

Summary
Paradoxically, many microRNAs appear to exhibit entirely opposite functions when placed in different contexts. For example, miR-125b has been shown to be pro-apoptotic in some studies, but anti-apoptotic in others. To investigate this phenomenon, we combine computational modeling with experimental approaches to examine how the function of miR-125b in apoptosis varies with respect to the expression levels of its pro-apoptotic and anti-apoptotic targets. In doing so, we elucidate a general trend that miR-125b is more pro-apoptotic when its anti-apoptotic targets are overexpressed, whereas it is more anti-apoptotic when its pro-apoptotic targets are overexpressed. We show that it is possible to completely reverse miR-125b′s function in apoptosis by modifying the expression levels of its target genes. Furthermore, miR-125b′s function may also be altered by the presence of anticancer drugs. These results suggest that the function of a microRNA can vary substantially and is dependent on its target gene expression levels.
Subject areas: Molecular biology, Molecular mechanism of gene regulation, Cell biology, Computational bioinformatics
Graphical abstract
Highlights
-
•
Many miRNAs exhibit entirely opposite functions when placed in different contexts
-
•
miR-125b can be pro- or anti-apoptotic depending on target gene expression levels
-
•
The function of a miRNA can be reversed by altering target gene expression levels
-
•
The presence of anticancer drugs can also alter a miRNA's function
Molecular biology; Molecular mechanism of gene regulation; Cell biology; Computational bioinformatics.
Introduction
MicroRNAs (miRNAs) are 18-25 nucleotide-long, noncoding RNA molecules that negatively regulate the expression of target genes by inhibiting mRNA function and facilitating mRNA degradation. They are of great interest as therapeutic targets for cancer therapy, as they are highly dysregulated in nearly all cancers. However, to maximize the therapeutic benefits and minimize adverse effects of miRNA-modulating cancer therapies, it is important to have a clear understanding of the mechanisms underlying the oncogenic or tumor suppressive properties of miRNAs. More and more, it is becoming apparent that the simple classification of a miRNA as oncogenic miRNA (oncomiR) or tumor suppressor can be misleading, as many miRNAs have been demonstrated to be oncogenic in certain contexts but tumor suppressive in others (Svoronos et al., 2016). For example, miR-155 is a potent oncomiR in many cancers and is even capable of generating an aggressive disseminated lymphoma when overexpressed in mouse lymphoid tissues (Babar et al., 2012), yet it has also been found to act as a tumor suppressor in gastric cancer, ovarian-cancer initiating cells, and a subset of acute myeloid leukemia through the suppression of key oncogenes (Li et al., 2012; Palma et al., 2014; Qin et al., 2013). Similarly, although miR-125b typically serves as an oncomiR in hematologic cancers, it acts as either an oncomiR or a tumor suppressor in many solid tumors (Sun et al., 2013). The interested reader is referred to Svoronos et al. (2016) for several other examples of this phenomenon. Notably, this phenomenon likely applies to many, if not most, miRNAs. As a precursor to the present study, we analyzed the literature of twenty randomly-selected, well-studied miRNAs in terms of their individual functions with respect to three processes in cancer development (apoptosis, cell proliferation, and invasion/metastasis). We found that, for 85% of the miRNAs, there were simultaneously both reports indicating a promoting effect and reports indicating a suppressive effect in one or more of the three processes (Table S1).
Although the mechanisms behind this phenomenon have yet to be studied in detail, one likely explanation is that an individual miRNA can have tens to hundreds of target genes, some of which may be oncogenic and others of which may be tumor suppressive. Depending on the specific cancer and which oncogenic/tumor suppressive pathways are up/downregulated, the miRNA may have an overall net oncogenic or net tumor suppressive effect. Consequently, it may be possible for a miRNA's function to change when the expression levels of its target genes change.
To examine this hypothesis, we generated a computational model of the effect of miRNA-125b (miR-125b) on the apoptosis pathway. MiR-125b is a classic example of a miRNA that is oncogenic in some contexts but tumor suppressive in others, as it targets several genes with opposing functions in cell proliferation, apoptosis, and metastasis (Shaham et al., 2012; Sun et al., 2013). We focus on miR-125b′s role in apoptosis, as its targets include several directly interacting pro- and anti-apoptotic members of the pathway, which can be modeled using a system of differential equations. It is therefore an opportune system for examining the expression-level hypothesis. From the model, we find that miR-125b is more pro-apoptotic when its anti-apoptosis targets are overexpressed and/or its pro-apoptosis targets are underexpressed, and vice versa. We provide experimental evidence to support the model's conclusions, and we find that exposure of cells to various anticancer drugs also influences whether miR-125b is tumor suppressive or oncogenic. Our results suggest that miRNA function is not static. Rather, a miRNA's function can be completely altered by changing the expression levels of its target genes.
Results
Design of a mathematical model to predict the effect of miR-125b on the apoptosis pathway
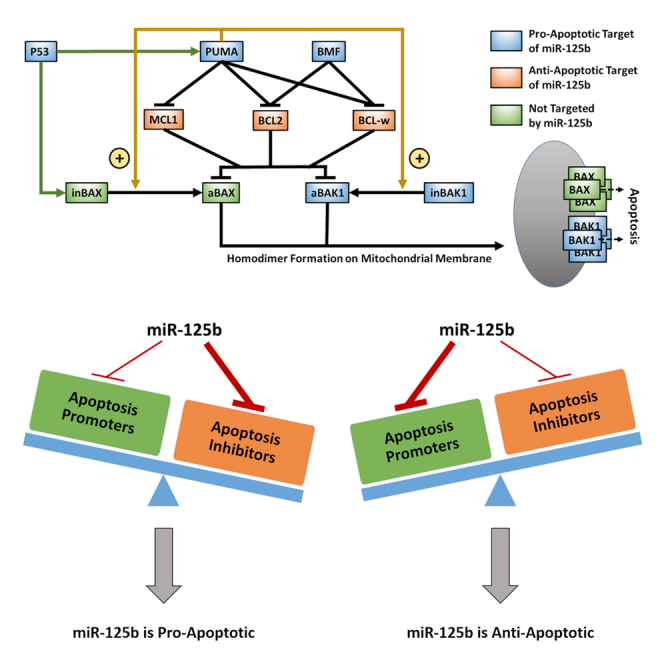
We constructed a mathematical model to describe the portion of the apoptosis pathway affected by miR-125b (Figure 1A). Within the apoptosis pathway, miR-125b directly targets the pro-apoptosis genes TP53 (P53), BBC3 (PUMA), BMF, and BAK1 and the anti-apoptosis genes MCL1, BCL2, and BCL2L2 (BCL-w) (Fan et al., 2018; Gong et al., 2013; Le et al., 2009; Shaham et al., 2012; Shi et al., 2007, 2011; Sun et al., 2013; Willimott and Wagner, 2012; Wu et al., 2013; Xia et al., 2009; Zhao et al., 2012; Zhou et al., 2010). It also indirectly influences the pro-apoptosis protein BAX through modulation of P53 (which transcriptionally activates BAX along with PUMA), MCL1, BCL2, and BCL-w expression levels. Within the modeled pathway, the most downstream effectors of apoptosis are BAX and BAK1. These proteins ultimately form homodimers on the mitochondrial membrane, which are in turn linked together to form pores that promote the release of mitochondrial contents, including cytochrome c, into the cytoplasm (Korsmeyer et al., 2000). Cytochrome c in turn activates the caspase cascade, resulting in cell apoptosis. MCL1, BCL2, and BCL-w inhibit BAX and BAK1 via directly binding them and forming a complex, which inhibits their further function (Westphal et al., 2014). PUMA and BMF in turn inhibit the inhibitors, thereby promoting apoptosis, by forming alternate complexes with MCL1, BCL2, and BCL-w. Of note, in the literature, there are currently two proposed models of the apoptosis pathway, referred to as the direct activation model and the indirect activation model, respectively (Westphal et al., 2014). In the indirect activation model, BAX and BAK1 are constitutively active unless bound by anti-apoptosis proteins. PUMA and BMF therefore enable apoptosis to occur by reducing this binding. In the direct activation model, PUMA has the additional function of activating BAX and BAK1 from inactive to active states. In their inactive states, they remain unbound. Once activated by PUMA, BAX and BAK1 are held in check by the anti-apoptosis proteins, which PUMA and BMF can competitively bind with to result in the release of activated BAX and BAK1. In the present study, we focus on the direct activation version of the pathway, as there is generally more support for it in the literature, but our conclusions are unaltered by this choice, as shown in the supplement. Of note, there are several other proteins in the pathway with functions redundant to the abovementioned proteins, but which are not regulated by miR-125b. Examples include BIM (redundant to PUMA), BAD (redundant to BMF), and BCL2L1 (redundant to MCL1, BCL2, and BCL-w). We do not consider these proteins here, as our goal is solely to investigate trends in the overall function of miR-125b in the apoptosis pathway, which should be unaffected by functionally redundant proteins. In addition, we do not aim for quantitative accuracy in terms of the model's predictions.
Figure 1.

Computational model of miR-125b-modulated portion of the apoptosis pathway
(A) Diagram of the modeled portion of the apoptosis pathway. Blue and orange boxes represent pro-apoptotic and anti-apoptotic members of the pathway that are targeted by miR-125b, respectively. Green boxes represent closely interacting members of the pathway that are not targeted by miR-125b. Green arrowheads from P53 represent transcriptional activation. Gold arrowheads represent activation of inactivated BAX/BAK1 (inBAX/inBAK1) to the form of activated BAX/BAK1 (aBAX/aBAK1). Hammerheads represent inhibition by competitive binding. Activated BAX and BAK1 form homodimers on the mitochondrial membrane, which are in turn linked together to form pores that result in increased mitochondrial outer membrane permeability (MOMP) and apoptosis.
(B) Model-predicted MOMP as a function of time for varying degrees of fractional inhibition of miR-125b′s targets in the apoptosis pathway.
(C) Steady State MOMP as a function of the relative inhibition of miR-125b′s targets in the apoptosis pathway. For (B) and (C), the model was run with the default parameter values and 400 nM baseline activator.
The mathematical model consists of a series of ordinary differential equations (ODEs) (Table S2), which were derived using standard dynamical modeling principles. The model calculates the concentration vs. time of each state of each member of the apoptosis pathway both before and after the induction of apoptotic stress via P53 activation, given a set of predetermined rate constants, which define the rates of production, degradation, and, where applicable, binding, unbinding, activation, and deactivation of each protein, given a user-defined miR-125b level. All parameter values in the model were either directly acquired or calculated from real, measured values in the literature. The main output of the model is the sum of BAX and BAK1 homodimers, which serve as a proxy for the mitochondrial outer membrane permeability (MOMP), which in turn represents the likelihood that apoptosis will occur. To account for the effects of miR-125b, the production rate of each component of the pathway targeted by miR-125b was reduced by a user-defined percentage. Notably, this percentage was assumed to be the same for each of miR-125b′s targets, meaning that miR-125b was assumed to have the same affinity for each. In reality, variations in the miR-125b target site sequence and mRNA secondary structure may cause differences in the affinity of miR-125b for its target genes. However, we emphasize that this model is not meant to be quantitatively predictive. Rather, it is meant solely to be qualitatively predictive and thereby be useful for elucidating overall trends and how various factors affect them. Furthermore, it is important to keep in mind that our model is limited to the effects of miR-125b on the apoptosis pathway. Because of miR-125b′s effects on cell proliferation, metastasis, and differentiation, miR-125b′s effects on apoptosis are not necessarily indicative of whether miR-125b exerts a net oncogenic or net tumor suppressive effect. All of the model equations, the methodology by which they were derived, all parameter values, and the code used to run the model are presented in the STAR Methods and supplemental information (Tables S2–S5, Data S1).
Each time the model was run, all concentrations were allowed to equilibrate to steady-state, after which P53 activation was induced and a new steady-state was reached. Predicted protein concentrations during a typical model run are shown in Figure S1. To assess the dependence of apoptosis on miR-125b level, the model was run with the default parameters and varying degrees of fractional inhibition by miR-125b, ranging from 0 to 1 (Figures 1B and 1C). As can be seen, the steady state MOMP experiences an overall decrease with increasing miR-125b. Thus, with the default parameters, miR-125b exerts an overall net anti-apoptotic effect.
Effect of common cancer mutations on the apoptoticity of miR-125b
The modeled portion of the apoptosis pathway includes genes which are frequently mutated in cancer. We hence used the model to investigate how common cancer mutations affect the apoptoticity of miR-125b (i.e., the average slope of the curve for MOMP vs. miR-125b level, which is a measure of how pro-apoptotic (more positive slope) vs. anti-apoptotic (more negative slope) miR-125b is). With the default parameters, our model predicts that miR-125b exerts a net anti-apoptotic effect. However, miR-125b is tumor suppressive in many cancers, and we thus postulated that, given the right parameters, miR-125b could be driven to exert a net pro-apoptotic effect. In chronic lymphocytic leukemia (CLL), BCL2 is characteristically overexpressed, and inhibition of BCL2 potently suppresses the cancer (Roberts et al., 2012). In addition, Tili et al. demonstrated that miR-125b is tumor suppressive in CLL, but they did not link this to the effect of miR-125b on BCL2 expression (Tili et al., 2012). This is in contrast to the vast majority of hematologic malignancies, for which miR-125b acts as an oncomiR (Shaham et al., 2012). We thus hypothesized that inhibition of BCL2 by miR-125b could account, at least partially, for the tumor suppressive effect of miR-125b in CLL. We postulated that when BCL2 is highly overexpressed, as it is in CLL, miR-125b could reverse its role so as to produce a net pro-apoptotic effect. To test this, we ran our model with varying degrees of BCL2 overexpression, manifested by increasing the BCL2 production rate constant (Figure 2A). As can be seen, the apoptoticity of miR-125b increases with increasing BCL2 expression. Our results therefore support that miR-125b exerts a pro-apoptotic effect in cancers that overexpress BCL2, which can at least partially account for miR-125b′s tumor suppressive effect in CLL.
Figure 2.

Effect of common cancer mutations on miR-125b apoptoticity
(A and B) Steady State MOMP vs. miR-125b level for when the computational model was run with varying degrees of BCL2 overexpression (A) or P53 repression (B). Note that the average slope of the line (i.e., the apoptoticity of miR-125b) becomes increasingly positive with increasing BCL2 expression and decreasing P53 expression.
(C and D) Kaplan-Meier analysis of The Cancer Genome Atlas (TCGA) pan-cancer overall survival data. Patients were first stratified by BCL2 (C) or P53 (D) tumor expression level (low vs. high), and then further subclassified by miR-125b expression level.
We also examined the effect of P53 loss on miR-125b apoptoticity. We ran the model with varying degrees of P53 loss and found a corresponding increase in the apoptoticity of miR-125b (Figure 2B). These results suggest that miR-125b is more likely to be tumor suppressive in a cancer with P53 loss. Interestingly, miR-125b predominantly acts as a tumor suppressor in the five cancers with the highest rates of P53 mutation (ovarian, colorectal, esophageal, head and neck, and laryngeal cancer) (Fan et al., 2018; Guan et al., 2011; Hui et al., 2018; Nakanishi et al., 2014; Olivier et al., 2010; Yang et al., 2019). Conversely, in the vast majority of hematological cancers, which exhibit low rates of P53 mutation (Olivier et al., 2010; Peller and Rotter, 2003), miR-125b is oncogenic (Shaham et al., 2012; Sun et al., 2013).
To investigate our model's predictions further, we analyzed pan-cancer data from The Cancer Genome Atlas (TCGA) (Cancer Genome Atlas Research Network et al., 2013). We found that miR-125b expression is associated with enhanced overall survival in patients exhibiting tumors with either high BCL2 or low P53, respectively, but not in patients with tumors exhibiting low BCL2 or high P53 (Figures 2C and 2D). Hence, the TCGA data suggest that miR-125b is more likely to be tumor suppressive in cancers overexpressing BCL2 or underexpressing P53, consistent with the results of our model.
Effect of a 10-fold increase or decrease in expression of each apoptosis pathway component on miR-125b apoptoticity
To determine the effect of the expression of each component of the model on the apoptoticity of miR-125b, we ran the model with either 10-fold increased expression or 10-fold reduced expression of each component (Figure 3). This analysis elucidated a general pattern for miR-125b apoptoticity: miR-125b was more anti-apoptotic when the expression of its pro-apoptotic targets was increased, while miR-125b was more pro-apoptotic when the expression of its anti-apoptotic targets was increased. The same pattern also held true when the respective expression levels were decreased, and analogous results were found when the model was modified to reflect the indirect activation version of the apoptosis pathway (Figure S2). Together, these data constitute an important finding, as they reveal a general mechanism underpinning the diversity of results seen in miRNA studies. Essentially, when a miRNA inhibits genes of opposing functions, the overall net effect it has is dependent on the balance of expression of its target mRNAs. In the case of cancer, when a miRNA's tumor suppressive targets are more highly expressed, the miRNA is more likely to have a net oncogenic effect, whereas it is more likely to be a tumor suppressor when the expression levels of its oncogenic targets are high.
Figure 3.

MiR-125b function is altered in opposite directions by changing the expression levels of its pro-apoptotic and anti-apoptotic targets, respectively
Difference in apoptoticity (average slope of curve for MOMP vs. miR-125b) compared to default parameter values for each miR-125b-targeted component of the model when the respective basal production rate was increased 10-fold (black) or decreased 10-fold (gray). MiR-125b is more anti-apoptotic left of the vertical axis and more pro-apoptotic right of the vertical axis.
Experimental reversal of the apoptoticity of miR-125b
The above finding suggests that it is possible to change whether a miRNA is tumor suppressive or oncogenic by modulating the expression levels of its target genes. For instance, by inhibiting the expression of the pro-survival proteins, it should be possible to change miR-125b from being pro-apoptotic to anti-apoptotic. We therefore decided to test this directly in vitro with siRNA in cultured cells. We co-transfected human lung adenocarcinoma NCI-H23 cells with a miR-125b mimic and an siRNA against MCL1, BCL2, or BCL-w, respectively, or the respective controls, and measured the proportion of apoptotic cells 72 h later. As expected, while miR-125b increased the rate of apoptosis on its own (in the presence of control siRNA), it significantly reduced apoptosis levels in the presence of reduced MCL1 expression from MCL1 siRNA (Figures 4A and 4B). The pro-apoptotic effects of miR-125b were also abrogated when BCL-w expression was inhibited. BCL2 siRNA had no effect (data not shown). However, BCL2 siRNA also did not change the rate of apoptosis compared to the control, indicating that it is likely not significantly expressed or has negligible function with respect to apoptosis in NCI-H23 cells. The results of this experiment make sense in the context of our model. Intrinsically, miR-125b promotes apoptosis, largely by reducing MCL1 and BCL-w expression. However, when these genes are already inhibited by a siRNA, the pro-survival functions of miR-125b dominate, and a net anti-apoptotic effect is produced. This experiment yields strong support for our model. By changing miR-125b from pro-apoptotic to pro-survival, we validate our model's main conclusion: that the net effect of miR-125b on the apoptosis pathway is a function of the expression levels of its pro- and anti-apoptotic targets.
Figure 4.

Complete reversal of the function of miR-125b by modulation of target gene expression or treatment with anticancer drugs
(A and B) Flow cytometric analysis of apoptosis of NCI-H23 cells after co-transfection with miR-125b and MCL1 or BCL-w siRNA, or the respective controls, followed by Annexin V/propidium iodide staining. The percent differences in the rate of apoptosis for miR-125b-transfected cells relative to their respective controls (i.e., miR-control + the indicated siRNA) were quantified (n = 3).
(C) Cell viability of HCT116 cells after transfection with miR-125b or control, followed by treatment with the indicated drug. The results are the averages of four independent experiments.
Data are mean ± S.E.; ∗p < 0.01; ∗∗p < 0.05.
Reversal of miR-125b from tumor suppressive to oncogenic in the presence of anticancer drugs
The expression levels of various genes, especially those pertaining to apoptosis and survival, can be radically impacted by exposure to anticancer therapies. We hence investigated if treating cells with various anticancer drugs could also change miR-125b from tumor suppressive to oncogenic. We transfected HCT116 cells, for which miR-125b had previously been demonstrated to act as a tumor suppressor (Gong et al., 2013; Yang et al., 2019), with miR-125b mimic or control, followed by exposure to TW-37, ABT-263, doxorubicin, camptothecin, or 5-fluorouracil, respectively, and measured the effects on cell viability. Importantly, we found that treatment with each of these drugs resulted in a reversal in the tumor suppressive nature of miR-125b (Figure 4C). Although miR-125b on its own reduced cell viability by ∼30%, it had the perverse effect of significantly improving viability while in the presence of anticancer drugs. Together with our model, these results highlight the importance of considering miRNA-drug interactions when searching for therapeutic miRNA targets in cancer. Elevating miR-125b levels may initially appear to be beneficial for the treatment of certain cancers, but can actually be detrimental when accounting for interactions with other anticancer therapies.
Discussion
We developed a model of the effect of miR-125b on the apoptosis pathway in order to investigate how target mRNA expression levels can affect miRNA function. In so doing, we found that miR-125b is more likely to be pro-apoptotic when its anti-apoptotic targets are overexpressed and/or its pro-apoptotic targets are underexpressed, whereas it is more likely to be anti-apoptotic when cells overexpress the miRNA's pro-apoptotic targets and/or underexpress its anti-apoptotic targets. Hence, the net impact of miR-125b with respect to apoptosis is a function of the balance of the expression levels of its pro- and anti-apoptotic targets, and, by altering these expression levels, it is possible to reverse the role of miR-125b in apoptosis.
From these studies, we have deduced a general mechanism by which a miRNA can produce opposite effects in different settings. Importantly, our results suggest that miRNA function is malleable and can be governed by the expression levels of the miRNA's target mRNAs. This finding contributes to the growing body of literature on how miRNAs may be counter-regulated by their target binding sites. For instance, certain miRNA binding sites may facilitate miRNA degradation by trapping the miRNA regulatory complex in a conformation that exposes the 3′ end of the miRNA to enzymatic attack, a process termed target-directed miRNA degradation (Ameres et al., 2010; Sheu-Gruttadauria et al., 2019). In addition, many non-coding RNAs have been found to act as miRNA sponges, thereby sequestering miRNAs from interacting with other RNAs. Together with targeted mRNAs, these non-coding RNAs may act as competing endogenous RNAs (ceRNAs), which regulate each other by altering the availability of shared miRNAs (Tay et al., 2014). In essence, our finding may be viewed as an extension of the ceRNA phenomenon, in which ceRNAs not only modify miRNA availability, but also alter miRNA function. Importantly, this principle likely applies to many, if not most, miRNAs, as our literature analysis (Table S1) suggests, in which 85% of randomly selected miRNAs were found to have seemingly conflicting reports that attribute opposite functions to the miRNA, albeit in different contexts.
The reasons why such miRNA systems would evolve warrants further investigation. Notably, when a miRNA simultaneously inhibits two genes or sets of genes with opposing functions, a network motif called an ‘incoherent feedforward loop’ is formed. Incoherent feedforward loops appear to have important roles in modulating variability and noise within systems, which could be critical for a number of physiologic processes, such as cell development. For instance, miR-181a has been shown to reduce cell-to-cell variability in Cd69 expression in developing mouse thymocytes by simultaneously inhibiting and stimulating Cd69 expression through direct inhibition of the Cd69 mRNA and stimulation of TCR signaling, which upregulates Cd69, respectively (Blevins et al., 2015). Similarly, miR-34a simultaneously inhibits and stimulates Notch, a key determinant of colon cancer stem cell fate, by directly targeting both Notch itself and Numb, an inhibitor of Notch, respectively (Bu et al., 2016). This results in a bimodal distribution for Notch expression, in which Notch levels are insensitive to variations and increases in miR-34a concentration up to a sharp transition point, beyond which Notch levels are low. Hence, the miR-34a-Numb-Notch network creates a robust bimodal switch for Notch levels, which in turn enforces the stem cell vs. non-stem cell fate decision (Bu et al., 2016). Together with our own results, such findings suggest that the fundamental role of miRNAs is not regulation of gene expression, but rather the modulation of systems-level network properties.
For the investigation of network properties, computational studies tend to be particularly useful. We therefore encourage more miRNA studies along these lines. Notably, our computational model of miR-125b is limited in the sense that it incorporates only the effects of miR-125b (by reducing the production rate of each targeted gene by a user-defined percentage), as opposed to miR-125b itself (i.e., the concentration of miR-125b is not a variable in the model). As a result, our model is limited to qualitative prediction; it cannot be quantitatively predictive. It would be extremely interesting to investigate such miRNA systems in more detail by modeling the miRNA directly. Notably, variations in the miRNA target site sequence and mRNA secondary structure may cause differences in both the miRNA's affinity for and the kinetics of its interactions with its target mRNAs. Modeling the miRNA directly would allow for analysis of how different kinetics of the miRNA-mRNA interaction, including nonlinear kinetics, affect the system as a whole. A detailed understanding of the kinetics of these interactions would be crucial for quantitative prediction of systems-level outcomes, and our study therefore motivates the need for such investigations.
On a clinical level, our findings have particularly important implications for the use of miRNA-modulating therapies in cancer. Owing to the variability of miRNA function, a precision medicine approach may often be critical for successful therapy. Such precision medicine approaches would ideally involve not only cancer gene expression profiling to determine the expression levels of the miRNA's targets, but also an analysis of how extrinsic factors, such as the presence of drugs and other therapies, affect the miRNA's function. As our results with miR-125b demonstrate, treating a cancer with exogenous miRNA may produce the opposite of the original effect when anticancer drugs are present. In some cases, this may result in unexpected negative consequences. However, the opposite is also true; in other cases, the presence of anticancer drugs may reinforce or synergize with the effect of a miRNA-modulating therapy. For instance, if a miR-125b inhibitor were used to treat a cancer in which miR-125b acts as an oncomiR, then the miR-125b inhibitor would likely work synergistically with apoptosis-inducing drugs by blocking miR-125b-mediated downregulation of pro-apoptosis factors, thereby further reducing cancer cell survival. In fact, it may be possible to take advantage of this phenomenon by using drugs to influence the expression levels of a miRNA's targets so as to push the miRNA's function in one direction over another. This might allow a miRNA-modulating therapy to produce a constant effect regardless of the context, thereby eliminating the need for a precision medicine approach. Ultimately, although the malleability of miRNA function necessitates caution in the use of miRNA-modulating therapies, it may also be possible to leverage this phenomenon for enhanced therapeutic efficacy. Hence, when investigating the therapeutic potential of a miRNA, we suggest that a holistic approach be taken, which accounts for all of the miRNA's known targets, as well as how various extrinsic factors, such as the presence of other treatments, affect them.
Limitations of study
As previously mentioned, our computational model has several limitations which prevent it from being quantitatively predictive; it is meant solely to be qualitatively predictive, such that overall trends/patterns may be elucidated. In addition, the model is limited to miR-125b′s effect on the apoptosis pathway. It does not extend to cover miR-125b′s effect on other core processes, such as cell proliferation and invasion/metastasis. Furthermore, we provide experimental evidence for the conclusions of our model with in vitro experiments. In vivo experiments with actual tumors would be valuable for further support of the model. Lastly, our experiments and modeling are limited to miR-125b. Although our literature analysis of random miRNAs (Table S1) suggests that most other microRNAs also simultaneously inhibit genes or sets of genes with opposing functions, this must still be verified with additional studies.
Supporting citations
Chen et al., 2005; Dai et al., 2009; Dai et al., 2011; Ferrer et al., 2012; Fricker et al., 2010; Gaglia et al., 2013; Grespi et al., 2010; Ku et al., 2011; Lindner et al., 2013; Ma et al., 2005; Maltzman and Czyzyk, 1984; Rooswinkel et al., 2014; Stewart et al., 2010; Wang et al., 2010.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| RPMI-1640 Medium | Gibco | Cat #: 11,875-093 |
| Dulbecco's Modified Eagle Medium (DMEM) | Gibco | Cat #: 11995-065 |
| Fetal Bovine Serum | Gibco | Cat #: 16140-071 |
| Penicillin-Streptomycin | Gibco | Cat #: 15140122 |
| Lipofectamine RNAiMAX | ThermoFisher Scientific | Cat #: 13778030 |
| Andy Fluor 647 Annexin V | ABP Biosciences | Cat #: A038 |
| Annexin V Binding Buffer | BioLegend | Cat #: 422201 |
| Propidium Iodide | MilliporeSigma | Cat #: P4170 |
| TRIzol | ThermoFisher Scientific | Cat #: 15596026 |
| mirVANA miR-125b mimic | Invitrogen | Cat #: 4464066-MC10148 |
| mirVANA miRNA mimic negative control | Invitrogen | Cat #: 4464058 |
| TW-37 | Selleck Chemicals | Cat #: S1121 |
| ABT-263 (Navitoclax) | Selleck Chemicals | Cat #: S1001 |
| Doxorubicin hydrochloride | MilliporeSigma | Cat #: D1515 |
| Camptothecin | MilliporeSigma | Cat #: C9911 |
| 5-Fluorouracil | MilliporeSigma | Cat #: F6627 |
| Critical commercial assays | ||
| High-Capicity cDNA Reverse Transcription Kit | Applied Biosystems | Cat #: 4368814 |
| Power SYBR Green PCR Master Mix | Applied Biosystems | Cat #: 4367659 |
| CellTiter-Glo 2.0 Assay | Promega | Cat #: G9242 |
| MycoAlert Plus Mycoplasma Detection Kit | Lonza | Cat #: LT07-701 |
| Deposited data | ||
| The Cancer Genome Atlas (TCGA) | National Cancer Institute | Data Release 23.0 |
| Experimental models: Cell lines | ||
| NCI-H23 cells | American Type Culture Collection (ATCC) | Cat #: CRL-5800 |
| HCT116 cells | American Type Culture Collection (ATCC) | Cat #: CCL-247 |
| Oligonucleotides | ||
| miR-125b miRNA mimic | GenePharma | double strand microRNA mimics (hsa-miR-125b-5p) |
| miRNA mimic negative control | GenePharma | microRNA mimics double strand negative control |
| MCL1 Forward Primer | Yale Keck Oligo Synthesis Resource | 5’-TGCTTCGGAAACTGGACATCA-3’ |
| MCL1 Reverse Primer | Yale Keck Oligo Synthesis Resource | 5’-TAGCCACAAAGGCACCAAAAG-3’ |
| BCL2 Forward Primer | Yale Keck Oligo Synthesis Resource | 5′-TCGCCCTGTGGATGACTGA-3′ |
| BCL2 Reverse Primer | Yale Keck Oligo Synthesis Resource | 5′-CAGAGACAGCCAGGAGAAATCA-3′ |
| BCL-w Forward Primer | Yale Keck Oligo Synthesis Resource | 5’-GCGGAGTTCACAGCTCTATAC-3’ |
| BCL-w Reverse Primer | Yale Keck Oligo Synthesis Resource | 5’-AAAAGGCCCCTACAGTTACCA-3’ |
| GAPDH Forward Primer | Yale Keck Oligo Synthesis Resource | 5’-GGCCTCCAAGGAGTAAGACC-3’ |
| GAPDH Reverse Primer | Yale Keck Oligo Synthesis Resource | 5’-AGGGGTCTACATGGCAACTG-3’ |
| Software and algorithms | ||
| MATLAB R2018a | MathWorks | https://www.mathworks.com/products/matlab.html |
| Computational model of the effect of miR-125b on the apoptosis pathway | supplemental information of this paper | Supplemental information of this paper |
| UCSC Xena Functional Genomics Explorer | Goldman et al., 2020 | https://xena.ucsc.edu |
| Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Donald Engelman (donald.engelman@yale.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
NCI-H23 and HCT116 cells were obtained from the American Type Culture Collection (ATCC). For growth and maintenance, NCI-H23 and HCT116 cells were cultured in RPMI-1640 Medium and Dulbecco's Modified Eagle Medium (DMEM), respectively, supplemented with 10% fetal bovine serum (Gibco), 1% penicillin, and 1% streptomycin in a 37°C, 5% CO2 humidified incubator. All cells were cultured for fewer than 20 passages before use. During maintenance, cells were periodically verified to be mycoplasma negative using the MycoAlert Mycoplasma Detection kit (Lonza).
Method details
MicroRNA literature analysis
Twenty random, well-studied miRNAs (defined as being mentioned in 50 or more published research articles) were selected for analysis using a random number generator. More specifically, MATLAB was used to generate random whole numbers between 1 and 500. For each random number, the corresponding miRNA (e.g., miR-451 for the number 451) was chosen for analysis. If there were multiple miRNAs corresponding to a single number (e.g., miR-30a, miR-30b, miR-30c), then the ‘a’ version (i.e., miR-30a) was chosen. The PubMed search engine (National Center for Biotechnology Information, National Institutes of Health) was used to search for each selected miRNA. If there were fewer than 50 search results, then the miRNA was excluded from the analysis. For each miRNA with 50 or more search results, the corresponding literature was analyzed for the presence or absence of studies that found a promoting or suppressive function of the miRNA in terms of apoptosis, cell proliferation, and invasion/metastasis, respectively. The results of the analysis are shown in Table S1.
Construction and implementation of mathematical model to predict the effect of miR-125b on the apoptosis pathway
The mathematical model we constructed consists of a set of ordinary differential equations (ODEs) which describe the concentration vs. time of each state (i.e., free, bound, activated, inactivated) of each member of the modeled portion of the apoptosis pathway. All model equations were derived from standard dynamical modeling principles and are listed in Table S2 of the supplemental information. First, each protein in the pathway with multiple binding or activation states was assigned a mass-balance equation that describes the total, bound, active, and inactive concentrations of the protein. For example:
where [inBak1] is the inactive BAK1 concentration, [Bak1] is the concentration of activated BAK1, [Bakdimer] is the concentration of homodimerized active BAK1, and [Bak1.Mcl1], [Bak1.Bcl2], and [Bak1.Bclw] are the concentrations of activated BAK1 bound by MCL1, BCL2, and BCL-w, respectively. In turn, the change in total concentration with time was described by a production rate with rate constant k and the sum of the degradation rates with rate constant d, of each respective state n of the protein:
To account for the effects of miR-125b, the production rate of each component of the pathway targeted by miR-125b was modulated by the term (1 – f), where f is the fractional inhibition of the protein's expression by miR-125b. For instance, if 30% of the target's mRNA were bound and inhibited by miR-125b, then f = 0.3, resulting in a 30% decrease in the target's production rate. Thus, rather than directly modeling the concentration of miR-125b, we modeled the degree of target inhibition by miR-125b. Of note, f was assumed to be constant regardless of target mRNA levels (i.e., the number of miR-125b binding sites). This assumption is consistent with the results of Denzler et al., who demonstrated that the number of additional miRNA binding sites needed to influence a miRNA's target expression levels is very large, far higher than physiologic mRNA levels for a single gene (Denzler et al., 2014). In addition, f was assumed to be the same for each of miR-125b′s targets, meaning that miR-125b was assumed to have the same affinity for each. Hence, it was unnecessary to include actual target mRNA concentrations within the model.
To model the inhibition of a protein by direct binding to other proteins in the apoptosis pathway, ODEs that describe the following reactions were generated:
Of note, proteins within a complex were assumed to degrade independently, resulting in the release of the other protein in the complex. This is consistent with reports that the proteasome degrades individual proteins within a complex while sparing others (Prakash et al., 2009). In addition, the half-life of the active form of the pro-apoptosis protein BID was found to be independent of the expression of proteins which it forms complexes with, such as BCL2 (Breitschopf et al., 2000). We hence assumed the same for other proteins within the apoptosis pathway. The ODEs we generated describe the change in concentration with time of both the free and the bound states of each protein, respectively. The change in concentration with time of the free protein was taken to be a function of its production and degradation, modified by binding and unbinding to the protein's inhibitors (including via degradation of the inhibitor). For instance:
where b and u are binding and unbinding rate constants, respectively. The corresponding rates of change in the concentrations of bound protein were described by the binding and unbinding rates, together with the degradation rates of each protein within the complex:
In the case where a component is transcriptionally activated by P53, the component's corresponding mRNA molecule was also modeled with a production rate dependent on the P53 concentration, [P53]. In its inactive state, P53 exists predominantly as a homodimer with KD < 1 nM (Rajagopalan et al., 2011), and hence the P53 dimer concentration could be approximated as simply [P53]/2. However, when activated, P53 forms tetramers on its targeted DNA response elements, thereby resulting in transcriptional activation of the target genes. The DNA response elements themselves catalyze tetramerization by binding two P53 dimers, thereby closely associating them and facilitating tetramer formation (Natan et al., 2009; Weinberg et al., 2004). Hence, transcriptional activation by P53 could be modeled as a function of the P53 dimer concentration, [P53]/2, and the KD for binding of P53 dimers to the gene's DNA response elements using the Hill equation. The Hill coefficient for this interaction is 1.8, and, for BAX, the KD is 73 nM, whereas PUMA possesses two P53 DNA response elements with KD's of 7.1 nM and 260 nM, respectively, which together yield an overall KD of 6.9 nM (Weinberg et al., 2005). Therefore:
Of note, for both BAX and PUMA, the rate constants for mRNA production, kmRNA, and mRNA degradation, dmRNA, were set to values of 1 arbitrary unit/min and 1 min−1, respectively, as modeling realistic mRNA concentrations was beyond the scope of our model. Hence, in the absence of miR-125b (f = 0) and with the default P53 concentration (140 nM), the steady state concentration of each mRNA was equal to ∼1 arbitrary unit. As can be seen from the above equations, the P53 concentration was assumed to account for 100% and 50% of PUMA and BAX mRNA production, respectively. These values were estimated from the results of Nakano and Vousden (2001) (Nakano and Vousden, 2001). The corresponding protein production rate was then modeled as a function of the mRNA concentration, as follows:
In addition, our model was constructed so as to generate a dynamic response to P53 activation. This was achieved by modeling the PUMA and BAX mRNA production rates as if the P53 concentration were zero up until a user-defined time point, at which P53 activation occurs. At this point, the PUMA and BAX mRNA production rates were increased according to the P53 concentration in the above equations.
We constructed two versions of our model, which correspond to the direct activation model (Figure 1A) and the indirect activation model (Figure S2A) of the apoptosis pathway, respectively. In the indirect activation model, BAX and BAK1 are constitutively active, but are prevented from oligomerizing and inducing apoptosis by BCL2, MCL1, and BCL-w. In the direct activation model, which we focus on in our study, BAX and BAK1 must first be activated by an activator protein before forming protein complexes and oligomerizing. Known BAX/BAK1 activators include PUMA, BIM, and the truncated form of BID. Of these, only PUMA is included in our model, as it is the only one regulated by miR-125b. However, without the other activators, the concentrations of activated BAX and BAK1 would be zero without PUMA (e.g., if PUMA production were 100% inhibited by miR-125b). We hence included the effects of these other activators in the form of a baseline level of activation of BAX and BAK1. This baseline activation was implemented as a user-defined concentration of baseline additional activator, AddAct. Hence, BAX and BAK1 activation were modeled to occur with Rate = kact(AddAct + [Puma]), where kact is the activation rate constant. BAX and BAK1 deactivation were modeled to occur with a rate constant of kdeact.
After activation, BAX and BAK1 form homodimers. The dimers then link together to form oligomeric pores of various sizes on the mitochondrial outer membrane (Martinez-Caballero et al., 2009). Since the sizes of these pores are variable and difficult to predict, we did not attempt to model pore formation. Rather, we took the total concentration of BAX/BAK1 dimers as a proxy for the mitochondrial outer membrane permeability (MOMP) of the cell and its relative level of apoptotic stress.
The default values for all of the model's parameters were adapted from published literature values. Each protein's rate constant for degradation, d, was determined from its respective half-life, t1/2, using the equation d = ln(2)/t1/2. The corresponding production rate constant, k, was determined by multiplying the steady-state concentration of the protein, C, by the degradation rate constant, or k = Cd. The steady-state concentration of each protein was based off measured values in the literature. The rate constants of protein association (binding, b) and dissociation (unbinding, u) were determined from published KD values and dissociation rates and were related to each other by the equation KD = u/b. All model parameters, their values, and the literature sources of their values are listed in Tables S3–S5.
The model was implemented into MATLAB R2018a, and the ode15s solver was used for all ODE calculations. The MATLAB code used to run the model is available for download in the supplemental information (Data S1). Data was generated by running the model with inactive P53 (i.e., no transcriptional activation of PUMA or BAX by P53) until all individual protein state concentrations had equilibrated to steady-state values. Afterward, to simulate P53 activation, the model was set to continue running, except now with P53 contributing to PUMA and BAX production, resulting in increased apoptotic stress and elevated MOMP. The model was allowed to continue running until steady-state was again reached. The effect of miR-125b on apoptosis was investigated by running the model for varying values of the parameter f and observing the impact on the final steady-state MOMP. The results of a typical model run are shown in Figure S1.
The Cancer Genome Atlas (TCGA) analysis
TCGA pan-cancer gene expression, miRNA expression, and overall survival data were downloaded using the UCSC Xena Functional Genomics Explorer (Goldman et al., 2020). Data was sorted and matched by TCGA sample barcode. Samples taken from normal tissue or which lacked gene expression, miRNA expression, or survival data were excluded. When multiple samples were associated with a single patient, their data were averaged. Samples were stratified by target gene expression using a 25th percentile high/low cutoff and then further subclassified by miR-125b expression. Kaplan-Meier curves were then generated and compared. Statistical significance was assessed using the log rank test.
Cell experiments
NCI-H23 and HCT116 cells were obtained from the American Type Culture Collection (ATCC). For growth and maintenance, NCI-H23 and HCT116 cells were cultured in RPMI-1640 Medium and Dulbecco's Modified Eagle Medium (DMEM), respectively, supplemented with 10% fetal bovine serum (Gibco), 1% penicillin, and 1% streptomycin in a 37°C, 5% CO2 humidified incubator. All cells were cultured for fewer than 20 passages before use. During maintenance, cells were periodically verified to be mycoplasma negative using the MycoAlert Mycoplasma Detection kit (Lonza).
Transfections were performed using Lipofectamine RNAiMAX according to the manufacturer's reverse transfection protocol. For siRNA experiments, NCI-H23 cells were co-transfected with 50 nM miR-125b mimic or miRNA mimic negative control (GenePharma) and 20 nM MCL1, BCL2, or BCL-w Silencer Select siRNA (ThermoFisher) and incubated for 72 h. They were then trypsinized, resuspended in Annexin V binding buffer, and stained with Andy Fluor 647 Annexin V (ABP Biosciences) and propidium iodide according to the manufacturer's protocol. Apoptosis was then analyzed by flow cytometry using a BD Accuri C6 flow cytometer. The efficiency of siRNA knockdown was verified by transfecting additional cell samples in parallel, which underwent TRIzol (ThermoFisher) RNA extraction according to the manufacturer's protocol. The isolated RNA was then reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (ThermoFisher), and RT-qPCR was performed using Power SYBR Green Master Mix (ThermoFisher), the appropriate forward and reverse primers (Yale Keck Oligo Synthesis Resource), and a StepOnePlus Real-Time PCR machine (Applied Biosystems), all according to the manufacturer's protocol. Relative expression levels were determined via the ΔΔCt method with normalization to GAPDH mRNA (Figure S3).
For drug treatment experiments, HCT116 cells were transfected with 50 nM mirVANA miR-125b mimic or negative control (Invitrogen) for 16-20 h. The media was then replaced twice with fresh media, and the cells were allowed to incubate for an additional 5 h. Afterward, the appropriate drugs were added at the following concentrations: 10 μM TW-37, 10 μM ABT-263, 1 μM doxorubicin, 1 μM camptothecin, 10 μM 5-fluorouracil. The cells were then incubated for an additional 48 h, and cell viability was analyzed using the CellTiter-Glo 2.0 assay (Promega) according to the manufacturer's protocol. A cell standard was generated in conjunction with each experiment, and relative cell numbers were determined by comparing CellTiter-Glo luminescence to the cell standard.
Quantification and statistical analysis
The computational model was implemented in MATLAB R2018a. Therefore, all quantification of model-generated data was performed using MATLAB. For survival analysis of TCGA data, statistical significance was assessed via the log rank test using GraphPad Prism 8 software. Two-sample t-tests were used to assess statistical significance for all cell experiments. The statistical parameters of the experiments are reported in the figure legends or on the figures themselves. Statistical significance was defined as a p value of less than 0.05.
Acknowledgments
We thank J. Steitz and J. Howard for valuable discussions and thoughtful suggestions. Work in the lab of D.M.E. is supported by NIH grant R01-GM073857. A.A.S. was additionally supported by an NIH National Research Service Award (F30-CA196020) and Yale University's NIH Medical Scientist Training Program grant (T32-GM007205).
Author contributions
A.A.S. conceived the project. A.A.S., S.G.C., and D.M.E. designed the research and developed the methodology. A.A.S. and S.G.C. wrote the code for the model. A.A.S. performed the experiments. A.A.S. and D.M.E. wrote the paper.
Declaration of interests
The authors declare no competing interests.
Published: October 22, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103208.
Contributor Information
Alexander A. Svoronos, Email: alexsvoronos@gmail.com.
Donald M. Engelman, Email: donald.engelman@yale.edu.
Supplemental information
Data and code availability
All original code has been included in the supplemental information and is available for download. Any additional information required to analyze the data reported in this paper is available from the lead contact upon request.
References
- Ameres S.L., Horwich M.D., Hung J.H., Xu J., Ghildiyal M., Weng Z., Zamore P.D. Target RNA-directed trimming and tailing of small silencing RNAs. Science. 2010;328:1534–1539. doi: 10.1126/science.1187058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babar I.A., Cheng C.J., Booth C.J., Liang X., Weidhaas J.B., Saltzman W.M., Slack F.J. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc. Natl. Acad. Sci. U S A. 2012;109:E1695–E1704. doi: 10.1073/pnas.1201516109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blevins R., Bruno L., Carroll T., Elliott J., Marcais A., Loh C., Hertweck A., Krek A., Rajewsky N., Chen C.Z. microRNAs regulate cell-to-cell variability of endogenous target gene expression in developing mouse thymocytes. PLoS Genet. 2015;11:e1005020. doi: 10.1371/journal.pgen.1005020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitschopf K., Zeiher A.M., Dimmeler S. Ubiquitin-mediated degradation of the proapoptotic active form of bid. A functional consequence on apoptosis induction. J. Biol. Chem. 2000;275:21648–21652. doi: 10.1074/jbc.M001083200. [DOI] [PubMed] [Google Scholar]
- Bu P., Wang L., Chen K.Y., Srinivasan T., Murthy P.K., Tung K.L., Varanko A.K., Chen H.J., Ai Y., King S. A miR-34a-Numb Feedforward loop triggered by inflammation regulates asymmetric stem cell division in intestine and colon cancer. Cell Stem Cell. 2016;18:189–202. doi: 10.1016/j.stem.2016.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network, Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M. The cancer Genome Atlas pan-cancer analysis project. Nat. Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Willis S.N., Wei A., Smith B.J., Fletcher J.I., Hinds M.G., Colman P.M., Day C.L., Adams J.M., Huang D.C. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- Dai H., Meng X.W., Lee S.H., Schneider P.A., Kaufmann S.H. Context-dependent Bcl-2/Bak interactions regulate lymphoid cell apoptosis. J. Biol. Chem. 2009;284:18311–18322. doi: 10.1074/jbc.M109.004770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H., Smith A., Meng X.W., Schneider P.A., Pang Y.P., Kaufmann S.H. Transient binding of an activator BH3 domain to the Bak BH3-binding groove initiates Bak oligomerization. J. Cell Biol. 2011;194:39–48. doi: 10.1083/jcb.201102027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzler R., Agarwal V., Stefano J., Bartel D.P., Stoffel M. Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Mol. Cell. 2014;54:766–776. doi: 10.1016/j.molcel.2014.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y.X., Bian X.H., Qian P.D., Chen Z.Z., Wen J., Luo Y.H., Yan P.W., Zhang Q. MicroRNA-125b inhibits cell proliferation and induces cell apoptosis in esophageal squamous cell carcinoma by targeting BMF. Oncol. Rep. 2018;40:61–72. doi: 10.3892/or.2018.6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer P.E., Frederick P., Gulbis J.M., Dewson G., Kluck R.M. Translocation of a Bak C-terminus mutant from cytosol to mitochondria to mediate cytochrome C release: implications for Bak and Bax apoptotic function. PLoS One. 2012;7:e31510. doi: 10.1371/journal.pone.0031510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricker M., O'Prey J., Tolkovsky A.M., Ryan K.M. Phosphorylation of Puma modulates its apoptotic function by regulating protein stability. Cell Death Dis. 2010;1:e59. doi: 10.1038/cddis.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaglia G., Guan Y., Shah J.V., Lahav G. Activation and control of p53 tetramerization in individual living cells. Proc. Natl. Acad. Sci. U S A. 2013;110:15497–15501. doi: 10.1073/pnas.1311126110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman M.J., Craft B., Hastie M., Repecka K., McDade F., Kamath A., Banerjee A., Luo Y., Rogers D., Brooks A.N. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020;38:675–678. doi: 10.1038/s41587-020-0546-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J., Zhang J.P., Li B., Zeng C., You K., Chen M.X., Yuan Y., Zhuang S.M. MicroRNA-125b promotes apoptosis by regulating the expression of Mcl-1, Bcl-w and IL-6R. Oncogene. 2013;32:3071–3079. doi: 10.1038/onc.2012.318. [DOI] [PubMed] [Google Scholar]
- Grespi F., Soratroi C., Krumschnabel G., Sohm B., Ploner C., Geley S., Hengst L., Hacker G., Villunger A. BH3-only protein Bmf mediates apoptosis upon inhibition of CAP-dependent protein synthesis. Cell Death Differ. 2010;17:1672–1683. doi: 10.1038/cdd.2010.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y., Yao H., Zheng Z., Qiu G., Sun K. MiR-125b targets BCL3 and suppresses ovarian cancer proliferation. Int. J. Cancer. 2011;128:2274–2283. doi: 10.1002/ijc.25575. [DOI] [PubMed] [Google Scholar]
- Hui L., Zhang J., Guo X. MiR-125b-5p suppressed the glycolysis of laryngeal squamous cell carcinoma by down-regulating hexokinase-2. Biomed. Pharmacother. 2018;103:1194–1201. doi: 10.1016/j.biopha.2018.04.098. [DOI] [PubMed] [Google Scholar]
- Korsmeyer S.J., Wei M.C., Saito M., Weiler S., Oh K.J., Schlesinger P.H. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000;7:1166–1173. doi: 10.1038/sj.cdd.4400783. [DOI] [PubMed] [Google Scholar]
- Ku B., Liang C., Jung J.U., Oh B.H. Evidence that inhibition of BAX activation by BCL-2 involves its tight and preferential interaction with the BH3 domain of BAX. Cell Res. 2011;21:627–641. doi: 10.1038/cr.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le M.T., Teh C., Shyh-Chang N., Xie H., Zhou B., Korzh V., Lodish H.F., Lim B. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009;23:862–876. doi: 10.1101/gad.1767609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.L., Nie H., Wang M., Su L.P., Li J.F., Yu Y.Y., Yan M., Qu Q.L., Zhu Z.G., Liu B.Y. microRNA-155 is downregulated in gastric cancer cells and involved in cell metastasis. Oncol. Rep. 2012;27:1960–1966. doi: 10.3892/or.2012.1719. [DOI] [PubMed] [Google Scholar]
- Lindner A.U., Concannon C.G., Boukes G.J., Cannon M.D., Llambi F., Ryan D., Boland K., Kehoe J., McNamara D.A., Murray F. Systems analysis of BCL2 protein family interactions establishes a model to predict responses to chemotherapy. Cancer Res. 2013;73:519–528. doi: 10.1158/0008-5472.CAN-12-2269. [DOI] [PubMed] [Google Scholar]
- Ma L., Wagner J., Rice J.J., Hu W., Levine A.J., Stolovitzky G.A. A plausible model for the digital response of p53 to DNA damage. Proc. Natl. Acad. Sci. U S A. 2005;102:14266–14271. doi: 10.1073/pnas.0501352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltzman W., Czyzyk L. UV irradiation stimulates levels of p53 cellular tumor antigen in nontransformed mouse cells. Mol. Cell Biol. 1984;4:1689–1694. doi: 10.1128/mcb.4.9.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Caballero S., Dejean L.M., Kinnally M.S., Oh K.J., Mannella C.A., Kinnally K.W. Assembly of the mitochondrial apoptosis-induced channel, MAC. J. Biol. Chem. 2009;284:12235–12245. doi: 10.1074/jbc.M806610200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi H., Taccioli C., Palatini J., Fernandez-Cymering C., Cui R., Kim T., Volinia S., Croce C.M. Loss of miR-125b-1 contributes to head and neck cancer development by dysregulating TACSTD2 and MAPK pathway. Oncogene. 2014;33:702–712. doi: 10.1038/onc.2013.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano K., Vousden K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- Natan E., Hirschberg D., Morgner N., Robinson C.V., Fersht A.R. Ultraslow oligomerization equilibria of p53 and its implications. Proc. Natl. Acad. Sci. U S A. 2009;106:14327–14332. doi: 10.1073/pnas.0907840106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier M., Hollstein M., Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010;2:a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma C.A., Al Sheikha D., Lim T.K., Bryant A., Vu T.T., Jayaswal V., Ma D.D. MicroRNA-155 as an inducer of apoptosis and cell differentiation in Acute Myeloid Leukaemia. Mol. Cancer. 2014;13:79. doi: 10.1186/1476-4598-13-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peller S., Rotter V. TP53 in hematological cancer: low incidence of mutations with significant clinical relevance. Hum. Mutat. 2003;21:277–284. doi: 10.1002/humu.10190. [DOI] [PubMed] [Google Scholar]
- Prakash S., Inobe T., Hatch A.J., Matouschek A. Substrate selection by the proteasome during degradation of protein complexes. Nat. Chem. Biol. 2009;5:29–36. doi: 10.1038/nchembio.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W., Ren Q., Liu T., Huang Y., Wang J. MicroRNA-155 is a novel suppressor of ovarian cancer-initiating cells that targets CLDN1. FEBS Lett. 2013;587:1434–1439. doi: 10.1016/j.febslet.2013.03.023. [DOI] [PubMed] [Google Scholar]
- Rajagopalan S., Huang F., Fersht A.R. Single-Molecule characterization of oligomerization kinetics and equilibria of the tumor suppressor p53. Nucleic Acids Res. 2011;39:2294–2303. doi: 10.1093/nar/gkq800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A.W., Seymour J.F., Brown J.R., Wierda W.G., Kipps T.J., Khaw S.L., Carney D.A., He S.Z., Huang D.C., Xiong H. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 2012;30:488–496. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooswinkel R.W., van de Kooij B., de Vries E., Paauwe M., Braster R., Verheij M., Borst J. Antiapoptotic potency of Bcl-2 proteins primarily relies on their stability, not binding selectivity. Blood. 2014;123:2806–2815. doi: 10.1182/blood-2013-08-519470. [DOI] [PubMed] [Google Scholar]
- Shaham L., Binder V., Gefen N., Borkhardt A., Izraeli S. MiR-125 in normal and malignant hematopoiesis. Leukemia. 2012;26:2011–2018. doi: 10.1038/leu.2012.90. [DOI] [PubMed] [Google Scholar]
- Sheu-Gruttadauria J., Pawlica P., Klum S.M., Wang S., Yario T.A., Schirle Oakdale N.T., Steitz J.A., MacRae I.J. Structural basis for target-directed MicroRNA degradation. Mol. Cell. 2019;75:1243–1255 e1247. doi: 10.1016/j.molcel.2019.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X.B., Xue L., Ma A.H., Tepper C.G., Kung H.J., White R.W. miR-125b promotes growth of prostate cancer xenograft tumor through targeting pro-apoptotic genes. The Prostate. 2011;71:538–549. doi: 10.1002/pros.21270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X.B., Xue L., Yang J., Ma A.H., Zhao J., Xu M., Tepper C.G., Evans C.P., Kung H.J., deVere White R.W. An androgen-regulated miRNA suppresses Bak1 expression and induces androgen-independent growth of prostate cancer cells. Proc. Natl. Acad. Sci. U S A. 2007;104:19983–19988. doi: 10.1073/pnas.0706641104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart D.P., Koss B., Bathina M., Perciavalle R.M., Bisanz K., Opferman J.T. Ubiquitin-independent degradation of antiapoptotic MCL-1. Mol. Cell Biol. 2010;30:3099–3110. doi: 10.1128/MCB.01266-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y.M., Lin K.Y., Chen Y.Q. Diverse functions of miR-125 family in different cell contexts. J. Hematol. Oncol. 2013;6:6. doi: 10.1186/1756-8722-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoronos A.A., Engelman D.M., Slack F.J. OncomiR or tumor suppressor? The duplicity of MicroRNAs in cancer. Cancer Res. 2016;76:3666–3670. doi: 10.1158/0008-5472.CAN-16-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay Y., Rinn J., Pandolfi P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344–352. doi: 10.1038/nature12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tili E., Michaille J.J., Luo Z., Volinia S., Rassenti L.Z., Kipps T.J., Croce C.M. The down-regulation of miR-125b in chronic lymphocytic leukemias leads to metabolic adaptation of cells to a transformed state. Blood. 2012;120:2631–2638. doi: 10.1182/blood-2012-03-415737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Sun S.Y., Khuri F., Curran W.J., Deng X. Mono- or double-site phosphorylation distinctly regulates the proapoptotic function of Bax. PLoS One. 2010;5:e13393. doi: 10.1371/journal.pone.0013393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg R.L., Veprintsev D.B., Bycroft M., Fersht A.R. Comparative binding of p53 to its promoter and DNA recognition elements. J. Mol. Biol. 2005;348:589–596. doi: 10.1016/j.jmb.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Weinberg R.L., Veprintsev D.B., Fersht A.R. Cooperative binding of tetrameric p53 to DNA. J. Mol. Biol. 2004;341:1145–1159. doi: 10.1016/j.jmb.2004.06.071. [DOI] [PubMed] [Google Scholar]
- Westphal D., Kluck R.M., Dewson G. Building blocks of the apoptotic pore: how Bax and Bak are activated and oligomerize during apoptosis. Cell Death Differ. 2014;21:196–205. doi: 10.1038/cdd.2013.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willimott S., Wagner S.D. miR-125b and miR-155 contribute to BCL2 repression and proliferation in response to CD40 ligand (CD154) in human leukemic B-cells. J. Biol. Chem. 2012;287:2608–2617. doi: 10.1074/jbc.M111.285718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N., Lin X., Zhao X., Zheng L., Xiao L., Liu J., Ge L., Cao S. MiR-125b acts as an oncogene in glioblastoma cells and inhibits cell apoptosis through p53 and p38MAPK-independent pathways. Br. J. Cancer. 2013;109:2853–2863. doi: 10.1038/bjc.2013.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H.F., He T.Z., Liu C.M., Cui Y., Song P.P., Jin X.H., Ma X. MiR-125b expression affects the proliferation and apoptosis of human glioma cells by targeting Bmf. Cell. Physiol. Biochem. 2009;23:347–358. doi: 10.1159/000218181. [DOI] [PubMed] [Google Scholar]
- Yang M., Tang X., Wang Z., Wu X., Tang D., Wang D. miR-125 inhibits colorectal cancer proliferation and invasion by targeting TAZ. Biosci. Rep. 2019;39 doi: 10.1042/BSR20190193. BSR20190193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao A., Zeng Q., Xie X., Zhou J., Yue W., Li Y., Pei X. MicroRNA-125b induces cancer cell apoptosis through suppression of Bcl-2 expression. J. Genet. Genomics. 2012;39:29–35. doi: 10.1016/j.jgg.2011.12.003. [DOI] [PubMed] [Google Scholar]
- Zhou M., Liu Z., Zhao Y., Ding Y., Liu H., Xi Y., Xiong W., Li G., Lu J., Fodstad O. MicroRNA-125b confers the resistance of breast cancer cells to paclitaxel through suppression of pro-apoptotic Bcl-2 antagonist killer 1 (Bak1) expression. J. Biol. Chem. 2010;285:21496–21507. doi: 10.1074/jbc.M109.083337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All original code has been included in the supplemental information and is available for download. Any additional information required to analyze the data reported in this paper is available from the lead contact upon request.