

Abstract
The DNA polymerase zeta (Polζ) plays a critical role in bypassing DNA damage. REV3L, the catalytic subunit of Polζ, is also essential in mouse embryonic development and cell proliferation for reasons that remain incompletely understood. In this study, we reveal that REV3L protein interacts with heterochromatin components including repressive histone marks and localizes in pericentromeric regions through direct interaction with HP1 dimer. We demonstrate that Polζ/REV3L ensures progression of replication forks through difficult‐to‐replicate pericentromeric heterochromatin, thereby preventing spontaneous chromosome break formation. We also find that Rev3l‐deficient cells are compromised in the repair of heterochromatin‐associated double‐stranded breaks, eliciting deletions in late‐replicating regions. Lack of REV3L leads to further consequences that may be ascribed to heterochromatin replication and repair‐associated functions of Polζ, with a disruption of the temporal replication program at specific loci. This is correlated with changes in epigenetic landscape and transcriptional control of developmentally regulated genes. These results reveal a new function of Polζ in preventing chromosome instability during replication of heterochromatic regions.
Keywords: DNA replication, heterochromatin, replication timing, REV3L, TLS polymerase
Subject Categories: Chromatin, Transcription & Genomics; DNA Replication, Recombination & Repair
HP1‐mediated targeting of REVL3, the catalytic subunit of translesion synthesis Polζ, influences replication fork progression and epigenetic and transcriptional landscapes of developmentally‐regulated genes.
Introduction
It is well established that DNA polymerase delta (Polδ) and epsilon (Polϵ) catalyze the high‐fidelity duplication of the genome (reviewed in Burgers & Kunkel, 2017). However, an emerging concept is that translesion synthesis (TLS) DNA polymerases, known for their error‐prone lesion‐bypass properties, can also facilitate synthesis of non‐damaged DNA. TLS polymerases may be employed when replicative DNA polymerases are not able to pass through stalled sites of replication at structured DNA sequences and/or hard‐to‐replicate genomic regions (reviewed in Tsao & Eckert, 2018). For example, DNA polymerase eta (Polη) and zeta (Polζ) are required to maintain common fragile site stability (CFS) in human cells (Bergoglio et al, 2013; Bhat et al, 2013; Despras et al, 2016). It is not known whether specialized DNA polymerases are recurrently recruited to DNA more broadly during S‐phase to assist replicative DNA polymerases for replicating unconventional DNA structures preventing thus genome instability at the cost of increased point mutations. Intriguingly, growing evidence suggests that replication timing influences genomic mutation rates with an increasing gradient of single‐nucleotide substitutions that correlate with late‐replicating regions (Stamatoyannopoulos et al, 2009; Koren et al, 2012; Polak et al, 2015), but the causative underlying mechanisms remain elusive.
Polζ is a TLS polymerase complex in eukaryotes, consisting of four subunits: Rev3, the catalytic subunit, Rev7 that enhances Rev3 activity, and two subunits shared with the replicative polymerase Polδ (Baranovskiy et al, 2012; Johnson et al, 2012; Makarova et al, 2012; Lee et al, 2014). In contrast to other TLS enzymes that belong to the Y‐family of polymerases, Polζ belongs to the B‐family that includes the highly accurate replicative DNA Polδ and Polϵ (Gan et al, 2008). Polζ lacks an intrinsic 3′–5′ exonuclease activity, making this TLS polymerase error‐prone with a spontaneous mutation rate that is 10‐ to 100‐fold greater than that of replicative DNA polymerases in yeast (McCulloch & Kunkel, 2008; Stone et al, 2009). Polζ is a key player in translesion DNA synthesis by elongating primer termini that are positioned opposite base damage and non‐instructional lesions (Johnson et al, 2000). This DNA polymerase has been extensively characterized in the budding yeast S. cerevisiae, showing that spontaneous as well as damage‐induced mutagenesis is largely dependent on Polζ (Makarova & Burgers, 2015).
Counterparts of the yeast REV genes have been identified in other eukaryotes. Mouse and human Rev3‐like (Rev3l) orthologs have a large extra segment which is not conserved in yeast REV3 and are thus about twice the size of the 173‐kDa yeast Rev3 (350 and 353 kDa, respectively). Despite the established participation of Rev3l in important cellular processes (Martin & Wood, 2019), the role of REV3L protein is incompletely understood and studies have been hampered by the inability to detect this large protein in cells. Polζ is unique among TLS polymerases in mammalian cells, because inactivation of Rev3l gene leads to embryonic lethality in mice (Esposito et al, 2000; Wittschieben et al, 2000; Van Sloun et al, 2002). Rev3l −/− mouse embryonic stem cells are not viable, and primary mouse embryonic fibroblasts (MEFs) obtained from conditional Rev3l knockouts show genome instability and growth defects without external damage to DNA (Wittschieben et al, 2006; Lange et al, 2012). All these data underscore an essential role of Polζ in mammalian cells.
In this study, we investigated the biological function of Polζ during S‐phase under normal growth conditions. We show that Polζ/REV3L is able to interact with heterochromatin through direct interaction with HP1 dimer and ensures progression of replication forks through the hard‐to‐replicate pericentromeric heterochromatin, thus preventing spontaneous DNA and chromosome break formation. We also find that lack of REV3L compromises heterochromatin‐associated DSB repair and shows increased number of genomic deletions in late‐replicating regions. Lack of REV3L leads to further consequences that may be ascribed to heterochromatin replication‐associated functions of Polζ, with a disruption of the temporal replication program at specific loci associated with changes in epigenetic and transcriptional landscape. These results show that Polζ helps maintain genome stability by contributing to heterochromatin replication.
Results
S‐phase progression is altered in Rev3l −/− MEFs
Polζ is critical for proliferation of normal primary cells, suggesting that Polζ participates in unchallenged DNA replication (Lange et al, 2012). To gain cellular and molecular insights into its function during DNA replication, we first investigated the dynamics of S‐phase progression in the absence of REV3L, the catalytic subunit of Polζ. Unchallenged Rev3l −/− and Rev3l +/+ mouse embryonic fibroblasts (MEFs) (Lange et al, 2012) were pulse‐labeled with the thymidine analog 5′‐bromo‐2′‐deoxyuridine (BrdU) for 15 min, released into fresh medium, and harvested at the indicated time points. FACS analysis showed that cell cycle progression was altered in Rev3l −/− MEFs (Fig 1A). Indeed, Rev3l −/− cells exhibited a significant higher percentage in late S/G2‐phase at 4, 6, and 8 h as compared to control cells. This is mirrored by a depletion of BrdU‐positive cells in G1/early S at the same time periods (Fig 1A, see red arrows in upper panel and histograms in lower panel). Moreover, analysis of BrdU‐negative cells showed that Rev3l −/− cells tend to accumulate in G2‐phase as compared to Rev3l +/+ (Fig 1A, see green arrows in upper panel). Similar observations were obtained using REV3L‐depleted human cells (Fig EV1). These results indicate that cell cycle progression in Rev3l −/− cells is impaired in late S‐phase and G2/M‐phase and prompted us to investigate the patterns of spatio‐temporal replication in Rev3l null MEFs.
Figure 1. S‐phase progression is impaired in Rev3l −/− MEFs.
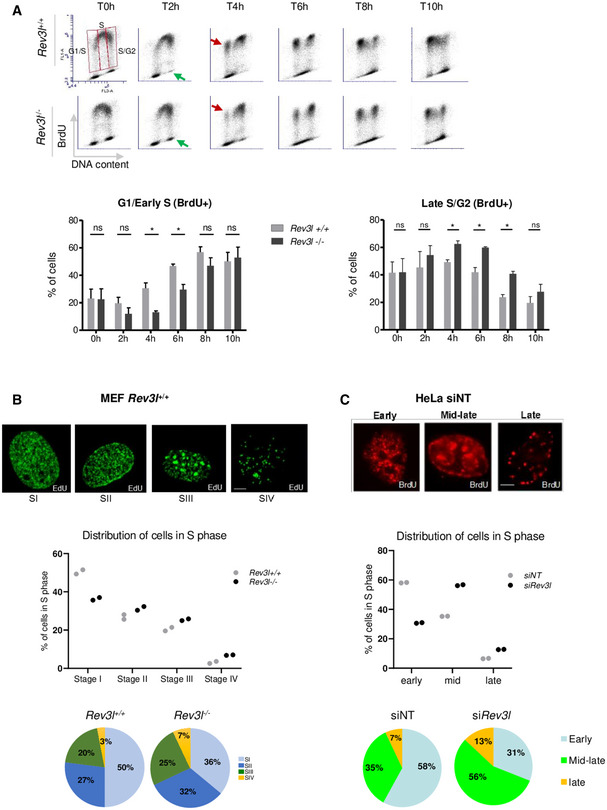
- Rev3l +/+ and Rev3l −/− MEFs were pulse‐labeled with BrdU prior to harvesting and analyzed by flow cytometry at different time points. The analysis was focused on S‐phase divided into three parts: G1/early S, middle S, and late S/G2 BrdU+ cells. Red arrow represents BrdU+ cells that re‐entry in S‐phase, and green arrow shows BrdU− cells in G2‐phase. Histograms represent the percentage of cells in G1/early and late S/G2‐phase after BrdU pulse. Error bars represent standard error for three independent experiments. ns: not significant, *P < 0.05 by Student’s t‐test.
- Rev3l +/+ and Rev3l −/− MEFs were pulse‐labeled with EdU for 15 min, permeabilized, fixed, and stained for EdU incorporation (green). S‐phase sub‐stages from I to IV were evaluated by visual inspection of the cycling population (> 300 EdU+ cells, top panel). Scale bar = 5 μm. Dot plots and pie charts show the relative proportion (percentage of total S) of each sub‐stage from I to IV (middle and bottom panel, respectively). Each dot represents the mean of two technical replicates.
- Seventy‐two hours after transfection with non–targeting (NT) or Rev3l siRNA, HeLa cells were pulse‐labeled with BrdU for 15 min, permeabilized, fixed, and stained for BrdU (red). As in (B), S‐phase sub‐stages were evaluated by visual inspection of the cycling population (> 300 BrdU+ cells, top panel). Scale bar = 5 μm. Dot plots and pie charts show the relative proportion (percentage of total S) of early, middle–late, and late S‐phase (middle and bottom panel, respectively). Each dot represents the mean of two technical replicates.
Figure EV1. S‐phase progression is impaired in human cells after down‐regulation of REV3L.
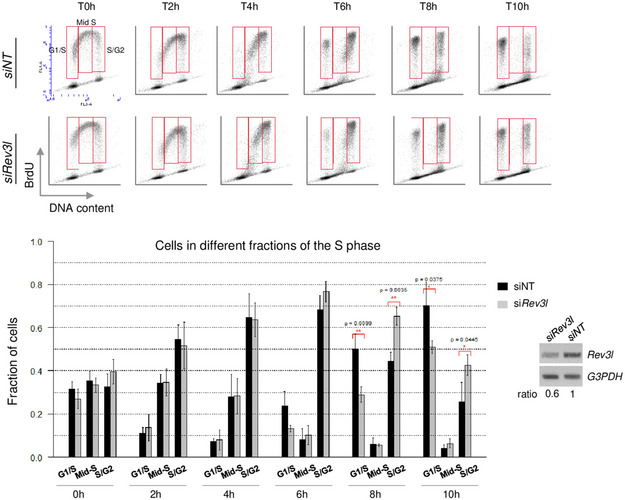
Forty‐two hours after transfection with non‐targeting (NT) siRNA or Rev3l siRNA, HeLa cells were pulse‐labeled with BrdU prior to harvest and analyzed by flow cytometry at different time points (top panel). Analysis was focused on S‐phase divided into three parts: G1/early S, middle S, and late S/G2 BrdU+ cells. Histograms represent the percentage of cells in G1/early and late S/G2‐phase after the BrdU pulse (bottom panel). Error bars indicate standard error of the mean from three independent experiments (Student’s t‐test). RT–PCR was performed to verify the down‐regulation of Rev3l mRNA level in HeLa cells.
S‐phase can be divided into different stages by immunofluorescence observation of characteristic thymidine analog incorporation patterns corresponding to early, mid‐, or late S‐phase (Dimitrova & Berezney, 2002; Guenatri et al, 2004). To analyze the spatial organization of DNA replication in Rev3l −/− MEFs, cells were pulsed‐labeled with EdU and the replication patterns typically observed in mouse cells were identified and quantified (Guenatri et al, 2004). Representative replication patterns for each category are represented: early S‐phase pattern with a high density of small foci distributed throughout the nucleus (stage I), early–mid‐S‐phase pattern with foci which become bigger and less abundant than stage I (stage II), mid–late S‐phase pattern with a typical ring‐shaped labeling around pericentromeric heterochromatin domains (stage III), and late S‐phase pattern with a few big dots located mainly at the nuclear periphery and in heterochromatin area (stage IV) (Fig 1B, upper panel). We found that in the absence of REV3L, the proportion of cells with an early replication spatial pattern (stage I) decreased whereas the proportion of cells in mid‐ and late S‐phase increased (Fig 1B, middle and lower panels). A similar effect was found after depletion of REV3L in human cells (Fig 1C). These results suggest that lack of REV3L could affect the temporal control of DNA replication.
Loss of REV3L delays replication timing in specific genomic loci
We thus examined the replication timing in Rev3l +/+ and Rev3l −/− cells after a few (p5 and p7 in Rev3l +/+ and Rev3l −/− cells, respectively) or serial passages (p60) in cell culture (Fig EV2A). To perform genome‐wide profiling of replication timing (RT), cells were pulse‐labeled with BrdU and sorted into early and late S‐phase fractions by flow cytometry (see methods (Fernandez‐Vidal et al, 2014)). Newly synthesized DNA of each fraction was BrdU‐immunoprecipitated and specifically labeled (Cy3 for early fraction and Cy5 for late fraction) before co‐hybridization on microarrays. The replication timing of genomic domains was obtained by measuring the log2‐ratio of early versus late fractions and analyzed using the START‐R program (Hadjadj et al, 2020). We first compared the replication timing obtained at different passages for each cell line. For Rev3l +/+ MEF, there was a 1.9% difference between p5 and p60. For Rev3l −/− cells, we observed only 0.2% difference between p7 and p60, suggesting that the replication timing remains stable during cell culture passages in both cell lines, and can be considered as two independent replicates.
Figure EV2. Loss of REV3L disrupts replication timing in specific genomic loci.
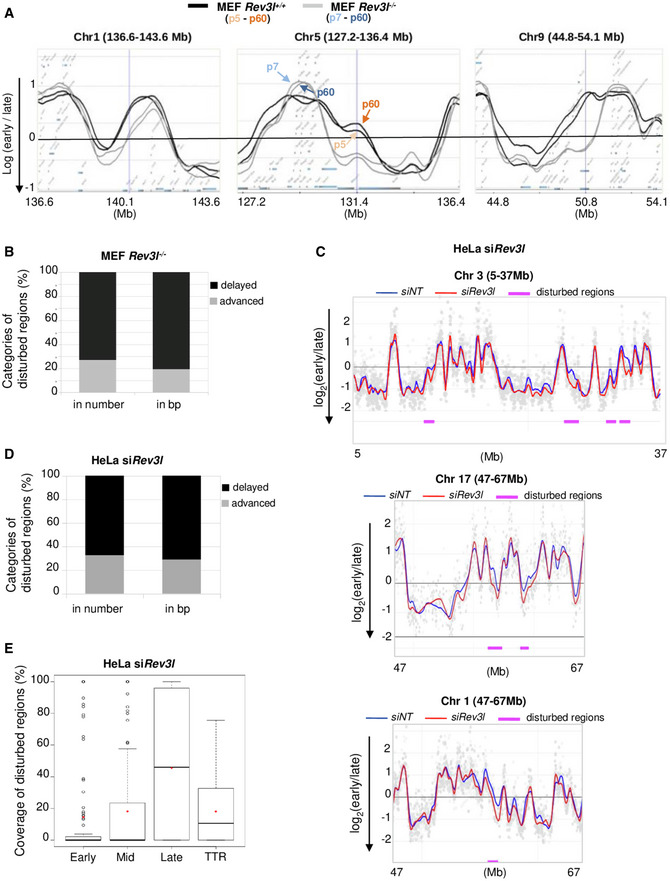
- Cells were pulse‐labeled with BrdU for 1.5 h and sorted by flow cytometry in two fractions, S1 and S2, corresponding to early and late S‐phase fractions, respectively. Neo‐synthesized DNA was immunoprecipitated with BrdU antibodies. Early and late neo‐synthesized DNAs were labeled with Cy3 and Cy5, respectively, and hybridized on microarrays. After processing analysis, replication‐timing profiles were obtained. Shown are the zoomed microarray profiles of the timing of replication on chromosome 1 (136.6–143.6 Mb), chromosome 5 (127.2–136.4 Mb), and chromosome 9 (44.8–54.1 Mb) from Rev3l +/+ and Rev3l −/− MEFs overlaid. Black lines represent replication timing from Rev3l +/+ MEFs, and gray lines represent Rev3l −/− MEFs. For each group, replication timing at early passage (p5 or p7) and late passage (p60) was analyzed and shown a great conservation of replication timing over serial passages (double line on zoomed microarray profiles for each cell line, see corresponding arrow).
- Fraction of advanced and delayed regions in the disturbed replication timing in Rev3l −/− MEFs. The proportions were calculated either in the percentage of numbers of advanced and delayed regions, or the size of advanced and delayed regions (in base pairs).
- HeLa cells were transfected with non‐targeting siRNA (siNT) or siRNA against Rev3l; then, 60 h later cells were pulse‐labeled with BrdU for 1.5 h and sorted by flow cytometry in two fractions, S1 and S2, corresponding to early and late S‐phase fractions, respectively. Neo‐synthesized DNA was immunoprecipitated with BrdU antibodies. Early and late neo‐synthesized DNAs were labeled with Cy3 and Cy5, respectively, and hybridized on microarrays. After processing analysis with the START‐R software, replication‐timing profiles were obtained. Shown are the zoomed microarray profiles of the timing of replication on chromosome 1 (47–67 Mb), chromosome 3 (5–37 Mb), and chromosome 17 (47–67 Mb) from HeLa siNT and siRev3l overlaid. Blue lines represent replication timing from HeLa siNT, and red lines represent replication timing from HeLa siRev3l. Genomic regions detected significantly different by START‐R are indicated by a pink line (P < 0.01).
- Fraction of advanced and delayed regions in the disturbed replication timing in HeLa siRev3l. The proportions were calculated either in the percentage of numbers of advanced and delayed regions or in the size of advanced and delayed regions (in base pairs).
- Boxplot of coverage of the disturbed regions in each category (early, mid, late, TTR). Bar in boxplot represents the median, and red points represent the average. The limit of the boxes corresponds to the 0.25–0.75 quartiles with whiskers extending to the maximum value of 1.5 times the interquartile range. Graphs show data from one biological experiment.
We next compared the replication timing between Rev3l +/+ and Rev3l −/− MEF at early passage. The START‐R analysis identified a set of genomic compartments that changed replication timing in response to Rev3l inactivation (Fig 2A). About 5.7% of the whole genome was affected; 19.2% (in bp) of these regions were advanced in timing, and 80.8% of regions were delayed (Fig EV2B). A major effect of Rev3l loss was boundary shifts as exemplified in Fig 2A, corresponding to a delay in regions that lie between early‐ and late‐replicating domains called temporal transition regions (TTR) by Gilbert and colleagues (Hiratani et al, 2008). We observed that 67% of disturbed domains fall in TTR (corresponding to 83% of delayed regions).
Figure 2. Loss of REV3L disrupts the replication timing in specific genomic loci.
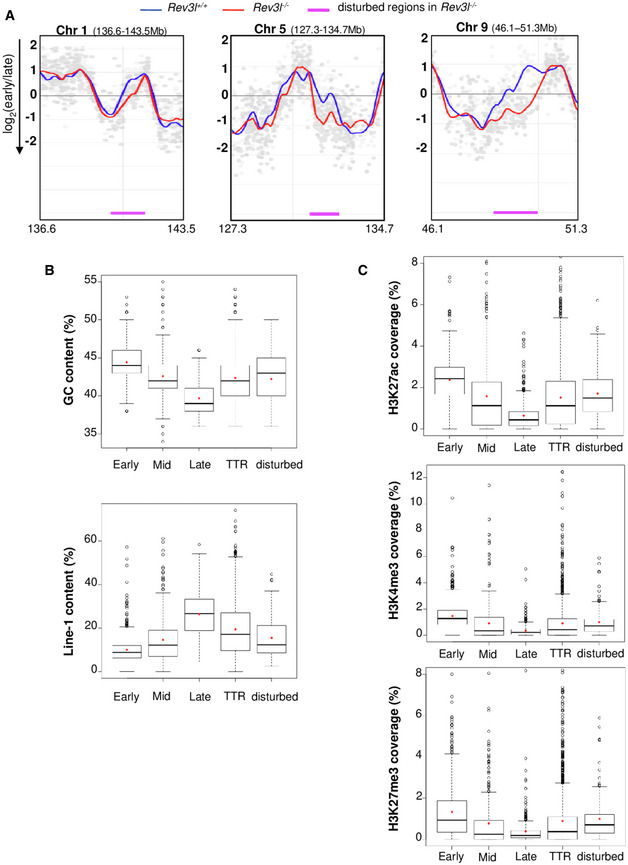
- Cells were pulse‐labeled with BrdU for 1.5 h and sorted by flow cytometry in two fractions, S1 and S2, corresponding to early and late S‐phase fractions, respectively. Neo‐synthesized DNA was immunoprecipitated with BrdU antibodies. Early and late neo‐synthesized DNAs were labeled with Cy3 and Cy5, respectively, and hybridized on microarrays. After processing analysis with the START‐R software, replication‐timing profiles were obtained from two replicates (see Fig EV2A). Shown are the zoomed microarray profiles of the timing of replication on chromosome 1 (136.6–143.5 Mb), chromosome 5 (127.3–134.7 Mb), and chromosome 9 (46.1–51.3 Mb) from Rev3l +/+ and Rev3l −/− MEFs overlaid. Blue lines represent replication timing from Rev3l +/+ MEFs, and red lines represent replication timing from Rev3l −/− MEFs. Genomic regions displaying significant difference between Rev3l +/+ and Rev3l −/− MEFs by START‐R are indicated by a pink line (P < 0.01).
- Analysis of GC and Line‐1 content in early, mid, late, and TTR compared with disturbed replicating regions found in Rev3l −/− MEFs. Bar in boxplot represents the median, and red points represent the mean. The limit of the boxes corresponds to the 0.25–0.75 quartiles with whiskers extending to the maximum value of 1.5 times the interquartile range. Graphs show data from two biological experiments.
- Analysis of the active marks H3K27ac and H3K4me3 and facultative heterochromatin mark H3K27me3 content in early, mid, late, and TTR compared with disturbed replicating regions found in Rev3l −/− MEFs. Bar in boxplot represents the median, and red points represent the mean. The limit of the boxes corresponds to the 0.25–0.75 quartiles with whiskers extending to the maximum value of 1.5 times the interquartile range. Graphs show data from two biological experiments.
We then explored the correlation between genomic regions that changed replication timing in Rev3l −/− cells (called disturbed regions) and the coverage for GC and LINE‐1 contents (Fig 2B) and also for H3K27ac, H3K4me3, and H3K27me3 epigenetic marks (Fig 2C). For each parameter, we observed a molecular signature intermediate between that of early and late domains, strengthening our hypothesis that these disturbed regions correspond to TTR. In Rev3l −/− cells, 18.7% of TTR were disturbed. These results reveal that Rev3l inactivation in MEFs induces changes in the temporal replication program, especially in specific genomic regions located in TTR and suggest that REV3L/Polζ might contribute to replicate these specific loci.
To confirm these observations, we examined the replication timing in human cells depleted for REV3L. For that, HeLa cells were transfected with non‐targeting siRNA (siNT) or siRNA against Rev3l. Sixty hours later, cells were pulse‐labeled with BrdU for 1.5 h and processed as for MEFs. Analysis by the START‐R program revealed 116 disturbed regions in REV3L‐depleted cells, corresponding to approximatively 5.4% of the whole genome with more than 70% of delayed regions (Fig EV2C and D). We found that depletion of REV3L in human cells affected mostly genomic domains that replicate late in S‐phase (Fig EV2E). Thus, the specific replicating domains disturbed after REV3L depletion are not exactly the same in MEFs and HeLa genomes (TTR versus Late, respectively).
Rev3l inactivation results in epigenetic changes with down‐regulation of numerous developmentally regulated genes
Links between transcription and replication timing have been well documented (Hiratani et al, 2008; Sima et al, 2019). Genome‐wide analyses have identified a strong correlation between early replication and high transcriptional potential (MacAlpine et al, 2004; Farkash‐Amar et al, 2008; Hiratani et al, 2008). We reasoned that changes in replication timing in the absence of Rev3l may impact the transcriptional program. We therefore performed microarray‐based transcriptome profiling from Rev3l +/+ and Rev3l −/− MEFs (Fig 3A). Genes altered by at least threefold between conditions were considered (FDR < 0.05). Analysis of microarray data indicated a total of 317 genes with mRNA expression altered by ≥ 3‐fold in Rev3l‐deficient cells as compared to control cells, with 112 genes up‐regulated and 205 genes that were down‐regulated. Genes displaying a down‐regulation in Rev3l −/− MEFs had significant enrichment for Gene Ontology terms related to organ and system development (Fig 3B). Prominent repressed genes are in the HoxB gene cluster, including Hoxb2 to Hoxb9 as well as two HoxA genes (Fig 3A and C). Rev3l inactivation also resulted in the decrease in expression of approximately 14 genes from imprinted loci (corresponding to 6.8% of down‐regulated genes,). These included paternally expressed genes Dlk1, Snrpn, Peg3, and Mest (Fig 3A, blue) and maternally expressed genes H19, Dcn, Meg3, and Cdkn1c (Fig 3A, pink). WT1 is a well‐characterized developmental gene that is mutated in Wilms' tumor and was identified as being imprinted (Schwienbacher et al, 2000). This gene was significantly down‐regulated by at least 50‐fold in Rev3l −/− MEFs as compared to control cells. qRT–PCR from independent cell cultures of Rev3l −/− and Rev3l +/+ MEFs confirmed the down‐regulation of multiple HoxB and imprinted genes (Fig 3D and E).
Figure 3. Inactivation of Rev3l impairs expression of numerous developmentally and imprinted genes.
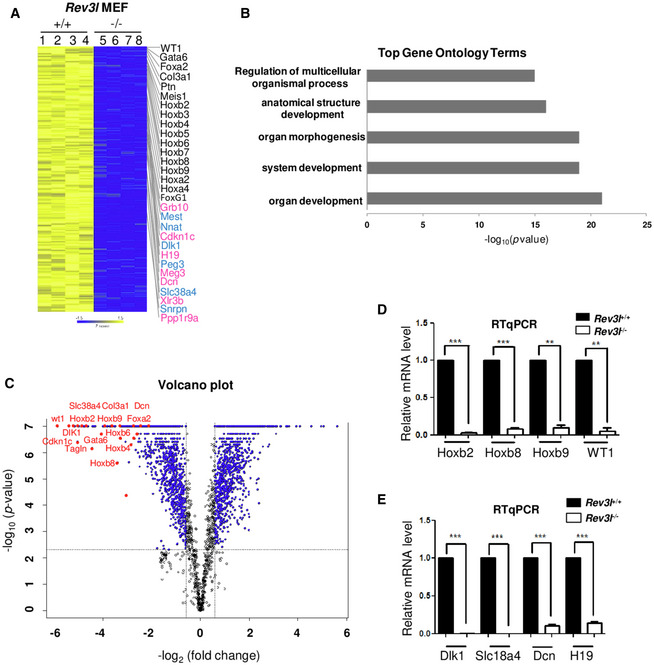
- Heat map showing log2 fold change in differentially expressed genes in Rev3l +/+ and Rev3l −/− MEFs from 2 independent biological experiments (Rev3l+/+: samples 1 and 2, Rev3l−/−: samples 5 and 6) with 2 technical replicates (samples 3,4,7,8: technical repeats from samples 1,2,5,6, respectively). Several developmentally regulated genes and imprinting genes (paternally in blue and maternally in pink) down‐regulated in Rev3l −/− MEFs are indicated on the right. Yellow and blue indicate high and low mRNA expression levels, respectively.
- Top five biological process gene ontology (GO) terms of genes found down‐regulated in Rev3l −/− MEFs transcriptome analysis.
- Volcano plot shows in red genes involved in development and imprinting with high fold changes ≥ 3 (FDR < 0.05).
- Relative mRNA levels of four genes involved in development (Hoxb2, Hoxb8, Hoxb9, and WT1) were validated through qRT–PCR from Rev3l +/+ and Rev3l −/− MEF samples. The data were normalized to the amount of GAPDH mRNA. Error bars indicate standard error of the mean for three independent experiments. ***P < 0.001 and **P < 0.005 by Student’s t‐test.
- Relative mRNA levels of genes involved in imprinting (paternal expressed genes: Dlk1 and Slc18a4 and maternal expressed genes: Dcn1 and H19) were validated through qRT–PCR from Rev3l +/+ and Rev3l −/− MEF samples. The data were normalized to the amount of GAPDH mRNA. Error bars indicate standard error of the mean for three independent experiments. ***P < 0.001 by Student’s t‐test.
Source data are available online for this figure.
We next asked whether genes showing altered mRNA expression in the absence of REV3L are located in genomic regions that displayed a RT delay. For this, we integrated the data on gene expression and genome‐wide profiling of replication timing. We found that of the 24 genes (corresponding to 7.6% of deregulated genes) located in these specific genomic domains (Table 1), all showed less expression in REV3L‐defective cells. These observations are consistent with the fact that late‐replicating genes are often silenced. However, the majority of deregulated genes do not fall within disturbed regions of replication timing, indicating that down‐regulation of genes might be a secondary consequence of REV3L inactivation. Nevertheless, these data suggest that loss of Rev3l is correlated to dysregulation of genes involved in growth and development.
Table 1.
Loci displaying both delay in RT and misregulation in Rev3l −/− MEFs.
| Domain | Chr | Size (bp) | Replication timing domain | Gene | Misregulation (fold change a ) |
|---|---|---|---|---|---|
| 1 | 1 | 1,042,476 | TTR | Pax3 | −3.57 |
| 2 | 2 | 2,573,606 | TTR | Plcb4 | −4.17 |
| 3 | 6 | 6 628 128 | TTR | Frmd4b | −3.13 |
| 4 | 8 | 671,566 | TTR | Hand2 | −7.69 |
| 5 | 8 | 641,165 | TTR | Lpl | −27.03 |
| 6 | 10 | 567,222 | TTR | Ptprb | −5.56 |
| 6 | 10 | 567,222 | TTR | Tspan8 | −7.14 |
| 7 | 11 | 920,184 | TTR | Meis1 | −5.56 |
| 8 | 12 | 487,988 | TTR | Foxg1 | −20.41 |
| 9 | 12 | 548,987 | TTR | Dlk1 | −6.25 |
| 9 | 12 | 548,987 | TTR | Meg3 | −5.26 |
| 9 | 12 | 548,987 | TTR | Mirg | −6.67 |
| 10 | 12 | 1,372,498 | TTR | Mycn | −5.88 |
| 11 | 13 | 1,179,925 | TTR | Sfrp4 | −6.25 |
| 12 | 14 | 671,779 | TTR | Wnt5a | −4.55 |
| 13 | 15 | 1,054,278 | TTR | Fbxl7 | −8.33 |
| 14 | 15 | 1,145,389 | TTR | Has2 | −4.55 |
| 15 | 17 | 808,766 | TTR | Rftn1 | −12.35 |
| 16 | 18 | 402,349 | TTR | Gata6 | −12.82 |
| 17 | 18 | 539,392 | TTR | Pcdhb20 | −3.45 |
| 17 | 18 | 539,392 | TTR | Pcdhb21 | −5.56 |
| 18 | 18 | 1,232,897 | TTR | Ppp2r2b | −8.33 |
| 19 | 19 | 509,162 | TTR | Aldh1a1 | −11.33 |
| 19 | 19 | 509,162 | TTR | Aldh1a7 | −3.57 |
FDR < 0.05.
In addition to gene expression, various genomic features are linked to replication timing. These include histone modification, DNA methylation, DNA repeat sequences, ordered chromatin structure, and nuclear compartmentalization (Aladjem, 2007; Hiratani et al, 2008). Changes in replication timing as well as dysregulation of gene expression in response to Rev3l inactivation prompted us to assess whether these modifications were associated with variations in global levels of histone modifications. Total histones from Rev3l +/+ and Rev3l −/− MEFs were screened by immunoblotting with a panel of histone modification‐specific antibodies. Rev3l inactivation resulted in a fourfold up‐regulation of histone H3K27 tri‐methylation (Fig 4A) and a more than two‐fold increase in H3K9me3 and H3K4me3. While H3K27me3 and H3K9me3 are repressive marks, H3K4me3 is associated with active genes.
Figure 4. Loss of REV3L results in epigenetic changes.
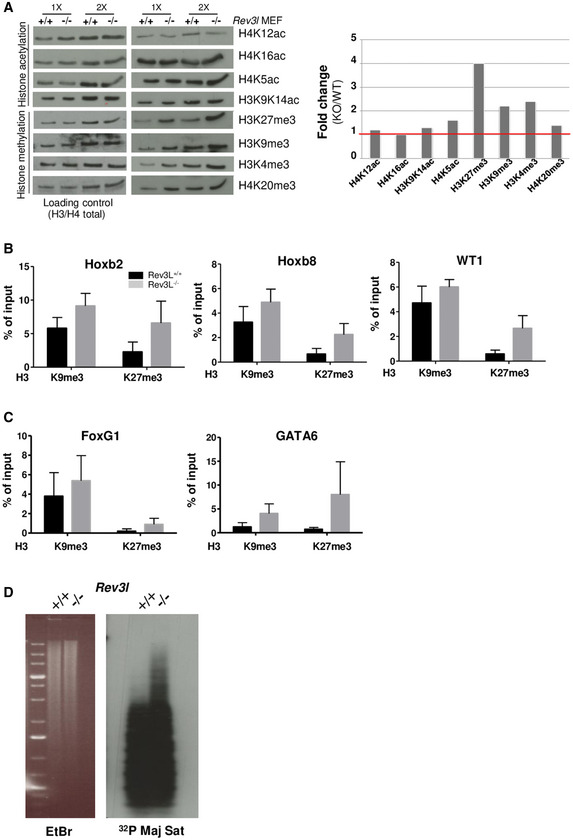
- Total histones were acid extracted from Rev3l +/+ and Rev3l −/− MEFs. Samples were analyzed by Western blot using indicated antibodies specific to histone marks (right panel). An anti‐pan H4 or H3 was used as loading control (left panel). Histograms show WT/KO Rev3l fold change relative to the immunoblot shown on the top panel (using 1× sample intensity).
- Histone mark levels were examined at selected genes repressed in Rev3l −/− MEFs (Hoxb2, Hoxb8, WT1). Histograms represent enrichment of H3K9me3 and H3K27me3 at indicated loci assessed by ChIP‐qPCR in Rev3l +/+ and Rev3l −/− MEFs. Error bars indicate standard error of the mean for three independent experiments.
- H3K9me3 and H3K27me3 mark levels were evaluated at selected genes that localized in genomic loci displaying replication timing delay and down‐regulated in Rev3l −/− MEFs (FoxG1, GATA6). Histograms represent enrichment of H3K9me3 and H3K27me3 assessed by ChIP‐qPCR at indicated loci in Rev3l +/+ and Rev3l −/− MEFs. Error bars indicate standard error of the mean for three independent experiments.
- Southern blot analysis of genomic DNA extracted from Rev3l +/+ and Rev3l −/− MEFs and digested with the CpG methylation‐sensitive enzyme MaeII (5′ ‐ACGT‐ 3′). The membrane was hybridized with radiolabeled probes specific to major satellites. The presented data are representative of two repeats (see Source Data file for this figure).
Source data are available online for this figure.
We then examined repressive histone modifications (H3K27me3 and H3K9me3) at gene loci down‐regulated in Rev3l −/− MEFs (including Hoxb2, Hoxb8, WT1). By ChIP‐qPCR, we found that H3K27me3 and H3K9me3 levels were higher in Rev3l −/− than in Rev3l +/+ MEFs (Fig 4B). In contrast, we observed a trend toward reduced active marks H3K9ac and H3K27ac in Rev3l −/− cells (Appendix Fig S1). We also observed enrichment of repressive histone marks in gene loci that showed a delay in replication timing in Rev3l −/− cells such as FoxG1 and GATA6 genes (Fig 4C), supporting the idea that the down‐regulation of gene expression observed in Rev3l −/− cells might be caused by epigenetic silencing.
Constitutive heterochromatin exhibits a chromatin landscape marked by high levels of H3K9me3, DNA methylation, and histone hypoacetylation. The pericentromere is a heterochromatic domain that provides a structural scaffold for centromere formation and plays a crucial role in genome stability (Allshire & Karpen, 2008). In mouse cells, pericentromeric heterochromatin consists of ~105 major satellite DNA repeats that are methylated, decorated by H3K9me3, and enriched in the heterochromatin protein 1 alpha (HP1α) (Maison et al, 2002). Since we observed an up‐regulation of H3K9me3 levels in Rev3l‐deficient cells, we next investigated the DNA methylation pattern at major satellite repeats. Genomic DNA was extracted from Rev3l +/+ and Rev3l −/− MEFs, digested with methylation‐sensitive restriction enzymes MaeII, and DNA blots hybridized with major satellite probes. In Rev3l‐deficient cells, a fraction of DNA satellite was resistant to enzymatic digestion, indicating an increase in DNA methylation in the absence of REV3L (Fig 4D). Therefore, our data suggest that REV3L can play a role in the duplication of DNA repeat‐rich pericentromeric regions known to be replicated in mid‐to‐late S‐phase in mouse cells (Guenatri et al, 2004). Loss of REV3L results in modification of epigenetic marks in such heterochromatic regions.
Direct interaction of REV3L with HP1 dimer allows REV3L recruitment at pericentromeric regions
We reasoned that if REV3L is implicated in heterochromatin replication, it may interact with heterochromatin components. As no commercially available antibodies can detect endogenous REV3L, we generated a MEF cell line with an alteration at the genomic Rev3l locus so that endogenous REV3L protein harbors a 3X‐Flag tag (Flag‐REV3L) (Fig EV3A). We first verified that we were able to detect endogenous Flag‐REV3L protein in this cell line (Fig EV3B and C). We then used co‐immunoprecipitation to examine interaction of REV3L with heterochromatin proteins. We found that both HP1α and ATRX, a chromatin remodeling protein known to localize in heterochromatin (Eustermann et al, 2011), co‐immunoprecipitated with REV3L. As positive control, we confirmed that REV7, the regulatory subunit of Polζ, was also co‐immunoprecipitated (Fig 5A).
Figure EV3. REV3L interacts with heterochromatin components.
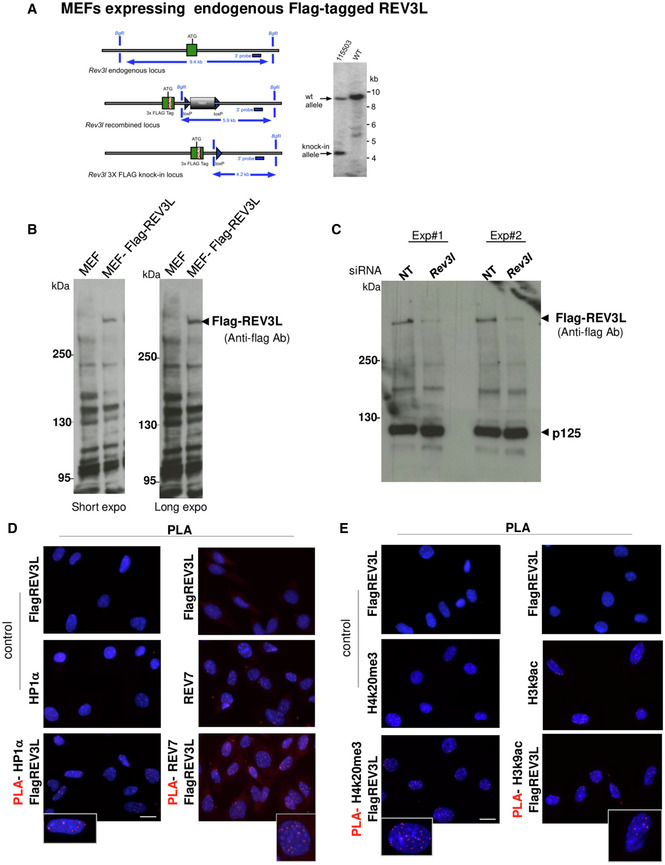
- Diagram of the mouse Rev3l 3x‐FLAG knock‐in allele. The wild‐type (WT) locus is shown at the top (left panel). A green rectangle shows the first exon with the initiator ATG sequence. The gray line represents chromosomal sequence. The middle diagram shows the targeted locus after homologous recombination. The neomycin‐positive selection cassette (neo) is shown in gray. The cassette was excised by breeding mosaic animals with C57BL/6 Cre deleter mice, to produce the 3x‐FLAG‐tagged knock‐in allele shown at bottom. Example of Southern blot analysis of the knock‐in locus. Genomic DNA of the tested animals (number 115503 shown here) was compared with C57BL/6 wild‐type genomic DNA (WT). BglII digested DNA was blotted on a nylon membrane and hybridized with the external 3′ probe with the position shown at the left. Restriction fragments of 9.4 and 4.2 kb were observed for the wild‐type, and knock‐in locus, respectively. Genomic DNA was further analyzed extensively and confirmed by specific PCR assays and complete DNA sequencing as described in the Materials and Methods (right panel).
- Total proteins were extracted from MEFs or MEFs expressing endogenous Flag‐REV3L; then, REV3L protein was detected by immunoblotting using anti‐Flag (M2) antibody. Specific signal above 250 kDa is detected only in MEF Flag‐REV3L.
- Specificity of Flag‐REV3L detection was confirmed by down‐regulating the REV3L protein. MEF Flag‐REV3L was transfected with non‐targeting siRNA (NT) or siRNA targeting mRev3l. Seventy‐two hours later, cells were lysed; then, proteins were analyzed by immunoblotting using anti‐Flag (M2) antibody or anti‐p125 antibody as loading control. The experiment was repeated twice (Exp#1 and #2).
- Asynchronous MEFs expressing Flag‐tagged REV3L were subjected to in situ proximity ligation assay (PLA) to test the interactions REV3L‐REV7 and REV3L‐HP1α as indicated in Fig 5B. Representative images from each PLA condition with controls are shown. Scale bar: 20 μm.
- Asynchronous MEFs expressing Flag‐tagged REV3L were subjected to PLA to test the interactions REV3L‐H4K20me3, REV3L‐H3K9me3, REV3L‐H3K27me3, REV3L‐H3K4me3, and REV3L‐H3K9ac as indicated in Fig 5C. Representative images from PLA REV3L‐H4K20me3 and REV3L‐H3K9ac with their controls are shown. Note the high background for H3K9ac control. Scale bar: 20 μm.
Figure 5. REV3L localizes in heterochromatin through a direct interaction with HP1 dimer.
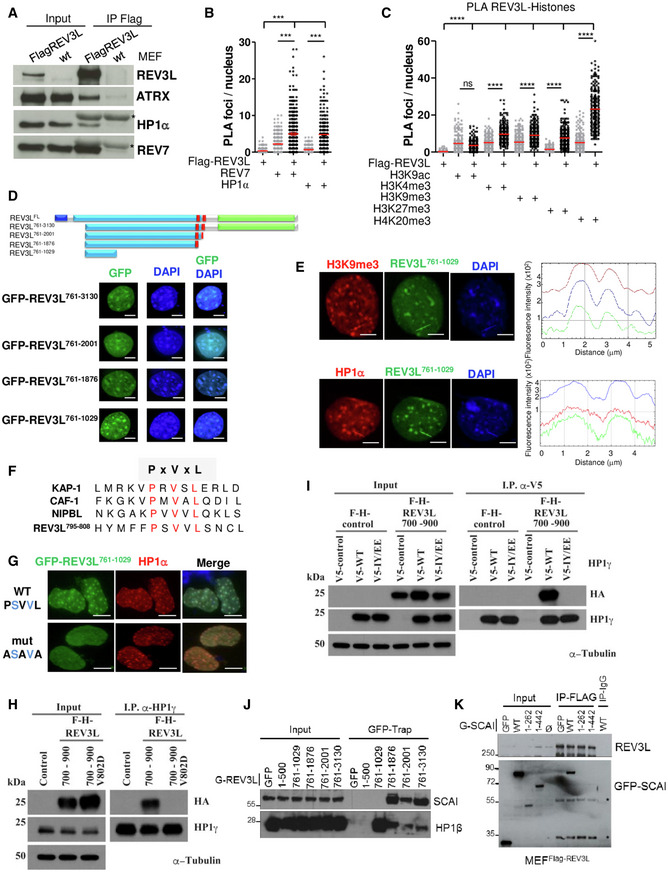
- Asynchronous MEFs expressing Flag‐tagged REV3L from the endogenous locus were lysed, and REV3L was immunoprecipitated using anti‐Flag (M2) antibodies. Co‐immunoprecipitated proteins were analyzed by immunoblotting using the indicated antibodies. *: IgG light chain. This experiment was repeated 2–4 times.
- Asynchronous MEFs expressing Flag‐tagged REV3L were subjected to in situ proximity ligation assay (PLA) to test the interactions REV3L‐REV7 and REV3L‐HP1α. Nuclear foci were quantified (more than 150 nuclei for each condition were counted). Reactions omitting one of the primary antibodies were used as negative controls. Horizontal bars show the mean. Mann–Whitney test, ns: not significant, ***P < 0.001. Experiments were repeated three times.
- Asynchronous MEFs expressing Flag‐tagged REV3L were subjected to PLA to test the interactions REV3L‐H4K20me3, REV3L‐H3K9me3, REV3L‐H3K27me3, REV3L‐H3K4me3, and REV3L‐H3K9ac. Nuclear foci were quantified as in (B). Horizontal bars show the mean. Mann–Whitney test, ns: not significant, ****P < 0.0001. Three independent experiments were performed.
- Schematic representation of human REV3L and truncated constructs. Conserved domains between yeast and human REV3L are in black blue (the N‐terminal domain, 1–333 aa) and in green (the catalytic domain, 2,276–3,130 aa), the REV7 interacting domains are in red (1,880–2,001 aa). A large region in royal blue not found in yeast protein is almost exclusively encoded by exon 14. All the truncated constructs lack the NTD domain and were fused to GFP. MEFs cells were transiently transfected with various GFP‐REV3L constructs and fixed with 4% formaldehyde. The distribution of the GFP‐REV3L mutants was detected by autofluorescence, and nuclei were visualized using DAPI staining. Scale bar = 5 μm.
- MEFs cells were transiently transfected with GFP‐REV3L761–1,029 and fixed with 4% formaldehyde. The distribution of GFP‐ REV3L761–1,029 was detected by autofluorescence (green), chromocenters were visualized by H3K9me3 immunostaining (red, top panel) or HP1α (red, bottom panel), and DNA was counterstained with DAPI. Line scans represent the colocalization of proteins within each image (right panels). Scale bar = 5 μm.
- Sequence alignment of proteins containing the PxVxL motif important for an interaction with HP1, with canonical residues shown in red.
- MEFs cells were transfected with GFP‐REV3L761–1,029 WT PSVVL or ASAVA mutant and fixed with 4% formaldehyde. The distribution of GFP‐REV3L constructs was detected by autofluorescence (green), chromocenters were visualized by HP1α immunostaining (red), and DNA was counterstained with DAPI. Scale bar = 10 μm.
- Human 293T cells were transfected with either FH‐REV3L700–900, mutant V802D F‐H‐REV3L700–900, or empty vector. Forty‐eight hours after transfection, cell lysates were made and used for immunoprecipitation with HP1 antibody. Western blot was processed, and membranes were immunoblotted with the indicated antibodies.
- 293T cells were co‐transfected with F‐H‐REV3L700–900 or empty vector and V5‐HP1γ, mutant IY/EE V5‐HP1γ or empty vector. Forty‐eight hours after transfection, cell lysates were made and used for immunoprecipitation with α‐V5 antibody. After electrophoresis, samples were immunoblotted with anti‐HA, anti‐HP1γ, or anti‐α‐Tubulin as indicated.
- 293 cells were transfected with various GFP‐REV3L constructs or empty vector (GFP). Twenty‐four hours after transfection, cell lysates were made and GFP‐REV3L was affinity‐purified on GFP‐Trap beads. After electrophoresis, samples were analyzed by immunoblotting with antibodies against SCAI or HP1β as indicated.
- MEFFlag‐REV3L were transfected with various GFP‐SCAI constructs, empty vector (GFP), or not transfected (Ø). Twenty‐four hours after transfection, cell lysates were made and Flag‐REV3L was affinity‐purified on M2 beads. After electrophoresis, samples were analyzed by immunoblotting with antibodies against GFP or Flag (M2). *: non‐specific bands.
Source data are available online for this figure.
To strengthen these observations, we performed a proximity ligation assay (PLA) that detects protein associations within 40 nm. As expected, we observed specific PLA signals between REV7 and Flag‐REV3L in the nucleus. We also detected PLA signals between HP1α and Flag‐REV3L (Figs 5B and EV3D). We then tested whether REV3L was spatially close to particular modified histone marks. We found REV3L enriched in proximity to H3K9me3, H3K27me3, H3K4me3, H4K20me3, but not to H3K9ac (Figs 5C and EV3E). Together, these results show that REV3L localizes in heterochromatin regions and interacts with heterochromatin proteins and repressive histone marks.
To better characterize the molecular mechanisms linking Polζ/REV3L to heterochromatin, we examined the subcellular localization of REV3L. In mouse cells, pericentromeric heterochromatin (PHC) domains are easily identified by their intense DNA staining with the dye DAPI. They are clearly discernible during interphase when major satellites from different chromosomes associate with form clusters (so called chromocenters) that co‐localize with HP1α (Maison et al, 2002). To explore REV3L subcellular localization, we generated a panel of eGFP‐tagged REV3L truncated proteins (Fig 5D, upper panel). All constructs were detected mainly in the nucleus and localized into chromocenters, visualized by dense DAPI staining (Fig 5D, lower panel). Interestingly, the smaller REV3L construct (REV3L761–1,029) consisting of 268 residues was sufficient to target most, if not all the fusion protein into the nucleus, suggesting that this polypeptide fragment contains a putative nuclear localization signal (NLS); the amino acid sequence KSRKRRKMSKKLPP at position 960–973 is a good candidate (Nguyen & Lavenier, 2009). More importantly, this small construct is also sufficient to target fusion protein into chromocenters. We then confirmed that location in PHC by visualizing the colocalization of REV3L761–1,029 with HP1α and H3K9me3 by immunofluorescence (Fig 5E). In the mouse, the association of HP1α at pericentromeric heterochromatin is severely compromised in cells lacking the H3 Lys9 methyltransferases, Suv39h1 and Suv39h2 (Peters et al, 2001). We therefore investigated REV3L localization in Suv39h double‐null (dn) cells. HP1α was no longer enriched at PHC in the Suv39h dn cells as previously reported (Maison et al, 2002). Likewise, REV3L761–1,029 displayed a diffuse staining throughout the nucleus (Fig EV4A). We also observed that REV3L761–1,029 staining was disrupted when cells were treated with trichostatin A (TSA), a histone deacetylase inhibitor which reversibly increases the acetylation level of histone H3K9 at pericentromeric regions. In TSA‐treated cells, HP1α dissociates from heterochromatic domains (Taddei et al, 2001), and REV3L761–1,029 followed this distribution (Fig EV4B). The localization to chromocenters was re‐established rapidly after drug removal. Collectively, these results strongly suggest that HP1α is required for targeting REV3L to pericentromeric heterochromatin.
Figure EV4. PxVxL motif targets REV3L to pericentromeric heterochromatin through a direct interaction with HP1.
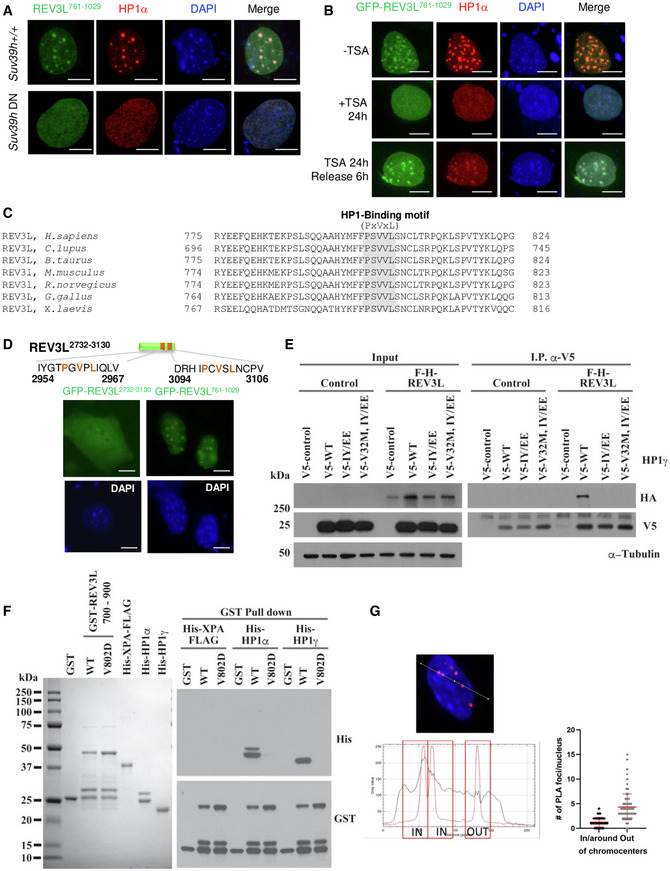
- Wild‐type cells and MEFs lacking Suv39h (Suv39h dn) were transiently transfected with GFP‐REV3L761–1,029 construct then fixed with 4% formaldehyde after 0.5% Triton X‐100 pre‐extraction. The distribution of GFP‐REV3L761–1,029 was detected by autofluorescence, HP1α immunodetection was processed using specific antibodies (red), and nuclei were visualized by DAPI staining. Scale bar = 10 μm.
- Wild‐type MEFs were transiently transfected with GFP‐REV3L761–1,029 construct and treated or not for 24 h with trichostatin A (TSA). Cells were released or not with fresh medium during 6 h and fixed with 4% formaldehyde after 0.5% Triton X‐100 pre‐extraction. The distribution of GFP‐REV3L761–1,029 was detected by autofluorescence, HP1α was immunodetected using antibodies (red) and nuclei were visualized using DAPI staining. Scale bar = 10 μm.
- Sequence alignment of REV3L protein among different species containing the PxVxL motif with canonical residues shown in blue.
- Schematic representation of REV3L2,732–3,130 construct. MEF cells were transiently transfected with GFP‐REV3L2,732–3,130 or GFP‐REV3L761–1,029 constructs and fixed with 4% formaldehyde. The distribution of GFP‐REV3L constructs was detected by autofluorescence and nuclei with DAPI staining. Scale bar = 10 μm.
- 293T cells were co‐transfected with F‐H‐REV3L full‐length or empty vector and V5‐HP1γ, mutant IY165 168EE V5‐HP1γ, mutant V32 M IY165 168EE V5‐HP1γ or empty vector. Forty‐eight hours after transfection, cell lysates were made and used for immunoprecipitation with V5 antibody. After electrophoresis, samples were immunoblotted with anti‐HA, anti‐HP1γ, or anti‐α–Tubulin as indicated.
- GST, GST‐REV3L700–900, mutant V802D GST‐REV3L700–900, His‐XPA‐FLAG, His‐HP1α, and His‐HP1γ were purified from E. coli and stained with Coomassie Brilliant Blue. These were used with glutathione beads for GST pulldown experiments. After electrophoresis, samples were immunoblotted with anti‐His or anti‐GST as indicated.
- Images from REV3L‐HP1α PLA experiment described in EV3D were analyzed for the localization of PLA signals in the nucleus by distinguishing PLA foci In/around chromocenters and PLA foci out of chromocenters using ImageJ tools. Chromocenters were identified by the DAPI density. One hundred fifteen nuclei have been analyzed. Red lines indicate the mean values ± SD.
Source data are available online for this figure.
We therefore assessed whether REV3L directly interacts with HP1α. The chromoshadow domain of HP1α interacts with proteins containing a pentapeptide motif, PxVxL (Murzina et al, 1999; Thiru et al, 2004). Such a motif is present at position 800–804 in the 268‐amino acid polypeptide (REV3L761–1,029) that can target pericentromeric heterochromatin (Fig 5F). The PxVxL sequence and the putative NLS in this region are both completely conserved in vertebrate REV3L [(Fig EV4C) and Appendix Fig S1 in Lange et al, 2016)]. We tested whether this motif was critical for REV3L localization into chromocenters by introducing point mutations in the PSVVL motif. Localization of a transfected mutant GFP fusion protein (e.g., ASAVA) was strongly impaired (Fig 5G). Two alternative candidate PxVxL motifs are present near the C‐terminus of REV3L. The expression of a REV3L construct containing these two motifs (GFP‐REV3L2,732–3,130) in mouse cells showed a diffuse staining in both nucleus and cytoplasm (Fig EV4D), indicating that these motifs alone cannot target REV3L at PHC. Taken together, these data reveal that the PxVxL motif at position 800–804 is important for REV3L localization at pericentromeric heterochromatin.
To confirm a direct interaction between REV3L and HP1, we performed a GST pull‐down assay by using fusion polypeptides purified from E. coli. GST‐XPA was used as negative control. While GST‐REV3L700–900 constructs bound to His‐HP1α and His‐HP1γ, the V802D mutant REV3L700–900 did not (Fig EV4E), demonstrating that REV3L directly interacts with HP1 via its PxVxL motif at position 800–804. We confirmed these results by transfecting 293T cells with plasmids expressing F‐H‐REV3L700–900, V802D F‐H‐REV3L700–900, or empty vector. WT REV3L700–900 but not the mutant co‐immunoprecipitated using HP1γ antibody (Fig 5H).
Homodimerization of HP1 through its chromoshadow domain is critical for HP1 binding of H3K9me3 chromatin and chromatin condensation (Hiragami‐Hamada et al, 2016). To gain insight into the molecular interaction between REV3L and HP1, we asked whether HP1 dimerization is mandatory for REV3L interaction with HP1. 293T cells were co‐transfected with F‐H‐REV3L700–900 or empty vector and V5‐HP1γ, IY165‐168EE V5‐HP1γ mutant (unable to dimerize), or empty vector. We found that dimerization of HP1 is required for its interaction with REV3L (Fig 5I). We confirmed this result using F‐H‐tagged full‐length REV3L (Fig EV4F). These results suggest that REV3L binds HP1 when HP1 forms condensed H3K9me3‐modified chromatin.
We then evaluated at the endogenous level whether REV3L interacts with HP1 only at chromocenters, or also in heterochromatic regions dispersed in the genome. For that, we used images acquired for REV3L‐HP1 PLA quantification (Figs 5B and EV3D) and determined the localization of each signal in the nucleus (Fig EV4G). Given that replication of pericentromeric regions occurs mainly at the surface of the chromocenters where PCNA is located (Quivy et al, 20042004), we distinguished foci “In/around” of chromocenters with foci “Out” of chromocenters. We used DAPI density to recognize these structures. We determined the localization of REV3L‐HP1 PLA signals in 115 nuclei. We observed that 41/115 cells (36%) have ≥ 2 REV3L‐HP1 PLA foci in/around chromocenters, suggesting that REV3L interacts with HP1 in pericentromeric regions in one third of cells which probably undergo DNA replication. The mean of REV3L‐HP1 PLA foci in and out of chromocenters/nucleus was 1.25 and 4.4, respectively (Fig EV4G), consistent with the idea that REV3L interacts with HP1 in pericentromeric heterochromatin, but also in other heterochromatic regions localized throughout the genome. To further explore specific interactions of REV3L with heterochromatin, we examined the HP1‐binding protein SCAI (suppressor of cancer cell invasion; C9orf126) (Nozawa et al, 2010). SCAI functions in connection with 53BP1 to mediate ATM‐dependent DSB signaling in heterochromatin (Hansen et al, 2016). A mass spectrometry‐based approach identified REV3L as a high‐confidence SCAI interactor (Appendix Fig S1 in Isobe et al, 2017). We validated the REV3L‐SCAI interaction by co‐immunoprecipitation with GFP‐tagged REV3L constructs (Fig 5J). These experiments showed that SCAI‐REV3L interaction is mediated by a region in REV3L between amino acids 1,030–1,876. HP1β was used as positive control for its interaction with residues 800–804 in REV3L. Further, full‐length SCAI, but not SCAI1–262 or SCAI1–442, can be co‐immunoprecipitated with endogenous REV3L (Fig 5K), indicating an interaction of REV3L with the C‐terminal part of SCAI.
REV3L facilitates replication through pericentromeric heterochromatin, preventing DNA double‐stranded breaks
These observations prompted us to investigate whether Polζ/REV3L may facilitate DNA replication at pericentromeric heterochromatin. DNA combing coupled with PNA probe hybridization was performed to specifically analyze pericentromeric fibers independently from bulk DNA fibers by using pericentromeric Satellite III (SatIII) repeats as probe (5′‐TGGAA). We found that depletion of REV3L in HeLa cells resulted in shorter nascent DNA tracks and an increased fork ratio at SatIII regions, indicating that REV3L loss impairs fork progression at pericentromeric regions (Fig 6A). In contrast, REV3L depletion does not have a major effect on genome replication dynamics globally. However, a slight but significant increase in fork ratio was observed at globally after REV3L depletion, suggesting that Polζ/REV3L might assist replication of genomic regions in addition to pericentromeric heterochromatin.
Figure 6. REV3L facilitates replication in pericentromeric heterochromatin, preventing DNA double‐stranded breaks.
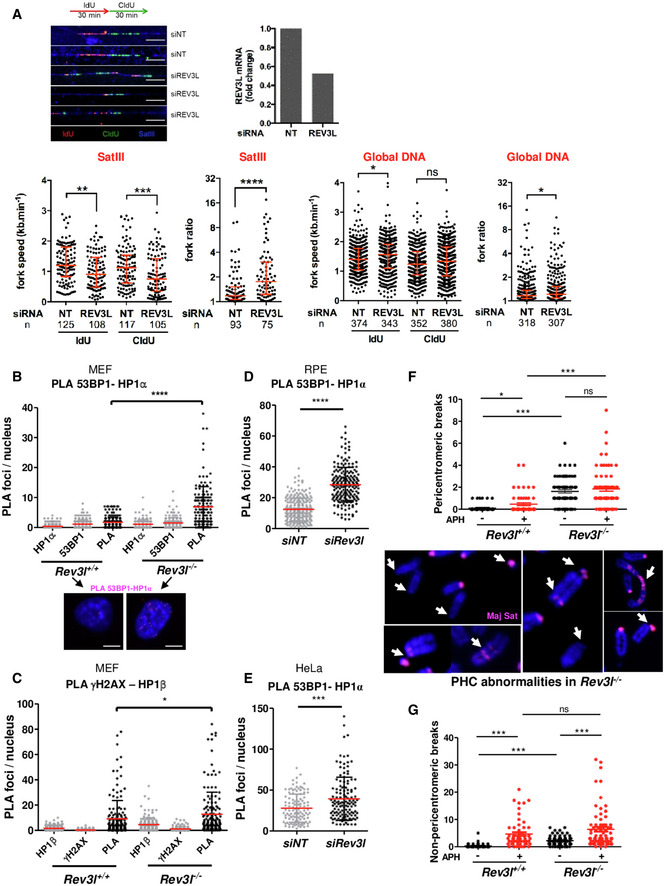
- Representative fibers of newly synthesized DNA labeled with IdU (red) for 30 min and CldU (green) for 30 min in HeLa cells transfected with non‐targeting siRNA (siNT) or siRNA against Rev3l. SatIII probe is visualized in blue. Scale bar: 10 μm = 20 kb. Relative Rev3l mRNA level normalized to GAPDH mRNA level is shown (top panel). Distribution of fork speeds (kb/min) and fork ratios (IdU/CldU track length) are shown in dot plots for SatIII and global DNA (bottom panel). The number of fibers analyzed is indicated in (n). Bars represent the median ± interquartile range (Mann–Whitney test. ns: not significant, *P < 0.05; **P < 0.01, ***P < 0.001 and ****P < 0.0001). The presented data are representative of three biological repeats.
- Asynchronous Rev3l +/+ and Rev3l −/− MEFs were fixed with PFA then subjected to in situ proximity ligation assay (PLA) using 53BP1 and HP1α antibodies; then, PLA foci were counted in both cell lines (more than 150 nuclei for each condition were counted). P‐values were calculated by Mann–Whitney test (****P < 0.0001). Red lines indicate the mean values. Error bars: SEM. Controls with a single antibody are also shown. Experiments were repeated three times. Representative images are shown. Scale bar = 5 μm.
- Asynchronous Rev3l +/+ and Rev3l −/− MEFs were subjected to PLA as by using γH2AX and HP1β antibodies and processed as in (B). P‐values were calculated by Mann–Whitney test (*P < 0.05). Red lines indicate the mean values. Error bars: SEM. Controls with a single antibody are also shown. Three independent experiments were performed.
- RPE cells were transfected with non‐targeting siRNA (siNT) or siRNA against Rev3l; then, 72 h later cells were subjected to PLA by using 53BP1 and HP1α antibodies and processed as in (B). P‐values were calculated by Mann–Whitney test (****P < 0.0001). Red lines indicate the mean values. Error bars: SEM. Three independent experiments were performed.
- HeLa cells were transfected with non‐targeting siRNA (siNT) or siRNA against Rev3l; then, 72 h later cells were subjected to PLA by using 53BP1 and HP1α antibodies and processed as in (B). P‐values were calculated by Mann–Whitney test (***P < 0.001). Red lines indicate the mean values. Error bars: SEM. Three independent experiments were performed.
- Rev3l +/+ and Rev3l −/− MEFs were incubated with or without 0.23 μM aphidicolin for 24 h before metaphase spreading. FISH was performed using major satellite probe to quantify breaks in pericentromeric regions (F). Representative images of chromosomes showing abnormalities (see arrows) in pericentromeric regions from Rev3l −/− MEFs. Chromosomes were labeled with DAPI, and breaks in non‐pericentromeric regions were quantified (G). Error bars indicate standard error of the mean for three independent experiments. Mann–Whitney test (ns: not significant, *P < 0.05; ***P < 0.001).
Given that REV3L loss leads to genomic instability and chromosome breaks (Wittschieben et al, 2000), we asked whether DNA double‐stranded breaks (DSBs) occur in heterochromatin regions. We first used PLA to examine whether DSB markers 53BP1 and γH2AX were spatially close to the heterochromatin‐associated factor HP1 in Rev3l −/− cells. We observed that specific PLA signals between 53BP1 and HP1α (Fig 6B) and γH2AX and HP1β (Fig 6C) were higher in Rev3l −/− cells than in Rev3l +/+ cells in unchallenged conditions. We confirmed these observations in human cells after depleting REV3L in RPE and HeLa cells (Fig 6D and E, respectively). These results suggest that DNA replication forks in heterochromatic regions might be prone to collapse in the absence of Polζ/REV3L.
Thus, we explored the consequences of REV3L depletion on PHC stability. DNA fluorescence in situ hybridization was performed on metaphase chromosome spreads using major satellite DNA as probe. For spontaneous breaks (in the absence of aphidicolin), a Rev3l defect elevates the frequency of breaks ~20‐fold in pericentromeric regions (Fig 6F, upper panel), and about 10‐fold in non‐pericentromeric regions (Fig 6G). The consequence of Rev3l disruption is thus highly biased toward pericentromeric breaks, indicating an especially important function of Pol ζ in PHC. A variety of abnormalities were observed in PHC, including breaks/gaps, loss, duplication, and rearrangement (Fig 6F, lower panel). Exposure to aphidicolin (causing fork slowing) increases breaks in all regions. In PHC, the increase is Rev3l‐dependent (Fig 6F), indicating that pol ζ is also particularly important in PHC for limiting the frequency of aphidicolin‐stimulated breaks. In non‐pericentromeric regions, Rev3l status did not affect the aphidicolin‐stimulated frequency, showing that other mechanisms are effective globally in preventing the additional breaks initiated by aphidicolin.
Rev3l‐deficient cells show increased number of genomic deletions
To gain deeper insight into the role of Polζ/REV3L in preventing DSBs and genomic instability at the genome‐wide level in mammalian cells, we characterized structural variations that arise upon loss of Rev3l. Whole‐genome sequencing from subclones of Rev3l‐deficient MEFs was analyzed (Fig 7A). We identified in total 102 structural variants, including 66 deletions, 13 inversions, 10 duplications, and 13 complex events from 4 samples (Fig 7B), using both GRIDSS2 and Manta algorithms (see Materials and Methods). The mean deletion size was 2,092 bp and ranged from 55 to 9,115 bp. Importantly, we found 61 deletions in Rev3l −/− samples and only 5 in Rev3l +/+ samples (95%CI = 7.6–13.4, P‐value < 2.2e‐16, Poisson test) and the number of deletions was consistent between replicates (Fig 7C). Note that none deletion was shared between samples, suggesting that they occurred during independent cell cultures. Moreover, the size of the deletions that accumulate in Rev3l −/− cells was within a rather narrow range, with 21 deletions (34%) smaller than 1,000 bp.
Figure 7. Rev3l‐deficient cells show numerous genomic deletions.
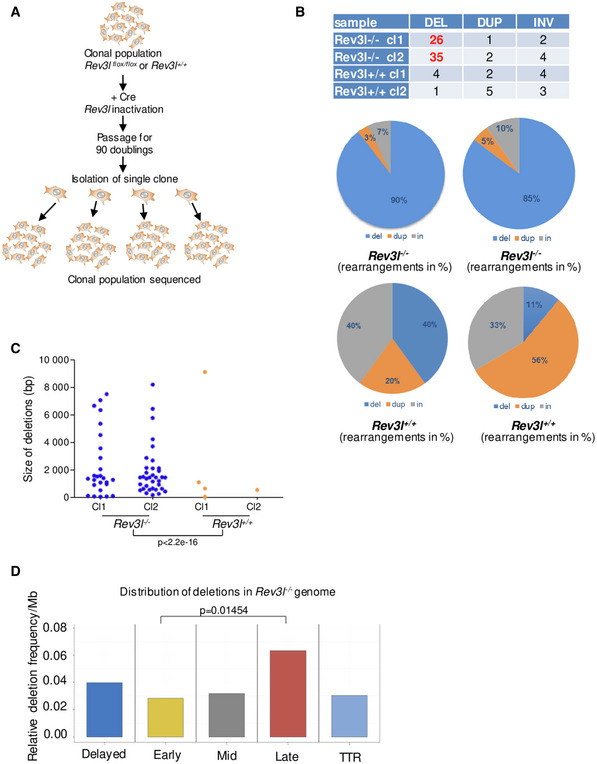
- Experimental design for the whole genome sequencing of Rev3l‐deficient cells. Populations of mouse embryonic fibroblasts Rev3l +/+ and Rev3l −/− were isolated from Rev3l +/+ and Rev3l flox/flox littermates, and Rev3l was excised using CRE recombinase. The resulting Rev3l‐deficient cells and Rev3l +/+ cells were expanded for 90 doublings. Rev3l +/+ and Rev3l −/− subclones were then isolated for deep sequencing.
- Percentage of rearrangements (deletions, duplications, and inversions) identified in Rev3l +/+ and Rev3l −/− subclones.
- Size distribution of the deletions detected in Rev3l +/+ and Rev3l −/− subclones. Poisson test. Deletions were counted from four independent clones.
- Distribution of the deletions detected in Rev3l −/− subclones related to the replication timing in deletions per megabase. Difference between late and early regions is statistically significant (P = 0.01454, Poisson test with the correction for different length of intervals through the ratio). Data were calculated from two independent clones.
We then correlated deletions with replication timing observed in Rev3l −/− cells and found that deletions were preferentially localized in late‐replicating regions as well as in domains for which replication timing was disturbed in the absence of REV3L (Fig 7D). Given that heterochromatic regions (facultative and constitutive) replicate in mid‐ and late S‐phase, we assume that deletions found in Rev3l −/− mouse cells might be initiated by replication‐associated DSBs that arise when replication forks stall and collapse in difficult‐to‐replicate heterochromatic regions.
Loss of REV3L compromises heterochromatin‐associated DSB repair
The results above imply that spontaneous DSBs originate in Rev3l‐deficient cells, at least partly, as a consequence of inefficient replication fork progression through heterochromatin. However, it has been proposed that Polζ/REV3L can also facilitate DSB repair by homologous recombination (Sharma et al, 2012). Therefore, the high level of DSBs in the absence of Polζ could be due to a combination of increased replication‐associated DSBs and inefficient repair of those breaks.
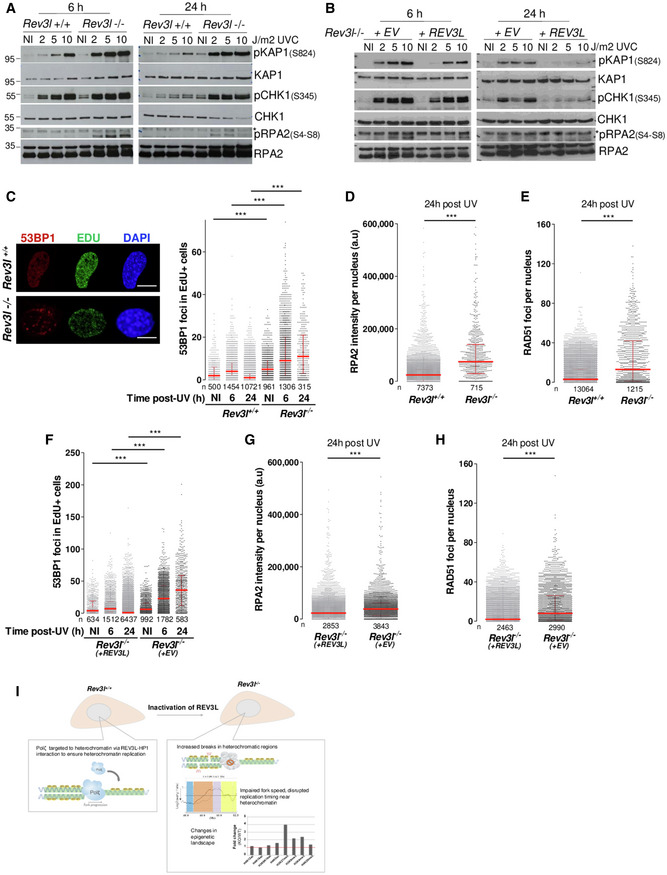
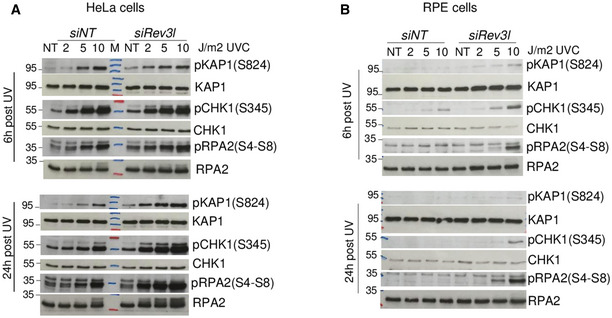
To investigate the effect of Polζ/REV3L in regulating repair of heterochromatin‐associated DSBs, we challenged MEFs with UV radiation. This should lead to enhanced replication‐associated DSBs in Rev3l −/− cells (Sonoda et al, 2003). DSB signaling and repair were monitored in several ways. Phosphorylation of the Kruppel‐associated box (KRAB)‐associated co‐repressor, KAP1 by ATM in response to DSBs, occurs during DNA repair in heterochromatin (Goodarzi et al, 2008). There was a marked increase in S824‐phosphorylated KAP1 from 2 J/m2 in Rev3l −/− MEFs (Fig 8A and B) which remained elevated 24 h after UV irradiation. In contrast, induction of pKAP1 required higher doses in Rev3l +/+ MEFs and Rev3l −/− cells complemented with REV3L, and largely disappeared after 24 h. This suggests that heterochromatin‐associated DSBs accumulate in UV‐irradiated cells when Polζ is missing. Moreover, Rev3l −/− cells had increased activation of phosphorylated (S4‐S8) RPA2 (an indicator of resection at DSBs), and a persistent level of pCHK1 (Fig 8A and B). Similar results were obtained in human cells after down‐regulating REV3L in HeLa and RPE cells (Fig EV5).
Figure 8. REV3L loss compromises heterochromatin‐associated DSB repair.
-
AAsynchronous Rev3l +/+ and Rev3l −/− MEFs were UV‐irradiated at the indicated doses and harvested 6 or 24 h later. Cell lysates were analyzed by immunoblotting with indicated antibodies.
-
BAsynchronous Rev3l −/− MEFs complemented with empty vector (EV) or REV3L were UV‐irradiated at the indicated doses and harvested 6 or 24 h later. Cell lysates were analyzed by immunoblotting with indicated antibodies.
-
C–EAsynchronous Rev3l +/+ and Rev3l −/− MEFs were pulse‐labeled for 15 min using 10 μM EdU then UV‐irradiated at 4 J/m2. Cells pre‐treated with CSK buffer were fixed, and 53BP1, RPA, and RAD51 were detected by immunofluorescence. Representative images in non‐irradiated cells (NI) and quantification of 53BP1 foci in EdU‐positive cells at different time points are shown (C). Scale bar = 10 μm. Quantification of the intensity of chromatin‐bound RPA2 (D) and RAD51 foci (E) 24 h after UV irradiation was performed. The number of cells analyzed is indicated in (n) on each graph. ***P < 0.001 (Kruskal–Wallis test). The presented data are representative of 2 repeats. Bars represent the median ± interquartile range.
-
F–HAsynchronous Rev3l −/− MEFs complemented with empty vector (EV) or REV3L were processed as in (C). Quantification of 53BP1 foci in EdU‐positive cells at different time points is shown (F). Quantification of the intensity of chromatin‐bound RPA2 (G) and RAD51 foci (H) 24 h after UV irradiation was performed. The number of cells analyzed is indicated in (n) on each graph. ***P < 0.001 (Kruskal–Wallis test). The presented data are representative of two repeats. Bars represent the median ± interquartile range.
-
IModel that summarizes the results obtained in this study.
Source data are available online for this figure.
Figure EV5. REV3L loss impacts on heterochromatin‐associated DSB repair in human cells.
-
A, BHeLa cells (A) or RPE cells (B) were transfected with non‐targeting siRNA (siNT) or siRNA against Rev3l; then, 72 h later cells were UV‐irradiated at the indicated doses and harvested 6 or 24 h post‐irradiation. Cell lysates were analyzed by immunoblotting with indicated antibodies.
Source data are available online for this figure.
We further examined the dynamics of DSB formation and repair by single‐cell immunofluorescence. Following irradiation with 4 J/m2, the levels of 53BP1 foci in EdU+ cells slightly increased 6 h after UV exposure in Rev3l‐proficient cells and returned to a basal level 24 h later (Fig 8C and F). In Rev3l −/− cells in S‐phase, 53BP1 foci accumulated at 5.6‐fold higher levels 24 h post‐UV.
To assess a potential defect in Rev3l‐deficient cells to complete DNA repair by homologous recombination (HR), we quantified the level of resection indirectly by measuring levels of chromatin‐bound RPA2 in individual cell nuclei 24 h after UV irradiation. In agreement with pRPA2 immunoblots, we observed that chromatin‐bound RPA2 was 3.6‐fold higher in cells lacking REV3L than in wild‐type cells (Fig 8D and G). We also evaluated RAD51 focus formation, an indicator of HR initiation. Twenty‐four hours after UV exposure, RAD51 foci were elevated 3‐fold in Rev3l‐deficient MEFs compared with wild‐type cells (Fig 8E and H). This suggests that Polζ/REV3L may be required for efficient completion of DSB repair by HR.
Discussion
Polζ contributes to heterochromatin replication and repair
This study unveils a direct role for mammalian Polζ/REV3L in replication and repair in heterochromatin. Several lines of evidence point toward this conclusion, summarized in Fig 8I. First, inactivation of Rev3l drives an elevated number of breaks and structural variations in heterochromatic regions, particularly deletions that likely arise as a consequence of replication fork stalling or collapse occurring during DNA replication. Second, Polζ/REV3L influences replication fork progression in pericentromeric heterochromatin. REV3L‐compromised cells exhibited a marked decrease in fork speed at pericentromeres which contain highly repeated DNA sequences embedded into compacted chromatin, making them difficult to replicate. A disruption of replication timing at specific loci mainly located in TTR may also be ascribed to heterochromatin replication and repair‐associated functions of Polζ. Third, we show that Polζ directly interacts with specific components of heterochromatin including HP1 and SCAI. Fourth, we show that REV3L disruption is correlated with changes in epigenetic landscape and transcriptional control of developmentally regulated genes.
Polζ/REV3L interacts with heterochromatin proteins
We found that REV3L directly interacts, via a central PxVxL motif, with the heterochromatin component HP1. Dimerization of HP1 is necessary for interaction with REV3L, indicating that such binding occurs only when HP1 is engaged on chromatin through its reader interaction with H3K9me3 (Hiragami‐Hamada et al, 2016). The PxVxL motif located at positions 800–804 in the human REV3L sequence is embedded in a larger region that is well‐conserved in vertebrates but absent in invertebrate and fungal genomes. This indicates that the heterochromatin targeting of Polζ described in this study evolved in concert with larger genomes having more complex controls. The REV7 subunit also is associated with HP1α (Tomida et al, 2018), suggesting another mode of interaction of Polζ with heterochromatin. It is not yet known whether Polζ associates with the replisome to duplicate heterochromatin regions. The replisome may change throughout the S‐phase to assist conventional DNA polymerases to duplicate challenging areas of the genome such as compacted heterochromatin. Potentially, targeting of REV3L to heterochromatin could facilitate a proposed catalytic subunit switch with Polδ, mediated by PCNA and the shared POLD2 and POLD3 subunits. The interaction of the DONSON protein and the FANCM translocase with the replisome complex is dependent on replication timing and chromatin environment (Zhang et al, 2020). Further, progression of replication forks through pericentromeric heterochromatin is also facilitated by the shelterin subunit TRF2, in association with the helicase RTEL1 (Mendez‐Bermudez et al, 2018). Investigation is warranted to define whether REV3L/Polζ works together with TRF2 and RTEL1 to facilitate fork progression in heterochromatic regions.
Polζ limits heterochromatin‐associated DSBs
Our data also show that Rev3l‐deficient MEFs exhibit an increased incidence of deletions, preferentially in late‐replicating regions. This is consistent with the observed ~10‐fold increase in the frequency of DNA double‐stranded breaks and chromosome rearrangements in such MEFs (Lange et al, 2013, 2016), similar to the spectrum of changes in HR‐deficient MEFs (Wittschieben et al, 2006). The present study shows that many spontaneous DSBs originate during replication of pericentromeric heterochromatic regions. We also provide evidence that Rev3l inactivation compromises repair of heterochromatin‐associated DSBs. Low doses of UV radiation provoke replication‐associated DSBs, leading to a massive increase of sustained 53BP1 foci in S‐phase, but also markers of HR including pRPA2 and RAD51 foci. One hypothesis to explain the high level of these breaks can be the delay in repair caused by saturation of the repair machinery in Rev3l KO cells. Alternatively, Polζ/REV3L might be required for efficient HR‐associated DNA synthesis to complete DSB repair (Sharma et al, 2012). There is evidence that Polζ‐mediated DNA synthesis occurs during DSB repair in some settings in different organisms (Kane et al, 2012) (Martin & Wood, 2019). The contribution of specific DNA polymerases in HR‐associated DNA synthesis may be subject to temporal control of replication and to chromatin composition. Indeed, our results showed that Rev3l‐deficient cells fail to efficiently resolve DSBs in heterochromatin, with a pronounced persistence of S824‐phosphorylated KAP1, known to facilitate chromatin decondensation and allows efficient DSB repair in heterochromatin (Goodarzi et al, 2008). Consistent with this, we demonstrated that REV3L interacts with the SCAI protein, a mediator of 53BP1‐dependent repair of heterochromatin‐associated DSBs (Hansen et al, 2016). By interacting with 53BP1, SCAI counteracts RIF1 function, facilitating BRCA1‐mediated repair (Isobe et al, 2017). Future studies may determine the mechanisms by which Polζ/REV3L is involved in the repair of heterochromatin‐associated DSBs by interaction with SCAI.
Absence of Polζ disrupts replication timing at specific loci
We found that REV3L loss perturbs the temporal replication program in mouse and human cells, which might be a consequence of fork progression alterations. The replication‐timing defect affects only specific areas, mainly in temporal transition regions, corresponding to 5.7% of the whole genome in Rev3l‐deficient mouse cells. This is a contrast to the change in replication patterns following inactivation of RIF1, with a shift in replication timing in over 40% of all replication segments (Cornacchia et al, 2012; Hayano et al, 2012). Our data indicate an involvement of REV3L/Polζ in the replication of particular TTRs that lie between early‐replicating DNA and late‐replicating DNA and, therefore, often replicate in mid‐to‐late S‐phase. Consistent with this, pericentromeric heterochromatin is replicated in mid‐to‐late S‐phase in mouse cells (Guenatri et al, 2004; Natale et al, 2018). Replication in TTR is achieved by either sequential activation of a series of origins (Guilbaud et al, 2011) or sequential activation of single long unidirectional fork that initiates at an adjacent early origin (Norio et al, 2005; Hiratani et al, 2008; Schultz et al, 2010). It has been reported that the replication machinery propagates at a slower velocity in some TTRs (Farkash‐Amar et al, 2008; Desprat et al, 2009; Donley & Thayer, 2013), suggesting that one major consequence is an increase in the probability of replication fork stalling and breaks as has been proposed for fragile sites (Watanabe & Maekawa, 2010; Letessier et al, 2011; Ozeri‐Galai et al, 2014). Consistent with this, Rev3l‐deficient MEFs exhibit genomic deletions in perturbed TTR.
Potential contribution of REV3L/Polζ to the mutation rate in late‐replicating heterochromatic regions
A growing body of evidence suggests that the replication‐timing program strongly influences the spatial distribution of mutagenic events during both species and cancer evolution with an increasing gradient of single‐nucleotide substitutions that correlate with late‐replicating heterochromatic regions (Stamatoyannopoulos et al, 2009; Koren et al, 2012; Schuster‐Bockler & Lehner, 2012). Given the error‐prone activity of Polζ, our results suggest that REV3L/Polζ may contribute to this increased mutation rate in heterochromatic regions which are relatively poor in transcribed genes. This is consistent with a bioinformatic analysis showing that mutations related to Polζ signature increase in late‐replicating regions of the human genome (Seplyarskiy et al, 2015). Moreover, experiments in yeast established that late‐replicating regions of the genome also have higher rates of spontaneous mutagenesis than early‐replicating regions. Deletion of yRev1 significantly lowers mutation rate specifically in these late‐replicating regions, suggesting that Rev1/Polζ complex is involved in the gradient of mutagenesis in lower eukaryotes (Lang & Murray, 2011).
REV3L loss impacts the epigenetic landscape and transcriptional program
The majority of disturbed regions in Rev3l‐deficient mouse cells (> 80%) replicate later than in wild‐type cells, suggesting heterochromatinization of these regions that shifts their replication timing. We further found that the inactivation of Rev3l affects epigenetic landscape and transcriptional program in mouse cells, with a substantial increase in H3K27me3 levels and, to a lesser extent, H3K9me3 and H3K4me3. This may be linked to DSB repair processes. Heterochromatin factors including KAP1, HP1, and the H3K9 methyltransferase Suv39h1 accumulate rapidly and transiently at DNA damage sites, in both euchromatic and heterochromatic regions (Lemaitre & Soutoglou, 2014; Nikolov & Taddei, 2016). Moreover, polycomb proteins involved in gene silencing at facultative heterochromatin are also recruited to DSB sites, where they are thought to switch off transcription to facilitate DSB repair (Vissers et al, 2012; Ui et al, 2015). Thus, continued DSB formation in Rev3l‐deficient cells might result in a “heterochromatinization” at particular genomic regions. Interestingly, the TLS polymerase REV1, a key partner of Polζ, also influences the epigenetic landscape. During DNA replication, REV1 is required for the maintenance of repressive chromatin marks and gene silencing in the vicinity of G4 structure in DT40 chicken cells (Sarkies et al, 2010). Moreover, REV7 has been shown to function in epigenetic reprogramming by interacting with the G9a/G9a‐like protein (GLP) histone lysine methyltransferase complex which catalyzes H3K9 mono‐ and dimethylation (Pirouz et al, 2013).
Our transcriptome analysis revealed that Rev3l‐deficient cells repress numerous genes, known to contain bivalent promoters such as multiple Hox genes which are regulated by H3K27me3/H3K4me3 (Sachs et al, 2013). Whether REV3L/Polζ contributes to H3K27me3 regulation during DNA replication is largely unexplored. Of note, the large majority of the genes whose expression is down‐regulated by Rev3l deletion do not fall within disturbed regions of replication timing. One possibility is that gene repression is due to the heterochromatinization of the loci that undergo continued DSBs repair. This implies that DSBs induced by Rev3l inactivation occur not only in heterochromatin, but also in euchromatin. We also found that several imprinted genes are down‐regulated in the absence of REV3L. Rajewsky and colleagues suggested that the overall phenotype of Rev3l −/− embryos appears comparable to androgenetic embryos (Fundele & Surani, 1994), raising the possibility that Polζ participates in imprinting establishment (Esposito et al, 2000). Thus, our results on the transcriptional program in Rev3l‐deficient mouse cells may partially explain the embryonic lethality observed in Rev3l KO mice.
Conclusion
Unique among translesion synthesis DNA polymerases, Polζ is essential during embryogenesis and cell proliferation (Wittschieben et al, 2000; Lange et al, 2012, 2018). However, the mechanisms of essentiality under normal growth conditions have not been clear. This study shows that Polζ prevents spontaneous chromosome break formation, rearrangements, and deletions in pericentromeric regions by facilitating fork progression, and may also operate in heterochromatin‐associated DSB repair. Altogether, these results reveal a new function of Polζ in preventing chromosome instability during replication of heterochromatic regions possibly at the expense of increased mutations in late‐replicating regions found in numerous tumors.
Materials and Methods
Cell culture and treatment
Immortalized mouse embryonic fibroblast (MEF) Rev3l −/− and Rev3l +/+ were previously described (Lange et al, 2012) and were cultivated in D‐MEM (Dulbecco’s modified Eagle medium; Gibco) containing 100 U/ml penicillin, 100 µg/ml streptomycin, and 10% fetal calf serum (FCS) in an atmosphere containing 5% CO2. MEFs expressing functional REV3L with a 3X‐Flag epitope tag at the endogenous locus were derived from knock‐in mice as described below. Rev3l −/− MEFs complemented with POZ empty vector (clone 4–5 POZN Cl2) or POZ‐hREV3L (clone 4–5 POZRev3l2 Cl H11) have been previously described (Lange et al, 2016). Suv39h double‐null MEFs kindly provided by T. Jenuwein and NIH3T3 cells (ATCC) were cultivated in D‐MEM supplemented with 10% FCS, 100 U/ml penicillin, and 100 µg/ml streptomycin. hTERT RPE‐1 cells (ATCC) were grown in D‐MEM/F‐12 (Gibco) supplemented with 10% FCS, 100 U/ml penicillin, and 100 µg/ml streptomycin. HeLa cells (ATCC) were cultivated in D‐MEM containing sodium pyruvate, penicillin/streptomycin, and 10% FCS. All cell lines were incubated at 37°C in a 5% CO2 atmosphere and were regularly tested for mycoplasma.
For UVC irradiation (254 nm), cells were rinsed in pre‐heated phosphate buffer saline (PBS) and irradiated without any medium at a fluency of 0.65 J/m2/s. Aphidicolin (Sigma) stock solutions was at 3 mM in DMSO, and trichostatin A (Sigma) stock solution was at 2 mg/ml in methanol.
Generation of knock‐in mice expressing 3x‐FLAG tag REV3L
Ethics statement
All animal work in this study was done according to The University of Texas, MD Anderson Cancer Center Institutional Animal Care and Use Committee guidelines, and approved by the MD Anderson Animal Care and Use Committee (IACUC).
Construction of the targeting vector
The targeting vector construction was designed and performed by genOway (Lyon, France). The Rev3l targeting vector was constructed from 129 Sv/Pas mouse strain genomic DNA with a long (5.8 kb) homology arm upstream of exon 1, and a short (1.5 kb) homology arm downstream of exon 1. A 3x‐FLAG peptide sequence was inserted in frame with the ATG codon. A positive selection neomycin gene was flanked by loxP sites. The targeting vector also incorporated a diphtheria toxin‐negative selection cassette.
Screening of Rev3l targeted knock‐in ES cell clones
Linearized targeting vector was transfected into 129 Sv ES cells (genOway, Lyon, France) according to genOway's electroporation procedures (i.e., 5 × 106 ES cells with 40 µg of linearized plasmid, 260 V, 500 µF). Positive selection started 48 h after electroporation in medium containing 200 µg/ml of G418 (150 µg/ml of active component, Life Technologies, Inc.). Rev3l‐resistant clones were isolated and amplified in 96‐well plates. Duplicates of 96‐well plates were made. The set of plates containing ES cell clones amplified on gelatin were genotyped by both PCR and Southern blot analysis.
For PCR analysis, one primer pair was designed to validate the presence of the 3xFLAG tag within the 5′ homology arm. This primer pair was designed to specifically amplify the targeted locus:
Forward (Neo cassette): 5′‐CCTGCTCTTTACTGAAGGCTCTTTACTATTGC‐3′
Reverse: 5′‐GGAACCCACAGTGGTTGTCCTAGTGC‐3′
PCR products were sequenced in order to validate the presence of all elements. The targeted locus was confirmed by Southern blotting using a 3′ probe (Fig EV4A). Six clones were identified as correctly targeted and containing the 3x‐FLAG tag at the Rev3l locus.
Generation of mosaic mice and breeding scheme
Clones were microinjected into C57BL/6J blastocysts and gave rise to male mosaics with a significant ES cell contribution (as determined by agouti coat color). Mice were bred to C57BL/6 mice expressing the Cre recombinase to remove the Neo cassette.
Genotyping of the knock‐in mouse line
The following primers were used to monitor the Cre‐mediated excision event and served for genotyping.
Forward: 5′‐ACGAGTTCGCGGGTCCTTAGAGGTC‐3′
Reverse: 5′‐ACTTTCTACAGCCACAGCATCTCCGG‐3′
The wild‐type allele generates a product of 195 bp, the recombined allele 1,958 bp, and the Cre‐excised allele 289 bp.
Heterozygous mutant mice (which were maintained as pure C57BL/6 mice) were used to produce MEFs from embryos isolated at days 13.5 dpc. Seven heterozygous clones were obtained and T‐antigen immortalized as described (Lange et al, 2012).
Plasmid construction
The human REV3L full‐length cDNA was kindly provided by C. Lawrence (University of Rochester Medical Center, Rochester, NY, USA). To generate the series of GFP‐Rev3l plasmids, constructs were obtained by polymerase chain reaction (PCR) amplification from Rev3l cDNA as a XhoI–EcoRI fragment and cloned into peGFP‐C3 expression vector (Clontech). Point mutants were generated by PCR‐based methods using a QuikChange® site‐directed mutagenesis kit (Stratagene) according to the manufacturer's instructions.
Full‐length REV3L with an N‐terminal FLAG‐HA dual‐epitope tag was constructed in the pOZN vector as described previously (Tomida et al, 2015).
The cDNA encoding REV3L amino acids 700–900 was PCR amplified from REV3L/pETDuet‐1 (Tomida et al, 2015) as a XhoI–NotI fragment with 5′ REV3L (XhoI) primer and 3′ REV3L (NotI) primers (5′‐CCGCTCGAGATGAATACATTGGGCAAAAATTCTTTC and 5′‐TAAAAGCGGCCGCTTATCCAAAGTGACAGTCTATAAAAC) to clone into pGEX6P‐1 (GE Healthcare). The V802D mutation was introduced into pGEX6P‐1 using a site‐directed mutagenesis kit. The GST fusion protein was expressed and purified from E. coli as described (Tomida et al, 2015).
The HP1α (CBX5) and HP1γ (CBX3) cDNAs were obtained from Thermo Fisher Scientific. The cDNAs were cloned into vectors pETDuet‐1 and pCDH‐EF1α‐V5‐MCS‐IRES‐Puro for His and V5‐tagging, respectively. pCDH‐EF1α‐V5‐MCS‐IRES‐Puro was cloned into pCDH‐EF1α‐V5‐REV7‐IRES‐Puro from pRSFDuet‐1 (Novagen) XhoI–NotI fragment. pCDH‐EF1α‐V5‐REV7‐IRES‐Puro was initially PCR amplified from pOZN REV7 (Tomida et al, 2018) with 5′ V5‐1st primer and 3′ pOZ 3′ primers (5′‐ATTCTACGCTCGAGATGACCACGCTCACACGACAAG and 5′‐CGGAATTGATCCGCTAGAG). Three subsequent rounds of PCR used the following primer sets: 5′ V5 2nd primer and 3′ pOZ 3′ primers (5′‐CTCTCCTCGGTCTCGATTCTACGCTCGAGATGACCACGC and 5′‐CGGAATTGATCCGCTAGAG); 5′ V5 3rd primer and 3′ pOZ 3′ primers (5′‐AGCCTATCCCTAACCCTCTCCTCGGTCTCGATTCTAC and 5′‐CGGAATTGATCCGCTAGAG); 5′ V5 4th primer (EcoRI) and 3′ pOZ 3′ primers (5′‐GGAATTCATGGGTAAGCCTATCCCTAACCCTCTCCTCG and 5′‐CGGAATTGATCCGCTAGAG). The resulting V5‐REV7 fragment (XhoI–NotI) was cloned into pCDH‐EF1α‐MCS‐IRES‐Puro (System Biosciences). Mutations were introduced using a site‐directed mutagenesis kit (Stratagene). His‐XPA was as described (Manandhar et al, 2017). All constructs were verified by DNA sequencing. Details on all constructs are available upon request.
The cDNA encoding SCAI was cloned from HeLa mRNA by RT–PCR using the following primers: 5′‐ATGCATCTCGAGGTCAGAGGAGCCCGGCAGCCCCAGCAGC‐3′ and 5′‐ATGCATGAATTCTTAATAGTCATCAATGGTATTCTCAAAGAA‐3′. The resulting SCAI construct (XhoI–EcoRI) was verified by Sanger sequencing then cloned into peGFP‐C3 (Clontech).
Transfection
For transient transfection, plasmids were transfected using jetPEI (Polyplus), according to the manufacturer’s instructions, and cells were processed 24–48 h later.
siRNAs were purchased from Dharmacon: siRNA mRev3l (ON‐TARGETplus Smart pool), non‐targeting siRNA (ON‐TARGETplus non target pool). Cells were transfected with 30 nM of siRNAs using INTERFERin (Polyplus) according to the manufacturer’s instructions and were processed 72 h later.
Protein extraction, Immunoprecipitation, and Western blotting
Histone extraction was carried out in 10 volumes of CSK buffer (200 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 10 mM Tris–HCl pH 7, 1 mM EGTA, 0.5% Triton X‐100, 1 mM PMSF, 10 mM Na‐Butyrate) during 5 min at 37°C. After centrifugation, the pellet was resuspended in 0.4 mM H2SO4 supplemented with Na‐Butyrate and PMSF for 1 h, then centrifuged before adding acetone to supernatant, and incubated overnight in cold room. After centrifugation, the pellet was resuspended in distilled water and histone extracts were kept at −80°C.
For immunoprecipitation, cells were lysed in NETN buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5% NP‐40, protease inhibitor cocktail and anti‐phosphatases) for 30 min on ice and sonicated twice at 29% amplitude for 10 s. Samples were cleared by centrifugation for 5 min at 4°C. Immunoprecipitations were performed with Anti‐FLAG M2 Agarose Beads (Sigma‐Aldrich) overnight at 4°C on a wheel. Beads were extensively washed in NETN, with 300 mM NaCl for the final wash, and denatured in 2× Laemmli buffer.
For GFP‐Trap, cells transfected with plasmids expressing GFP, GFP‐REV3L mutants, GFP‐SCAIwt, or GFP‐SCAI mutants were collected in NETN buffer, incubated for 30 min on ice, sonicated twice at 29% Amplitude for 10 s then centrifuged at 4°C. The supernatant was incubated with GFP‐Trap beads (Chromotek) for 2 h at 4°C under rotation. Beads were washed and eluted in 2× Laemmli buffer.
Proteins were separated on 5, 8, or 15% acrylamide SDS–PAGE, transferred on Nylon membrane (GE Healthcare), and detected with the indicated antibodies and ECL reagents (Thermo Fisher Scientific).
GST pull‐down assay
GST‐tagged REV3L fragments and His‐tagged test proteins were incubated together with Glutathione‐Sepharose 4B (GS4B) beads (GE Healthcare) at 4°C for 4 h in 500 μl of buffer 0.1B (100 mM KCl, 20 mM Tris–HCl [pH 8.0], 5 mM MgCl2, 10% glycerol, 1 mM PMSF, 0.1% Tween‐20, 10 mM 2‐mercaptoethanol). The beads were washed three times with 0.1B and eluted with 30 μl of 2× SDS loading buffer (100 mM Tris–HCl [pH 6.8], 4% SDS, 0.2% bromophenol blue, 20% glycerol, 200 mM DTT).
Flow cytometry
In order to study the S‐phase progression in Rev3L −/− and Rev3L +/+ fibroblasts, cells were incubated with 50 µM bromodeoxyuridine (BrdU) for 15 min prior to be harvested at different time points. Cells were collected, washed in PBS, and fixed in 80% ice‐cold ethanol overnight at −20°C. Total DNA was stained in 2.5 µg propidium iodide (IP) and 50 µg/µl RNAse (Sigma) in PBS for 30 min at room temperature. Samples were analyzed on a C6 flow cytometer using the C6 Flow software (BD Accuri).
Immunofluorescence
For the visualization of DNA replication sites, asynchronous cells seeded on glass coverslips were incubated with 30 µM of thymidine analog (EdU or BrdU) for 15 min, pre‐extracted with CSK100 buffer (100 mM NaCl, 300 mM sucrose, 10 mM PIPES [pH 6.8], 3 mM MgCl2, 1 mM EGTA, 0.5% Triton X‐100, protease inhibitors) for 5 min at 4°C and fixed with 4% paraformaldehyde. For BrdU detection, cells were treated with 2 M HCl at 37°C for 20 min and then neutralized in 0.1 M borate buffer (pH 8.5). After washing in PBS, cells were incubated for 1 h at room temperature with anti‐BrdU mAb (347580, BD Biosciences). After washes, secondary antibodies coupled to Alexa Fluor were incubated for 30 min at room temperature. After washing three times with PBS, cells were mounted with mounting medium (Dako) supplemented with DAPI (Sigma). For EdU‐mediated foci visualization, EdU was detected using the Click‐iT EdU Alexa Fluor 488 Imaging Kit (Life Technologies) according to manufacturer’s instructions.
For detection of heterochromatin‐associated proteins and repressive histone marks, cells were pre‐extracted or not with CSK100 buffer as described above for 5 min on ice with gentle agitation then fixed with 4% paraformaldehyde. GFP‐REV3L mutants were detected by autofluorescence, and interesting proteins were visualized by immunodetection with indicated primary antibodies for 1 h then processed as described above. Images were acquired using Axio Imager Z1 microscope (Zeiss) or confocal Leica SPE.
For quantification of immunofluorescence in UV‐irradiated cells, MEFs growing on coverslips were incubated with 30 μM EdU for 15 min just before UV irradiation at 4 J/m2. The soluble proteins were extracted by incubating cells in CSK100 for 5 min then fixed with 4% paraformaldehyde. After washes in PBS, EdU was detected using the Click‐iT EdU Alexa Fluor 488 Imaging Kit (Life Technologies) according to the manufacturer’s instructions. Subsequently, coverslips were blocked for 30 min with 3% BSA in PBS and then incubated 1 h 30 at room temperature with indicated primary antibodies in PBS containing 3% BSA, 0.1% Tween‐20. The coverslips were further washed with PBS and incubated with Alexa Fluor 555 for 1 h at room temperature. The DNA was stained with DAPI, and the coverslips were mounted in fluorescent mounting medium (DAKO). Images were acquired on an Axio Imager Z1 microscope equipped with a motorized stage, EC Plan‐Neofluar x20 dry objective, and a digital monochrome Hamamatsu ORCA‐ER camera. For each condition, 20–50 images were acquired under non‐saturating conditions. Identical settings were applied to all samples within one experiment. Images were analyzed with the Axio Vision software (Zeiss). Specific module was generated for nuclei segmentation based on DAPI signal according to intensity threshold, generating a mask that identified each individual nucleus as an individual object. Focus segmentation for 51BP1 and RAD51 was performed using an integrated spot‐detection module. After segmentation and pixel quantification, the quantified values for each cell/foci (mean and total intensities, area, number of foci) were extracted and exported to a home‐made software as well as to CellProfiler software.
DNA FISH on metaphase chromosome spreads
A mouse major satellite repeat probe was generated by PCR with 5′‐ATATGTTGAGAAAACTGAAAATCACG‐3′ and 5′‐CCTTCAGTGTGCATTTCTCATTTTTCAC‐3′ primers and murine genomic DNA as a template. The probe was labeled by nick translation with biotin‐16‐dUTP. Rev3l −/− and Rev3l +/+ MEFs were treated with 0.15 μg/ml of colcemid for 3 h before fixation to arrest cells in metaphase. Trypsinized cells were rinsed in PBS and incubated in 75 mM KCl for 15 min then fixed with ethanol: acetic acid (3:1 ratio). The cells were then dropped onto glass slides and left to dry. Samples were incubated with 0.1 mg/ml RNase A solution at 37°C for 1 h in humid chamber then washed once for 5 min in 2× SSC. Slides were dehydrated in 70, 85, and 100% (v/v) ethanol for 5 min each and then air‐dried. Metaphase chromosome spreads were hybridized with mouse major satellite probe in hybridization buffer (10% dextran sulfate, 50% formamide, 2× SSC, 1% Tween‐20), denatured on a hot plate at 72°C for 4 min and then incubated overnight at 37°C in a humid chamber. Following hybridization, slides were washed with 50% formamide and 2× SSC pH 7 solution then washed in 2× SSC and incubated in BlockAid blocking solution (Thermo Fisher Scientific, B10710) and 0.05% Tween‐20. Immunodetection was performed by successive incubation (30 min at 37°C in BlockAid solution) with Alexa Fluor 555 streptavidin (S32355, Thermo Fisher Scientific), rabbit anti‐streptavidin biotin conjugated followed by Alexa Fluor 555 streptavidin. Samples were washed three times with 1× PBS after each incubation. After final wash, samples were mounted with VectaShield (Vector Laboratories, H‐1000). Images were acquired using Axio Imager Z1 microscope (Zeiss).
DNA combing‐coupled FISH
HeLa cells transfected with non‐targeting siRNA (siNT) or siRNA against Rev3l were pulse‐labeled for 30 min with IdU followed by 30 min with CldU (100 μM final concentration), and a final 10‐min incubation with 1 mM thymidine. Genomic DNA was combed onto in‐house silanized coverslips using a Genomic Vision apparatus. FISH and immunostaining of analogues were performed essentially as described in Mendez‐Bermudez et al (2018). Slides were denatured in 1 N NaOH for 6 min at room temperature, rinsed in 1× PBS at 4°C for 5 min, and dehydrated in 70% ethanol (v/v) for 5 min at −20°C, followed by 85 and 100% ethanol for 5 min each at room temperature.
For SatIII hybridization, coverslips were incubated overnight at 37°C with the PNA probe (SatIII: Biotin‐O‐TTCCATTCCATTCCATTCCA; Eurogentec) diluted in hybridization mix (10% dextran sulfate, 50% formamide, 2× SSC, 1% Tween‐20) to a final concentration of 1 μM and denatured for 5 min at 95°C. Coverslips were washed three times in 50% formamide, 2× SSC pH 7, three times in 2× SSC and one time in PBS, before blocking for 30 min at 37°C in BlockAid blocking solution (Thermo Fisher Scientific, B10710). Immunodetection was performed by successive incubations (30 min at 37°C in BlockAid solution) with: (i) Alexa Fluor 488 streptavidin (Thermo Fisher Scientific, S32354, 1/100), (ii) rabbit anti‐streptavidin‐biotin conjugated (Rockland, 200‐406‐095, 1/50), (iii) Alexa Fluor 488 streptavidin (1/100) + mouse anti‐BrdU (BD Biosciences, 347580, 1/5) + rat anti‐BrdU (Abcam Ab6326, 1/50), and (iv) goat anti‐mouse Cy5.5 (Abcam ab6947, 1/100) + goat anti‐rat Alexa Fluor 555 (Thermo Fisher Scientific, A21434, 1/100). Coverslips were washed three times with 1× PBS after each incubation, with the addition of a 6‐min wash in NaCl 0.5 M, Tris 20 mM pH 7.8, Tween‐20 0.5% after step 3. After final wash, samples were mounted with VectaShield (Vector Laboratories, H‐1000).
For global replication analysis, coverslips were blocked in BlockAid solution and incubated with: (i) mouse anti‐BrdU + rat anti‐BrdU, (ii) goat anti‐mouse Alexa Fluor 488 (Thermo Fisher Scientific, A11029, 1/50) + goat anti‐rat Alexa Fluor 555 (Thermo Fisher Scientific, A21434, 1/50), (iii) mouse anti‐ssDNA (DSHB, AB10805144, 1/25), (iv) goat anti‐mouse Cy5.5 (Abcam ab6947, 1/100), and (v) donkey anti‐goat Cy5.5 (Abcam ab6951, 1/100).
Images were acquired on an epifluorescence microscope Axio Imager.Z2(Zeiss) equipped with a motorized stage and a 63× objective lens (PL APO, NA 1.4 Oil DIC M27) connected to a charge‐coupled device camera (Cool‐SNAP HQ2; Roper Scientific). MetaMorph software (Roper Scientific) was used for image acquisition and analysis.
Only unbroken replication signals, as evidenced by embedment in total DNA staining or SatIII FISH signal, were taken into consideration. Fork speed (in kb/min) was calculated by divided the length of the IdU or CldU track (in kb) by the labeling time (in min). Fork ratio was calculated for each unbroken bicolor replication signal as the ratio between max (IdU length, CldU length) and min (IdU length, CldU length). Statistical analysis was performed with the nonparametric Mann–Whitney rank‐sum test using GraphPad Prism 6 software.
Proximity Ligation Assay (PLA)
Mouse embryonic fibroblast (MEF) Flag‐REV3L were seed on coverslips for 24 h and washed with PBS; then, protein pre‐extraction was carried out with CSK50 (50 mM NaCl for PLA Flag‐Histones) or CSK100 (100 mM NaCl for PLA DSB markers) for 5 min on ice, fixed with 4% paraformaldehyde, and blocked in BSA‐PBS 3% for 30 min. Coverslips were incubated with primary antibodies for 1 h at RT. Proximity ligation was performed using the Duolink® In Situ Red Starter Kit Mouse/Rabbit (Sigma‐Aldrich) according to the manufacturer’s protocol. The oligonucleotides and antibody‐nucleic acid conjugates used were those provided in the Sigma‐Aldrich PLA kit (DUO92101). Images were quantified by counting the number of foci per nucleus using ImageJ software. Statistical analysis was performed with the nonparametric Mann–Whitney rank‐sum test using GraphPad Prism 5 software.
Southern blotting
DNA methylation status at major satellites was performed on 5 μg of genomic DNA (gDNA) extracted from Rev3L −/− and Rev3L +/+ fibroblasts and digested with the methylation‐sensitive enzyme MaeII (NEB) overnight at 65°C. After precipitation with EtOH, the digested gDNA was run on an agarose gel overnight and successively depurinated with a 250 mM HCl solution, denatured with a solution containing 0.5 M NaOH and 1.5 M NaCl. After neutralization, the gDNA was transferred onto a Hybond‐N+ membrane (GE Healthcare Amersham) overnight. The membrane was prehybridized with hybridization buffer (6× SSC, 5× Denhardt’s, 0.5% SDS) containing 100 μg/ml salmon sperm DNA for 1 h at 65°C and incubated overnight at 65°C with the hybridization buffer containing radiolabeled probes against major satellites (5′‐GTGAAATATGGCGAGGAAAACT‐3′). The membrane was washed once with 5× SSC, 0.1% SDS at 65°C for 10 min, twice with 3× SSC, 0.1% SDS at 65°C for 10 min and twice with 2× SSC, 0.1% SDS at room temperature for 10 min before visualization using X‐ray film.
RNA extraction and RT–qPCR
Total RNAs were extracted from cell cultures and purified using RNeasy Mini Kit (QIAGEN), and concentration was measured with a Qubit Fluorometer (Invitrogen, Carlsbad, CA, USA). Total RNAs were then reverse transcribed using Superscript III (Invitrogen). cDNAs were used for quantitative PCR using TaqMan Gene Expression Assays (Applied Biosystems) on an ABI Prism 7500 apparatus (Applied Biosystems). GAPDH was used as an internal control. The comparative threshold (ΔCt) method was used.
ChIP‐qPCR
Rev3L −/− and Rev3L +/+ fibroblasts were seeded at 1.5 × 106 and 106 cells per dish, respectively. After 24 h, cells were cross‐linked with 1% formaldehyde for 10 min at RT and formaldehyde was quenched with Glycine 0.1 M for 5 min. Cells were washed twice with PBS + protease inhibitor, scraped in PBS + protease inhibitor, and then centrifuged at 800 × g –4°C for 5 min. Cell pellets were lysed in 1 ml of Cell Lysis Buffer (20 mM Tris–HCl [pH 8], 85 mM KCl, 0.5% NP‐40) for 15 min on ice and centrifuged at 800 × g at 4°C for 5 min. Pellets were then resuspended in 1 ml of Nuclei Lysis Buffer (Tris–HCl pH 8 50 mM, EDTA pH 8 10 mM, SDS 1%), sonicated using QSonica, Amplitude 40% for 10 cycles of 15 s ON/45 s OFF, and samples were centrifuged at 10,000 × g –4°C for 10 min. For each IP, supernatants were diluted 10 times in Dilution Buffer (0.01% SDS, 1.1% Triton X‐100, 1.2 mM EDTA, 16.7 mM Tris–HCl [pH 8], 150 mM NaCl) and incubated with 5 μg antibody, 20 μl Dynabeads Magnetic beads and 1% protease inhibitor cocktail overnight at 4°C with rotation. After magnetic separation, supernatant was removed and beads were washed successively with Low Salt Immune Complex Wash Buffer, High Salt Immune Complex Wash Buffer, and LiCl Immune Complex Wash Buffer followed by magnetic separation. The last wash was performed in 200 μl TE Buffer for 3min at 4°C with rotation; then, beads were resuspended in 100 μl TE Buffer. Samples were then deproteinated by adding Proteinase K and incubated at 55°C overnight with shaking. After proteinase K inactivation, beads were removed by magnetic separation to keep the supernatant. DNA purification was performed by phenol/chloroform extraction followed by ethanol precipitation and DNA resuspension in EB buffer. DNA samples were analyzed by quantitative real‐time PCR on the Applied Biosystems Real Time PCR system (7500 system) using the Maxima SYBR Green/Rox qPCR Mastermix (Thermo Fisher Scientific). The primers used are listed below.
Replication‐timing experiments and microarrays
Cells were incubated with 50 µM of BrdU for 1.5 h and collected, washed three times with DPBS (Dulbecco's phosphate‐buffered saline, Gibco Life Technologies), and then fixed in ethanol 75%. Cells were resuspended in DPBS with RNAse (0.5 mg/ml) and then with propidium iodide (50 µg/ml) followed by incubation in the dark at room temperature for 30 min with low agitation. Two fractions of 100,000 cells, S1 and S2 corresponding to early and late S‐phase fractions, respectively, were sorted by flow cytometry using INFLUX 500 (Cytopeia BD Biosciences). Whole DNA was extracted with a lysis buffer (50 mM Tris [pH 8], 10 mM EDTA, 300 mM NaCl, 0.5% SDS) and 0.2 mg/ml of Proteinase K for 2 h at 65°C. Neo‐synthesized DNA was immunoprecipitated with BrdU antibodies (Anti‐BrdU Pure, BD Biosciences, # 347580) as previously described (Fernandez‐Vidal et al, 2014). To control the quality of enrichment of early and late fractions in S1 and S2, qPCR was performed with BMP1 oligonucleotides (early control) and with Dppa2 oligonucleotides (late control; data not shown, (Hiratani et al, 2008)). Microarray hybridization requires a minimum of 1,000 ng of DNA. To obtain sufficient specific immunoprecipitated DNA for this hybridization step, whole genome amplification was conducted (WGA, Sigma) on immunoprecipitated DNA. A post‐WGA qPCR was performed to preserve specific enrichment in both S1 and S2 fractions. Early and late amplified neo‐synthesized DNAs were then labeled with Cy3 and Cy5 ULS molecules, respectively (Genomic DNA labeling Kit, Agilent). The hybridization was performed according to the manufacturer instructions on 4×180K mouse microarrays (SurePrint G3 Mouse CGH Microarray Kit, 4x180K, AGILENT Technologies, reference genome: mm9). Microarrays were scanned with an Agilent High‐Resolution C Scanner using a resolution of 3 µm and the autofocus option. Feature extraction was performed with the Feature Extraction 9.1 software (Agilent Technologies). For each experiment, the raw data sets were automatically normalized by the Feature extraction software. Analysis was performed with the Agilent Genomic Workbench 5.0 software. The log2‐ratio timing profiles were smoothed using the Triangular Moving Average option of the Agilent Genomic Workbench 5.0 software with the linear algorithm and 500 kb windows.
Identification of replication domains and changes in replication‐timing profiles
To determine the replication domains in control cells and in Rev3L −/− MEFs, the algorithms from the STAR‐R software were exploited as previously described (Hadjadj et al, 2020). A comparison was conducted between early and late domains from both cell lines in order to determine segments where replication‐timing changes.
GC content
For the GC content, two steps were required: (i) Loading of the DNA sequence from interval files was performed with the Extract Genomic DNA tool with the mm9 genome annotation and (ii) calculation of the GC content for each interval by the GeeCee EMBOSS tool installed on the GALAXY Web site.
Intersection and coverage with H3K27ac, H3K4me3, and H3K27me3 marks
Data are loaded from the UCSC Web site. Peak coordinates of H3K27me3 come from the Broad Institute in the mm8 genome annotation (http://genome‐euro.ucsc.edu/cgi‐bin/hgTrackUi?hgsid=199709818_uvGUfB28yATavzb9eAlxuHR5KGbQ&c=chr12&g=broadChromatinChIPSeq). The conversion of coordinates to mm9 genome annotation was performed with the Lift‐over tool available in the GALAXY Web site (https://usegalaxy.org/). The peak coordinates of H3K4me3 and H3K27ac were obtained from the Ludwig Institute for Cancer Research via the UCSC Web site (http://genome‐euro.ucsc.edu/cgi‐bin/hgTrackUi?hgsid=208559602_SARngKJBy54VXgefrlgnd8rJaWFD&c=chr12&g=wgEncodeLicrHistone) in MEF mouse cell line and then used knowingly for intersection and coverage with the Intersection and Coverage tools installed on the GALAXY Web site. The statistical analysis was performed with the R package.
LINE‐1 content
We obtained the line1 coordinates from the RepeatMasker track via the UCSC Web site. Intersection and coverage were performed with the Intersection and Coverage tools installed on the GALAXY Web site.
Structural variant analysis
Single cells from Rev3L −/− and Rev3L +/+ fibroblasts were isolated, amplified, and subjected to whole genome sequencing. The genomes were sequenced according to the manufacturer protocols (BGI Tech solutions, Hong Kong, Co., Ltd) with a mean coverage of 30× using 150 bp paired‐end reads with BGISEQ‐500 sequencer. Reads were then aligned to the mm9 mouse genome using BWA mem software (Li & Durbin, 2009) and sorted with SAMtools (Li et al, 2009). Then, we removed duplicates from the BAM file according to the GATK Best Practices pipeline (Van der Auwera et al, 2013). To identify structural variants (SV) from genomic data, we used GRIDSS2 (Cameron et al, 2017) and Manta (Chen et al, 2016) software and compared replicates 1 and 2 between each other for Rev3l +/+ and Rev3l −/− samples. Resulting structural variants from two methods were intersected using BEDTools for each sample (Quinlan, 2014) with overlap at least 90% and used for further analysis.
Gene expression profiling data analysis
RNA was extracted using Qiagen column (RNeasy kit) according to the manufacturer’s protocols. The quantity and purity of the total RNA were evaluated using a NanoDrop spectrophotometer. Quality of RNA samples was assessed using Lab‐on‐a‐chip Bioanalyser 2000 technology (Agilent Technologies), based on the 28S/18S ribosomal RNAs ratio. All samples included in this study displayed a ratio of ribosomal RNAs between 1.5 and 2.
Labeling of RNA samples was done according to Agilent oligo Cy5 or Cy3 probes labeling protocol using the Agilent Low Input QuickAmp labeling kit (ref 5190‐2306) adapted for small amount of total RNA (100 ng total RNA per reaction). Hybridization was performed using the Agilent Hybridization Protocol (Gene expression Hyb kit Large ‐ ref 5188‐5280). Scanning was performed with an Agilent G2505C DNA Microarray scanner using default parameters: 20 bits mode, 3 µm resolution, at 20 C in low ozone concentration environment. Microarray images were analyzed by using Feature extraction software version 10.7.3.1 from Agilent technologies and Agilent normalization protocol GE2_107_Sep09 with 028005_D_F_20130207 as design. Default settings are used. A quantile array normalization was performed on raw MedianSignal values with in‐house scripts using Bioconductor LIMMA package. Controls and flags spots were filtered. Missing values were completed with KNN imputation method. Then, the median of all probes for a given transcript was computed to summarize the data. Normalized data (intensities in log2) were imported in BrB Arrays Tools (https://brb.nci.nih.gov/BRB‐ArrayTools/). Class Comparison was performed as weighted t‐test and an FDR adjustment (< 10−5) for multiple testing between Rev3l wt and KO samples (Class Comparison in BrB Arrays Tools 4.6.0) [https://brb.nci.nih.gov/BRB‐ArrayTools]. Gene expressions altered by at least threefold between conditions were considered.
Antibodies
| Antibody | Source | Identifier |
|---|---|---|
| Mouse Monoclonal anti‐Flag M2 | Sigma‐Aldrich | F1804 |
| Mouse Monoclonal anti‐flag M2 affinity gel | Sigma‐Aldrich | A2220 |
| Rabbit Polyclonal anti‐Flag | Sigma‐Aldrich | F7425 |
| Rabbit Polyclonal anti‐Histone H3K4me3 | Abcam | ab8580 |
| Goat anti‐Histone H3 | Abcam | ab12079 |
| Rabbit polyclonal anti‐Histone H4 | Active Motif | 39269 |
| Rabbit Polyclonal anti‐ATRX | Santa Cruz | sc‐15408 |
| Mouse Monoclonal anti‐BrdU (for FACS) BU20a | Dako | MO744 |
| Purified Mouse Anti‐BrdU (for IF) clone B44 | BD Biosciences | 347580 |
| Rabbit Polyclonal anti‐Histone H3K9me3 | Abcam | ab8898 |
| Rabbit Polyclonal anti‐acetyl Histone H3 (Lys 9/14) | Cell Signaling technology | 9677 |
| Mouse Monoclonal anti‐Histone H3K27me3 | Abcam | ab6002 |
| Mouse Monoclonal anti‐Histone H4K20me3 | Active Motif | 39672 |
| Rabbit polyclonal anti‐Histone H4 (acetyl K12) | Abcam | ab46983 |
| Rabbit monoclonal anti‐Histone H4 (acetyl K16) | Abcam | ab109463 |
| Rabbit monoclonal anti‐Histone H4 (acetyl K5) | Abcam | ab51997 |
| Mouse monoclonal anti‐HP1α | Millipore | 05‐689 |
| Mouse monoclonal anti‐HP1β | Abcam | ab10478 |
| Mouse Monoclonal anti‐phospho Histone H2A.X (ser139) | Millipore | 05‐636 |
| Rabbit Polyclonal anti‐53BP1 | Abcam | ab21083 |
| Rabbit monoclonal anti‐HA Tag | Cell Signaling Technologies | 3724 |
| Mouse monoclonal anti‐His | Cell Signaling Technologies | 2366 |
| Mouse monoclonal anti‐V5 | Invitrogen | R962‐25 |
| Mouse monoclonal anti‐HP1γ | Millipore Sigma | MAB3450 |
| Anti‐GST‐HRP conjugate | Millipore Sigma | RPN1236 |
| Rabbit monoclonal anti‐Mad2L2/REV7 | Abcam | ab180579 |
| Mouse monoclonal anti‐Chk1 (G‐4) | Santa Cruz | sc‐8408 |
| Rabbit anti‐pChk1 S345 (133D3) | Cell Signaling | 2348S |
| GFP‐Trap | Chemotek | gta‐20 |
| Rabbit anti‐RPA2 | Bethyl | A300‐244A |
| Rabbit anti‐pRPA2 S4/S8 | Bethyl | A300‐245A |
| Rabbit anti‐KAP1 | Abcam | ab10484 |
| Rabbit anti‐pKAP1 (Ser824) | Bethyl | A300‐767A |
| Rabbit anti‐SCAI antibody EPR4128 | Abcam | 124688 |
| Rabbit anti‐Rad51 antibody EPR4030 | Abcam | ab133534 |
| Alexa Fluor 555 streptavidin | Thermo Fisher Scientific | S32355 |
| rabbit anti‐streptavidin biotin conjugated | Rockland | 200‐406‐095 |
| Alexa Fluor 488 streptavidin | Thermo Fisher Scientific | S32354 |
| rat anti‐BrdU | Abcam | ab6326 |
| goat anti‐mouse Cy5.5 | Abcam | ab6947 |
| anti‐rat Alexa Fluor 555 | Thermo Fisher Scientific | A21434 |
| anti‐ssDNA | DSHB | AB10805144 |
| anti‐goat Cy5.5 | Abcam | ab6951 |
Primers
| Gene | Forward | Reverse |
|---|---|---|
| Primers for ChIP‐qPCR | ||
| Hoxb2 | CCGAGTGAGTCCGTTTGGTT | GACTCCAGCCATGAGTAGCC |
| Hoxb8 | GCCGGATGCAAAATACCGAC | GAGAAGATGTGGGGTGGGTG |
| Hoxb9 | CCCTTTCACAGGTGAGTCCC | GGTGTCCACAGGAAGAGCAA |
| WT1 | GAGGGAGGGAGATGAGAGGC | CCTGCATCTCAGGGCACTTT |
| FoxG1 | TGAGGACAGGCCAGGAAAAC | GTCAAGGCTTCCATGTGTGC |
| Gata6 | CGACTTGGGAGGACCTGTTG | TGGTACATTTCCTCCGGCTG |
| Primers for screening KI mouse line | ||
| Rev3l | ACGAGTTCGCGGGTCCTTAGAGGTC | ACTTTCTACAGCCACAGCATCTC |
| Primers for major satellite probe (FISH) | ||
| Maj sat | ATATGTTGAGAAAACTGAAAATCACG | CCTTCAGTGTGCATTTCTCATTTTTCAC |
List of TaqMan Gene Expression Assays used in this study.
(TaqMan Gene Expression Assays consist of forward and reverse primers with TaqMan minor groove binder probe for each gene).
| Gene | Catalog # |
|---|---|
| Hoxb2 | Mm04209931‐m1 |
| Hoxb8 | Mm00439368‐m1 |
| Hoxb9 | Mm01700220‐m1 |
| WT1 | Mm01337048‐m1 |
| Dlk1 | Mm00494477‐m1 |
| Slcl8a4 | Mm0045056‐m1 |
| Dcn | Mm0051435‐m1 |
| H19 | Mm011567221‐g1 |
| GAPDH | Mm999999915‐g1 |
Author contributions
BBY performed experiments for protein interaction (co‐IP and PLA) and ChIP‐qPCR; SA‐S performed GFP‐fused protein localization experiments and FACS analyses. QD performed FISH on metaphase spread, and ED carried out experiments with DNA combing. JG and RC performed immunofluorescence analysis on UV‐irradiated cells. JCC, ND, GB, and DG designed experiments for replication timing, and JCC performed genome‐wide analysis. PD performed transcriptome analysis. AY, SN, and PLK designed experiments for whole genome sequencing and AY and SN performed mutational analysis. JT and SB carried out GST pull‐down and Co‐IP experiments involving HP1 protein. SSL designed the endogenous tagged Rev3l mouse and derived all MEF cell lines used in this research. MTM‐G helped in the design of cellular studies, and CP brought technical support in many experiments. PLK and RDW wrote the manuscript and all authors reviewed it. PLK, RDW, JCC, and JT conceived and planned the study.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Source Data for Figure 3B
Source Data for Figure 4AE
Source Data for Figure 5A
Source Data for Figure 8AB
Acknowledgements
We thank T. Jenuwein (Max Planck Institute, Germany) for providing Suv39h dn cells. We are also grateful to Niels de Wind (Leiden University, Netherland) and Julien Duxin (Copenhagen University, Denmark) for sharing unpublished results on HP1 and SCAI, respectively. P.L.K. and E.D. express their gratitude to Michelle Debatisse and Stephane Koundrioukoff for their help and valuable advice on the application of the DNA combing‐FISH technique to study replication dynamics in pericentromeric heterochromatin. We thank Nikola Djordjevic, Charlène Garandeau, Loélia Babin, and Kei‐ichi Takata for experimental help and all members of the Kannouche lab for helpful discussions, especially Said Aoufouchi. This work was performed thanks to Gustave Roussy core facilities (Cell imaging, Genomics and Bio‐informatics). The P.L.K lab was supported by La Ligue Nationale contre le Cancer (Equipe labellisée). This work was supported by grants from Institut National du Cancer (INCa) PLBIO16‐011 (P.L.K. and J‐C.C), INCa‐DGOS‐Inserm 12551, ARC‐ARCPGA12019120001055_1578, and ANR‐14‐CE10‐0008‐01 (P.L.K. and J‐C.C). S.A.S received support from the Ministère de l'Enseignement Supérieur et de la Recherche and the Foundation ARC and B.B received support from the Ministère de l'Enseignement Supérieur et de la Recherche and the Foundation pour la Recherche Médicale. The J‐C.C lab is supported by the IdEX Université de Paris ANR‐18‐IDEX‐0001. R.D.W. and J.T were funded by National Institutes of Health grant CA193124, Department of Defense grant W81XWH‐17‐10239, and the J. Ralph Meadows Chair in Carcinogenesis Research.
The EMBO Journal (2021) 40: e104543.
Data availability
Microarray data: EMBL‐EBI E‐MTAB‐8338 (https://www.ebi.ac.uk/arrayexpress/experiments/E‐MTAB‐8338/).
Replication‐timing data: GSE178927 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE178927).
References
- Aladjem MI (2007) Replication in context: dynamic regulation of DNA replication patterns in metazoans. Nat Rev Genet 8: 588–600 [DOI] [PubMed] [Google Scholar]
- Allshire RC, Karpen GH (2008) Epigenetic regulation of centromeric chromatin: old dogs, new tricks? Nat Rev Genet 9: 923–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranovskiy AG, Lada AG, Siebler HM, Zhang Y, Pavlov YI, Tahirov TH (2012) DNA polymerase delta and zeta switch by sharing accessory subunits of DNA polymerase delta. J Biol Chem 287: 17281–17287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergoglio V, Boyer AS, Walsh E, Naim V, Legube G, Lee MY, Rey L, Rosselli F, Cazaux C, Eckert KA et al (2013) DNA synthesis by Pol eta promotes fragile site stability by preventing under‐replicated DNA in mitosis. J Cell Biol 201: 395–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat A, Andersen PL, Qin Z, Xiao W (2013) Rev3, the catalytic subunit of Polzeta, is required for maintaining fragile site stability in human cells. Nucleic Acids Res 41: 2328–2339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgers PMJ, Kunkel TA (2017) Eukaryotic DNA replication fork. Annu Rev Biochem 86: 417–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron DL, Schroder J, Penington JS, Do H, Molania R, Dobrovic A, Speed TP, Papenfuss AT (2017) GRIDSS: sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly. Genome Res 27: 2050–2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Schulz‐Trieglaff O, Shaw R, Barnes B, Schlesinger F, Kallberg M, Cox AJ, Kruglyak S, Saunders CT (2016) Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32: 1220–1222 [DOI] [PubMed] [Google Scholar]
- Cornacchia D, Dileep V, Quivy JP, Foti R, Tili F, Santarella‐Mellwig R, Antony C, Almouzni G, Gilbert DM, Buonomo SB (2012) Mouse Rif1 is a key regulator of the replication‐timing programme in mammalian cells. EMBO J 31: 3678–3690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despras E, Sittewelle M, Pouvelle C, Delrieu N, Cordonnier AM, Kannouche PL (2016) Rad18‐dependent SUMOylation of human specialized DNA polymerase eta is required to prevent under‐replicated DNA. Nat Commun 7: 13326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desprat R, Thierry‐Mieg D, Lailler N, Lajugie J, Schildkraut C, Thierry‐Mieg J, Bouhassira EE (2009) Predictable dynamic program of timing of DNA replication in human cells. Genome Res 19: 2288–2299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova DS, Berezney R (2002) The spatio‐temporal organization of DNA replication sites is identical in primary, immortalized and transformed mammalian cells. J Cell Sci 115: 4037–4051 [DOI] [PubMed] [Google Scholar]
- Donley N, Thayer MJ (2013) DNA replication timing, genome stability and cancer: late and/or delayed DNA replication timing is associated with increased genomic instability. Semin Cancer Biol 23: 80–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G, Godindagger I, Klein U, Yaspo ML, Cumano A, Rajewsky K (2000) Disruption of the Rev3l‐encoded catalytic subunit of polymerase zeta in mice results in early embryonic lethality. Curr Biol 10: 1221–1224 [DOI] [PubMed] [Google Scholar]
- Eustermann S, Yang JC, Law MJ, Amos R, Chapman LM, Jelinska C, Garrick D, Clynes D, Gibbons RJ, Rhodes D et al (2011) Combinatorial readout of histone H3 modifications specifies localization of ATRX to heterochromatin. Nat Struct Mol Biol 18: 777–782 [DOI] [PubMed] [Google Scholar]
- Farkash‐Amar S, Lipson D, Polten A, Goren A, Helmstetter C, Yakhini Z, Simon I (2008) Global organization of replication time zones of the mouse genome. Genome Res 18: 1562–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Vidal A, Guitton‐Sert L, Cadoret JC, Drac M, Schwob E, Baldacci G, Cazaux C, Hoffmann JS (2014) A role for DNA polymerase theta in the timing of DNA replication. Nat Commun 5: 4285 [DOI] [PubMed] [Google Scholar]
- Fundele RH , Fundele RH, Surani MA (1994) Experimental embryological analysis of genetic imprinting in mouse development. Dev Genet 15: 515–522 [DOI] [PubMed] [Google Scholar]
- Gan GN, Wittschieben JP, Wittschieben BO, Wood RD (2008) DNA polymerase zeta (pol zeta) in higher eukaryotes. Cell Res 18: 174–183 [DOI] [PubMed] [Google Scholar]
- Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Lobrich M, Jeggo PA (2008) ATM signaling facilitates repair of DNA double‐strand breaks associated with heterochromatin. Mol Cell 31: 167–177 [DOI] [PubMed] [Google Scholar]
- Guenatri M, Bailly D, Maison C, Almouzni G (2004) Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J Cell Biol 166: 493–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilbaud G, Rappailles A, Baker A, Chen CL, Arneodo A, Goldar A, d'Aubenton‐Carafa Y, Thermes C, Audit B, Hyrien O (2011) Evidence for sequential and increasing activation of replication origins along replication timing gradients in the human genome. PLoS Comput Biol 7: e1002322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjadj D, Denecker T, Guerin E, Kim SJ, Fauchereau F, Baldacci G, Maric C, Cadoret JC (2020) Efficient, quick and easy‐to‐use DNA replication timing analysis with START‐R suite. NAR Genom Bioinform 2: lqaa045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen RK, Mund A, Poulsen SL, Sandoval M, Klement K, Tsouroula K, Tollenaere MA, Raschle M, Soria R, Offermanns S et al (2016) SCAI promotes DNA double‐strand break repair in distinct chromosomal contexts. Nat Cell Biol 18: 1357–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayano M, Kanoh Y, Matsumoto S, Renard‐Guillet C, Shirahige K, Masai H (2012) Rif1 is a global regulator of timing of replication origin firing in fission yeast. Genes Dev 26: 137–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiragami‐Hamada K, Soeroes S, Nikolov M, Wilkins B, Kreuz S, Chen C, De La Rosa‐Velazquez IA, Zenn HM, Kost N, Pohl W et al (2016) Dynamic and flexible H3K9me3 bridging via HP1beta dimerization establishes a plastic state of condensed chromatin. Nat Commun 7: 11310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratani I, Ryba T, Itoh M, Yokochi T, Schwaiger M, Chang CW, Lyou Y, Townes TM, Schubeler D, Gilbert DM (2008) Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol 6: e245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isobe SY, Nagao K, Nozaki N, Kimura H, Obuse C (2017) Inhibition of RIF1 by SCAI allows BRCA1‐mediated repair. Cell Rep 20: 297–307 [DOI] [PubMed] [Google Scholar]
- Johnson RE, Prakash L, Prakash S (2012) Pol31 and Pol32 subunits of yeast DNA polymerase delta are also essential subunits of DNA polymerase zeta. Proc Natl Acad Sci USA 109: 12455–12460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RE, Washington MT, Haracska L, Prakash S, Prakash L (2000) Eukaryotic polymerases iota and zeta act sequentially to bypass DNA lesions. Nature 406: 1015–1019 [DOI] [PubMed] [Google Scholar]
- Kane DP, Shusterman M, Rong Y, McVey M (2012) Competition between replicative and translesion polymerases during homologous recombination repair in Drosophila . PLoS Genet 8: e1002659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A, Polak P, Nemesh J, Michaelson JJ, Sebat J, Sunyaev SR, McCarroll SA (2012) Differential relationship of DNA replication timing to different forms of human mutation and variation. Am J Hum Genet 91: 1033–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GI, Murray AW (2011) Mutation rates across budding yeast chromosome VI are correlated with replication timing. Genome Biol Evol 3: 799–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange SS, Bedford E, Reh S, Wittschieben JP, Carbajal S, Kusewitt DF, DiGiovanni J, Wood RD (2013) Dual role for mammalian DNA polymerase zeta in maintaining genome stability and proliferative responses. Proc Natl Acad Sci USA 110: E687–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange SS, Bhetawal S, Reh S, Powell KL, Kusewitt DF, Wood RD (2018) DNA polymerase zeta deficiency causes impaired wound healing and stress‐induced skin pigmentation. Life Sci Alliance 1: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange SS, Tomida J, Boulware KS, Bhetawal S, Wood RD (2016) The polymerase activity of mammalian DNA Pol zeta is specifically required for cell and embryonic viability. PLoS Genet 12: e1005759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange SS, Wittschieben JP, Wood RD (2012) DNA polymerase zeta is required for proliferation of normal mammalian cells. Nucleic Acids Res 40: 4473–4482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Gregory MT, Yang W (2014) Human Pol zeta purified with accessory subunits is active in translesion DNA synthesis and complements Pol eta in cisplatin bypass. Proc Natl Acad Sci USA 111: 2954–2959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaitre C, Soutoglou E (2014) Double strand break (DSB) repair in heterochromatin and heterochromatin proteins in DSB repair. DNA Repair 19: 163–168 [DOI] [PubMed] [Google Scholar]
- Letessier A, Millot GA, Koundrioukoff S, Lachages AM, Vogt N, Hansen RS, Malfoy B, Brison O, Debatisse M (2011) Cell‐type‐specific replication initiation programs set fragility of the FRA3B fragile site. Nature 470: 120–123 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAlpine DM, Rodriguez HK, Bell SP (2004) Coordination of replication and transcription along a Drosophila chromosome. Genes Dev 18: 3094–3105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maison C, Bailly D, Peters AH, Quivy JP, Roche D, Taddei A, Lachner M, Jenuwein T, Almouzni G (2002) Higher‐order structure in pericentric heterochromatin involves a distinct pattern of histone modification and an RNA component. Nat Genet 30: 329–334 [DOI] [PubMed] [Google Scholar]
- Makarova AV, Burgers PM (2015) Eukaryotic DNA polymerase zeta. DNA Repair 29: 47–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova AV, Stodola JL, Burgers PM (2012) A four‐subunit DNA polymerase zeta complex containing Pol delta accessory subunits is essential for PCNA‐mediated mutagenesis. Nucleic Acids Res 40: 11618–11626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manandhar M, Lowery MG, Boulware KS, Lin KH, Lu Y, Wood RD (2017) Transcriptional consequences of XPA disruption in human cell lines. DNA Repair 57: 76–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SK, Wood RD (2019) DNA polymerase zeta in DNA replication and repair. Nucleic Acids Res 47: 8348–8361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch SD, Kunkel TA (2008) The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res 18: 148–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez‐Bermudez A, Lototska L, Bauwens S, Giraud‐Panis MJ, Croce O, Jamet K, Irizar A, Mowinckel M, Koundrioukoff S, Nottet N et al (2018) Genome‐wide control of heterochromatin replication by the telomere capping protein TRF2. Mol Cell 70: 449–461.e5 [DOI] [PubMed] [Google Scholar]
- Murzina N, Verreault A, Laue E, Stillman B (1999) Heterochromatin dynamics in mouse cells: interaction between chromatin assembly factor 1 and HP1 proteins. Mol Cell 4: 529–540 [DOI] [PubMed] [Google Scholar]
- Natale F, Scholl A, Rapp A, Yu W, Rausch C, Cardoso MC (2018) DNA replication and repair kinetics of Alu, LINE‐1 and satellite III genomic repetitive elements. Epigenet Chromatin 11: 61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen VH, Lavenier D (2009) PLAST: parallel local alignment search tool for database comparison. BMC Bioinformatics 10: 329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolov I, Taddei A (2016) Linking replication stress with heterochromatin formation. Chromosoma 125: 523–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norio P, Kosiyatrakul S, Yang Q, Guan Z, Brown NM, Thomas S, Riblet R, Schildkraut CL (2005) Progressive activation of DNA replication initiation in large domains of the immunoglobulin heavy chain locus during B cell development. Mol Cell 20: 575–587 [DOI] [PubMed] [Google Scholar]
- Nozawa RS, Nagao K, Masuda HT, Iwasaki O, Hirota T, Nozaki N, Kimura H, Obuse C (2010) Human POGZ modulates dissociation of HP1alpha from mitotic chromosome arms through Aurora B activation. Nat Cell Biol 12: 719–727 [DOI] [PubMed] [Google Scholar]
- Ozeri‐Galai E, Tur‐Sinai M, Bester AC, Kerem B (2014) Interplay between genetic and epigenetic factors governs common fragile site instability in cancer. Cell Mol Life Sci 71: 4495–4506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters AH, O'Carroll D, Scherthan H, Mechtler K, Sauer S, Schofer C, Weipoltshammer K, Pagani M, Lachner M, Kohlmaier A et al (2001) Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107: 323–337 [DOI] [PubMed] [Google Scholar]
- Pirouz M, Pilarski S, Kessel M (2013) A critical function of Mad2l2 in primordial germ cell development of mice. PLoS Genet 9: e1003712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak P, Karlic R, Koren A, Thurman R, Sandstrom R, Lawrence M, Reynolds A, Rynes E, Vlahovicek K, Stamatoyannopoulos JA et al (2015) Cell‐of‐origin chromatin organization shapes the mutational landscape of cancer. Nature 518: 360–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR (2014) BEDTools: the swiss‐army tool for genome feature analysis. Curr Protoc Bioinformatics 47: 11–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quivy J‐P, Roche D, Kirschner D, Tagami H, Nakatani Y, Almouzni G (2004) A CAF‐1 dependent pool of HP1 during heterochromatin duplication. EMBO J 23: 3516–3526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs M, Onodera C, Blaschke K, Ebata KT, Song JS, Ramalho‐Santos M (2013) Bivalent chromatin marks developmental regulatory genes in the mouse embryonic germline in vivo. Cell Rep 3: 1777–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkies P, Reams C, Simpson LJ, Sale JE (2010) Epigenetic instability due to defective replication of structured DNA. Mol Cell 40: 703–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz SS, Desbordes SC, Du Z, Kosiyatrakul S, Lipchina I, Studer L, Schildkraut CL (2010) Single‐molecule analysis reveals changes in the DNA replication program for the POU5F1 locus upon human embryonic stem cell differentiation. Mol Cell Biol 30: 4521–4534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster‐Bockler B, Lehner B (2012) Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature 488: 504–507 [DOI] [PubMed] [Google Scholar]
- Schwienbacher C, Angioni A, Scelfo R, Veronese A, Calin GA, Massazza G, Hatada I, Barbanti‐Brodano G, Negrini M (2000) Abnormal RNA expression of 11p15 imprinted genes and kidney developmental genes in Wilms' tumor. Cancer Res 60: 1521–1525 [PubMed] [Google Scholar]
- Seplyarskiy VB, Bazykin GA, Soldatov RA (2015) Polymerase zeta activity is linked to replication timing in humans: evidence from mutational signatures. Mol Biol Evol 32: 3158–3172 [DOI] [PubMed] [Google Scholar]
- Sharma S, Hicks JK, Chute CL, Brennan JR, Ahn JY, Glover TW, Canman CE (2012) REV1 and polymerase zeta facilitate homologous recombination repair. Nucleic Acids Res 40: 682–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sima J, Chakraborty A, Dileep V, Michalski M, Klein KN, Holcomb NP, Turner JL, Paulsen MT, Rivera‐Mulia JC, Trevilla‐Garcia C et al (2019) Identifying cis elements for spatiotemporal control of mammalian DNA replication. Cell 176: 816–830.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda E, Okada T, Zhao GY, Tateishi S, Araki K, Yamaizumi M, Yagi T, Verkaik NS, van Gent DC, Takata M et al (2003) Multiple roles of Rev3, the catalytic subunit of polzeta in maintaining genome stability in vertebrates. EMBO J 22: 3188–3197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatoyannopoulos JA, Adzhubei I, Thurman RE, Kryukov GV, Mirkin SM, Sunyaev SR (2009) Human mutation rate associated with DNA replication timing. Nat Genet 41: 393–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JE, Kissling GE, Lujan SA, Rogozin IB, Stith CM, Burgers PM, Kunkel TA (2009) Low‐fidelity DNA synthesis by the L979F mutator derivative of Saccharomyces cerevisiae DNA polymerase zeta. Nucleic Acids Res 37: 3774–3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei A, Maison C, Roche D, Almouzni G (2001) Reversible disruption of pericentric heterochromatin and centromere function by inhibiting deacetylases. Nat Cell Biol 3: 114–120 [DOI] [PubMed] [Google Scholar]
- Thiru A, Nietlispach D, Mott HR, Okuwaki M, Lyon D, Nielsen PR, Hirshberg M, Verreault A, Murzina NV, Laue ED (2004) Structural basis of HP1/PXVXL motif peptide interactions and HP1 localisation to heterochromatin. EMBO J 23: 489–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomida J, Takata K, Lange SS, Schibler AC, Yousefzadeh MJ, Bhetawal S, Dent SY, Wood RD (2015) REV7 is essential for DNA damage tolerance via two REV3L binding sites in mammalian DNA polymerase zeta. Nucleic Acids Res 43: 1000–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomida J, Takata KI, Bhetawal S, Person MD, Chao HP, Tang DG, Wood RD (2018) FAM35A associates with REV7 and modulates DNA damage responses of normal and BRCA1‐defective cells. EMBO J 37: e99543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao WC, Eckert KA (2018) Detours to replication: functions of specialized DNA polymerases during oncogene‐induced replication stress. Int J Mol Sci 19: 1–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ui A, Nagaura Y, Yasui A (2015) Transcriptional elongation factor ENL phosphorylated by ATM recruits polycomb and switches off transcription for DSB repair. Mol Cell 58: 468–482 [DOI] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy‐Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J et al (2013) From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43: 11 10 11–11 10 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Sloun PP, Varlet I, Sonneveld E, Boei JJ, Romeijn RJ, Eeken JC, De Wind N (2002) Involvement of mouse Rev3 in tolerance of endogenous and exogenous DNA damage. Mol Cell Biol 22: 2159–2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers JH, van Lohuizen M, Citterio E (2012) The emerging role of Polycomb repressors in the response to DNA damage. J Cell Sci 125: 3939–3948 [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Maekawa M (2010) Spatiotemporal regulation of DNA replication in the human genome and its association with genomic instability and disease. Curr Med Chem 17: 222–233 [DOI] [PubMed] [Google Scholar]
- Wittschieben JP, Reshmi SC, Gollin SM, Wood RD (2006) Loss of DNA polymerase zeta causes chromosomal instability in mammalian cells. Cancer Res 66: 134–142 [DOI] [PubMed] [Google Scholar]
- Wittschieben J, Shivji MK, Lalani E, Jacobs MA, Marini F, Gearhart PJ, Rosewell I, Stamp G, Wood RD (2000) Disruption of the developmentally regulated Rev3l gene causes embryonic lethality. Curr Biol 10: 1217–1220 [DOI] [PubMed] [Google Scholar]
- Zhang J, Bellani MA, James RC, Pokharel D, Zhang Y, Reynolds JJ, McNee GS, Jackson AP, Stewart GS, Seidman MM (2020) DONSON and FANCM associate with different replisomes distinguished by replication timing and chromatin domain. Nat Commun 11: 3951 [DOI] [PMC free article] [PubMed] [Google Scholar]