplasmic processing machinery. Genes Dev 31: 1858–1869. 10.1101/gad.303214.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryazansky S, Radion E, Mironova A, Akulenko N, Abramov Y, Morgunova V, Kordyukova MY, Olovnikov I, Kalmykova A. 2017. Natural variation of piRNA expression affects immunity to transposable elements. PLoS Genet 13: e1006731. 10.1371/journal.pgen.1006731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Inagaki S, Mituyama T, Kawamura Y, Ono Y, Sakota E, Kotani H, Asai K, Siomi H, Siomi MC. 2009. A regulatory circuit for piwi by the large Maf gene traffic jam in Drosophila. Nature 461: 1296–1299. 10.1038/nature08501 [DOI] [PubMed] [Google Scholar]
- Sarot E, Payen-Groschêne G, Bucheton A, Pélisson A. 2004. Evidence for a piwi-dependent RNA silencing of the gypsy endogenous retrovirus by the Drosophila melanogaster flamenco gene. Genetics 166: 1313–1321. 10.1534/genetics.166.3.1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi MC, Sato K, Pezic D, Aravin AA. 2011. PIWI-interacting small RNAs: the vanguard of genome defence. Nat Rev Mol Cell Biol 12: 246–258. 10.1038/nrm3089 [DOI] [PubMed] [Google Scholar]
- Slotkin RK, Martienssen R. 2007. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet 8: 272–285. 10.1038/nrg2072 [DOI] [PubMed] [Google Scholar]
- Stein CB, Genzor P, Mitra S, Elchert AR, Ipsaro JJ, Benner L, Sobti S, Su Y, Hammell M, Joshua-Tor L, et al. 2019. Decoding the 5′ nucleotide bias of PIWI-interacting RNAs. Nat Commun 10: 828. 10.1038/s41467-019-08803-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagih O. 2017. ggseqlogo: a versatile R package for drawing sequence logos. Bioinformatics 33: 3645–3647. 10.1093/bioinformatics/btx469 [DOI] [PubMed] [Google Scholar]
- Wasik KA, Tam OH, Knott SR, Falciatori I, Hammell M, Vagin VV, Hannon GJ. 2015. RNF17 blocks promiscuous activity of PIWI proteins in mouse testes. Genes Dev 29: 1403–1415. 10.1101/gad.265215.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weick EM, Miska EA. 2014. piRNAs: from biogenesis to function. Development 141: 3458–3471. 10.1242/dev.094037 [DOI] [PubMed] [Google Scholar]
- Wickham H. 2016. ggplot2: elegant graphics for data analysis. Springer-Verlag, New York. https://ggplot2.tidyverse.org. [Google Scholar]
- Williams Z, Morozov P, Mihailović A, Lin C, Puvvula PK, Juranek S, Rosenwaks Z, Tuschl T. 2015. Discovery and characterization of piRNAs in the human fetal ovary. Cell Rep 13: 854–863. 10.1016/j.celrep.2015.09.030 [DOI] [PubMed] [Google Scholar]
- Wu P-H, Fu Y, Cecchini K, Özata DM, Arif A, Yu T, Colpan C, Gainetdinov I, Weng Z, Zamore PD. 2020. The evolutionarily conserved piRNA-producing locus pi6 is required for male mouse fertility. Nat Genet 52: 728–739. 10.1038/s41588-020-0657-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin T, Cook D, Lawrence M. 2012. ggbio: an R package for extending the grammar of graphics for genomic data. Genome Biol 13: R77. 10.1186/gb-2012-13-8-r77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Koppetsch BS, Pagliarani S, Johnston S, Silverstein NJ, Luban J, Chappell K, Weng Z, Theurkauf WE. 2019. The piRNA response to retroviral invasion of the koala genome. Cell 179: 632–643.e12. 10.1016/j.cell.2019.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Pointer B, Kelleher ES. 2020. Rapid evolution of piRNA-mediated silencing of an invading transposable element was driven by abundant de novo mutations. Genome Res 30: 566–575. 10.1101/gr.251546.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.