

Summary
Neoadjuvant PD-1 blockade may be efficacious in some individuals with high-risk, resectable oral cavity head and neck cancer. To explore correlates of response patterns to neoadjuvant nivolumab treatment and post-surgical recurrences, we analyzed longitudinal tumor and blood samples in a cohort of 12 individuals displaying 33% responsiveness. Pretreatment tumor-based detection of FLT4 mutations and PTEN signature enrichment favors response, and high tumor mutational burden improves recurrence-free survival. In contrast, preexisting and/or acquired mutations (in CDKN2A, YAP1, or JAK2) correlate with innate resistance and/or tumor recurrence. Immunologically, tumor response after therapy entails T cell receptor repertoire diversification in peripheral blood and intratumoral expansion of preexisting T cell clones. A high ratio of regulatory T to T helper 17 cells in pretreatment blood predicts low T cell receptor repertoire diversity in pretreatment blood, a low cytolytic T cell signature in pretreatment tumors, and innate resistance. Our study provides a molecular framework to advance neoadjuvant anti-PD-1 therapy for individuals with resectable head and neck cancer.
Keywords: neoadjuvant anti-PD-1/L1 therapy; head-and-neck cancer/oral-cavity SCC; response, resistance, and recurrence; FLT4/PTEN/PPARG/CDKN2A/YAP1/JAK2; T cell repertoire; T regulatory to Th17 ratio; tumor mutational burden; recurrence-free survival; tumor phylogeny; multiplex immunofluorescence
Graphical abstract
Highlights
-
•
FLT4 nsSNVs favor response; CDKN2A nsSNVs favor non-response; high TMB improves RFS
-
•
Recurrences select for CN loss in PTEN, JAK2 and/or gain in YAP1, MDM2, PPARG
-
•
T cell clones (blood) diversify and preexisting clones (tumors) expand with response
-
•
High ratios of TREG/Th17 in pretreatment blood predict innate resistance
Liu et al. nominate multi-omic correlates of response, recurrence, and survival in individuals treated with neoadjuvant anti-PD-1 therapy for resectable, locally advanced, oral cavity SCC. Future validation of blood- and tumor-based biomarkers and mechanistic insights have implications for neoadjuvant and adjuvant management of individuals with systemic treatment-naive resectable disease.
Introduction
Since its first clinical testing,1 anti-PD-1 immune checkpoint blockade (ICB) has revolutionized management of individuals with advanced malignancies and is poised to re-shape multidisciplinary treatment of those with earlier-stage but high-risk malignancies. Deployment of anti-PD-1 therapy may be relatively more effective against earlier-stage (versus advanced metastatic stage) cancers because of a less evolved cancer and less suppressed immune system. Preclinical experiments support anti-PD-1 therapy in the neoadjuvant (before surgery) compared with the adjuvant (after surgery) setting,2 presumably because bulk tumor presence during therapy is critical for therapy-induced antitumor T cell persistence and activity. Clinically, in palpable stage III melanoma, where neoadjuvant versus adjuvant combined ICB was compared,3 T cell expansion was more vigorous in the neoadjuvant setting.
Head and neck squamous cell carcinoma (HNSCC) ranks sixth among common epithelial malignancies worldwide.4 Human papillomavirus (HPV)-positive and -negative categories of HNSCC have distinct multi-omic, clinical, and therapeutic response characteristics, with HPV-negative HNSCC accounting for 75% of all HNSCCs and portending a far worse prognosis.5 Historically, over a third of individuals, in particular those with HPV-negative HNSCC, relapse despite intensive postoperative (adjuvant) chemoradiotherapy.6 Compared with HPV-positive HNSCC, HPV-negative HNSCC harbors more mutations and displays heightened chromosome instability.7 Oral cavity squamous cell carcinoma (OCSCC) as a subsite consists of mostly HPV-negative HNSCC.
Anti-PD-1 therapy with nivolumab or pembrolizumab improves the overall survival of individuals with platinum-resistant recurrent and metastatic HNSCC, including OCSCC,8, 9, 10 with ∼20% response rates and a survival benefit compared with chemotherapy. The potential clinical benefit of neoadjuvant anti-PD-1 therapy has been explored in small cohorts with resectable, locally advanced, HPV-negative HNSCC.11,12 In one correlative study using PD-L1 immunofluorescence, pretreatment PD-L1 was not correlated with volumetric or pathologic response among 12 individuals who received two doses of neoadjuvant nivolumab.11 In another correlative study using genomic techniques (whole-exome sequencing [WES] and RNA sequencing [RNA-seq]), pretreatment tumor mutational burden (TMB) did not correlate with the extent of pathologic response among 24 individuals who received one dose of neoadjuvant pembrolizumab.12
Here, using longitudinal blood and tumor tissues obtained from 12 individuals with newly diagnosed, locally invasive OCSCC who were treated with neoadjuvant anti-PD-1 therapy,13 we generate hypotheses regarding tumor cell-intrinsic and immunological mechanisms of response patterns, survival, and post-operative recurrence. By analyzing multi-omic and multiplex data, we dissect temporal relationships between mutational and transcriptomic alterations as well as systemic and intratumoral immunity. This study provides insights into tumor and immune cell co-evolution in OCSCC treated with neoadjuvant anti-PD-1 therapy and identifies potential predictive biomarkers and mechanisms of response, resistance, and post-surgical recurrence.
Results
Clinical characteristics associated with tissues
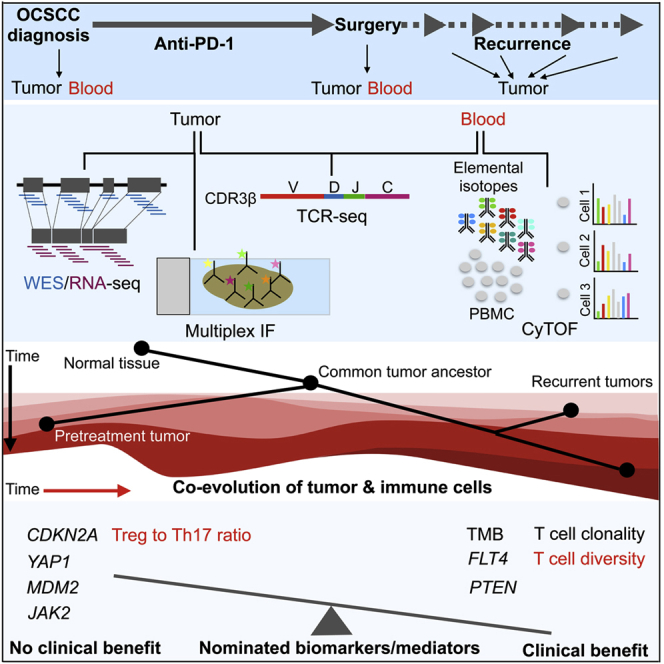
We obtained tissues from individuals (n = 11) who enrolled in a single-arm, investigator-initiated, single-institution phase II clinical trial (NCT03021993) of OCSCC and from one additional individual (individual 8) (total n = 12) who fell out of eligibility because of rapid progression. Detailed eligibility and inclusion/exclusion criteria are described in a related manuscript.13 Individual and disease characteristics are summarized in Table S1. Enrollment of subjects in the trial and participation in tissue biopsies and analyses were approved by the local institutional review board. Briefly, we performed (1) time-of-flight mass cytometry (CyTOF) on pre- and post-treatment peripheral blood mononuclear cells (PBMCs); (2) WES on subject-matched normal tissues, pretreatment tumors, and, when applicable, post-operative recurrent tumors; (3) RNA-seq on subject-matched pre- and post-treatment tumors and, when applicable, post-operative recurrent tumors; and (4), genomic DNA (gDNA)-based T cell receptor (TCR)-seq on subject-matched pre- and post-treatment PBMCs and tumors in a subset of responders and non-responders.
Design of the trial and tissue collection is schematized in Figure S1. Briefly, primary tumors were required to be from individuals who were newly diagnosed with systemic and radiation treatment-naive stage II–IVA OCSCC and whose tumors could be accurately assessed clinically and radiographically. Individuals included in this study received 3–4 biweekly doses of 3 mg/kg nivolumab (except one individual who received only 2 biweekly doses), followed by surgical resection with curative intent. Radiographic tumor size was defined as the greatest cross-sectional dimension of the tumor on the enrollment imaging study, and post-treatment size was the greatest cross-sectional dimension of the tumor on surgical pathology. Interval radiographic evaluation occurred after a total of three doses of nivolumab and between days 28–35. Disease progression (>20% increase in tumor size) determined at interval radiographic evaluation was treated with definitive surgical resection between days 36–42. In the event of stable disease or response, individuals received a fourth dose of nivolumab on day 43 ± 1, followed by definitive surgical resection on days 50–56. The objective response rate was defined as pathologic complete + pathologic partial response (>30% reduction in tumor size of the surgical specimen). Change in size was calculated by comparing the pre-nivolumab radiographic measurement (single greatest dimension) with the final pathologic measurement (Table S2). Given the short duration of nivolumab therapy, responders were defined as individuals who derived clinical benefit (complete response, partial response, and stable disease per response evaluation criteria in solid tumors [RECIST] 1.1), and non-responders were defined as individuals who derived no clinical benefit (progression per RECIST 1.1). At the cutoff of this study, the median follow-up was 2.05 years. Subject-matched and longitudinal tumor and PBMC tissues analyzed by multi-omics are summarized in Table S3.
Genomic features of response patterns and survival in pretreatment tumors
We analyzed WES from 16 tumors (12 pretreatment and 4 recurrence) and matched blood samples from 12 individuals. The average number of mutations per tumor was 347 (range, 31–559), corresponding to a mean TMB of 5.79 mutations/megabase (MB) (range, 0.52–9.32), which is typical for HNSCC (Table S4). We observed no significant difference in the TMBs of pretreatment tumors from responders versus non-responders (median, 6.63 versus 5.98 single-nucleotide variants (SNVs)/MB, respectively; p = 0.4676, Wilcoxon rank-sum one-sided test; Figure 1A). Among responsive pretreatment tumors, individual 12’s tumor harbored a very low TMB (0.52 SNVs/MB), although its response was categorized as stable disease, and the recurrence-free survival (RFS) of individual 12 was less than 0.3 years (Table S2; overall survival (OS), not available because of loss of follow-up). In contrast, high TMBs (defined as the upper half of the median) in the pretreatment tumors were significantly associated with improved RFS (Figure 1B) but not with improved OS (Figure 1C) (median follow-up of 2.05 years). Moreover, tumor response did not associate with improved RFS (Figure S2A) or OS (Figure S2B). Neither pretreatment tumor purity nor ploidy was correlated with a pathologic response (Figure S2C).
Figure 1.

Genomic correlates of innate tumor sensitivity versus resistance and survival in pretreatment tumors
(A) TMBs in responders (n = 7) versus non-responders (n = 5); p value, Wilcoxon rank-sum test. Red dots, median values.
(B and C) Kaplan-Meier curves of RFS (B) and OS (C) comparing tumors with high TMB (≥ median TMB, n = 6) versus tumors with a low TMB (< median TMB, n = 5); two-sided log rank test. The tumor from individual 12, who was lost to follow-up, was excluded.
(D) Genes with recurrent somatic mutations (responders, n = 7; non-responders, n = 5). Recurrence was defined as non-synonymous mutations in 2 or more individuals and CN alterations in 7 or more individuals. Indel, insertion or deletion; amp, amplification; del, deletion. The status of subject-matched recurrent tumors is shown but not counted toward recurrence.
(E) Ratios of variant versus normal allele frequencies in CDKN2A detected in one responder and three non-responders. The CN of CDKN2A is labeled on top.
(F) Infiltration levels of CD8+ T, TREG, and resting NK cells in FLT4WT (n = 508) versus FLT4Mut (n = 14) clinical HNSCC tumors from a public dataset in cBioPortal; p values, Wilcoxon rank-sum test. ∗p < 0.05, ∗∗∗p < 0.001.
See also Figures S1 and S2, Tables S1–S4, and STAR Methods.
HLA-I homozygosity was correlated with poor response and reduced overall survival in advanced melanoma and non-small cell lung carcinoma individuals treated with immunotherapies.14 We observed a non-significant association in the opposite direction; HLA-I (HLA-A, HLA-B, and HLA-C) homozygosity in at least one locus was of a higher proportion in the responder group (4 of 7 in responders versus 0 of 4 in non-responders; Fisher’s exact test, p = 0.0808; Figure S2D). HNSCC has been shown to harbor relatively high levels of somatic changes in HLA class I genes,15 and hotspot mutations in HLA I genes have been associated with upregulation of signatures of effector T cell cytolytic signatures.16 However, in this study of locally advanced OCSCC, HLA-I mutations were rare and detected in a single progressive tumor pretreatment (Figure S2D).
Next we identified non-synonymous (missense, nonsense, and frameshift) mutations, splice site mutations, in-frame insertions or deletions (indels), as well as amplifications and deletions. We then visualized the genes affected mostly recurrently by these mutations among the pretreatment tumors (Figure 1D). We observed well-known significantly mutated genes in HNSCC, including TP53 (75%), CDKN2A (33.3%), and CREBBP (25%). We also identified frequently amplified genes; e.g., CCND1 (75%), MAP3K13, PIK3CA, EGFR, and SOX2 (58.3%). Non-synonymous mutations in CDKN2A were detected in three of five non-responding tumors (H83Y in individual 2, R80∗ in individual 3, and splice site mutation in individual 8), in contrast to one of seven responsive tumors (in-frame deletion in individual 14) (Figure 1D). This mutation frequency (60%) in the non-responders is higher compared with the background mutation rate of 20.32% (291 of 1,452 HNSCC tumors in cBioPortal; Fisher’s exact test, p = 0.0590; Benjamini-Hochberg-adjusted p = 0.0861). Also, the ratios of variant to normal allele frequencies of CDKN2A are elevated among the non-responders, driven in part by deletion of the wild-type copy (individuals 3 and 8) and selective amplification of the mutant copy (individual 2) (Figure 1E). Interestingly, FLT4 was mutated exclusively in responsive tumors (2 of 7 tumors) (Figure 1D). Given the background mutation rate of 2.20% (32 of 1,452 HNSCC tumors in cBioPortal), FLT4 was mutated more frequently than expected in responders (Fisher’s exact test, p = 0.0103; Benjamini-Hochberg-adjusted p = 0.0515). We estimated immune cell proportions from a public RNA-seq dataset of HNSCC in cBioPortal. We identified gene expression specific to three immune cell types to be significantly differentially expressed between FLT4MUT and FLT4WT tumors. Notably, in FLT4MUT (versus FLT4WT) tumors, CD8+ T cells and resting natural killer (NK) cell levels were elevated, whereas TREG levels were lower (Figure 1F).
Clonal evolution of recurrence after neoadjuvant anti-PD-1 therapy and surgery
We exploited WES data to retrace the evolutionary trajectories of OCSCC from normal epithelial cells to malignant tumors before treatment with neoadjuvant nivolumab and then to recurrent tumors after neoadjuvant therapy and after surgery. Phylogenetic trees for two responders and one non-responder were constructed (Figure 2A). In individual 1, neoadjuvant PD-1 blockade (four doses) elicited a 45% reduction in tumor size (Table S2) despite very low TMB pretreatment (Figure 1A). However, the individual’s tumor recurred (in the lungs) 0.58 year after surgical excision. We compared WES data from this recurrent tumor versus the pretreatment tumor and subject-matched normal tissue and found that the pretreatment and recurrent tumors evolved in a branched manner. In addition, chromosome 10, where PTEN resides, was amplified because of arm-sized duplication before nivolumab treatment. However, in the recurrent tumor, PTEN copy number (CN) was neutral, indicating a loss relative to the pretreatment tumor. Moreover, in the recurrent tumor, we observed CN losses of CDKN2A, CDKN2B, and JAK2 (Figure 2A). However, only PTEN and JAK2 displayed concordant DNA and RNA loss in the recurrent tumor (PTEN, ∼4-fold; JAK2, ∼2-fold) (Figure 2B). By immunofluorescence (IF), we corroborated PTEN and JAK2 protein-level reduction specifically in tumor cells of the recurrent tumor (Figure 2C). Given emerging reports of mechanistic links between PTEN loss and innate anti-PD-1 resistance,17, 18, 19 we speculate that PTEN CN gain pretreatment may contribute to innate responsiveness of this tumor despite its low TMB and that PTEN CN loss may promote tumor recurrence in the lungs of this individual after neoadjuvant anti-PD-1 therapy and surgery.
Figure 2.

Evolution of post-operative recurrent tumors
(A) Phylogenetic relationships of subject-specific normal tissue, pretreatment, and recurrent tumors in two responders (individuals 1 and 6) and one non-responder (individual 7). Phylogenetic distances between germline gDNA, most recent common tumor ancestor, pretreatment tumor, and recurrent tumor(s) reflect the number of SNVs and small indels. Select driver genes and their mutations are shown for each evolutionary trajectory.
(B) Expression levels of PTEN and JAK2 in pretreatment and recurrent tumors of individual 1.
(C) Representative immunofluorescent images merging (1) DAPI (nuclei), pan-cytokeratin (panCK), and PTEN or JAK2 signals from post-treatment and recurrent tumors (individual 1); (2) DAPI (nuclei), panCK, and YAP1 or MDM2 signals from post-treatment and two recurrent tumors (individual 6); and (3) DAPI (nuclei), panCK, and YAP1 signals from post-treatment and recurrent tumors of individual 7 as well as post-treatment tumors (controls) of individuals 9 and 10. Scale bars represent 50 microns, except for MDM2 images (20 μm).
(D) Quantification of mIF across whole tissue sections comparing post-treatment versus recurrent tumors in individuals 1, 6, and 7.
(E) Images representative of mIF quantifications in (D). Scale bar, 50 μm.
See also Figure S1 and Tables S1–S4.
In another responsive individual (individual 6), neoadjuvant PD-1 blockade elicited a 30% reduction in tumor size (Table S2). After the residual tumor was excised, the individual relapsed in 1.91 years with two recurrent tumors. As in the case of individual 1, evolution of pretreatment and recurrent tumors followed a branched pattern, where the ancestral clone harbored the same TP53 mutation (Figure 2A). Notably, both recurrent tumors originated from this ancestral clone with shared hits; namely, YAP1 and MDM2 amplification. YAP1 post-transcriptional upregulation and nuclear translocation in tumor cells have been implicated in immune evasion during mitogen-activated protein kinase (MAPK)-targeted and anti-PD-1 therapies.20, 21, 22 Also, MDM2 amplification, which has been linked to hyperprogression on anti-PD-1 therapy,23 can be targeted by small-molecule inhibitors to improve anti-PD-1 responsiveness and T cell killing of cancer cells.24,25 Concordant with these gDNA amplification events, YAP1 and MDM2 protein levels were elevated in tumor cells of recurrent (versus post-treatment) tumors, with YAP1 protein upregulation being cytoplasmic and nuclear in recurrent tumor 1 and largely nuclear in recurrent tumor 2 (Figure 2C). Also relevant to recurrence may be loss of the FLT4 mutation in both recurrent tumors (Figure 1D), suggesting that the missense FLT4 mutations that are enriched in responders may be gain-of-function mutations.
In a non-responsive individual (individual 7), the tumor recurred 0.73 years after neoadjuvant nivolumab and surgery (Table S2). Individual 7 was deceased within 1.5 months after clinical relapse. Despite a 26% increase in tumor size after neoadjuvant nivolumab therapy, the tumor that recurred after definitive surgery followed a branched evolutionary pattern, suggesting that some level of immune editing occurred despite the lack of radiographic and pathologic response. As shown in Figure 2A, YAP1 amplification predated the most recent ancestral tumor clone, suggesting a role in innate resistance. Consistent with preexisting YAP1 amplification, the YAP1 protein level was elevated in tumor cells of post-treatment and recurrent tumors in individual 7, in contrast to YAP1 levels in the post-treatment tumors of individuals 9 and 10 (which served as controls) (Figure 2C). Interestingly, PPARG amplification was private to the recurrent tumor (Figure 2A). Given that amplification-driven overexpression of PPARG, in concert with RXRα activation, has been shown to confer partial resistance to immunotherapy by impairing CD8+ T cell infiltration in muscle-invasive bladder cancer,26 PPARG amplification may complement YAP1 amplification to tip the balance toward immune evasion.
Using multiplex IF (mIF), we histologically characterized concurrent evolution of the tumor immune microenvironment (quantification in Figure 2D and representative images in Figure 2E). The panel consisted of antibodies against pan-cytokeratin (panCK), CD3, CD8, CD68, PD-L1, and granzyme B (GzmB). Based on whole-tissue quantification, we observed, as expected, that panCK+ tumor cells (per mm2 of tissue) increased in the recurrent tumor of individual 1 and both recurrent tumors of individual 6 (versus matched post-treatment surgical tumors) because individuals 1 and 6 were responders (Figure 2D). In contrast, there was little change in the density of panCK+ tumor cells in the recurrent tumor of individual 7, a non-responder (Figure 2D). In subject-matched comparisons, all recurrent tumors displayed a significant decrease in total CD3+ T cells, which corresponded to a decrease in CD4+ and CD8+ T cells (Figures 2D and 2E). This observation further supports the aforementioned notion that some level of immune editing occurred despite the lack of radiographic and pathologic response in individual 7.
We observed additional recurrence-specific features compared with tumors on anti-PD-1 neoadjuvant therapy (Figures 2D and 2E). In individual 1, recurrence was associated with a loss of CD8+ T cell cytolytic activity, as defined by a reduced GzmB level, and an increase in CD68+ macrophages. There was minimal change in PD-L1 expression in tumor cells and macrophages. In individual 6, recurrence was associated with a gain in the level of CD8+ GzmB+ T cells. However, there was a concurrent increase in the levels of PD-L1+ tumor cells and macrophages. Thus, in individual 6, the combination of reduced overall T cell infiltration and increased PD-L1 immune checkpoint expression may have resulted in relapses. In individual 7, recurrence was depleted of immune cells, suggestive of an immune desert.
Pre- and post-treatment transcriptional signatures of response patterns
We analyzed RNA-seq data generated from 11 pairs of subject-matched pre- and post-treatment tumors for statistically significant differential enrichment of 10,401 gene sets (molecular signature database [MSigDB]) between the responsive and non-responsive tumors before or after neoadjuvant nivolumab therapy. Among the two groups of pretreatment tumors, two processes were differentially enriched (Figure 3A). First, genes downregulated in the intestine after tissue-specific knockout of PTEN (HE_PTEN_TARGETS_DN) were negatively enriched among the non-responsive pretreatment tumors, suggesting lower PTEN gene dosage or expression with innate anti-PD-1 resistance. Second, responsive pretreatment tumors were positively enriched for PPARG pathway genes. PPARγ signaling increases PTEN activity.27,28 We detected a positive correlation of enrichment scores between these two gene sets (Figure 3B). Furthermore, two gene sets related to de-differentiation and cancer stemness (IIZUKA_LIVER_CANCER_PROGRE-SSION_G1_G2_UP29 and REACTOME_INTERLEUKIN_6_SIGNALING30) were enriched in responders’ post-treatment tumors (Figure 3C).
Figure 3.

Transcriptomic features of response in pre- and post-treatment tumors
(A) Heatmap showing the top gene sets differentially enriched in responsive versus non-responsive pretreatment tumors (n = 11; one pretreatment tumor was excluded because of RNA degradation of its matched post-treatment tumor).
(B) Pearson correlation of enrichment scores between PTEN_DN and PPARG signatures in pretreatment tumors (n = 11).
(C) Heatmap showing top gene sets differentially enriched in responsive versus non-responsive post-treatment tumors (n = 11).
We next investigated gene signatures reported previously to be associated with ICB responsiveness in our cohort. Among the pretreatment tumors, we observed that the enrichment of such signatures (effector T cell signature31, a six-gene IFNγ signature [IFNγ-6],32 and a cytolytic activity signature33) and PD-L1 RNA levels decreased from pretreatment tumors with partial responses to those displaying stable disease and then to those displaying progressive disease, although the differences were not statistically significant (Figure S3A). Enrichment levels of effector T cell signatures and PD-L1 RNA levels in the pretreatment tumor were negatively correlated with pathology-based changes in tumor sizes after treatment, but these negative correlations were not significant (Figure S3B). Based on RNA-seq data, we then estimated the subtypes of infiltrating immune cells by CIBERSORTX and observed that pretreatment CD8+ T cell infiltration levels were negatively correlated with pathology-based changes in tumor sizes after treatment, although this negative correlation was not significant (Figure S3C). Additionally, levels of pretreatment enrichment of a PTEN signature and intratumoral CD8+ T cells estimated by CIBERSORTX did not associate with improved RFS and OS (Figures S3D and S3E). Moreover, enrichment levels of a PTEN signature among the pretreatment tumors were not correlated with CD8+ T cell infiltration levels (Pearson correlation, R = 0.06, p > 0.87) or enrichment of effector T cell signature (Pearson correlation, R = 0.09, p > 0.80).
Relationships between TCRβ clonotypes and tumor response
To understand how TCRβ clonotypes (pre- and post-nivolumab treatment in peripheral blood and tumor) track with response patterns, we selected available subject-matched PBMCs and tumors from three responders versus three non-responders (Tables S2 and S3) to generate gDNA-based TCRβ sequencing. As expected, we observed greater overlaps of productive CDR3 amino acid sequences among subject-matched samples (Figure S4). Clonality, characterized by the Gini index, was not different in tumors of responders versus non-responders before or after neoadjuvant nivolumab therapy (Figure 4A). However, within PBMCs, T cell clonality was significantly elevated in non-responders after neoadjuvant nivolumab therapy (Figure 4A). Consistently, T cell clonality in PBMCs after treatment was positively correlated (p = 0.013) with changes in tumor size (Figure 4B). From an analysis of subject-matched TCRβ clonotypes pre- versus post-nivolumab treatment, we noted opposite patterns in the tumors versus PBMCs of responders versus non-responders (Figure 4C). Responsive tumors maintained (2 of 3) or harbored (1 of 3) increased TCRβ clonality after treatment, whereas 2 of 3 non-responsive tumors lost TCRβ clonality after treatment (Figure 4C). In contrast, PBMCs of responders (3 of 3) lost T cell clonality, whereas PBMCs of non-responders (3 of 3) gained T cell clonality (Figure 4C). Data (Figures 4A–4C) supportive of intratumoral T cell clonal expansion upon therapy-induced tumor shrinkage led us to investigate the clonal origins of expanded clones. 52.83% of the preexisting intratumoral TCRβ clones (repertoires shared by pre- and post-tumors) and 22.18% of novel intratumoral clonotypes (clones specific to post-tumors) were detectable in pretreatment PBMCs across individuals, but these detection rates of intratumoral T cell clonotypes within pretreatment PBMCs were not significantly different in responders versus non-responders. Importantly, the clone sizes of preexisting clones in post-treatment tumors were significantly and negatively correlated with changes in tumor size, suggesting that preexisting intratumoral T cell clones that expanded in response to neoadjuvant nivolumab treatment led to tumor shrinkage (Figure 4D).
Figure 4.

Post-treatment elevation in systemic TCR diversity and tumoral TCR clonality reflects responsiveness
(A) Gini indices of TCRβ clones in tumors (left) and PBMCs (right) before or after neoadjuvant nivolumab treatment (red dots, average values; n = 3 per group). Pairwise comparisons by Student’s t test, ∗p < 0.05.
(B) Pearson correlations of pathologic responses and Gini indices detected in pre- and post-treatment tumors (top) and PBMCs (bottom).
(C) Temporal changes in Gini indices within longitudinal tumors (top) or PBMCs (bottom) of each individual (n = 3 responders, n = 3 non-responders).
(D) Pearson correlation of pathologic responses and total clone sizes of preexisting TCR clonotypes in post-treatment tumors.
TREG versus Th17 imbalance in pretreatment PBMCs predicts tumor progression
To evaluate differences in immune cell populations in the peripheral blood between responders (n = 5) versus non-responders (n = 4), we used CyTOF to analyze PBMCs collected before and after treatment (at the time of surgery) and PBMCs from healthy donors (n = 4). By clustering analysis, we identified 18 immune cell populations (Figures 5A and 5B) consisting of three CD8+ T cell subpopulations (naive T [TN] cells, T effector memory [TEM] cells, and T terminally differentiated [TTD] cells), seven CD4+ T cell subpopulations (TN cells, T central memory [TCM] cells, TEM cells, regulatory T [TREG] cells, T helper 2 [Th2] cells, T helper 17 [Th17] cells, and TTDcells), one gamma delta [γδ] T cell subpopulation, three monocyte (major histocompatibility complex [MHC] class II+ classical, MHC class II− classical, and non-classical monocytes), two NK cell subpopulations (NK-1, CD62L− and NK-2, CD62L+), one B cell subpopulation, and one dendritic cell (DC) subpopulation. T cells (CD4+ and CD8+ subsets) were most abundant in healthy donors’, responders’ and non-responders’ PBMCs (Figure S5A). Moreover, after neoadjuvant nivolumab treatment, the DC subpopulation was greatly compromised in the non-responder (versus responder) group. Before treatment, the level of B cells was significantly higher in the non-responder group (versus healthy donors) (Figure S5A).
Figure 5.

Elevated ratio of TREG to Th17 cells in peripheral blood as a pretreatment marker of non-response
(A) t-distribution stochastic neighbor embedding (t-SNE) map of live cell clusters and immune subpopulations in pre- and post-treatment PBMCs analyzed by CyTOF (n = 5 responders, n = 4 non-responders, n = 4 healthy donors).
(B) Heatmap showing the expression values of immune phenotypic protein markers normalized to the maximum mean value across subpopulations.
(C) Frequencies of CD4+ T cell subpopulations in the total T cell population in responders versus non-responders before or after neoadjuvant nivolumab therapy. p value, Student’s t test; ∗∗p < 0.01.
(D) Ratios of frequencies of TREG versus Th17 cells. p value, Student’s t test; ∗p < 0.05.
(E) Pearson correlations of the pretreatment PBMC TREG/Th17 cell ratios with pretreatment intratumoral levels of CD8+ T cells, cytolytic activity signature enrichment, effector T cell signature enrichment, IFNG-6 genes signature enrichment, PD-L1 expression, and Gini indices of TCRβ clonotypes in pretreatment PBMCs or post-treatment tumors.
We then evaluated differences in the most abundant T cell subpopulations (Figures 5C and S5B). Importantly, we observed a significantly higher level of CD4+ TREG cells in the pretreatment blood of non-responders (versus those in healthy donors, pretreatment responders, or post-treatment non-responders) (Figure 5C). In this context, the level of FOXP3+ TREG cells was significantly higher in the pretreatment peripheral blood of non-responders (versus responders) to PD-1 blockade in individuals with non-small cell lung carcinoma.34 Interestingly, after neoadjuvant nivolumab treatment, TREG cell levels increased in the responders but decreased in the non-responders. Contrary to TREG cells, CD4+ Th17 cells trended higher in pretreatment PBMCs of responders (versus non-responders), but this difference was not significant. Considering their opposing functional effects on antitumor CD8+ T cell immunity, we calculated the TREG/Th17 cell ratio for each group and treatment time point. Notably, a 6-fold higher TREG/Th17 cell ratio was observed in pretreatment PBMCs of the non-responder (versus responder) group (Figure 5D). Also, pretreatment PBMC TREG/Th17 cell ratios were negatively correlated with cytolytic activity and effector T cell signatures in pretreatment tumors, positively correlated with TCRβ clonality in pretreatment blood, but negatively correlated with TCRβ clonality in post-treatment tumors (Figure 5E). Furthermore, individuals with lower pretreatment blood TREG/Th17 cell ratios tended to display improved RFS and OS, although the differences did not reach statistical significance (Figure S5C). Hence, elevation of the pretreatment peripheral blood TREG/Th17 cell ratio may be predictive of innate resistance and reduced survival after neoadjuvant anti-PD-1 therapy.
Discussion
Early studies to understand innate response or resistance to anti-PD-1 therapy in advanced human malignancies pointed to the importance of TMB,35,36 T cell infiltration into tumor cores,37 intratumoral immune-suppressive processes, as well as cellular differentiation states.35 In individuals with HPV-negative, locally advanced, treatment-naive OCSCC treated with neoadjuvant nivolumab, we identified TMB, mutations in specific genes (CDKN2A, FLT4, and YAP1), intratumoral PPARG/PTEN signatures, and peripheral blood TREG/Th17 cell ratio as putative pretreatment predictive biomarkers.
TMBs were not different between responders and non-responders. However, a higher TMB was predictive of improved RFS. Two individuals (individuals 1 and 12) with low-TMB pretreatment tumors displayed tumor responses. Individual 12 was lost to follow-up after 0.28 years. However, individual 1 relapsed within 0.58 years after treatment but remains alive as of the most recent follow-up after 3.2 years, suggesting that other factors, such as a high pretreatment PTEN gene dosage, could have compensated for the low TMB. Consistent with this hypothesis, a recurrent tumor from individual 1 displayed loss of PTEN CN, transcript, and protein levels. Moreover, CDKN2A loss-of-function mutations were observed at a higher-than-expected frequency among non-responders. In advanced melanoma, innate resistance to anti-PD-1 therapy trended with CCND1 CN gain and CDKN2A CN loss.38 However, within subsets (acral melanoma and melanoma of unknown primary), this association (between innate anti-PD-1 resistance and CCND1 CN gain or CDKN2A CN loss) was significant. In clinical melanoma, progressive tumors on ICB lose senescence-inducing genes such as CDKN2A.39 Mechanistically, in preclinical models, Cdkn2a deletion abrogated ICB-elicited tumor control, suggesting that induction of tumor cell senescence may be important to prevent progression of tumor clones that escape immune-mediated tumor cell cytotoxicity.39 It is currently unclear whether FLT4 mutations enriched in the pretreatment tumors of responders are gain- or loss-of-function mutations. Lack of detection of the FLT4 mutant allele in both recurrent tumors, from a responder whose pretreatment tumor harbored a FLT4 mutation, supports FLT4 missense mutations as gain-of-function mutations. FLT4 (VEGFR3) promotes lymphangiogenesis, although little is known regarding effects of its mutations on cancer hallmarks. Recent studies of clinical colorectal carcinoma and clinical melanoma have correlated lymphatic vessel density and lymphatic gene expression to cytotoxic T cell density and immune infiltration, respectively.40,41 In mice lacking dermal lymphatics, fewer immune cells infiltrate melanoma.42 Thus, tumor-elicited lymphangiogenesis may promote immune infiltration, perhaps by increasing trafficking of tumor antigens and antigen-presenting cells to draining lymph nodes and facilitating T cell priming. Furthermore, the finding of co-enriched PPARG/PTEN gene sets in responsive pretreatment tumors implicates COX-2 as a co-target because PPARγ serves to adaptively temper COX-2-mediated inflammation.43 The action of PPARγ may be mediated, at least in part, by PTEN upregulation27,28, which is supported here by the positive correlation between PPARG and PTEN signature enrichments among pretreatment OCSCC tumors.
Despite our small sample size, retracing the evolutionary histories of several individuals’ disease provided clues to gene alterations potentially driving the patterns of initial responses and subsequent post-surgical recurrences. Several observations link determinants of initial response patterns with those of recurrence. As examples, CDKN2A deletions were enriched among initial non-responders and detected in a recurrent tumor. A PTEN signature was enriched among responders and its deletion detected in a recurrent tumor. YAP1 amplification occurred in the pretreatment tumor of a non-responder; its amplification characterized two recurrent tumors in an initially responding individual. FLT4 missense mutations were enriched among initial responders. Among one of these responders, whose pretreatment tumor carried a FLT4 mutation, both matched recurrent tumors had lost the FLT4 mutant allele.
Systemic TCR repertoire diversification before and especially after neoadjuvant nivolumab treatment in OCSCC is associated with tumor response and may be predictive of improved survival. In this context, high pretreatment diversity of TCR clones in the peripheral blood has been associated with improved outcomes in individuals with melanoma treated with anti-PD-1 or anti-CTLA-4 therapy.44,45 In a recent study of individuals with non-small cell lung carcinoma (NSCLC) treated with anti-PD-L1 therapy, induction of TCR diversity in circulating T cells on day 15 was significantly associated with improved OS.46 Moreover, expansion of the preexisting intratumoral TCR repertoire after neoadjuvant nivolumab treatment was positively correlated with tumor shrinkage. We also identified the level of FOXP3+ TREG cells to be significantly higher in the pretreatment peripheral blood of non-responders (versus responders). Because PD-1 signaling restrains the suppressive activity of TREG cells,47 this pattern suggests that TREG cell co-targeting may improve responsiveness to neoadjuvant anti-PD-1 therapy. In addition, determining the pretreatment ratio of TREG/Th17 cells in peripheral blood may be an important component of pretreatment analytics to stratify individuals for neoadjuvant anti-PD-1 therapy as well as for adjuvant treatment intensification versus de-escalation.
Here we used response versus non-response status based on tumor size changes measured radiographically before nivolumab treatment and pathologically after nivolumab treatment. Although there was a short time interval between post-nivolumab radiographic and pathologic assessment (Figure S1), 0 of 12 individuals had a partial response based on the former, but 4 of 12 had a partial response based on the latter (Table S2). This type of discrepancy has been noted before. For example, in individuals with early-stage NSCLC, only two experienced a radiographic partial response despite a high rate of major pathologic response, and two tumors that had increased in size after treatment harbored minimal residual tumor in the surgical specimen.48 All tumors in this study that responded pathologically also displayed a reduction in tumor size radiographically. Our pathology-enhanced response criteria facilitated this correlative analysis.
In three small cohorts, including the current cohort, variations in the efficacy of neoadjuvant PD-1 blockade in resectable OCSCC are likely due to variations in treatment (one to four doses of nivolumab or pembrolizumab) and evaluation protocols. One study (nivolumab, 2 doses)11 reported 13% response based on RECIST and 54% pathologic responses, with one of 12 individuals displaying a major pathologic response (>90%). Another study (pembrolizumab, 1 dose)12 reported 44% pathologic response ≥10%, with no major pathologic response observed. In the clinical trial related to this study, we observed a 33% overall response rate based on pathology-enhanced RECIST. These early clinical findings warrant larger studies that should conform treatment and evaluation standards to facilitate validation of the molecular biomarkers nominated here.
Limitations of study
More mature follow-up of survival data and larger cohorts are needed to improve identification of significant associations and detect/validate the predictive capabilities of key correlates of RFS and OS. Functional analyses of nominated pathways using in vivo models of OCSCC are needed to add mechanistic insights to this correlative study.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD26 (BA5b) | BioLegend | Cat# 302702, RRID:AB_314286 |
| CD4 (RPA-T4) | Fluidigm | Cat# 3145001B, RRID:AB_2661789 |
| CCR6 (G034E3) | Fluidigm | Cat# 3141003A, RRID:AB_2687639 |
| CD11a (HI111) | Fluidigm | Cat# 3142006B, RRID:AB_2877095 |
| CD45RA (HI100) | Fluidigm | Cat# 3143006B, RRID:AB_2651156 |
| CD11c (Bu15) | Fluidigm | Cat# 3147008B, RRID:AB_2687850 |
| CD16 (3G8) | Fluidigm | Cat# 3148004B, RRID:AB_2661791 |
| CD62L (DREG56) | Fluidigm | Cat# 3153004B, RRID:AB_2810245 |
| TIM3 (F38-2E2) | Fluidigm | Cat# 3154010B, RRID:AB_2893002 |
| CXCR3 (G025H7) | Fluidigm | Cat# 3156004B, RRID:AB_2687646 |
| CCR4 (L291H4) | Fluidigm | Cat# 3158032A, RRID:AB_2893003 |
| CCR7 (G043H7) | Fluidigm | Cat# 3159003A, RRID:AB_2714155 |
| CD28 (CD28.2) | Fluidigm | Cat# 3160003B, RRID:AB_2868400 |
| CTLA4 (14D3) | ThermoFisher | Cat# 14-1529-82, RRID:AB_467512 |
| FoxP3 (PCH101) | Fluidigm | Cat# 3162011A, RRID:AB_2687650 |
| CD45RO (UCHL1) | Fluidigm | Cat# 3165011B, RRID:AB_2756423 |
| CD57 (HCD57) | Fluidigm | Cat# 3172009B, RRID:AB_2888930 |
| HLA-DR (L243) | Fluidigm | Cat# 3173005B, RRID:AB_2810248 |
| CD94 (HP3D9) | Fluidigm | Cat# 3174015B, RRID:AB_2756429 |
| CD127 (A019D5) | Fluidigm | Cat# 3176004B, RRID:AB_2687863 |
| CD27 (L128) | Fluidigm | Cat# 3155001B, RRID:AB_2687645 |
| CD44 (BJ18) | Fluidigm | Cat# 3166001B, RRID:AB_2744692 |
| CD11b (ICRF44) | Fluidigm | Cat# 3209003B, RRID:AB_2687654 |
| CD38 (HIT2) | Fluidigm | Cat# 3167001B, RRID:AB_2802110 |
| Ki-67 (B56) | Fluidigm | Cat# 3168007B, RRID:AB_2800467 |
| PD-1 (EH12.2H7) | Fluidigm | Cat# 3175008B, RRID:AB_2687629 |
| ICOS (C398.4A) | Fluidigm | Cat# 3168024B, RRID:AB_2858237 |
| Pan Cytokeratin antibody [AE1/AE3] | Abcam | Cat# ab27988, RRID:AB_777047 |
| PTEN antibody | GeneTex | Cat# GTX101025, RRID:AB_1241223 |
| JAK2 antibody [EPR108(2)] | Abcam | Cat# ab108596, RRID:AB_10865183 |
| YAP1 antibody [EP1674Y] | Abcam | Cat# ab52771, RRID:AB_2219141 |
| MDM2 (D1V2Z) antibody | Cell Signaling Technology | Cat# 86934, RRID:AB_2784534 |
| Goat Anti-Mouse IgG (H+L) Highly Cross-adsorbed Antibody, Alexa Fluor 488 Conjugated | Molecular Probes | Cat# A-11029, RRID:AB_138404 |
| Goat Anti-Rabbit IgG (H+L) Highly Cross-adsorbed Antibody, Alexa Fluor 555 Conjugated | Molecular Probes | Cat# A-21429, RRID:AB_2535850 |
| DISC. OmniMap ANTI-MS HRP RUO | Roche | Cat# 760-4310, RRID:AB_2885182 |
| DISC. OmniMap ANTI-Rb HRP RUO | Roche | Cat# 760-4311, RRID:AB_2811043 |
| CD3 | Roche | Cat# 790-4341, RRID:AB_2335978 |
| CD8 | Leica | Cat# CD8-4B11-L-CE, AB_10555292 |
| Granzyme B | Leica | Cat# NCL-L-GRAN-B, RRID:AB_563751 |
| FOXP3 | Cell Signaling Technology | Cat# 98377S, RRID:AB_2747370 |
| Ki-67 | Agilent | Cat# M724029-2, RRID:AB_2893005 |
| Pan Cytokeratin antibody [AE1/AE3] | Roche | Cat# 760-2135, RRID:AB_2810237 |
| Biological samples | ||
| Patient-derived tissues | Table S3 | Table S3 |
| Chemicals, peptides, and recombinant proteins | ||
| Discovery Inhibitor | Roche | Cat# 760-4840 |
| Citrate Buffer, pH 6.0, 10x, Antigen Retriever | Sigma Aldrich | C9999-1000ML |
| Critical commercial assays | ||
| AllPrep DNA/RNA Mini Kit | QIAGEN | Cat# 80204 |
| mirVana miRNA Isolation Kit, with phenol | Thermo Fischer Scientific | Cat# AM1560 |
| QIAamp DNA FFPE Tissue Kit | QIAGEN | Cat# 56404 |
| FlexiGene DNA Kit | QIAGEN | Cat# 51206 |
| Deposited data | ||
| WES of tumor tissues and matched normal tissues | This Paper | SRA: PRJNA744256 |
| RNA-seq of tumor tissues | This Paper | GEO: GSE179730 |
| Mass cytometry data of patient- and healthy donor-derived PBMCs | This Paper | FlowRepository: FR-FCM-Z475 |
| TCR-seq of matched patient-derived tumors and PBMCs | This Paper | https://clients.adaptivebiotech.com/pub/liu-2021-crm |
| Software and algorithms | ||
| cytofkit | Bioconductor | Version: 3.7 |
| R software | CRAN | Version: 3.5.1 |
| GraphPad Prism | https://swcstore.oit.ucla.edu/secure/browse_vendors.php | Version: 7 |
| Cytobank | https://www.cytobank.org/ | N/A |
| tcR | CRAN | Version: 2.2.4.1 |
| BWA | http://bio-bwa.sourceforge.net/ | Version: 0.7.15 |
| Picard | https://broadinstitute.github.io/picard/ | Version: 1.141 |
| gatk | https://gatk.broadinstitute.org/hc/en-us | Version: 3.8 |
| Samtools | http://www.htslib.org/ | Version: 0.1.19 |
| MuTect | https://software.broadinstitute.org/cancer/cga/mutect | Version: 1.1.7 |
| VarScan2 | http://varscan.sourceforge.net/ | Version: 2.4.3 |
| Oncotator | https://software.broadinstitute.org/cancer/cga/oncotator | Version: 1.9.9.0 |
| Sequenza | http://www.cbs.dtu.dk/biotools/sequenza/ | Version: 2.1.2 |
| PHYLIP | https://evolution.genetics.washington.edu/phylip.html | Version: 3.698 |
| POLYSOLVER | https://software.broadinstitute.org/cancer/cga/polysolver_run | Version: 4.2 |
| HISAT2 | https://ccb.jhu.edu/software/hisat2/manual.shtml | Version: 2.0.6 |
| HTSeq | https://htseq.readthedocs.io/en/release_0.11.1/count.html | Version: 0.5.4 |
| GeoTcgaData | CRAN | Version: 0.2.5 |
| GSVA | Bioconductor | Version: 1.34.0 |
| CIBERSORTx | https://cibersortx.stanford.edu/ | N/A |
| survival | CRAN | Version: 3.1.8 |
| Phenochart viewer | Akoya Biosciences | Version 1.0.12 |
| inForm software | Akoya Biosciences | Version 2.4.4 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Roger S. Lo (rlo@mednet.ucla.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Human subjects
Patients were screened and/or enrolled in a clinical trial with informed consents obtained from all patients and participation approved by institutional review board (Pro00062193). We collected tissue (peripheral blood, tumor) samples from 12 patients with OCSCC who were treated with neoadjuvant nivolumab therapy. Informed consent for tissue samples used in this paper was obtained from all patients. PBMCs from peripheral blood were analyzed directly, without maintenance or expansion in culture. The clinical characteristics as well as radiographic and pathological measurements of tumor sizes are in Tables S1 and S2. The designs of associated clinical trial and this correlative study are shown in Figure S1. Table S3 displays the list of tissues collected and multi-omic analyses performed in this study. Given the short duration of nivolumab therapy, responders were defined as patients who derived clinical benefit (complete response, partial response, and stable disease per RECIST 1.1), and non-responders were defined as patients who derived no clinical benefit (progression per RECIST 1.1).
Method details
WES and RNA-seq data generation
gDNAs and total RNAs were extracted from snap-frozen tumor tissues using the QIAGEN AllPrep DNA/RNA Mini Kit and the Ambion mirVana miRNA Isolation Kit. Formalin-fixed paraffin-embedded (FFPE) tumor tissues were extracted for gDNA using the QIAGEN QIAamp DNA FFPE Tissue Kit. Patient-matched normal gDNA from viably frozen PBMCs were extracted using the QIAGEN FlexiGene DNA Kit. Frozen tissue-derived and FFPE tissue-derived gDNA libraries were constructed using the Roche Kapa HyperPlus Library Preparation Kit. Briefly, after enzymatic fragmentation of gDNAs, libraries were constructed by end-repairing and A-tailing the fragmented gDNAs, ligation of adapters, and PCR amplification. After library construction, indexed frozen tissue-derived and FFPE tissue-derived libraries were separately pooled and then hybridized using SeqCap EZ HyperCap Workflow v2.1 and Kapa HyperCap Workflow v3.0, respectively, followed by PCR amplification. Finally, indexed DNA libraries were quantified for equal molar pooling and paired-end sequenced with a read length of 2x150 bp on the Illumina NovaSeq 6000 S4 platform. RNA libraries were constructed using the NuGEN Universal Plus mRNA-Seq with NuQuant Library Preparation Kit to enrich for all poly(A) transcripts within the transcriptome. Briefly, after RNA fragmentation, double-stranded cDNAs were generated using a mixture of random and oligo(dT) priming. Then the libraries were constructed by end-repairing the cDNAs to generate blunt ends, ligation of unique dual index (UDI) adapters, followed by strand selection and PCR amplification. Finally, indexed cDNA libraries were quantified for equal molar pooling and paired-end sequenced with a read length of 2x150 bp on the Illumina NovaSeq 6000 S4 platform. In total, 16 tumors from 12 patients and patient-matched normal PBMC samples were subjected to WES, and 23 tumors from 11 patients were subjected to RNA-seq.
WES and RNA-seq data processing
Somatic mutation calling, copy-number analysis, and phylogeny
We conducted somatic variant calling for single-nucleotide variants (SNVs) and small insertion-deletions (INDELs) as we previously reported.20,35,49, 50, 51, 52 Mutations were then annotated by using Oncotator.53 Tumor purity, ploidy, and somatic copy-number alterations (CNAs) were detected by Sequenza.54 Characteristics of WES data are summarized in Table S4. Phylogenetic analyses were performed using the PHYLIP program with the parsimony algorithm, as we previously reported.51
HLA genotyping and mutation calling
HLA typing for each patient was inferred based on normal blood WES data using the POLYSOLVER algorithm16. HLA mutation calling for HLA-A, HLA-B, and HLA-C genes was performed using the POLYSOLVER-based mutation detection pipeline from the Broad Institute’s Polysolver Docker container (https://software.broadinstitute.org/cancer/cga/polysolver_run).
RNA-seq data analysis
We analyzed paired-end 2x150bp RNA-seq data according to the pipeline we reported recently.49 Briefly, paired-end transcriptome reads were mapped to the Genome Reference Consortium Human Build 37 (GRCh37) reference genome using HISAT2,55 and then gene-level counts were estimated by the htseqcount program,56 The normalized expression level of each gene, TPM, was calculated by the R package GeoTcgaData. By using the R package GSVA,57 we performed single-sample gene set enrichment analysis to generate absolute enrichment scores of the collections of gene sets from the Broad Institute’s Molecular Signatures Database (C2 oncogenic gene sets and C7 immunologic gene sets) and gene signatures previously reported to be associated with ICB response. TPM values were used as input into the GSVA program using the default ‘kcdf = Gaussian’ option. Differentially enriched gene sets between the responder versus non-responders, pre- and post-treatment samples, were defined by the sum of differences in enrichment scores being greater than 0.3 and a t test p value being less than 0.05. CIBERSORTx58 was used in the ‘absolute mode’ to estimate infiltration levels of 22 immune cell types with TPM values as the input.
Analysis of public genomic datasets
We downloaded the normalized gene expression levels of a RNA-seq dataset from HNSCC patients (Head and Neck Squamous Cell Carcinoma; TCGA, Firehose Legacy) from cBioPortal. CIBERSORTx was used to estimate the abundance of 22 immune cell types with the normalized expression levels as input. FLT4 genotypes (FLT4Mut or FLT4WT) in each patient was obtained from cBioPortal and then mapped to patient IDs in the RNA-seq dataset. Group comparison between FLT4Mut versus FLT4WT patients was performed using the enrichment level of each immune cell type with the Wilcoxon rank sum test.
CyTOF data generation from PBMCs
After thawing, PBMCs were live/dead stained with 200 μM Rh-103 (Fluidigm) for 2 min at room temperature. To achieve increased throughput and homogeneous staining, metal cell barcoding against human immune CD45+ cells was used. The metal isotopes (Trace Sciences International, Richmond Hill, ON, Canada) used for barcoding were: 105Pd, 106Pd, 108Pd, 111In, 115In, 194Pt, 195Pt, 196Pt, and 198Pt. Metal barcoding reagents were prepared by combining 2 molar equivalents of isothiocyanobenzyl-EDTA (Dojindo Molecular Technologies, Rockville, MD) with 1 molar equivalent of metal chloride in ammonium acetate buffer (20 mM, pH 6.0). Chelated metal solutions were immediately lyophilized and dissolved in DMSO at 10 mM final concentration for long-term storage at −20°C. Pd-loaded SCN-Bn-EDTA stock was thawed and 6.4 μL were added to 100 μg of the anti-human CD45 antibody (clone: HI30) dissolved in a total of 313 μL PBS, mixed by pipetting and incubated for 1 h at 37°C. The conjugate was washed at least three times with 300 μL PBS over a 50 kDa spin filter for 10 min at 4°C and 12,500 x g, then transferred to a 1.6 mL microcentrifuge tube. Protein concentration was quantified by Nanodrop (Thermo Fisher, Waltham, MA, USA) at 280 nm; antibody stabilizer (Candor Biosciences, Wangen, Germany) was added to the preparation at a 1:1 ratio; and antibodies were kept at 4°C. Barcoding reagents were titrated to achieve optimal labeling. A unique-to-each-sample combination of exactly 3 metal cell barcoding reagents diluted in 300 uL PBS was added and then incubated for 20 min at room temperature. Cells were washed twice with 1 mL PBS at 4°C. Barcoded cells were then combined in a single tube and washed with cell staining buffer (CSB, PBS + 0.5% BSA + 2 mM EDTA). Surface proteins were stained with antibodies at 37°C for 20 min and for an additional 10 min at 4°C. Cells were washed in CSB and incubated overnight with 250 nM iridium intercalator (Fluidigm) in Maxpar cell fix/perm buffer (Fluidigm) to label cellular DNA. Subsequently, cells were washed with PBS followed by distilled water and resuspended in 10% EQ beads (Fluidigm) in distilled water. Mass cytometry acquisition was performed on a CyTOF2.1 (Helios) mass cytometer (Fluidigm).
CyTOF data analysis
Mass cytometry flow cytometry standard (FCS) data files were concatenated, bead-normalized, and debarcoded using Helios software (Fluidigm). Data were then exported into individual files for each sample. Total live cell populations were manually identified and exported using negative and positive gating strategies in Cytobank.59 We applied Cytofkit60 to perform the t-Distribution Stochastic Neighbor Embedding (t-SNE) analysis separately on the manually gated live-cell populations. We selected 5,000 in each sample to ensure equal representation of cells across samples. All the cell lineage markers in the immune panel were used in clustering analysis. We chose 3,000 iterations, perplexity of 30, and theta of 0.5 as the standard t-SNE parameters. Mean intensity values of markers in each cluster were calculated and visualized via heatmaps. Cells were assigned to different functional populations on the basis of the local gradient expression of known cell lineage markers. Based on expression of known marker genes, clusters were annotated as MHC II- classical monocytes (CD14+CD11b+CD16-HLA-DR-), MHC II+ classical monocytes (CD14+CD11b+CD16-HLA-DR+), non-classical monocytes (CD14+CD11b+CD16+), dendritic cells or DCs (CD33+CD11c+HLA-DR+), B cells (CD19+), T cell subsets (naive or TN, CD45RA+CD62L+CCR7+CD45RO-; effector memory or TEM, CD45RA-CCR7-CD45RO+; central memory or TCM, CD45RA-CCR7+CD45RO+; T terminally differentiated or TTD, CD45RA+CCR7-CD27-CD28-; regulatory T or TREG, CD4+FOXP3+; T helper 2 or Th2, CD4+CCR4+; T helper 17 or Th17, CD4+CD26hi; Gamma delta or γδTC, CD3+TCRgd+), NK1 (CD3+CD94+CD16+CD62L-) and NK2 (CD3+CD94+CD16+CD62L+). The percentages of different immune cell subsets were calculated for each sample. We defined a TREG/Th17 ratio as the fold change of frequencies between TREG and Th17 cells.
Generation and analysis of TCR-seq data
gDNAs were isolated from patient-matched PBMCs and tumor tissues using Maxwell RSC DNA from Cells and DNA from Tissue kits, respectively (Promega, Madison, WI). TCRβ libraries were prepared using the ImmunoSeq hsTCRβ kit (Adaptive Biotechnologies, Seattle, WA) according to manufacturer’s instructions. Briefly, TCRβ libraries were generated from PBMC gDNA samples (480 ng input DNA except for matched samples from Pt7 at 310.4 ng input DNA) for deep sequencing (6 replicates per sample, except for Pt7 post-treatment, for which 5 replicates were generated due to limited gDNA recovery) and from tumor gDNA samples (4.8 μg input DNA except for matched samples from Pt4 at 1.44 μg input DNA and matched samples from Pt9 at 1.96 μg input DNA) for survey sequencing (2 replicates per sample). Final libraries were pooled at a concentration of 3 nM and sequenced on an Illumina NovaSeq 6000 S4 flow cell at VANTAGE (Vanderbilt University, Nashville, TN).
We performed pre-processing and quality control of the raw data by using the immunoSEQ analyzer (Adaptive Biotechnologies, Inc.). We then exported measurement metrics of processed data into the tsv file. Only productive rearrangements and corresponding productive CDR3 amino acid sequences were considered for downstream analysis. Clonotypes were defined by unique CDR3 amino acid sequences. The clonality of TCR repertoires was estimated through calculating the Gini-Simpson index by the R package tcR.61
Survival analysis
Survival analyses for RFS and OS were carried out via the two-sided log-rank test by the R package survival. We compared RFS and OS in responders versus non-responders, and patients with high-levels versus low-levels of a certain factor. High- or low-levels of a certain factor were defined, respectively, by the values of the factor ≥ median value or values of the factor < median value across the cohorts.
Immunofluorescence (IF) analysis
Tumor tissues were fixed in formalin followed by paraffin-embedding. After deparaffinization and rehydration, tissue sections were antigen-retrieved by heat. Permeabilization and blocking were followed by overnight incubation with primary antibodies [pan-cytokeratin (Abcam, ab27988), PTEN (Genetex, GTX101025)], JAK2 (Abcam, ab108596), YAP1 (Abcam, ab52771), and MDM2 (Cell Signaling Technology, 86934). IF was performed with Alexa Fluor–conjugated secondary antibodies (Life Technologies, A-11029, A-21429). Nuclei were counterstained by DAPI. Signals were captured with a Zeiss microscope (AXIO Imager A1) mounted with a charge-coupled device camera (Retiga EXi QImaging), and the images captured by Image-Pro plus 6.0. Representative images are shown. Digitized images of whole-slide stains are available upon request.
Multiplex IF analysis
mIF was performed utilizing Ventana Discovery Ultra (Roche) and Opal fluorophores (Akoya Biosciences). Five micrometer-thick tissue sections on Superfrost microscopic slides (VWR International) were deparaffinized using EZ-Prep reagent (Roche) followed by antigen retrieval in CC1 buffer (pH 9, 95°C; Roche). Discovery Inhibitor (Roche) was applied to inhibit enzymatic activities followed by 6 sequential rounds of staining. Each round included the addition of a primary antibody followed by detection using the OmniMap secondary antibody (Roche). Signal amplification was performed utilizing Opal fluorophores in the conditions suggested by the manufacturer. Between rounds of staining the tissue sections underwent heat-induced epitope retrieval to remove the primary-secondary-HRP antibody complexes before staining with the subsequent antibody. The primary antibodies and corresponding fluorophores are PanCK (DAKO) in Opal 480; PD-L1 (Cell Signaling) in Opal 520; CD68 (DAKO) in Opal 570; Granzyme B (Leica) in Opal 620; CD8 (Leica) in Opal 690, and CD3 (Roche) in Opal 780. The slides were then counterstained with Spectral DAPI (Akoya Biosciences) and mounted with ProLong Diamond antifade mounting medium (Thermo Fisher Scientific).
Stained slides were imaged using the Vectra Polaris imaging system (Akoya Biosciences). A whole slide scan was acquired with 20x resolution. Following image capture, regions of interest (ROIs) were selected on each slide using the Phenochart viewer (Akoya Biosciences) and imported into the inForm software (Akoya Biosciences) followed by unmixing the spectral libraries, cell segmentation and cell phenotyping. ROIs corresponding to whole tumor regions from each slide were then analyzed to identify and characterize the cells. Data were then exported and graphed with Prism (GraphPad). Representative images were exported using inForm software following spectral umixing.
Quantification and statistical analysis
Pretreatment tumor TMBs (responsive versus non-responsive) and infiltration levels of CD8+ T, TREG and resting NK cells (FLT4WT versus FLT4Mut in HNSCC tumors from a public dataset in cBioPortal) were compared using the Wilcoxon rank-sum test. Group differences of HLA-I homozygosity (responsive versus non-responsive tumors) and mutation frequencies of FLT4 (responsive pretreatment tumors versus HNSCC tumors in cBioPortal) and CDKN2A (non-responsive pretreatment tumors versus HNSCC tumors in cBioPortal) were tested by using Fisher’s exact test. The mutation frequencies were then subjected to multiple testing corrections with the Benjamin-Hochberg method. Student’s t test was used to compare differences in Gini indices of TCRβ clones in tumors and PBMCs before or after treatment, frequencies in T cell functional subpopulations and ratios of frequencies of TREG versus Th17 cells. Statistical associations between any two variables (e.g., TREG /Th17 ratios and TCRβ Gini indices) in this study were measured by the Pearson correlation coefficient. In the aforementioned tests, P values < 0.05 were considered statistically significant. Statistical details of each experiment are reported in the respective text (methods and/or Results section) and figure legends. Statistical analyses were performed using R version 3.5.1.
Additional resources
ClinicalTrials.gov Identifier: NCT03021993
Acknowledgments
We thank all members of the Lo Laboratory for critical comments. This research was supported by grants (to R.S.L.) from the National Institutes of Health (NIH) (1R01CA176111A1, 1R21CA215910-01, R21CA255837-01, and 1P01CA168585), the Melanoma Research Alliance (MRA) (Team Science Award), and the V Foundation for Cancer Research (Translational Award). Additional funding was provided by the MRA Dermatology Fellows Award (to S.L. and Z.Y.); a Jonsson Comprehensive Cancer Center (JCCC) postdoctoral seed grant (to Z.Y.); JCCC postdoctoral fellowships (to S.L. and Z.Y.); the NIH T32CA009120 Tumor Immunology Postdoctoral Fellowship (to A.H.); NIH F30243307 and the American Head and Neck Society Pilot Award (to H.M.K.); NIH 5K08DE026542-03, SCRT UL1TR001450, and Bristol Myers Squibb CA209-831 (to D.M.N.); NIH R01CA175061 and R01CA208514 (to C.M.P.), and 1R21DE029592-01 (to C.M.P. and D.M.N). Additional support came from Mary Tanner and Maurizio Grimaldi and the Ressler Family Foundation. We thank Xinmin Li, PhD (Director) and the Technology Center for Genomics and Bioinformatics at UCLA and the Translational Science Shared Resource, Hollings Cancer Center, Medical University of South Carolina (P30 CA138313) for excellent technical support.
Author contributions
Conceptualization, S.L. and R.S.L.; methodology, S.L., H.M.K., S.H.L., A.H., M.R., Z.Y., R.J.L., Y.W., C.D., K.K., C.T., M.J.R., C.K., E.C.O., J.D.H., S.M.D., C.M.P., D.M.N., and R.S.L; formal analysis, S.L., A.H., Z.Y., R.J.L., and R.S.L.; investigation, S.L., H.M.K., S.H.L., A.H., Z.Y., R.J.L., K.K., S.M.D., C.M.P., D.M.N., and R.S.L.; resources, H.M.K., C.M.P., D.M.N., and R.S.L.; data curation, S.L. and R.S.L.; writing – original draft, S.L. and R.S.L.; writing – review & editing, S.L., H.M.K., S.H.L., A.H., Z.Y., R.J.L., Y.W., C.M.P., D.M.N., and R.S.L.; visualization, S.L., A.H., Z.Y., R.J.L., and R.S.L.; supervision, S.L. and R.S.L.; project administration, R.S.L.; funding acquisition, C.M.P., D.M.N., and R.S.L.
Declaration of interests
R.S.L. receives research or clinical trial support from Merck, Pfizer, BMS, and OncoSec. C.M.P. is a co-founder of Ares Immunotherapy.
Published: October 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100411.
Supplemental information
Data and code availability
Raw sequencing files of WES data are deposited at the SRA with accession number PRJNA744256. RNA-seq data are available at Gene Expression Omnibus (GSE179730). Mass cytometry data are deposited at FlowRepository (http://flowrepository.org/) using the experiment ID FR-FCM-Z475. T cell receptor sequencing (TCR-seq) data are available at ImmuneACCESS with the DOI link: https://clients.adaptivebiotech.com/pub/liu-2021-crm.
There are no original codes generated in this paper.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
References
- 1.Topalian S.L., Hodi F.S., Brahmer J.R., Gettinger S.N., Smith D.C., McDermott D.F., Powderly J.D., Carvajal R.D., Sosman J.A., Atkins M.B. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu J., Blake S.J., Yong M.C., Harjunpää H., Ngiow S.F., Takeda K., Young A., O’Donnell J.S., Allen S., Smyth M.J., Teng M.W. Improved Efficacy of Neoadjuvant Compared to Adjuvant Immunotherapy to Eradicate Metastatic Disease. Cancer Discov. 2016;6:1382–1399. doi: 10.1158/2159-8290.CD-16-0577. [DOI] [PubMed] [Google Scholar]
- 3.Blank C.U., Rozeman E.A., Fanchi L.F., Sikorska K., van de Wiel B., Kvistborg P., Krijgsman O., van den Braber M., Philips D., Broeks A. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat. Med. 2018;24:1655–1661. doi: 10.1038/s41591-018-0198-0. [DOI] [PubMed] [Google Scholar]
- 4.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 5.Leemans C.R., Snijders P.J.F., Brakenhoff R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer. 2018;18:269–282. doi: 10.1038/nrc.2018.11. [DOI] [PubMed] [Google Scholar]
- 6.Cooper J.S., Pajak T.F., Forastiere A.A., Jacobs J., Campbell B.H., Saxman S.B., Kish J.A., Kim H.E., Cmelak A.J., Rotman M., Radiation Therapy Oncology Group 9501/Intergroup Postoperative concurrent radiotherapy and chemotherapy for high-risk squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2004;350:1937–1944. doi: 10.1056/NEJMoa032646. [DOI] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas N., Cancer Genome Atlas Network Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–582. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burtness B., Harrington K.J., Greil R., Soulières D., Tahara M., de Castro G., Jr., Psyrri A., Basté N., Neupane P., Bratland Å., KEYNOTE-048 Investigators Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet. 2019;394:1915–1928. doi: 10.1016/S0140-6736(19)32591-7. [DOI] [PubMed] [Google Scholar]
- 9.Ferris R.L., Blumenschein G., Jr., Fayette J., Guigay J., Colevas A.D., Licitra L., Harrington K., Kasper S., Vokes E.E., Even C. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016;375:1856–1867. doi: 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seiwert T.Y., Burtness B., Mehra R., Weiss J., Berger R., Eder J.P., Heath K., McClanahan T., Lunceford J., Gause C. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016;17:956–965. doi: 10.1016/S1470-2045(16)30066-3. [DOI] [PubMed] [Google Scholar]
- 11.Schoenfeld J.D., Hanna G.J., Jo V.Y., Rawal B., Chen Y.H., Catalano P.S., Lako A., Ciantra Z., Weirather J.L., Criscitiello S. Neoadjuvant Nivolumab or Nivolumab Plus Ipilimumab in Untreated Oral Cavity Squamous Cell Carcinoma: A Phase 2 Open-Label Randomized Clinical Trial. JAMA Oncol. 2020;6:1563–1570. doi: 10.1001/jamaoncol.2020.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uppaluri R., Campbell K.M., Egloff A.M., Zolkind P., Skidmore Z.L., Nussenbaum B., Paniello R.C., Rich J.T., Jackson R., Pipkorn P. Neoadjuvant and Adjuvant Pembrolizumab in Resectable Locally Advanced, Human Papillomavirus-Unrelated Head and Neck Cancer: A Multicenter, Phase II Trial. Clin. Cancer Res. 2020;26:5140–5152. doi: 10.1158/1078-0432.CCR-20-1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knochelmann H.M., Horton J.D., Liu S., Armeson K., Kaczmar J.M., Wyatt M., Richardson M., Lomeli S.H., Xiong Y., Graboyes E.M. Neoadjuvant presurgical PD-1 inhibition in oral cavity squamous cell carcinoma. Cell Reports Medicine. 2021;2:100426-1–100426-10. doi: 10.1016/j.xcrm.2021.100426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chowell D., Morris L.G.T., Grigg C.M., Weber J.K., Samstein R.M., Makarov V., Kuo F., Kendall S.M., Requena D., Riaz N. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. 2018;359:582–587. doi: 10.1126/science.aao4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stransky N., Egloff A.M., Tward A.D., Kostic A.D., Cibulskis K., Sivachenko A., Kryukov G.V., Lawrence M.S., Sougnez C., McKenna A. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shukla S.A., Rooney M.S., Rajasagi M., Tiao G., Dixon P.M., Lawrence M.S., Stevens J., Lane W.J., Dellagatta J.L., Steelman S. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat. Biotechnol. 2015;33:1152–1158. doi: 10.1038/nbt.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.George S., Miao D., Demetri G.D., Adeegbe D., Rodig S.J., Shukla S., Lipschitz M., Amin-Mansour A., Raut C.P., Carter S.L. Loss of PTEN Is Associated with Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity. 2017;46:197–204. doi: 10.1016/j.immuni.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peng W., Chen J.Q., Liu C., Malu S., Creasy C., Tetzlaff M.T., Xu C., McKenzie J.A., Zhang C., Liang X. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6:202–216. doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao J., Chen A.X., Gartrell R.D., Silverman A.M., Aparicio L., Chu T., Bordbar D., Shan D., Samanamud J., Mahajan A. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019;25:462–469. doi: 10.1038/s41591-019-0349-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hugo W., Shi H., Sun L., Piva M., Song C., Kong X., Moriceau G., Hong A., Dahlman K.B., Johnson D.B. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell. 2015;162:1271–1285. doi: 10.1016/j.cell.2015.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim M.H., Kim C.G., Kim S.K., Shin S.J., Choe E.A., Park S.H., Shin E.C., Kim J. YAP-Induced PD-L1 Expression Drives Immune Evasion in BRAFi-Resistant Melanoma. Cancer Immunol. Res. 2018;6:255–266. doi: 10.1158/2326-6066.CIR-17-0320. [DOI] [PubMed] [Google Scholar]
- 22.Yu M., Peng Z., Qin M., Liu Y., Wang J., Zhang C., Lin J., Dong T., Wang L., Li S. Interferon-γ induces tumor resistance to anti-PD-1 immunotherapy by promoting YAP phase separation. Mol. Cell. 2021;81:1216–1230.e9. doi: 10.1016/j.molcel.2021.01.010. [DOI] [PubMed] [Google Scholar]
- 23.Kato S., Goodman A., Walavalkar V., Barkauskas D.A., Sharabi A., Kurzrock R. Hyperprogressors after Immunotherapy: Analysis of Genomic Alterations Associated with Accelerated Growth Rate. Clin. Cancer Res. 2017;23:4242–4250. doi: 10.1158/1078-0432.CCR-16-3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fang D.D., Tang Q., Kong Y., Wang Q., Gu J., Fang X., Zou P., Rong T., Wang J., Yang D., Zhai Y. MDM2 inhibitor APG-115 synergizes with PD-1 blockade through enhancing antitumor immunity in the tumor microenvironment. J. Immunother. Cancer. 2019;7:327. doi: 10.1186/s40425-019-0750-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sahin I., Zhang S., Navaraj A., Zhou L., Dizon D., Safran H., El-Deiry W.S. AMG-232 sensitizes high MDM2-expressing tumor cells to T-cell-mediated killing. Cell Death Discov. 2020;6:57. doi: 10.1038/s41420-020-0292-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korpal M., Puyang X., Jeremy Wu Z., Seiler R., Furman C., Oo H.Z., Seiler M., Irwin S., Subramanian V., Julie Joshi J. Evasion of immunosurveillance by genomic alterations of PPARγ/RXRα in bladder cancer. Nat. Commun. 2017;8:103. doi: 10.1038/s41467-017-00147-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonofiglio D., Gabriele S., Aquila S., Catalano S., Gentile M., Middea E., Giordano F., Andò S. Estrogen receptor alpha binds to peroxisome proliferator-activated receptor response element and negatively interferes with peroxisome proliferator-activated receptor gamma signaling in breast cancer cells. Clin. Cancer Res. 2005;11:6139–6147. doi: 10.1158/1078-0432.CCR-04-2453. [DOI] [PubMed] [Google Scholar]
- 28.Patel L., Pass I., Coxon P., Downes C.P., Smith S.A., Macphee C.H. Tumor suppressor and anti-inflammatory actions of PPARgamma agonists are mediated via upregulation of PTEN. Curr. Biol. 2001;11:764–768. doi: 10.1016/s0960-9822(01)00225-1. [DOI] [PubMed] [Google Scholar]
- 29.Iizuka N., Oka M., Yamada-Okabe H., Mori N., Tamesa T., Okada T., Takemoto N., Sakamoto K., Hamada K., Ishitsuka H. Self-organizing-map-based molecular signature representing the development of hepatocellular carcinoma. FEBS Lett. 2005;579:1089–1100. doi: 10.1016/j.febslet.2004.10.113. [DOI] [PubMed] [Google Scholar]
- 30.Iliopoulos D., Hirsch H.A., Wang G., Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc. Natl. Acad. Sci. USA. 2011;108:1397–1402. doi: 10.1073/pnas.1018898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bolen C.R., McCord R., Huet S., Frampton G.M., Bourgon R., Jardin F., Dartigues P., Punnoose E.A., Szafer-Glusman E., Xerri L. Mutation load and an effector T-cell gene signature may distinguish immunologically distinct and clinically relevant lymphoma subsets. Blood Adv. 2017;1:1884–1890. doi: 10.1182/bloodadvances.2016000786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ayers M., Lunceford J., Nebozhyn M., Murphy E., Loboda A., Kaufman D.R., Albright A., Cheng J.D., Kang S.P., Shankaran V. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 2017;127:2930–2940. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson B.J., Costelloe E.O., Fitzpatrick D.R., Haanen J.B., Schumacher T.N., Brown L.E., Kelso A. Single-cell perforin and granzyme expression reveals the anatomical localization of effector CD8+ T cells in influenza virus-infected mice. Proc. Natl. Acad. Sci. USA. 2003;100:2657–2662. doi: 10.1073/pnas.0538056100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kagamu H., Kitano S., Yamaguchi O., Yoshimura K., Horimoto K., Kitazawa M., Fukui K., Shiono A., Mouri A., Nishihara F. CD4(+) T-cell Immunity in the Peripheral Blood Correlates with Response to Anti-PD-1 Therapy. Cancer Immunol. Res. 2020;8:334–344. doi: 10.1158/2326-6066.CIR-19-0574. [DOI] [PubMed] [Google Scholar]
- 35.Hugo W., Zaretsky J.M., Sun L., Song C., Moreno B.H., Hu-Lieskovan S., Berent-Maoz B., Pang J., Chmielowski B., Cherry G. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rizvi N.A., Hellmann M.D., Snyder A., Kvistborg P., Makarov V., Havel J.J., Lee W., Yuan J., Wong P., Ho T.S. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tumeh P.C., Harview C.L., Yearley J.H., Shintaku I.P., Taylor E.J., Robert L., Chmielowski B., Spasic M., Henry G., Ciobanu V. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu J., Yan J., Guo Q., Chi Z., Tang B., Zheng B., Yu J., Yin T., Cheng Z., Wu X. Genetic Aberrations in the CDK4 Pathway Are Associated with Innate Resistance to PD-1 Blockade in Chinese Patients with Non-Cutaneous Melanoma. Clin. Cancer Res. 2019;25:6511–6523. doi: 10.1158/1078-0432.CCR-19-0475. [DOI] [PubMed] [Google Scholar]
- 39.Brenner E., Schörg B.F., Ahmetlić F., Wieder T., Hilke F.J., Simon N., Schroeder C., Demidov G., Riedel T., Fehrenbacher B. Cancer immune control needs senescence induction by interferon-dependent cell cycle regulator pathways in tumours. Nat. Commun. 2020;11:1335. doi: 10.1038/s41467-020-14987-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mlecnik B., Bindea G., Kirilovsky A., Angell H.K., Obenauf A.C., Tosolini M., Church S.E., Maby P., Vasaturo A., Angelova M. The tumor microenvironment and Immunoscore are critical determinants of dissemination to distant metastasis. Sci. Transl. Med. 2016;8:327ra26. doi: 10.1126/scitranslmed.aad6352. [DOI] [PubMed] [Google Scholar]
- 41.Cancer Genome Atlas Network Genomic Classification of Cutaneous Melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lund A.W., Wagner M., Fankhauser M., Steinskog E.S., Broggi M.A., Spranger S., Gajewski T.F., Alitalo K., Eikesdal H.P., Wiig H., Swartz M.A. Lymphatic vessels regulate immune microenvironments in human and murine melanoma. J. Clin. Invest. 2016;126:3389–3402. doi: 10.1172/JCI79434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Korbecki J., Bobiński R., Dutka M. Self-regulation of the inflammatory response by peroxisome proliferator-activated receptors. Inflamm. Res. 2019;68:443–458. doi: 10.1007/s00011-019-01231-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hogan S.A., Courtier A., Cheng P.F., Jaberg-Bentele N.F., Goldinger S.M., Manuel M., Perez S., Plantier N., Mouret J.F., Nguyen-Kim T.D.L. Peripheral Blood TCR Repertoire Profiling May Facilitate Patient Stratification for Immunotherapy against Melanoma. Cancer Immunol. Res. 2019;7:77–85. doi: 10.1158/2326-6066.CIR-18-0136. [DOI] [PubMed] [Google Scholar]
- 45.Postow M.A., Manuel M., Wong P., Yuan J., Dong Z., Liu C., Perez S., Tanneau I., Noel M., Courtier A. Peripheral T cell receptor diversity is associated with clinical outcomes following ipilimumab treatment in metastatic melanoma. J. Immunother. Cancer. 2015;3:23. doi: 10.1186/s40425-015-0070-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Naidus E., Bouquet J., Oh D.Y., Looney T.J., Yang H., Fong L., Standifer N.E., Zhang L. Early changes in the circulating T cells are associated with clinical outcomes after PD-L1 blockade by durvalumab in advanced NSCLC patients. Cancer Immunol. Immunother. 2021;70:2095–2102. doi: 10.1007/s00262-020-02833-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tan C.L., Kuchroo J.R., Sage P.T., Liang D., Francisco L.M., Buck J., Thaker Y.R., Zhang Q., McArdel S.L., Juneja V.R. PD-1 restraint of regulatory T cell suppressive activity is critical for immune tolerance. J. Exp. Med. 2021;218:e20182232. doi: 10.1084/jem.20182232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Forde P.M., Chaft J.E., Pardoll D.M. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N. Engl. J. Med. 2018;379:e14. doi: 10.1056/NEJMc1808251. [DOI] [PubMed] [Google Scholar]
- 49.Hong A., Piva M., Liu S., Hugo W., Lomeli S.H., Zoete V., Randolph C.E., Yang Z., Wang Y., Lee J.J. Durable Suppression of Acquired MEK Inhibitor Resistance in Cancer by Sequestering MEK from ERK and Promoting Antitumor T-cell Immunity. Cancer Discov. 2021;11:714–735. doi: 10.1158/2159-8290.CD-20-0873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moriceau G., Hugo W., Hong A., Shi H., Kong X., Yu C.C., Koya R.C., Samatar A.A., Khanlou N., Braun J. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell. 2015;27:240–256. doi: 10.1016/j.ccell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi H., Hugo W., Kong X., Hong A., Koya R.C., Moriceau G., Chodon T., Guo R., Johnson D.B., Dahlman K.B. Acquired Resistance and Clonal Evolution in Melanoma during BRAF Inhibitor Therapy. Cancer Discov. 2014;4:80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shi H., Moriceau G., Kong X., Lee M.K., Lee H., Koya R.C., Ng C., Chodon T., Scolyer R.A., Dahlman K.B. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat. Commun. 2012;3:724. doi: 10.1038/ncomms1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramos A.H., Lichtenstein L., Gupta M., Lawrence M.S., Pugh T.J., Saksena G., Meyerson M., Getz G. Oncotator: cancer variant annotation tool. Hum. Mutat. 2015;36:E2423–E2429. doi: 10.1002/humu.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Favero F., Joshi T., Marquard A.M., Birkbak N.J., Krzystanek M., Li Q., Szallasi Z., Eklund A.C. Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann. Oncol. 2015;26:64–70. doi: 10.1093/annonc/mdu479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anders S., Pyl P.T., Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Newman A.M., Steen C.B., Liu C.L., Gentles A.J., Chaudhuri A.A., Scherer F., Khodadoust M.S., Esfahani M.S., Luca B.A., Steiner D. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019;37:773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kotecha N., Krutzik P.O., Irish J.M. Web-based analysis and publication of flow cytometry experiments. Curr. Protoc. Cytom. 2010 doi: 10.1002/0471142956.cy1017s53. Chapter 10, Unit 10.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen H., Lau M.C., Wong M.T., Newell E.W., Poidinger M., Chen J. Cytofkit: A Bioconductor Package for an Integrated Mass Cytometry Data Analysis Pipeline. PLoS Comput. Biol. 2016;12:e1005112. doi: 10.1371/journal.pcbi.1005112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nazarov V.I., Pogorelyy M.V., Komech E.A., Zvyagin I.V., Bolotin D.A., Shugay M., Chudakov D.M., Lebedev Y.B., Mamedov I.Z. tcR: an R package for T cell receptor repertoire advanced data analysis. BMC Bioinformatics. 2015;16:175. doi: 10.1186/s12859-015-0613-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing files of WES data are deposited at the SRA with accession number PRJNA744256. RNA-seq data are available at Gene Expression Omnibus (GSE179730). Mass cytometry data are deposited at FlowRepository (http://flowrepository.org/) using the experiment ID FR-FCM-Z475. T cell receptor sequencing (TCR-seq) data are available at ImmuneACCESS with the DOI link: https://clients.adaptivebiotech.com/pub/liu-2021-crm.
There are no original codes generated in this paper.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.