

Abstract
The core structure of the extracellular basement membrane is made up of self-assembling networks of collagen and laminin which associate with each other through the bridging adapter proteins including the sulfated monomeric glycoprotein nidogen. While collagen and laminin are known to support platelet adhesion and activation via β1 integrins and glycoprotein (GP) VI, respectively, whether nidogen contributes to platelet activation and hemostasis is unknown. In this study we demonstrate that recombinant human nidogen-1 supports platelet adhesion and stimulates platelet activation in a phospholipase-C γ−2 (PLCγ2), Src and Syk kinase-dependent manner downstream. Platetet adhesion to nidogen-1 was inhibited by blocking the platelet receptors GPVI and β1 integrins. Platelet adhesion to nidogen-1 activated the IκB kinase (IKK) complex, while pharmacological inhibition of IKK blocked platelet spreading on nidogen. Taken together our results suggest that nidogen may play a redundant role in hemostasis by activating platelets downstream of GPVI.
Keywords: platelet, nidogen, hemostasis, extracellular matrix proteins
Introduction
The major constituents of the subendothelial matrix include various variants of collagen and laminin, which assembly to form two independent networks[1]. Collagen plays a major role in providing structural stability while laminin is essential for the initial assembly of the basement membrane. As these networks have only a weak affinity for each other, the matrix protein nidogen acts as an integrating element for basement membrane assembly by promoting noncovalent molecular connections between laminin and collagen IV[2]. Indeed, in mice, nidogen deficiency causes impaired lung and heart development leading to perinatal lethality[3]. Studies including those in C. elegans revealed that nidogen may play other nonstructural roles including synaptic transmission and axonal pathfinding[4, 5]. As collagen and laminin are known to play nonstructural roles in hemostasis through activation of blood platelets and coagulation factors, we designed the current study to investigate whether nidogen-1 likewise contributes to hemostasis by supporting platelet activation.
Hemostasis is dependent upon concomitant activation of the blood coagulation cascade and blood platelet adhesion to and activation by subendothelial extracellular matrix (ECM) proteins at sites of vascular injury[6]. The adhesive protein von Willebrand factor (VWF) binds to collagen to facilitate recruitment of platelets from the blood stream in a glycoprotein (GP) Ib-dependent manner[7]. β1 integrin-dependent adhesion to collagen mediates firm adhesion while platelets are rapidly activated by the platelet Immunoglobulin superfamily receptor, GPVI. Crosslinking of GPVI induces Src kinase-dependent tyrosine phosphorylation of the FcR γ-chain immunoreceptor tyrosine-based activation motif (ITAM)[8, 9]. This initiates a Syk-dependent signaling cascade that leads to formation of the LAT signalosome and activation of one of the major effector enzymes in the GPVI signaling cascade, phospholipase C (PLC) γ2, which triggers intracellular calcium mobilization, liberation of the second messengers 1,2-diacylglycerol and inositol 1,4,5 trisphosphate and granule release[10]. Activated platelets subsequently flip their membrane to expose phosphatidylserine and catalyze local thrombin generation and fibrin formation to rapidly form a hemostatic plug[11].
The discovery that the ECM protein laminin likewise binds and activates platelets in a GPVI-dependent manner to support thrombus formation under flow brought to light GPVI as more than a faithful platelet receptor for collagen[12]. Rather, GPVI is a promiscuous receptor for a growing number of ligands including adhesive proteins fibrin, fibrinogen, fibronectin and vitronectin acting in concert to support thrombus growth and stabilization[12–16]. Herein this study suggests that nidogen-1 may be added to the growing list of ligands that bind and activate platelets in part through GPVI, providing further evidence that a cacophony of redundant mechanisms have evolved to activate GPVI to maintain hemostasis[17].
Materials and Methods
Reagents.
Reagents were purchased from Sigma-Aldrich (St Louis, MO, USA) unless specified otherwise. Recombinant human nidogen-1 was obtained from R&D Systems (Minneapolis, MN, USA), soluble collagen from Corning (Corning, NY, USA), fibrillar collagen from Chrono-Log (Havertown, PA, USA), Collagen-related peptide (CRP-XL) was from R. Farndale (Cambridge University, UK) and human fibrinogen from Enzyme Research (South Bend, IN, USA). U73122 and U73343 were obtained from Tocris (Bristol, UK). Anti-GPVI ACT017 blocking antibody was donated by Acticor Biotech (Paris, France). Anti–β1 (clone: AIIB2) blocking antibody was purchased from Millipore (Burlington, MA, USA).
Isolation of human washed platelets.
Platelets were isolated from human venous blood drawn from healthy volunteers by venipuncture into 3.8% sodium citrate (1:9; v/v), in accordance with an Institutional Review Board-approved protocol at Oregon Health & Science University as previously described. Briefly, anticoagulated blood was centrifuged (200 × g, 20 min) to obtain platelet-rich plasma (PRP). PRP was centrifuged (1000 × g, 10 min) in the presence of prostacyclin (0.1 μg/mL) to obtain a platelet pellet. The platelet pellet was resuspended in modified HEPES/Tyrode buffer (129 mM NaCl, 0.34 mM Na2HPO4, 2.9 mM KCl, 12 mM NaHCO3, 20 mM HEPES, 5 mM glucose, 1 mM MgCl2; pH 7.3) and washed once via centrifugation at 1000 × g for 10 min in modified HEPES/Tyrode buffer in the presence of prostacyclin (0.1 μg/mL). Purified platelets were resuspended in modified HEPES/Tyrode buffer at the indicated concentrations.
Static platelet adhesion and spreading assay.
Platelet adhesion and spreading assay was carried out as previously described. Briefly, glass coverslips were coated with human fibrinogen, soluble collagen or recombinant nidogen-1. All proteins were coated at a concentration of 50 μg/mL unless indicated otherwise, followed by surface blocking with bovine serum albumin (BSA) (5 mg/mL). Inhibitors or vehicle were added to platelets in solution (5 × 107/mL) for 15 minutes before exposure to indicated immobilized surfaces. After 45 minutes, nonadherent platelets were discarded and surface-bound platelets were washed 3 times with PBS. Platelets were imaged using Kohler illuminated Nomarski differential interference contrast (DIC) optics with a Zeiss 63x oil immersion 1.40 NA plan-apochromat lens on a Zeiss Axio Imager M2 microscope.
Western blot analysis.
Western blot experiments were carried out as previously described[18]. Briefly, platelet solutions were denatured in an equal volume of Laemmli sample buffer (Biorad, Hercules, CA) with 0.5 M dithiothreitol (100°C, 5 min), separated by SDS-PAGE, transferred to polyvinylidene difluoride membranes, and blotted with indicated antibodies and horseradish peroxidase-conjugated secondary antibodies. Protein was detected using ECL (Thermo Scientific).
Statistical analysis.
Data were analyzed using GraphPad PRISM 5.0 software (San Diego, CA, USA). To determine statistical significance, Student’s paired t-test was used for comparison between treatment and control, while one way-ANOVA was performed with Dunnet’s multiple comparison test for experiments with multiple treatments. Results are expressed as the mean ± standard error of the means (SEM). Differences were considered significant at p < 0.05.
Results
Nidogen-1 supports platelet adhesion and activation
We initially investigated the ability of immobilized recombinant nidogen-1 to support platelet adhesion and spreading as compared to fibrinogen and collagen. As shown in Fig.1A–B, an increasing degree of platelet binding was observed on surfaces coated with an increasing concentration of recombinant human nidogen-1, fibrinogen or soluble collagen. Moreover, platelets fully spread to form lamellipodia on immobilized nidogen-1, fibrinogen and soluble collagen. In contrast, a minimal number of platelets bound to a BSA-coated surface which served as a negative control.
Figure 1. Adhesion and spreading of human platelets on nidogen-1.
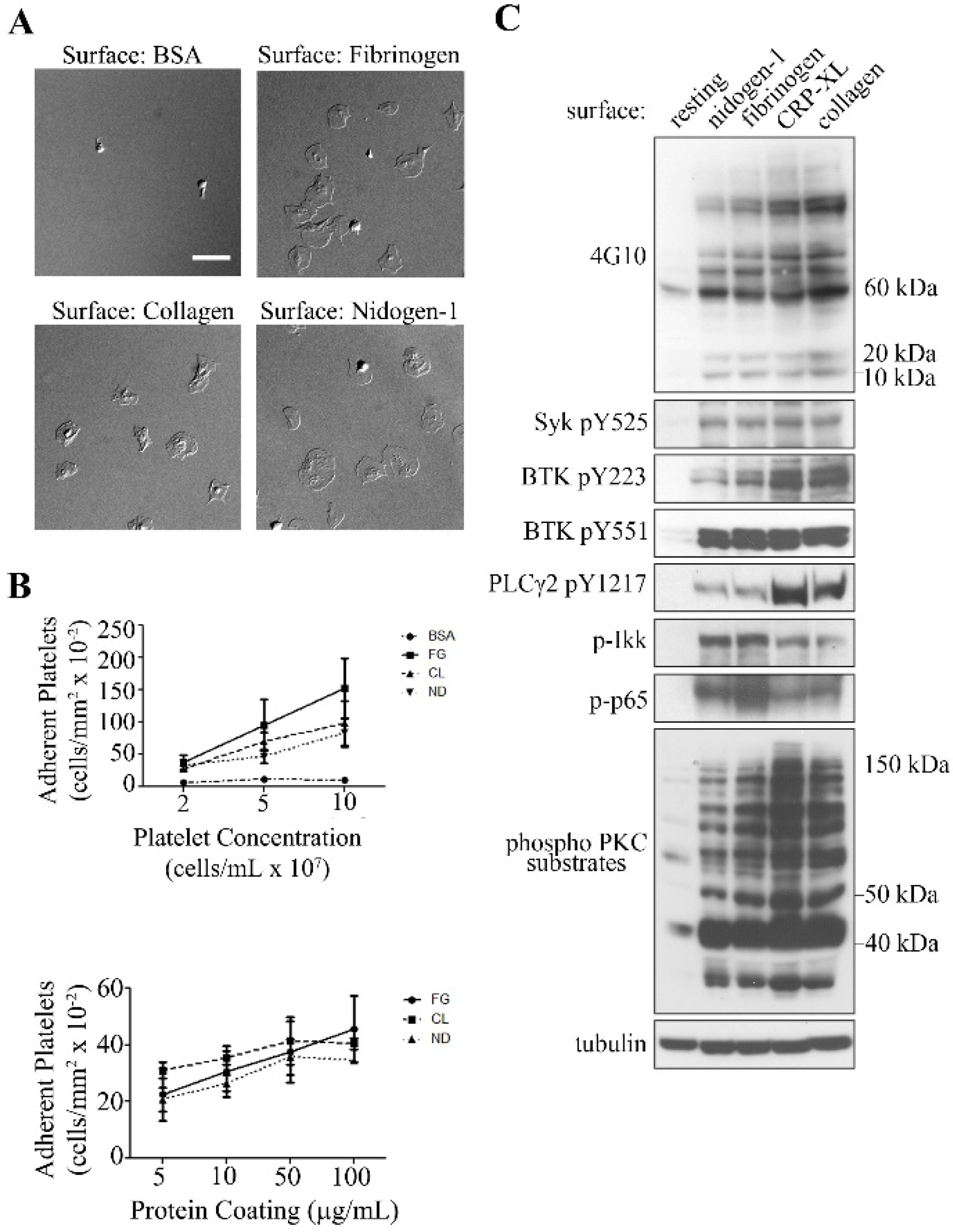
A. Human washed platelets (2 × 107/mL) were placed on coverslips coated with BSA (5 mg/mL), fibrinogen (100 μg/mL), soluble collagen (50 μg/mL), or recombinant human nidogen-1 (100 μg/mL) for 45 min at 37° C and imaged using differential interference contrast (DIC) microscopy. Images are representative of 3 independent experiments. Scale bar, 10 μm. B. The number of adherent platelets on BSA, fibrinogen (FG), soluble collagen (CL) and nidogen-1 (ND) for increasing platelet concentrations and increasing protein concentrations were recorded for 3 fields of view and expressed as mean ± SEM from at least 3 different experiments. C. Lysates from washed human platelets seeded on coverslips coated with recombinant nidogen-1, fibrinogen, CRP-XL (collagen related peptide), fibrillar collagen, or quiescent platelets (resting) in solution were analyzed for total phosphoprotein content with 4G10 or phosphorylation of Syk pY525, BTK pY223 (Bruton’s tyrosine kinase), BTK pY551, activation of PKC (protein kinase C), PLCγ2 pY1217 (phospholipase Cγ2), or activation of NF-κB through the analysis of IKK and p65 phosphorylation by western blotting (WB). Protein molecular markers on the right.
We investigated the intracellular signaling cascades activated in platelets following adhesion to nidogen-1 relative to binding to fibrinogen, the GPVI-agonist collagen-related peptide (CRP-XL), or fibrillar collagen. Platelet adhesion on nidogen-1 promoted the activation of tyrosine kinase signaling events, as determined by Western blotting for tyrosine phosphorylated proteins with 4G10 antisera, phosphorylated Syk or for protein kinase C (PKC) activation as determined by phosphorylation of PKC substrates in a manner similar to that observed for platelets on fibrinogen, CRP-XL, or fibrillar collagen (Fig. 1C). We also detected phosphorylation of proteins of the size of the FcRγ chain (10 kDa in 4G10 section of Fig. 1C), suggesting ITAM-mediated signaling in platelets activated by nidogen-1. Relative to CRP-XL- or collagen-bound platelets, platelets adherent to nidogen-1 showed less phosphorylation of PLCγ2, a critical regulator of secretion and activation. Other critical mediators of platelet secretion in the NF-kB system were phosphorylated at levels comparable to CRP-XL[19]. Interestingly, we also detected phosphorylated IKK in platelets adherent on nidogen-1, fibrinogen, CRP-XL and collagen (Fig. 1C), suggesting the nongenomic role of IKK in platelets may extend to regulating platetet signaling and activation.
Next, we sought to investigate mechanisms of platelet adherence and spreading on nidogen-1 surfaces using pharmacological inhibitors of key signaling proteins in platelet activation programs. As shown in Fig. 2A,B, platelet spreading on nidogen-1 was significantly reduced in the presence of a Src family kinase inhibitor (PP2), a Syk-specific inhibitor (BAY 61–3606), or the PI3K inhibitor wortmannin. Inhibition of PLCγ2 with the broad-spectrum PLC inhibitor U73122 but not the inactive analog U73343 dramatically reduced both platelet adhesion and spreading on nidogen-1 (Fig. 2C–D). In parallel experiments and in accord with previous reports, we found that PP2, BAY 61–3606 and wortmannin significantly decreased platelet spreading on fibrinogen- and soluble collagen-coated surfaces (data not shown)[18]. We next examined the role of the transcription factor NF-κB, which has recently been demonstrated to play non-genomic roles in platelet activation, secretion and aggregation[19]. Inhibition of IκB kinase (IKK), an activator of NF-κB, with IKK-16 decreased the number of adherent platelets and significantly reduced platelet spreading (Fig. 2C–D), which is consistent with the phosphorylation of IKK detected in western blot (Fig. 1C). Additionally, inhibition of IKK decreased platelet adhesion and prevented platelet spreading on fibrinogen and soluble collagen (data not shown).
Figure 2. Effect of platelet inhibitors on platelet adhesion and spreading on nidogen-1.
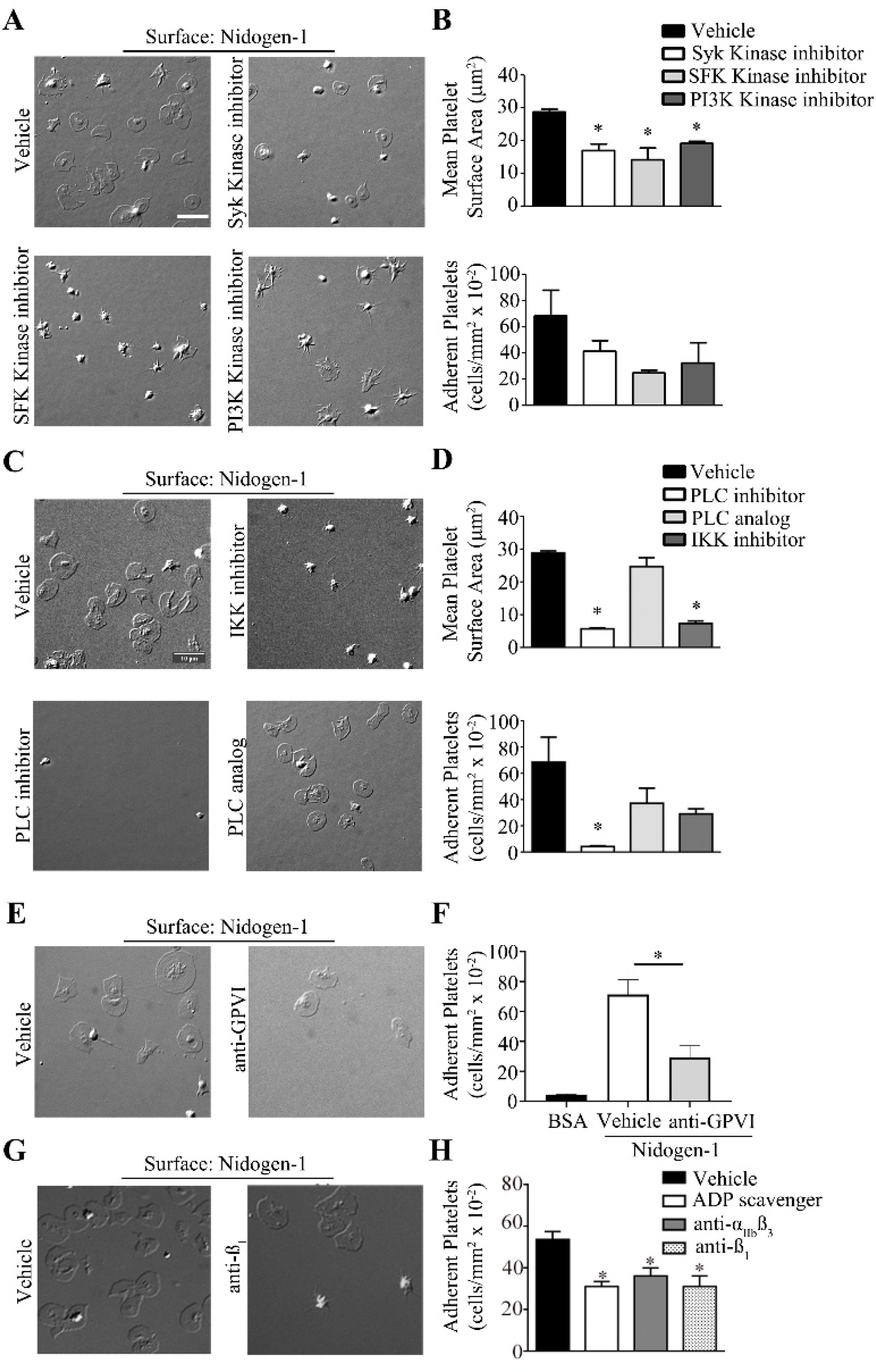
Human washed platelets (2 × 107/mL) were pre-treated with A. vehicle (DMSO), Syk inhibitor (Bay-61–3606, 5 μM), SFK inhibitor (PP2, 10 μM), PI3K inhibitor (wortmannin, 100 nM), C. PLC inhibitor (U73122, 10 μM), PLC inhibitor analog (U73343,10 μM) or IKK inhibitor (IKK-16, 10 μM) E. vehicle (DMSO) or anti-GPVI (ACT017 GPVI inhibitor, 40 μg/mL) H. vehicle (DMSO), anti- β1 (AIIB2, 20 μg/mL), anti-αIIbβ3 (integrillin, 20 μg/mL) (data not shown) or ADP scavenger (apyrase, 2U/ml) for 15 min prior to seeding on coverslips coated with recombinant nidogen-1 (100 μg/mL) for 45 min at 37° C and imaged using differential interference contrast (DIC) microscopy. Images are representative of at least 3 independent experiments. B and D. Number of adherent platelets and mean surface area of platelet on nidogen-1 were recorded for 3 fields of view for each condition and expressed as mean ± SEM. F and H. Number of adherent platelets was recorded for 3 fields of view for each condition and expressed as mean ± SEM. * P < 0.05 with respect to platelet adhesion in the absence of inhibitors. Scale bar, 10 μm.
Together, Western blot and inhibitor studies suggest that both nidogen-1 and collagen share a common platelet receptor and signaling responses consistent with an ITAM-based signaling cascade. We therefore next tested the hypothesis that platelet activation by nidogen-1 was mediated by the ITAM receptor GPVI. Our results show that inhibition of GPVI with ACT017, a blocking antibody specific to GPVI, resulted in a significant decrease in the degree of adhesion of human platelets on nidogen-1. Consistent with previous studies, blockade of GPVI with ACT017 reduced the ability of platelets to spread on soluble collagen-coated surfaces (data not shown)[20, 21]. The degree of platelet adhesion and spreading onto nidogen-1 was significantly reduced by the presence of the ADP inhibitor apyrase, αIIbβ3 inhibitor eptifibatide, and AIIB2, a β1 integrin blocking antibody (Fig. 2E–H). Apyrase hydrolyses the ADP secreted by activated platelets into AMP (Adenosine Mono Phosphate) preventing secondary activation of platelets through ADP mediated receptors such as P2Y12. The reduction in number of platelets adherent to nidogen-1 with apyrase suggests nidogen-1 is likely a weak agonist of GPVI predisposing platelets to require secondary activation from ADP to produce full platelet activation. In addition, reduction in platelet adhesion in the presence of eptifibatide could be the consequence of release of αIIbβ3 ligands upon activation from the platelet α-granules and might not directly indicate the involvement of αIIbβ3 as a receptor [22]. Platelet spreading was also reduced on fibrinogen and soluble collagen by apyrase and eptifibatide, while AIIB2 only reduced platelet adhesion and spreading on soluble collagen (data not shown). Taken together these observations suggest that nidogen-1 may be a ligand for GPVI and β1 integrin which stimulates downstream tyrosine kinase signaling pathways including Src, Syk, PLC and PKC activation.
Discussion
Here we report a potential hemostatic role for the extracellular matrix protein nidogen-1 in supporting platelet adhesion and activation. Moreover, our data adds nidogen-1 to the triumvirate of GPVI ligands present in the ECM including collagen and laminin. Nidogen-1 exhibits a modular structure containing three globular domains, G1–3, separated by a linker region between G1 and G2 and a longer rod-like region located between G2 and G3[23]. Common to other ECM proteins, nidogen-1 contains an epidermal growth factor-like (EGF) module crosslinked to a β-barrel domain within the G2 globule; this complex is responsible for mediating interactions with both perlecan and collagen type IV[1]. The rod domain between G2 and G3 contains another four EGF-like repeats, the first of which contains an RGD binding motif known for potentiating integrin interactions. The six LDL receptor LY modules present in the G3 globule of nidogen mediate interactions with laminin via its single laminin γ1 EGF-like repeat III4. The laminin-nidogen complex and nidogen alone but not laminin alone are known to bind collagen, resulting in ternary complex formation, permitted by the fact that the G2 domain of nidogen contains the binding site for nidogen-collagen interactions[24]. The binding site for nidogen is predominantly located within the triple helix region of collagen; it is the triple helical structure of collagen that is thought to promote the dimerization of the platelet receptor GPVI to induce signaling through receptor tyrosine kinases. An alternative mechanism for GPVI-mediated dimerization and activation may be higher-order receptor clustering as a result of increased ligand density[25]. This may underlie the mechanism by which the polymers of fibrin and laminin and nidogen dimerize GPVI to induce platelet activation[26]. How these ligands are recognized by GPVI remains to be established. Yet, the fact that soluble laminin and nidogen-1 are incapable of activating platelets via GPVI in solution but rather require immobilization lends credence to the concept that the ECM microenvironment plays a critical role in congregating GPVI ligands to ensure hemostasis at sites of vascular injury[12]. Nidogen-1 and nidogen-2 are homologous proteins found in the basement membrane and share similar interactions with ECM proteins laminin and collagen and display redundant physiological functions[27]. Both nidogen-1 and nidogen-2 contain the G2 and G3 domains responsible for their binding to collagen and laminin respectively. Although we investigated only nidogen-1 with respect to platelet interactions in our current study, we hypothesize that nidogen-2 will have similar interactions with platelets due to the structural similarity in these proteins. Our preliminary studies with whole blood flow on nidogen-1 found limited platelet adhesion (data not shown), likely due to the fact that nidogen-1 is a weak activator of platelets. Even with the addition of vWF to nidogen-1, our platelet adhesion data from flow experiments was within the signal to noise ratio (data not shown), although it must be noted that our flow studies did not consider the three dimensional structure of nidogen-1 and its structural binding with ECM proteins laminin and collagen that may affect the role of nidogen-1 in supporting platelet adhesion and aggregation under flow. Taken together, our study suggests that nidogen-1 should be considered as an addition to the growing list of hemostatic proteins of the ECM.
Acknowledgements
This work was supported by grants from the National Institutes of Health (R01HL146549 to J. Aslan and R01HL101972, R01HL144113 and R01GM116184 to O. McCarty) and the American Heart Association (18UFEL33960363 to O. McCarty). A. Sepp and A. Khader are Oregon State University Johnson Scholars. R. Thompson is an AHA Undergraduate Research Fellow.
Abbreviations
- BSA
bovine serum albumin
- CRP
collagen-related peptide
- GP
glycoprotein
- PBS
phosphate buffered saline
- PFA
paraformaldehyde
- PRP
platelet-rich plasma
- SDS
sodium dodecyl sulfate
Footnotes
Disclosures
MJP is a cofounder of Acticor Biotech, owns shares of Acticor Biotech. The other authors declare no conflict of interest.
References
- [1].Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci 2010;123:4195–4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Breitkreutz D, Koxholt I, Thiemann K, Nischt R. Skin basement membrane: the foundation of epidermal integrity--BM functions and diverse roles of bridging molecules nidogen and perlecan. Biomed Res Int 2013;2013:179784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bader BL, Smyth N, Nedbal S, Miosge N, Baranowsky A, Mokkapati S, Murshed M, Nischt R. Compound genetic ablation of nidogen 1 and 2 causes basement membrane defects and perinatal lethality in mice. Mol Cell Biol 2005;25:6846–6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ackley BD, Kang SH, Crew JR, Suh C, Jin Y, Kramer JM. The basement membrane components nidogen and type XVIII collagen regulate organization of neuromuscular junctions in Caenorhabditis elegans. J Neurosci 2003;23:3577–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kim S, Wadsworth WG. Positioning of longitudinal nerves in C. elegans by nidogen. Science 2000;288:150–154. [DOI] [PubMed] [Google Scholar]
- [6].Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev 2013;93:327–358. [DOI] [PubMed] [Google Scholar]
- [7].Naimushin YA, Mazurov AV. Von Willebrand factor can support platelet aggregation via interaction with activated GPIIb-IIIa and GPIb. Platelets 2004;15:419–425. [DOI] [PubMed] [Google Scholar]
- [8].Kuriri FA, O’Malley CJ, Jackson DE. Molecular mechanisms of immunoreceptors in platelets. Thromb Res 2019;176:108–114. [DOI] [PubMed] [Google Scholar]
- [9].Senis YA, Mazharian A, Mori J. Src family kinases: at the forefront of platelet activation. Blood 2014;124:2013–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Watson SP, Auger JM, McCarty OJ, Pearce AC. GPVI and integrin alphaIIb beta3 signaling in platelets. J Thromb Haemost 2005;3:1752–1762. [DOI] [PubMed] [Google Scholar]
- [11].Aslan JE, Itakura A, Gertz JM, McCarty OJ. Platelet shape change and spreading. Methods Mol Biol 2012;788:91–100. [DOI] [PubMed] [Google Scholar]
- [12].Inoue O, Suzuki-Inoue K, McCarty OJ, Moroi M, Ruggeri ZM, Kunicki TJ, Ozaki Y, Watson SP. Laminin stimulates spreading of platelets through integrin alpha6beta1-dependent activation of GPVI. Blood 2006;107:1405–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Alshehri OM, Hughes CE, Montague S, Watson SK, Frampton J, Bender M, Watson SP. Fibrin activates GPVI in human and mouse platelets. Blood 2015;126:1601–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mangin PH, Onselaer MB, Receveur N, Le Lay N, Hardy AT, Wilson C, Sanchez X, Loyau S, Dupuis A, Babar AK, et al. Immobilized fibrinogen activates human platelets through glycoprotein VI. Haematologica 2018;103:898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mammadova-Bach E, Ollivier V, Loyau S, Schaff M, Dumont B, Favier R, Freyburger G, Latger-Cannard V, Nieswandt B, Gachet C, et al. Platelet glycoprotein VI binds to polymerized fibrin and promotes thrombin generation. Blood 2015;126:683–691. [DOI] [PubMed] [Google Scholar]
- [16].Schonberger T, Ziegler M, Borst O, Konrad I, Nieswandt B, Massberg S, Ochmann C, Jurgens T, Seizer P, Langer H, et al. The dimeric platelet collagen receptor GPVI-Fc reduces platelet adhesion to activated endothelium and preserves myocardial function after transient ischemia in mice. Am J Physiol Cell Physiol 2012;303:C757–766. [DOI] [PubMed] [Google Scholar]
- [17].Grover SP, Bergmeier W, Mackman N. Platelet Signaling Pathways and New Inhibitors. Arterioscler Thromb Vasc Biol 2018;38:e28–e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Aslan JE, Tormoen GW, Loren CP, Pang J, McCarty OJ. S6K1 and mTOR regulate Rac1-driven platelet activation and aggregation. Blood 2011;118:3129–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Beaulieu LM, Freedman JE. NFkappaB regulation of platelet function: no nucleus, no genes, no problem? J Thromb Haemost 2009;7:1329–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lebozec K, Jandrot-Perrus M, Avenard G, Favre-Bulle O, Billiald P. Design, development and characterization of ACT017, a humanized Fab that blocks platelet’s glycoprotein VI function without causing bleeding risks. MAbs 2017;9:945–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Voors-Pette C, Lebozec K, Dogterom P, Jullien L, Billiald P, Ferlan P, Renaud L, Favre-Bulle O, Avenard G, Machacek M, et al. Safety and Tolerability, Pharmacokinetics, and Pharmacodynamics of ACT017, an Antiplatelet GPVI (Glycoprotein VI) Fab. Arterioscler Thromb Vasc Biol 2019;39:956–964. [DOI] [PubMed] [Google Scholar]
- [22].Thornber K, McCarty OJ, Watson SP, Pears CJ. Distinct but critical roles for integrin alphaIIbbeta3 in platelet lamellipodia formation on fibrinogen, collagen-related peptide and thrombin. FEBS J 2006;273:5032–5043. [DOI] [PubMed] [Google Scholar]
- [23].Pozzi A, Yurchenco PD, Iozzo RV. The nature and biology of basement membranes. Matrix Biol 2017;57–58:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Reinhardt D, Mann K, Nischt R, Fox JW, Chu ML, Krieg T, Timpl R. Mapping of nidogen binding sites for collagen type IV, heparan sulfate proteoglycan, and zinc. J Biol Chem 1993;268:10881–10887. [PubMed] [Google Scholar]
- [25].Jung SM, Moroi M, Soejima K, Nakagaki T, Miura Y, Berndt MC, Gardiner EE, Howes JM, Pugh N, Bihan D, et al. Constitutive dimerization of glycoprotein VI (GPVI) in resting platelets is essential for binding to collagen and activation in flowing blood. J Biol Chem 2012;287:30000–30013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Induruwa I, Moroi M, Bonna A, Malcor JD, Howes JM, Warburton EA, Farndale RW, Jung SM. Platelet collagen receptor Glycoprotein VI-dimer recognizes fibrinogen and fibrin through their D-domains, contributing to platelet adhesion and activation during thrombus formation. J Thromb Haemost 2018;16:389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bechtel M, Keller MV, Bloch W, Sasaki T, Boukamp P, Zaucke F, Paulsson M, Nischt R. Different domains in nidogen-1 and nidogen-2 drive basement membrane formation in skin organotypic cocultures. FASEB J 2012;26:3637–3648. [DOI] [PubMed] [Google Scholar]