

Abstract
Neutrophils are critical components of the body's immune response to infection, being loaded with a potent arsenal of toxic mediators and displaying immense destructive capacity. Given the potential of neutrophils to impart extensive tissue damage, it is perhaps not surprising that when augmented these cells are also implicated in the pathology of inflammatory diseases. Prominent neutrophilic inflammation is a hallmark feature of patients with chronic lung diseases such as chronic obstructive pulmonary disease, severe asthma, bronchiectasis and cystic fibrosis, with their numbers frequently associating with worse prognosis. Accordingly, it is anticipated that neutrophils are central to the pathology of these diseases and represent an attractive therapeutic target. However, in many instances, evidence directly linking neutrophils to the pathology of disease has remained somewhat circumstantial and strategies that have looked to reduce neutrophilic inflammation in the clinic have proved largely disappointing. We have classically viewed neutrophils as somewhat crude, terminally differentiated, insular and homogeneous protagonists of pathology. However, it is now clear that this does not do the neutrophil justice, and we now recognize that these cells exhibit heterogeneity, a pronounced awareness of the localized environment and a remarkable capacity to interact with and modulate the behaviour of a multitude of cells, even exhibiting anti‐inflammatory, pro‐resolving and pro‐repair functions. In this review, we discuss evidence for the role of neutrophils in chronic lung disease and how our evolving view of these cells may impact upon our perceived assessment of their contribution to disease pathology and efforts to target them therapeutically.
Keywords: asthma, COPD, lung, neutrophil
Prominent neutrophilic inflammation is a hallmark feature of patients with chronic lung diseases such as chronic obstructive pulmonary disease, severe asthma, bronchiectasis and cystic fibrosis, with their numbers frequently associating with worse prognosis. We have classically viewed neutrophils as somewhat crude, terminally differentiated, insular and homogeneous protagonists of pathology, and thus, they have been viewed as attractive therapeutic targets in chronic lung diseases. However, given our evolving view of neutrophils, it is critical that we re‐evaluate their perceived contribution to disease pathology and reconsider strategies to target them therapeutically.
OUR EVOLVING VIEW OF NEUTROPHILS
Neutrophils have classically been considered to be a short‐lived, terminally differentiated, phenotypically homogeneous population of cells whose main task is pathogen killing. Accordingly, they are equipped with a vast arsenal of toxic mediators to facilitate rapid clearance of invading microorganisms. However, owing to the capacity of their payload to also impart significant bystander host tissue damage, neutrophilic inflammation needs to be tightly regulated. Overzealous or persistent neutrophilic inflammation, as often seen in chronic lung diseases, has traditionally been assumed to be largely detrimental, and neutrophils have been regarded as an attractive therapeutic target. However, it is increasingly apparent that neutrophils are far more sophisticated than previously anticipated, and thus, our perception of these cells in the context of disease pathology needs to be re‐evaluated.
We now recognize that neutrophils are highly responsive to environmental cues, exhibiting plasticity and a capacity to modulate their transcriptional profile to instigate a substantial de novo biosynthetic programme receptive to their specific needs [1]. This ability to integrate environmental signals and translate them into distinct effector programmes is accentuated by recognition that neutrophils can exhibit a far greater life span than originally anticipated [2]. The realization of neutrophil plasticity has subsequently led to the concept of neutrophil heterogeneity, whereby we now appreciate that not all neutrophils display comparable phenotype or function. Such heterogeneity is likely not solely driven by variable transcriptional profiles in response to microenvironmental cues, but also by the stage of neutrophil maturity, as the phenotype and function of circulating neutrophils vary considerably over the course of their life span [3]. Studies in mice and humans have demonstrated that senescent neutrophils are characterized by elevated CXCR4 expression, reduced size, hypersegmented nuclei and proclivity to undergo activation and neutrophil extracellular trap (NET) formation (NETosis) [4]. Accordingly, inflammatory, pro‐survival signals in the context of chronic inflammatory diseases may prolong neutrophil life span, whereby ‘aged’ neutrophils may subsequently contribute to disease pathology. Supportive of this concept, the frequency of CXCR4hi neutrophils is increased in patients with asthma, chronic obstructive pulmonary disease (COPD) and cystic fibrosis (CF) [5]. Conversely, emergency granulopoiesis in response to sustained inflammation can result in the release of immature neutrophils into the circulation. These are characterized by reduced expression of CD16, CD10 and CXCR2, band‐shaped nuclei and reduced granule content [3]. Immature neutrophil populations have been described both in asthma [6] and in COPD [7], but their functional significance in the context of chronic lung diseases is still unclear. Reflective of a growing recognition of neutrophil heterogeneity, discrete neutrophil subsets with divergent phenotypes and functionality have now been reported under both physiological and pathological conditions (Table 1) [3, 8]. To what extent these subsets represent bona fide distinct populations as opposed to a function of variable plasticity and maturity is yet to be clearly defined, but such studies highlight the necessity to no longer consider neutrophils as a sole, uniform population.
TABLE 1.
Neutrophil subsets and their relevance in homeostatic and pathological settings
| Neutrophil subset | Frequently co‐expressed markers and/or morphological features | Known functions of the markers defining the subset | Prevalence and role in health and disease |
|---|---|---|---|
| CD177+ | Proteinase 3 (PR3). |
|
Humans: 45%–65% of circulating neutrophils during homeostasis.
|
| OLFM4+ | _ |
|
Humans: 20%–25% of circulating neutrophils during homeostasis.
|
| CXCR4hi CD62Llow | CD11bhi, CD49dhi; hypersegmented nucleus and reduced size (in senescent neutrophils). |
|
Mouse: 55 ± 14% of circulating neutrophils in ZT5 (i.e. 5 h post‐initiation of the light period). Humans: ~60% of circulating neutrophils at night‐time.
|
| CD63+ (otherwise known as A2 or ‘GRIM’ neutrophils in CF airways) |
CD11bhi, CD66hi, MHC II+, CD16low, CD14low ↑ Arginase 1 [ARG1] (in CF patients) |
|
|
| CD64+ | PD‐L1 (in COVID‐19 patients) |
|
|
| CD49d+ |
Cysteinyl leukotriene receptor 1 [CysLTR1] (in patients with viral respiratory tract infections, including IAV, IBV, RSV HRV, HMPV and HPIV). Can be co‐expressed with CXCR4 (possible link to intrinsic ageing?). |
|
Mouse:
Humans:
|
|
CD49dhiCXCR4hi VEGFR1hi |
↑MMP‐9 |
|
|
| ICAM‐1hiCXCR1low | ↑ROS production |
|
|
| Low‐density neutrophils (LDNs), multiple subsets |
CD15hi, CD14low, CD10variable, CD16variable; CD11bhi, CD66bhi (in SLE, HIV, asthma and cancer); CD33hi (in cancer). |
|
|
|
PMN‐I and PNM‐II (in the context of infection by methicillin‐resistant Staphylococcus aureus [MRSA]) |
PMN‐I: CD49d+, CD11b−, TRL5+, TRL8+, secrete IL‐12 and CCL3, drive classical activation of macrophages. PMN‐II: CD49d−, CD11b+, TRL7+, TRL9+, secrete IL‐10 and CCL2, drive alternative activation of macrophages. |
_ |
Mouse:
|
Abbreviations: HBV, hepatitis B virus; HCV, hepatitis C virus; HMPV, human metapneumovirus; HPIV, human parainfluenza virus; HRV, human rhinovirus; IAV, influenza A virus; IBD, irritable bowel disease; IBV, infectious bronchitis virus; ICAM‐1, intercellular adhesion molecule‐1; OLFM4, olfactomedin 4; PMN, polymorphonuclear cells; RSV, respiratory syncytial virus; VCAM‐1, vascular cell adhesion molecule‐1; VEGF, vascular endothelial growth factor; VEGFR1, vascular endothelial growth factor receptor 1; VLA‐4, very late antigen‐4.
Another concept that has been debunked is that neutrophils are insular cells, which act swiftly and then disappear without impacting on the immune response beyond the acute phase of inflammation. It is now clear that neutrophils exhibit pleiotropic roles in shaping the phenotype and scale of an elicited immune response and ensuing repair programmes through engaging in complex crosstalk with both immune and structural cells via cell–cell contact and release of soluble mediators [9] (Figure 1). The depth of such interactions is beyond the scope of this review, but we highlight specific examples to demonstrate the marked capacity of neutrophils to modulate diverse and disparate components of the host's response. As frequent first responders, neutrophils are a rich source of the chemokines CCL2, CCL3 and CCL20, which operate to potentiate ensuing monocyte influx [10]. Neutrophil‐derived granule proteins have subsequently been shown to augment macrophage phagocytosis [11] and cytokine secretion [12], whilst their cathelicidins have been shown to be potent inducers of NLRP3 inflammasome activation and IL‐1β cytokine production by macrophages [13]. Moreover, neutrophil‐derived IL‐18 has been demonstrated to promote NK cell activation and IFN‐γ production [14]. Neutrophils can also profoundly potentiate facets of the host's adaptive response. Neutrophil‐derived products have been demonstrated to facilitate recruitment of dendritic cells, whilst additionally promoting dendritic cell maturation and antigen presentation [9]. Neutrophils themselves can even upregulate MHC II and costimulatory molecules and operate as antigen‐presenting cells [15, 16], whilst also acting as a source of signals that drive the recruitment, activation and differentiation of Th1 and Th17 cells [17, 18, 19]. Neutrophils have additionally been shown to exhibit profound effects on stromal cells, with their products capable of modifying epithelial/endothelial barrier function and cytokine production [20, 21, 22], fibroblast proliferation, differentiation and extracellular matrix (ECM) generation [23, 24]. Thus, it is clear that overexuberant neutrophilia, as seen in chronic lung diseases, can potentiate pathological inflammation and tissue remodelling.
FIGURE 1.
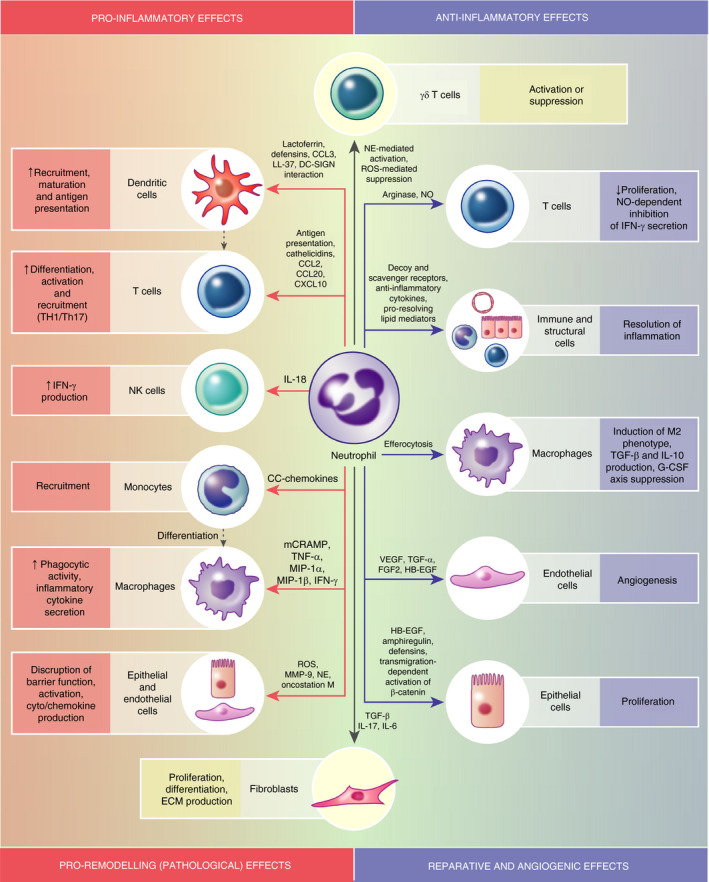
Neutrophil interactions with immune and structural cells. Neutrophils can engage in complex crosstalk with structural and immune cells, either by directly interacting with them via cell surface molecules or by secreting a variety of mediators. This figure provides examples whereby neutrophils modulate the phenotype and function of different cell types. Depending on variable microenvironmental cues, age and maturity, neutrophils can exhibit divergent roles in regulating both the initiation and the resolution of inflammation, as well as driving tissue repair or pathological tissue remodelling. DC‐SIGN, dendritic cell‐specific intercellular adhesion molecule‐3 grabbing non‐integrin; FGF2, fibroblast growth factor 2; HB‐EGF, heparin binding‐epidermal growth factor; mCRAMP, murine cathelicidin‐related antimicrobial peptide; MIP, macrophage inflammatory protein; NO, nitric oxide; VEGF, vascular endothelial growth factor
However, it is increasingly recognized that neutrophils are not solely pro‐inflammatory but also exhibit extensive anti‐inflammatory, pro‐resolving and repair functions. Neutrophils have been described as a source of anti‐inflammatory cytokines TGF‐β and IL‐10, whilst also operating to scavenge pro‐inflammatory cytokines/chemokines through the production of decoy receptors or receptor antagonists [25]. Whilst neutrophils have the capacity to promote adaptive immunity, they also have the potential to suppress lymphocyte responses, such as through their ability to deplete extracellular L‐arginine and reduce T‐cell proliferation [26]. Persistent neutrophilia is not the norm, and endogenous feedback loops function to suppress their own activation and limit the toxicity of their most potent effector mechanisms, as well as dictating the production of pro‐resolving lipid mediators that retard further neutrophil recruitment [27]. Moreover, neutrophils are inherently programmed to undergo apoptosis within a defined period and, upon their phagocytosis by tissue‐resident macrophages, can impart negative feedback regulatory mechanisms to suppress granulocyte colony‐stimulating factor (G‐CSF)‐mediated neutrophil granulopoiesis [28], whilst also reprogramming macrophages to a reparative, M2‐like phenotype characterized by production of IL‐10 [29]. A role for neutrophils in promoting tissue repair has become increasingly apparent, and they can operate as a potent source of growth factors and pro‐angiogenic mediators [25, 30]. Given our new appreciation of the multifaceted roles of neutrophils, it is important to revisit our view of these cells in the pathophysiology of chronic lung diseases. This will aid in the design of improved therapeutic strategies, which consider the nuances of their behaviour rather than assuming their contribution is solely destructive.
CHRONIC OBSTRUCTIVE PULMONARY DISEASE
COPD is a progressive inflammatory disease of the lungs and a leading cause of morbidity and mortality worldwide [31]. Cigarette smoking remains the most definitive risk factor for COPD onset in developed countries [31, 32], with prolonged exposure to noxious agents, primarily associated with biomass smoke, largely responsible for disease inception in non‐smokers from low‐ and middle‐income countries [33]. Once established, COPD is characterized by irreversible airflow limitation owing to small airway fibrosis (obstructive bronchiolitis) and alveolar destruction (emphysema) [34], which, in conjunction with acute bacterial‐ and viral‐induced exacerbations [35], culminate in patient dyspnoea, persistent cough, excessive sputum production, significantly impaired quality of life and death [36].
Neutrophils are recognized as the central cellular mediator in COPD pathogenesis. Irrespective of underlying clinical phenotype [37], the airways of patients with COPD are dominated by neutrophilic inflammation, directly correlating with severity of airway obstruction and accelerated decline in lung function based on forced expiratory volume in 1 second (FEV1) [38]. Inhalation of noxious agents and acute exacerbations trigger the NF‐κB and p38 MAPK‐dependent release of neutrophil chemotactic mediators by activated airway epithelial cells and macrophages [39], collectively promoting neutrophil mobilization, recruitment and activation within the airways. Ensuing degranulation results in the accumulation of serine proteases, myeloperoxidase (MPO) and matrix metalloproteinase (MMP)‐8 and MMP‐9 [40, 41, 42], which consequently drive pathological features of COPD by triggering proteolytic degradation of the airway ECM and driving emphysematous disease. Enzymatic breakdown of the ECM additionally liberates the neutrophil chemo‐attractant proline–glycine–proline (PGP) [43] within the airways of patients with COPD, acting in synergy with cigarette smoke to perpetuate a vicious cycle of CXCR2‐mediated neutrophilic chemotaxis and ensuing clinical disease (Figure 2) [44, 45]. It is also noteworthy that early‐life NF‐κB‐induced activation of neutrophils and NE accumulation within the lungs of pre‐clinical models contribute towards lifelong impairments in lung architecture and predisposition to COPD development during adulthood [46].
FIGURE 2.
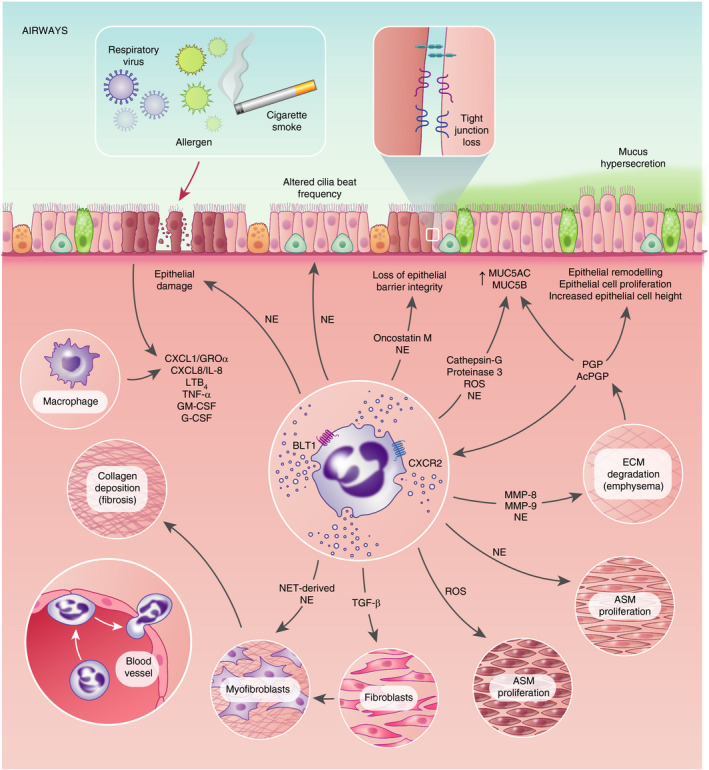
Putative mechanisms by which neutrophils and their products may directly drive pathological features of chronic lung disease. Neutrophils and their products are considered major drivers of pathology in multiple chronic lung diseases. Direct evidence demonstrating a role for neutrophils and their products in driving tissue destruction and hallmark features of pathological remodelling in patients is often lacking. Accordingly, putative pathways defined within this figure are based, in part, upon circumstantial data or extrapolated from in vitro studies or a specific disease state – but could conceivably have relevance to multiple chronic lung diseases. AcPGP, acetylated proline–glycine–proline; ASM, airway smooth muscle; BLT1, high‐affinity LTB4 receptor; MUC5AC, mucin 5AC; MUC5B, mucin 5B
In parallel with conventional pathways, the role of NETosis is an evolving mechanism in COPD pathogenesis. Elevated concentrations of NETs are observed in sputum from stable and acutely exacerbating COPD (AECOPD) patients, positively correlating with severity of airflow limitation and exacerbation frequency [47]. Moreover, NETosis induced by Haemophilus influenzae (a key pathogen involved in stable colonization and AECOPD [35]) amplifies pro‐inflammatory IL‐6 trans‐signalling within the airways, driving persistent sputum neutrophilia within a subset of patients with COPD [48]. Interestingly, this highlights a potential causal relationship between NET formation and Haemophilus dominance within the COPD lung microbiome [47].
Despite their enhanced activity, neutrophils from patients with COPD demonstrate multiple features of impaired functionality. Sapey and colleagues [49] first identified this compromised state whereby peripheral neutrophils from patients with COPD exhibit aberrant chemotactic responses towards an IL‐8 or GROα gradient, likely associated with enhanced phosphoinositide 3‐kinase (PI3K; an enzyme vital in regulating chemotactic directionality) signalling [50]. These defective migratory dynamics may explain the protease‐mediated collateral tissue damage caused upon neutrophil recruitment to the lungs of patients with COPD following upregulation of adhesion molecules [51]. Compounding this is the impaired phagocytic activity of neutrophils from patients with COPD [47], although conflicting results are reported based on bacterial species [52]. Nevertheless, direct inhibition of phagocytosis appears to be a consequence of elevated NET complexes within COPD sputum [47] and is exaggerated by the ability of cigarette smoke to suppress neutrophil phagocytosis via the disruption of caspase‐3‐mediated homeostatic regulation [53], likely contributing towards the persistent bacterial colonization and increased risk for exacerbation‐induced morbidity and mortality [54].
The centrality of neutrophils in COPD pathogenesis has prompted a concerted effort to therapeutically target these effector cells, albeit largely disappointingly. Inhaled corticosteroids (ICS) are widely prescribed during both stable disease and episodic exacerbations, yet ICS prolong neutrophil survival [55] and have limited capacity to reduce exacerbation frequency in patients with COPD [56]. NETosis is additionally resistant to corticosteroid treatment, with suggestions the common ICS fluticasone propionate may inadvertently increase sputum NETosis and further diminish phagocytic capacity [47]. Such caveats are compounded by the heightened risk of secondary bacterial infection and airway mucus hypersecretion in patients with COPD prescribed ICS during respiratory viral‐induced exacerbations [57], collectively highlighting ICS as a modestly effective treatment for all disease states.
Small molecule compounds targeting neutrophil signalling pathways are being investigated as a more precise means of therapeutic intervention (Table 2). CXCR2 antagonists routinely limit neutrophil infiltration in the lungs and periphery of patients with moderate‐to‐severe COPD [58, 59] although improvements in FEV1 are infrequent and protection is not always observed in a clinical setting following initial pre‐clinical promise [60]. Moreover, increased exacerbation risk, neutropenia and pneumonia are reported in an unacceptable proportion of patients following intervention [58, 59, 60], thus mitigating clinical benefit. Likewise, inhibitory compounds targeting neutrophil elastase (NE) [61], TNF‐α [62] and leukotriene B4 (LTB4) synthesis [63] repeatedly show limited capacity for clinically meaningful protection in patients with COPD, raising several questions regarding dose, intervention time‐points and the appropriate use of pre‐clinical models in determining novel therapeutic efficacy. Recent pre‐clinical studies targeting NETosis employ the use of mucolytics (deoxyribonuclease I) for degrading cigarette smoke‐induced NETs within the airways [64]. Murine COPD models of cigarette smoke‐ and non‐typeable H. influenza‐induced respiratory inflammation have additionally identified anti‐PD‐1 treatment as a strategy to limit airway neutrophilic influx and alveolar destruction [65]. Whilst these novel approaches support effective disease mitigation, the aforementioned studies suggest a high degree of scrutiny concerning immunosuppression should be applied if progression to clinical trials ensues. Restoring neutrophil chemotactic accuracy via the selective inhibition of hyperactive PI3K signalling is being explored as a low toxicity alternative to the mitigation of pro‐inflammatory activity, and shows initial promise without altering migratory speed [50]. PI3Kδ inhibition further limits MMP‐9 and reactive oxygen species (ROS) secretion from stable and AECOPD neutrophils [66], although the beneficial impact appears most prominent in stable patients [67, 68].
TABLE 2.
Clinical trials targeting neutrophilic inflammation in chronic lung disease
| Treatment | Target | Stratified on neutrophils | Patients | Duration of treatment | Outcomes | Ref. |
|---|---|---|---|---|---|---|
| COPD | ||||||
| MK‐7123 | CXCR2 | No |
GOLD stage II‐III FEV1 30%−70% predicted FEV1/FVC ≤70% Stable |
≥26 weeks (primary analysis). ≥52 weeks (optional safety extension). |
Reduced sputum neutrophils at 3 months. Reduced sputum MMP‐9. Reduced blood neutrophils. Reduced plasma MMP‐9, MPO and CXCL5. Neutropenia in 2·6%–18·4% of treatment groups. Improved FEV1 |
[58] |
| AZD5069 | CXCR2 | No |
GOLD stage II‐III FEV1 ≥30% and <80% predicted FEV1/FVC <70% Stable |
4 weeks |
Reduced peripheral blood neutrophils. Neutropenia in 4%−10% of treatment groups. No improvement in clinical end‐points. |
[59] |
| GKS1325756 (Danirixin) | CXCR2 | No |
FEV1 >40% predicted FEV1/FVC <70% |
24 weeks |
No change in blood neutrophils. No improvement in clinical end‐points. |
[60] |
| AZD9668 | NE | No | GOLD stage II‐III | 12 weeks |
No change in urine desmosine (biomarker for lung matrix degradation) concentration. No improvement in clinical end‐points. |
[61] |
| Infliximab | TNF‐α | No |
GOLD stage I‐IV Stable |
24 weeks |
No treatment benefits. Higher rate of pneumonia in the treatment group. No improvement in clinical end‐points. |
[62] |
| BAYx1005 | LTB4 | No |
FEV1 <65% predicted Stable |
2 weeks |
Reduced sputum LTB4 Clinical outcomes not assessed. |
[63] |
| GSK045 | PI3Kδ | No |
GOLD stage II–III FEV1/FVC <70% Stable and AECOPD |
In vitro stimulation |
Reduced ROS release from sputum neutrophils in stable patients. Reduced MMP‐9 and ROS release from blood neutrophils in stable and AECOPD patients. Clinical outcomes not assessed. |
[66] |
| BIRB796 | P38 MAPK | No |
GOLD stage II–III FEV1/FVC <70% Stable and AECOPD |
In vitro stimulation |
Reduced MMP‐9 release from blood neutrophils in stable patients. Clinical outcomes not assessed. |
[66] |
| GSK2269557 (Nemiralisib) | PI3Kδ | No |
GOLD stage II‐III FEV1 40%−80% predicted FEV1/FVC <70% Stable |
2 weeks |
No change in sputum neutrophils. Reduced sputum IL‐6 and IL−8. No improvement in clinical end‐points. |
[67] |
| GSK2269557 (Nemiralisib) | PI3Kδ | No |
GOLD stage II‐III FEV1 ≤80% predicted FEV1/FVC <70% Stable and AECOPD |
2 weeks |
Upregulation of genes associated with sputum neutrophil migration in stable patients only. Inhibition of NE and ROS from stimulated blood neutrophils from AECOPD patients. Improved FEV1 |
[68] |
| Asthma | ||||||
| SCH527123 | CXCR2 | Yes |
Severe asthma >40% sputum neutrophils |
4 weeks |
Reduced sputum and blood neutrophils. Fewer mild exacerbations. No improvement in ACQ or FEV1. No change in sputum MPO, IL‐8 or elastase. |
[111] |
| AZD5069 | CXCR2 | Yes |
Uncontrolled, persistent asthma Blood neutrophil counts greater than 2·7 × 10⁹/L |
6 months |
Reduced peripheral blood neutrophils. No improvement in clinical end‐points. |
[112] |
| LY293111 | LTB4 | No | Atopic asthma | 7 days (allergen challenge model) |
Reduced BAL neutrophils. No improvement in clinical end‐points. |
[113] |
| MK‐0633 | LTB4/Cysteinyl LTs | No |
Chronic asthma FEV1 >45 and <85% predicted |
6 weeks |
Modest improvement in baseline FEV1. Changes in neutrophil numbers not reported. Elevated aspartate aminotransferase and alanine aminotransferase. Benefit–risk ratio did not support clinical use. |
[114] |
| GSK2190915 | LTB4/Cysteinyl LTs | Yes |
Asthma Raised sputum neutrophils at 2 visits (>50% and >45%) |
14 days |
Reduced sputum LTB4. No effect on sputum neutrophils. No improvement in clinical end‐points. |
[115] |
| JNJ‐40929837 | LTB4 | No | Mild, atopic asthmatics | 7 days (bronchial allergen challenge model) |
Reduced sputum LTB4. Changes in neutrophil numbers not reported. No improvement in clinical end‐points. |
[116] |
| AMG 827 (Brodalumab) | IL‐17RA | No | Inadequately controlled moderate‐to‐severe asthma | 12 weeks |
Changes in neutrophil numbers not reported. No improvement in clinical end‐points. |
[117] |
| Etanercept | TNF‐α | No | Moderate‐to‐severe persistent asthma | 12 weeks |
Changes in neutrophil numbers not reported. No improvement in clinical end‐points. |
[118] |
| Golimumab | TNF‐α | No | Uncontrolled, severe persistent asthma | 24 weeks |
Changes in neutrophil numbers not reported. No improvement in clinical end‐points. Unfavourable risk–benefit profile led to early discontinuation. |
[119] |
| Bronchiectasis | ||||||
| Brensocatib | DPP1 | No |
Bronchiectasis Frequent exacerbations |
24 weeks |
Increased time to first exacerbation. Reduced sputum NE. |
[128] |
| AZD9668 | NE | No | Bronchiectasis | 4 weeks |
No change in sputum neutrophils. Improved FEV1 by 100 mL. |
[129] |
| BAY 85‐8501 | NE | No | Bronchiectasis | 4 weeks |
Reduced NE activity. No improvement in clinical end‐points. |
[130] |
| Cystic fibrosis | ||||||
| AZD9668 | NE | No | Cystic fibrosis | 4 weeks |
No effect on sputum neutrophils or NE. Reduced sputum cytokines. No improvement in clinical end‐points. |
[139] |
| Alpha1‐antitrypsin | Protease/antiprotease balance | No | Cystic Fibrosis | 4 weeks |
Decreased levels of elastase activity. Decreased neutrophils and pro‐inflammatory cytokines. |
[140] |
| BIIL 284 BS | LTB4 | No | Cystic Fibrosis | 24 weeks |
Increased pulmonary exacerbations. No clinical benefit. |
[141] |
Abbreviation: FVC, forced vital capacity.
We now understand that neutrophils are a heterogeneous population with a repertoire of variable immune modulatory and tissue remodelling functions, and this therefore needs to be considered when evaluating neutrophils and their role in COPD pathogenesis. The development of new biologics targeting specific neutrophil subsets and their respective functions/products, and subsequently stratifying patient groups on neutrophil composition, may thereby enable a personalized medicine approach to disease mitigation, whilst circumventing many of the off‐target effects of current strategies.
ASTHMA
Asthma is a chronic respiratory disease characterized by airway inflammation and pathological airway remodelling (subepithelial fibrosis, increased smooth muscle mass, goblet cell and mucous gland hyperplasia, neovascularization and epithelial alterations), resulting in variable symptoms of wheeze, shortness of breath, chest tightness and cough. Subacute flare‐ups of symptoms termed exacerbations represent a major source of disease morbidity and a significant economic burden. Asthma is an extremely common condition affecting 358 million people worldwide and resulting in 495 000 annual deaths [69]. It has classically been considered an allergic, Th2‐driven pathology characterized by airway eosinophilia [70]. However, it is now recognized that asthma is a heterogeneous disease with as many as 50% of asthmatic patients presenting with non‐Th2 inflammation [71]. Accordingly, variable symptomatic burden, airway inflammation and remodelling have led to the realization of distinct asthma endotypes whereby specific pathological mechanisms are clustered.
Increasingly, the concept of a ‘neutrophilic endotype’ has gained traction, whereby elevated numbers of neutrophils in patient sputum, peripheral blood, bronchoalveolar lavage and lung biopsies frequently associate with severe, corticosteroid‐refractory disease, exacerbations, patient hospitalizations, comorbidities and fatalities [72, 73, 74, 75, 76]. Whilst patients presenting with severe neutrophilic disease are often considered non‐atopic, neutrophils are still largely present in the airways of allergic ‘eosinophilic’ patients, where they are readily increased in number during symptomatic manifestations, such as allergen challenge, viral‐induced exacerbation or nocturnal crisis [77]. Neutrophils are capable of displaying marked plasticity, and those present in asthmatic patients have been reported to exhibit an altered phenotype, and increased activation and functionality [78]. Given our growing awareness of neutrophil heterogeneity and plasticity, more precise phenotyping of neutrophils in asthma patients will be important in delineating potentially variable contributions of neutrophil subsets to disease pathologies and in identifying novel therapeutic strategies. A recent study assessing in‐depth single‐cell analysis of granulocytes in the airway of asthmatic patients and healthy controls highlighted the likely presence of distinct neutrophil subsets [79].
Elevated neutrophil numbers in asthmatic patients are associated with an increase in classical pro‐neutrophilic factors such as CXCL8 and LTB4 [80, 81]. Transcripts of IL17A, a classical inducer of CXCL8, were found to be elevated in sputum of asthmatic patients, correlating with CXCL8 transcripts, neutrophilic inflammation and disease severity [82]. Accordingly, studies in patients and mice have highlighted a role for both Th1/IFN‐γ and Th17/IL‐17A responses in severe asthma and associated airway neutrophilia [82, 83, 84, 85]. Elevated TNF‐α levels have also been reported in the airways of asthmatic patients associating with neutrophils [86], with hierarchical clustering analysis of sputum transcriptomics showing a neutrophil cluster to be associated with expression of IFN and TNF family members [87]. Accumulating evidence highlights a strong association between airway infection and the presence of neutrophils in asthmatic patients. It has been postulated that neutrophilic asthma may manifest as a consequence of a dysbiosis of the airway microbiome whereby there are increased levels of bacteria but reduced bacterial diversity [88]. Similarly, respiratory viral infections, such as rhinovirus (RV), are a frequent cause of asthma exacerbations and concomitant increases in airway neutrophilia, whilst fungal pathogens also associate with neutrophilic, severe asthma [88]. Other factors have also been associated with the appearance of a neutrophilic endotype of asthma, including environmental factors such as cigarette smoke and pollution [89, 90, 91], and comorbid factors such as obesity and gastroesophageal reflux disease [92, 93]. Given the capacity of corticosteroids to inhibit neutrophil apoptosis and promote their survival [55], debate has arisen as to whether neutrophilic asthma reflects a true endotype of disease or is merely a consequence of the treatment itself [94]. However, others argue that treatment strategies do not ubiquitously underlie this endotype and neutrophils can still be observed in the absence of corticosteroid therapy [95, 96].
Given the destructive capacity of neutrophils, it is perhaps not surprising that they are considered to be protagonists of pathology in severe asthma. Manipulation of neutrophils in animal models of asthma has gone some way to support a role in disease progression [97, 98, 99, 100, 101]—though these limited studies have sometimes been confounded by a lack of physiological relevance to the disease they are seeking to recapitulate and/or the specificity of neutrophil targeting strategies. Moving forward, it is critical that the pre‐clinical animal models utilized to understand the pathogenesis of ‘neutrophilic asthma’ and test novel therapeutic strategies recapitulate the key aforementioned aetiological, clinical, inflammatory and remodelling aspects of disease observed in these patients. As such, many of the models utilized to date do not capture the steroid‐resistant, severe neutrophilic disease with low Th2 inflammation seen in a frequently non‐atopic patient group. Assertions as to how neutrophil products may participate in pathological processes in asthma are frequently based upon circumstantial data, or extrapolated from in vitro studies or other disease states, and their direct relevance to asthma pathogenesis should therefore be viewed with caution. MMPs, such as MMP‐8 and MMP‐9, are elevated in asthmatic patients, associating with neutrophilic inflammation and poor prognosis [102, 103, 104, 105]. Accordingly, mice deficient in MMP‐9 show ameliorated inflammation, and bronchial hyper‐responsiveness following allergen exposure [106]. NE is also elevated in the airways of asthmatic patients, associating with adverse outcomes. Largely indirect evidence highlights the capacity of NE to evoke pathological features of asthma [78], whilst chronic exposure of mice to NE results in pulmonary eosinophilia and mucus cell metaplasia [107]. Peripheral blood neutrophils derived from asthmatics produce enhanced levels of superoxide upon stimulation, with levels correlating with airway hyper‐responsiveness and inversely with FEV1, whilst ROS have been shown to be capable of inducing airway smooth muscle contraction, goblet cell metaplasia and mucus hypersecretion [78]. Neutrophils have also been proposed as a source of pro‐fibrotic TGF‐β [108], and epithelial barrier modulating cytokine oncostatin M (Figure 2) [109], whilst NETs have been purported to be essential to the pathology of RV‐induced exacerbations in a murine model [110].
Much of the data supporting a pathological role for neutrophils in asthma remains primarily circumstantial with a lack of convincing evidence directly demonstrating the mechanisms by which they participate in pathological processes, leading to counterarguments that their presence is inconsequential or even beneficial, and that strategies that seek to broadly block their infiltration carry inherent risks. Strategies that have sought to manipulate neutrophils in a clinical setting in asthmatic patients have been largely unsuccessful in ameliorating disease [111, 112, 113, 114, 115, 116, 117, 118, 119] (Table 2), and thus have done little to support the notion that neutrophils are truly pathological and a viable therapeutic target. Whilst, in some instances, suboptimal patient enrolment could have contributed to a lack of efficacy in these trials, it could also suggest that neutrophils are inconsequential in the context of asthma pathology. Conversely, given our evolving view of neutrophils, is it feasible that these cells, or at least a subset of these cells, could actually be beneficial in the context of asthma and that their presence is reflective of an effort to control inflammation or promote tissue repair? The presence of neutrophil chemokines in a subset of patients with severe asthma is associated with better clinical outcome [98], whilst increased intraepithelial neutrophils in the lungs of children with severe therapy‐resistant asthma correlated with better lung function [120]. Depletion of neutrophils in a mouse model of allergic airway disease inadvertently augmented type 2 inflammation [121]. In this context, neutrophil ablation resulted in a perturbation of neutrophil‐mediated regulatory pathways in the periphery culminating in G‐CSF accumulation, which in turn boosted type 2 innate lymphoid cell (ILC2) function [121]. Similarly, adoptive transfer of neutrophils has also been demonstrated to ameliorate Th2 inflammation in murine models. Intriguingly, biologics shown to ameliorate neutrophilic inflammation in asthmatic patients can cause increased G‐CSF [122, 123] and percentage sputum eosinophils [111]. Similarly, whilst some studies have suggested key pathological roles for neutrophil‐derived products in asthma, others have demonstrated a putative beneficial role for MMP‐9 in limiting and resolving allergic airway inflammation [124] and for NE in destroying mucus plugs [98] that cause airway obstruction.
BRONCHIECTASIS
Bronchiectasis is a heterogeneous chronic lung condition with multiple aetiological causes. Disease pathogenesis is believed to be driven by microbial dysbiosis and chronic infection associated with impaired mucociliary clearance. This leads to persistent airway neutrophilic inflammation, which contributes to airway tissue damage through release of proteases. Numerous defects in neutrophil function have been reported in the bronchiectasis literature including impaired bacterial phagocytosis and killing [125], greater presence of necrotic neutrophils in the airways (suggesting perturbed resolution of inflammation) [126], spontaneous neutrophil apoptosis [125] and increased NETosis [127]. It remains unclear as to whether these defects are inherent to bronchiectasis or whether neutrophil dysfunction occurs secondary to airway inflammatory milieu. The importance of neutrophil serine proteases as a driver of pathology in bronchiectasis was recently confirmed by results from the WILLOW phase 2 clinical trial, which evaluated brensocatib, a dipeptidyl peptidase‐1 (DPP1) inhibitor [128] (Table 2). This drug acts to prevent combined activation of NE, proteinase 3 and cathepsin G and demonstrated the beneficial clinical effect of significantly prolonging time to next exacerbation compared with placebo. Conversely, other smaller studies that have evaluated more selective NE inhibitors have reported lack of effects on a range of clinical outcomes [129, 130]. Larger independent trials are now needed to further investigate the effects of therapies targeting neutrophil dysfunction in bronchiectasis.
CYSTIC FIBROSIS
CF is an important genetic cause of bronchiectasis due to defects in the cystic fibrosis transmembrane conductance regulator (CFTR) protein. Chronic infection and inflammation induce lung damage in patients with CF with changes beginning early in life. The CF airway is dominated by a neutrophil‐mediated inflammatory process. A range of neutrophil functional abnormalities are recognized in CF. As for bronchiectasis, neutrophils from patients with CF have reduced phagocytic capacity, potentially due to diminished surface expression of key phagocytosis receptors CD14 and CD16 [131, 132], and also demonstrate greater oxidative burst and MPO release [133, 134]. These functional attributes contribute in part to the classification of two distinct neutrophil populations observed in the CF airways, namely the CD63loCD16hi A1 and CD63hiCD16lo A2 (otherwise known as the ‘GRIM’ phenotype due to their granule releasing, immunoregulatory and metabolic attributes [135]) subsets, based primarily on low and high levels of NE‐rich primary granule exocytosis, respectively [131, 136]. MPO concentrations correlate with lung function decline and CF disease severity [137]. Neutrophil proteases including NE and MMPs are also markedly upregulated in the CF airway with MMP‐9 previously shown to correlate with lung function and airway inflammation [138]. Clinical trials of neutrophil‐focussed therapies (Table 2) have largely been unsuccessful in CF, including inhaled NE inhibitors and recombinant human alpha‐1 antitrypsin therapy, which both showed no effect on lung function or clinical outcomes [139, 140]. An LTB4 receptor antagonist BIIL 284 BS has actually been shown to have detrimental effects including increased exacerbation frequency and worsened lung function [141]. Conversely, nebulized DNase is a widely used efficacious therapy that cleaves DNA released from neutrophils, reduces mucous viscosity and improves lung function. Several trials have proven its efficacy in patients with mild and more advanced disease [142]. More recently, CFTR modulator therapy has been added to the therapeutic repertoire and emerging evidence indicates that these drugs can modulate neutrophil function including beneficial changes in markers of activation [143] and increased bacterial killing [144].
IDIOPATHIC PULMONARY FIBROSIS
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, fibrotic lung disease of unknown cause. It is irreversible and responsible for ~1 in every 100 deaths each year in the UK. In early studies, neutrophil numbers [145], the neutrophil chemokine CXCL8/IL‐8 [146] and the neutrophil regulatory cytokine G‐CSF [147] have all been reported to be increased in bronchoalveolar lavage (BAL) fluid from subjects with IPF. BAL neutrophil numbers also independently predict mortality associated with the disease [145] providing suggestive evidence for a role in disease pathogenesis. This may occur through augmented release of proteases such as NE, which has been shown to degrade a range of ECM proteins. In bleomycin models, mice deficient in NE [148] or treated with a NE inhibitor [149] are protected from lung fibrosis. There is also evidence from animal studies that aged neutrophils can cause fibrotic interstitial lung disease if not restrained by B cells, which ordinarily act to limit their capacity to drive lung pathology [150]. Neutrophil MMP‐2, MMP‐8 and MMP‐9 can degrade all components of the ECM and other non‐matrix proteins. A range of MMPs are reported to be upregulated in IPF [151, 152, 153], and gene‐targeted deletion of Mmp3 [154], Mmp7 [155] and Mmp8 [156] reduces development of pulmonary fibrosis in bleomycin mouse models. Further work is needed to define methods of targeting MMPs as existing MMP inhibitors have had poor efficacy in lung cancer trials and are associated with dose‐limiting side‐effects such as musculoskeletal pain [157].
CONCLUSIONS
It is clear that neutrophils are a prominent feature of the airway inflammation observed in multiple chronic lung diseases, frequently associating with a worse prognosis. However, neutrophil targeting biologics entering clinical trials have yielded largely disappointing results to date. Accordingly, it is clear that in many instances there remains much still to learn about what underlies the influx of neutrophils, how precisely neutrophils are contributing to disease pathogenesis, and how, or even if, we should be targeting these cells therapeutically. Moving forward, it is important that we incorporate better pre‐clinical animal models that truly reflect the pathology we are seeking to simulate, and that we are rigorous in our patient selection for clinical trial enrolment. Increasingly, we are recognizing the heterogeneity within neutrophil populations, and the important immune modulatory, regulatory and repair roles of these cells. It is also clear that neutrophils must be considered in the context of comorbidities and intrinsic and extrinsic confounders. It is feasible, and perhaps probable, that the neutrophil exhibits pleiotropic and potentially conflicting roles in defining pathophysiology of chronic lung disease—some almost certainly detrimental and some potentially even beneficial—that may be variable in a context‐ and patient‐specific setting. More precise phenotyping of neutrophils in patients and a greater awareness of their heterogeneity, complexity and plasticity will be instrumental in defining more targeted therapeutic approaches for manipulating neutrophils and their effector functions in patients. Accordingly, therapeutic manipulation of neutrophils with a broad sword approach may not be the answer. Rather, we should seek to understand their complex and multifaceted roles in the disease state and target them within clearly stratified patient populations with the same subtleties and specificity that they themselves exhibit.
CONFLICT OF INTERESTS
The authors confirm they have no competing interests.
AUTHOR CONTRIBUTIONS
KTM, NB, AS and RJS contributed equally to the planning and writing of the manuscript. KTM and NB prepared figures and tables within the manuscript.
PERMISSION TO REPRODUCE
The authors provide full permission to reproduce content of the article where relevant.
Mincham KT, Bruno N, Singanayagam A, Snelgrove RJ. Our evolving view of neutrophils in defining the pathology of chronic lung disease. Immunology. 2021;164:701–721. 10.1111/imm.13419
Funding information
RJS is a Wellcome Trust Senior Research Fellow in Basic Biomedical Sciences (209458/Z/17/Z).
OTHER ARTICLES PUBLISHED IN THIS REVIEW SERIES
We need to talk about neutrophils. Immunology 2021, 164: 655‐656.
Neutrophil‐T cell crosstalk in inflammatory bowel disease. Immunology 2021, 164: 657‐664.
Neutrophils in pregnancy: New insights into innate and adaptive immune regulation. Immunology 2021, 164: 665‐676.
Neutrophils in secondary lymphoid organs. Immunology 2021, 164: 677‐688.
Crosstalk between B cells and neutrophils in rheumatoid arthritis. Immunology 2021, 164: 689‐700.
REFERENCES
- 1. Silvestre‐Roig C, Hidalgo A, Soehnlein O. Neutrophil heterogeneity: Implications for homeostasis and pathogenesis. Blood. 2016;127:2173–81. [DOI] [PubMed] [Google Scholar]
- 2. Pillay J, Den Braber I, Vrisekoop N, Kwast LM, de Boer RJ, Borghans JAM, et al. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood. 2010;116:625–7. [DOI] [PubMed] [Google Scholar]
- 3. Ng LG, Ostuni R, Hidalgo A. Heterogeneity of neutrophils. Nat Rev Immunol. 2019;19:255–65. [DOI] [PubMed] [Google Scholar]
- 4. Nicolás‐Ávila JÁ, Adrover JM, Hidalgo A. Neutrophils in homeostasis, immunity, and cancer. Immunity. 2017;46:15–28. [DOI] [PubMed] [Google Scholar]
- 5. Hartl D, Krauss‐Etschmann S, Koller B, Hordijk PL, Kuijpers TW, Hoffmann F, et al. Infiltrated neutrophils acquire novel chemokine receptor expression and chemokine responsiveness in chronic inflammatory lung diseases. J Immunol. 2008;181:8053–67. [DOI] [PubMed] [Google Scholar]
- 6. Lokwani R, Wark PA, Baines KJ, Fricker M, Barker D, Simpson JL. Blood neutrophils in COPD but not asthma exhibit a primed phenotype with downregulated CD62L expression. Int J Chron Obstruct Pulmon Dis. 2019;14:2517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hughes M, Mciver W, Mcgettrick H, et al. Phenotyping neutrophils in COPD through surface proteins. Eur Respir J. 2019;54:PA4086. [Google Scholar]
- 8. Silvestre‐Roig C, Fridlender ZG, Glogauer M, Scapini P. Neutrophil diversity in health and disease. Trends Immunol. 2019;40:565–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mayadas TN, Cullere X, Lowell CA. The multifaceted functions of neutrophils. Annu Rev Pathol. 2014;9:181–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tecchio C, Micheletti A, Cassatella MA. Neutrophil‐derived cytokines: facts beyond expression. Front Immunol. 2014;5:508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Soehnlein O, Kai‐Larsen Y, Frithiof R, Sorensen OE, Kenne E, Scharffetter‐Kochanek K, et al. Neutrophil primary granule proteins HBP and HNP1‐3 boost bacterial phagocytosis by human and murine macrophages. J Clin Invest. 2008;118:3491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Prame Kumar K, Nicholls AJ, Wong CHY. Partners in crime: neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res. 2018;371:551–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peiró T, Patel DF, Akthar S, Gregory LG, Pyle CJ, Harker JA, et al. Neutrophils drive alveolar macrophage IL‐1β release during respiratory viral infection. Thorax. 2018;73(6):546–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Spörri R, Joller N, Hilbi H, Oxenius A. A novel role for neutrophils as critical activators of NK cells. J Immunol. 2008;181:7121–30. [DOI] [PubMed] [Google Scholar]
- 15. Mudzinski SP, Christian TP, Guo TL, et al. Expression of HLA‐DR (major histocompatibility complex class II) on neutrophils from patients treated with granulocyte‐macrophage colony‐stimulating factor for mobilization of stem cells. Blood. 1995;86:2452–3. [PubMed] [Google Scholar]
- 16. Gosselin EJ, Wardwell K, Rigby WF, et al. Induction of MHC class II on human polymorphonuclear neutrophils by granulocyte/macrophage colony‐stimulating factor, IFN‐gamma, and IL‐3. J Immunol. 1993;151:1482–90. [PubMed] [Google Scholar]
- 17. Krishnamoorthy N, Douda DN, Brüggemann TR, et al. Neutrophil cytoplasts induce TH17 differentiation and skew inflammation toward neutrophilia in severe asthma. Sci Immunol. 2018;3:eaao4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Minns D, Smith KJ, Alessandrini V, Hardisty G, Melrose L, Jackson‐Jones L, et al. The neutrophil antimicrobial peptide cathelicidin promotes Th17 differentiation. Nat Commun. 2021;12:1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Trentini MM, de Oliveira FM, Kipnis A, Junqueira‐Kipnis AP. The role of neutrophils in the induction of specific Th1 and Th17 during vaccination against tuberculosis. Front Microbiol. 2016;7:898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sun S, Sursal T, Adibnia Y, Zhao C, Zheng Y, Li H, et al. Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways. PLoS One. 2013;8:e59989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Di Gennaro A, Kenne E, Wan M, et al. Leukotriene B4‐induced changes in vascular permeability are mediated by neutrophil release of heparin‐binding protein (HBP/CAP37/azurocidin). FASEB J. 2009;23:1750–7. [DOI] [PubMed] [Google Scholar]
- 22. McHugh BJ, Wang R, Li H‐N, Beaumont PE, Kells R, Stevens H, et al. Cathelicidin is a “fire alarm”, generating protective NLRP3‐dependent airway epithelial cell inflammatory responses during infection with Pseudomonas aeruginosa . PLoS Pathog. 2019;15:e1007694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chrysanthopoulou A, Mitroulis I, Apostolidou E, et al. Neutrophil extracellular traps promote differentiation and function of fibroblasts. J Pathol. 2014;233(3):294–307. [DOI] [PubMed] [Google Scholar]
- 24. Gregory AD, Kliment CR, Metz HE, et al. Neutrophil elastase promotes myofibroblast differentiation in lung fibrosis. J Leukoc Biol. 2015;98:143–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mantovani A, Cassatella MA, Costantini C, et al. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011;11:519. [DOI] [PubMed] [Google Scholar]
- 26. Munder M, Schneider H, Luckner C, et al. Suppression of T‐cell functions by human granulocyte arginase. Blood. 2006;108:1627–34. [DOI] [PubMed] [Google Scholar]
- 27. Snelgrove RJ, Patel DF, Patel T, Lloyd CM. The enigmatic role of the neutrophil in asthma: friend, foe or indifferent? Clin Exp Allergy. 2018;48:1275–85. [DOI] [PubMed] [Google Scholar]
- 28. Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL‐23 and IL‐17. Immunity. 2005;22:285–94. [DOI] [PubMed] [Google Scholar]
- 29. Filardy AA, Pires DR, Nunes MP, et al. Proinflammatory clearance of apoptotic neutrophils induces an IL‐12(low)IL‐10(high) regulatory phenotype in macrophages. J Immunol. 2010;185:2044–50. [DOI] [PubMed] [Google Scholar]
- 30. Phillipson M, Kubes P. The healing power of neutrophils. Trends Immunol. 2019;40(7):635–47. [DOI] [PubMed] [Google Scholar]
- 31. Soriano JB, Kendrick PJ, Paulson KR, Gupta V, Abrams EM, Adedoyin RA, et al. Prevalence and attributable health burden of chronic respiratory diseases, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Respir Med. 2020;8(6):585–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vestbo J, Hurd SS, Agustí AG, Jones PW, Vogelmeier C, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187(4):347–65. [DOI] [PubMed] [Google Scholar]
- 33. Salvi SS, Barnes PJ. Chronic obstructive pulmonary disease in non‐smokers. Lancet. 2009;374(9691):733–43. [DOI] [PubMed] [Google Scholar]
- 34. McDonough JE, Yuan R, Suzuki M, Seyednejad N, Elliott WM, Sanchez PG, et al. Small‐airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med. 2011;365(17):1567–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Papi A, Bellettato CM, Braccioni F, Romagnoli M, Casolari P, Caramori G, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med. 2006;173(10):1114–21. [DOI] [PubMed] [Google Scholar]
- 36. Singanayagam A, Schembri S, Chalmers JD. Predictors of mortality in hospitalized adults with acute exacerbation of chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2013;10(2):81–9. [DOI] [PubMed] [Google Scholar]
- 37. Brightling CE, Monteiro W, Ward R, Parker D, Morgan MDL, Wardlaw AJ, et al. Sputum eosinophilia and short‐term response to prednisolone in chronic obstructive pulmonary disease: a randomised controlled trial. Lancet. 2000;356(9240):1480–5. [DOI] [PubMed] [Google Scholar]
- 38. Donaldson GC, Seemungal TA, Patel IS, Bhowmik A, Wilkinson TMA, Hurst JR, et al. Airway and systemic inflammation and decline in lung function in patients with COPD. Chest. 2005;128(4):1995–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138(1):16–27. [DOI] [PubMed] [Google Scholar]
- 40. Keatings VM, Barnes PJ. Granulocyte activation markers in induced sputum: comparison between chronic obstructive pulmonary disease, asthma, and normal subjects. Am J Respir Crit Care Med. 1997;155(2):449–53. [DOI] [PubMed] [Google Scholar]
- 41. Vernooy JH, Lindeman JH, Jacobs JA, Hanemaaijer R, Wouters EFM, et al. Increased activity of matrix metalloproteinase‐8 and matrix metalloproteinase‐9 in induced sputum from patients with COPD. Chest. 2004;126(6):1802–10. [DOI] [PubMed] [Google Scholar]
- 42. Thulborn SJ, Mistry V, Brightling CE, Moffitt KL, Ribeiro D, Bafadhel M, et al. Neutrophil elastase as a biomarker for bacterial infection in COPD. Respir Res. 2019;20(1):170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gaggar A, Jackson PL, Noerager BD, O'Reilly PJ, McQuaid DB, Rowe SM, et al. A novel proteolytic cascade generates an extracellular matrix‐derived chemoattractant in chronic neutrophilic inflammation. J Immunol. 2008;180(8):5662–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Snelgrove RJ, Jackson PL, Hardison MT, Noerager BD, Kinloch A, Gaggar A, et al. A critical role for LTA4H in limiting chronic pulmonary neutrophilic inflammation. Science. 2010;330(6000):90–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wells JM, O’Reilly PJ, Szul T, et al. An Aberrant Leukotriene A4 hydrolase–proline‐glycine‐proline pathway in the pathogenesis of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2014;190(1):51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Benjamin JT, Plosa EJ, Sucre JM, van der Meer R, Dave S, Gutor S, et al. Neutrophilic inflammation during lung development disrupts elastin assembly and predisposes adult mice to COPD. J Clin Invest. 2021;131(1):139481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dicker AJ, Crichton ML, Pumphrey EG, Cassidy AJ, Suarez‐Cuartin G, Sibila O, et al. Neutrophil extracellular traps are associated with disease severity and microbiota diversity in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2018;141(1):117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Winslow S, Odqvist L, Diver S, et al. Multi‐omics links IL‐6 trans‐signalling with neutrophil extracellular trap formation and Haemophilus infection in COPD. Eur Respir J. 2021; PMID: 33766947. Online ahead of print. [DOI] [PubMed] [Google Scholar]
- 49. Sapey E, Stockley JA, Greenwood H, Ahmad A, Bayley D, Lord JM, et al. Behavioral and structural differences in migrating peripheral neutrophils from patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;183(9):1176–86. [DOI] [PubMed] [Google Scholar]
- 50. Sapey E, Greenwood H, Walton G, Mann E, Love A, Aaronson N, et al. Phosphoinositide 3‐kinase inhibition restores neutrophil accuracy in the elderly: toward targeted treatments for immunosenescence. Blood. 2014;123(2):239–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mallia P, Message SD, Contoli M, Gray KK, Telcian A, Laza‐Stanca V, et al. Neutrophil adhesion molecules in experimental rhinovirus infection in COPD. Respir Res. 2013;14(1):72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Donnelly LE, Barnes PJ. Defective phagocytosis in airways disease. Chest. 2012;141(4):1055–62. [DOI] [PubMed] [Google Scholar]
- 53. Stringer KA, Tobias M, O'Neill HC, Franklin CC. Cigarette smoke extract‐induced suppression of caspase‐3‐like activity impairs human neutrophil phagocytosis. Am J Physiol Lung Cell Mol Physiol. 2007;292(6):L1572–9. [DOI] [PubMed] [Google Scholar]
- 54. Patel IS, Seemungal TAR, Wilks M, et al. Relationship between bacterial colonisation and the frequency, character, and severity of COPD exacerbations. Thorax. 2002;57(9):759–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cox G. Glucocorticoid treatment inhibits apoptosis in human neutrophils. Separation of survival and activation outcomes. J Immunol. 1995;154(9):4719–25. [PubMed] [Google Scholar]
- 56. Wedzicha JA, Calverley PMA, Seemungal TA, Hagan G, Ansari Z, Stockley RA. The prevention of chronic obstructive pulmonary disease exacerbations by salmeterol/fluticasone propionate or tiotropium bromide. Am J Respir Crit Care Med. 2008;177(1):19–26. [DOI] [PubMed] [Google Scholar]
- 57. Singanayagam A, Glanville N, Girkin JL, Ching YM, Marcellini A, Porter JD, et al. Corticosteroid suppression of antiviral immunity increases bacterial loads and mucus production in COPD exacerbations. Nat Commun. 2018;9(1):2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rennard SI, Dale DC, Donohue JF, Kanniess F, Magnussen H, Sutherland ER, et al. CXCR2 antagonist MK‐7123. A phase 2 proof‐of‐concept trial for chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;191(9):1001–11. [DOI] [PubMed] [Google Scholar]
- 59. Kirsten AM, Förster K, Radeczky E, Linnhoff A, Balint B, Watz H, et al. The safety and tolerability of oral AZD5069, a selective CXCR2 antagonist, in patients with moderate‐to‐severe COPD. Pulm Pharmacol Ther. 2015;31:36–41. [DOI] [PubMed] [Google Scholar]
- 60. Lazaar AL, Miller BE, Donald AC, Keeley T, Ambery C, Russell J, et al. CXCR2 antagonist for patients with chronic obstructive pulmonary disease with chronic mucus hypersecretion: a phase 2b trial. Respir Res. 2020;21(1):149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kuna P, Jenkins M, O'Brien CD, Fahy WA. AZD9668, a neutrophil elastase inhibitor, plus ongoing budesonide/formoterol in patients with COPD. Respir Med. 2012;106(4):531–9. [DOI] [PubMed] [Google Scholar]
- 62. Rennard SI, Fogarty C, Kelsen S, Long W, Ramsdell J, Allison J, et al. The safety and efficacy of infliximab in moderate to severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175(9):926–34. [DOI] [PubMed] [Google Scholar]
- 63. Gompertz S, Stockley RA. A randomized, placebo‐controlled trial of a leukotriene synthesis inhibitor in patients with COPD. Chest. 2002;122(1):289–94. [DOI] [PubMed] [Google Scholar]
- 64. King PT, Sharma R, O’Sullivan KM, Callaghan J, Dousha L, Thomas B, et al. Deoxyribonuclease 1 reduces pathogenic effects of cigarette smoke exposure in the lung. Sci Rep. 2017;7(1):12128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ritzmann F, Borchardt K, Vella G, Chitirala P, Angenendt A, Herr C, et al. Blockade of PD‐1 decreases neutrophilic inflammation and lung damage in experimental COPD. Am J Physiol Lung Cell Mol Physiol. 2021;320(5):L958–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gupta V, Khan A, Higham A, Lemon J, Sriskantharajah S, Amour A, et al. The effect of phosphatidylinositol‐3 kinase inhibition on matrix metalloproteinase‐9 and reactive oxygen species release from chronic obstructive pulmonary disease neutrophils. Int Immunopharmacol. 2016;35:155–62. [DOI] [PubMed] [Google Scholar]
- 67. Cahn A, Hamblin JN, Begg M, Wilson R, Dunsire L, Sriskantharajah S, et al. Safety, pharmacokinetics and dose‐response characteristics of GSK2269557, an inhaled PI3Kδ inhibitor under development for the treatment of COPD. Pulm Pharmacol Ther. 2017;46:69–77. [DOI] [PubMed] [Google Scholar]
- 68. Begg M, Hamblin JN, Jarvis E, Bradley G, Mark S, Michalovich D, et al. Exploring PI3Kδ molecular pathways in stable COPD and following an acute exacerbation, two randomized controlled trials. Int J Chron Obstruct Pulmon Dis. 2021;16:1621–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Global, regional, and national deaths, prevalence, disability‐adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990‐2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Respir Med. 2017;5(9):691–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lloyd CM, Snelgrove RJ. Type 2 immunity: expanding our view. Sci Immunol. 2018;3(25):eaat1604. [DOI] [PubMed] [Google Scholar]
- 71. Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR, Ellwanger A, et al. T‐helper type 2‐driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180(5):388–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Fahy JV, Kim KW, Liu J, Boushey HA. Prominent neutrophilic inflammation in sputum from subjects with asthma exacerbation. J Allergy Clin Immunol. 1995;95(4):843–52. [DOI] [PubMed] [Google Scholar]
- 73. Green RH, Brightling CE, Woltmann G, et al. Analysis of induced sputum in adults with asthma: identification of subgroup with isolated sputum neutrophilia and poor response to inhaled corticosteroids. Thorax. 2002;57(10):875–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Moore WC, Hastie AT, Li X, Li H, Busse WW, Jarjour NN, et al. Sputum neutrophil counts are associated with more severe asthma phenotypes using cluster analysis. J Allergy Clin Immunol. 2014;133(6):1557–63.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sur S, Crotty TB, Kephart GM, et al. Sudden‐onset fatal asthma. A distinct entity with few eosinophils and relatively more neutrophils in the airway submucosa? Am Rev Respir Dis. 1993;148(3):713–9. [DOI] [PubMed] [Google Scholar]
- 76. Wenzel SE, Szefler SJ, Leung DY, Sloan SI, Rex MD, Martin RJ. Bronchoscopic evaluation of severe asthma. Persistent inflammation associated with high dose glucocorticoids. Am J Respir Crit Care Med. 1997;156(3 Pt 1):737–43. [DOI] [PubMed] [Google Scholar]
- 77. Radermecker C, Louis R, Bureau F, Marichal T. Role of neutrophils in allergic asthma. Curr Opin Immunol. 2018;54:28–34. [DOI] [PubMed] [Google Scholar]
- 78. Monteseirín J. Neutrophils and asthma. J Investig Allergol Clin Immunol. 2009;19(5):340–54. [PubMed] [Google Scholar]
- 79. Scapini P, Marini O, Tecchio C, Cassatella MA. Human neutrophils in the saga of cellular heterogeneity: insights and open questions. Immunol Rev. 2016;273(1):48–60. [DOI] [PubMed] [Google Scholar]
- 80. Gibson PG, Simpson JL, Saltos N. Heterogeneity of airway inflammation in persistent asthma: evidence of neutrophilic inflammation and increased sputum interleukin‐8. Chest. 2001;119(5):1329–36. [DOI] [PubMed] [Google Scholar]
- 81. Patel DF, Peiró T, Shoemark A, Akthar S, Walker SA, Grabiec AM, et al. An extracellular matrix fragment drives epithelial remodeling and airway hyperresponsiveness. Sci Transl Med. 2018;10(455):eaaq0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bullens DM, Truyen E, Coteur L, Dilissen E, Hellings PW, Dupont LJ, et al. IL‐17 mRNA in sputum of asthmatic patients: linking T cell driven inflammation and granulocytic influx? Respir Res. 2006;7(1):135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Raundhal M, Morse C, Khare A, Oriss TB, Milosevic J, Trudeau J, et al. High IFN‐γ and low SLPI mark severe asthma in mice and humans. J Clin Invest. 2015;125(8):3037–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. McKinley L, Alcorn JF, Peterson A, et al. TH17 cells mediate steroid‐resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol. 2008;181(6):4089–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Agache I, Ciobanu C, Agache C, Anghel M. Increased serum IL‐17 is an independent risk factor for severe asthma. Respir Med. 2010;104(8):1131–7. [DOI] [PubMed] [Google Scholar]
- 86. Bradding P, Roberts JA, Britten KM, Montefort S, Djukanovic R, Mueller R, et al. Interleukin‐4, ‐5, and ‐6 and tumor necrosis factor‐alpha in normal and asthmatic airways: evidence for the human mast cell as a source of these cytokines. Am J Respir Cell Mol Biol. 1994;10(5):471–80. [DOI] [PubMed] [Google Scholar]
- 87. Kuo CS, Pavlidis S, Loza M, Baribaud F, Rowe A, Pandis I, et al. T‐helper cell type 2 (Th2) and non‐Th2 molecular phenotypes of asthma using sputum transcriptomics in U‐BIOPRED. Eur Respir J. 2017;49(2):1602135. [DOI] [PubMed] [Google Scholar]
- 88. Crisford H, Sapey E, Rogers GB, Taylor S, Nagakumar P, Lokwani R, et al. Neutrophils in asthma: the good, the bad and the bacteria. Thorax. 2021;76(8):835–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Siew LQC, Wu SY, Ying S, Corrigan CJ. Cigarette smoking increases bronchial mucosal IL‐17A expression in asthmatics, which acts in concert with environmental aeroallergens to engender neutrophilic inflammation. Clin Exp Allergy. 2017;47(6):740–50. [DOI] [PubMed] [Google Scholar]
- 90. Lazarus SC, Chinchilli VM, Rollings NJ, Boushey HA, Cherniack R, Craig TJ, et al. Smoking affects response to inhaled corticosteroids or leukotriene receptor antagonists in asthma. Am J Respir Crit Care Med. 2007;175(8):783–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wooding DJ, Ryu MH, Li H, Alexis NE, Pena O, Carlsten C. Acute air pollution exposure alters neutrophils in never‐smokers and at‐risk humans. Eur Respir J. 2020;55(4):1901495. [DOI] [PubMed] [Google Scholar]
- 92. Scott HA, Gibson PG, Garg ML, Wood LG. Airway inflammation is augmented by obesity and fatty acids in asthma. Eur Respir J. 2011;38(3):594–602. [DOI] [PubMed] [Google Scholar]
- 93. Simpson JL, Baines KJ, Ryan N, et al. Neutrophilic asthma is characterised by increased rhinosinusitis with sleep disturbance and GERD. Asian Pac J Allergy Immunol. 2014;32(1):66–74. [DOI] [PubMed] [Google Scholar]
- 94. Nair P, Surette MG, Virchow JC. Neutrophilic asthma: misconception or misnomer? Lancet Respir Med. 2021;9(5):441–3. [DOI] [PubMed] [Google Scholar]
- 95. Berry M, Morgan A, Shaw DE, Parker D, Green R, Brightling C, et al. Pathological features and inhaled corticosteroid response of eosinophilic and non‐eosinophilic asthma. Thorax. 2007;62(12):1043–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Maneechotesuwan K, Essilfie‐Quaye S, Kharitonov SA, Adcock IM, Barnes PJ, et al. Loss of control of asthma following inhaled corticosteroid withdrawal is associated with increased sputum interleukin‐8 and neutrophils. Chest. 2007;132(1):98–105. [DOI] [PubMed] [Google Scholar]
- 97. Nabe T, Hosokawa F, Matsuya K, Morishita T, Ikedo A, Fujii M, et al. Important role of neutrophils in the late asthmatic response in mice. Life Sci. 2011;88(25–26):1127–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Manni ML, Trudeau JB, Scheller EV, Mandalapu S, Elloso MM, Kolls JK, et al. The complex relationship between inflammation and lung function in severe asthma. Mucosal Immunol. 2014;7(5):1186–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. De Vooght V, Smulders S, Haenen S, Belmans J, Opdenakker G, Verbeken E, et al. Neutrophil and eosinophil granulocytes as key players in a mouse model of chemical‐induced asthma. Toxicol Sci. 2013;131(2):406–18. [DOI] [PubMed] [Google Scholar]
- 100. O'Byrne PM, Walters EH, Gold BD, Aizawa HA, Fabbri LM, Alpert SE, et al. Neutrophil depletion inhibits airway hyperresponsiveness induced by ozone exposure. Am Rev Respir Dis. 1984;130(2):214–9. [DOI] [PubMed] [Google Scholar]
- 101. Hosoki K, Aguilera‐Aguirre L, Brasier AR, Kurosky A, Boldogh I, Sur S. Facilitation of allergic sensitization and allergic airway inflammation by pollen‐induced innate neutrophil recruitment. Am J Respir Cell Mol Biol. 2016;54(1):81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Prikk K, Maisi P, Pirilä E, Reintam MA, Salo T, Sorsa T, et al. Airway obstruction correlates with collagenase‐2 (MMP‐8) expression and activation in bronchial asthma. Lab Invest. 2002;82(11):1535–45. [DOI] [PubMed] [Google Scholar]
- 103. Cundall M, Sun Y, Miranda C, Trudeau JB, Barnes S, Wenzel SE. Neutrophil‐derived matrix metalloproteinase‐9 is increased in severe asthma and poorly inhibited by glucocorticoids. J Allergy Clin Immunol. 2003;112(6):1064–71. [DOI] [PubMed] [Google Scholar]
- 104. Wenzel SE, Balzar S, Cundall M, Chu HW. Subepithelial basement membrane immunoreactivity for matrix metalloproteinase 9: association with asthma severity, neutrophilic inflammation, and wound repair. J Allergy Clin Immunol. 2003;111(6):1345–52. [DOI] [PubMed] [Google Scholar]
- 105. Kelly EA, Busse WW, Jarjour NN. Increased matrix metalloproteinase‐9 in the airway after allergen challenge. Am J Respir Crit Care Med. 2000;162(3 Pt 1):1157–61. [DOI] [PubMed] [Google Scholar]
- 106. Cataldo DD, Tournoy KG, Vermaelen K, Munaut C, Foidart JM, Louis R, et al. Matrix metalloproteinase‐9 deficiency impairs cellular infiltration and bronchial hyperresponsiveness during allergen‐induced airway inflammation. Am J Pathol. 2002;161(2):491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Voynow JA, Fischer BM, Malarkey DE, Burch LH, Wong T, Longphre M, et al. Neutrophil elastase induces mucus cell metaplasia in mouse lung. Am J Physiol Lung Cell Mol Physiol. 2004;287(6):L1293–302. [DOI] [PubMed] [Google Scholar]
- 108. Chu HW, Trudeau JB, Balzar S, et al. Peripheral blood and airway tissue expression of transforming growth factor beta by neutrophils in asthmatic subjects and normal control subjects. J Allergy Clin Immunol. 2000;106(6):1115–23. [DOI] [PubMed] [Google Scholar]
- 109. Pothoven KL, Norton JE, Suh LA, et al. Neutrophils are a major source of the epithelial barrier disrupting cytokine oncostatin M in patients with mucosal airways disease. J Allergy Clin Immunol. 2017;139(6):1966–78.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Toussaint M, Jackson DJ, Swieboda D, Guedán A, Tsourouktsoglou TD, Ching YM, et al. Host DNA released by NETosis promotes rhinovirus‐induced type‐2 allergic asthma exacerbation. Nat Med. 2017;23(6):681–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Nair P, Gaga M, Zervas E, Alagha K, Hargreave FE, O'Byrne PM, et al. Safety and efficacy of a CXCR2 antagonist in patients with severe asthma and sputum neutrophils: a randomized, placebo‐controlled clinical trial. Clin Exp Allergy. 2012;42(7):1097–103. [DOI] [PubMed] [Google Scholar]
- 112. O'Byrne PM, Metev H, Puu M, Richter K, Keen C, Uddin M, et al. Efficacy and safety of a CXCR2 antagonist, AZD5069, in patients with uncontrolled persistent asthma: a randomised, double‐blind, placebo‐controlled trial. Lancet Respir Med. 2016;4(10):797–806. [DOI] [PubMed] [Google Scholar]
- 113. Evans DJ, Barnes PJ, Spaethe SM, van Alstyne El, Mitchell MI, O'Connor BJ. Effect of a leukotriene B4 receptor antagonist, LY293111, on allergen induced responses in asthma. Thorax. 1996;51(12):1178–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wasfi YS, Villarán C, de Tilleghem CB, Smugar SS, Hanley WD, Reiss TF, et al. The efficacy and tolerability of MK‐0633, a 5‐lipoxygenase inhibitor, in chronic asthma. Respir Med. 2012;106(1):34–46. [DOI] [PubMed] [Google Scholar]
- 115. Chaudhuri R, Norris V, Kelly K, Zhu C‐Q, Ambery C, Lafferty J, et al. Effects of a FLAP inhibitor, GSK2190915, in asthmatics with high sputum neutrophils. Pulm Pharmacol Ther. 2014;27(1):62–9. [DOI] [PubMed] [Google Scholar]
- 116. Barchuk W, Lambert J, Fuhr R, Jiang JZ, Bertelsen K, Fourie A, et al. Effects of JNJ‐40929837, a leukotriene A4 hydrolase inhibitor, in a bronchial allergen challenge model of asthma. Pulm Pharmacol Ther. 2014;29(1):15–23. [DOI] [PubMed] [Google Scholar]
- 117. Busse WW, Holgate S, Kerwin E, Chon Y, Feng JY, Lin J, et al. Randomized, double‐blind, placebo‐controlled study of brodalumab, a human anti‐IL‐17 receptor monoclonal antibody, in moderate to severe asthma. Am J Respir Crit Care Med. 2013;188(11):1294–302. [DOI] [PubMed] [Google Scholar]
- 118. Holgate ST, Noonan M, Chanez P, Busse W, Dupont L, Pavord I, et al. Efficacy and safety of etanercept in moderate‐to‐severe asthma: a randomised, controlled trial. Eur Respir J. 2011;37(6):1352–9. [DOI] [PubMed] [Google Scholar]
- 119. Wenzel SE, Barnes PJ, Bleecker ER, et al. A randomized, double‐blind, placebo‐controlled study of tumor necrosis factor‐alpha blockade in severe persistent asthma. Am J Respir Crit Care Med. 2009;179(7):549–58. [DOI] [PubMed] [Google Scholar]
- 120. Andersson CK, Adams A, Nagakumar P, Bossley C, Gupta A, De Vries D, et al. Intraepithelial neutrophils in pediatric severe asthma are associated with better lung function. J Allergy Clin Immunol. 2017;139(6):1819–29.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Patel DF, Peiró T, Bruno N, Vuononvirta J, Akthar S, Puttur F, et al. Neutrophils restrain allergic airway inflammation by limiting ILC2 function and monocyte–dendritic cell antigen presentation. Sci Immunol. 2019;4(41):eaax7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Jurcevic S, Humfrey C, Uddin M, Warrington S, Larsson B, Keen C. The effect of a selective CXCR2 antagonist (AZD5069) on human blood neutrophil count and innate immune functions. Br J Clin Pharmacol. 2015;80(6):1324–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Watz H, Uddin M, Pedersen F, Kirsten A, Goldmann T, Stellmacher F, et al. Effects of the CXCR2 antagonist AZD5069 on lung neutrophil recruitment in asthma. Pulm Pharmacol Ther. 2017;45:121–3. [DOI] [PubMed] [Google Scholar]
- 124. McMillan SJ, Kearley J, Campbell JD, Zhu X‐W, Larbi KY, Shipley JM, et al. Matrix metalloproteinase‐9 deficiency results in enhanced allergen‐induced airway inflammation. J Immunol. 2004;172(4):2586–94. [DOI] [PubMed] [Google Scholar]
- 125. Bedi P, Davidson DJ, McHugh BJ, Rossi AG, Hill AT. Blood neutrophils are reprogrammed in bronchiectasis. Am J Respir Crit Care Med. 2018;198(7):880–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Watt AP, Brown V, Courtney J, et al. Neutrophil apoptosis, proinflammatory mediators and cell counts in bronchiectasis. Thorax. 2004;59(3):231–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Keir HR, Shoemark A, Dicker AJ, Perea L, Pollock J, Giam YH, et al. Neutrophil extracellular traps, disease severity, and antibiotic response in bronchiectasis: an international, observational, multicohort study. Lancet Respir Med. 2021. [DOI] [PubMed] [Google Scholar]
- 128. Chalmers JD, Haworth CS, Metersky ML, Loebinger MR, Blasi F, Sibila O, et al. Phase 2 trial of the DPP‐1 inhibitor brensocatib in bronchiectasis. N Engl J Med. 2020;383(22):2127–37. [DOI] [PubMed] [Google Scholar]
- 129. Stockley R, De Soyza A, Gunawardena K, Perrett J, Forsman‐Semb K, Entwistle N, et al. Phase II study of a neutrophil elastase inhibitor (AZD9668) in patients with bronchiectasis. Respir Med. 2013;107(4):524–33. [DOI] [PubMed] [Google Scholar]
- 130. Watz H, Nagelschmitz J, Kirsten A, Pedersen F, van der Mey D, Schwers S, et al. Safety and efficacy of the human neutrophil elastase inhibitor BAY 85–8501 for the treatment of non‐cystic fibrosis bronchiectasis: A randomized controlled trial. Pulm Pharmacol Ther. 2019;56:86–93. [DOI] [PubMed] [Google Scholar]
- 131. Tirouvanziam R, Gernez Y, Conrad CK, Moss RB, Schrijver I, Dunn CE, et al. Profound functional and signaling changes in viable inflammatory neutrophils homing to cystic fibrosis airways. Proc Natl Acad Sci USA. 2008;105(11):4335–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Morris MR, Doull IJ, Dewitt S, Hallett MB. Reduced iC3b‐mediated phagocytotic capacity of pulmonary neutrophils in cystic fibrosis. Clin Exp Immunol. 2005;142(1):68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Witko‐Sarsat V, Allen RC, Paulais M, et al. Disturbed myeloperoxidase‐dependent activity of neutrophils in cystic fibrosis homozygotes and heterozygotes, and its correction by amiloride. J Immunol. 1996;157(6):2728–35. [PubMed] [Google Scholar]
- 134. Van Der Vliet A, Nguyen MN, Shigenaga MK, Eiserich JP, Marelich GP, Cross CE. Myeloperoxidase and protein oxidation in cystic fibrosis. Am J Physiol Lung Cell Mol Physiol. 2000;279(3):L537–46. [DOI] [PubMed] [Google Scholar]
- 135. Forrest OA, Ingersoll SA, Preininger MK, et al. Pathological conditioning of human neutrophils recruited to the airway milieu in cystic fibrosis. J Leukoc Biol. 2018;104(4):665–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Makam M, Diaz D, Laval J, Gernez Y, Conrad CK, Dunn CE, et al. Activation of critical, host‐induced, metabolic and stress pathways marks neutrophil entry into cystic fibrosis lungs. Proc Natl Acad Sci USA. 2009;106(14):5779–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Witko‐Sarsat V, Delacourt C, Rabier D, Bardet J, Nguyen AT, Descamps‐Latscha B. Neutrophil‐derived long‐lived oxidants in cystic fibrosis sputum. Am J Respir Crit Care Med. 1995;152(6 Pt 1):1910–6. [DOI] [PubMed] [Google Scholar]
- 138. Sagel SD, Kapsner RK, Osberg I. Induced sputum matrix metalloproteinase‐9 correlates with lung function and airway inflammation in children with cystic fibrosis. Pediatr Pulmonol. 2005;39(3):224–32. [DOI] [PubMed] [Google Scholar]
- 139. Elborn JS, Perrett J, Forsman‐Semb K, Marks‐Konczalik J, Gunawardena K, Entwistle N. Efficacy, safety and effect on biomarkers of AZD9668 in cystic fibrosis. Eur Respir J. 2012;40(4):969–76. [DOI] [PubMed] [Google Scholar]
- 140. Griese M, Latzin P, Kappler M, Weckerle K, Heinzlmaier T, Bernhardt T, et al. alpha1‐Antitrypsin inhalation reduces airway inflammation in cystic fibrosis patients. Eur Respir J. 2007;29(2):240–50. [DOI] [PubMed] [Google Scholar]
- 141. Konstan MW, Doring G, Heltshe SL, Lands LC, Hilliard KA, Koker P, et al. A randomized double blind, placebo controlled phase 2 trial of BIIL 284 BS (an LTB4 receptor antagonist) for the treatment of lung disease in children and adults with cystic fibrosis. J Cyst Fibros. 2014;13(2):148–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Yang C, Montgomery M. Dornase alfa for cystic fibrosis. Cochrane Database Syst Rev. 2021;3(3):CD001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Hardisty GR, Law SM, Carter S, Grogan B, Singh PK, McKone EF, et al. Ivacaftor modifies cystic fibrosis neutrophil phenotype in subjects with R117H residual function CFTR mutations. Eur Respir J. 2020;57(1):2002161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Pohl K, Hayes E, Keenan J, Henry M, Meleady P, Molloy K, et al. A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by CFTR potentiator therapy. Blood. 2014;124(7):999–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Kinder BW, Brown KK, Schwarz MI, Ix JH, Kervitsky A, King TE. Baseline BAL neutrophilia predicts early mortality in idiopathic pulmonary fibrosis. Chest. 2008;133(1):226–32. [DOI] [PubMed] [Google Scholar]
- 146. Xaubet A, Agusti C, Luburich P, Barberá JA, Carrión M, Ayuso MC, et al. Interleukin‐8 expression in bronchoalveolar lavage cells in the evaluation of alveolitis in idiopathic pulmonary fibrosis. Respir Med. 1998;92(2):338–44. [DOI] [PubMed] [Google Scholar]
- 147. Ashitani J, Mukae H, Taniguchi H, Ihi T, Kadota J‐I, Kohno S, et al. Granulocyte‐colony stimulating factor levels in bronchoalveolar lavage fluid from patients with idiopathic pulmonary fibrosis. Thorax. 1999;54(11):1015–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Chua F, Dunsmore SE, Clingen PH, Mutsaers SE, Shapiro SD, Segal AW, et al. Mice lacking neutrophil elastase are resistant to bleomycin‐induced pulmonary fibrosis. Am J Pathol. 2007;170(1):65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Takemasa A, Ishii Y, Fukuda T. A neutrophil elastase inhibitor prevents bleomycin‐induced pulmonary fibrosis in mice. Eur Respir J. 2012;40(6):1475–82. [DOI] [PubMed] [Google Scholar]
- 150. Kim JH, Podstawka J, Lou Y, Li L, Lee EKS, Divangahi M, et al. Aged polymorphonuclear leukocytes cause fibrotic interstitial lung disease in the absence of regulation by B cells. Nat Immunol. 2018;19(2):192–201. [DOI] [PubMed] [Google Scholar]
- 151. Henry MT, McMahon K, Mackarel AJ, Prikk K, Sorsa T, Maisi P, et al. Matrix metalloproteinases and tissue inhibitor of metalloproteinase‐1 in sarcoidosis and IPF. Eur Respir J. 2002;20(5):1220–7. [DOI] [PubMed] [Google Scholar]
- 152. McKeown S, Richter AG, O'Kane C, McAuley DF, Thickett DR. MMP expression and abnormal lung permeability are important determinants of outcome in IPF. Eur Respir J. 2009;33(1):77–84. [DOI] [PubMed] [Google Scholar]
- 153. Rosas IO, Richards TJ, Konishi K, Zhang Y, Gibson K, Lokshin AE, et al. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med. 2008;5(4):e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Yamashita CM, Dolgonos L, Zemans RL, Young SK, Robertson J, Briones N, et al. Matrix metalloproteinase 3 is a mediator of pulmonary fibrosis. Am J Pathol. 2011;179(4):1733–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Zuo F, Kaminski N, Eugui E, Allard J, Yakhini Z, Ben‐Dor A, et al. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc Natl Acad Sci USA. 2002;99(9):6292–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Garcia‐Prieto E, Gonzalez‐Lopez A, Cabrera S, Astudillo A, Gutiérrez‐Fernández A, Fanjul‐Fernandez M, et al. Resistance to bleomycin‐induced lung fibrosis in MMP‐8 deficient mice is mediated by interleukin‐10. PLoS One. 2010;5(10):e13242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Drummond AH, Beckett P, Brown PD, Bone EA, Davidson AH, Galloway WA, et al. Preclinical and clinical studies of MMP inhibitors in cancer. Ann N Y Acad Sci. 1999;878:228–35. [DOI] [PubMed] [Google Scholar]