

Abstract
Bruton's tyrosine kinase (BTK) is a TEC kinase with a multifaceted role in B‐cell biology and function, highlighted by its position as a critical component of the B‐cell receptor signalling pathway. Due to its role as a therapeutic target in several haematological malignancies including chronic lymphocytic leukaemia, BTK has been gaining tremendous momentum in recent years. Within the immune system, BTK plays a part in numerous pathways and cells beyond B cells (i.e. T cells, macrophages). Not surprisingly, BTK has been elucidated to be a driving factor not only in lymphoproliferative disorders but also in autoimmune diseases and response to infection. To extort this role, BTK inhibitors such as ibrutinib have been developed to target BTK in other diseases. However, due to rising levels of resistance, the urgency to develop new inhibitors with alternative modes of targeting BTK is high. To meet this demand, an expanding list of BTK inhibitors is currently being trialled. In this review, we synopsize recent discoveries regarding BTK and its role within different immune cells and pathways. Additionally, we discuss the broad significance and relevance of BTK for various diseases ranging from haematology and rheumatology to the COVID‐19 pandemic. Overall, BTK signalling and its targetable nature have emerged as immensely important for a wide range of clinical applications. The development of novel, more specific and less toxic BTK inhibitors could be revolutionary for a significant number of diseases with yet unmet treatment needs.
Keywords: autoimmunity, Bruton's tyrosine kinase, BTK inhibitor, chronic lymphocytic leukaemia, ibrutinib, infections, lymphoproliferative disorders
The role of BTK in the immune system and disease: structure, function and inhibition.
Abbreviations
- AF
atrial fibrillation
- APC
antigen‐presenting cell
- BCR
B‐cell receptor
- BMX
bone marrow‐expressed kinase
- BTK
Bruton's tyrosine kinase
- BTKi
BTK inhibitor
- CLL
chronic lymphocytic leukaemia
- DC
dendritic cells
- DLBCL
diffuse large B‐cell lymphoma
- ITK
interleukin‐2‐inducible T‐cell kinase
- MCL
Mantle cell lymphoma
- MS
multiple sclerosis
- NLC
nurse‐like cells
- PAMP
pathogen‐associated molecular patterns
- PH
Pleckstrin
- PV
pemphigus vulgaris
- RA
rheumatoid arthritis
- SH
SRC homology (SH) domains
- SLE
systemic lupus erythematosus
- TEC
tyrosine kinase expressed in hepatocellular carcinoma
- TLR
Toll‐like receptor
- TXK
tyrosine protein kinase
- WM
Waldenström's macroglobulinaemia
- XID
X‐linked immunodeficiency
- XLA
X‐linked agammaglobulinaemia
INTRODUCTION
Bruton's tyrosine kinase (BTK) is a non‐receptor Tec kinase that has been gaining momentum since the approval of BTK‐targeted therapies as a first‐line treatment for B‐cell malignancies. The exponential expansion of the number of publications relating to BTK over the past decade can be attributed to the enhanced understanding of the multifaceted role it plays within the immune system and the advent of novel more specific BTK inhibitors. Historically, it all started in 1952 when Ogden Bruton reported the first case of the later termed X‐linked agammaglobulinaemia (XLA) [1]. In 1993, a BTK mutation was identified by two independent groups to be causally linked to XLA. XLA patients experience a blockage on B‐cell development in the bone marrow, which, in severely affected patients, leads to little/no functional serum immunoglobulins and therefore increased disease susceptibility [2, 3] (Figure 1). Subsequently, BTK has been shown to play a crucial role in several biological processes including B‐cell differentiation. BTK was shown to be phosphorylated in vitro upon B‐cell antigen receptor (BCR) stimulation, followed by higher levels of kinase activity. These events lead to the enlisting of BTK as a member of the BCR signalling pathway. This was further established in BTK‐deficient mouse studies. Through blocking the expression of BTK, Khan et al. [4] showed a decrease in mature B cells and a deficiency of immunoglobulins in the serum of BTK‐deficient mice, underlying the indispensable nature of BTK for B‐cell differentiation. A milder disease form, harbouring a point mutation in BTK, is known as X‐linked immunodeficiency (XID). The defects present in XID are minor in comparison with XLA, but the B cells present within patients have disrupted BCR signalling [5].
FIGURE 1.
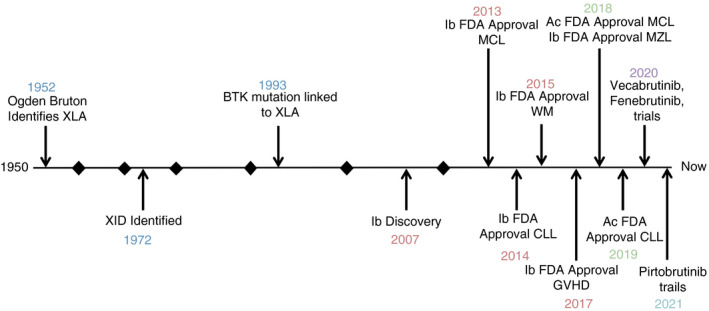
Timeline for the discovery of BTK highlighting key milestones in BTK‐related research and therapies. Each section represents a decade. Ac, acalabrutinib; CLL, chronic lymphocytic leukaemia; GVHD, graft‐versus‐host disease; Ib, ibrutinib; MCL, mantle cell lymphoma; MZL, marginal zone lymphoma; WM, Waldenström's macroglobulinaemia; XID, X‐linked immunodeficiency; XLA, X‐linked agammaglobulinaemia
Given the emerging wider interest for BTK signalling across disciplines, we will review the key research over the last four years with regard to (i) the role of BTK in different signalling and immune cells, (ii) the advances in our understanding of its role in immunity, autoimmunity and infection, (iii) BTK signalling in lymphoproliferative disorders and (iv) current therapeutic options for BTK inhibition and ongoing clinical trials looking into novel therapeutics and resistance mechanisms.
THE STRUCTURE OF BTK
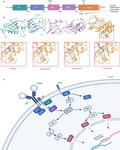
The structure of BTK has been extensively documented since the 1990s, and nowadays, we have achieved a clear view on this aspect. A review by Hendriks group summarizes excellent work with regard to BTK structure. In short, BTK (together with bone marrow‐expressed kinase [BMX], interleukin‐2‐inducible T‐cell kinase [ITK], tyrosine kinase expressed in hepatocellular carcinoma [TEC] and tyrosine protein kinase [TXK]) belongs to the second‐largest non‐receptor tyrosine kinase family, namely TEC. BTK consists of (i) two SRC homology (SH) domains (SH2 and SH3), (ii) a kinase domain, (iii) an N‐terminal pleckstrin (PH) domain and (iv) a TEC homology domain (Figure 2a). Unlike SRC, BTK is a cytoplasmic protein and only is present on the membrane upon recruitment. Once at the cell membrane, BTK can become activated by the interaction with SRC kinases and phosphorylation occurs at the 551 position of the kinase. This can also occur through interaction with a SYK kinase [6]. The SH2 domain and kinase domain together form an allosteric interface, which has been reported to be critical for the activation of BTK. Interestingly, Duarte et al. [7] have recently demonstrated that this interface can contain SH2 mutations in XLA, which dramatically impacts BTK activation. The cysteine residue at position 481 of the kinase domain (C481) of BTK is a region of particular interest due to it being a target of many BTK inhibiting drugs. However, a cysteine residue is also present at the 481 position of several other kinases including the previously mentioned BMX, TEC and ITK. This overlap leads to several off‐target toxicities. Of note, the off‐target inhibition of TEC is believed to be the cause of the bleeding risk associated with the BTK inhibitor ibrutinib [6].
FIGURE 2.
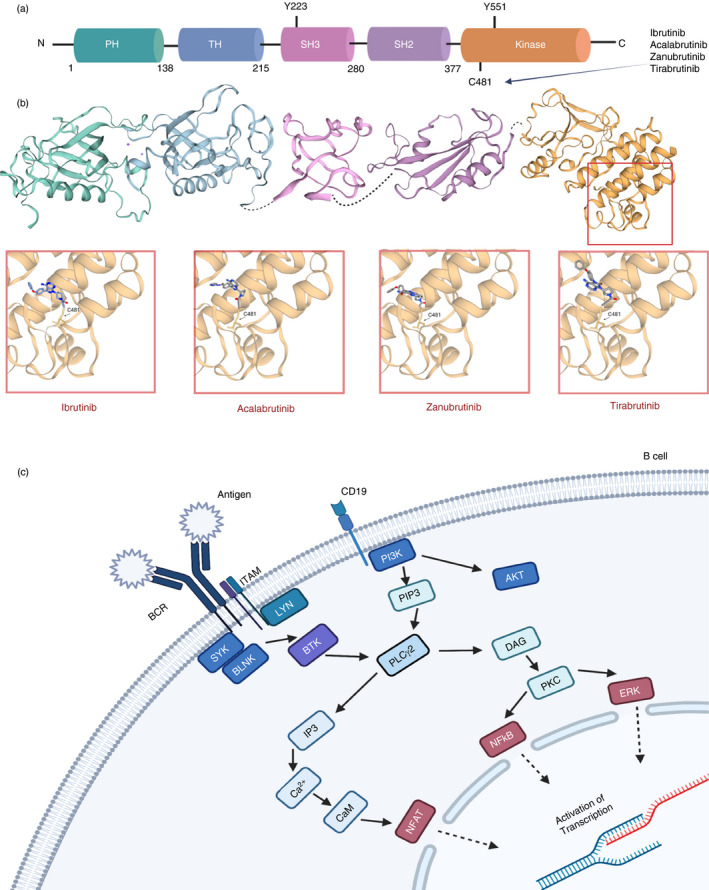
Structural overview of BTK and BTK inhibitors and its position within the B‐cell receptor signalling pathway. (a) The 77 kDa BTK protein consists of 659 amino acids, which make up the five domains for protein interaction. The 2 critical sites within the protein are Y223 and (SH3 domain) and Y551 (kinase domain: orange/yellow domain). (b) BTK inhibitors act through the binding to one of the proteins interacting domains and blocking BTK’s catalytic action. The main site of binding for current covalent inhibitors is the C481 residue within the kinase domain. This includes ibrutinib and second‐generation inhibitors acalabrutinib, zanubrutinib and tirabrutinib as depicted at the 3D models (data obtained from SWISS‐MODEL repository by the Swiss Institute of Bioinformatics [101] for the crystal structures of each BTK domain and then NCBI's PubChem database for the line structures for each inhibitor [102]). (c) A simplified version of B‐cell receptor signalling pathway and BTK position within it [103]
BTK IN SIGNALLING PATHWAYS
B‐cell receptor signalling pathway
Firstly, shortly after its discovery, BTK was positioned within BCR signalling downstream of the antigen receptor [8] (Figure 2b). BCR activation generates a cascade of events that amass in the activation of the B cell, enabling B‐cell differentiation and proliferation. IgM and heterodimer form the BCR; they act as a unit for antigen binding on the surface of the B cell. The Ig‐α and Ig‐β’s tail carries an immuno‐receptor tyrosine‐based activation motif (ITAM), which contains a sequence with two tyrosine residues. Upon BCR stimulation, BTK activation starts with PH domain‐mediated plasma membrane association and transphosphorylation at Y551 within the BTK catalytic domain, by SYK, LYN or SRC kinases. Y551 promotes the catalytic activity of BTK and results in its autophosphorylation at position Y223 in the SH3 domain [9]. BTK inhibitors such as ibrutinib block the full activation of BTK by inhibiting also its autophosphorylation at Y223. Upon BTK activation, the protein kinases SRC, SYK and LYN regulate the signalling of pathways further downstream. BTK is responsible for the phosphorylation of phospholipase C‐γ (PLCγ2), which together stimulate a positive feedback loop. Two second messengers, diacylglycerol (DAG) and inositol triphosphate (IP3), can be cleaved by PLCγ2, which regulate downstream mediators such as the MAPK family and activate transcription of nuclear factor of activated T cells (NFAT) through calmodulin [10]. Without BTK, B cells fail to reach functional maturity, which highlights the indispensable nature of BTK [4]. Further insights on the role of BTK within BCR signalling have recently arisen from studies in lymphoma. Autoantigens have been shown to drive BCR‐dependent activation of NF‐κB through a cascade that includes BTK, SYK and PKCβ and promote the assembly of the CARD11‐BCL10‐MALT1 (CBM) adaptor complex that recruits and activates IκB kinase [11].
Toll‐like receptors
Toll‐like receptors (TLRs) are pivotal for the recognition of pathogens through the detection of pathogen‐associated molecular patterns (PAMPs). Expressed mainly on antigen‐presenting cells (APCs), TLRs bring about the induction of the adaptive immune response [12]. Together, BCR and TLRs carefully control immunity against pathogens via linking the innate and adaptive axes of immune response. TLR signalling in B cells depends on the recruitment of MYD88. Although MYD88 signalling is not fully required for the T‐cell‐dependent humoral immunity, it can affect it through controlling major processes such as germinal centre reaction, differentiation into plasma cells and memory B‐cell class switching recombination. BTK interacts with MYD88 and other proteins downstream within the TLR signalling pathway such as MAL and IRAK1 [13].
TLR is another potential pathway that malignant T cells can exploit for their survival upon BTK inhibitory treatment. It has been reported that chronic lymphocytic leukaemia (CLL) cells within the lymph node exhibit increased TLR activation compared with circulating cells, as well as increased levels of NF‐κB phosphorylation. Due to TLR’s role in CLL pathogenesis, combinatorial targeting of BTK and TLR is seen as a potential beneficial therapeutic approach [14]. The crosstalk of BTK with TLRs has also been reported in autoimmune disorders, where both of these signalling pathways seem to contribute to disease pathogenesis [15]. In addition to BTK mutations, resistance to BTK‐targeted therapies can emerge from downstream targets or other pathways entirely. TLR signalling aberrations have recently been identified in Waldenström's macroglobulinaemia (WM), specifically in regulators of MYD88/NF‐κB. The identification of these mutations is crucial for the correct treatment of patients with WM and other related diseases [16].
Chemokine receptors
It has been well established that BTK participates in chemokine receptor signalling pathways. For instance, SDF‐1 is known to activate BTK and is essential for CXCR4‐regulated B‐cell functions such as B‐cell trafficking from the peripheral bloodto lymphoid organs [17]. More recently, CX3CR1 has been linked to immunosuppression. CX3CR1 knockout mice are characterized by impaired BCR signalling, and BTK expression was shown to be caused by defects in actin remodelling, which is normally controlled by CX3CR1 [18].
THE ROLE OF BTK IN THE NUCLEUS
It has been shown that BTK is present in small quantities in the nucleus, even though it is primarily a cytoplasmic protein. The mechanisms behind the regulation of BTK in the nucleus and cytoplasmic shuttling are still ambiguous. BAM11 binds to BTK’s PH domain and inhibits both in vivo and in vitro BTK activity. BTK acts as a transcriptional regulator for BAM11 in B cells [19]. TFII‐I has been reported to be one of BTK’s targets. Novina et al. reported that BTK increases the transcriptional activity of TFII‐I by mediating signals in B‐cell‐specific pathways. In BTK‐mutant cell TFII‐I‐dependent transcription was reduced and B‐cell development impaired [20].
Several proteins have been reported to modulate the nucleocytoplasmic shuttling of BTK. Lyn‐interacting ankyrin repeat protein (LIAR) has been suggested to regulate BTK trafficking via BTK phosphorylation [21]. More recently, Gustafsson et al. [22] reported that the ANKRD54 modulates the shuttling of BTK to the nucleus.
Overexpression and knockdown models have identified BTK to be essential for TNF expression through the stimulation of cytokines, activating TLR4 and TLR7/8 signalling. In macrophages, BTK has been reported to enter the nucleus through mediating the phosphorylation of Ser‐536 in p65 RelA, part of the TLR7/8 pathway. BTK can regulate TNF, induced by TLR7/8, through the promoter sites of NF‐κB and also downstream gene regions. This study detailed BTK control of the production of TNF and suggested this pathway could have the potential to be exploited for an anti‐inflammatory effect [23]. Despite the research so far, the relevance of BTK in the nucleus still needs systematic investigation in order to obtain conclusive evidence [24].
BTK AND IMMUNITY
Beyond B cells
BTK’s roles within cells are both diverse and numerous. Aside from B cells, the kinase is found to be highly expressed in mast cells, macrophages and dendritic cells; immune cells are found to be involved in the elimination of pathogens [25]. In addition to B cells, BTK’s function in T cells has also been explored. Nowadays, BTK is considered a vital protein expressed by immunocompetent cells of both innate and adaptive immunity (Figure 3).
FIGURE 3.
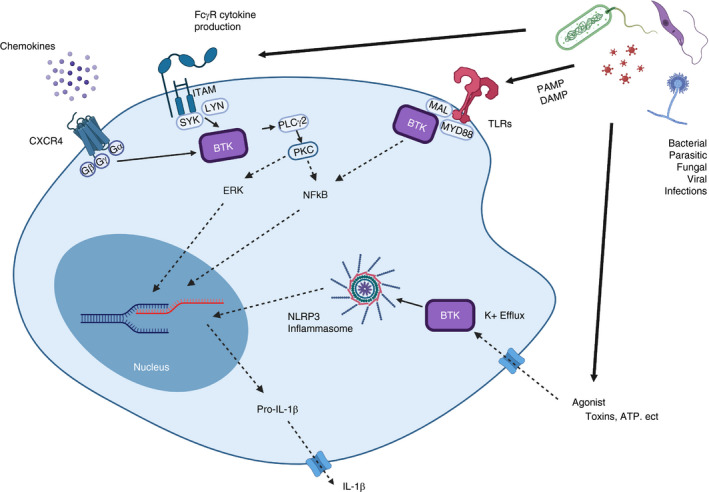
Overview of the various roles of Bruton's tyrosine kinase (BTK) in innate immunity. Chemokine receptor (CXCR4) activation upon chemokine binding leads to the dissociation of G proteins made up of Gα, Gβ and Gy subunits and downstream activation of BTK. ITAM‐containing (and also ITIM‐containing) Fc receptor crosslinking leads to activation of SYK and in turn BTK. Toll‐like receptors (TLRs) are activated by pathogen‐associated molecular patterns (PAMPs) and‐damage associated molecular patterns (DAMPs). Activation of TLRs is followed by recruitment of MYD88. BTK in turn interacts with MYD88 leading to activation of transcription factors such as NF‐кB. BTK is a direct regulator in the activation of the NLRP3 inflammasome. Efflux of K+ into the cell leads to phosphorylation of BTK, most likely by SYK, followed by activation. This phosphorylation promotes assembly of the inflammasome and leads to the cleavage and secretion of IL‐1β [103]
T Lymphocytes
BTK inhibition in vivo has been reported to increase the persistence of activated T cells, decrease the Treg /CD4+ T‐cell ratio and diminish the immune‐suppressive properties of CLL cells via BTK‐dependent and BTK‐independent mechanisms [26]. Characterization of T‐cell compartment in CLL patients upon ibrutinib therapy showed elevated CD4+ and CD8+ T‐cell numbers and T‐related cytokine levels upon therapy [27]. Ibrutinib has been shown to enhance the antitumor properties of the T‐cell compartment suggesting a rationale for immunotherapeutic combinatory treatments [28]. The immunomodulatory effects of ibrutinib and the therapeutic potential of its combination with immune checkpoint inhibitors were also highlighted in a recent study, in which ibrutinib, together with blocking antibodies targeting PD‐1/PD‐L1 axis, improved CD8+ T‐cell effector function and control of CLL [29].
Cells of myeloid origin
Macrophages, originating from monocytes, function in pathogen detection and phagocytosis and together with dendritic cells play a special role linking the innate and adaptive immune systems through antigen presentation [30]. Ren et al. showed that although BTK inhibition with ibrutinib did not affect monocyte FcγR‐mediated phagocytosis, it did suppress FcγR‐mediated cytokine production. This effect could be rescued by IFN priming when monocytes were co‐cultured in vitro with NK cells, suggesting that combining ibrutinib with monoclonal antibody therapy could enhance tumour killing without affecting macrophage effector function [31]. Interestingly, an immunomodulatory action of BTK inhibition on monocyte/macrophage population has been reported in CLL. Specifically, Ibrutinib targets BTK in nurse‐like cells (NLCs), which leads to reduced phagocytic ability and enhanced immunosuppression related to NLCs’ expression of M2 markers [32]. Additional evidence for the role of BTK in cells of myeloid origin arose from transcriptomic analysis of XLA‐derived monocytes, which revealed downregulation of several innate immunity genes in parallel with upregulation of oxidative phosphorylation and apoptotic pathways. These findings suggest that BTK mutations may significantly impair the innate axis of immunity and indicate a vital role of BTK in innate immune responses [33]. However, another study reported no impact of BTK mutation on monocytes and PMN functions in XLA [34]. Further investigation of the role of BTK in monocytes is needed to provide a better understanding of the events occurring in these studies.
Neutrophils are yet another immune cell type in which BTK has been shown to play a key role [35]. For instance, a neutrophil‐BTK‐signalosome was reported to selectively activate Mac‐1 and thereby enhance neutrophil recruitment during inflammation [35]. In CLL, BTK inhibition suppresses FcγR‐mediated neutrophil functions during the early phase of treatment and potentially in a clinically relevant way. The reported short‐term neutrophil impairment upon ibrutinib treatment could translate to additional infection risk for CLL patients under ibrutinib [36]. Furthermore, ibrutinib has been reported to inhibit γδT‐cell activation and CD107a degranulation and affect neutrophils by reducing NET formation, ROS production and bacteria‐killing capacity [37].
Mast cells have been investigated in association with BTK. Zorn et al. [38] reported that BTK plays a role in FcεRI‐mediated signal transduction and effector functions in SHIP1‐deficient mast cells and that reduced activation can be tackled with BTK inhibitors.
Inflammasomes
The NLRP3 inflammasome is an essential inflammatory complex for various human disorders linked to the activity of IL‐1 cytokine. The identification of BTK as a positive regulator, directly acting on the NLRP3 inflammasome, has further shown the significant role this kinase plays in immunity [39]. BTK binds to the caspase recruitment domain (ASC) of NLRP3, ASC oligomerization is induced, and caspase‐1 is thereby activated. Pharmacologic ablation and genetic BTK ablation severely diminish NLRP3 activation suggesting a therapeutic opportunity for inflammatory conditions [40]. Additionally, BTK is required for NLRP3 tyrosine phosphorylation and IL‐1β release. The BTK‐mediated phosphorylation of multiple NLRP3 tyrosine residues can serve as a molecular switch, which could be therapeutically exploited [41].
BTK in infections
The multifaceted role of BTK in immunity, as summarized above, underscores its importance for a wide variety of immune functions, including responses against pathogens. A large body of evidence offers insights into the role of BTK signalling in fungal, bacterial and viral infections, including the SARS‐CoV‐2 virus and the COVID‐19 pandemic.
Fungal infections
Activation of calcineurin–NFAT in macrophages occurs via a phagocytic TLR9‐dependent and BTK‐dependent pathway in the context of Aspergillus fumigatus infection. Calcineurin inhibition leads to impaired pathogen clearance in the airway due to diminished macrophage inflammatory responses and neutrophil recruitment. In line with that, defecting BTK signalling in macrophages has been associated with susceptibility to pulmonary aspergillosis [42], whereas BTK depletion significantly impaired human macrophage NFAT and NF‐κB responses [43]. Furthermore, ibrutinib treatment has been linked to a high incidence of invasive aspergillosis in lymphoma [44].
Apart from macrophages, neutrophils are part of the first line of defence against fungal infections. Patients on ibrutinib due to lymphoid malignancies are characterized by significant functional defects in their neutrophil compartment that impair their response against A. fumigatus [45], whereas data from CLL patients on ibrutinib indicate an increased risk of Pneumocystis jirovecii pneumonia [46].
Bacterial and parasitic infections
Ibrutinib induces changes in gene expression and phenotype in macrophages and severely impairs the macrophage and γδT‐cell responses to Mycobacterium tuberculosis [47]. Ibrutinib has been reported to have the potential to inhibit inflammation caused by bacterial infection. Reports have shown that ibrutinib can inhibit the acute lung inflammation associated with pneumococcal pneumonia infections. BTK inhibition through Ibrutinib diminishes myeloid cell activation and migration during lung inflammation and has been identified as a possible therapy for resolving acute lung inflammation during pneumococcal pneumonia [48].
By employing a murine platelet‐specific BTK‐deficient pneumosepsis model (PF4creBtkfl/Y), Streptococcus pneumoniae and Klebsiella pneumoniae infections were investigated, revealing a role of BTK in maintaining vascular integrity in the lung. However, this mechanism is pathogen‐dependent and the platelet BTK is not crucial for antibacterial defence in pneumosepsis [49]. Recently, Nguyen et al reported on another aspect of K. pneumoniae infection using in vivo and in vitro models to show that SKAP2‐dependent signalling in neutrophils is important for ROS activation and promotion of bacterial clearance during infection. Interestingly, among the key molecules that were identified for K. pneumoniae‐induced neutrophil ROS response was BTK [50].
BTK inhibition has been proposed to confer protection against Leishmania infection via promoting host immunity. In a mouse model for visceral leishmaniasis, ibrutinib treatment was shown to have a protective effect via increasing cytokines’ production, NK T cells’ number in the liver and spleen and granuloma formation [51].
Viral infections
Studies have described in‐depth the crucial role of BTK expressed by innate cells in viral infections [25]. In macrophages, TLRs recognize single‐stranded RNA from viruses and initiate signalling through BTK‐dependent activation of NF‐κB, triggering the production of multiple inflammatory cytokines and chemokines, as well as phagocytosis [52]. The latest experimental evidence indicates that BTK is involved in Influenza A virus (IAV) infection. Specifically, it was found that BTK expressed in neutrophils plays a substantial role in regulating inflammation in the respiratory region during acute lung injury in mice. Inhibition of the kinase activity reduced weight loss, increased survival and minimized morphological changes in IAV infection showing a protective effect in the lung during influenza‐induced inflammation [53].
COVID‐19 pandemic
With the recent emergence of the novel coronavirus, COVID‐19, the need for new therapeutic targets has surged as the severity of the pandemic increases. In severe cases of COVID‐19, high levels of activation of macrophages have been identified as a cause of the hyperinflammatory immune response seen in these patients. As previously mentioned, BTK regulates the activity of macrophages, prompting the concept that the inhibition of BTK could be used as a therapeutic option for COVID‐19 patients. So far, preliminary data have shown promising results. Based on this knowledge of the role of BTK in innate immune cells, COVID‐19 patients exhibiting increasing oxygen requirements and hyperinflammation were treated with the second‐generation BTKi acalabrutinib. The majority of patients’ conditions rapidly improved upon treatment with increased oxygenation and reduced inflammation. The patients from this study exhibited significantly elevated BTK phosphorylation in monocytes indicating the improved conditions were an on‐target effect of BTK inhibition [54].
From another point of view, we must be wary of the fact that BTK inhibition impairs various functions of the innate immunity and increases the susceptibility to infections or impaired humoral immunity in patients on BTKi. Awareness of these issues during the current COVID‐19 pandemic is essential as weakened immune states increase susceptibility to infection. However, following the rationale stated by Chong et al., the risk of depriving cancer patients of treatment and dulling the hyperactive macrophage outweighs the potential dampened immune response. Furthermore, long‐term BTKi therapy may allow for meaningful recovery of humoral immune function, ultimately leading to decreased infection rates [55] and potentially protecting against lung injury in COVID‐19 patients [56].
Taking into account the full effect of BTK inhibition in the setting of treating COVID‐19‐infected B‐cell lymphoma patients, a recent controversial debate in the discontinuation of BTK inhibitors to those patients has been brought to attention [57]. Two pilot studies published the clinical characteristics and progress of 6 CLL and 8 WM patients, respectively, with COVID‐19 infection who continued or held the BTKi therapy. The authors of both studies suggested that BTK inhibition may indeed have protective effects against SARS‐CoV‐2 virulence. The small cohort of patients evaluated is a limitation to support the therapeutic approach of BTKi, but ideally, two clinical trials assessing the effect of second‐generation BTK inhibitors in hospitalized COVID‐19 patients will shed light on that setting [58, 59].
BTK in autoimmune diseases
Since its discovery in XLA, the link of BTK with autoimmune phenomena is strengthened by numerous studies in various autoimmune diseases and mechanistic insights concerning BTK’s contribution in driving autoimmune pathogenesis. For instance, a driving factor for autoimmunity is that increased levels of BTK support autoantibody production [60].
Systemic lupus erythematous
Systemic lupus erythematosus (SLE) is characterized by the secretion of autoantibodies. These autoreactive B cells exhibit increased levels of BTK expression [61]. Murine studies suggest that inhibition of BTK can control the autoreactive B cells with potential therapeutic implications [62]. In line with that, the covalent BTK inhibitor, evobrutinib, has shown efficacy in murine models for SLE, rheumatoid arthritis and cutaneous anaphylaxis. In vivo studies have demonstrated evobrutinib to be potent and highly selective for BTK, showing high BTK occupancy. Current clinical trials are ongoing for its efficacy in the treatment of SLE, among other autoimmune diseases [63, 64].
Rheumatoid arthritis
Rheumatoid arthritis (RA) is characterized by autoantibodies causing chronic inflammation and joint pain [65]. Interest in BCR‐targeted therapies has been increasing recently, especially BTK due to its role in controlling the Fcγ receptor downstream pathway among others.
Jansson et al. reported a gene on the murine X chromosome linked to susceptibility for developing arthritis. XID mouse models harbouring BTK mutations showed that in the absence of BTK, there are lower chances of developing arthritis [66]. From this development, many BTK inhibitors have been produced and investigated for their use in RA. For instance, the reversible BTK inhibitor 7H‐pyrrolo[2,3‐d] pyrimidine‐4‐amine derivative has been shown to have an anti‐arthritic effect. Despite initial promising in vivo results, a select few have seen successful developments and been moved onto clinical trials [67].
Currently in phase II clinical trials, branebrutinib is an irreversible, covalent BTK inhibitor that has been reported to be highly selective and efficient even in low doses. Branebrutinib has demonstrated a high BTK occupancy, and it has been suggested this inhibitor would be effective for the treatment of autoimmune diseases, in particular RA and SLE [68, 69]. Fenebrutinib, a reversible and non‐covalent BTK inhibitor, has previously been shown to be effective in patients with CLL and is now under investigation for its efficacy in RA and SLE [70].
Pemphigus vulgaris
The rare chronic autoimmune disease pemphigus vulgaris (PV) is identified by characteristic blistering on the skin and mucous membranes. The pathogenesis of PV involves the IgG autoantibodies attacking the desmoglein 3 glycoprotein within the desmosome [71]. T‐follicular helper cells have been reported to have a role in PV pathogenesis. Increased BTK expression has been shown to induce the differentiation of these T cells [72]. Studies have shown the use of BTK inhibitors in cases of PV patients who also presented with B‐cell lymphomas showed promising results for both diseases. From here, clinical trials of the BTK inhibitor PRN1008 are underway for PV therapy [73]. Studies have been looking to target the BCR‐identified BTK as a potential target for PV therapies [74].
Multiple sclerosis
Recent work has highlighted the potential of blocking BTK activity as a therapeutic option to improve anti‐CD20 therapies such as rituximab for multiple sclerosis (MS) patients. Trials using a BTK inhibitor with a brain‐penetrating property, tolebrutinib, have shown promising results. These trials have reported that BTK inhibition can halt the engulfing of myelin sheaths by microglia and prevent demyelination. Tolebrutinib has a favourable outcome in MS patients compared with other BTK inhibitors such as ibrutinib. The off‐target effects of ibrutinib make it unsuitable for the treatment of diseases outside of malignancies [75].
BTK in lymphoproliferative disorders
Chronic lymphocytic leukaemia
BTK has been established as an attractive mark for targeted therapies for the B‐cell malignancy chronic lymphocytic leukaemia (CLL) due to its upregulated expression. Through targeting BTK, cell death has been established as a direct consequence due to the blockage of signalling pathways and impeding cell proliferation. Reports of BTK inhibition influencing downstream targets MAPK and NF‐κB have only heightened interest in its impact in CLL [76].
The BTK inhibitor ibrutinib has already proved to be highly effective in the treatment of CLL not only through affecting BTK but also through the inhibition of other kinases and growth factors; it has caused a shift in the paradigm of treatment. Despite the success of Ibrutinib, the emerging resistance clones are leading to fewer patients achieving complete remission, whereas discontinuation of therapy due to off‐target effect hampers further drug efficiency. New inhibitors with higher levels of specificity and efficacy have been gaining momentum in the hopes of overcoming the rising levels of resistance [77].
Mantle cell lymphoma
Another lymphoproliferative disorder highly dependent on BCR signalling and BTK is mantle cell lymphoma (MCL). BTK has been identified as a possible target for the treatment of this aggressive malignancy. MCL cells overexpress BTK, which seems essential for its pathogenesis. Ibrutinib has been shown to be effective in the treatment of MCL in many studies, and it has been approved for treating relapsed/refractory (R/R) patients [78, 79].
Although the use of BTK inhibitors has made considerable improvements to the overall outcome of MCL, it is still largely an incurable disease; there is a need for novel targeted therapies. Combination therapies are an attractive option for overcoming resistance and improving overall outcomes such as BTK inhibition and venetoclax (BCL2 inhibitor) treatment. Matsumura‐Kimoto et al. demonstrated the potential of a combination of the serine/threonine kinase ribosomal protein S6 kinase (RSK2) and the BTK inhibitor ibrutinib. The inhibition of RSK2 was reported to affect downstream proteins involved in the BCR signalling pathway such as BLNK and CD19, as well as proteins from other pathways, blocking the B‐cell pathogenesis [80].
Waldenström's macroglobulinaemia
BTK has been reported to be constitutively activated in the less common haematological malignancy Waldenström's macroglobulinaemia (WM). Characterized by the excessive secretion of monoclonal IgM antibodies, WM is defined and diagnosed by a MYD88L265P somatic mutation [81]. BTK is a downstream component that is affected by the mutation MYD88L265P and leads to activation of NF‐κB. In WM, a higher level of phosphorylated BTK has been observed than in healthy counterparts with the preferential formation of a complex consisting of phosphorylated BTK and MYD88L265P [82].
Recent studies have identified MYD88 mutations in a complex with the protein kinase SYK, a component of the BCR signalling pathway upstream of BTK. Munshi et al. reported the inhibition of both BTK and SYK had a synergistic effect and caused an increased level of cell death than either treatment alone. This was due to BTK and SYK having different pathways for pro‐survival signalling. This combination of Ibrutinib and the SYK inhibitor could be a promising target for future therapies of mutated WM [83].
Other, less common, mutations identified in WM include the C‐X‐C chemokine receptor type 4 (CXCR4), present in around 40% of patients. CXCR4 mutations have been reported to cause shorter and decreased response rates for WM patients under ibrutinib. This emphasizes the importance of a clear understanding of the genetic landscape when treating a disease, as it has been shown here mutations can effect on BTK and its inhibition in patients with WM [84].
Diffuse large B‐cell lymphoma
Diffuse large B‐cell lymphoma (DLBCL) is an aggressive B‐cell malignancy divided into distinct molecular subtypes gene expressional profiling. BCR signalling has been identified to be upregulated in DLBCL, and DLBCL tumours are dependent on this signalling, but this differs between subtypes. Due to this BCR dependency, BCR‐inhibitory therapies have received a large amount of interest, including BTK and SYK inhibitors. Studies have reported that DLBCL cell lines can be sensitized to venetoclax, through treatment with ibrutinib or the SYK inhibitor, fostamatinib, due to a shift in the binding of BIM and MCL1 [85].
BTK INHIBITORS
BTK inhibitors (BTKi) have revolutionized the treatment of haematological disorders such as CLL. With recent studies suggesting a therapeutic potential for BTK inhibitors in autoimmunity and infection, the interest in these inhibitors is only increasing. Multiple new BTKi have been developed offering more selective and efficient targeting of BTK, and there are many ongoing clinical trials. Here, we will discuss some of the key inhibitors available and in trials (Table 1).
TABLE 1.
Targeting Bruton's tyrosine kinase through the use of covalent and non‐covalent inhibitors
| Inhibitor | Binding mechanism | Present use | Stage | Reference |
|---|---|---|---|---|
| Ibrutinib |
Covalent Irreversible |
Lymphoproliferative disorders SARS‐CoV‐2 |
FDA Approved Phase III trials |
[100] |
| Acalabrutinib |
Covalent Irreversible |
Lymphoproliferative disorders |
FDA Approved Phase III trials |
[90] |
| Zanubrutinib |
Covalent Irreversible |
Lymphoproliferative disorders |
FDA Approved Phase III trials |
[93] |
| Evobrutinib |
Covalent Irreversible |
MS, SLE | Phase III trials | [63] |
| Fenebrutinib |
Non‐covalent Reversible |
CLL, DLBCL, MS | Phase III trials | [70] |
| Branebrutinib |
Covalent Irreversible |
RA, SLE, Sjögren's syndrome | Phase II trials | [68] |
| Tolebrutinib |
Covalent Irreversible |
MS | Phase III trials | [75] |
| Tirabrutinib |
Covalent Irreversible |
R/R lymphoproliferative disorders | Phase II trials | [94] |
| Remibrutinib |
Covalent Irreversible |
Urticaria | Phase II trials | [98] |
| Pirtobrutinib |
Non‐covalent Reversible |
Lymphoproliferative disorders C481‐mutant CLL |
Phase I trials Phase II trials |
[99] |
Abbreviations: CLL, chronic lymphocytic leukaemia; DLBCL, diffuse large B‐cell lymphoma; MS, multiple sclerosis; R/R, relapsed/refractory; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus.
Ibrutinib
Ibrutinib transformed the treatment of CLL as the pioneering BTKi, which is now FDA‐approved for the treatment of CLL, naïve and R/R, as well as MCL, WM, MZL and small lymphocytic lymphoma. Acting irreversibly, ibrutinib binds to the C481 of the kinase domain (Figure 2a), arresting its enzymatic activity [76, 86]. Advani et al. were the first to report that patients, particularly those with R/R disease, tolerated Ibrutinib very well. They demonstrated the clinical safety and high potential of this inhibitor as a key targeted therapy for CLL [87]. Compared with the efficacy and side effects associated with standard chemotherapeutic agents, these developments in novel targeted therapies have significantly improved patient outcomes.
Despite the high success rate of ibrutinib, there are several complications associated with the BTKi. Firstly, ibrutinib is associated with off‐target toxicities as it also binds and inhibits other members of the TEC family kinases. Secondly, ibrutinib treatment has been associated with adverse cardiac side effects including atrial fibrillation (AF) and more commonly bleeding, upper respiratory infections and fatigue [88]. These complications can have serious ramifications in elderly patients with underlying health problems who do not tolerate the drug well. Finally, there is the problem of rising levels of resistance to ibrutinib, particularly in R/R patients. The most frequent cause of resistance seen is the cysteine‐to‐serine mutation at the C481 residue where Ibrutinib binds to BTK. PLCγ2 gain‐of‐function mutations are next in line for causing resistance, further downstream of BTK [89].
Second‐generation inhibitors – irreversible inhibitors
Alternative inhibitors are currently undergoing clinical trials to address and improve upon the downfalls of ibrutinib. These new inhibitors aim to offer higher selectivity with fewer off‐target toxicities. Following ibrutinib, the next BTKi to be FDA‐approved (CLL and MCL) was acalabrutinib. Acting in a similar method to ibrutinib by covalently binding to the C481 site of BTK’s kinase domain (Figure 2a), acalabrutinib exhibits a favourable safety profile and less off‐target effects on other TEC kinases. Overall, acalabrutinib has been reported to have a high response rate, especially for CLL patients who have previously received ibrutinib treatment but have become intolerant [90, 91].
More recently, inhibitors zanubrutinib and tirabrutinib have shown to be highly selective in irreversibly binding to BTK through targeting the C481 residue (Figure 2a), again showing fewer off‐target toxicities. In the first study in humans, Zanubrutinib was well tolerated, with patients experiencing a favourable safety profile over ibrutinib. Further trials are ongoing to evaluate its efficacy in WM [92, 93].
Tirabrutinib recently received approval in 2020 for use in Japan for R/R primary central nervous system lymphoma and is under investigation for WM and CLL. It acts through binding to BTK covalently and irreversibly to block BCR signalling in lymphoproliferative disorders and autoimmune diseases (PV and RA) [94].
Alternative inhibitors – reversible inhibitors
Ongoing clinical trials are currently proceeding for new agents, which inhibit BTK through novel mechanisms [95]. These alternative inhibitors differ from the previously mentioned therapies as they are reversible, bind to various regions of BTK other than the C481 and can interact with other kinases such as LYN and MEK1. These properties have the potential to overcome the resistance caused by mutations [96]. Teng et al. tested 20 novel BTKi containing a 1,3,5‐triazine core. Among them, the compound B8 exerted promising activity in vitro and exhibited a favourable safety profile with low off‐target toxicities [97].
For the treatment of Sjögren's syndrome, a BTKi with an even higher specificity is required for improved clinical outcomes. Remibrutinib (LOU064), which has been developed for this purpose, acts through a new mode: through interacting with Tyr511 of BTK and binding to the cyclopropyl phenyl group, preventing its phosphorylation [98]. Despite these measures, resistance still occurs in ways that are yet understood.
Pirtobrutinib (LOXO‐305) is a next‐generation, reversible, highly selective BTK inhibitor, which seems to inhibit both WT and C481S mutant BTK with minimal off‐target inhibition. Pirtobrutinib potently inhibits BCR signalling and cell survival in treatment‐naïve and BTK C481‐mutant CLL cells in vitro. The trial BRUIN evaluates pirtobrutinib in patients with previously treated B‐cell malignancies is currently ongoing. Preliminary data suggest that pirtobrutinib was effective in patients with heavily pretreated CLL/SLL and NHLs, including ibrutinib‐ and venetoclax‐resistant cases [99].
CONCLUSIONS
BTK is a vital component of the immune system not only in the BCR signalling pathway but also in a diverse range of cells and pathways. The potential therapeutic applications of BTK inhibition span from B‐cell malignancies, for which these inhibitors were originally created, to autoimmune disorders and COVID‐19 infection. Covalent BTK inhibitors such as ibrutinib and acalabrutinib have already revolutionized the treatment of CLL and MCL. With the development of new non‐covalent inhibitors including pirtobrutinib improving both the specificity and toxicity profiles, these build upon the foundations set up by first‐ and second‐generation inhibitors while overcoming mechanisms of resistance. Further understanding of the underlying mechanisms behind BTK and its inhibition has the potential to remodel traditional treatment schemes, to enhance combinatorial therapies and to improve patient outcomes in a wide range of diseases.
CONFLICT OF INTEREST
The authors of this review article declare that they have no conflicts of interest to disclose.
AUTHOR CONTRIBUTIONS
C. McDonald contributed to the development of the review and authored the overall review. C. Xanthopoulos authored the sections on BTK and immunity: Beyond B cells and BTK in Infections. E. Kostareli directly and substantially contributed through the supervision in writing and the editing of the overall review.
ACKNOWLEDGEMENT
We would like to thank Dr. Christine Van Duyn from the VCU Institute for Engineering and Medicine for her support with designing Figure 2.
McDonald C, Xanthopoulos C, Kostareli E. The role of Bruton's tyrosine kinase in the immune system and disease. Immunology. 2021;164:722–736. 10.1111/imm.13416
Funding information
This work has been supported by the Academy of Medical Sciences, Springboard Programme and Leukaemia and Lymphoma NI Funding (to EK, SBF005\1113 & R2740CEM). CMD is funded by a PhD Fellowship from the Department of Economy (DfE, N. Ireland).
REFERENCES
- 1. Bruton OC. Agammaglobulinemia. Pediatrics. 1952;9(6):722–728. [PubMed] [Google Scholar]
- 2. Vetrie D, Vořechovský I, Sideras P, Holland J, Davies A, Flinter F, et al. The gene involved in X‐linked agammaglobulinaemia is a member of the src family of protein‐tyrosine kinases. Nature. 1993;361(6409):226–33. [DOI] [PubMed] [Google Scholar]
- 3. Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I, et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X‐linked agammaglobulinemia. Cell. 1993;72(2):279–90. [DOI] [PubMed] [Google Scholar]
- 4. Khan WN, Alt FW, Gerstein RM, Malynn BA, Larsson I, Rathbun G, et al. Defective B cell development and function in Bfk‐deficient mice. Immunity. 1995;3:283–99. [DOI] [PubMed] [Google Scholar]
- 5. Rawlings DJ, Saffran DC, Tsukada S, Largaespada DA, Grimaldi JC, Cohen L, et al. Mutation of unique region of Bruton’s tyrosine kinase in immunodeficient XID mice. Science. 1993;261(5119):358–61. [DOI] [PubMed] [Google Scholar]
- 6. Pal Singh S, Dammeijer F, Hendriks RW. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol Cancer. 2019;18(1):79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Duarte DP, Lamontanara AJ, La Sala G, Jeong S, Sohn YK, Panjkovich A, et al. Btk SH2‐kinase interface is critical for allosteric kinase activation and its targeting inhibits B‐cell neoplasms. Nat Commun. 2020;11(1):2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Du Z & Lovly CM Mechanisms of receptor tyrosine kinase activation in cancer. [cited 2020 Jun 11]. 10.1186/s12943-018-0782-4 [DOI] [PMC free article] [PubMed]
- 9. Corneth OBJ, Wolterink RGJK, Hendriks RW. BTK signaling in B cell differentiation and autoimmunity. Curr Top Microbiol Immunol. 2016;393:67–105. [DOI] [PubMed] [Google Scholar]
- 10. Burger JA, Wiestner A. Targeting B cell receptor signalling in cancer: preclinical and clinical advances. Nat Rev Cancer. 2018;18(3):148–67. [DOI] [PubMed] [Google Scholar]
- 11. Phelan JD, Young RM, Webster DE, Roulland S, Wright GW, Kasbekar M, et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature. 2018;560(7718):387–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Takeda K, Kaisho T, Akira S. Toll‐like receptors. Annu Rev Immunol. 2003;21(1):335–76. [DOI] [PubMed] [Google Scholar]
- 13. Rawlings DJ, Schwartz MA, Jackson SW, Meyer‐Bahlburg A. Integration of B cell responses through Toll‐like receptors and antigen receptors. Nat Rev Immunol. 2012;12(4):282–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dadashian EL, McAuley EM, Liu D, Shaffer AL, Young RM, Iyer JR, et al. TLR signaling is activated in lymph node–resident CLL cells and is only partially inhibited by ibrutinib. Cancer Res. 2019;79(2):360–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rip J, de Bruijn MJW, Appelman MK, Pal Singh S, Hendriks RW, Corneth OBJ. Toll‐like receptor signaling drives Btk‐mediated autoimmune disease. Front Immunol. 2019;10:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jiménez C, Chan GG, Xu L, Tsakmaklis N, Kofides A, Demos MG, et al. Genomic evolution of ibrutinib‐resistant clones in Waldenström macroglobulinaemia. Br J Haematol. 2020;189:1165–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. de Gorter DJJ, Beuling EA, Kersseboom R, Middendorp S, van Gils JM, Hendriks RW, et al. Bruton’s Tyrosine kinase and phospholipase Cγ2 mediate chemokine‐controlled B cell migration and homing. Immunity. 2007;26(1):93–104. [DOI] [PubMed] [Google Scholar]
- 18. Li N, Jiang P, Chen A, Luo X, Jing Y, Yang L, et al. CX3CR1 positively regulates BCR signaling coupled with cell metabolism via negatively controlling actin remodeling. Cell Mol Life Sci. 2020;77:4379–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hirano M, Kikuchi Y, Nisitani S, Yamaguchi A, Satoh A, Ito T, et al. Bruton’s tyrosine kinase (Btk) enhances transcriptional co‐activation activity of BAM11, a Btk‐associated molecule of a subunit of SWI/SNF complexes. Int Immunol. 2004;16(5):747–57. [DOI] [PubMed] [Google Scholar]
- 20. Novina CD, Kumar S, Bajpai U, Cheriyath V, Zhang K, Pillai S, et al. Regulation of nuclear localization and transcriptional activity of TFII‐I by Bruton’s Tyrosine Kinase. Mol Cell Biol. 1999;19(7):5014–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gustafsson MO, Hussain A, Mohammad DK, Mohamed AJ, Nguyen V, Metalnikov P, et al. Regulation of nucleocytoplasmic shuttling of Bruton’s Tyrosine Kinase (Btk) through a Novel SH3‐dependent interaction with ankyrin repeat domain 54 (ANKRD54). Mol Cell Biol. 2012;32(13):2440–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gustafsson MO, Mohammad DK, Ylösmäki E, Choi H, Shrestha S, Wang Q, et al. ANKRD54 preferentially selects Bruton’s Tyrosine Kinase (BTK)from a Human Src‐Homology 3 (SH3) Domain library. PLoS One. 2017;12(4):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Page TH, Urbaniak AM, Espirito Santo AI, Danks L, Smallie T, Williams LM, et al. Bruton’s tyrosine kinase regulates TLR7/8‐induced TNF transcription via nuclear factor‐κB recruitment. Biochem Biophys Res Commun. 2018;499(2):260–6. 10.1016/j.bbrc.2018.03.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pontoriero M, Fiume G, Vecchio E, de Laurentiis A, Albano F, Iaccino E, et al. Activation of NF‐κB in B cell receptor signaling through Bruton’s tyrosine kinase‐dependent phosphorylation of IκB‐α. J Mol Med. 2019;97(5):675–90. [DOI] [PubMed] [Google Scholar]
- 25. Ye B, Zhou C, Guo H, Zheng M. Effects of BTK signalling in pathogenic microorganism infections. J Cell Mol Med. 2019;23(10):6522–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Long M, Beckwith K, Do P, Mundy BL, Gordon A, Lehman AM, et al. Ibrutinib treatment improves T cell number and function in CLL patients. J Clin Invest. 2017;127(8):3052–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yin Q, Sivina M, Robins H, Yusko E, Vignali M, O’Brien S, et al. Ibrutinib therapy increases T cell repertoire diversity in patients with chronic lymphocytic leukemia. J Immunol. 2017;198(4):1740–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mhibik M, Wiestner A, Sun C. Harnessing the effects of BTKi on T cells for effective immunotherapy against CLL. Int J Mol Sci. 2019;21(1):68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hanna BS, Yazdanparast H, Demerdash Y, Roessner PM, Schulz R, Lichter P, et al. Combining ibrutinib and checkpoint blockade improves CD8+ T‐cell function and control of chronic lymphocytic leukemia in Em‐TCL1 mice. Haematologica. 2020;106:968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat Rev Immunol. 2014;14(8):571–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ren L, Campbell A, Fang H, Gautam S, Elavazhagan S, Fatehchand K, et al. Analysis of the effects of the Bruton’s tyrosine kinase (Btk) inhibitor ibrutinib on monocyte Fcγ receptor (FcγR) function. J Biol Chem. 2016;291(6):3043–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fiorcari S, Maffei R, Audrito V, Martinelli S, Hacken E, Zucchini P, et al. Ibrutinib modifies the function of monocyte/macrophage population in chronic lymphocytic leukemia. Oncotarget. 2016;7(40):65968–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mirsafian H, Ripen AM, Leong WM, Chear CT, Bin Mohamad S, Merican AF. Transcriptome profiling of monocytes from XLA patients revealed the innate immune function dysregulation due to the BTK gene expression deficiency. Sci Rep. 2017;7(1):6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cavaliere FM, Prezzo A, Bilotta C, Iacobini M, Quinti I. The lack of BTK does not impair monocytes and polymorphonuclear cells functions in X‐linked agammaglobulinemia under treatment with intravenous immunoglobulin replacement. . PLOS ONE. 2017;12(4):e0175961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Volmering S, Block H, Boras M, Lowell CA, Zarbock A. The Neutrophil Btk signalosome regulates integrin activation during sterile inflammation. Immunity. 2016;44(1):73–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Prezzo A, Cavaliere FM, Bilotta C, Pentimalli TM, Iacobini M, Cesini L, et al. Ibrutinib‐based therapy impaired neutrophils microbicidal activity in patients with chronic lymphocytic leukemia during the early phases of treatment. Leuk Res. 2019;87:106233. [DOI] [PubMed] [Google Scholar]
- 37. Risnik D, Elías EE, Keitelman I, Colado A, Podaza E, Cordini G, et al. The effect of ibrutinib on neutrophil and γδ T cell functions. Leuk Lymphoma. 2020;61(10):2409–18. [DOI] [PubMed] [Google Scholar]
- 38. Zorn CN, Simonowski A, Huber M. Stimulus strength determines the BTK‐dependence of the SHIP1‐deficient phenotype in IgE/antigen‐triggered mast cells. Sci Rep. 2018;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Weber ANR. Targeting the NLRP3 Inflammasome via BTK. Front Cell Dev Biol. 2021;9:630479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu X, Pichulik T, Wolz OO, Dang TM, Stutz A, Dillen C, et al. Human NACHT, LRR, and PYD domain–containing protein 3 (NLRP3) inflammasome activity is regulated by and potentially targetable through Bruton tyrosine kinase. J Allergy Clin Immunol. 2017;140(4):1054–67.e10. [DOI] [PubMed] [Google Scholar]
- 41. Bittner Z, Liu X, Shankar S, Tapia‐Abellán A, Kalbacher H, Andreeva L, et al. BTK operates a phospho‐tyrosine switch to regulate NLRP3 inflammasome activity. bioRxiv. 2019;864702. 10.1101/864702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Herbst S, Shah A, Mazon Moya M, Marzola V, Jensen B, Reed A, et al. Phagocytosis‐dependent activation of a TLR9–BTK–calcineurin–NFAT pathway co‐ordinates innate immunity to Aspergillus fumigatus . EMBO Mol Med. 2015;7(3):240–58. 10.15252/emmm.201404556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bercusson A, Colley T, Shah A, Warris A, Armstrong‐James D. Ibrutinib blocks Btk‐dependent NF‐ĸB and NFAT responses in human macrophages during Aspergillus fumigatus phagocytosis. Blood. 2018;132(18):1985–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lionakis MS, Dunleavy K, Roschewski M, Widemann BC, Butman JA, Schmitz R, et al. Inhibition of B cell receptor signaling by Ibrutinib in primary CNS lymphoma. Cancer Cell. 2017;31(6):833–43.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Blez D, Blaize M, Soussain C, Boissonnas A, Meghraoui‐Kheddar A, Menezes N, et al. Ibrutinib induces multiple functional defects in the neutrophil response against Aspergillus fumigatus . Haematologica. 2020;105(2):478–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ahn IE, Jerussi T, Farooqui M, Tian X, Wiestner A, Gea‐Banacloche J. Atypical Pneumocystis jirovecii pneumonia in previously untreated patients with CLL on single‐agent ibrutinib. Blood. 2016;128(15):1940–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Colado A, Genoula M, Cougoule C, Marín Franco JL, Almejún MB, Risnik D, et al. Effect of the BTK inhibitor ibrutinib on macrophage‐ and γδ T cell‐mediated response against Mycobacterium tuberculosis . Blood Cancer J. 2018;8(11):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. de Porto AP, Liu Z, de Beer R, Florquin S, de Boer OJ, Hendriks RW, et al. Btk inhibitor ibrutinib reduces inflammatory myeloid cell responses in the lung during murine pneumococcal pneumonia. Mol Med. 2019;25(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. De Porto APNA, Claushuis TAM, Van Der Donk LEH, De Beer R, De Boer OJ, Florquin S, et al. Platelet Btk is required for maintaining lung vascular integrity during murine pneumococcal pneumosepsis. Thromb Haemost. 2019;119(6):930–40. [DOI] [PubMed] [Google Scholar]
- 50. Nguyen GT, Shaban L, Mack M, Swanson KD, Bunnell SC, Sykes DB, et al. Skap2 is required for defense against k. Pneumoniae infection and neutrophil respiratory burst. eLife. 2020;9:56656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Varikuti S, Volpedo G, Saljoughian N, Hamza OM, Halsey G, Ryan NM, et al. The potent ITK/BTK inhibitor Ibrutinib is effective for the treatment of experimental visceral Leishmaniasis caused by Leishmania donovani . J Infect Dis. 2019;219(4):599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Page TH, Urbaniak AM, Espirito Santo AI, Danks L, Smallie T, Williams LM, et al. Bruton’s tyrosine kinase regulates TLR7/8‐induced TNF transcription via nuclear factor‐κB recruitment. Biochem Biophys Res Commun. 2018;499(2):260–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Florence JM, Krupa A, Booshehri LM, Davis SA, Matthay MA, Kurdowska AK. Inhibiting bruton’s tyrosine kinase rescues mice from lethal influenza‐induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2018;315(1):L52–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Roschewski M, Lionakis MS, Sharman JP, Roswarski J, Goy A, Monticelli MA, et al. Inhibition of Bruton tyrosine kinase in patients with severe COVID‐19. Sci Immunol. 2020;5(48):eabd0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chong EA, Roeker LE, Shadman M, Davids MS, Schuster SJ, Mato AR. BTK inhibitors in cancer patients with COVID‐19: “The Winner Will be the One Who Controls That Chaos” (Napoleon Bonaparte). Clin Cancer Res. 2020;26(14):3514–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Treon SP, Castillo JJ, Skarbnik AP, Soumerai JD, Ghobrial IM, Guerrera ML, et al. The BTK inhibitor ibrutinib may protect against pulmonary injury in COVID‐19 infected patients. Blood. 2020;135:1912–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Koffman B, Mato A, Byrd JC, Danilov A, Hedrick B, Ujjani C, et al. Management of CLL patients early in the COVID‐19 pandemic: An international survey of CLL experts. Am J Hematol. 2020;95(8):E199–203. 10.1002/ajh.25851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Treon SP, Castillo J, Skarbnik AP, Soumerai JD, Ghobrial IM, Guerrera ML, et al. The BTK‐inhibitor ibrutinib may protect against pulmonary injury in COVID‐19 infected patients. Blood. 2020;135(21):1912–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Thibaud S, Tremblay D, Bhalla S, Zimmerman B, Sigel K, Gabrilove J. Protective role of Bruton tyrosine kinase inhibitors in patients with chronic lymphocytic leukaemia and COVID‐19. Br J Haematol. 2020;190(2):e73–6. 10.1111/bjh.16863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Crofford LJ, Nyhoff LE, Sheehan JH, Kendall PL. The role of Bruton’s tyrosine kinase in autoimmunity and implications for therapy. Expert Rev Clin Immunol. 2016;12(7):763–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dörner T, Furie R. Novel paradigms in systemic lupus erythematosus. Lancet. 2019;393(10188):2344–58. [DOI] [PubMed] [Google Scholar]
- 62. Satterthwaite AB. Bruton’s tyrosine kinase, a component of B cell signaling pathways, has multiple roles in the pathogenesis of lupus. Frontiers in Immunology. 2018;8:1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Caldwell RD, Qiu H, Askew BC, Bender AT, Brugger N, Camps M, et al. Discovery of Evobrutinib: an oral, potent, and highly selective, covalent Bruton’s Tyrosine Kinase (BTK) inhibitor for the treatment of immunological diseases. J Med Chem. 2019;62:7643–55. [DOI] [PubMed] [Google Scholar]
- 64. Haselmayer P, Camps M, Liu‐Bujalski L, Nguyen N, Morandi F, Head J, et al. Efficacy and pharmacodynamic modeling of the BTK inhibitor Evobrutinib in autoimmune disease models. J Immunol. 2019;202(10):2888–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cheung TT, McInnes IB. Future therapeutic targets in rheumatoid arthritis? Semin Immunopathol. 2017;39(4):487–500. 10.1007/s00281-017-0623-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jansson L, Holmdahl R. Genes on the X chromosome affect development of collagen‐induced arthritis in mice. Clin Exp Immunol. 1993;94(3):459–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang C, Pei H, He J, Zhu J, Li W, Niu T, et al. Design, synthesis and evaluation of novel 7H‐pyrrolo[2,3‐d]pyrimidin‐4‐amine derivatives as potent, selective and reversible Bruton’s tyrosine kinase (BTK) inhibitors for the treatment of rheumatoid arthritis. Eur J Med Chem. 2019;169:121–43. [DOI] [PubMed] [Google Scholar]
- 68. Watterson SH, Liu Q, Beaudoin Bertrand M, Batt DG, Li L, Pattoli MA, et al. Discovery of Branebrutinib (BMS‐986195): a strategy for identifying a highly potent and selective covalent inhibitor providing rapid in vivo inactivation of Bruton’s Tyrosine Kinase (BTK). J Med Chem. 2019;62(7):3228–50. [DOI] [PubMed] [Google Scholar]
- 69. Catlett IM, Nowak M, Kundu S, Zheng N, Liu A, He B, et al. Safety, pharmacokinetics and pharmacodynamics of branebrutinib (BMS‐986195), a covalent, irreversible inhibitor of Bruton’s tyrosine kinase: Randomised phase I, placebo‐controlled trial in healthy participants. Br J Clin Pharmacol. 2020;86(9):1849–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chan P, Yu J, Chinn L, Prohn M, Huisman J, Matzuka B, et al. Population pharmacokinetics, efficacy exposure‐response analysis. Pharm Res. 2020;37(2):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kridin K. Pemphigus group: overview, epidemiology, mortality, and comorbidities. Immunol Res. 2018;66:255–70. [DOI] [PubMed] [Google Scholar]
- 72. Corneth OBJ, de Bruijn MJW, Rip J, Asmawidjaja PS, Kil LP, Hendriks RW. Enhanced expression of Bruton’s tyrosine kinase in B cells drives systemic autoimmunity by disrupting T cell homeostasis. J Immunol. 2016;197(1):58–67. [DOI] [PubMed] [Google Scholar]
- 73. Musette P, Bouaziz JDB. Cell modulation strategies in autoimmune diseases: new concepts. Front Immunol. 2018;9:622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Didona D, Maglie R, Eming R, Hertl M. Pemphigus: current and future therapeutic strategies. Front Immunol. 2019;10:1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dolgin E. BTK blockers make headway in multiple sclerosis. Nat Biotechnol. 2021;39(1):3–5. [DOI] [PubMed] [Google Scholar]
- 76. Herman SEM, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI‐32765. Blood. 2011;117(23):6287–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bond DA, Woyach JA. Targeting BTK in CLL: beyond Ibrutinib. Curr Hematol Malign Rep. 2019;14(3):197–205. [DOI] [PubMed] [Google Scholar]
- 78. Maddocks K. Update on mantle cell lymphoma. Blood. 2018;132(16):1647–56. [DOI] [PubMed] [Google Scholar]
- 79. Rule S, Dreyling M, Goy A, Hess G, Auer R, Kahl B, et al. Ibrutinib for the treatment of relapsed/refractory mantle cell lymphoma: Extended 3.5‐year follow up from a pooled analysis. Haematologica. 2019;104(5):e211–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Matsumura‐Kimoto Y, Tsukamoto T, Shimura Y, Chinen Y, Tanba K, Kuwahara‐Ota S, et al. Serine‐227 in the N‐terminal kinase domain of RSK2 is a potential therapeutic target for mantle cell lymphoma. Cancer Med. 2020;9(14):5185–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Dimopoulos MA, Kastritis E. How I treat Waldenström macroglobulinemia. Blood. 2019;134(23):2022–35. [DOI] [PubMed] [Google Scholar]
- 82. Yu X, Li W, Deng Q, Li L, Hsi ED, Young KH, et al. MYD88 L265P mutation in lymphoid malignancies. Cancer Res. 2018;78:2457–62. [DOI] [PubMed] [Google Scholar]
- 83. Munshi M, Liu X, Chen JG, Xu L, Tsakmaklis N, Demos MG, et al. SYK is activated by mutated MYD88 and drives pro‐survival signaling in MYD88 driven B‐cell lymphomas. Blood Cancer J. 2020;10(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Castillo JJ, Xu L, Gustine JN, Keezer A, Meid K, Dubeau TE, et al. CXCR4 mutation subtypes impact response and survival outcomes in patients with Waldenström macroglobulinaemia treated with ibrutinib. Br J Haematol. 2019;187(3):356–63. [DOI] [PubMed] [Google Scholar]
- 85. Sasi BK, Martines C, Xerxa E, Porro F, Kalkan H, Fazio R, et al. Inhibition of SYK or BTK augments venetoclax sensitivity in SHP1‐negative/BCL‐2‐positive diffuse large B‐cell lymphoma. Leukemia. 2019;33(10):2416–28. [DOI] [PubMed] [Google Scholar]
- 86. Pan Z, Scheerens H, Li S‐J, Schultz BE, Sprengeler PA, Burrill LC, et al. Discovery of selective irreversible inhibitors for Bruton’s Tyrosine Kinase. ChemMedChem. 2007;2(1):58–61. 10.1002/cmdc.200600221 [DOI] [PubMed] [Google Scholar]
- 87. Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI‐32765) has significant activity in patients with relapsed/refractory B‐cell malignancies. J Clin Oncol. 2013;31(1):88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Tang CPS, McMullen J, Tam C. Cardiac side effects of bruton tyrosine kinase (BTK) inhibitors. Leukemia Lymphoma. 2018;59(7):1554–64. [DOI] [PubMed] [Google Scholar]
- 89. Burger JA, Landau DA, Taylor‐Weiner A, Bozic I, Zhang H, Sarosiek K, et al. Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat Commun. 2016;7:11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Byrd JC, Harrington B, O’Brien S, Jones JA, Schuh A, Devereux S, et al. Acalabrutinib (ACP‐196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Awan FT, Schuh A, Brown JR, Furman RR, Pagel JM, Hillmen P, et al. Acalabrutinib monotherapy in patients with chronic lymphocytic leukemia who are intolerant to ibrutinib. Blood Adv. 2019;3(9):1553–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Tam CS, Trotman J, Opat S, Burger JA, Cull G, Gottlieb D, et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B‐cell malignancies and safety and efficacy evaluation in CLL. Blood. 2019;134(11):851–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Syed YY. Zanubrutinib: first approval. Drugs. 2020;80(1):91–7. [DOI] [PubMed] [Google Scholar]
- 94. Dhillon S. Tirabrutinib: first approval. Drugs. 2020;80(8):835–40. [DOI] [PubMed] [Google Scholar]
- 95. Gu D, Tang H, Wu J, Li J, Miao Y. Targeting Bruton tyrosine kinase using non‐covalent inhibitors in B cell malignancies. J Hematol Oncol. 2021;14(1):40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Iovino L, Shadman M. Novel therapies in chronic lymphocytic leukemia: a rapidly changing landscape. Curr Treat Options Oncol. 2020;21(4):1–16. [DOI] [PubMed] [Google Scholar]
- 97. Teng Y, Lu X, Xiao M, Li Z, Zou Y, Ren S, et al. Discovery of potent and highly selective covalent inhibitors of Bruton’s tyrosine kinase bearing triazine scaffold. Eur J Med Chem. 2020;199:112339. [DOI] [PubMed] [Google Scholar]
- 98. Gabizon R, London N. A fast and clean BTK inhibitor. J Med Chem. 2020;63:5100–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Mato AR, Shah NN, Jurczak W, Cheah CY, Pagel JM, Woyach JA, et al. Pirtobrutinib in relapsed or refractory B‐cell malignancies (BRUIN): a phase 1/2 study. Lancet. 2021;397(10277):892–901. [DOI] [PubMed] [Google Scholar]
- 100. Molica S, Matutes E, Tam C, Polliack A. Ibrutinib in the treatment of chronic lymphocytic leukemia: 5 years on. Hematol Oncol. 2019;38:129–36. [DOI] [PubMed] [Google Scholar]
- 101. Bienert S, Waterhouse A, de Beer TAP, Tauriello G, Studer G, Bordoli L, et al. The SWISS‐MODEL Repository—new features and functionality. Nucleic Acids Res. 2017;45(D1):D313–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2021;49(D1):D1388–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Biorender [Internet]. [cited 2021 Aug 30]. https://app.biorender.com/