

Abstract
IL‐20 is a proinflammatory cytokine of the IL‐10 family and involved in several diseases. However, the regulatory role of IL‐20 in obesity is not well understood. We explored the function of IL‐20 in the pathogenesis of obesity‐induced insulin resistance by ELISA, Western blotting and flow cytometry. The therapeutic potential of IL‐20 monoclonal antibody 7E for ameliorating diet‐induced obesity was analysed in murine models. Higher serum IL‐20 levels were detected in obese patients. It was upregulated in leptin‐deficient (ob/ob), leptin‐resistant (db/db) and high‐fat diet (HFD)‐induced murine obesity models. In vitro, IL‐20 regulated the adipocyte differentiation and the polarization of bone marrow‐derived macrophages into proinflammatory M1 type. It also caused inflammation and macrophage retention in adipose tissues by upregulating TNF‐α, monocyte chemotactic protein 1 (MCP‐1), netrin 1 and unc5b (netrin receptor) expression in macrophages and netrin 1, leptin and MCP‐1 in adipocytes. IL‐20 promoted insulin resistance by inhibiting glucose uptake in mature adipocytes through the SOCS‐3 pathway. In HFD‐induced obesity in mice, 7E treatment reduced body weight and improved glucose tolerance and insulin sensitivity; it also reduced local inflammation and the number of M1‐like macrophages in adipose tissues. We have identified a critical role of IL‐20 in obesity‐induced inflammation and insulin resistance, and we conclude that IL‐20 may be a novel target for treating obesity and insulin resistance in patients with metabolic disorders.
Keywords: cytokines, inflammation, insulin resistance, interleukin 20, obesity
IL‐20 acts as an important regulator in obesity‐related adipose tissue inflammation and insulin resistance by regulating adipocyte differentiation and affecting macrophage recruitment and polarization.
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate transaminase
- ATMs
adipose tissue macrophages
- GM‐CSF
granulocyte–macrophage colony‐stimulating factor
- HFD
high‐fat diet
- IL‐20
interleukin 20
- LFD
low‐fat diet
- MCP‐1
monocyte chemotactic protein 1
- M‐CSF
macrophage colony‐stimulating factor
- MSCs
mesenchymal stem cells
- SVF
stromal vascular fractions
- T2DM
type 2 diabetes mellitus
- TG
triglyceride
- WAT
white adipose tissue
INTRODUCTION
The worldwide prevalence of overweight and obesity has risen remarkably over the last 30 years (WHO, 2013). Excess adiposity is a risk factor for metabolic diseases including insulin resistance, type 2 diabetes mellitus (T2DM), hypertension and nonalcoholic fatty liver disease and for several types of cancer [1, 2. Obesity is the most frequent cause of insulin resistance in humans. Consequently, the obesity epidemic is driving a parallel rise in the incidence of T2DM [3, 4.
Hypertrophied adipocytes and adipose tissue‐resident immune cells (primarily lymphocytes and macrophages) both contribute to higher proinflammatory cytokine production in the obese. The obesity‐associated state of chronic low‐grade systemic inflammation is considered a key step in the progression of insulin resistance and T2DM in humans and murine animal models [5, 6, 7, 8.
Adipose tissue macrophages (ATMs) infiltrate white adipose tissue and contribute to insulin resistance. ATMs consist of at least two different phenotypes: classically activated M1 macrophages and alternatively activated M2 macrophages. M1 ATMs produce proinflammatory cytokines, such as TNF‐α, IL‐6 and monocyte chemotactic protein 1 (MCP‐1), which promote insulin resistance [9, 10, 11. M2 ATMs are the major resident macrophages in lean adipose tissue and are characterized by the relatively high expression of CD206, arginase‐1, Mgl1 and IL‐10. Therefore, M2 ATMs are suggested to contribute in the repair and remodelling of tissue [9, 10, 11, 12, 13. Recent studies [14, 15 have identified that multiple macrophage populations existed in obese adipose tissues. These distinct populations express specific surface markers and have unique transcriptional profiles and biological functions [16, 17.
Macrophage recruitment to white adipose tissue (WAT) during the development of obesity is an important step for systemic inflammation. In vivo, the MCP‐1/CCR2 (C‐C chemokine receptor type 2) signal is important for macrophage infiltration into WAT. CCR2‐deficient mice are protected against obesity‐induced inflammation and insulin resistance [3, 12. Studies [18, 19 have reported that netrin 1 is highly expressed in human and mouse obese but not lean adipose tissue, where it directs the retention of macrophages. Netrin 1 acts as a macrophage retention signal via its receptor unc5b in obese adipocytes and induces chronic inflammation and insulin resistance.
The pleiotropic inflammatory cytokine IL‐20, a member of the IL‐10 family [20, 21, is expressed in monocytes, epithelial cells and endothelial cells. IL‐20 affects multiple cell types by activating a heterodimer receptor complex: IL‐20R1/IL‐20R2 or IL‐22R1/IL‐20R2· [22] Other studies [23, 24, 25, 26, 27 have reported that IL‐20 and its receptors are expressed on osteoclasts, osteoblasts, hepatocytes, rheumatoid synovial fibroblasts, proximal tubular epithelial cells, breast cancer cells, bladder cancer cells and oral cancer cells. IL‐20 is involved in various inflammatory diseases [28], such as psoriasis [20, 29, atherosclerosis, [28] rheumatoid arthritis [24], osteoporosis [30] and renal diseases [25, 31, 32.
We previously [24] showed that IL‐20 is regulated by hypoxia and inflammatory stimuli such as IL‐1β and LPS [33, 34. IL‐20 induces synovial fibroblasts to secrete MCP‐1, IL‐6 and IL‐8, and it acts as a proinflammatory cytokine [24]. However, little is known about the function of IL‐20 in obesity. In the present study, we investigated whether IL‐20 is involved in obesity‐induced adipose tissue inflammation and whether it promotes insulin resistance. In addition, we explored the therapeutic potential of IL‐20 monoclonal antibody for ameliorating inflammation and insulin sensitivity in obesity.
METHODS
Human participants
We recruited 105 hundred people (age range: 25–81 years old) participating in a community‐based chronic disease prevention study conducted by the Department of Family Medicine, National Cheng Kung University Hospital. Exclusion criteria were known metabolic bone diseases, taking any medications likely to affect bone mineral density (BMD), using steroids, being bedridden, being alcohol dependent or having a history of osteoporosis, hypertension, stroke, arthrosclerosis, renal disease or cancer. Twenty‐one healthy individuals (BMI: 18·5–24·9; age range: 25–49 years old), 19 patients with overweight (BMI: 25–29·9; age range: 47–77 years old), 21 patients with obese class I (BMI: 30–34·9; age range: 38–68 years old) and 19 patients with obese class II (BMI: 35–39·9; age range: 41–73) were included in the analysis. Written informed consent was obtained. The National Cheng Kung University Hospital Institutional Review Board approved the study (A‐ER‐107–377). The study was done in accordance with approved guidelines. Blood samples were collected from all participants.
Antibody preparation
Anti‐IL‐20 monoclonal antibody (7E) was generated and prepared as previously described [20, 29. Its specificity was determined as previously described [30].
Mice
Six‐week‐old C57BL/6, ob/ob and db/db mice were purchased from the National Laboratory Animal Center, Tainan, Taiwan, and kept on a 12‐hour light–dark cycle at 22 ± 2℃. All animal experiments were done in accordance with the approved guidelines of the Taiwan National Institutes of Health (Taipei), standards and guidelines for the care and use of experimental animals. The Animal Ethics Committee of National Cheng Kung University approved the research procedures (IACUC Approval No. 108089). The study was done in accordance with approved guidelines. To measure serum IL‐20 levels, C57BL/6 mice were fed a standard chow (low‐fat) diet until they turned 6 weeks old. Subsequently, they were assigned randomly to either the low‐fat diet (LFD)‐fed (10% kcal derived from fat) or HFD‐fed (60% kcal derived from fat) group (n = 6 mice/group) for 16 weeks. The ob/ob and db/db mice (n = 6 mice/group) were an LFD for 16 and 8 weeks, respectively. To evaluate the beneficial effects of 7E, C57BL/6 mice were randomly assigned to either an LFD‐fed or an HFD‐fed group for 16 weeks. The experiments began 1 week after the HFD began, and then, the mice were divided into three groups (n = 10 mice/group): HFD mice without treatment, HFD mice treated with 10 mg mIgG/kg/3 days and HFD mice treated with 10 mg 7E/kg/3 days. Body weight changes were measured weekly. At the end of the study, all the mice were anaesthetized with an overdose of pentobarbital. Serum was collected from blood that had been centrifuged at 2000 rpm for 10 min at 4℃.
Measuring biomarker
IL‐20 and leptin in human serum and the conditioned medium of SGBS cells were measured using a human IL‐20 ELISA kit (PeproTech) and a human leptin ELISA kit (R&D Systems). IL‐20 and leptin in mouse serum were determined using mouse IL‐20 and mouse leptin ELISA kits (R&D Systems). In serum, alanine aminotransferase (ALT), aspartate transaminase (AST) and triglyceride (TG) were measured using an automatic analyser (Elecsys 2010; Roche Diagnostics, Mannheim, Germany) and an electrochemiluminescent immunoassay.
RT‐qPCR analysis
Reverse transcription was done using reverse transcriptase (Clontech, BD Biosciences, Palo Alto, CA, USA). IL‐20, IL‐20R1, IL‐20R2, IL‐22R1, TNF‐α, MCP‐1, netrin 1, unc5b, DCC, leptin, MIF, FABP‐4, C/EBPα, PPARγ and SREBP‐1C expression was then amplified on a thermocycler (LC 480; Roche Diagnostics, Roche Applied Science, Indianapolis, IN, USA), with SYBR Green (Roche Diagnostics) as the interaction agent. Quantification analysis of messenger RNA (mRNA) was normalized with β‐actin, which was used as the housekeeping gene. Relative multiples of changes in mRNA expression were determined by calculating 2–ΔΔCt.
Immunohistochemistry
The white adipose tissue from HFD‐fed mice was excised, fixed in formalin overnight, embedded in paraffin and then sectioned. The immunofluorescence analyses of IL‐20, FABP‐4 (Proteintech) and F4/80 (BioLegend) were done after the adipose tissue samples had been deparaffinized, as previously described [32]. Secondary antibodies (Alexa Fluor 488, Alexa Fluor 594 and Alexa Fluor 647; Invitrogen) were then applied, and the samples were finally mounted on slides with ProLong Gold Antifade containing DAPI (Molecular Probe, Invitrogen). The procedure for immunohistochemical (IHC) staining IL‐20 has also been previously described [30]. The working concentration for primary antibody and for the control mouse IgG1 was 3 μg/ml.
Isolating adipocytes, ATMs and peritoneal exudate macrophage
The fat pads of all the mice were excised and minced in Hanks’ balanced salt solution (HBSS). Tissue suspensions were centrifuged to remove erythrocytes and free leucocytes. Thermolysin (Liberase™; Roche Applied Science) was added (1 mg/ml), and suspensions were incubated at 37℃ for 60 min with shaking. The cell suspension was filtered through a 100‐μm filter and then centrifuged to separate floating adipocytes from the stromal vascular fraction (SVF) pellet. CD11b+ ATMs were positively selected from the total SVF using the MACS Microbeads according to the manufacturer's instructions (Miltenyi Biotec, Bergisch‐Gladbach, Germany). To ensure proper isolation, adipocyte fractions were examined under a microscope before and after they were plated on plastic to detect adherent cells. Samples were digested until adipocyte fractions were free of adherent cells to ensure the recovery of the majority of the SVF population. Murine adipocytes were cultured in Dulbecco's modified Eagle's medium (DMEM)/F12 medium with 10% fetal bovine serum (FBS). The peritoneal cells collected using lavage were seeded onto 24‐well plates in RPMI‐1640 medium with 5% FBS for 4 h to allow the macrophages to adhere to the plates. Nonadherent cells were subsequently removed by washing with RPMI‐1640 medium, and the adherent macrophages were refed with RPMI‐1640 medium with 5% FBS. Macrophages were used for experiments immediately after they had been isolated.
Cell culture
Adipocytes and peritoneal exudate macrophages were isolated from LFD‐ and HFD‐fed mice. Adipocytes were incubated with IL‐20 (200 ng/ml) in serum‐free DMEM/F12 medium for 1 to 5 h. Peritoneal macrophages were incubated with IL‐20 (200 ng/ml) in serum‐free RPMI‐1640 medium for 6 h. For macrophage differentiation, primary bone marrow‐derived monocytes were prepared by flushing the bone marrow of the tibia and then culturing it for 3 h. The attached monocyte‐enriched cells were cultured in RPMI‐1640 medium supplemented with 10% FBS, penicillin (100 units/ml) and streptomycin (100 μg/ml). For M1 polarization, bone marrow‐derived monocytes treated for 4 days with granulocyte–macrophage colony‐stimulating factor (GM‐CSF) (100 ng/ml) (BD Pharmingen) were then treated with GM‐CSF (100 ng/ml), lipopolysaccharide (LPS) (100 ng/ml) and IL‐20 (200 ng/ml) or 7E (2 μg/ml) for another 3 days. For M2 polarization, bone marrow‐derived monocytes treated for 4 days with macrophage colony‐stimulating factor (M‐CSF) (100 ng/ml) (PeproTech) were then treated with M‐CSF (100 ng/ml), IL‐4 (10 ng/ml) and IL‐20 (200 ng/ml) or 7E (2 μg/ml) for another 3 days. For monocyte chemotaxis, Tamm–Horsfall protein (THP)‐1 monocytes were treated with IL‐20 (50–200 ng/ml), isotype control antibody mIgG (2 μg/ml), 7E (2 μg/ml), IL‐20 (200 ng/ml) plus mIgG (2 μg/ml) or IL‐20 (200 ng/ml) plus 7E (2 μg/ml) for 6 h and assayed using a 48‐well modified Boyden chamber housing a polycarbonate filter with 8‐μm pores (Nucleopore, Cabin John, MD) as previously described [27]. The number of migrated cells was counted. The results are expressed as a mean of 12 randomly selected fields. For adipocyte differentiation, SGBS preadipocytes (a gift kindly provided by Prof Dr Martin Wabitsch, University of Ulm, Germany) were differentiated into mature adipocytes as previously described [35]. SGBS preadipocytes were incubated with IL‐20 (200–800 ng/ml) in differentiation medium for 14 days, and then, the leptin level was measured. For hypoxia experiments, mature SGBS adipocytes were cultured under hypoxia conditions as previously described [33]. Mature SGBS adipocytes were treated with phosphate‐buffered saline (PBS), mIgG (2 μg/ml) or 7E (2 μg/ml) under hypoxic conditions for 96 h.
Oil Red O staining
Oil Red O stock solution (Sigma‐Aldrich) was diluted in distilled water (3:2 v/v) before staining the adipocyte samples. The cells were washed with PBS, fixed with buffered 4% formaldehyde for 20 min, rinsed with distilled water and stained with Oil Red O solution for 20 min. The cells were then washed twice with water. Pictures were taken using an Olympus microscope (Tokyo, Japan). The stain was extracted for quantification 5 min after isopropanol had been added. Equal amounts of elution from each well were transferred to a 96‐well plate, and, using a microplate reader (Multiskan Spectrum; Thermo Scientific, Vantaa, Finland), the absorbance values were measured at a wavelength of 492‐nm.
Western blotting
SGBS mature adipocytes were stimulated with IL‐20 (200 ng/ml) for the indicated time‐points. Cell lysates were prepared using cell lysis buffer (Cell Signaling Technology, Beverly, MA, USA) and separated using SDS–PAGE. Western blotting was done with antibodies specific for phosphor‐ERK1/2, SOCS‐1, SOCS‐3, phosphor‐AKT, AKT (Cell Signaling Technology), FABP‐4, C/EBPα and PPARγ (Proteintech). β‐Actin (GeneTex) was used as a loading control and detected using a β‐actin‐specific antibody.
Flow cytometry
Single‐cell suspensions were prepared in ice‐cold PBS containing 5% FBS and 2 mM EDTA. Cells (1 × 106) were labelled with the indicated antibodies (FITC‐conjugated CD206; Bio‐Rad); (PE‐conjugated CD11c; BD Biosciences); (APC‐conjugated F4/80 and PE‐conjugated CD80; Biolegend) at 4℃ for 30 min. The cells were washed twice and analysed using a flow cytometer (FACSCalibur; BD Biosciences). WinMDI 2·8 software was used for data acquisition and analysis.
Glucose and insulin tolerance test
HFD‐fed mice (n = 10) were given a glucose tolerance test was done at week 15 after mIgG or 7E treatment. The mice were intraperitoneally injected with a 2% glucose solution (2 g/kg body weight). Blood samples were taken from the tail veins at 0 (before the glucose injection), 30, 60, 90 and 120 min after the injection, and blood glucose was measured using ACCU‐CHEK® (Roche Diagnostics). For the insulin tolerance test, the mice fasted for 12 h, and were then intraperitoneally injected with recombinant insulin (1 unit/kg). Blood glucose was similarly measured.
Glucose uptake assay
Glucose uptake was measured using a colorimetric assay according to the manufacturer's instructions (Glucose Uptake Colorimetric Assay Kit; BioVision, Milpitas, CA, USA). Briefly, differentiated mature SGBS adipocytes were starved and incubated with IL‐20 (200 ng/ml) in serum‐free medium for 16 h. They were then washed with PBS, and glucose uptake was initiated by incubating them in Krebs–Ringer–phosphate–HEPES (KRPH) buffer containing 2% BSA for 40 min, stimulated or not with 1 μM of insulin for 20 min to activate glucose transporter, 10 μl of 10 mM 2‐deoxyglucose (2‐DG) was added, and then, the cells were incubated for 20 min. Using a microplate reader, the absorbance values were measured at 37℃ at a wavelength of 492 nm.
Generating stable cell lines
To generate a stable clone of mouse bone marrow mesenchymal stem cell (MSC) expressing IL‐20 mAb 7E, heavy‐chain (HC) and light‐chain (LC) fragments of 7E were constructed into pLAS5w. PeGFP‐I2‐Bsd and pLAS5w. PtRFP‐I2‐Puro lentivirus vector (National RNAi Core Facility, Taiwan), respectively. The 7E‐HC and 7E‐LC plasmids were, respectively, co‐transfected with packing plasmids pMD.G and pCMVΔR8·91 into 293T cells using lipofectamine 2000 (Thermo Fisher Scientific) to produce virus‐containing medium. Virus particles were collected from the medium 48 h after the transfection. MSCs were infected with virus particles carrying 7E‐HC and 7E‐LC and then selected for monoclonal cells with GFP and RFP signals after two weeks of blasticidin and puromycin selection. ELISA and Western blotting confirmed that MSC‐expressing 7E stable clones recognized human, mouse and rat IL‐20.
Statistical analysis
The correlation between IL‐20 and leptin was analysed using SPSS 15·0 for Windows. Prism 7·0 (GraphPad Software, San Diego, CA, USA) was also used for the statistical analysis. A one‐way analysis of variance (ANOVA) nonparametric Kruskal–Wallis test was used to compare the data between groups. Post hoc comparisons were done using Dunn's multiple comparison test. Significance was set at p < 0·05.
RESULTS
Higher serum IL‐20 levels in obese patients and in obese mice
We analysed the IL‐20 serum levels in obese patients and compared them with those of healthy controls. Twenty‐one healthy controls (BMI: 18·5–24·9; age range: 25–49 years old), 19 overweight patients (BMI: 25–29·9; age range: 47–77 years old), 21 obese class I patients (BMI: 30–34·9; age range: 38–68 years old) and 19 obese class II patients (BMI: 35–39·9; age range: 41–73 years old) were included in the analysis. The serum IL‐20 level in overweight and obese patients was significantly higher than that in healthy controls (p < 0·05 and p < 0·01, respectively) (Figure 1a). The mean levels of serum IL‐20 were 99·9 pg/ml in the healthy controls, and 653·7 pg/ml in the overweight, 916·8 pg/ml in the obese class I, and 1275·2 pg/ml in the obese class II patients. To clarify the clinical correlation between IL‐20 and leptin in obese patients, we also analysed the serum leptin level, which was significantly higher in overweight and obese patients than those of healthy controls (p < 0·01) (Figure 1b). In addition, linear regression analysis showed that serum IL‐20 was positively correlated with leptin in obese patients (r = 0·7195, p < 0·01) (Figure 1c).
FIGURE 1.
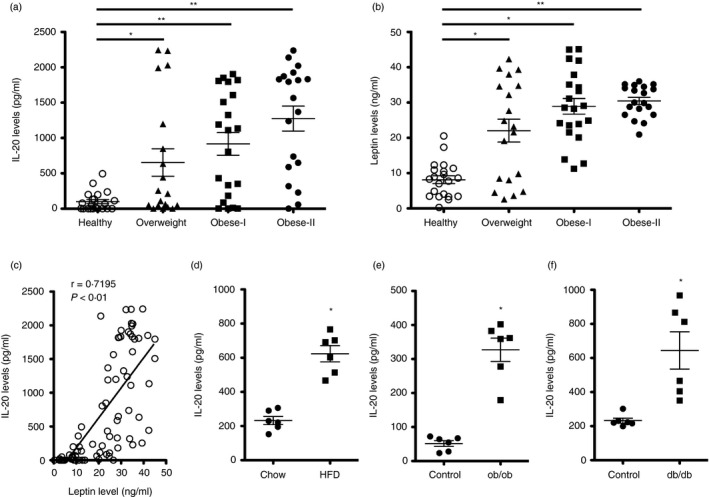
Higher serum IL‐20 levels in obese patients and obese mice. (a‐b) Serum IL‐20 and leptin levels from 21 healthy controls (BMI: 18·5–24·9; age range: 25–49 years), 19 overweight patients (BMI: 25–29·9; age range: 47–77 years), 21 class I obese patients (BMI: 30–34·9; age range: 38–68 years) and 19 class II obese patients (BMI: 35–39·9; age range: 41–73 years) were analysed using ELISA. Horizontal lines represent means. Data are expressed as mean ±SD of triplicate samples from a single experiment and are representative of three independent experiments. *p < 0·05, **p < 0·01 compared with healthy controls. (c) Linear regression analysis of the correlation of serum IL‐20 and leptin levels in obese patients (r = 0·7195, p < 0·01). Data are expressed as mean ±SEM. (d) Serum IL‐20 levels in low‐fat diet (LFD) mice (fed standard mouse chow) (n = 6) or a high‐fat diet (HFD (n = 6) for 16 weeks. (e‐f) Serum IL‐20 levels in leptin (ob/ob)‐ and leptin receptor (db/db)‐deficient LFD mice (n = 6 mice per group) for 16 and 8 weeks, respectively. Horizontal lines represent means. Data are expressed as mean ±SD and are representative of three independent experiments. *p < 0·05 compared with control mice
Based on the results from studies of obese patients, we investigated whether IL‐20 was also involved in diet‐induced obese of high‐fat diet (HFD)‐fed (60% kcal derived from fat) C57BL/6 mice and in genetically obese leptin (ob/ob)‐ and leptin receptor (db/db)‐deficient mice. ELISA showed that the serum IL‐20 level of HFD‐fed mice was significantly higher than those fed a normal chow diet: low‐fat diet (LFD) (10% kcal derived from fat)‐fed mice (p < 0·01) (Figure 1d). Leptin‐deficient (ob/ob) mice spontaneously became obese, even when kept on an LFD; we used them as a model for genetic obesity. ELISA showed that serum IL‐20 level was significantly higher in ob/ob mice than in age‐matched control mice (p < 0·05) (Figure 1e). Leptin receptor (db/db)‐deficient mice developed T2DM and obesity. In this genetic background, serum IL‐20 level was also significantly higher in db/db mice than in age‐matched control mice (p < 0·05) (Figure 1f).
IL‐20 expression increased in the adipocytes and ATMs of obese mice
To identify IL‐20 and its receptors (IL‐20R1, IL‐20R2 and IL‐22R1) regulated by obesity, we used reverse transcription quantitative real‐time PCR (RT‐qPCR) to profile the gene expression of adipose tissue from LFD‐ and HFD‐fed C57BL/6 mice for 16 weeks. IL‐20 mRNA expression was significantly (p < 0·05) higher in visceral white adipose tissue (WAT) from HFD‐fed obese mice than in that from LFD‐fed healthy controls (Figure 2a). In addition, the expression of IL‐20R2 mRNA was significantly (p < 0·0.5) higher, and IL‐22R1 was significantly lower in the adipose tissue of obese mice compared with that of lean mice, but IL‐20R1 expression was not significantly different between groups (Figure 2a). To clarify the cellular compartment expressing IL‐20, we isolated mRNA from the adipocyte, stromal vascular fractions (SVF) and adipose tissue macrophages (ATMs) of the visceral WAT of chow‐ and HFD‐fed mice and did an RT‐qPCR analysis. The expression of IL‐20 was higher in HFD‐fed mice than in that from LFD‐fed mice (p < 0·01) (Figure 2b). Consistent with this, immunofluorescence staining of the visceral WAT from HFD‐fed mice showed, in adipocytes, IL‐20 reactivity (green) that colocalized with staining for the adipocyte marker fatty acid binding protein 4 (orange; FABP‐4) (Figure 2c). Furthermore, IL‐20 (green) was also strongly stained in crown‐like structures that colocalized with staining for the macrophage marker F4/80 (red) (Figure 2d). We did not detect IL‐20 staining in visceral WAT from LFD‐fed mice (Figure S1).
FIGURE 2.
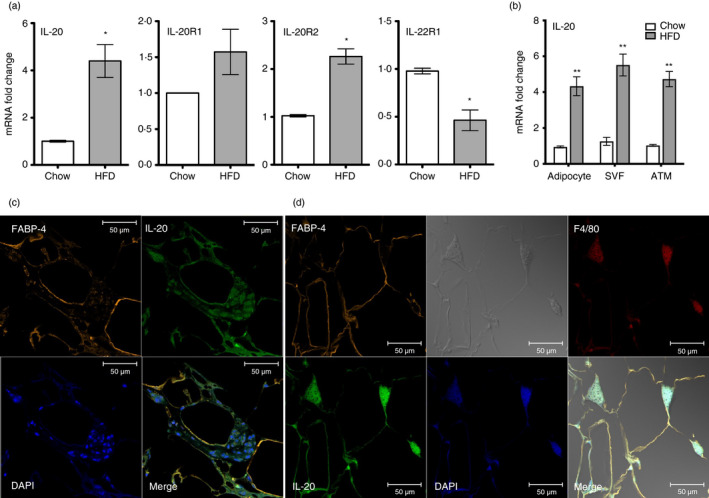
IL‐20 was upregulated in visceral white adipose tissue and macrophages in obese mice. (a) RT‐qPCR analysis of IL‐20, IL‐20R1, IL‐20R2 and IL‐22R1 mRNA expression changes in the visceral white adipose tissue (WAT) of HFD mice (n = 6) for 16 weeks and LFD mice (n = 6) for 16 weeks. (b) Relative mRNA expression of IL‐20 in the adipocytes and stromal vascular fractions (SVFs) isolated from the visceral WAT of LFD mice (n = 5) and HFD mice (n = 5) for 16 weeks. (c) Immunofluorescence staining for the adipocyte markers FABP‐4 (orange), IL‐20 (green) and DAPI (blue) in the visceral WAT of HFD mice. Co‐localization of IL‐20 with FABP‐4 is shown in yellow in the merged image. Scale bars =50 μm. (d) Immunofluorescence staining for the adipocyte markers FABP‐4 (orange), IL‐20 (green), the macrophage marker F4/80 (red) and DAPI (blue) in the visceral WAT of HFD mice. Co‐localization of IL‐20 with FABP‐4 and F4/80 is shown in yellow in the merged image. Scale bars =50 μm. Data in (a) and (b) are expressed as mean ±SEM *p < 0·05, **p < 0·01. Data are representative of three independent experiments
IL‐20 enhanced macrophage retention and monocyte chemotaxis
In obesity, cytokine and chemotactic signals originating from inflamed adipose tissue lead to monocyte infiltration, polarization of proinflammatory macrophages, tissue inflammation and insulin resistance [36]. IL‐20 was highly expressed in adipocytes and macrophages of inflamed adipose tissue in obese mice. To investigate the effect of IL‐20 on macrophages in obesity, we collected peritoneal exudate macrophages isolated from LFD‐ and HFD‐fed mice and performed RT‐qPCR analysis after IL‐20 treatment. TNF‐α and MCP‐1 mRNA expression was significantly (p < 0·05) higher in HFD‐fed mice (Figure 3a,b), whose gene products were recently reported to promote monocyte recruitment into adipose tissue during HFD feeding [7]. IL‐20 expression was also significantly (p < 0·05) upregulated after IL‐20 treatment but observed only in the peritoneal exudate macrophage isolated from HFD‐fed mice (Figure 3c). Furthermore, RT‐qPCR analysis showed that the mRNA expression of netrin 1 and its chemorepulsive receptor unc5b was significantly (p < 0·05) higher in IL‐20‐treated peritoneal exudate macrophage of obese mice than in lean mice (Figure 3d,e). The expression of DCC, netrin 1’s chemoattractive receptor, was also significantly (p < 0·01) higher in the peritoneal exudate macrophage of HFD‐fed mice (Figure 3f).
FIGURE 3.
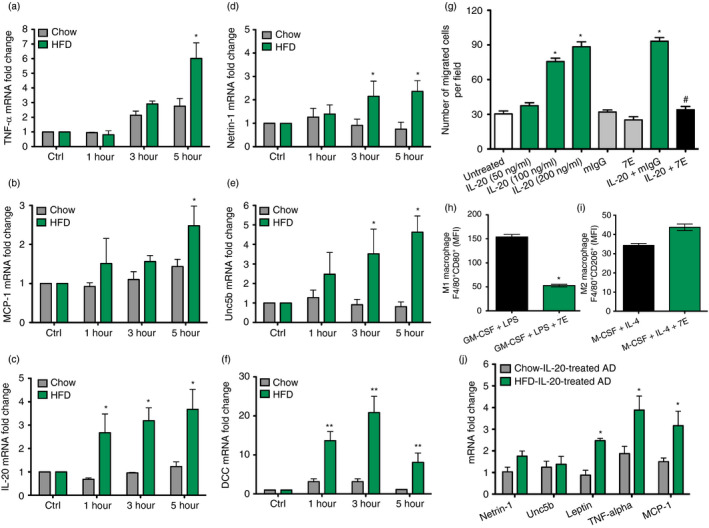
IL‐20 enhanced macrophage retention and monocyte chemotaxis. (a‐f) The peritoneal exudate macrophages were isolated after 16 weeks from LFD and HFD mice (n = 6 mice per group). Peritoneal exudate macrophages were treated with IL‐20 (200 ng/ml) for the indicated time periods. TNF‐α, MCP‐1, IL‐20, netrin 1, unc5b and DCC mRNA expression changes were analysed using RT‐qPCR with specific primers. *p < 0·05, **p < 0·01 (g) The migration of THP‐1 monocytes treated with IL‐20 (50–200 ng/ml), isotype control antibody mIgG (2 μg/ml), 7E (2 μg/ml), IL‐20 (200 ng/ml) plus mIgG (2 μg/ml) or IL‐20 (200 ng/ml) plus 7E (2 μg/ml) was analysed using a modified Boyden chamber. The number of migrated cells was counted. The results are expressed as a mean of 12 randomly selected fields. *p < 0·05 compared with untreated controls, #p < 0·05 compared with IL‐20 plus mIgG‐treated group. (h‐i) Bone marrow cells were incubated with GM‐CSF/LPS plus 7E for M1 macrophage differentiation or incubated with M‐CSF/IL‐4 plus 7E for M2 macrophage differentiation. The M1 type (F4/80+CD80+) and M2 type (F4/80+CD206+) macrophages were measured in mean fluorescence intensity (MFI) units using FACS analysis. *p < 0·05 compared with GM‐CSF plus LPS‐treated group. (j) The adipocytes were isolated after 16 weeks from the visceral WAT of LFD and HFD mice (n = 6 mice per group). The isolated adipocytes were treated with IL‐20 (200 ng/ml) for 6 h. Relative mRNA expression of netrin 1, unc5b, leptin, TNF‐α and MCP‐1 was analysed using RT‐qPCR with specific primers. *p < 0·05. Data are expressed as mean ±SEM and are representative of three independent experiments
Monocytes are circulatory precursor cells from myeloid origin that can develop into macrophages upon migration from the blood stream to tissues. To determine whether IL‐20 affected monocyte chemotaxis, we treated THP‐1 cells with IL‐20 to evaluate migration ability. A Boyden chamber assay showed that THP‐1 cell migration was significantly (p < 0·05) dose dependently higher in IL‐20‐treated cells but inhibited in 7E‐treated cells (Figure 3g).
M1 polarization of macrophages was suppressed in IL‐20‐depleted cells
We next investigated the function of IL‐20 in macrophage cell differentiation. Bone marrow cells from WT mice were incubated with GM‐CSF in vitro for 7 days. The cells were activated with LPS plus IL‐20 for M1 macrophage differentiation and examined for the F4/80+CD80+ cells using FACS analysis. We also incubated bone marrow cells with M‐CSF and then activated with IL‐4 plus IL‐20 for M2 macrophage differentiation and examined them for the F4/80+CD206+ cells. M1 and M2 macrophage polarization was not significantly affected by IL‐20 treatment (Figure S2). We hypothesized that an endogenously expressed basal level of IL‐20 may be sufficient to affect macrophage polarization. To test whether IL‐20 depletion modulates macrophage polarization, we treated bone marrow cells with GM‐CSF and then activated them with LPS in 7E‐treated cells. FACS analysis showed that M1 macrophage polarization was significantly (p < 0·05) inhibited (Figure 3h) and that M2 macrophage polarization was insignificantly affected (Figure 3i). In GM‐CSF+LPS‐induced M1 macrophage polarization, we further confirmed that most of cells were alive and F4/80‐positive (more than 90%) after 7E treatment. However, they did not express CD80 and/or CD206 marker, which suggested that these F4/80+ cells did not differentiate into M1‐like or M2‐like macrophages (Figure S3a). RT‐qPCR also showed that IL‐20 level was inhibited in GM‐CSF+LPS+7E‐treated group compared with GM‐CSF+LPS‐treated group (Figure S3b).
Leptin, TNF‐α and MCP‐1 levels were higher in IL‐20‐treated inflamed adipocytes
To investigate the role of IL‐20 on the adipocytes in the local inflammation in the obese state, the adipocytes isolated from visceral WAT from LFD‐ and HFD‐fed mice were incubated with IL‐20. RT‐qPCR showed significantly (p < 0·05) higher levels of leptin, TNF‐α and MCP‐1 mRNA expression in HFD‐fed mice than in LFD‐fed mice (Figure 3j).
IL‐20 expression in hypoxic adipocytes
We cultured differentiated mature Simpson–Golabi–Behmel syndrome (SGBS) adipocytes under normoxic and hypoxic conditions for 24 to 96 h and analysed IL‐20 protein levels. Western blotting and immunohistochemical (IHC) staining showed significantly greater IL‐20 production in mature SGBS cells under hypoxic conditions (Figure 4a,b), which was confirmed using ELISA (Figure 4c).
FIGURE 4.
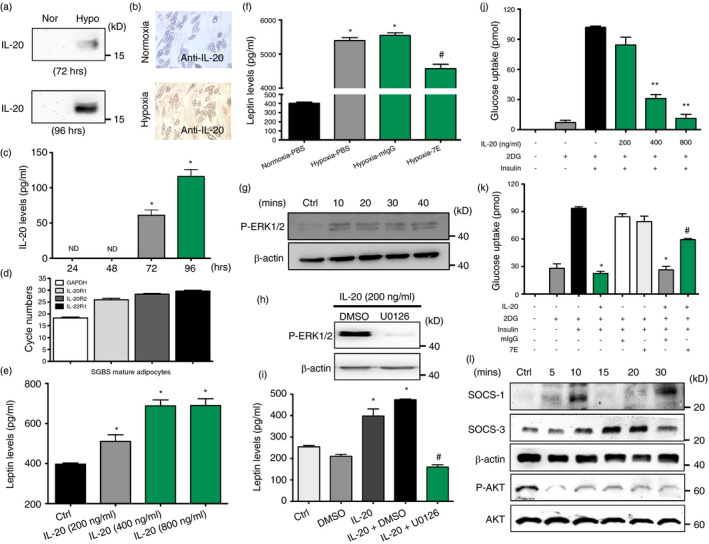
Functions of IL‐20 in SGBS adipocytes. (a) Western blotting of IL‐20 in the conditioned medium under normoxic and hypoxic conditions for the indicated time periods. (b) IHC staining of IL‐20 in mature SGBS cells under hypoxic conditions for 4 days. (c) The level of IL‐20 in mature SGBS cells under hypoxic conditions for 24–96 h was measured using ELISA. *p < 0·05 compared with hypoxia 24 h. ND =nondetectable. (d) The expression of IL‐20R1, IL‐20R2 and IL‐22R1 mRNA in mature SGBS adipocytes was analysed using RT‐qPCR. (e) The level of leptin in IL‐20‐treated SGBS cells (200–800 ng/ml for 14 days in differentiation medium) was measured using ELISA. *p < 0·05 compared with untreated controls. (f) The level of leptin in mIgG‐ or 7E‐treated mature SGBS cells under hypoxic conditions for 96 h was measured using ELISA. *p < 0·05 compared with normoxic controls, #p < 0·05 compared with mIgG‐treated group. (g) Mature SGBS cells were incubated with IL‐20 (200 ng/ml) for the indicated time periods, and then, cell lysates were collected and analysed using immunoblotting with specific antibodies against phospho‐ERK1/2. β‐Actin was an internal control. (h) Western blot of phospho‐ERK1/2 and β‐actin in mature SGBS cells treated with IL‐20 (200 ng/ml) in the presence of the ERK1/2 inhibitor U0126 (10 μM) or vehicle (DMSO). (i) Leptin level in culture supernatants of mature SGBS cells treated with IL‐20 (200 ng/ml) in the presence of the ERK1/2 inhibitor U0126 (10 μM) or vehicle (DMSO). *p < 0·05 compared with untreated controls, #p < 0·05 compared with IL‐20 plus DMSO‐treated group. (j‐k) The differentiated SGBS mature adipocytes were incubated with IL‐20 (200–800 ng/ml), mIgG (8 μg/ml), 7E (8 μg/ml), IL‐20 (800 ng/ml) plus mIgG (8 μg/ml) or IL‐20 (800 ng/ml) plus 7E (8 μg/ml) in serum‐free medium for 24 h, and then, 2‐deoxyglucose uptake was assessed using glucose uptake assay. *p < 0·05, **p < 0·01 compared with untreated controls, #p < 0·05 compared with IL‐20 plus mIgG‐treated group. (l) Western blot of SOCS‐1, SOCS‐3, β‐actin phospho‐AKT and AKT in mature SGBS cells treated with IL‐20 (200 ng/ml) for the indicated time periods. Data in (c), (e), (f), (i), (j) and (k) are expressed as mean ±SEM and are representative of three independent experiments
Leptin secretion was regulated in IL‐20‐treated adipocytes
RT‐qPCR showed that all three IL‐20 receptors (IL‐20R1, IL‐20R2 and IL‐22R1) were expressed in mature adipocytes (Figure 4d). We incubated SGBS preadipocytes with IL‐20 for 14 days in adipocyte differentiation medium. ELISA showed that leptin production significantly (p < 0·05) increased in IL‐20‐treated SGBS adipocytes (Figure 4e). We investigate whether leptin regulates the expression of IL‐20 and found that leptin did not regulate IL‐20 level in SGBS adipocytes (Figure S4). To explore whether IL‐20 promotes leptin secretion in an autocrine manner, we incubated mature 7E‐treated SGBS adipocytes under hypoxic conditions. ELISA showed that leptin secretion was lower after 7E treatment (Figure 4f). To determine the mechanism used to regulate leptin secretion, we assessed the ERK1/2 signal molecule. Western blotting showed that ERK1/2 was activated in IL‐20‐treated cells; the activity was inhibited in cells pretreated with U0126 (1,4‐diamino‐2,3‐dicyano‐1,4‐bis[2‐aminophenylthio] butadiene), an ERK1/2 inhibitor (Figure 4G‐H g‐h). ELISA also confirmed that leptin secretion was completely abolished in cells pretreated with U0126 (Figure 4i).
Insulin‐stimulated glucose uptake was negatively modulated in IL‐20‐treated adipocytes
To determine whether IL‐20 modulates insulin‐stimulated glucose transport using an autocrine/paracrine mechanism, we performed glucose uptake assay. SGBS adipocytes were incubated with IL‐20 and measured the uptake of 2‐deoxyglucose in the basal state and after insulin stimulation. A glucose uptake assay showed that IL‐20 dose dependently inhibited insulin‐stimulated glucose uptake (Figure 4j). Glucose uptake was not inhibited in cells treated with IL‐20 and 7E, but it was inhibited in cells treated with IL‐20 and mIgG (Figure 4k). These results clearly demonstrated that IL‐20 regulates insulin sensitivity and glucose uptake in the SGBS adipocytes in autocrine/paracrine manner.
To determine whether IL‐20 increases insulin‐stimulated glucose uptake by regulating SOCS expression and AKT pathway, mature SGBS adipocytes were treated with IL‐20, and cell lysate was used for Western blotting with an antibody specifically against SOCS‐1, SOCS‐3 and AKT. The production of SOCS‐3 was higher in IL‐20‐treated cells. The phosphorylation of AKT was decreased at 5 min after IL‐20 treatment (Figure 4l).
Adipogenesis was higher in IL‐20‐treated SGBS cells
To explore the effect of IL‐20 on adipogenesis, SGBS preadipocytes were differentiated with IL‐20 (200 to 800 ng/ml) for 14 days (from day 0 to day 14) in addition to the standard differentiation factors. Adipocyte differentiation was dose dependently induced by IL‐20 (Figure 5a). The OD value of Oil Red O eluted solution was higher in IL‐20‐treated than in PBS‐treated cells (Figure 5b). To further delineate the role of IL‐20 in adipogenesis, we used mesenchymal stem cells (MSCs) isolated from mouse bone marrow to evaluate whether neutralization of IL‐20 regulates the differentiation of adipocytes from multipotent stem cells. RT‐qPCR showed that IL‐20 and its 3 receptors were expressed in MSCs (Figure S5). We generated two MSC stable clones constantly expressing IL‐20 mAb 7E using a lentivirus delivery system (Figure 5c). MSCs, MSC transfected with control empty vector and two MSC clones expressing 7E were cultured under adipocyte differentiation conditions for 14 days. Oil Red O staining showed that the adipocyte differentiation was significantly inhibited in two MSC clones expressing 7E compared with the control MSC clone (Figure 5d). These results demonstrated that IL‐20 promoted adipocytes differentiation which was potently inhibited by 7E.
FIGURE 5.
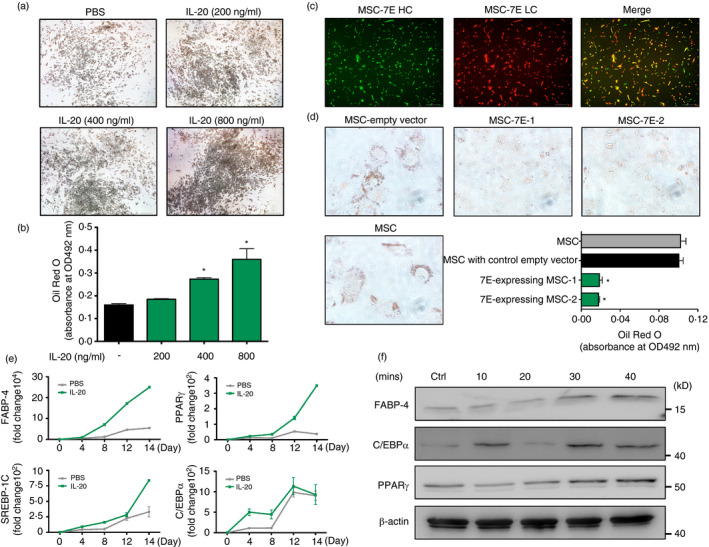
Adipogenesis was higher in IL‐20‐treated SGBS cells. (a) SGBS preadipocytes were incubated for 14 days in differentiation medium with IL‐20 (200–800 ng/ml). The lipid droplets were stained with Oil Red O. (b) The OD value of Oil Red O eluted solution, which represents lipid droplet accumulation in the cytoplasm. *p < 0·05 compared with untreated controls. (c) Expression of 7E heavy chain (HC) (green) and 7E light chain (LC) (red) in mouse mesenchymal stem cells (MSCs) using a lentivirus delivery system. Co‐expression of 7E‐HC and 7E‐LC is shown in yellow in the merged image. (d) Oil Red O staining of MSC/vector, MSC and MSC/7E under adipocyte differentiation medium for 14 days. OD value of Oil Red O eluted solution, which represents lipid droplet accumulation in the cytoplasm. *p < 0·05 compared with MSC/vector. (e) SGBS preadipocytes were incubated for 14 days in differentiation medium with PBS or IL‐20 (200 ng/ml). The mRNA of the treated cells was isolated and analysed using RT‐qPCR with specific primers. Data are representative of three independent experiments. (f) Mature SGBS cells were incubated with IL‐20 (200 ng/ml) for the indicated time periods, and then, cell lysates were collected and analysed using immunoblotting with specific antibodies against FABP‐4, C/EBPα and PPARγ. β‐actin was an internal control. Data in (b) and (d) are expressed as mean ±SD and are representative of three independent experiments
Transcription factors associated with adipogenesis were regulated in IL‐20‐treated adipocytes
We next tested whether IL‐20 promoted adipogenesis‐related factors. SGBS preadipocyte cells were incubated with IL‐20 for 14 days under standard adipocyte differentiation conditions. RT‐qPCR showed that FABP‐4, PPARγ, SREBP‐1C and C/EBPα transcripts were significantly higher in IL‐20‐treated cells at different time‐points (Figure 5e), which indicated that IL‐20 regulated adipogensis‐related factors during adipocyte maturation. We used Western blotting to confirm FABP‐4, PPARγ and C/EBPα (Figure 5f).
Obesity‐induced insulin resistance was ameliorated in 7E‐treated mice
We generated HFD‐induced obese mice to examine the beneficial effects of 7E on obesity‐induced inflammation and insulin resistance. C57BL/6 male mice were fed LFD until they were 6 weeks old. Subsequently, they were randomly assigned to the LFD or HFD group for 16 weeks. The treatment began after the 1‐week HFD, at which time the mice were divided into three groups: (i) HFD mice without treatment, (ii) HFD mice treated with 10 mg of nonspecific mIgG/kg/3 d and (iii) HFD mice treated with 10 mg of 7E/kg/3 d. Changes in body weight were measured weekly. Body weights were not significantly different between groups at the beginning, but after 16 weeks, body weight was significantly lower in the 7E‐treated mice than in the mIgG‐treated mice (Figure 6a), despite equivalent total caloric intakes (Figure 6b). Moreover, glucose and insulin tolerance tests showed that 7E‐treated mice had improved glucose homeostasis and insulin responsiveness than did mIgG‐treated mice (Figure 6c,d), and lower IL‐20 and leptin concentrations (Figure 6e,f). Fat pad weight (Figure 6g) and serum triglyceride, ALT and AST were also lower in the 7E‐treated mice (Figure 6h‐j). Haematoxylin and eosin staining showed that hepatic steatosis was also lower in the 7E‐treated mice (Figure S6). RT‐qPCR showed that IL‐20 expression was upregulated in liver, kidney and especially in adipose tissue of the mIgG‐treated mice but significantly downregulated in the 7E‐treated mice (Figure 6k). These data also suggested that the highest levels of IL‐20 were produced in adipose tissue based on our RT‐qPCR analysis.
FIGURE 6.
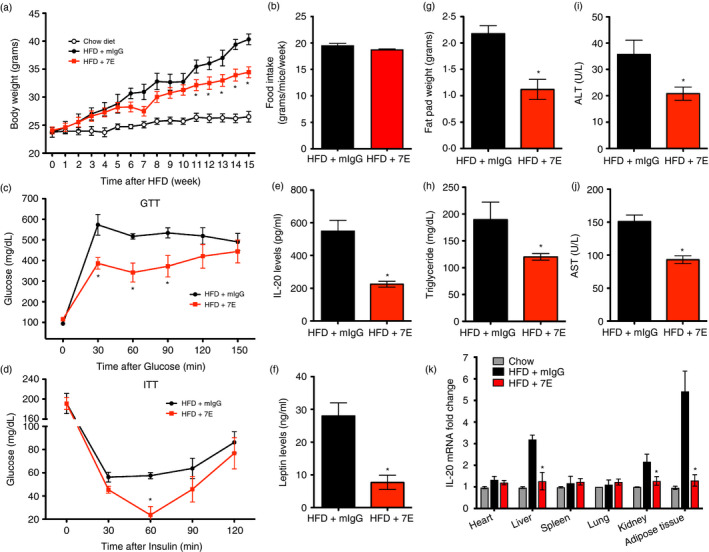
Obesity‐induced insulin resistance was ameliorated in 7E‐treated mice. Male C57BL/6 mice were randomly assigned to the LFD (10% kcal derived from fat) or the HFD (60% kcal derived from fat) group for 16 weeks. The experiments began 1 week after HFD and the mice were divided into three groups (n = 10 mice per group): HFD mice without treatment, and HFD mice treated with 10 mg mIgG/kg/3 d, or 10 mg 7E/kg/3 d. (a‐b) Body weight and food intake were measured weekly. (c) A glucose tolerance test (GTT) and (d) an insulin tolerance test (ITT) were performed at week 15 after mIgG or 7E treatment. The fat pad weight (g), serum IL‐20 (e), leptin (f), triglyceride (h), ALT (i) and AST (j) levels of mIgG‐ and 7E‐treated HFD mice were measured. (k) The expression of IL‐20 in different organs and adipose tissue of mIgG‐ and 7E‐treated HFD mice was analysed using RT‐qPCR. Data are expressed as mean ±SEM and are representative of three independent experiments. *p < 0·05 compared with mIgG‐treated HFD mice
Adipose tissue inflammation was ameliorated in 7E‐treated mice
We analysed the macrophages isolated from adipose tissue of 7E‐treated mice. The number of total macrophages (F4/80+) was lower in 7E‐treated mice (Figure 7a), and FACS analysis confirmed fewer M1 macrophage (F4/80+CD11c+CD206−) (Figure 7b), but there was no change in the number of M2 macrophages (F4/80+CD11c−CD206+) in 7E‐treated mice compared with mIgG‐treated mice (Figure 7c). The inflammation reduction in the adipose tissue of the 7E‐treated mice was confirmed using RT‐qPCR analysis (Figure 7d), and IHC staining showed fewer IL‐20‐positive cells and crown‐like structures in the adipose tissue of the 7E‐treated mice (Figure 7e).
FIGURE 7.
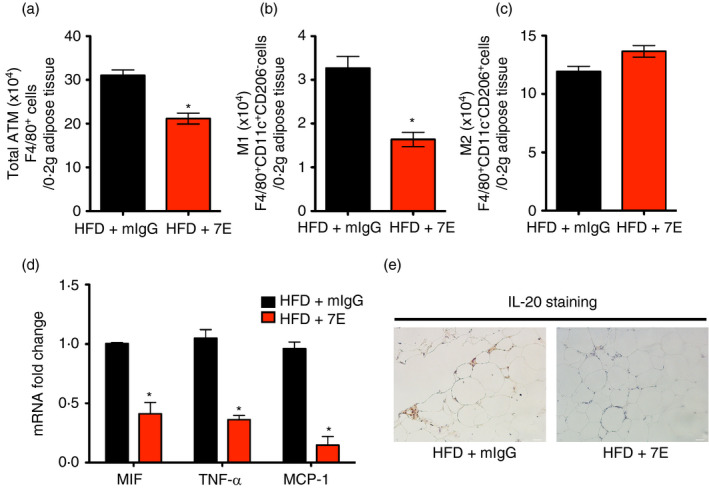
Obesity‐induced adipose tissue inflammation was reduced in 7E‐treated mice. Male C57BL/6 mice were randomly assigned to the LFD (10% kcal derived from fat) or the HFD (60% kcal derived from fat) group for 16 weeks. The experiments began 1 week after HFD and the mice were divided into three groups (n = 10 mice per group): HFD mice without treatment, and HFD mice treated with 10 mg mIgG/kg/3 d or 10 mg 7E/kg/3 d. The number of ATMs (a), M1 macrophages (b) and M2 macrophages (c) in adipose tissue isolated from mIgG‐treated and 7E‐treated HFD mice were calculated using FACS analysis. (d) RT‐qPCR analysis of MIF, TNF‐α and MCP‐1 mRNA expression in adipose tissue isolated from mIgG‐treated and 7E‐treated HFD mice. (e) IHC staining of IL‐20 in adipose tissue section from mIgG‐treated and 7E‐treated HFD mice. Data are expressed as mean ±SEM and are representative of three independent experiments. *p < 0·05 compared with mIgG‐treated HFD mice
DISCUSSION
Adipose tissue inflammation is a key process in the development of obesity‐induced insulin resistance. We demonstrate here that IL‐20 was upregulated in ATMs and adipocytes of the visceral WAT in obese patients. IL‐20 promoted adipose tissue inflammation, macrophage chemotaxis, and systemic insulin resistance. Moreover, IL‐20 mAb 7E treatment in vivo reduced local and systemic inflammation, insulin resistance, and ATM accumulation in visceral WAT. Collectively, these findings suggest that IL‐20 secretion by ATMs promotes their retention in visceral WAT and fosters the chronic inflammation that leads to metabolic disorder.
We showed that IL‐20 was higher in obese patients, which is consistent with the study [37] reporting that IL‐20 levels are higher in premenopausal obese women and are associated with body weight and waist–hip ratio. We also found that IL‐20 was positively correlated with leptin in obese patients. The level of IL‐20 was higher in leptin‐deficient (ob/ob), leptin‐resistant (db/db) and HFD‐induced murine models of obesity. These findings indicated that IL‐20 is involved in obesity by overnutrition through HFD feeding and it is associated with genetically‐induced obesity. One study [38] reported that the level of leptin in the serum and adipose tissues increased in response to TNF and LPS. We found that IL‐20 tightly regulated the leptin level. Overeating and obesity lead to hypothalamic inflammation, which, in turn, causes the dysfunction of hypothalamic neurons [39, 40. Recent studies [41, 42 reported that hypothalamic inflammation leads to experimental obesity, resistance to leptin, peripheral insulin resistance, and dysregulation of food intake and energy expenditure.
Obesity is associated with the accumulation of macrophages in adipose tissue. These cells secrete proinflammatory molecules—TNF‐α, IL‐1β and MCP‐1—that contribute both to local and systemic inflammation, thus potentiating insulin resistance. We found that the cellular sources of IL‐20 were ATM and adipocytes, which are mediated under hypoxic conditions in obese organisms. IL‐20 promoted IL‐20, TNF‐α and MCP‐1 expression in peritoneal exudate macrophages isolated from HFD, but not LFD mice; IL‐20 increased monocyte chemotaxis in vitro. Taken together, these findings suggest that IL‐20 is chemotactic for macrophage accumulation in obese organisms. Additionally, IL‐20 increased the expression of netrin 1 and unc5b expression, which contributed to macrophage retention in adipose tissues in obese mice.
Despite the importance of ATMs in adipose tissue inflammatory responses and systemic insulin sensitivity, the mechanisms underlying M1 versus M2 macrophage polarization are still poorly understood. In our bone marrow‐derived macrophage in vitro culture system, 7E significantly suppressed M1 macrophage polarization, which indicated that IL‐20 is involved in regulating macrophage polarization. 7E treatment had the same effects in obese HFD mice, which provides additional support that IL‐20 regulates the cellular balance between the M1 and M2 macrophages in obese mice model. Furthermore, MCP‐1 binding to its receptor, CCR2, plays an important in a variety of infectious and inflammatory diseases [43]. The MCP‐1/CCR2 system contributes to monocyte migration into adipose tissue. We found that 7E inhibited MCP‐1 expression in the adipose tissue of obese mice. Previous study [44] also reported that the expression level of CCR2b on F4/80+ macrophages was decreased after blocking of IL‐20’s function. These data suggested that 7E will suppress the migration of monocytes through the MCP‐1‐CCR2 axis in obesity.
Both the recruitment and the proinflammatory polarization of ATMs are required for the development of insulin resistance. Activated M1 ATMs are a prominent source of proinflammatory cytokines like TNF‐α and IL‐6, which block insulin activity in adipocytes, thereby causing systemic insulin resistance. We found that IL‐20 alone induced the expression of several cytokines in macrophages and adipocytes isolated from HFD mice, which suggested that IL‐20 and TNF‐α, IL‐6 and MCP‐1 synergistically mediate the initial inflammatory response in adipose tissue. IL‐20 inhibited insulin‐stimulated glucose uptake in adipocytes in vitro; 7E treatment improved glucose homeostasis and insulin responsiveness in obese HFD mice. Therefore, IL‐20 might directly affect local inflammation and insulin resistance and indirectly affect these activities by inducing other mediators via autocrine/paracrine signalling.
Recent studies [14, 15 reported that multiple macrophage populations existed in obese adipose tissues. It will be very interesting to determine the heterogeneity of the macrophage populations in adipose tissue after IL‐20 antibody treatment and such alterations in macrophage population may have impact to the pathogenesis of metabolic disorder caused by adipose tissue. The single‐cell RNA sequencing to elucidate the molecular mechanism of IL‐20 in the regulation of macrophage differentiation and the macrophage phenotype changes in HFD mice awaits further investigation.
No significant difference in body weight was observed in 7E‐treated HFD mice for the initial 11 weeks. However, body weight was significantly lower in 7E‐treated HFD mice than in mIgG‐treated HFD mice during weeks 11–15. We hypothesized that other cytokines compensated for the activity of IL‐20 during the early acute inflammatory response. IL‐20 might be more critical for macrophage accumulation and regulating glucose homeostasis in the chronic inflammatory state and contribute to metabolic dysfunction. IL‐20 signals through the type I (IL‐20R1/IL‐20R2) or type II (IL‐22R1/IL‐20R2) receptor complexes. IL‐22R1‐deficient HFD mice are prone to developing metabolic disorders. [45] This finding raised the possibility that IL‐20R1/IL‐20R2 signals promote obesity and that IL‐22R1/IL‐20R2 signals inhibit it. It is noteworthy that IL‐22R1 not only pairs with IL‐10R2 to form a functional IL‐22 receptor complex, but also couples with IL‐20R2 to form a receptor for IL‐20 and IL‐24· [21] The discrepancy in the HFD‐induced obese mouse model observed between neutralizing IL‐20 and IL‐22R1‐deficient mice suggests that other IL‐22R1 ligands may also regulate metabolic dysfunction. Additional studies are needed to explore the detailed molecular mechanism of modulation of obesity by these cytokines.
A chronic JAK‐STAT3‐SOCS‐3 pathway in an obese organism impairs normal leptin and insulin activities [46]. Serum IL‐20 level was higher in patients with diabetes mellitus than in healthy controls [32]. In the present study, we found that IL‐20 impaired insulin‐mediated glucose uptake and that IL‐20 stimulated SOCS‐3 expression in adipocytes. IL‐20 has been associated with pathways that regulate TLR4 expression. The expression of TLR4 was reduced after inhibiting IL‐20[44]. Activating TLR4 signalling in adipocytes activated JNK and NFκB signalling, which upregulated the expression of proinflammatory cytokines, which in turn increased SOCS‐3 production [47]. Higher SOCS‐3 expression in adipose tissue impairs insulin signals, which dysregulates glucose transport and homeostasis. Therefore, IL‐20 might regulate glucose homeostasis through these signal pathways.
MSCs differentiate into osteoblasts, adipocytes, chondrocytes and myoblasts. It has been suggested that a reciprocal relationship exists between the differentiation of MSCs into osteoblasts and adipocytes. We found that IL‐20 increased adipocyte differentiation in SGBS cells and the differentiation was inhibited in 7E‐expressing MSCs. Furthermore, IL‐20 inhibited osteoblast differentiation and maturation [48]. These findings suggest that IL‐20 is a critical regulator for determining the differentiation of MSCs into osteoblasts and adipocytes. It is necessary to further investigate the mechanism that IL‐20 uses to regulate the differentiation of MSCs into osteoblast and adipocytes. It will also be interesting to explore the mechanism in obese organisms to maintain the balance between adipogenesis and osteoblastogenesis.
Visceral WAT expands via two distinct mechanisms: hypertrophy (increase in the size of adipocytes) and hyperplasia (increase in the number of adipocytes). Hyperplasia is correlated more strongly with the severity of obesity and is most marked in the severely obese [49]. Prolonged periods of weight gain in adulthood increase the number of adipocytes [50]. We found significantly higher serum levels of IL‐20 in patients with moderate‐to‐severe obesity than in mildly obese patients and in healthy controls. In addition, IL‐20 modulated adipogenesis and adipocyte differentiation by upregulating PPARγ and C/EBPα. IL‐20 did not promote adipocyte hypertrophy. IL‐20 only increased the number of adipocytes which provides more evidence that IL‐20 contributes to maintaining a long‐term, low‐grade chronic inflammatory state in obesity. In summary, our findings demonstrate that IL‐20 is an important regulator for adipose tissue inflammation and adipose tissue macrophage chemotaxis. IL‐20 affected macrophage polarization and promoted insulin resistance in obese humans and mice. Therefore, we have identified a critical role of IL‐20 in obesity‐induced inflammation and insulin resistance. We conclude that IL‐20 is a novel target for treating obesity and insulin resistance in patients with metabolic disorders.
CONFLICT OF INTEREST
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AUTHOR CONTRIBUTIONS
M.S. Chang conceived and supervised the study. Y.H. Hsu and Y.C. Chang designed the experiments, interpreted the results and generated the figures. Y.C. Chang and C.H. Wu collected the clinical samples and analysed the data. Y.H. Hsu wrote the manuscript. Y.H. Hsu, C.J. Chiu, W.T. Chen and Y.C. Chang performed the experiments and analysed the data. All authors discussed the data and commented on the manuscript before submission.
ETHICAL APPROVAL
For human study, the National Cheng Kung University Hospital Institutional Review Board approved the study (A‐ER‐107–377). The study was done in accordance with approved guidelines. Written informed consent was obtained. For mouse study, all animal experiments were done in accordance with the approved guidelines of the Taiwan National Institutes of Health (Taipei), standards and guidelines for the care and use of experimental animals. The Animal Ethics Committee of National Cheng Kung University approved the research procedures (IACUC Approval No. 108089). The study was done in accordance with approved guidelines.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Professor Martin Wabitsch (University of Ulm, Germany) for kindly providing SGBS preadipocyte cell line.
Hsu Y‐H, Wu C‐H, Chiu C‐J, Chen W‐T, Chang Y‐C, Wabitsch M, et al. IL‐20 is involved in obesity by modulation of adipogenesis and macrophage dysregulation. Immunology. 2021;164:817–833. 10.1111/imm.13403
Funding information
This work was supported by the Ministry of Science and Technology of Taiwan (MOST‐109‐2628‐B‐006‐019 and MOST‐110‐2628‐B‐006‐023).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Hotamisligil GS, Erbay E. Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol. 2008;8:923–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Makki K, Froguel P, Wolowczuk I. Adipose tissue in obesity‐related inflammation and insulin resistance: cells, cytokines, and chemokines. ISRN Inflamm. 2013;2013:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lackey DE, Olefsky JM. Regulation of metabolism by the innate immune system. Nat Rev Endocrinol. 2016;12:15–28. [DOI] [PubMed] [Google Scholar]
- 4. Despres JP, Lemieux I. Abdominal obesity and metabolic syndrome. Nature. 2006;444:881–7. [DOI] [PubMed] [Google Scholar]
- 5. Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–7. [DOI] [PubMed] [Google Scholar]
- 6. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–45. [DOI] [PubMed] [Google Scholar]
- 8. Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11:85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. [DOI] [PubMed] [Google Scholar]
- 10. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–64. [DOI] [PubMed] [Google Scholar]
- 11. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–86. [DOI] [PubMed] [Google Scholar]
- 12. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lumeng CN, DelProposto JB, Westcott DJ, Saltiel AR. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes. 2008;57:3239–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, et al. Lipid‐associated macrophages control metabolic homeostasis in a trem2‐dependent manner. Cell. 2019;178:686–698.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weinstock A, Brown EJ, Garabedian ML, Pena S, Sharma M, Lafaille J, et al. Single‐cell RNA sequencing of visceral adipose tissue leukocytes reveals that caloric restriction following obesity promotes the accumulation of a distinct macrophage population with features of phagocytic cells. Immunometabolism. 2019;1:e190008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hill DA, Lim HW, Kim YH, Ho WY, Foong YH, Nelson VL, et al. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc Natl Acad Sci U S A. 2018;115:E5096–E105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Russo L, Lumeng CN. Properties and functions of adipose tissue macrophages in obesity. Immunology. 2018;155:407–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gurses KM, Ozmen F, Kocyigit D, Yersal N, Bilgic E, Kaya E, et al. Netrin‐1 is associated with macrophage infiltration and polarization in human epicardial adipose tissue in coronary artery disease. J Cardiol. 2016;69:851–858. [DOI] [PubMed] [Google Scholar]
- 19. Ramkhelawon B, Hennessy EJ, Menager M, Ray TD, Sheedy FJ, Hutchison S, et al. Netrin‐1 promotes adipose tissue macrophage retention and insulin resistance in obesity. Nat Med. 2014;20:377–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Blumberg H, Conklin D, Xu WF, Grossmann A, Brender T, Carollo S, et al. Interleukin 20: discovery, receptor identification, and role in epidermal function. Cell. 2001;104:9–19. [DOI] [PubMed] [Google Scholar]
- 21. Pestka S, Krause CD, Sarkar D, Walter MR, Shi Y, Fisher PB. Interleukin‐10 and related cytokines and receptors. Annu Rev Immunol. 2004;22:929–79. [DOI] [PubMed] [Google Scholar]
- 22. Dumoutier L, Leemans C, Lejeune D, Kotenko SV, Renauld JC. Cutting edge: STAT activation by IL‐19, IL‐20 and mda‐7 through IL‐20 receptor complexes of two types. J Immunol. 2001;167:3545–9. [DOI] [PubMed] [Google Scholar]
- 23. Hsieh MY, Chen WY, Jiang MJ, Cheng BC, Huang TY, Chang MS. Interleukin‐20 promotes angiogenesis in a direct and indirect manner. Genes Immun. 2006;7:234–42. [DOI] [PubMed] [Google Scholar]
- 24. Hsu YH, Li HH, Hsieh MY, Liu MF, Huang KY, Chin LS, et al. Function of interleukin‐20 as a proinflammatory molecule in rheumatoid and experimental arthritis. Arthritis Rheum. 2006;54:2722–33. [DOI] [PubMed] [Google Scholar]
- 25. Li HH, Hsu YH, Wei CC, Lee PT, Chen WC, Chang MS. Interleukin‐20 induced cell death in renal epithelial cells and was associated with acute renal failure. Genes Immun. 2008;9:395–404. [DOI] [PubMed] [Google Scholar]
- 26. Hsu YH, Hsing CH, Li CF, Chan CH, Chang MC, Yan JJ, et al. Anti‐IL‐20 monoclonal antibody suppresses breast cancer progression and bone osteolysis in murine models. J Immunol. 2012;188:1981–91. [DOI] [PubMed] [Google Scholar]
- 27. Hsu YH, Wei CC, Shieh DB, Chan CH, Chang MS. Anti‐IL‐20 monoclonal antibody alleviates inflammation in oral cancer and suppresses tumor growth. Mol Cancer Res. 2012;10:1430–9. [DOI] [PubMed] [Google Scholar]
- 28. Wei CC, Hsu YH, Li HH, Wang YC, Hsieh MY, Chen WY, et al. IL‐20: biological functions and clinical implications. J Biomed Sci. 2006;13:601–12. [DOI] [PubMed] [Google Scholar]
- 29. Wei CC, Chen WY, Wang YC, Chen PJ, Lee JY, Wong TW, et al. Detection of IL‐20 and its receptors on psoriatic skin. Clin Immunol. 2005;117:65–72. [DOI] [PubMed] [Google Scholar]
- 30. Hsu YH, Chen WY, Chan CH, Wu CH, Sun ZJ, Chang MS. Anti‐IL‐20 monoclonal antibody inhibits the differentiation of osteoclasts and protects against osteoporotic bone loss. J Exp Med. 2011;208:1849–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wei CC, Li HH, Hsu YH, Hsing CH, Sung JM, Chang MS. Interleukin‐20 targets renal cells and is associated with chronic kidney disease. Biochem Biophys Res Commun. 2008;374:448–53. [DOI] [PubMed] [Google Scholar]
- 32. Hsu YH, Li HH, Sung JM, Chen WY, Hou YC, Weng YH, et al. Interleukin‐20 targets podocytes and is upregulated in experimental murine diabetic nephropathy. Exp Mol Med. 2017;49:e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen WY, Chang MS. IL‐20 is regulated by hypoxia‐inducible factor and up‐regulated after experimental ischemic stroke. J Immunol. 2009;182:5003–12. [DOI] [PubMed] [Google Scholar]
- 34. Otkjaer K, Kragballe K, Johansen C, Funding AT, Just H, Jensen UB, et al. IL‐20 Gene expression is induced by IL‐1beta through mitogen‐activated protein kinase and NF‐kappaB‐dependent mechanisms. J Invest Dermatol 2007;127:1326–36. [DOI] [PubMed] [Google Scholar]
- 35. Fischer‐Posovszky P, Newell FS, Wabitsch M, Tornqvist HE. Human SGBS cells ‐ a unique tool for studies of human fat cell biology. Obes Facts. 2008;1:184–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Surmi BK, Hasty AH. Macrophage infiltration into adipose tissue: initiation, propagation and remodeling. Future Lipidol. 2008;3:545–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maiorino MI, Schisano B, Di Palo C, Vietri MT, Cioffi M, Giugliano G, et al. Interleukin‐20 circulating levels in obese women: effect of weight loss. Nutr Metab Cardiovasc Dis. 2010;20:180–5. [DOI] [PubMed] [Google Scholar]
- 38. Paz‐Filho G, Mastronardi C, Franco CB, Wang KB, Wong ML, Licinio J. Leptin: molecular mechanisms, systemic pro‐inflammatory effects, and clinical implications. Arq Bras Endocrinol Metabol. 2012;56:597–607. [DOI] [PubMed] [Google Scholar]
- 39. Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF‐kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135:61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest. 2012;122:153–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Arruda AP, Milanski M, Coope A, Torsoni AS, Ropelle E, Carvalho DP, et al. Low‐grade hypothalamic inflammation leads to defective thermogenesis, insulin resistance, and impaired insulin secretion. Endocrinology. 2011;152:1314–26. [DOI] [PubMed] [Google Scholar]
- 42. Milanski M, Arruda AP, Coope A, Ignacio‐Souza LM, Nunez CE, Roman EA, et al. Inhibition of hypothalamic inflammation reverses diet‐induced insulin resistance in the liver. Diabetes. 2012;61:1455–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Preobrazhensky AA, Dragan S, Kawano T, Gavrilin MA, Gulina IV, Chakravarty L, et al. Monocyte chemotactic protein‐1 receptor CCR2B is a glycoprotein that has tyrosine sulfation in a conserved extracellular N‐terminal region. J Immunol. 2000;165:5295–303. [DOI] [PubMed] [Google Scholar]
- 44. Cucak H, Hoj Thomsen L, Rosendahl A. IL‐20 contributes to low grade inflammation and weight gain in the Psammomys obesus. Int Immunopharmacol. 2017;45:53–67. [DOI] [PubMed] [Google Scholar]
- 45. Wang X, Ota N, Manzanillo P, Kates L, Zavala‐Solorio J, Eidenschenk C, et al. Interleukin‐22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature. 2014;514:237–41. [DOI] [PubMed] [Google Scholar]
- 46. Wunderlich CM, Hovelmeyer N, Wunderlich FT. Mechanisms of chronic JAK‐STAT3‐SOCS3 signaling in obesity. JAKSTAT. 2013;2:e23878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid‐induced insulin resistance. J Clin Invest. 2006;116:3015–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hsu YH, Chiu YS, Chen WY, Huang KY, Jou IM, Wu PT, et al. Anti‐IL‐20 monoclonal antibody promotes bone fracture healing through regulating IL‐20‐mediated osteoblastogenesis. Sci Rep. 2016;6:24339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hirsch J, Batchelor B. Adipose tissue cellularity in human obesity. Clin Endocrinol Metab. 1976;5:299–311. [DOI] [PubMed] [Google Scholar]
- 50. Moreno‐Navarrete JM, Fernández‐Real JM. Adipocyte Differentiation In: Adipose Tissue Biology. New York: Springer; 2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.