

Abstract
Cytokines are immunoregulatory proteins involved in many pathological states with promising potential as therapeutic agents. A diverse array of cytokines have been studied in preclinical disease models since the 1950s, some of which became successful biopharmaceutical products with the advancement of recombinant protein technology in the 1980s. However, following these early approvals, clinical translation of these natural immune signaling molecules has been limited due to their pleiotropic action in many cell types, and the fact that they have evolved to act primarily locally in tissues. These characteristics, combined with poor pharmacokinetics, have hindered the delivery of cytokines via systemic administration routes due to dose-limiting toxicities. However, given their clinical potential and recent clinical successes in cancer immunotherapy, cytokines continue to be extensively pursued in preclinical and clinical studies, and a range of molecular and formulation engineering strategies are being applied to reduce treatment toxicity while maintaining or enhancing therapeutic efficacy. This review provides a brief background on the characteristics of cytokines and their history as clinical therapeutics, followed by a deeper discussion on the engineering strategies developed for cytokine therapies with a focus on the translational relevance of these approaches.
Keywords: cytokines, clinical trials, immunotherapies, drug delivery, therapeutics
Graphical Abstract
1. Introduction
The term cytokine originates from the combination of the Greek words “cyto” and “kine” which translates to “cellular movement”. Coined by Stanley Cohen in 1974, it was used to describe cellular substances that induced immune cell-directed migration (chemotaxis) and activation.[1] Since that time, there have been many new insights into these molecules and cytokines are now defined as regulatory proteins that modulate the immune system and inflammation.[2] As immune regulators, cytokines have a major role as signaling molecules in response to danger, tissue damage, or injury.[3] Importantly, the vital role of the immune system in many pathologies make cytokines promising therapeutics for many disease states.
However, clinical use of cytokines has been restricted. This clinical translational challenge comes from two major characteristics of cytokines: 1) they are highly pleiotropic and 2) in normal physiology, they are generally produced and act very locally in tissues. Accordingly, systemic administration can lead to severe side effects. Given this challenge, cytokines present a promising opportunity for molecular and formulation engineering to improve their safety and therapeutic efficacy. Many of the drug delivery platforms presented here have been reviewed for general immunotherapy applications—focusing on the delivery of checkpoint inhibitors, engineered T cells, co-stimulatory receptor agonists, and cancer vaccines.[4–11] Here, we focus on the specific challenges presented by cytokine therapeutics, discuss the basic biology and clinical applications of key immunomodulatory cytokines, and review engineering strategies developed to increase their utility in therapeutic applications. To limit the scope of this review we focus on recombinant cytokine delivery; strategies involving cytokine-secreting cells (cellular vehicles) and gene therapy are only briefly introduced and cytokine inhibitor therapies are not covered here (we refer the interested reader to a number of recent reviews on these approaches[12–19]).
2. Cytokine Classification and Characteristics
Cytokines are cell-signaling proteins that perform their biological function via extracellular cell-membrane receptors.[2,20] This biological function may act on the cell that produces them (autocrine signaling) or on a different cell (paracrine signaling).[20] Accordingly, cytokines have similar characteristics to other soluble factors such as hormones, but some key differentiating factors of cytokines include local production and expression in response to specific stimuli.[2,20] Importantly, cytokines act primarily on the immune system while hormones primarily modulate the endocrine system.[21] Lastly, unlike hormones, baseline levels of cytokines in the circulation are typically low at steady state.[2] However, exceptions exist as some cytokines can act at distant sites (endocrine signaling) and some hormone-like substances—such as growth hormone, erythropoietin, thrombopoietin, and leptin can be categorized as cytokines.[20]
Albeit imperfect, there are various classifications of cytokines which aim to functionally distinguish these pleiotropic proteins. Some of the early functional classifications led to the naming of interleukins, which were thought to originate from and act on leukocytes[20]. Other functional groupings include colony-stimulating factors (GM-CSF, G-CSF) and interferons (IFN-α, IFN-β, IFN-γ).[22,23] However, these early historical classifications have become outdated as these factors are now known to be produced by many cell populations and have pleiotropic effects on various cell types[20,21]. For example, some cytokines (e.g., TNF-α, IL-1β, TGF-β, IL-6) are produced by or act on non-immune cells (e.g., fibroblasts, epithelial cells, and cancer cells). [24–26]
Cytokines may also be functionally classified as either pro-inflammatory or anti-inflammatory. Pro-inflammatory cytokines (e.g., IL-1α/β, TNF-α/β, IL-6, IL-11, IL-18, IFN-γ) up-regulate inflammatory reactions (i.e. tissue’s catabolic reactions against pathogens including immune cell recruitment, infiltration, and stimulation), while anti-inflammatory cytokines (e.g., IL-10, IL-6, TGF-β, IL-27, IL-35) down-regulate inflammatory responses and promote tissue healing.[21,27] However, the cytokine-induced inflammatory response is highly context-dependent with the same cytokine inducing either pro- or anti-inflammatory reactions depending on factors such as the target cell, concentration, and presence of other cytokines.[28] A common example for the varying inflammatory response of cytokines is IL-6 which, in addition to its major role in initializing inflammatory responses (along with TNF-α and IL-1), is a potent stimulant of acute-phase proteins in hepatocytes (an anti-inflammatory effect) and can inhibit TNF-α and IL-1 expression.[28].
More recent classifications have originated from the relation of cytokines to T cell responses. T cells can be biased into different functional states characterized by the production of certain groups of cytokines, e.g., Th1 cytokines (type 1 cytokines) such as IL-2, IL-12, and IFN-γ; Th2 cytokines (type 2 cytokines) such as IL-4, IL-5, IL-6, IL-10 and IL-13; or regulatory cytokines such as IL-10 and TGF-β[29]. Although other Th subsets exist, these three major classes provide a basic framework to understand diseases and potential therapeutic opportunities within them. In general, type 1 cytokines mediate the development of a strong cellular immune response while type 2 cytokines favor a strong humoral immune response.[30] Conversely, regulatory cytokines promote immune homeostasis, prevent autoimmunity, and moderate inflammation.[31] Importantly, cytokine groups exhibit cross-regulatory properties in which they not only favor a functional state but also suppresses the alternative states. Accordingly, high levels of one class of cytokines is indicative of a type of immune environment which could be reprogramed with cytokine therapies from other subsets. For example, in cancer, tumors are typically associated with tolerogenic and immunosuppressive microenvironments in which cytokine-mediated therapies primarily have aimed to deliver type 1 cytokines to stimulate an anti-cancer cellular immune response. On the other hand, some vaccine-based therapies may prefer type 2 cytokines as adjuvants base on their role in B cell maturation, while autoimmune conditions could benefit from regulatory cytokines. In many instances, however, these distinctions are insufficient to classify cytokines since their effects are highly context dependent. For example, IL-18 in isolation can promote Th2-biased cytokine production from T cells, but in the presence of IL-15 or IL-12, IL-18 leads to potent Th1-biased cytokine production.[29] Moreover, type 1 cytokines are not restricted to cellular immune responses as they aid in the development of certain antibody classes and functional differentiation of B cells. [32,33]
Lastly, more objective classifications exist such as those based on structural or receptor homology. This grouping includes type I cytokines (consisted of four α-helical bundle structures with an ‘up-up-down-down’ configuration) signaling through class I cytokine receptors (IL-6, IL-2, IL-4, IL-12, GM-CSF), type II cytokines signaling through class II cytokine receptors (IFNs, IL-10 family, IL-19 family), cytokines signaling through immunoglobulin superfamily cytokine receptors (MCSF, IL-1, IL-16) and the TNF family signaling through TNF receptors family (TNF-α/β).[20,21] However, the wide-range of functions of cytokines within the same group make this classification less practical; therefore, it is less commonly used.
In addition to immunomodulatory substances, cytokines are sometimes defined to include growth factors (PDGF, EGF, FGF, NGF, IGFs, TGF (α/β), BMPs, and CNTF) and chemokines (IL-8, MIP, MRO, IP-10)[22]. Growth factors are primarily molecules regulating embryogenesis, tissue repair, and wound healing, while chemokines are primarily molecules directing cell migration[21]. Yet, these substances can also modulate immune cells and immune responses, leading to an overlap in classifications. For example, the growth factor TGF-β is commonly classified as an anti-inflammatory cytokine or T regulatory cytokine. IL-8, in addition to serving as a chemokine, has inflammatory effects on immune cells.[34]
With more than 300 known cytokines, chemokines, and growth factors, here we restrict our discussions to immunomodulatory cytokines with a focus on cytokines tested in clinical studies.[23] A diagram depicting the major immune cell targets of cytokines that have or are being evaluated in clinical trials is shown in Figure 1. Other cytokines, their targets, and functions can be found in the literature[23,29].
Figure 1.

Major targets of cytokines used in clinical trials directed at immune cells involved in adaptive and innate immunity. Blue arrows indicate recruitment and differentiation. Red arrows indicate activation and expansion. Gray arrows indicate inhibition. IL-22 has been excluded here as its clinical trials have targeted its growth-factor properties and not its immunostimulant properties. TNF-related apoptosis-inducing ligand (TRAIL) is also excluded as its non-apoptotic role in immune-cells is not clearly understood.[35] The effects of IFN-λ are primarily on epithelial cells.[36]
Although not a comprehensive description of the effect of cytokines on immune cells, Figure 1 clearly illustrates their pleiotropic nature. Other features of cytokines contributing to their complex activity include context dependency, cascading, antagonism, and feedback control. As previously mentioned, context-dependency implies that the settings of cytokine stimulus can regulate its responses such as the presence of cytokine combinations. Moreover, the cytokine concentration can alter their effects as shown by IL-2 where low doses preferentially stimulate regulatory T cells, while high doses activate CD8+ T cells and NK cells.[37] The class of target cells, its environment, and time of cytokine activation are also contextual factors that can lead to altered responses of the same cytokine.[28] In addition to context-dependency, cytokines can have multiple effects based on the cascade of downstream cytokines produced in response to their stimulus. One such example is IFN-γ-mediated stimulation of activated macrophages leading to the release of IL-12 and TNF-α, both of which induce their own cascades.[32] As will be discussed in the Clinical Perspective section, this cytokine cascade effect is a crucial characteristic that hinders clinical translation as it expands the potential side effects of cytokine treatment. Conversely, antagonism refers to the property that a cytokine can restrict the effect or production of another such as IL-10-mediated inhibition proinflammatory cytokine production by macrophages or dendritic cells.[38] Finally, cytokine stimulation can be further enhanced (or downregulated) through feedback control mechanisms which can occur directly (e.g., macrophage auto-stimulation through TNF-α production) or indirectly (e.g., IFN-γ-mediated macrophage production of IL-12 leading to IFN-γ production via Th1 differentiated CD4+ T cells).
3. History of Cytokines as Therapeutics
The first steps of cytokine discovery were taken in the 1920s, when it was shown that the mechanism underlying ‘bacterial allergies’ differed from protein anaphylaxis and that the supernatants of tissue sensitized to tuberculin amplified the reaction to old tuberculin[39,40]. Approximately thirty years later, the fundamental class of proteins leading to these phenomena started to be unraveled when the first individual cytokines were discovered as the ‘endogenous pyrogen’ (later classified as IL-1) and ‘interferon’ (later termed IFN-α) were isolated[39,41]. The potential clinical use of these substances was clear as ‘interferon’ interfered with viral infection. Furthermore, the ‘endogenous pyrogen’ was directly related to pathology as its effect was to induce fever in animals (pyrogen comes from the Greek word pyro meaning heat and gen meaning generating).[41]
By the 1970s and ‘80s, interest in therapeutic applications in cytokines increased as more cytokines were identified and the generation of recombinant proteins became possible.[29] During this time, interferon was found to have many other effects beyond preventing viral infection such as enhancement of cell function, immune system modulation, and inhibition of cell division with antitumor activity in vivo.[42] Based on these and other findings, the primary applications of cytokines began as immunostimulatory molecules (such as IL-1, IL-2 TNF-α) for immunodeficiencies (mainly AIDS), infections, and cancer treatment.[29] Since the initial burst of research, although many other cytokines and their mechanism have been identified and explored for therapeutic applications, only a select few have received regulatory approval as therapeutics. The cytokines currently approved as recombinant protein therapeutics, their indications, and administration routes are summarized in Table 1.
Table 1.
Clinically approved recombinant cytokine therapies for in vivo immunomodulation.
| Cytokine | Approval Indication (Year) | Other Indications | Adm. Route* | Ref |
|---|---|---|---|---|
|
| ||||
| IFN-α IFN-α2a; IFN-α2b; peginterferon α2a; peginterferon α2b IFN-αn1; IFN-αn3 |
Hairy cell leukemia (1986) | Karposi’s sarcoma, chronic myelogenous leukemia, metastatic malignant melanoma, follicular lymphoma hepatitis B and C, condyloma acuminate, labial and genital herpes, rhinoviruses | s.c. >TIM (Q1W PEG) |
[43,44] |
| IFN-β IFN-β1a; IFN-β1b; peginterferon β1a; IFN-β (Soluferon) |
Relapsing–remitting multiple sclerosis (1993) | - | s.c./i.m. QOD/Q1W (Q2W PEG) | [44,45] |
| IFN-γ IFN-γ1b |
Chronic granulomatous disease (1990) | Malignant osteopetrosis | s.c. TIW |
[41,46] |
| TNF-α tasonermin |
Sarcoma (Europe, 1998) – application via isolated limb perfusion | Non-melanoma skin cancer– application via isolated limb perfusion | i.v. | [47,48] |
| IL-2 adesleukin |
Metastatic renal cell carcinoma (1992) | Metastatic melanoma | i.v. Q8H |
[41,49] |
| IL-11 oprelvekin |
Chemotherapy-induced thrombocytopenia (1997) | - | s.c. QD |
[50] |
| G-CSF filgrastim; lenograstim; pegfilgrastim |
Prophylaxis of febrile neutropenia in patients receiving myelosuppressive chemotherapy (1991) | Accelerating neutrophil recovery after bone marrow transplantation, mobilizing peripheral-blood progenitor cells, and shortening the duration of neutropenia in patients receiving induction chemotherapy for acute myelogenous leukemia / reduce the incidence and sequelae of neutropenia in symptomatic patients with congenital, cyclic, or idiopathic neutropenia | i.v./s.c. QD (PEG s.c. >Q1W) |
[51] |
| GM-CSF sargramostim; molgramostim |
Accelerate myeloid recovery after autologous bone marrow transplantation and delayed or failed engraftment after allogeneic or autologous bone marrow transplantation (1991) | Accelerating neutrophil recovery after bone marrow transplantation, mobilizing peripheral-blood progenitor cells, and shortening the duration of neutropenia in patients receiving induction chemotherapy for acute myelogenous leukemia | i.v./s.c. QD |
[51] |
| EPO epoetin alfa |
Anemia associated with chronic renal failure (1989) | Anemia from Zidovudine used in HIV-infection, from myelosuppressive chemotherapy / reduction of allogenic red-blood cell transfusion in patients undergoing elective, noncardiac, nonvascular surgery | s.c./i.v. TIW or Q1W (PEG Q2W) |
[52,53] |
Primary administration (Adm.) routes, some indications may differ. PEG – polyethylene glycol; Parenthesis indicates any changes on the PEG-conjugated form of the cytokine
Abbreviations: s.c. - subcutaneous; i.m. – intramuscular; i.v. – intravenous; QD – every day; Q1W – every week; TIM – three times a week; Q8H – every eight hours; Q2W – every two weeks
As shown in Table 1, the approved indications for cytokine therapies have been cancer, viral infections (IFN-α), and immunodeficiencies (IFN-γ). Moreover, IFN-β is indicated as an anti-inflammatory agent for autoimmunity. The successful development of these treatments belies challenges even for these approved drugs. For example, IL-2 approval came even when >90% of patients had doses withheld during treatment due to toxicity.[54] Further, even though high-dose IL-2 therapy is still the first-line therapy for metastatic renal cell carcinoma, it is only applicable in selected patients (good organ function and performance status) and requires proper monitoring and management of the side-effects to reduce risks associated with the treatment.[55] As a second example, the approved TNF-α treatment in Europe for skin cancer is dosed via isolated limb perfusion to reduce systemic exposure to the drug.[48]
No regulatory approval for new cytokines has been made since the initial burst in the early 1990s. The chief advance since that time has been the approval of new forms of IFN-α, IFN-β, and G-CSF surface conjugated with polyethylene glycol (PEG) to extend their circulation half-life.[41,56] Despite these modifications, toxicity limitations have continued to restrict the expansion of already approved cytokines to new indications. For example, Type I IFN (α/β) could be clinically used in many other infections, but strategies to minimize systemic toxicity are needed.[44] Many more preclinically promising therapeutic cytokines have not yet been clinically translated for similar reasons.[57] Nonetheless, great interest in expanding the use of cytokines as therapeutics remains, as indicated by the number of ongoing clinical trials (Table 2, compiled from clinicaltrials.gov).
Table 2.
Current cytokine therapy clinical trials (parenthesis indicates number of trials still ongoing) and indications of active clinical trials (excluding approved indications).
| Cytokine | Clinical Trial Phase |
Total | |||
|---|---|---|---|---|---|
| no record | 1 | 2 | 3 | ||
|
| |||||
| EPO | 51 (6) | 36 (3) | 179 (18) | 214 (18) | 480 |
| Indic | Phase 1: healthy subjects, autoimmune hepatitis || Phase 2: bipolar disorder/unipolar depression/cognitive impairment, mantle cell lymphoma, premature infant, asthma, amyotrophic lateral sclerosis, eosinophilia/angioedema || Phase 3: anemia, chronic kidney disease, erythroblastosis fetalis, hypoxic-ischemic encephalopathy, intraventricular hemorrhage of prematurity, traumatic optic neuropathy, myelodysplastic syndromes | ||||
| G-CSF | 24 (3) | 28 (3) | 97 (19) | 51 (14) | 200 |
| Indic. | Phase 1: solid tumor/ NSCLC/SCLC, advanced pancreatic cancer, postmenopausal symptoms || Phase 2: neurological diseases, multiple myeloma, lymphoma/leukemia, Fanconi anemia, early stage BC, Crohn’s disease, heart failure || Phase 3: prostate cancer, liver cirrhosis/malnutrition, decompensated liver cirrhosis | ||||
| GM-CSF | 18 (3) | 43 (7) | 130 (13) | 18 (1) | 209 |
| Indic. | Phase 1: prostate cancer, metastatic breast cancer || Phase 2: squamous cell carcinoma of the oral cavity, neuroblastoma, glioblastoma/gliosarcoma, recurrent neuroblastoma, colon cancer || Phase 3: pulmonary alveolar proteinosis | ||||
| IFNa) | 15 (4) | 9 (1) | 59 (7) | 29 (1) | 112 |
| Indic. | Phase 1: various tumor malignancies || Phase 2: renal cell carcinoma/melanoma, lymphomatoid granulomatosis, chronic myeloid leukemia, breast cancer || Phase 3: malignant pleural mesothelioma | ||||
| IFN-α | 45 (9) | 82 (6) | 360 (20) | 259 (11) | 746 |
| Indic. | Phase 1: Triple-negative BC || Phase 2: adverse effects of immunotherapy, squamous cell carcinoma, prostate cancer, recurrent ovarian cancer, myeloproliferative disorders, metastatic liver carcinoma, lymphoma, leukemia || Phase 3: polycythemia vera, melanoma, chronic myeloid leukemia, hepatitis, COVID-19 | ||||
| IFN-β | 4 | 30 (5) | 40 (3) | 49 (12) | 123 |
| Indic. | Phase 1: solid tumor/NSCLC/SCLC, malignant solid tumors, endometrial clear cell adenocarcinoma, stage III melanoma, hepatocellular carcinoma || Phase 3: relapsing/remitting multiple sclerosis, COVID-19, MERS-CoV | ||||
| IFN-γ | 8 (1) | 16 (1) | 40 (1) | 15 | 79 |
| Indic. | Phase 1: ovarian cancer || Phase 2: breast cancer | ||||
| IFN-λ | 1 | 7 (2) | 7 | 15 | |
| Indic. | Phase 2: hepatitis D, COVID-19 | ||||
| IL-1 | 4 | 1 | 2 | 7 | |
| IL-10 | 2 | 1 | 1 | 1 | 5 |
| IL-11 | 1 | 8 | 3 (1) | 12 | |
| Indic. | Phase 3: nasopharyngeal carcinoma | ||||
| IL-12 | 4 | 51 (5) | 24 (1) | 79 | |
| Indic. | Phase 1: HIV, malignant epithelial tumors, solid tumors, acute myeloid leukemia || Phase 2: TNBC | ||||
| IL-13 | 1 | 1 | |||
| IL-15 | 11 (4) | 2 | 13 | ||
| Indic. | Phase 1: Relapsed T cell lymphoma, peripheral T cell lymphoma, metastatic solid tumors, leukemia | ||||
| IL-18 | 1 | 1 | 2 | ||
| IL-2 | 16 (1) | 95 (25) | 233 (62) | 27 (4) | 371 |
| Indic. | Phase 1: ulcerative colitis, allotransplantation, solid tumors, recurrent or platinum resistant OC, metastatic colorectal carcinoma || Phase 2: type 1 diabetes, systemic lupus erythematosus, stage IV gastric carcinoma/stage IV nasopharyngeal carcinoma/lymphomas, autoimmune diseases, relapsing polychondritis, recurrent miscarriage, recurrent melanoma, recurrent acute myeloid leukemia, polymyalgia rheumatica, pleural mesothelioma, pemphigus vulgaris, NSCLC, metastatic OC, liver transplant, HIV, inflammatory myopathy, head and neck tumors, Crohn’s disease, chronic graft versus host disease, bone sarcoma, Behcet’s disease, amyotrophic lateral sclerosis, advanced plural mesothelioma, acute coronary syndromes || Phase 3: neuroblastoma | ||||
| IL-21 | 3 | 7 | 10 | ||
| IL-22 | 1 | 1 | |||
| IL-3 | 3 | 3 | |||
| IL-4 | 1 | 3 | 3 | 7 | |
| IL-6 | 2 | 2 | |||
| IL-7 | 6 (1) | 19 (5) | 25 | ||
| Indic. | Phase 1: acute myeloid leukemia || Phase 2: mycobacterium infections, metastatic bladder/renal urothelial carcinomas | ||||
| TNF-α | 10 | 7 | 17 | 6 | 40 |
| TRAIL | 2 (2) | 1 | 3 | ||
| Indic. | Phase 1: malignant pleural effusion, peritoneal carcinomatosis | ||||
|
| |||||
| Total | 202 (27) | 433 (63) | 1231 (151) | 679 (62) | 2545 |
Data obtained from searching interferon, interleukin, tumor necrosis factor and colony-stimulating factor in clinicaltrials.gov. Only interventional studies were included. Studies including the terms anti, antagonist, inhibitor or cell were excluded.
IFN type not specified in the intervention category for the clinical trial registry
Classification of early phase 1 as phase 1, phase1|phase2 as phase 2 and phase2|phase3 as phase 3
A total of 145 clinical trials used a combination of cytokines
Acronyms: BC – breast cancer; OC – ovarian cancer; TNBC – triple-negative breast cancer; NSCLC – non-small-cell lung carcinoma; SCLC – small cell lung cancer; HIV – human immunodeficiency virus; MERS-CoV – middle east respiratory syndrome coronavirus; COVID-19 – coronavirus disease 19
From Table 2, the major targeted indications for cytokine-based therapies continue to be cancer (~35%) and infections (~20%) with autoimmune conditions as the third most common class (~8%). Importantly, the use of cytokines in cancer has received renewed interest due to the success of checkpoint inhibitors (CPIs)[57]. The success of CPI treatment is highly dependent on immune infiltration of the tumor, which could be modulated via cytokine administration prior to or in combination with CPIs.[58] Combination treatments of highly immunostimulatory cytokines and CPIs are one of the main drivers for the recent increase in IL-2 clinical trials (indicated by the high fraction of active trials in Table 2). Furthermore, as will be noted in examples provided in this review, cytokine-based combination therapies also have therapeutic potential with radiotherapy, chemotherapy, cancer vaccines, and cellular therapies. [57,58]
In addition to the three major indication classes mentioned, there are many other potential applications for cytokine therapies. This is demonstrated by the various non-cancer indications in active clinical trials shown in Table 2 such as neurological diseases, inflammatory diseases, fibrotic disease, and wound healing.[59]
4. Clinical Perspective
The basis for using cytokine therapies in the clinic is understanding their mechanism of action and toxicities. Many of the side effects from cytokine administration originate from the downstream cytokines released (i.e., the cytokine cascade). In the case of interferons or interleukins, the cytokine cascade is the cause of the most common adverse reactions of flu-like symptoms (fever, chills, myalgia, headache, and nausea). [60] These occur from the downstream expression of IL-1, IL-6, and TNF-α which can stimulate the hypothalamus leading to the various observed autonomic and behavioral effects.[61] The cytokine cascade can also stimulate autoimmune conditions, affecting thyroid function and in some cases leading to psychiatric disorders such as depression. [61] Accordingly, combined with fatigue, even the more common systemic cytokine treatment toxicities can severely affect patient quality of life such that the therapeutic benefit is unsubstantiated. Further, under uncontrolled conditions, this cascade of cytokines released—termed ‘cytokine storm’—is one of the leading causes of the more severe toxicities seen in cytokine-based therapies.
Unfortunately, it is difficult to prevent immune activation without subsequent expression of downstream cytokines. For example, i.v. administration of IL-2 will directly activate peripheral blood mononuclear cells (PBMC) expressing the IL-2R of which natural killer (NK) cells (~10% of PBMC) are the main elements.[62] Upon IL-2 stimulation, NK cells secrete IFN-γ, GM-CSF, and TNF-α, which in turn have downstream effects on a broad range of cell types. Moreover, although the downstream cytokines may lead to toxicities, they may also participate in the therapeutic effect. For example, IL-12-mediated toxicities are correlated with induction of high systemic IFN-γ levels.[63] Although these toxicities may be reduced via co-treatment with anti-IFN-γ antibodies, a reduction in therapeutic effect may also be seen as IFN-γ is a major driver of cellular immunity and macrophage activation.[64]
In addition to cytokine storms, another main concern with cytokine therapies is increased microvascular permeability, leading to vascular leak syndrome (VLS).[65] Typically associated with sepsis, VLS is characterized by the retention of extravascular fluid, hypotension, and multi-organ dysfunction. Although the mechanisms triggering VLS are not fully understood, it is likely mediated by multiple factors including increased vascular adhesion of activated immune cells, alterations in the endothelium due to inflammatory cytokines such as IFN-γ or TNF-α, and increased nitric oxide production in response to cytokine stimuli.[66] Although most deeply studied in the context of IL-2 administration where VLS is a chief toxicity, VLS is also associated with the administration of many other cytokines.[67] [65]
Given the issues with systemic administration of cytokines, one key goal for clinical development of cytokine therapies has been to minimize plasma exposure while maintaining efficacy, as high blood levels correlate with higher toxicities.[68] Indeed, preclinical studies that led to approval of aldesleukin demonstrated this effect when optimizing the IL-2 formulation and dose regimen.[69] Formulation of aldesleukin with albumin or bolus administration compared to short (5 to 15 minute) infusion led to increased total plasma exposure. In the case of albumin formulation, there was a subsequent increase in toxicities and mortality in animals without improved therapeutic benefit. However, timing as well as peak plasma concentrations matter, as continuous dose regimens of IL-2 have a 10 fold lower maximum tolerated dose compared to a bolus i.v. administration.[62,70] Intriguingly, preclinical studies using bolus extended half-life IL-2 have indicated lower toxicities and improved efficacy compared to the native protein. [71,72]
Clinical development of cytokines is further hindered by limitations in preclinical animal models. For example, preclinical studies of cancer immunotherapies often require use of immunocompetent mouse models, but transplanted tumor models are characterized by very rapid tumor growth and the development of histopathology that often does not well replicate human cancer. These characteristics lead to different vascularization and immune infiltration which can have significant effects on the delivery or efficacy of cytokine treatment. Furthermore, the immune system of mice and humans has important differences in cellular makeup, receptor expression, and cytokine responses.[73] For example, human blood is neutrophil-rich (50–70% compared to 10–25% in mice). This difference can lead to an altered immune response from systemic cytokine administration since, for example, neutrophils play a major role in IL-2-induced capillary leak syndrome and IL-12 stimulation on human neutrophils leads to IFN-γ production.[74–76] Mice may lack a functioning intermediate IL-2Rβγ,[76] and this altered IL-2R biology could be a contributing reason for the increased toxicity noted when IL-2 immunocytokines were tested in humans compared to mouse models[77]. Importantly, these immunological differences are not restricted to mouse models as shown by the failed TGN1412 (a CD28 superagonist monoclonal antibody) Phase 1 trial in which differences in CD28 expression patterns between non-human primates and humans led to safe tests in cynomolgus and rhesus monkeys, but significant cytokine storms in humans.[78]
Some cytokine clinical trials have also failed to demonstrate clear relationships between administered dose and therapeutic effect.[79] This is likely due to marked patient-to-patient immune system heterogeneity. For example, in an IL-12 clinical trial, IL-12 failed to show a dose-response in IFN-γ induction.[79] Accordingly, understanding a patient’s individual immune profile and monitoring the changes associated with the cytokine therapy are likely necessities for successful clinical cytokine therapies.
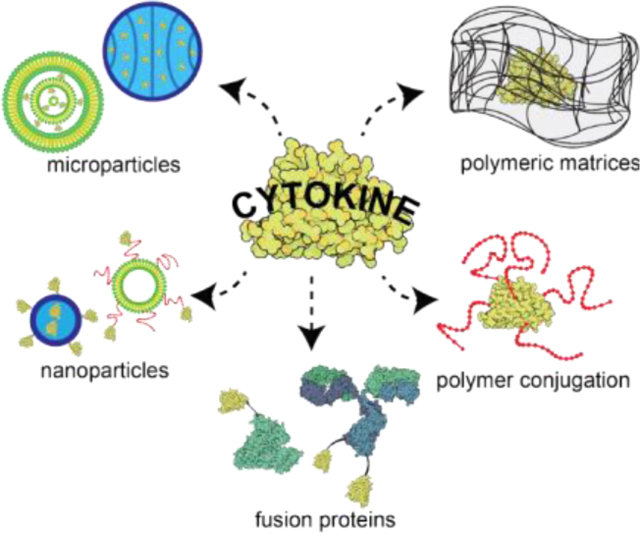
Given the pleiotropic characteristic of cytokines and their resulting clinical effects, the major challenges with the use of bolus or continuously-administered recombinant cytokines are their high toxicity when administered systemically and poor pharmacokinetic profile. As signaling molecules for damage or danger, cytokines can cause serious side effects when present systemically by over-activating the immune system. Further, cytokine therapy suffers from poor pharmacokinetics as the small size of the proteins facilitates rapid vascular extravasation and kidney excretion. Thus, for conventional intravenous administration, large and frequent dose regimens are often required to reach therapeutic efficacy. However, these dosing regimens come at the expense of high toxicity risks which can outweigh the therapeutic benefit. Together, these challenges motivate the application of drug delivery technologies to enable the safe and effective use of these proteins therapeutically. Accordingly, many approaches have been developed to address the above challenges including protein engineering, polyethylene glycol (PEG) conjugation, fusion proteins, polymeric matrices, and particle-mediated delivery (micro- and nanoparticles). These engineering strategies and their clinical utility are described in this review. Other approaches for cytokine delivery such as cytokine-secreting cells and gene therapy are briefly introduced, but these approaches present distinct challenges beyond the scope of this review.
5. Protein Engineering Strategies for Cytokine Delivery
5.1. Sequence Modifications
One of the first engineering strategies that enabled the clinical translation of cytokines was mutations or modifications to the protein sequences for improved biologic manufacturability. More recently, mutated cytokine versions are being developed to reduce the pleiotropic nature of cytokines. Accordingly, these mutations are used to either enhance or diminish binding to cellular receptors, allowing for a more selective effect of the desired cytokine.
5.1.1. Enhanced protein folding and stability
Many of the cytokines discussed here are complex eukaryotic glycoproteins. Thus, to improve manufacturing, point mutations have been introduced to the sequence of approved products. For example, aldesleukin (IL-2) has a mutation replacing the 125-cysteine with serine to facilitate the folding to the proper conformation without affecting its bioactivity (aldesleukin also does not contain the N-terminal alanine of IL-2).[80] Similarly, the approved IFN-β expressed in E. coli (IFN-β−1b) has a cysteine substituted for serine in position 17.[81] Further, substitution of hydrophobic residues for hydrophilic ones can improve bioavailability of non-glycosylated forms of these glycoproteins.[82]
5.1.2. Selective receptor binding
One of the first examples of sequence engineering to modulate cytokine receptor binding was the interferon alfacon-1. This synthetic protein contains the most common amino acids from the 14 IFN-α subtypes, yielding a non-natural protein with higher activity over IFN-α−2b (major subtype used in the clinic).[83] However, with the convenience of PEGylated IFN-α, this drug is currently restricted for treatment-failure patients dosed with peginterferon-α formulations.[84]
More recently, cytokine engineering has aimed to become more selective in modulating the receptor-binding properties of the cytokine. One of the most researched examples has been IL-2, which binds to two major forms of the IL-2 receptor (IL2R) in humans. One is an intermediate affinity receptor composed of IL2Rβ (CD122) and IL2Rγ (CD132) while the other a is high-affinity receptor containing IL2Rβ, IL2Rγ, and IL2Rα (CD25). Importantly, CD25 is upregulated on Tregs and activated T cells, while IL2Rβ and IL2Rγ are constitutively expressed in NK cells, macrophages, and T cells[85,86] Accordingly, there have been various engineered IL-2 muteins designed to preferentially bind to the IL2Rβγ complex and reduce Treg activation via IL-2Rα binding. For example, the IL-2v design employs three mutations in the CD25 binding interface, F42A, Y45A, and L72G, to restrict IL-2 binding to CD25.[87]
Interestingly, the opposite has also been attempted: a D20T mutation in IL-2 reduces intermediate affinity receptor binding but maintains high-affinity receptor (CD25) binding. This cytokine was designed to reduce systemic IL-2 toxicity by mutating a toxin-like motif involved in endothelial cell binding.[85] Reducing the affinity to the intermediate affinity receptor could also reduce systemic NK cell activation and its subsequent toxicities.[85] Although it could stimulate Tregs, it would serve to maintain activated T cells and was theorized to provide a potent anti-cancer response in combination with Treg depletion therapies.[85] However, this construct was discontinued for use in cancer treatment, potentially due to the low levels of CD25 expression in pulmonary endothelial cells, leading to toxicity or simply low potency in immune stimulation.[87] Importantly, although, selective CD25 binding IL-2 muteins have not to date had great success in cancer therapy, many of them remain under clinical investigation for immunodeficiencies (see Fusion Protein section for examples).
Modulation of cytokine-receptor interactions may also improve adoptive cell therapies. For example, an orthogonal pair of mutated IL-2 (orthoIL-2) and IL2Rβ (orthoIL2Rβ) were designed to have specific interactions without binding to the wild-type proteins.[88] When orthoIL2Rβ was expressed in engineered T cells, administration orthoIL-2 could selectively activate the transferred T cells without systemic immune activation and subsequent toxicity.[88]
5.2. Fusion Proteins
Fusion proteins consist of two or more proteins genetically linked to functionally enhance the resulting protein complex. The enhancement often originates from altered pharmacokinetics or targeted cytokine bioactivity. For pharmacokinetic alterations, cytokines fused to a partner protein have increased size, reducing kidney excretion and typically increasing the circulatory half-life of the protein. Moreover, the increased size can lower the interstitial transport rate, prolonging exposure once delivered to a site.[89] Cytokines can also be fused with targeting or functional proteins, to change their localization or introduce new functionalities to the protein. A variety of cytokine fusion protein constructs have reached clinical trials, including immunotoxins, tumor antigen cytokine fusion proteins, immunocytokines, non-antibody targeting motif fusion proteins, Fc-fusions, and albumin-fusions (Figure 2), These fusion proteins are discussed in this section.
Figure 2.

Illustration of major fusion proteins developed for cytokine delivery. The three-dimensional protein illustrations were generated in Qutemol[90] based on the Protein Data Bank structures of interleukin-2 (1M47), immunoglobin-G (1IGT), diabody scFv T84.66 (1MOE), scFv based on diabody structure (5GRV), human IgG1-Fc domain (5JII), diphtheria toxin (1F0L), prostatic acid phosphatase (1CVI), human serum albumin (1A06), and fibromodulin (5MX0).
5.2.1. Immunotoxins
Immunotoxins are fusions of cytokines with bacterial toxins designed to allow the specific killing of malignant cells that express a certain cytokine receptor. The first clinically approved immunotoxin consisted of IL-2 fused with the diphtheria toxin (DT) protein. This construct replaced the receptor-binding domain of diphtheria with IL-2, creating an IL-2 toxin directed at cells expressing the IL-2R (primarily T cells).[91] This IL-2 toxin (denileukin diftitox, Ontak) received regulatory approval from the FDA in 1999 for persistent or recurrent cutaneous T cell lymphoma.[92] However, the treatment had severe side effects and complicated manufacturing which led to its discontinuation from clinical use.[92] Notably, other cytokines such as GM-CSF and IL-3 were also fused with DT for hematological malignancies.[93] Although GM-CSF-DT failed in Phase 1 trials due to liver toxicity, the IL-3 receptor-targeted version (Elzonris) received FDA approval in 2018 for blastic plasmacytoid dendritic cell neoplasm and is currently being tested in other myeloma malignancies.[93] Lastly, an IL-13 immunotoxin has also re-entered clinical trials for gliomas after it reached Phase 3 clinical trials for recurrent glioblastoma, but did not show survival benefit over standard-of-care carmustine wafers.[94]
Importantly, although these constructs were not designed as immunomodulators the potential to deplete certain immune cells can be used to alter the immune profile of a disease. Accordingly, more recent clinical trials have used denileukin diftitox to deplete Treg cells in tumors as these cells have high expression of the high affinity IL-2 receptor.[95] A better understanding of the full effects of the therapy are still needed as conflicting and unexpected results have been reported.[95] With the discontinuation of Ontak, safer and more reliable formulations of the drug are needed for further studies.[92] However, although new constructs can be developed, a major limitation in these class of fusion proteins is the high immunogenicity of the toxin moiety, which can greatly affect the pharmacokinetics and efficacy of the therapeutics.[96]
5.2.2. Antigen-cytokine fusion proteins
Another cytokine fusion protein strategy is the linkage of cytokines to antigens to provide enhanced antigen immunogenicity. This class of fusion protein has been clinically developed for prostate cancer treatment (tradename Sipuleucel-T) and was approved in 2011 as the first cancer vaccine treatment.[97] In this therapy, the patient’s peripheral blood mononuclear cells (PBMCs) are treated ex-vivo with a fusion protein composed of prostatic acid phosphate (prostate cancer antigen) linked to GM-CSF. [97] Treatment with the fusion protein aims to enrich the autologous PBMC (mixture of lymphocytes, monocytes, and dendritic cells) with activated antigen-presenting cells (APCs) such as dendritic cells and monocytes. These fusion-protein-activated APCs are then reinjected into the patient to provide an immune response that prolonged survival by approximately 4 months in patients with asymptomatic or minimally symptomatic metastatic castrate resistant (hormone refractory) prostate cancer.[97] Importantly, the APC activation was enhanced with the fusion protein over its separated constituents, enabling the development of the therapy. However, Sipuleucel-T has been under-applied in the clinic as it has a high cost, and traditional biomarkers of disease progression such as serum levels of prostate-specific antigen do not track well with activity of the therapy.[98,99]
Although Sipuleucel-T delivers the cytokine fusion protein ex-vivo, preclinical studies have used the same concept for in vivo cytokine delivery. For example, cytokine-neuroantigen fusion proteins have been used to induce a tolerogenic response to the antigen in models of multiple sclerosis. Several cytokines including IL-2, GM-CSF, M-CSF, IL-16, and IFN-β have been tested in these constructs.[100] Notably, GM-CSF showed the greatest tolerogenic effect due to its role in active induction of myeloid APCs and the targeting of the antigen to GM-CSFR expressed by APCs. Importantly, this fusion protein further illustrates the pleiotropic activity of cytokines as GM-CSF alone is known to exacerbate experimental autoimmune encephalomyelitis (EAE) but showed a tolerogenic (anti-inflammatory) effect in the fusion protein for EAE treatment. This anti-inflammatory response is also seen when GM-CSF is used in other animal models of autoimmunity such as type 1 diabetes, thyroiditis, and graft versus host disease.[101] This function is thought to occur through support of regulatory myeloid DCs and enhanced antigen presentation to Tregs.[101]
5.2.3. Immunocytokines
Another class of cytokine fusion proteins developed in the early stages of cytokine therapies are cytokine-antibody conjugates. These fusion proteins, termed immunocytokines, combine the targeting specificity of antibodies with the potency of cytokines aiming to elicit a local immune response while also modulating the pharmacokinetics and pharmacodynamics of the drug molecule (Figure 2). Interestingly, tumor-cell targeting immunocytokines may also bridge tumor cells and leukocytes, acting similarly to bispecific antibodies.[102] Given their potential for a localized immune response and enhanced pharmacokinetics, these constructs have been heavily researched for cytokine delivery in oncology and non-oncological diseases such as autoimmune or inflammatory conditions.[103] Immunocytokine candidates are in clinical trials for delivery of a variety of cytokines including IL-2, IL-10, IL-12, and TNF-α[68]. These fusion proteins and their current clinical stage are summarized in Table 3. Other immunocytokines have been developed in preclinical studies that have been reviewed previously.[104]
Table 3.
Immunocytokines in clinical trials.
| Immuno-cytokine | Cytokine | Route | Target / Ab | Combination | Clinical Trial Phase (indication) | Ref. |
|---|---|---|---|---|---|---|
|
| ||||||
| NHS-IL12 | IL-12 | s.c. Q4W | necrosis (histones)/IgG | avelumab (anti-PD-L1) - | Phase 1 (solid tumors) Phase 2 (Kaposi Sarcoma) | [105] |
| Hu14.18-IL2 | IL-2 | i.v. 3xQ1D | cell surface (GD2) / IgG | - | Phase 2 (neuroblastoma/ melanoma) | [106] |
| FAP-IL2v (RG7461) | IL-2 (↓CD25) | i.v. Q1W | Cell surface (FAP) / IgG (↓FcγR ↓C1q) | tecetriq, avastin, pembrolizumab (anti-PD1) | Phase 1 (melanoma/ RCN) | [107] |
| IL12-F8-F8 (Dodekin) | IL-12 | - | vasculature / diabody | - | Phase 1 | [108] |
| L19-IL2 (Darleukin) | IL-2 | i.v. Q1W | vasculature (EDB) / diabody | SBRT | Phase 2 (NSCLC) | [108] |
| rituximab (anti-CD20) | Phase 1 (B cell lymphoma) | |||||
| L19-TNF (Fibromun) | TNF-α | i.v Q2D | vasculature (EDB) / scFv | doxorubicin / dacarbazine | Phase 3 EU / Phase 2 US (soft tissue sarcoma) | [108] |
| - | Phase 2 US (glioma) | |||||
| L19-IL2 + L19-TNF (Daromum) | IL-2 + TNF-α | i.t. Q1W | vasculature (EDB) | - | Phase 3 (Stage IIIB/C melanoma) | [108] |
| F16-IL2 (Teleukin) | IL-2 | i.v. Q1W | vasculature (tenascin-C)/ diabody | anti-CD33 antibody | Phase 1 (AML) | [108] |
| F8-IL10 (Dekavil) | IL-10 | s.c. Q1W | vasculature (EDA) / scFv | methotrexate | Phase 2 (RA and ulcerative colitis) | [108] |
| DI-Leu16-IL2 | IL2 | s.c. 3xQ1D | cell surface (CD20) / IgG | after rituximab (anti-CD20) | Phase 2 (Lymphoma) | [109] |
| CD20-IFNα (IGN002) | IFNα | i.v Q1W | cell surface (CD20) / IgG | - | Phase 1 (NHL) | [110] |
|
| ||||||
| Discontinued a) | ||||||
|
| ||||||
| NHS-IL2T (Selectkine) | IL-2 (↑CD25) | i.v. Q3W | necrosis (histones)/IgG | SBRT/ipilimumab (anti-CTLA-4) | Phase 2 (melanoma) | [111] |
| CEA-IL2v | IL-2 (↓CD25) | i.v. Q1–3W | cell surface (CEA) / IgG (↓FcγR ↓C1q) | atezolizumab (anti-PDL1) | Phase 1 (advanced and/or metastatic tumors) | [87] |
| huKS-IL2 | IL-2 | i.v. 3xQ1D | EpCAM / IgG | cyclophosphamide | Phase 2 (SCLC) | [112] |
| BC1-IL12 (AS1409) | IL-12 | i.v. Q1W | vasculature (B-FN) / IgG | - | Phase 1 (melanoma, RCN) | [113] |
no active trials and/or removed from companies’ pipeline
EDB: extra domain B of fibronectin; EDA: extra domain A of fibronectin; B-FN: isoform of fibronectin; FAP: Fibroblast activation protein alpha; CEA: Carcinoembryonic antigen; GD2: Disialoganglioside; RCN: renal cell carcinoma; SBRT: stereotactic body radiation therapy
As shown in Table 3, the antibody portion of the immunocytokine can be in various formats ranging from the full immunoglobulin G (IgG) to antibody fragments (Figure 2 depicts some of these constructs).[103] These structural configurations as well as glycosylation patterns can modulate the circulatory half-life and residence time at the disease site.[102] For example, full IgG constructs have higher circulatory half-life over fragments, but a smaller immunocytokine with fast extravasation may lead to a higher relative tumor to blood concentration (Figure 3), potentially minimizing systemic exposure while leading to similar tumor accumulation.[114]
Figure 3.

Selected formats of immunocytokines and their primary characteristics on vascular extravasation and tumor retention.
Although smaller immunocytokines can reduce systemic exposure, these fusion proteins need not be small to improve their therapeutic effect as size is only one of the factors that can alter its efficacy. For example, in addition to its structure, the site of linkage and type of linker can be used to modify pharmacokinetics and cytokine receptor binding properties.[115,116] Further, the use of an unmodified Fc region in immunocytokines mediates various effects such as antibody-dependent cell-mediated cytotoxicity, complement-dependent cytotoxicity, and extended half-life through the neonatal Fc receptor (FcRn) recycling.[114] Lastly, both the antibody and the cytokine may be mutated for altered receptor binding such as the FAP-IL2v which has a mutated IL-2 for lower CD25 binding and mutated IgG to restrict C1q and FcγR binding while maintaining FcRn recycling.[107] With these mutations, the therapy aims to prevent immunosuppression from IL-2-mediated stimulation of CD25hi Tregs, and prevents induction of myeloid phagocytosis via FcγR or complement binding via C1q of the drug and its target. In a number of the clinical trials noted in Table 3 the immunocytokine is given in combination with chemotherapy or CPIs. Promising results of combination therapies with CPIs have propelled research into immunocytokines targeting check-point markers such as fusion proteins of IL-15 and anti-PDL1 mAb (KD033)[117].
Although studied since the 1990s, most trials of immunocytokines are still in either Phase 1 or Phase 2. The greatest limitation with immunocytokines is the requirement of a tumor antigen to target. This requirement hinders the wide application of the therapy as bona fide tumor-specific antigens shared across patients are rare. Moreover, antigen loss by cancer cells can lead to tumor escape.[118] An alternative strategy has been to target tumor blood vessels or the tumor extracellular matrix, which is another major strategy currently used in clinical trials[102]. However, targeting of pro-inflammatory agents to blood vessels may lead to hypotension as a dose-limiting toxicity.[68] Moreover, for any of these antigen-targeted strategies, the therapy risks on-target toxicity. This occurs when these antigens are expressed at low levels in healthy tissues, promoting off-site targeting of the immune response. Further complicating their development is the observation that the cytokine payload of the fusion protein can compete with the variable region of the antibody by binding to cytokine receptors: rapid binding of the immunocytokine to cytokine receptor+ immune cells in the blood can dominate the biodistribution of the fusion protein, limiting tumor accumulation.[114]
One strategy to minimize unspecific binding and systemic toxicity is intratumoral administration (also called intralesional).[114] Intralesional treatments aim to exert their effect locally, reducing systemic toxicity. Indeed this approach has shown encouraging clinical success when delivering IL-2 with high response rates and low toxicity, but the laborious administration schedule tempered its utility.[119] Immunocytokines are thus promising candidates due to their high retention, which enables more practical dose scheduling. In this area, the most clinically advanced immunocytokine treatment is a combination of IL-2 and TNF-α immunocytokines (Daromum) which uses L-19 diabodies (L19IL-2 and L19TNF) to target the tumor vasculature. These fusion proteins are in Phase 3 clinical trials as a neoadjuvant treatment for stage IIIB/C melanoma via intralesional delivery.[119] In animal studies, they were shown to have longer residence time within the treated lesion over free cytokine. Further, Phase 2 clinical studies showed an overall response rate of 55%, with reduction of untreated lesions (“abscopal effect”) and minimal treatment-related toxicity. Notably, the low toxicity of Daromum makes it a promising candidate for neoadjuvant immunotherapy as previous clinical trials with CPIs have shown high response rates but with significant toxicities.[119]
5.2.4. Albumin-Cytokine Fusion Proteins
Many proteins may be used to modulate the pharmacokinetics of fusion proteins. Another major class of fusion protein for this effect are albumin fusions, which prolong the circulatory half-life of fusion partners. The fusion increases molecular size and, similar to IgG fusion proteins, albumin is recycled through the FcRn which further enhances the half-life of albumin fusion proteins.[71] However, unlike other strategies for half-life extension such as PEGylation of other materials-based approaches, albumin fusion proteins are more readily synthesized at large-scales, providing more cost-effective methods for improving cytokine pharmacokinetics.[120] Accordingly, IL-2 was fused with albumin (Albuleukin) for a prolonged serum half-life and targeted biodistribution to spleen, liver, and lymph nodes which prompted its testing in Phase 1 clinical trials.[121] However, after an acquisition of the developing company, the project ended. [121]
Notably, promising candidates from this class were albinterferon alfa-2b (tradename Albuferon) and albumin-G-CSF fusion which had prolonged half-life and reached Phase 3 clinical trials.[122] However, although both Albuferon (albumin-IFN-α) and albumin-G-GCF were noninferior to the approved PEGylated versions of the cytokines, they did not reach clinical approval. Albuferon have higher toxicities compared to PEG-IFN-α while albumin-G-CSF was discontinued by the developing company.[84,123,124]
Importantly, although albumin-fusion proteins failed to reach clinical approval, preclinical studies are still looking to use albumin fusion cytokines. For example, canine IFN-γ-albumin fusion protein is being developed for veterinary medicine.[120] Moreover, recently, an albumin fused to GM-CSF (albGM-CSF) was shown in mice to control tuberculosis by prolonging GM-CSF half-life, increasing its bioactivity, and targeting the cytokine to the draining lymph nodes and lungs.[125] The albGM-CSF treatment increased DC count in the draining lymph nodes and elicited an antitubercular effect ex vivo and in mouse lungs. Notably, these effects may enhance first-line regimens for tuberculosis treatment or serve as a new adjuvant in cancer prevention or therapeutic vaccines. Lastly, there have been promising results using IL-2 fused to albumin in mouse models in combination with anti-tumor antibodies.[71,72]
5.2.5. Fc Fusion Proteins
Fusion of cytokines with the Fc domain of antibodies can be used to both enhance the circulation lifetime of cytokines and introduce antibody effector functions. For example, in addition to prolonged half-life, IL-2-Fc fusion proteins have been shown to deplete Tregs via the FcγR[126]. This mechanism may even have higher antitumor effect over the immune activation of IL-2 as a standard IL-2-Fc overperformed a mutated IL-2 with limited binding to Tregs.[126] However, combining mutations that limited IL-2 interaction with Tregs and enhanced T cell and NK activation have been shown to have enhanced antitumor immune effects over native Fc-IL2.[127] Clinically, a few cytokine-Fc fusion proteins are under investigation. These include a Phase 1 trial using Fc-IL-2 (CC 92252[128] and AMG592[129]) designed to have preferential binding to Tregs for autoimmune diseases and Phase 2 trials using Fc-IL-22 fusion proteins (UTTR1147A[130] and F-652[131]) for inflammatory diseases. Further, a non-specific IgG-IL2 construct which has increased Treg activation is also being developed[107]. Like some immunocytokines in Table 3, the IgG-IL2 fusion protein is also mutated to remove FcγR and C1q binding while retaining FcRn binding.
Fc fusions are also being developed for treatment of cancer. Fc-scTRAIL (ABBV-621) is in Phase 1 trials for solid tumors or hematologic malignancies.[132] Also for cancer therapy, an IL-12-Fc construct (DF6002) has recently started Phase 1/2 clinical trials delivered subcutaneously with CPI (anti-PD1) in advanced solid tumors (NCT04423029). Another promising Fc fusion protein is an IL-15 superagonist, N-803 (previously known as ALT-803). This drug molecule consists of a mutated IL-15 for increased IL15Rβ binding complexed with IL15Rα-Fc protein.[133] The IL15:IL15Rα (hetIL-15) complex in itself also has increased bioavailability and half-life and is in Phase 1 clinical trials (NIZ985) administered subcutaneously for metastatic and advanced solid tumors in combination with anti-PD-1 CPI[134]. Based on promising Phase 1 clinical results, N-803 is currently in Phase 2 clinical trials for various indications as a monotherapy or in combination immunotherapies administered subcutaneously. [134] A related construct, an IL-15-IL-15Rα fusion protein (SO-C101) is also in Phase 1 clinical trials for metastatic and advanced solid tumors as monotherapy or combination with CPI (NCT04234113). The concept of fusing the cytokine with its receptor is also being tested with an IL2-IL-2Rα construct (ALKS 4230) which is designed to prevent IL-2 from interacting with the endogenous IL2Rα and also increase its half-life.[135] Currently, ALKS 4230 is under investigation in Phase 1/2 clinical trials in advanced cancers as monotherapy or in combination with CPIs.[135] The IL-15 superagonist N-803 has also been made into an immunocytokine by further fusion of anti-PDL1 scFv domains to the construct, yielding better results in animal models compared to N-803 in combination with anti-PDL1 mAb, which is currently being tested in the clinic.[136]
5.2.6. Non-antibody targeted fusion proteins
With the extensive experience and successes of monoclonal antibodies, immunocytokines have been the most studied class of targeted cytokine fusion proteins. However, in addition to antibodies, alternative mechanisms to enable targeted cytokine delivery have been developed including targeting peptides or targeting proteins. The most advanced and tested example of these constructs is the NGR-hTNF fusion proteins. This system uses the tumor vasculature targeting peptide Asn-Gly-Arg (NGR) to deliver TNF-α to tumor blood vessels.[137] The fusion with the targeting motif enabled a wider therapeutic window compared to the free cytokine.[137] Interestingly, it was observed that intermediate dose levels had diminished therapeutic efficacy compared to low or high dose treatments.[138] Mechanistic studies determined that the intermediate or high doses lead to the shedding of soluble TNFR which inhibited TNF-α activity. Thus, therapeutic efficacy was only seen when either low doses were administered to prevent soluble TNFR release or when high doses were used to overwhelm this counter-regulatory mechanism.[137] However, only low doses were likely to have selective interaction with tumor blood vessels as high doses lead to significant systemic toxicity. Importantly, TNF-α is not the only cytokine which can have its activity attenuated from the release of soluble receptors as it has been shown that IL-12 administration leads to an increase in soluble IFN-γ receptors.[79]
Currently, NGR-hTNF has completed various Phase 2 clinical trials with promising results in malignant mesothelioma and liver carcinoma. However, a Phase 3 clinical trial using NGR-hTNF as second line therapy for malignant plural mesothelioma did not reach its primary endpoint.[139] Interestingly, the results showed an overall survival and progression-free survival benefit for patients with short treatment-free intervals. Given that this patient subgroup had rapidly progressing tumors, this observation may be due to the high dependency of the proliferating cells on newly formed blood vessels which are the target of NRG-hTNF. [139] However, the data were not sufficient for EMA approval and NGR-hTNF needs to be further tested in clinical trials for mesothelioma.[140] Notably, NGR-hTNF has the potential to selectively increase vascular permeability as was demonstrated in a Phase 2 clinical trial in combination with an immune-chemotherapy treatment of primary central nervous system lymphoma.[141] In this trial, pre-treatment with NGR-hTNF was safe and enhanced vascular permeability of the blood-brain barrier with a targeted effect in the tumor and peritumoral areas. Importantly, there are other targeting peptide sequences such as RGD, isoDGR, or RGR which can also be fused to cytokines for targeting.[137]
Other promising tumor-targeting cytokine fusion protein constructs are under investigation in pre-clinical studies targeting the tumor extracellular matrix. For example, the fusion of IL-2 or anti-PDL1 CPI with the A3 collagen-binding domain (CBD) of von Willebrand factor can reduce toxicity and improve antitumor immunity compared to the unmodified forms.[142] Further, when IL-12 was fused with CBD, the construct showed a potent effect in immunologically cold tumors and synergized with CPIs even in CPI-unresponsive mouse models.[143] Interestingly, this construct has an even lower plasma half-life compared to IL-12, but its tumor localization properties allowed for an effective immune response while the low plasma retention prevented systemic exposure to the cytokine (similar to the antibody fragment fusion proteins of Figure 3). Moreover, the CBD fusion protein was shown to be more effective when administered i.v. versus peritumorally. On the other hand, fusion of cytokines with lumican, a collagen-binding protein, was shown to provide enhanced tumor retention from i.t. administration.[144] Importantly, the retention of lumican fusion protein was further increased by generating a three-protein fusion protein construct composed of the cytokine, lumican, and albumin. This design was used to generate IL-2 and IL-12 fusion proteins which, when co-administered, enhanced various immunotherapies such as chimeric antigen receptor T cells, vaccines, tumor-targeting antibodies, and CPI.
5.2.7. Other Fusion Proteins
In addition to the designs discussed above, there are many other potential fusion protein constructs for cytokine delivery.[145] Notably, there have been cytokine-cytokine fusion proteins such as Pixykine that also showed some clinical success. Pixykine, a fusion protein of IL-3 with GM-CSF was capable of eliciting a 10–20 fold higher potency than GM-CSF or IL-3 alone in vitro, reaching Phase 3 clinical trials for treating neutropenia and thrombocytopenia.[146] However, its use was discontinued when it was not found to have superior effects compared to GM-CSF therapy alone in its Phase 3 clinical trial.[147] Similarly, GM-CSF has been linked to various other interleukins (IL-2, IL-15, IL-21) in preclinical studies to simultaneously stimulate the myeloid and lymphoid immune system on an anticancer immune response.[146] Notably, GM-CSF-IL2 fusion proteins (GIFT-2) showed superior efficacy compared to the combination of individual cytokines while GIFT-15 showed an immune suppressive effect. Lastly, GIFT-21 had strong pro-inflammatory effects on monocytes, enabling DC differentiation ex-vivo which suppressed tumor growth in vivo when administered without antigen priming. [146]
Other interesting fusion protein concepts are cytokines linked to inhibitory molecules such as an scFv that prevents receptor binding of the cytokine.[148] In this construct, the linker between the cytokine to the inhibitory molecule is designed to be cleavable by enzymes overexpressed in some tumors such as matrix-metalloprotease 9. Accordingly, the cytokine is expected to remain inactive until it reaches the tumor microenvironment where the enzyme cleaves the linker, enabling the cytokine to elicit its effects. However, this technology has been primarily been tested in vitro, and future in vivo studies are needed to access its clinical potential.
5.3. Protein Engineering - Conclusion
Since its origins with sequence modifications for improved manufacturing, protein engineering has been continuously applied to improve cytokine-based therapies. As indicated by a late stage clinical trials and a number of on-going clinical trials, the field of cytokine fusion protein has many promising candidates. Further, many large biopharmaceutical companies are invested in immunocytokines or other types of fusion proteins.[149] As such, its real potential will be tested in the upcoming years and may lead to improved or new cytokine-based therapies.
6. Bioconjugation and Material-based Strategies for Cytokine Delivery
6.1. Polyethylene glycol (PEG) conjugation
Polyethylene glycol conjugation (PEGylation) is the most established technology to alter protein pharmacokinetics and pharmacodynamics.[150] Polyethylene glycol (PEG) is an amphiphilic non-ionic synthetic polymer that can be readily conjugated to cytokines. The hydrophilic nature of PEG facilitates the formation of an associated water layer around the polymer, which along with the entropic penalties of molecular adsorption to the PEG brush layer, reduces non-specific interactions of the conjugated protein with its surroundings.[151] Accordingly, PEGylation can shield the protein core from proteolytic enzymes, lower immunogenicity, increase stability and solubility, and prevent interactions with cell-surface proteins. [150] Moreover, the bound polymer and its water shell increase the hydrodynamic volume (i.e. size) of the resulting complex, preventing extravasation and kidney filtration, increasing the conjugated protein’s circulatory half-life.[150] Although there has been recent detection of anti-PEG antibodies in humans, PEG has no known biological receptors, allowing for modulation of the therapeutic properties with limited introduction of new side effects.
PEGylation may also lead to drug accumulation in tumors and at sites of inflammation due to the enhanced blood vessel permeability and tissue retention at these disease sites.[150] In cancer, this is termed the enhanced permeation and retention (EPR) effect, and is thought to occur due to alterations in tumor vasculature and tumor lymphatic drainage, leading to higher permeability and retention of macromolecules.[152] This is an important phenomenon that is commonly exploited for the delivery of nanosized molecules of particles in cancer, yet the actual mechanisms for accumulation and magnitude of enhancement are still debated.[153]
These collective properties have made PEGylation a common and viable method to enhance therapeutic efficacy and/or improve pharmacokinetics of cytokines. Notably, even without improvements in therapeutic efficacy, enhanced pharmacokinetics improves patient compliance and lowers administration costs as the patient needs to be dosed less frequently with therapy.[154] The first PEGylated protein was approved by the FDA in 1990, and research on its use with cytokines was not far behind, with the first approval of a PEGylated cytokine in 2000 (IFN-α2β, PegIntron). Notably, PegIntron and other PEGylated cytokines such as Pegasys (IFN-α2α), Mircera (erythropoietin), and Neulasta (G-CSF) have become blockbuster drugs.[155] These proteins showed great success due to their enhanced pharmacokinetic profiles. For example, PEGylation of IFN-α increased serum half-life (from 3–8 to 65 hrs) and lowered the clearance rate over 100-fold, eliciting the same response when administered once a week compared to thrice a week with unconjugated IFN-α in chronic hepatitis C[156,157] (other general changes in administration schedule on the PEGylated form of approved cytokines can be seen in Table 1).
Unfortunately, even on the approved PEGylated cytokines, there has not been a substantial reduction in toxicity. For example, the use of PEGylated IFN-α was approved due to its higher relapse-free survival (without significant improvement on overall survival), but treatment still leads to significantly decreased health-related quality of life due to its toxicity.[158,159] Further, although PEGylation provides a beneficial systemic administration pharmacokinetic profile, in general, PEGylation reduces protein activity by hindering binding to its receptor(s). However, the extent of activity loss has often been observed not to be significant enough to reduce the therapeutic value of PEGylation. For example, peginterferon α−2a (Pegasys) retains less than 10% of the original in vitro cytokine activity, but its in vivo efficacy and improved administration schedule justified replacement over the unconjugated cytokine.[155,160]
In addition to the approved PEGylated cytokines in Table 1, IL-2 PEGylation has been widely explored in the literature, making it an excellent case to highlight the pros and cons of PEGylated cytokines. Preclinical studies showed that PEGylation enhanced the stability and solubility of recombinant IL-2 and lowered its immunogenicity.[161] However, even though human clinical trials with PEG-IL-2 showed enhanced plasma retention (10–20 fold increase in half-life), decreased clearance, and similar biological activity,[162] efficacy did not show significant differences over treatment with unconjugated IL-2 when administered after a single high dose in metastatic renal cell carcinoma and melanoma.[163] Furthermore, a Phase 2 trial with PEG-IL-2 treatment as monotherapy in renal-cell carcinoma showed high toxicities with lower response rates than unconjugated IL-2.[164] PEG-IL-2 was also used at low doses in clinical trials for HIV-infected patients in combination with antiretroviral therapy, leading to enhanced CD4+ T cell counts, albeit not immediately restoring immune function.[165] However, this treatment also did not reach FDA approval likely due to the failure of the IL-2-mediated CD4+ T cell increases to show a clinical benefit in HIV-positive patients.[166]
Importantly, various other cytokines have been PEGylated aiming to improve the therapeutic efficacy.[167] Yet, even though these typically had increased half-life, the results did not lead to clinical translation. Like PEG-IL-2, this may be attributed to the high levels of toxicity of treatment or lack of improved efficacy compared to unconjugated cytokine or standard-of-care treatments. Of note were the recent clinical trials using Pegilodecakin (PEG-IL-10) for cancer immunotherapy, which aimed to enhance anticancer CD8+ T cell activity in immunologically “cold” cancers.[168] Although demonstrating promising preclinical and Phase 1 clinical results, PEG-IL-10 treatment for metastatic pancreatic cancer in combination with chemotherapy (FOLFOX) failed to promote overall survival in a Phase 3 clinical trial[169]. Moreover, the remaining two Phase 2 clinical trials that used PEG-IL-10 in combination with CPIs (pembrolizumab or nivolumab) for metastatic non-small cell lung cancer also did not reach their primary endpoints, ending a more than $1.5 billion dollar investment in the therapy.[170] While the full clinical data is still to be presented and more research is likely necessary to understand the failure of PEG-IL-10 despite its strong scientific rationale; the dose, activity or trafficking into the tumors may have hindered the efficacy of the treatment in these immunosuppressive cancers.[171] These challenges may have been noted had there been more careful clinical data prior to hastening the therapy into Phase 3 for metastatic pancreatic cancer and the two Phase 2 studies after early efficacy results based on results from less than thirty patients on Phase 1.[171,172]
Despite the disappointing results discussed above, studies of PEGylated cytokines have continued to generate interest in clinical trials. An example are ongoing trials using an engineered PEG-IL-2 cytokine (Bempegaldesleukin, NKTR-214).[86] This drug has approximately six PEG chains attached to IL2Rα binding region of the protein, creating an inactive prodrug of IL-2 (Figure 4). The cytokine becomes active as the PEG linkers are gradually cleaved by hydrolysis revealing active forms of IL-2. Importantly, the active forms of Bempegaldesleukin (containing one to two PEG-chains attached to the protein) bias IL-2 binding to the IL2Rβγ receptor which should favor CD8+ T cells and NK cell stimulation over Tregs.[173] Accordingly, preclinical results have shown that this drug can preferentially activate CD8+ T cells over Tregs in tumors of mouse models and synergize with CIP therapy.[86,174] Phase 1 clinical studies have suggested similar effects in humans[175] with encouraging objective response rates when combined with CPIs that were independent of baseline tumor PD-L1 expression[176]. Based on these results, various Phase 2 and Phase 3 clinical trials are ongoing using Bempegaldesleukin in combination with CPIs[177]. The PEGylation strategy is also being used in another PEG-IL-2 construct (NKTR-358) to maintain higher relative binding to the high-affinity receptor (IL2Rαβγ), preferentially activating Treg cells.[178] This engineered protein should serve as a treatment for immune-inflammatory disorders and is currently under Phase 1 clinical trials.
Figure 4.

Representation of Bempegaldesleukin and its biased IL2Rβγ receptor binding for enhanced IL-2-mediated immunotherapy of cancer. Reproduced under the terms of the CC BY 4.0 license. [173] Copyright 2017, Charych et. al.
Other more recent strategies for PEGylation have included the use of non-natural amino acids, enabling site-specific chemical modification such as a recent PEG-IFN-β1b tested in a Phase 1 clinical trial. [179] This concept is also being used for site-specific PEG conjugation in IL-2 for preferential IL-2Rβγ binding (THOR-707) which is currently in a Phase 1/2 clinical study or lowered IL-2Rβγ (THOR-809).[180] Site-specific PEG-conjugated IL-15 and IL-10 are also in preclinical studies.[180]
Although the clinical history of PEG makes it one of the most widely used polymers for surface conjugation, there have been increasing concerns in its use. One issue is the recent findings of anti-PEG antibodies present in the population or induced by the therapeutic administration.[151,179] When initially developed for bioconjugation, PEG was considered immunologically inert with 0.2% of the population possessing anti-PEG antibodies. However, recently, anti-PEG antibodies have been found in patients who became non-responsive to PEGylated forms of asparaginase and uricase.[179] Moreover, reassessment of the presence of anti-PEG antibodies in non-treated patients has shown a dramatic increase to more than 70% of the current population.[179] This rise in anti-PEG antibodies in treatment-naïve individuals has been attributed to the widespread use of PEG in household and hygiene products (shampoo, soap, toothpaste, lotion, etc.) and improved assay sensitivities.[151,179] As of yet, no direct negative relationship between the presence of anti-PEG antibodies and therapeutic efficacy with PEGylated cytokine therapies.[181] However, until recently, clinical trials with PEGylated therapeutics did not assay for the presence of anti-PEG antibodies, which may have obscured potential issues. A phase 1 study using PEG-IFN-β1b demonstrated a strong correlation between anti-PEG antibodies and accelerated blood clearance of the cytokine.[179] However, the approved formulation of PEG-IFN-β1a showed the development of neutralizing antibodies against the protein in only 1% of treated patients compared with 2.5% in the unconjugated protein.[181] Moreover, although total immunogenicity increased based on the development of anti-PEG antibodies in 7% of the patients, they were found to have no discernable impact on safety or clinical efficacy.[182]
Further hindering the use of PEG conjugates is their non-biodegradability which restricts elimination of the polymer to clearance via the kidneys.[183] This may lead to intracellular accumulation in the liver and tissue lysosomes, especially in chronically-administered PEGylated therapeutics. Accordingly, there have been various alternatives proposed. Of note have been the development in zwitterionic materials and use of polyaminoacids (polypeptides) as the conjugated polymer.[183] Notably, use of disordered biosynthetic polypeptides such as repeated proline, alanine, and/or serine (PAS) sequences may provide the benefits of PEG without the associated immunogenicity and non-degradability.[184]
Overall, after more than 20 years with PEGylation of cytokines, the results have shown some cases of enhanced pharmacokinetics with similar therapeutic efficacy and toxicity to that of unconjugated cytokines. However, PEGylation has not enabled clinical translation of new cytokines. While the reasons are not clear it may be that prolonged systemic exposure to the highly potent cytokines increases toxicity concurrently with the therapeutic efficacy, providing a similar therapeutic window range compared to bolus administration. Moreover, in a clinical setting, the prolonged serum half-life can become a liability since it precludes the rapid reversal of toxicity which is commonly required for IL-2 treatment.[163] Lastly, PEGylation can also have significant effects on biodistribution, which need to be carefully considered for the desired cytokine effect.[185] Thus, new and rational strategies for cytokine delivery with or without polymer-drug conjugates still need to be developed.
6.2. Polymeric matrices
Polymeric matrices consist of polymer chains capable of entrapping the desired drug molecule. In the clinic, the matrix may be deployed at the desired site (e.g., near or in a tumor) or implanted after surgery (e.g. tumor resection), and often employs biodegradable polymers that will dissolve by hydrolysis over time. When a drug is loaded into these polymeric matrices, the diffusional barrier or the matrix degradation rate modulate the drug release rate. These parameters can be designed based primarily on the (i) polymer composition, (ii) the resulting water content within the matrix, (iii) the type of crosslinker and drug conjugation (Figure 5). Notably, the matrix may protect the loaded proteins from enzyme degradation and promote or diminish cellular interactions. Furthermore, these systems can be either externally-controlled or responsive to the local environment, allowing for even higher-order control of the delivery system.[186] Lastly, by using biodegradable polymers, the administered matrix may not require removal after completion of therapy.
Figure 5.

Major design parameters of polymeric matrices used in cytokine theraapies and their applications. Polymeric matrices can have varied properties by altering (i) the polymer composition, (ii) the resulting water content, and (iii) the type of crosslinker and/or conjugation. These matrices have been primarly used as (a) depots, (b) to promote endogenous cell recruitment, and (c) as exogenous cell resevoirs. Red arrows indicate progression of polymeric matrix systems after administration.
Polymer matrices can be composed of solid polymers or hydrogels. Hydrogels are three-dimensional polymeric matrices comprised of hydrophilic polymers swollen with large amounts of water (generally more than 50%). These polymer networks have solid-like behavior on the macroscale (i.e. contain a definitive shape and do not flow) and solution-like behavior on the molecular scale (water-soluble molecules can diffuse through the hydrogel).[187] Accordingly, hydrophilic drugs are easily loaded into these matrices without the requirement of harsh processing conditions, making them promising vehicles for protein delivery. Moreover, the structural similarity to that of macromolecular-based scaffolds in the body make these hydrogels generally biocompatible.[187]
Based on the modular design space of polymeric matrices, they have been used to enhance the therapeutic efficacy of cytokines by providing a spatiotemporal control over their release. As will be seen, polymeric matrices have shown great promise as modular systems to enhance cancer vaccines by serving as cytokine depots (Figure 5A) and for cellular-based immunotherapies (Figure 5B-C).
Matrices releasing cytokines from a local site over prolonged periods have been extensively explored in applications for cancer vaccines or for intratumoral immunotherapy. To create enhanced cancer vaccines, one of the first approaches was based on the encapsulation and sustained release of tumor-associated antigens together with GM-CSF from hydrated polymer gel matrices derived from poly-N-acetyl glucosamine (p-GlcNAc).[188] Since its initial characterization, this gel matrix (F2 gel) has been used for not only combinations with tumor antigens for cancer vaccines, but also improved intratumoral cytokine monotherapy (GM-CSF, IL-2, IL-12, EPO).[189] Notably, co-formulation of cytokines with chitosan, a natural polymer composed of primarily deacetylated glucosamine polysaccharides, can also function as a controlled release platform due to the high viscosity of chitosan.[190] When IL-12 was formulated with chitosan, intravesical treatments were shown to eliminate up to 90% of tumors in orthotopic bladder cancer mouse models with induction of systemic immunity.[191,192] Furthermore, both the chitosan and p-GlcNAc matrices themselves are capable of inducing an immune response which may synergize with the cytokine effect. Importantly, p-GlcNAc and chitosan are biocompatible, biodegradable, and non-toxic with a p-GlcNAc-based hemostat already approved by the FDA.
A foundational study that propelled research into polymeric matrices for cytokine delivery was the use of a macroporous poly-lactide-co-glycolide (PLGA) scaffold to simultaneously deliver GM-CSF, innate immune “danger signals”, and cancer antigens (tumor cell lysate) for cancer vaccination.[193] This combination of molecules was chosen to mimic an infection when administered in which, a) the prolonged-release GM-CSF (~30 days) served as a cytokine gradient to attract dendritic cells, and b) the recruited dendritic cells were exposed to antigens and danger signals for antigen loading and maturation.[194] This concept is illustrated in Figure 5B. Notably, when implanted, GM-CSF released from the scaffold was shown to attract a similar number of dendritic cells to the scaffold as typical protocols for dendritic cell vaccines. The macroporous structure allowed for the dendritic cells to reside within the implant. Moreover, the design of the matrix could tune the number of dendritic cells recruited, activated and dispersed to local draining lymph nodes. Accordingly, this system could yield a robust immune response without the inherent difficulties with cellular therapeutics such as dendritic cell vaccines. In the poorly immunogenic B16-F10 murine melanoma model, the therapy yielded ~50% survival compared to 0% from GM-CSF-secreting tumor cells.[194] This therapeutic delivery system (termed WDVAX) is currently in Phase 1 clinical trials for melanoma treatment as the first-in-human biomaterial vaccine clinical trial. [194]
Notably, this concept of harboring and stimulating immune cells within a polymeric matrix via cytokines can also be used for enhanced cell therapies (Figure 5B).[195] For example, a macroporous alginate scaffold was integrated with silica microparticles conjugated with T cell stimulatory antibodies (anti-CD3, anti-CD28, anti-CD137) and loaded with soluble IL-15 superagonist. The alginate scaffold was functionalized with a synthetic collagen-mimetic peptide to enhance T cell migration within the matrix. [195] Accordingly, when loaded with tumor-specific T cells, this system is designed to provide a local and prolonged adoptive T cell therapy (ACT), improving the efficacy of ACT in solid tumors. Experiments in animal models showed ~100-fold higher T cell proliferation at the injected site compared to prestimulated T cells in both an unresectable ovarian cancer model and an incompletely-resected breast cancer model. This enhanced T cell therapy prevented recurrence in 100% of animals in the breast cancer model and had complete tumor clearance in 6/10 animals in the unresectable ovarian cancer model which was significantly better to the 0% prevention of recurrence and 0% of complete tumor clearance in animals receiving pre-stimulated T cells alone. [195]
Importantly, traditional implants with specific shapes typically have limited methods for dosing and administration. Accordingly, injectable matrices, which normally either undergo in situ chemical polymerization or sol-gel phase transitions, were developed and have been the main systems researched for polymeric matrix-mediated cytokine delivery.[196] These systems greatly facilitate clinical application as they do not require surgical application of the scaffold and can be designed to have similar performance to implantable materials. For example, prior to the WDVAX implant, it was shown that an injectable alginate gel loaded with CCL21 (a dendritic cell attractant cytokine) and activated dendritic cells was capable of sustained delivery of the chemokine and recruitment of the host dendritic cells and T cells into the injected matrix.[197] In this system, alginate microspheres containing calcium were mixed with an alginate solution to induce in situ gelation that occurred in ~60 min after s.c. administration. Alginate was chosen due to its biocompatibility and capability to electrostatically binding and retaining cytokines and chemokines that may be exogenously added or derived from immune cells. With this same in situ self-gelling system IL-2 and CpG oligonucleotides (immunostimulatory molecules) could be introduced into the injected matrix by electrostatically binding CpG to alginate microspheres and adding IL-2 to the alginate solution (Figure 6).[198] Peritumoral delivery of an IL-15 superagonist (see Fusion Protein section) combined with CpG via this matrix or two injections of IL-15 superagonist gel could elicit similar anti-tumor efficacy without exogeneous dendritic cells in the B16F10 melanoma mouse model.[199] In this construct the IL-15 superagonist was released in vivo over a period of a week with a ~40 fold higher peak cytokine concentration in the tumor and lower levels of the cytokine in circulation compared to systemic injection. Immune cell characterization revealed that the IL-15 superagonist recruited T cells into the matrix and tumor and reduced the relative frequency of regulatory T cells.
Figure 6.

Schematic of an alginate-based polymeric matrix designed for in situ gelation to enable prolonged and dual release of IL-2 and CpG as well as harbor and attract immune cells. Reproduced with permission. [198] Copyright 2009, Elsevier.
Another promising example of injectable materials for cytokine delivery are self-assembling mesoporous silica rods.[200] In this system, high aspect ratio silica rods loaded with GM-CSF enabled prolonged release of the cytokine over more than 35 days. One key point in this strategy is that the biodegradable amorphous silica is generally recognized as safe by the FDA, enabling the potential future clinical translation of these materials. In animal models, therapeutic doses of mesoporous silica rods did not elicit noticeable adverse effects and degraded to an unmeasurable size within 25 days.[200]
Polymeric matrix-assisted cancer vaccine systems can also be made into injectable biomaterials hosting the tumor cells. For example, the combination of GM-CSF with CpG has been used in injectable cryogel systems co-loaded with irradiated tumor cells.[201] This system was shown to out-perform bolus GM-CSF-secreting tumor cancer cells vaccination which enhanced spatiotemporal control of the vaccine delivery. However, the main limitation with such a system is the requirement of ex-vivo manipulations of autologous tumor cells which can alter their associated tumor antigen profile, as well as increased regulatory complexity of the therapy.
Injectable polymeric matrices have also been extensively used for as depots for cytokine monotherapy or combination therapies. [196] One of the first formulations for depot cytokine delivery was IL-2 loaded into a polyoxyethylene-polyoxypropylene block copolymer (F-127 gel).[202] The gel solidifies at physiological temperature providing prolonged released which prolonged survival in rat fibrosarcoma models.[202] In the case of combination therapies, an interesting example comprises the use of another thermo-sensitive polypeptide hydrogel for chemo-immunotherapy mediated by delivery of IL-2, doxorubicin, and IFN-γ:[203] This triple drug combination used poly(γ-ethyl-L-glutamate)-b-poly(ethylene glycol)-b-poly(γ-ethyl-L-glutamate) as a matrix which undergoes a thermally-induced sol-gel phase transition at body temperature, with good biocompatibility and biodegradability in mouse models. This scaffold yielded an improved anti-tumor response on the B16F10 melanoma mouse xenograft model over the free drug combination and had no observable systemic side-effects.
Lastly, in addition to depot function, engineered polymers can improve the therapeutic effect of cytokine-based therapies. One example was an injectable gel formed by a redox-active polyion complex which scavenges reactive oxygen species (ROS).[204] When delivered at the vicinity of tumors, the therapy elicited improved tumor growth-inhibition compared to IL-12 injection alone while also reducing toxicity from the IL-12 mediated ROS generation.
Fueled by a better understanding of immunology and chemistry of materials, there has been a recent surge in preclinical studies applying matrices for cytokine delivery. Some limitations encountered with clinical development of these scaffolds have been manufacturing, storage, regulatory complexity, and costs. Further, polymer degradation may yield unwanted effects such as acidification upon PLGA degradation. [205] However, a few hydrogel systems have recently been clinically translated, paving the way for future polymer-based drug delivery systems.[206]
6.3. Microparticles
Microparticles are large (>1 μm diameter) particles made of primarily polymers or lipids. In cytokine delivery, the most widely used microparticle system has been in the form of polymeric microparticles which, like the polymeric matrices, are used for spatiotemporal control of drug release. Similarly, lipid-based systems such as multilamellar vesicles (MLVs) have also been made into microparticles for cytokine delivery. MLVs are spherical particles composed of multiple lipid bilayers with aqueous internal cavities that can increase an encapsulated drug’s circulatory half-life and function as a depot for prolonged cytokine delivery.[207] These two main classes of microparticles are discussed in this section.
6.3.1. Lipid-based microparticles
Lipid-based microparticles were one of the first vehicles for cytokine delivery when macrophage-activating factor ‘lymphokines’ were encapsulated into large MLVs (average diameter of 1–2 μm) and shown to prevent pulmonary metastasis in mouse models of melanoma.[208,209] Later, more specific cytokine formulations containing IL-1α, TNF-α, IFN-α, and IL-2 were also tested in tumor models or as vaccine adjuvants.[210–212] Importantly, these formulations allowed for higher cytokine concentrations within tissues of the immune system such as lung, liver, spleen, and bone marrow over the free cytokine. Based on promising preclinical studies, IL-2 encapsulated in MLVs reached a preliminary Phase 1 clinical trial delivered i.v. to metastatic cancer patients, and showed immune activation with low toxicity in Phase 1 studies using aerosolized IL-2 MLVs for immunodeficiency and treatment of pulmonary metastases. [210,213,214] However, these MLV formulations did not process to further clinical trials, likely due to the fast clearance of these particles via the reticuloendothelial system (RES).
Notably, it was found that the interaction of IL-2 with small unilamellar vesicles (≤ 100 nm) of dimyristoylphophatigylcholine induced the formation of MLVs (> 1 μm). [215] This simple process allowed for >90% encapsulation of IL-2 and created large liposomal particles capable of targeting the immune cells of RES (primarily macrophages and monocytes) or to potentially serve as cytokine depots.[215] This IL-2 delivery vehicle is currently in clinical trials as a cancer vaccine by subcutaneously co-delivering autologous tumor lysates for lymphoma (Phase 2, NCT02194751) and leukemia (Phase 1, NCT01976520).
Although simple to fabricate, some of the processing conditions for these lipid-based microparticles such as heating, dehydration, or use of non-aqueous solvents can damage the activity of the loaded cytokine. Moreover, due to their fast RES clearance, MLVs tend to be limited for use as depots, but their high lipid contents lower the encapsulation efficiency of hydrophilic drugs. Lastly, lipid-based formations can be unstable in physiological conditions, can have difficult to control drug release rates and limited residence time of ~4 days as depots. [216] Thus, other depot systems such as polymeric gels or polymeric microparticles have typically prevailed as more efficient prolonged delivery systems. Notably, to address some of the limitations with MLVs, multivesicular liposomes were developed which had greater improved residence time to serve as a depo.[216] However, limited studies have assessed their therapeutic benefit for cytokine delivery.[216]
6.3.2. Solid Polymeric Microparticles
Biodegradable polymeric microspheres were one of the first drug delivery systems to enable controlled release of cytokine with a successful therapeutic effect in vivo.[217] This was accomplished using phase-inversion nanoencapsulation to load IL-2 into poly(lactic acid) biodegradable microspheres which showed better therapeutic effect over PEG-IL2 in a human tumor xenograft mouse model.[217] Compared to encapsulation methods used until that point, the phase-inversion nanoencapsulation process produced a limited size distribution range (0.1–10 μm diameter spheres), had limited exposure of the cytokine to non-aqueous solvents, and did not require vigorous stirring or sonication. Accordingly, more of the protein was encapsulated in a bioactive form. Later studies showed that using the same process to encapsulate IL-12 and TNF-α could induce both a local and systemic immune response in a weakly immunogenic primary breast cancer model whereas either agent alone or IL-12 and GM-CSF were not as effective.[218]
Importantly, since the initial studies, many new microsphere designs and processes have been developed and tested in preclinical studies for cytokine delivery.[219] These systems have focused on using methods to retain the cytokine bioactivity during processing. Some examples in early works include the use of gelatin, chitosan, and bovine serum albumin substrates which have rapid degradation over poly(lactic acid), but gentle encapsulation. [219] Moreover, using a poly(ether ester) multiblock copolymer microsphere, a Phase 2 clinical trial was performed for delivery of IFN-α (Locteron) in patients with chronic hepatitis C genotype 1.[220] This system aimed to deliver unmodified IFN-α for prolonged periods to compete with the PEG-IFN-α versions which led to significant reductions in cytokine bioactivity after PEGylation. Although the Phase 2 studies showed equal efficacy and lower adverse effects from two week dosing of Locteron over weekly PEG-IFN-α, the therapy never reached Phase 3 clinical trials, likely due to failure of the developing company.[221]
More recently, designs have focused on combination therapy approaches. For example, using IL-12 and GM-CSF encapsulated into PLGA microspheres, 44% of FVBneuN mice with advanced spontaneous mammary tumors were completely cured if cyclophosphamide (CY) was administered one day before cytokine treatment compared to 0% when only CY or microsphere-encapsulated cytokines were administered.[222] The cytotoxic CY chemotherapy was chosen due to its cytotoxic activity in Tregs which would enable a better anti-tumor immune response from the cytokine therapy. Notably, it was shown that the treatment yielded a 7-fold increase in CD8+ cytotoxic T cell (CTL) to Treg ratio, 3-fold increase in CTL cytotoxicity and extended the effector window for CD8+ T cell from 3 to 7 days. [222] In addition to immunochemotherapy approaches, the prolonged cytokine delivery from microspheres can be used in conjunction with radiation. Notably, the combination of stereotactic body radiation therapy (SBRT) with intratumorally administered IL-12 encapsulated in PLGA microspheres was capable of eliciting cures in preclinical mouse models of pancreatic ductal adenocarcinoma with abscopal effect.[223] In this study, SBRT was used to induce immune infiltration which, with the local and sustained IL-12 delivery, enabled the antitumor immune response.
Beyond simple slow-release matrices, more intricate delivery systems such as bio-responsive microspheres have also been developed. In depot formulations, these systems aim to have release of the therapeutic cytokine based on the degree of severity of the disease. For example, microspheres created by crosslinking of gelatin with genipin (a natural small molecule amine-amine crosslinker) yields negatively charged particles of ~10 μm on which positively charged cytokines such as IL-4, IL-10, and IL-13 are electrostatically bound for treatment of osteoarthritis (Figure 7).[224] Their bio-responsiveness originates from the degradability of the microsphere when exposed to enzymes characteristically expressed in osteoarthritis. Accordingly, the cytokine release rate is linearly correlated with the concentration of enzymes in solution.
Figure 7.

Bioresponsive polymeric microparticles for anti-inflammatory cytokine delivery to osteoarthritis. Reproduced with permission.[224] Copyright 2019, Wiley Periodicals, Inc.
Although most studies have been based on spherical microparticles, these need not be the only effective cytokine delivery vehicle; the shape is another physical property that can alter resulting in vivo effects. For example, soft discoidal microparticles were shown to provide prolonged in vivo cytokine exposure to phagocytic cell such as macrophages.[225] In this study, the discoidal microparticles were composed of an initial cell-adhesion layer followed by polyvinyl alcohol (PVA) layer in between PLGA layers. The hydrophilic PVA layer allowed for cytokine loading and PLGA provided structural support. The adhesive layer was synthesized via layer-by-layer assembly comprised of alternating layers of hyaluronic acid modified with aldehyde and poly(allylamine). This polyelectrolyte multilayer was designed to adhere to isolated macrophage cells. When loaded with IFN-γ, these cellular ‘backpacks’ were shown to alter macrophages to their antitumor phenotype (M1) in vitro and, more importantly, prevent differentiation from the M1 phenotype deep within the immunosuppressive environment of solid tumors from mouse 4T1 breast cancer model when delivered intratumorally.[225] Moreover, this treatment was shown to reprogram tumor-associated macrophages to the antitumor phenotype. Accordingly, treatment with the backpack-IFN-γ-loaded macrophages could slow tumor growth and reduce metastasis. Interestingly, the delivery of drug-loaded polymeric backpacks via monocytes has been previously shown to target inflamed tissues and may enable drug delivery further into the tumor cores.[226]
Although there has been great progress in the development of microparticles for drug delivery, they have not led to clinical translation of cytokine therapeutic. Some of the major limitations include ineffective drug loading, complex manufacturing, loss of cytokine bioactivity, biocompatibility and regulations.[219,227] However, these issues have begun to be addressed demonstrating in-vivo proof-of-concept of the effectiveness of these delivery vehicles.
6.4. Nanoparticles
Nanoparticles are small (typically 10–100 nm) particles that, due to their size, have special biophysical properties that enable the therapeutic enhancement of various drug molecules. Notably, nanoparticles have been extensively used to alter the pharmacokinetics and toxicity of drug agents, making them promising candidates for cytokine delivery.[10] Some of their benefits include increases in circulatory half-life and controlled drug release and/or activation. Moreover, either through the EPR effect or other properties, they can be used as targeting systems to the desired tissue or cell for targeted drug delivery. These benefits can be achieved through a variety of modifications including the material composition, charge, shape, and flexibility.[228] For example, nanoparticles in the ~10–100 nm size range show preferential accumulation in lymph nodes following parenteral administration[10]. Furthermore, surface modifications such as PEGylation or conjugation of targeting motifs are also commonly employed to enhance the therapeutic efficacy of the loaded drug as they can prevent unwanted interactions or target certain interactions, respectively. Lastly, especially in the delivery of cytokines, nanoparticles are amenable for drug delivery either through encapsulation within the particle core or attached to the particle surface. As will be presented, in cytokine delivery, this surface attachment allows for presentation of cytokines to their receptors while retaining pharmacokinetics governed by the particle itself. Over the years, there have been many different classes of nanoparticles developed for cytokine delivery. In this section, we will focus on lipid-based nanoparticles and polymeric nanoparticles which have been the most widely studied classes of nanoparticles for cytokine delivery.
Notably, the approved formulation of IL-2 (aldesleukin) can be considered the first cytokine nanoparticle to reach the clinic. Aldeslueukin contains nanometer-sized (11–13 nm) ‘microaggregates’ composed of an average of 27 non-covalently bound IL-2 molecules complexed with sodium-dodecyl sulfate (SDS).[229] Initially described as a surfactant to solubilize the hydrophobic IL-2 molecules which, based on its FDA labeling, “may have an effect on the kinetic properties” of aldesleukin,[230] it was later disclosed that the concentration and formulation process of the SDS detergent were vital for the therapeutic efficacy of IL-2 due to the formation of these ‘microaggregates’.[229] Importantly, the microaggregate formulation does not alter bioactivity of IL-2, but significantly alters the pharmacokinetics and biodistribution compared to monomeric IL-2. In rats, the clearance rate of IL-2 decreased ~30-fold, and the in vivo distribution altered from mainly the kidneys to lung, liver, and kidney. [229] Accordingly, when administered into animals, IL-2 microaggregates had superior efficacy at treating preventing lung metastasis compared to the monomeric cytokine. Importantly, this observation demonstrates the clinical potential of nanoparticle formulations for increased therapeutic efficacy. Moreover, likely due to the late disclosure, there have not been detailed studies on these IL-2 microaggregates in the literature and most IL-2 studies fail to discuss their significance. However, due to the altered clinical properties, new IL-2 therapeutics should be compared to the approved SDS/IL-2 microaggregates and not monomeric or other IL-2 formulations.
There are many techniques to synthesize lipid-based nanoparticles with controlled size and size distribution, making them attractive vehicles for nanomedicine. Notably, liposomes have been one of the most studies class nanoparticles for drug delivery, making them the most common class of FDA-approved nanoparticles.[231] As discussed in the Microparticles section, early formulations of lipid-based particles were typically microns in size which, although useful as a depot, have very short circulatory half-lives due to rapid RES capture. [210] Accordingly, smaller liposome formulations were developed using PEG-lipids for even longer circulatory half-life. In an early example of employing liposomes for cytokine delivery, PEGylated ‘stealth’ liposomes were used to encapsulate IL-2, increasing the protein stability and its plasma half-life by 10–30 times over free IL-2, and eliciting superior anti-tumor effect compared to free IL-2 in combination with chemotherapy. [210]
Since these promising developments, many preclinical studies have pursued liposomal cytokine delivery in nanoparticle formulation. For example, attachment of IL-2-Fc fusion protein and anti-CD137 antibodies to liposomes (Figure 8A) enabled systemic administration of this potent combination therapy, eliciting an effective antitumor immune response with minimal toxicity.[232] In this design, the conjugated IL-2-Fc maintained its in vitro activity while conjugated anti-CD137 antibodies showed enhanced immune stimulation over the soluble antibody (Figure 8B-D). Further, the conjugated proteins had reduced circulatory half-life as the EPR effect concentrated the nanoparticle in the tumor, lowering the overall systemic exposure in circulation to the immunostimulant. Notably, an earlier study showed that intratumoral administration of the free immunomodulatory agents was too toxic for therapeutic use, corroborating the importance of anchoring the cytokines to the liposomes to prevent systemic exposure.[233]
Figure 8.

Synthesis and bioactivity of liposomes surface conjugated with anti-CD137 antibody or IL2-Fc fusion protein. (A) schematic for synthesis of liposomes. (B) Flow cytometry of CD4+ and CD8+ T cells incubated with fluorescently-labeled liposomes (solid), unconjugated liposomes (dashed), or no liposomes (grey area). (C) In vitro T cell proliferation normalized to unstimulated cells. (D) IFN-γ production by polyclonal T cells. Reproduced with permission.[233] Copyright 2013, American Association for Cancer Research.
Notably, the surface attachment of cytokines in liposomes may be combined with the external coating of the nanoparticle via the layer-by-layer technique. This approach can reduce exposure of the immunostimulatory protein in circulation but maintain immune activation at the desired tumor site.[234] Accordingly, when negatively charged liposomes were surface conjugated with IL-12 followed by deposition of a positively charged poly-L-arginine (PLR) and a terminal negative poly-L-glutamic acid (PLE) layers (Figure 9), the IL-12 therapy showed reduced toxicity without loss of therapeutic efficacy in mouse models of ovarian (OV2944-HM-1) and colon (MC38) cancer when delivered intratumorally. In this system, the outer PLE layer was chosen due to its affinity for cancer cells with localization on the outer cellular membrane. Importantly, the modular nature of the layer-by-layer assembly tune the nanoparticle characteristics for improved therapeutic efficacy and equips the carrier for staged-release of combination treatments.[235]
Figure 9.

Cancer-cell targeting liposomal layer-by-layer nanoparticle containing surface conjugated IL-12. (A) Diagram for assembly of nanoparticle. (B) Cancer cell association and subsequent targeted immune activation. Adapted with permission.[234] Copyright 2020, American Chemical Society.
However, although liposomes have low toxicity and low immunogenicity, efficient protein encapsulation in liposomes can be challenging, and release rates of encapsulated therapeutics can be difficult to tailor. Accordingly, other nanoparticle formulations have been developed that aim to address these limitations.
Polymeric nanoparticles are a second important class of materials explored for cytokine delivery. In these systems, polymer composition dictates the drug release rate which is typically longer than that of liposome-based nanoparticles. One example of PLGA-modification for enhanced encapsulation is the use of avidin-conjugated PLGA nanoparticles for co-encapsulation of IL-2 and TGF-β.[236] The avidin conjugation is used to both facilitate encapsulation of hydrophilic drugs as all as enable for surface conjugation of the PLGA nanoparticles as the protein is primarily present within the particle surface. In the IL-2 and TGF-β example, the avidin was used to conjugate biotin-anti-CD4 antibodies. This CD4+ T cell-targeting construct was used to induce Treg differentiation mediated by the TGF-β followed by expansion of these regulatory cells via the IL-2. Accordingly, when administered in vivo these nanoparticles generated stable and functional Tregs for potential treatment of autoimmune disorders. Other approaches for chemoimmunotherapy using PLGA-based nanoparticle-mediated cytokine delivery include the use of “thermosponge” nanoparticles formulated from PEG-PLGA, PLGA, and F-127 gel. [237] In this system the hydrophobic PLGA core was used to load paclitaxel while a hydrophilic outer themosensitive F-127 shell was used for facile entrapment of IL-2 in an aqueous environment.[237]
Although PLGA is an attractive polymer for nanoparticles due to the history of use in FDA approved products, other polymers may be used with additional benefits. An example is pH-sensitive poly(β-amino ester)-based carriers that can exploit the lower pH of the tumor microenvironment for targeted release.[238] Using such a system for IL-12 delivery was shown to have anti-tumoral effects in B16F10 mouse model without signs of significant toxicity. [238] Notably, the antitumoral response was similar when the particles were injected i.t. or i.v. and attributable to the reprograming of tumor-associated macrophages from an M2 to M1 phenotype which is indicative of an anti-tumor immune response.
Importantly, the hydrophobicity of polymer-based nanoparticles can make encapsulation of hydrophilic proteins such as cytokines difficult. This challenge has prompted the development of nanogel-based delivery systems– hydrogels in the form of nano-sized particles. An interesting example of this class of nanomaterial employed reducible protein-based nanogels to create TCR-signaling-responsive delivery of IL-15 superagonist (N-803) to T cells during adoptive T cell therapy.[239] Notably, instead of using a polymer chain to yield the nanogels and carry the drug load, this design used a reducible small-molecule crosslinker connecting the IL-15 protein cargo itself to form the nanogel. Accordingly, compared to a traditional approach of encapsulating drug inside a lipid or polymeric nanoparticle, there was marked increase in drug loading. Moreover, by using a disulfide-containing cross-linkers, the nanogels had T cell activation responsive drug release due to the increased reduction rate of antigen-activated T cells over naïve T cells. Lastly, the nanogel was surface conjugated with PEG-b-poly(L-lysine) and anti-CD45 antibodies. The polymers provided a uniform positive charge that promoted an initial binding to cellular membrane while the anti-CD45 antibodies prevented intracellular uptake of the nanoparticles. Using this construct, a 16-fold increase in tumor T cell expansion was observed selectively in tumors, which enabled an increase in the therapeutic window of IL-15 superagonist adjuvant delivery in adoptive T cell therapy. [239]
There are many alternative nanoparticle formulations that may be used for cytokine delivery. Notably, the use of hybrid systems can enable for better controlled release of combination therapies by exploiting the properties of different materials. For example, the use of liposomal nanogels (nLG) enabled the co-delivery delivery of hydrophilic IL-2 hydrophilic and a hydrophobic small molecule TGF-β inhibitor while also incorporating the advantageous pharmacokinetic properties of PEGylated liposomes (Figure 10).[240] The liposomes also served as the molds for photo-crosslinking of the polymers to yield the hydrogel core which contained both IL-2 and the TGF-β inhibitor solubilized within methacrylate-conjugated β-cyclodextrins (Figure 10A). Using the combination therapy within the nLG for intratumoral treatment of metastatic melanoma, there was a marked increase in overall survival and reduction in tumor growth with 40% of animals having complete tumor regression (Figure 10B-D).[240] Importantly, neither the agents alone, their combination, nor the single agents in the nanoparticles yielded similar therapeutic efficacy. Moreover, a significant improvement in overall survival was also seen in when the combination therapy nLG were administered systemically (i.v.) with reduction in lung metastatic tumor burden.
Figure 10.

Schematic for synthesis of liposomal nanogel encapsulating a TGF-β inhibitor and IL-2 and its effects when intratumorally administered to a subcutaneous mouse metastatic melanoma model. (a) Components and final liposomal nanogel assembly. (b) Plot of tumor area versus time (day 0 was day of tumor inoculation). Red arrows indicate treatment. (c) Tumor masses after 7 days of treatment. (d) Survival plot of animals in (b). Complete tumor regression and survival was obtained in 40% of the group after 60 days (data not shown). Adapted with permission.[240] Copyright 2012, Nature Publishing Group.
Beyond degradable polymers and lipids, many other nanomaterials may be used for immunotherapy that enable different mechanisms to improve the therapeutic efficacy of cytokines. [6] For example, dimercaptosuccinic acid (DMSA)-coated magnetite nanoparticles may be used for magnetically guided delivery of cytokines.[241] The DMSA serves as a functional group for cytokine conjugation while the iron-based nanoparticle can have its biodistribution altered to accumulate in tumors via application of an external magnetic field.
Importantly, although nanoparticles have a highly modular assembly with many potential modifications, their clinical utility has been limited with no current clinical trials aiming at cytokine delivery.[231] This limited clinical success may be attributed to low targeting efficiency, manufacturing cost and complexity, and the currently limited design-space exploration.[153,242] Much of the fundamental understanding of physical parameters (e.g., size, shape, degradability, elasticity) still must be elucidated.[6] However, as demonstrated by the various examples presented here, nanoparticles have shown to be effective carriers for improvement of cytokine-based immunotherapy. Moreover, many of the other engineering strategies presented in this review can be combined with nanoparticle delivery as shown by Fc-fusion proteins or the antibody-mediated targeting. Accordingly, nanoparticles present themselves as one of the most promising approaches to enable future clinical translation of cytokines.
7. Other Cytokine Delivery Strategies
Other major classes of cytokine delivery strategies used in clinical trials have been mediated by advancements in genetic engineering. Initially, cytokine-secreting tumor cells were transfected ex-vivo then administered as a cancer vaccine, but had limited clinical success.[12] More recently, other cells such as mesenchymal stem cells (MSCs), dendritic cells, tumor infiltrating lymphocytes (TILs) and, chimeric antigen receptor T cell (CAR-T) have also been genetically modified for cytokine production with some clinical success.[14,243] However, cell-based therapies are still in early stages of clinical translation and have unique manufacturing and regulatory challenges prior to potential wide-scale use.[244]
One way to circumvent the limitations of cell-based therapies is use of gene therapy for in vivo transfection. However, gene therapy has been limited by efficient transfection of mammalian cells in vivo and control of dosing based on gene transcription[13]. Further, unlike the delivery of extracellular-targeting cytokine proteins, gene therapy requires the cargo to be intracellularly delivered for cytokine expression and subsequent secretion. Accordingly, there are many challenges with use of these system to which various technologies have been developed including viral and non-viral vectors systems as well as physical methods of gene insertion.[15] Although these three major gene therapy modalities have candidates in clinical trials, only an oncolytic viral vector encoding for GM-CSF has been approved.[245–248] The modified virus, termed Talimogene laherparepvec (T-VEC), yielded significant and durable improvements when intratumorally delivered in advanced melanoma patients, making T-VEC the first oncolytic virus to obtain approval in 2015.[248]
8. Conclusions and Future in Cytokine Therapeutics
As detailed in this review, there have been many engineering strategies designed to improve the therapeutic applications of cytokines. These systems have mainly attempted to modulate the pharmacokinetics and pharmacodynamics to elicit a targeted effect and, therefore, reduce overall toxicity. Most designs have aimed at the localized delivery of cytokines as systemic exposure to the therapeutic cause dose-limiting toxicities. Importantly, these localized treatments have only recently garnered acceptance in clinical and surgical oncology as intralesional treatment was not always possible and surgical removal of primary lesions were prioritized over drug treatments.[249] Further, reluctance existed due to concerns that local injection could aid in metastasis through the needle track and have minimal effect on distal sites due to low systemic drug levels.[249] More recently, with technological developments and various clinical studies demonstrating the benefits of intralesional treatments, these hesitancies have greatly decreased.[249,250] However, the challenge in demonstrating systemic effects beyond homogenous tumors from preclinical animal models remains. Accordingly, as new fusion protein and nanoparticle designs have continued to show promising preclinical data, systemically administered cytokine therapeutics may find their way into clinical applications.
As these designs are developed, it will become important to fully understand their mechanisms to modulate therapeutic effect and toxicity. For example, even intratumoral injection does not prevent systemic leakage of cytokines.[251] Therefore, targeted or locally delivery strategies need to monitor the systemic levels of characteristic cytokines potentially released into circulation.[251] Unfortunately, many preclinical studies have failed to monitor for systemic leakage of the cytokine or its downstream induced signals. Further, protein engineering strategies and biomaterials-based approaches need to ensure that processing of a cytokine or its modifications are carefully characterized, to understand potential undesirable changes or losses in cytokine function. The sophisticated chemistry protocols required for these designs may also hinder manufacturing at the clinical scale and require powerful analytical tools to assess product quality.
Design considerations for clinical translation will also need to account for the pharmaceutical formulation challenges required to maintain drug stability in storage and their ease of administration. Optimization of protein formulation can be complex with additional regulatory complexity for introduction of any new excipient. Implementation of new delivery vehicles further complicates the problem as the vehicle’s structure and composition also needs to be properly maintained. For example, lyophilization is employed in some approved cytokine formulations to maintain product stability, but could lead to changes in the physicochemical properties of liposome delivery systems.[252,253]
In addition to single-agent treatments, one of the more promising areas for cytokine therapeutics are combination therapies. These include use of cytokines as adjuvants for vaccines, CPIs, surgeries, chemotherapy or radiation therapy. Moreover, cytokine combinations with other immunostimulatory agents can also enable successful therapies. One example is the approval of dinutuximab in high-risk neuroblastoma in 2015 in which the anti-disialoganglioside GD2 antibody is combined with GM-CSF, IL-2 and isotretinoin. [254] Importantly, from a pharmaceutical perspective, the administration of two therapeutic agents may be cumbersome. Thus, fusion proteins or multi-therapeutic delivery systems to reduce toxicity ease the introduction of these combinations into the clinic. Moreover, whether in combination therapies or monotherapies, targeted delivery systems are demonstrating to be the necessary characteristics to mitigate the toxicities associated with cytokine delivery. Unfortunately, as the therapeutics become more complex, the more critical quality attributes are necessary for manufacturing, which delay development and increase costs of treatment.
In addition to the engineering design of the drug, the route and schedule of administration are major factors in the therapeutic efficacy. Variations in the administration route can dictate biodistributions, half-lives, and bioavailability.[255] Moreover, different administration schedules can elicit different immune responses due to varied concentration and duration of exposure of the immune cells to the therapeutics.[255] In the case of immunocytokines, there is initial clinical evidence that lower toxicities may be observed when administered at longer infusion rates, where the blood levels are sustained at a lower level.[68] These characteristics may be even more important in combination therapies as it has been shown that the sequence of combination therapies can be vital for their success. [256] For example, the administration IL-2 before IFN-α leads to severe toxicities that can be prevented via concurrent or after IFN-α.[257] These are crucial characteristics that need to be considered and better understood in the design of controlled release systems for multiple immunostimulatory agents. The sequence is also important when immunotherapy is used in combination with surgeries as recent reports indicate that improved efficacy is seen in a neoadjuvant-based cytokine schedule. [258]
Although the unfavorable pharmacokinetic profile of cytokine therapeutics is a major hindrance for their clinical translation, the failure of Albuferon signals another crucial consideration in development of cytokine therapies: they must be more tolerable or more efficacious than current therapies. Mere improvement in the dosing convenience will be difficult to justify regulatory approval, especially if the new therapy has an increase in adverse events. [84] Moreover, modifications that aim to alter the pharmacokinetics need to be carefully designed as they can also alter the biodistribution, potentially reducing the therapeutic effect of the cytokine.
Overall, there are various engineering strategies for cytokine-based therapeutics. Many of these have had successes in preclinical and early clinical studies; however, full implementation of cytokines in the clinic will require a better understanding of immunology and how these designs modulate it to better develop system that can effectively mimic or manipulate immune responses.
Acknowledgments
The authors thank Felice Frankel for her aid in designing figures. This work was supported by the U.S. Department of Defense Congressionally Directed Medical Research Programs (PTH, Teal Innovator Award W81XWH13-1-0151); the National Institutes of Health (PTH, DJI, Project# 1-R01- CA235375-01A1); the Bridge Project - a partnership between the Koch Institute for Integrative Cancer Research at MIT and the Dana-Farber/Harvard Cancer Center; and the Marble Center for Cancer Nanomedicine.
Cytokines have been extensively researched as immunomodulatory therapeutic proteins for a diverse set of diseases. This review briefly describes cytokines and their clinical history, followed by a deeper discussion on the engineering strategies developed improve their therapeutic efficacy. Focus is given to the clinical utility and perspectives of protein engineering, bioconjugation and biomaterials-based approaches for cytokine delivery.
BIOGRAPHIES:

Ivan S. Pires obtained his bachelor’s degree in Chemical Engineering with Honors from The Ohio State University. He is currently pursuing his Ph.D. in Chemical Engineering under the join supervision of Prof. Paula T. Hammond and Darrell J. Irvine. His research focuses on the design of electrostatically assembled layer-by-layer nanoparticles for cancer immunotherapy.

Paula T. Hammond is the David H. Koch Chair Professor of Engineering at the Massachusetts Institute of Technology, and the Head of the Department of Chemical Engineering. She is a member of MIT’s Koch Institute for Integrative Cancer Research, the MIT Energy Initiative, and a founding member of the MIT Institute for Soldier Nanotechnology. The core of her work is the use of electrostatics and other complementary interactions to generate functional materials with highly controlled architecture. Her research in nanomedicine encompasses the development of new biomaterials to enable drug delivery from surfaces with targeted spatio-temporal control.

Darrell J. Irvine is presently a Professor at the Massachusetts Institute of Technology and an Investigator of the Howard Hughes Medical Institute. He is also an Associate Director for the Koch Institute for Integrative Cancer Research at MIT and serves on the steering committee of the Ragon Institute of MGH, MIT, and Harvard. His research aims to engineer immunity through a fusion of immunology with biotechnology and materials chemistry, employing an engineering-centric approach to create new therapies based on the controlled modulation of the immune system. His lab has two primary research foci, developing technologies to enhance vaccine-elicited immunity and engineering cancer immunotherapies.
Contributor Information
Ivan S. Pires, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, 500 Main Street, Cambridge, Massachusetts 02142, United States Department of Chemical Engineering, Massachusetts Institute of Technology, 25 Ames Street, Cambridge, Massachusetts 02142, United States.
Prof. Paula T. Hammond, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, 500 Main Street, Cambridge, Massachusetts 02142, United States Department of Chemical Engineering, Massachusetts Institute of Technology, 25 Ames Street, Cambridge, Massachusetts 02142, United States; Institute for Soldier Nanotechnologies, Massachusetts Institute of Technology, 500 Technology Square, Cambridge, Massachusetts 02139, United States.
Prof. Darrell J. Irvine, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, 500 Main Street, Cambridge, Massachusetts 02142, United States Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA; Department of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA; Ragon Institute of Massachusetts General Hospital, Massachusetts Institute of Technology and Harvard University, Cambridge, MA 02139, USA; Howard Hughes Medical Institute, Chevy Chase, MD 20815, USA.
REFERENCES
- [1].Cohen S, Bigazzi PE, Yoshida T, Similarities of T cell function in cell-mediated immunity and antibody production, Vol. 12, Academic Press, 1974, pp. 150–159. [DOI] [PubMed] [Google Scholar]
- [2].Kronfol Z, Cytokines and mental health, Kluwer Academic Pub, 2003. [Google Scholar]
- [3].Caligiuri MA, Lotze MT, Cytokines in the genesis and treatment of cancer, Humana Press, 2007. [Google Scholar]
- [4].Wang C, Ye Y, Hu Q, Bellotti A, Gu Z, Tailoring Biomaterials for Cancer Immunotherapy: Emerging Trends and Future Outlook, Vol. 29, Wiley-VCH Verlag, 2017. [DOI] [PubMed] [Google Scholar]
- [5].Riley RS, June CH, Langer R, Mitchell MJ, Nat. Rev. Drug Discov 2019, 18, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shields CW, Wang LL, Evans MA, Mitragotri S, Adv. Mater 2020, 32, 1901633. [DOI] [PubMed] [Google Scholar]
- [7].Leach DG, Young S, Hartgerink JD, Acta Biomater. 2019, 88, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yang F, Shi K, peng Jia Y, Hao Y, rong Peng J, yong Qian Z, Advanced biomaterials for cancer immunotherapy, Springer Nature, 2020, pp. 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Goldberg MS, Improving cancer immunotherapy through nanotechnology, Vol. 19, Nature Publishing Group, 2019, pp. 587–602. [DOI] [PubMed] [Google Scholar]
- [10].Irvine DJ, Dane EL, Enhancing cancer immunotherapy with nanomedicine, Nature Research, 2020, pp. 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dellacherie MO, Seo BR, Mooney DJ, Macroscale biomaterials strategies for local immunomodulation, Vol. 4, Nature Publishing Group, 2019, pp. 379–397. [Google Scholar]
- [12].Tepper RI, Mulé JJ, Hum. Gene Ther 1994, 5, 153. [DOI] [PubMed] [Google Scholar]
- [13].Wysocki PJ, Karczewska-Dzionk A, Mackiewicz-Wysocka M, Mackiewicz A, Expert Opin. Biol. Ther 2004, 4, 1595. [DOI] [PubMed] [Google Scholar]
- [14].Dwyer CJ, Knochelmann HM, Smith AS, Wyatt MM, Rangel Rivera GO, Arhontoulis DC, Bartee E, Li Z, Rubinstein MP, Paulos CM, Front. Immunol 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bono N, Ponti F, Mantovani D, Candiani G, Pharmaceutics 2020, 12, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shirley SA, In Handbook of Electroporation, Springer International Publishing, 2017, pp. 1755–1768. [Google Scholar]
- [17].Sung YK, Kim SW, Recent advances in the development of gene delivery systems, Vol. 23, BioMed Central Ltd., 2019, pp. 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Liu L, Wang S, Shan B, Sang M, Liu S, Wang G, Advances in viral-vector systemic cytokine gene therapy against cancer, Vol. 28, Elsevier, 2010, pp. 3883–3887. [DOI] [PubMed] [Google Scholar]
- [19].Feldmann M, J. Clin. Invest 2008, 118, 3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Paul WE, Fundamental immunology, Wolters Kluwer/Lippincott Williams & Wilkins, 2008. [Google Scholar]
- [21].Morán GAG, Parra-Medina R, Cardona AG, Quintero-Ronderos P, Rodríguez ÉG, In Autoimmunity: From Bench to Bedside [Internet] (Eds.: Anaya J-M; Shoenfeld Y; Rojas-Villarraga A; Levy RA; Cervera R), El Rosario University Press, Bogota, Colombia, 2013. [PubMed] [Google Scholar]
- [22].Horowitz MC, The role of cytokines in bone remodeling, Vol. 1, Humana Press, 1998, pp. 187–198. [Google Scholar]
- [23].Turner MD, Nedjai B, Hurst T, Pennington DJ, Biochim. Biophys. Acta - Mol. Cell Res 2014, 1843, 2563. [DOI] [PubMed] [Google Scholar]
- [24].Zhang J-M, An J, Int. Anesthesiol. Clin 2007, 45, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Setrerrahmane S, Xu H, Tumor-related interleukins: Old validated targets for new anti-cancer drug development, Vol. 16, BioMed Central Ltd., 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Esquivel-Velázquez M, Ostoa-Saloma P, Palacios-Arreola MI, Nava-Castro KE, Castro JI, Morales-Montor J, Interf J. Cytokine Res. 2015, 35, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chung KF, In Asthma and COPD, Elsevier Ltd, 2009, pp. 327–341. [Google Scholar]
- [28].Cavaillon JM, Cell. Mol. Biol. (Noisy-le-grand) 2001, 47, 695. [PubMed] [Google Scholar]
- [29].Dinarello CA, Eur. J. Immunol 2007, 37, S34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lucey DR, Clerici M, Shearer GM, Clin. Microbiol. Rev 1996, 9, 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Vignali DAA, Collison LW, Workman CJ, Nat. Rev. Immunol 2008, 8, 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sompayrac L, How the immune system works, 6th ed., Wiley-Blackwell, 2019. [Google Scholar]
- [33].Lund FE, Randall TD, Nat. Rev. Immunol 2010, 10, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Baggiolini M, Clark-Lewis I, Interleukin-8, a chemotactic and inflammatory cytokine, Vol. 307, 1992, pp. 97–101. [DOI] [PubMed] [Google Scholar]
- [35].Sag D, Ayyildiz ZO, Gunalp S, Wingender G, Cancers (Basel). 2019, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kelm NE, Zhu Z, Ding VA, Xiao H, Wakefield MR, Bai Q, Fang Y, The role of IL-29 in immunity and cancer, Vol. 106, Elsevier Ireland Ltd, 2016, pp. 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kosmaczewska A, Low-dose interleukin-2 therapy: A driver of an imbalance between immune tolerance and autoimmunity, Vol. 15, MDPI AG, 2014, pp. 18574–18592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Asadullah K, Sterry W, Volk HD, Interleukin-10 therapy - Review of a new approach, Vol. 55, American Society for Pharmacology and Experimental Therapy, 2003, pp. 241–269. [DOI] [PubMed] [Google Scholar]
- [39].Dayer JM, From supernatants to cytokines: A personal view on the early history of IL-1, IL-1Ra, TNF and its inhibitor in rheumatology, Vol. 20, BioMed Central Ltd., 2018, p. 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zinsser H, Tamiya T, J. Exp. Med 1926, 44, 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Vilček J, Feldmann M, Trends Pharmacol. Sci 2004, 25, 201. [DOI] [PubMed] [Google Scholar]
- [42].Gresser I, On the varied biologic effects of interferon, Vol. 34, Academic Press, 1977, pp. 406–415. [DOI] [PubMed] [Google Scholar]
- [43].Ningrum RA, Scientifica (Cairo). 2014, 2014. [Google Scholar]
- [44].Pestka S, Krause CD, Walter MR, Immunol. Rev 2004, 202, 8. [DOI] [PubMed] [Google Scholar]
- [45].Jakimovski D, Kolb C, Ramanathan M, Zivadinov R, Weinstock-Guttman B, Interferon β for Multiple Sclerosis, Vol. 8, NLM (Medline), 2018, p. a032003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Spada S, Walsh G, Directory of Approved Biopharmaceutical Products, 1st ed., CRC PRESS, 2019. [Google Scholar]
- [47].Eggermont AMM, de Wilt JHW, ten Hagen TLM, Current uses of isolated limb perfusion in the clinic and a model system for new strategies, Vol. 4, Lancet Publishing Group, 2003, pp. 429–437. [DOI] [PubMed] [Google Scholar]
- [48].van Horssen R, ten Hagen TLM, Eggermont AMM, Oncologist 2006, 11, 397. [DOI] [PubMed] [Google Scholar]
- [49].Clement J, McDermott D, Clin. Genitourin. Cancer 2009, 7, E7. [DOI] [PubMed] [Google Scholar]
- [50].National Institute of Diabetes and Digestive and Kidney Diseases, Oprelvekin, National Institute of Diabetes and Digestive and Kidney Diseases, 2012. [Google Scholar]
- [51].Sylvester RK, Am. J. Heal. Pharm 2002, 59, S6. [DOI] [PubMed] [Google Scholar]
- [52].Fishbane S, Semin. Dial 2006, 19, 1. [DOI] [PubMed] [Google Scholar]
- [53].Blau CA, Stem Cells 2007, 25, 2094. [DOI] [PubMed] [Google Scholar]
- [54].Prometheus Laboratories, FDA, PROLEUKIN ® (aldesleukin) Injection Label, 2012. [Google Scholar]
- [55].Amin A, White RL, J. Kidney Cancer VHL 2014, 1, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Metcalf D, The colony-stimulating factors and cancer, Vol. 10, Nature Publishing Group, 2010, pp. 425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Berraondo P, Sanmamed MF, Ochoa MC, Etxeberria I, Aznar MA, Pérez-Gracia JL, Rodríguez-Ruiz ME, Ponz-Sarvise M, Castañón E, Melero I, Br. J. Cancer 2019, 120, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Galon J, Bruni D, Approaches to treat immune hot, altered and cold tumours with combination immunotherapies, Vol. 18, Nature Publishing Group, 2019, pp. 197–218. [DOI] [PubMed] [Google Scholar]
- [59].Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M, Wound Repair Regen. 2008, 16, 585. [DOI] [PubMed] [Google Scholar]
- [60].Fent K, Zbinden G, Trends Pharmacol. Sci 1987, 8, 100. [Google Scholar]
- [61].Sleijfer S, Bannink M, Van Gool AR, Kruit WHJ, Stoter G, Side effects of interferon-α therapy, Vol. 27, Springer, 2005, pp. 423–431. [DOI] [PubMed] [Google Scholar]
- [62].Smith K, Blood 1993, 81, 1414. [PubMed] [Google Scholar]
- [63].Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, Sosman JA, Dutcher JP, Vogelzang NJ, Ryan JL, Blood 1997, 90, 2541. [PubMed] [Google Scholar]
- [64].Lasek W, Zagożdżon R, Jakobisiak M, Cancer Immunol. Immunother 2014, 63, 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Baluna RG, In Cytokines in Human Health, Humana Press, Totowa, NJ, 2007, pp. 205–231. [Google Scholar]
- [66].Orucevic A, Lala PK, Role of nitric oxide in IL-2 therapy-induced capillary leak syndrome, Vol. 17, Kluwer Academic Publishers, 1998, pp. 127–142. [DOI] [PubMed] [Google Scholar]
- [67].Siegel JP, Puri RK, J. Clin. Oncol 1991, 9, 694. [DOI] [PubMed] [Google Scholar]
- [68].Hutmacher C, Neri D, Antibody-cytokine fusion proteins: Biopharmaceuticals with immunomodulatory properties for cancer therapy, Vol. 141, Elsevier B.V., 2019, pp. 67–91. [DOI] [PubMed] [Google Scholar]
- [69].U.S. Food and Drug Administration, Proleukin Toxicologist’s Review (PLA# 97–0501), 1998. [Google Scholar]
- [70].Lotze MT, Matory YL, Ettinghausen SE, Rayner AA, Sharrow SO, Seipp CA, Custer MC, Rosenberg SA, J. Immunol 1985, 135, 2865. [PubMed] [Google Scholar]
- [71].Zhu EF, Gai SA, Opel CF, Kwan BH, Surana R, Mihm MC, Kauke MJ, Moynihan KD, Angelini A, Williams RT, Stephan MT, Kim JS, Yaffe MB, Irvine DJ, Weiner LM, Dranoff G, Wittrup KD, Cancer Cell 2015, 27, 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Moynihan KD, Opel CF, Szeto GL, Tzeng A, Zhu EF, Engreitz JM, Williams RT, Rakhra K, Zhang MH, Rothschilds AM, Kumari S, Kelly RL, Kwan BH, Abraham W, Hu K, Mehta NK, Kauke MJ, Suh H, Cochran JR, Lauffenburger DA, Wittrup KD, Irvine DJ, Nat. Med 2016, 22, 1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Mestas J, Hughes CCW, J. Immunol 2004, 172, 2731. [DOI] [PubMed] [Google Scholar]
- [74].Ethuin F, Gérard B, Benna JE, Boutten A, Gougereot-Pocidalo MA, Jacob L, Chollet-Martin S, Lab. Investig 2004, 84, 1363. [DOI] [PubMed] [Google Scholar]
- [75].Li J, Gyorffy S, Lee S, Kwok CS, Inflammation 1996, 20, 361. [DOI] [PubMed] [Google Scholar]
- [76].Assier E, Jullien V, Lefort J, Moreau JL, Vargaftig BB, Jose Lapa JLE, Thèze J, Cytokine 2005, 32, 280. [DOI] [PubMed] [Google Scholar]
- [77].Perez Horta Z, Saseedhar S, Rakhmilevich AL, Carmichael L, Hank JA, Boyden M, Gillies SD, Sondel PM, Oncoimmunology 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Attarwala H, J. Young Pharm 2010, 2, 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Haicheur N, Escudier B, Dorval T, Negrier S, De Mulder PHM, Dupuy JM, Novick D, Guilloth T, Wolf S, Pouillart P, Fridman WH, Tartour E, Clin. Exp. Immunol 2000, 119, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Winkelhake JL, Gauny SS, Pharmacol. Rev 1990, 42. [PubMed] [Google Scholar]
- [81].Mark DF, Lu SD, Creasey AA, Yamamoto R, Lin LS, Proc. Natl. Acad. Sci. U. S. A 1984, 81, 5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Zídek Z, Anzenbacher P, Kmoníčková E, Br. J. Pharmacol 2009, 157, 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Melian EB, Plosker GL, Interferon alfacon-1: A review of its pharmacology and therapeutic efficacy in the treatment of chronic hepatitis C, Vol. 61, Adis International Ltd, 2001, pp. 1661–1691. [DOI] [PubMed] [Google Scholar]
- [84].Pockros PJ, Gastroenterology 2010, 139, 1084. [DOI] [PubMed] [Google Scholar]
- [85].Gillies SD, Lan Y, Hettmann T, Brunkhorst B, Sun Y, Mueller SO, Lo KM, Clin. Cancer Res 2011, 17, 3673. [DOI] [PubMed] [Google Scholar]
- [86].Sharma M, Khong H, Fa’ak F, Bentebibel SE, Janssen LME, Chesson BC, Creasy CA, Forget MA, Kahn LMS, Pazdrak B, Karki B, Hailemichael Y, Singh M, Vianden C, Vennam S, Bharadwaj U, Tweardy DJ, Haymaker C, Bernatchez C, Huang S, Rajapakshe K, Coarfa C, Hurwitz ME, Sznol M, Hwu P, Hoch U, Addepalli M, Charych DH, Zalevsky J, Diab A, Overwijk WW, Nat. Commun 2020, 11, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Klein C, Waldhauer I, Nicolini VG, Freimoser-Grundschober A, Nayak T, Vugts DJ, Dunn C, Bolijn M, Benz J, Stihle M, Lang S, Roemmele M, Hofer T, van Puijenbroek E, Wittig D, Moser S, Ast O, Brünker P, Gorr IH, Neumann S, de Vera Mudry MC, Hinton H, Crameri F, Saro J, Evers S, Gerdes C, Bacac M, van Dongen G, Moessner E, Umaña P, Oncoimmunology 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Sockolosky JT, Trotta E, Parisi G, Picton L, Su LL, Le AC, Chhabra A, Silveria SL, George BM, King IC, Tiffany MR, Jude K, Sibener LV, Baker D, Shizuru JA, Ribas A, Bluestone JA, Garcia KC, Science (80-. ). 2018, 359, 1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Dreher MR, Liu W, Michelich CR, Dewhirst MW, Yuan F, Chilkoti A, JNCI J. Natl. Cancer Inst 2006, 98, 335. [DOI] [PubMed] [Google Scholar]
- [90].Tarini M, Cignoni P, Montani C, IEEE Trans. Vis. Comput. Graph 2006, 12, 1237. [DOI] [PubMed] [Google Scholar]
- [91].Williams DP, Parker K, Bacha P, Bishai W, Borowski M, Genbauffe F, Strom TB, Murphy JR, Protein Eng. 1987, 1, 493. [DOI] [PubMed] [Google Scholar]
- [92].Cheung LS, Fu J, Kumar P, Kumar A, Urbanowski ME, Ihms EA, Parveen S, Bullen CK, Patrick GJ, Harrison R, Murphy JR, Pardoll DM, Bishai WR, Proc. Natl. Acad. Sci. U. S. A 2019, 116, 3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Alkharabsheh O, Frankel AE, Biomedicines 2019, 7, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Heiss JD, Jamshidi A, Shah S, Martin S, Wolters PL, Argersinger DP, Warren KE, Lonser RR, J. Neurosurg. Pediatr 2019, 23, 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Baur AS, Lutz MB, Schierer S, Beltrame L, Theiner G, Zinser E, Ostalecki C, Heidkamp G, Haendle I, Erdmann M, Wiesinger M, Leisgang W, Gross S, Pommer AJ, Kämpgen E, Dudziak D, Steinkasserer A, Cavalieri D, Schuler-Thurner B, Schuler G, Blood 2013, 122, 2185. [DOI] [PubMed] [Google Scholar]
- [96].Akbari B, Farajnia S, Ahdi Khosroshahi S, Safari F, Yousefi M, Dariushnejad H, Rahbarnia L, Int. Rev. Immunol 2017, 36, 207. [DOI] [PubMed] [Google Scholar]
- [97].Sims RB, Vaccine 2012, 30, 4394. [DOI] [PubMed] [Google Scholar]
- [98].Holl EK, McNamara MA, Healy P, Anand M, Concepcion RS, Breland CD, Dumbudze I, Tutrone R, Shore N, Armstrong AJ, Harrison M, Wallace JA, Wu Y, George DJ, Prostate Cancer Prostatic Dis. 2019, 22, 588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Pieczonka CM, Telonis D, Mouraviev V, Albala D, Rev. Urol 2015, 17, 203. [PMC free article] [PubMed] [Google Scholar]
- [100].Mannie MD, Blanchfield JL, Islam SMT, Abbott DJ, Front. Immunol 2012, 3, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Moorman CD, Bastian AG, DeOca KB, Mannie MD, J. Neuroinflammation 2020, 17, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Neri D, Sondel PM, Curr. Opin. Immunol 2016, 40, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Bootz F, Neri D, Drug Discov. Today 2016, 21, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Pasche N, Neri D, Immunocytokines: A novel class of potent armed antibodies, Vol. 17, Elsevier Current Trends, 2012, pp. 583–590. [DOI] [PubMed] [Google Scholar]
- [105].Strauss J, Heery CR, Kim JW, Jochems C, Donahue RN, Montgomery AS, McMahon S, Lamping E, Marte JL, Madan RA, Bilusic M, Silver MR, Bertotti E, Schlom J, Gulley JL, Clin. Cancer Res 2019, 25, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Yang RK, Kuznetsov IB, Ranheim EA, Wei JS, Sindiri S, Gryder BE, Gangalapudi V, Song YK, Patel V, Hank JA, Zuleger C, Erbe AK, Morris ZS, Quale R, Kim K, Albertini MR, Khan J, Sondel PM, Clin. Cancer Res 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Roche, Pipeline summary, 2020. [Google Scholar]
- [108].Philogen, PIPELINE - Philogen - innovating targeting. [Google Scholar]
- [109].Bachanova V, Lansigan F, Quick DP, Vlock D, Gillies S, Nakamura R, Blood 2015, 126, 1533. [Google Scholar]
- [110].Young PA, Dang NH, Nastoupil LJ, Minning D, Gresser MJ, Timmerman J, J. Clin. Oncol 2016, 34, TPS3109. [Google Scholar]
- [111].Kaufman HL, Mehnert JM, Cuillerot J-M, von Heydebreck A, Ott PA, Hodi FS, J. Clin. Oncol 2014, 32, TPS9107. [Google Scholar]
- [112].Gladkov O, Biakhov M, Ramlau R, Serwatowski P, Milanowski J, Tomeczko J, Komarnitsky PB, Bernard L, Kramer D, Krzakowski MJ, J. Clin. Oncol 2012, 30, 7090. [Google Scholar]
- [113].Rudman SM, Jameson MB, McKeage MJ, Savage P, Jodrell DI, Harries M, Acton G, Erlandsson F, Spicer JF, Clin. Cancer Res 2011, 17, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Tzeng A, Kwan BH, Opel CF, Navaratna T, Wittrup KD, Proc. Natl. Acad. Sci. U. S. A 2015, 112, 3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Gillies SD, Protein Eng. Des. Sel 2013, 26, 561. [DOI] [PubMed] [Google Scholar]
- [116].Gillies SD, Lo KM, Lan Y, Dahl T, Wong WK, Burger C, Clin. Cancer Res 2002, 8, 210. [PubMed] [Google Scholar]
- [117].Martomo S, Lu D, Polonskaya Z, Luna X, Miyara F, Patel J, Ann. Oncol 2019, 30, xi4. [Google Scholar]
- [118].Amos SM, Duong CPM, Westwood JA, Ritchie DS, Junghans RP, Darcy PK, Kershaw MH, Autoimmunity associated with immunotherapy of cancer, Vol. 118, American Society of Hematology, 2011, pp. 499–509. [DOI] [PubMed] [Google Scholar]
- [119].Miura JT, Zager JS, Futur. Oncol 2019, 15, 3665. [DOI] [PubMed] [Google Scholar]
- [120].Li B, Chen A, Zou S, Wu J, Wang H, Chen R, Luo M, Int. J. Pharm 2019, 558, 404. [DOI] [PubMed] [Google Scholar]
- [121].Skrombolas D, Frelinger JG, Expert Rev. Clin. Immunol 2014, 10, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Rustgi VK, Curr. Med. Res. Opin 2009, 25, 991. [DOI] [PubMed] [Google Scholar]
- [123].Huang YS, Wen XF, Yang ZY, Wu YL, Lu Y, Zhou LF, Khan RH, PLoS One 2014, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Gladkov O, Moiseyenko V, Bondarenko IN, Shparyk Y, Barash S, Adar L, Avisar N, Oncologist 2016, 21, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Chuang YM, He L, Pinn ML, Tsai YC, Cheng MA, Farmer E, Karakousis PC, Hung CF, Cell. Mol. Immunol 2020, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Vazquez-Lombardi R, Loetsch C, Zinkl D, Jackson J, Schofield P, Deenick EK, King C, Phan TG, Webster KE, Sprent J, Christ D, Nat. Commun 2017, 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Sun Z, Ren Z, Yang K, Liu Z, Cao S, Deng S, Xu L, Liang Y, Guo J, Bian Y, Xu H, Shi J, Wang F, Fu YX, Peng H, Nat. Commun 2019, 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Bristol Myers Squibb, Pharmaceutical R&D Pipeline, 2020. [Google Scholar]
- [129].Tchao N, Gorski K, Yuraszeck T, Sohn S, Ishida K, Wong H, Park K, Blood 2017, 130, 696.28798058 [Google Scholar]
- [130].Genentech, Genentech: Our Pipeline. [Google Scholar]
- [131].Generon, Generon Corporation (Shanghai) Ltd. [Google Scholar]
- [132].Tahir SK, Smith ML, Solomon LR, Zhang H, Xue JC, Xiao Y, Cheng D, Buchanan G, Morgan-Lappe S, Phillips DC, Blood 2017, 130, 2812. [Google Scholar]
- [133].Wrangle JM, Velcheti V, Patel MR, Garrett-Mayer E, Hill EG, Ravenel JG, Miller JS, Farhad M, Anderton K, Lindsey K, Taffaro-Neskey M, Sherman C, Suriano S, Swiderska-Syn M, Sion A, Harris J, Edwards AR, Rytlewski JA, Sanders CM, Yusko EC, Robinson MD, Krieg C, Redmond WL, Egan JO, Rhode PR, Jeng EK, Rock AD, Wong HC, Rubinstein MP, Lancet Oncol. 2018, 19, 694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Knudson KM, Hodge JW, Schlom J, Gameiro SR, Expert Opin. Biol. Ther 2020, 1. [DOI] [PubMed] [Google Scholar]
- [135].Lopes JE, Fisher JL, Flick HL, Wang C, Sun L, Ernstoff MS, Alvarez JC, Losey HC, J. Immunother. Cancer 2020, 8, e000673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Knudson KM, Hicks KC, Ozawa Y, Schlom J, Gameiro SR, J. Immunother. Cancer 2020, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Corti A, Curnis F, Rossoni G, Marcucci F, Gregorc V, BioDrugs 2013, 27, 591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Curnis F, Sacchi A, Corti A, J. Clin. Invest 2002, 110, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Gregorc V, Gaafar RM, Favaretto A, Grossi F, Jassem J, Polychronis A, Bidoli P, Tiseo M, Shah R, Taylor P, Novello S, Muzio A, Bearz A, Greillier L, Fontana F, Salini G, Lambiase A, O’Brien M, Lancet Oncol. 2018, 19, 799. [DOI] [PubMed] [Google Scholar]
- [140].CHMP, Withdrawal Assessment report - Zafiride, 2017. [Google Scholar]
- [141].Ferreri AJM, Calimeri T, Conte GM, Cattaneo D, Fallanca F, Ponzoni M, Scarano E, Curnis F, Nonis A, Lopedote P, Citterio G, Politi LS, Foppoli M, Girlanda S, Sassone M, Perrone S, Cecchetti C, Ciceri F, Bordignon C, Corti A, Anzalone N, Blood 2019, 134, 252. [DOI] [PubMed] [Google Scholar]
- [142].Ishihara J, Ishihara A, Sasaki K, Lee SS-Y, Williford J-M, Yasui M, Abe H, Potin L, Hosseinchi P, Fukunaga K, Raczy MM, Gray LT, Mansurov A, Katsumata K, Fukayama M, Kron SJ, Swartz MA, Hubbell JA, Sci. Transl. Med 2019, 11, eaau3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Mansurov A, Ishihara J, Hosseinchi P, Potin L, Marchell TM, Ishihara A, Williford JM, Alpar AT, Raczy MM, Gray LT, Swartz MA, Hubbell JA, Nat. Biomed. Eng 2020, 4, 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Momin N, Mehta NK, Bennett NR, Ma L, Palmeri JR, Chinn MM, Lutz EA, Kang B, Irvine DJ, Spranger S, Wittrup KD, Sci. Transl. Med 2019, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Strohl WR, Fusion Proteins for Half-Life Extension of Biologics as a Strategy to Make Biobetters, Vol. 29, Springer International Publishing, 2015, pp. 215–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Williams P, Galipeau J, J. Immunol 2011, 186, 5527. [DOI] [PubMed] [Google Scholar]
- [147].O’Shaughnessy J, Tolcher A, Riseberg D, Venzon D, Zujewski J, Noone M, Gossard M, Danforth D, Jacobson J, Chang V, Goldspiel B, Keegan P, Giusti R, Cowan K, Blood 1996, 87, 2205. [PubMed] [Google Scholar]
- [148].Skrombolas D, Sullivan M, Frelinger JG, J. Interf. Cytokine Res 2019, 39, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Neri D, Cancer Immunol. Res 2019, 7, 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Gupta V, Bhavanasi S, Quadir M, Singh K, Ghosh G, Vasamreddy K, Ghosh A, Siahaan TJ, Banerjee S, Banerjee SK, Protein PEGylation for cancer therapy: bench to bedside, Vol. 13, Springer; Netherlands, 2019, pp. 319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Yang Q, Lai SK, Wiley Interdiscip. Rev. Nanomedicine Nanobiotechnology 2015, 7, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Maeda H, Polymer therapeutics and the EPR effect, Vol. 25, Taylor and Francis Ltd, 2017, pp. 781–785. [DOI] [PubMed] [Google Scholar]
- [153].Sindhwani S, Syed AM, Ngai J, Kingston BR, Maiorino L, Rothschild J, MacMillan P, Zhang Y, Rajesh NU, Hoang T, Wu JLY, Wilhelm S, Zilman A, Gadde S, Sulaiman A, Ouyang B, Lin Z, Wang L, Egeblad M, Chan WCW, Nat. Mater 2020, 1. [DOI] [PubMed] [Google Scholar]
- [154].Bailon P, Won CY, PEG-modified biopharmaceuticals, Vol. 6, Taylor & Francis, 2009, pp. 1–16. [DOI] [PubMed] [Google Scholar]
- [155].JevsÌevar S, Kunstelj M, Porekar VG, Biotechnol. J 2010, 5, 113. [DOI] [PubMed] [Google Scholar]
- [156].Zeuzem S, Feinman SV, Rasenack J, Heathcote EJ, Lai M-Y, Gane E, O’Grady J, Reichen J, Diago M, Lin A, Hoffman J, Brunda MJ, Engl N. J. Med 2000, 343, 1666. [DOI] [PubMed] [Google Scholar]
- [157].Rajender Reddy K, Modi MW, Pedder S, Adv. Drug Deliv. Rev 2002, 54, 571. [DOI] [PubMed] [Google Scholar]
- [158].Bottomley A, Coens C, Suciu S, Santinami M, Kruit W, Testori A, Marsden J, Punt C, Salès F, Gore M, Mackie R, Kusic Z, Dummer R, Patel P, Schadendorf D, Spatz A, Keilholz U, Eggermont A, J. Clin. Oncol 2009, 27, 2916. [DOI] [PubMed] [Google Scholar]
- [159].Herndon TM, Demko SG, Jiang X, He K, Gootenberg JE, Cohen MH, Keegan P, Pazdur R, Oncologist 2012, 17, 1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Cao Z, Jiang S, Super-hydrophilic zwitterionic poly(carboxybetaine) and amphiphilic non-ionic poly(ethylene glycol) for stealth nanoparticles, Vol. 7, Elsevier, 2012, pp. 404–413. [Google Scholar]
- [161].V Katre N, J. Immunol 1990, 144, 209. [PubMed] [Google Scholar]
- [162].Menzel T, Schomburg A, Körfer A, Hadam M, Meffert M, Dallmann I, Casper S, Kirchner H, Poliwoda H, Atzpodien J, Cancer Biother. 1993, 8, 199. [DOI] [PubMed] [Google Scholar]
- [163].Yang JC, Topalian SL, Schwartzentruber DJ, Parkinson DR, Marincola FM, Weber JS, Seipp CA, White DE, Rosenberg SA, Cancer 1995, 76, 687. [DOI] [PubMed] [Google Scholar]
- [164].Bukowski RM, Young J, Goodman G, Meyers F, Issell BF, Sergi JS, McLain D, Fyfe G, Finke J, Invest. New Drugs 1993, 11, 211. [DOI] [PubMed] [Google Scholar]
- [165].Carr A, Emery S, Lloyd A, Hoy J, Garsia R, French M, Stewart G, Fyfe G, Cooper DA, J. Infect. Dis 1998, 178, 992. [DOI] [PubMed] [Google Scholar]
- [166].Onwumeh J, Okwundu CI, Kredo T, Interleukin-2 as an adjunct to antiretroviral therapy for HIV-positive adults, Vol. 2017, John Wiley and Sons Ltd, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Eliason JF, BioDrugs 2001, 15, 705. [DOI] [PubMed] [Google Scholar]
- [168].Naing A, Infante JR, Papadopoulos KP, Chan IH, Shen C, Ratti NP, Rojo B, Autio KA, Wong DJ, Patel MR, Ott PA, Falchook GS, Pant S, Hung A, Pekarek KL, Wu V, Adamow M, McCauley S, Mumm JB, Wong P, Van Vlasselaer P, Leveque J, Tannir NM, Oft M, Cancer Cell 2018, 34, 775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Hecht JR, Lonardi S, Bendell JC, Sim H-W, Macarulla T, Lopez CD, Van Cutsem E, Martin AJM, Park JO, Greil R, Lin Y, Rao S, Ryoo B-Y, J. Clin. Oncol 2020, 38, 637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Lilly Eli, TWENTY NINETEEN ELI LILLY AND COMPANY FINANCIAL REPORT, 2020. [Google Scholar]
- [171].Nyberg K, Pegilodecakin Fails to Improve Survival When Added to Second-Line FOLFOX in PDAC, 2020. [Google Scholar]
- [172].Autio K, Oft M, Pegylated Interleukin-10: Clinical Development of an Immunoregulatory Cytokine for Use in Cancer Therapeutics, Vol. 21, Current Medicine Group LLC; 1, 2019. [DOI] [PubMed] [Google Scholar]
- [173].Charych D, Khalili S, Dixit V, Kirk P, Chang T, Langowski J, Rubas W, Doberstein SK, Eldon M, Hoch U, Zalevsky J, PLoS One 2017, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Charych DH, Hoch U, Langowski JL, Lee SR, Addepalli MK, Kirk PB, Sheng D, Liu X, Sims PW, VanderVeen LA, Ali CF, Chang TK, Konakova M, Pena RL, Kanhere RS, Kirksey YM, Ji C, Wang Y, Huang J, Sweeney TD, Kantak SS, Doberstein SK, Clin. Cancer Res 2016, 22, 680. [DOI] [PubMed] [Google Scholar]
- [175].Bentebibel SE, Hurwitz ME, Bernatchez C, Haymaker C, Hudgens CW, Kluger HM, Tetzlaff MT, Tagliaferri MA, Zalevsky J, Hoch U, Fanton C, Aung S, Hwu P, Curti BD, Tannir NM, Sznol M, Diab A, Cancer Discov. 2019, 9, 711. [DOI] [PubMed] [Google Scholar]
- [176].Diab A, Tannir NM, Bentebibel S-E, Hwu P, Papadimitrakopoulou V, Haymaker C, Kluger HM, Gettinger SN, Sznol M, Tykodi SS, Curti BD, Tagliaferri MA, Zalevsky J, Hannah AL, Hoch U, Aung S, Fanton C, Rizwan A, Iacucci E, Liao Y, Bernatchez C, Hurwitz ME, Cho DC, Cancer Discov. 2020, 10, 1158. [DOI] [PubMed] [Google Scholar]
- [177].Doberstein SK, Expert Opin. Biol. Ther 2019, 19, 1223. [DOI] [PubMed] [Google Scholar]
- [178].Langowski JL, Kirk P, Addepalli M, Chang T, Dixit V, Kim G, Kirksey Y, Kuo P, Lee M, Maiti M, Rubas W, Sims P, Song Y, Tang Y, Vanderveen L, Zhang P, Doberstein SK, Zalevsky J, NKTR-358: a selective, first-in-class IL-2 pathway agonist, which increases number and suppressive function of regulatory T cells for the treatment of immune innammatory disorders. [Google Scholar]
- [179].Nairn NW, Grabstein K, Shanebeck KD, Li G, Wang A, In Polymer-Protein Conjugates, Elsevier, 2020, pp. 317–349. [Google Scholar]
- [180].Synthorx, Scientific Publications - Synthorx. [Google Scholar]
- [181].Mora JR, White JT, DeWall SL, AAPS J. 2020, 22, 35. [DOI] [PubMed] [Google Scholar]
- [182].CHMP, Committee for Medicinal Products for Human Use (CHMP) Assessment report Plegridy International non-proprietary name: peginterferon beta-1a, 2014. [Google Scholar]
- [183].Hoang Thi TT, Pilkington EH, Nguyen DH, Lee JS, Park KD, Truong NP, Polymers (Basel). 2020, 12, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Binder U, Skerra A, PASylation®: A versatile technology to extend drug delivery, Vol. 31, Elsevier Ltd, 2017, pp. 10–17. [Google Scholar]
- [185].Chen SA, Sawchuk RJ, Brundage RC, Horvath C, Mendenhall HV, Gunther RA, Braeckman RA, J. Pharmacol. Exp. Ther 2000, 293, 248. [PubMed] [Google Scholar]
- [186].Sershen S, West J, Adv. Drug Deliv. Rev 2002, 54, 1225. [DOI] [PubMed] [Google Scholar]
- [187].Lee KY, Yuk SH, Polymeric protein delivery systems, Vol. 32, Pergamon, 2007, pp. 669–697. [Google Scholar]
- [188].Cole DJ, Gattoni-Celli S, McClay EF, Metcalf JS, Brown JM, Nabavi N, Newton DA, Woolhiser CB, Wilson MC, Vournakis JN, Clin. Cancer Res 1997, 3, 867. [PubMed] [Google Scholar]
- [189].Salem ML, Gillanders WE, Kadima AN, El-Naggar S, Rubinstein MP, Demcheva M, Vournakis JN, Cole DJ, Interf J. Cytokine Res. 2006, 26, 593. [DOI] [PubMed] [Google Scholar]
- [190].Yang L, Zaharoff DA, Biomaterials 2013, 34, 3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Smith SG, prasanth Koppolu B, Ravindranathan S, Kurtz SL, Yang L, Katz MD, Zaharoff DA, Cancer Immunol. Immunother. 2015, 64, 689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Smith SG, Baltz JL, Koppolu BP, Ravindranathan S, Nguyen K, Zaharoff DA, Oncoimmunology 2017, 6, e1259050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Ali OA, Huebsch N, Cao L, Dranoff G, Mooney DJ, Nat. Mater 2009, 8, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Koshy ST, Mooney DJ, Biomaterials for enhancing anti-cancer immunity, Vol. 40, Elsevier Ltd, 2016, pp. 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Stephan SB, Taber AM, Jileaeva I, Pegues EP, Sentman CL, Stephan MT, Nat. Biotechnol 2015, 33, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Chao Y, Chen Q, Liu Z, Adv. Funct. Mater 2020, 30, 1902785. [Google Scholar]
- [197].Hori Y, Winans AM, Huang CC, Horrigan EM, Irvine DJ, Biomaterials 2008, 29, 3671. [DOI] [PubMed] [Google Scholar]
- [198].Hori Y, Winans AM, Irvine DJ, Acta Biomater. 2009, 5, 969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Hori Y, Stern PJ, Hynes RO, Irvine DJ, Biomaterials 2009, 30, 6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Kim J, Li WA, Choi Y, Lewin SA, Verbeke CS, Dranoff G, Mooney DJ, Nat. Biotechnol 2015, 33, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Bencherif SA, Sands RW, Ali OA, Li WA, Lewin SA, Braschler TM, Shih TY, Verbeke CS, Bhatta D, Dranoff G, Mooney DJ, Nat. Commun 2015, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Morikawa K, Okada F, Hosokawa M, Kobayashi H, Cancer Res. 1987, 47. [PubMed] [Google Scholar]
- [203].Lv Q, He C, Quan F, Yu S, Chen X, Bioact. Mater 2018, 3, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Ishii S, Kaneko J, Nagasaki Y, Biomaterials 2016, 84, 210. [DOI] [PubMed] [Google Scholar]
- [205].Sinha VR, Trehan A, Biodegradable microspheres for protein delivery, Vol. 90, Elsevier, 2003, pp. 261–280. [DOI] [PubMed] [Google Scholar]
- [206].Li J, Mooney DJ, Nat. Rev. Mater 2016, 1, 16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Anderson PM, Katsanis E, Sencer SF, Hasz D, Ochoa AC, Bostrom B, J. Immunother 1992, 12, 3. [PubMed] [Google Scholar]
- [208].Fidler IJ, Science (80-. ). 1980, 208, 1469. [DOI] [PubMed] [Google Scholar]
- [209].Fidler IJ, Raz A, Fogler WE, Poste G, In Cancer Chemo- and Immunopharmacology (Eds.: Mathé G; Muggia FM), Springer-Verlag Berlin Heidelberg, 1980, pp. 246–251. [Google Scholar]
- [210].Kedar E, Rutkowski Y, Braun E, Emanuel N, Barenholz Y, J. Immunother 1994, 16, 47. [DOI] [PubMed] [Google Scholar]
- [211].Babai I, Samira S, Barenholz Y, Zakay-Rones Z, Kedar E, Vaccine 1999, 17, 1223. [DOI] [PubMed] [Google Scholar]
- [212].Christian DA, Hunter CA, Immunotherapy 2012, 4, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Skubitz KM, Anderson PM, Anticancer. Drugs 2000, 11, 555. [DOI] [PubMed] [Google Scholar]
- [214].Ten RM, Anderson PM, Zein NN, Temesgen Z, Lou Clawson M, Weiss W, Int. Immunopharmacol 2002, 2, 333. [DOI] [PubMed] [Google Scholar]
- [215].Boni LT, Batenjany MM, Neville ME, Guo Y, Xu L, Wu F, Mason JT, Robb RJ, Popescu MC, Biochim. Biophys. Acta - Biomembr 2001, 1514, 127. [DOI] [PubMed] [Google Scholar]
- [216].Katre NV, Am. J. Drug Deliv 2004, 2, 213. [Google Scholar]
- [217].Egilmez NK, Jong YS, Iwanuma Y, Jacob JS, Santos CA, Chen FA, Mathiowitz E, Bankert RB, Cancer Immunol. Immunother 1998, 46, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Sabel MS, Skitzki J, Stoolman L, Egilmez NK, Mathiowitz E, Bailey N, Chang WJ, Chang AE, Ann. Surg. Oncol 2004, 11, 147. [DOI] [PubMed] [Google Scholar]
- [219].Egilmez NK, Immunol. Invest 2020, 1. [DOI] [PubMed] [Google Scholar]
- [220].Dzyublyk I, Yegorova T, Moroz L, Popovych O, Zaytsev I, Miroshnichenko V, Kromminga A, Wilkes MM, Van Hoogdalem EJ, Humphries JE, J. Viral Hepat 2011, 18, 271. [DOI] [PubMed] [Google Scholar]
- [221].Biolex, Locteron, controlled-release interferon alpha 2b, 3 studies at EASL 2010 Vienna - 3 company press releases, 2010. [Google Scholar]
- [222].Rowswell-Turner RB, Harden JL, Nair RE, Gu T, Kilinc MO, Egilmez NK, J. Immunol 2011, 187, 4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Mills BN, Connolly KA, Ye J, Murphy JD, Uccello TP, Han BJ, Zhao T, Drage MG, Murthy A, Qiu H, Patel A, Figueroa NM, Johnston CJ, Prieto PA, Egilmez NK, Belt BA, Lord EM, Linehan DC, Gerber SA, Cell Rep. 2019, 29, 406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Park E, Hart ML, Rolauffs B, Stegemann JP, Annamalai RT, J. Biomed. Mater. Res. Part A 2020, 108, 722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Wyatt Shields C, Evans MA, Wang LLW, Baugh N, Iyer S, Wu D, Zhao Z, Pusuluri A, Ukidve A, Pan DC, Mitragotri S, Sci. Adv 2020, 6, eaaz6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Anselmo AC, Gilbert JB, Kumar S, Gupta V, Cohen RE, Rubner MF, Mitragotri S, J. Control. Release 2015, 199, 29. [DOI] [PubMed] [Google Scholar]
- [227].Cleland JL, Daugherty A, Mrsny R, Emerging protein delivery methods, Vol. 12, Elsevier Ltd, 2001, pp. 212–219. [DOI] [PubMed] [Google Scholar]
- [228].Zhao Z, Ukidve A, Krishnan V, Mitragotri S, Adv. Drug Deliv. Rev 2019, 143, 3. [DOI] [PubMed] [Google Scholar]
- [229].Maninder C/O Chiron Corporation Hora, Preparing aldesleukin for pharmaceutical use, European Patent Office, 2006. [Google Scholar]
- [230].Geigert J, Solli N, Woehleke P, Vemuri S, In Stability and Characterization of Protein and Peptide Drugs (Eds.: Pearlman R; Wang YJ), Springer, 1993, pp. 249–262. [Google Scholar]
- [231].Anselmo AC, Mitragotri S, Bioeng. Transl. Med 2019, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Zhang Y, Li N, Suh H, Irvine DJ, Nat. Commun 2018, 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Kwong B, Gai SA, Elkhader J, Wittrup KD, Irvine DJ, Cancer Res. 2013, 73, 1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Barberio AE, Smith SG, Correa S, Nguyen C, Nhan B, Melo M, Tokatlian T, Suh H, Irvine DJ, Hammond PT, ACS Nano 2020, 14, 11238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Alkekhia D, Hammond PT, Shukla A, Annu. Rev. Biomed. Eng 2020, 22, 1. [DOI] [PubMed] [Google Scholar]
- [236].McHugh MD, Park J, Uhrich R, Gao W, Horwitz DA, Fahmy TM, Biomaterials 2015, 59, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Zhao Y, Song Q, Yin Y, Wu T, Hu X, Gao X, Li G, Tan S, Zhang Z, J. Control Release 2018, 269, 322. [DOI] [PubMed] [Google Scholar]
- [238].Wang Y, Lin YX, Qiao SL, An HW, Ma Y, Qiao ZY, Rajapaksha RPYJ, Wang H, Biomaterials 2017, 112, 153. [DOI] [PubMed] [Google Scholar]
- [239].Tang L, Zheng Y, Melo MB, Mabardi L, Castaño AP, Xie YQ, Li N, Kudchodkar SB, Wong HC, Jeng EK, Maus MV, Irvine DJ, Nat. Biotechnol 2018, 36, 707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Park J, Wrzesinski SH, Stern E, Look M, Criscione J, Ragheb R, Jay SM, Demento SL, Agawu A, Licona Limon P, Ferrandino AF, Gonzalez D, Habermann A, Flavell RA, Fahmy TM, Nat. Mater 2012, 11, 895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Mejías R, Pérez-Yagüe S, Gutiérrez L, Cabrera LI, Spada R, Acedo P, Serna CJ, Lázaro FJ, Villanueva Á, del P M. Morales, Barber DF, Biomaterials 2011, 32, 2938. [DOI] [PubMed] [Google Scholar]
- [242].Anselmo AC, Mitragotri S, Bioeng. Transl. Med 2016, 1, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Hombach AA, Geumann U, Günther C, Hermann FG, Abken H, Cells 2020, 9, 873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Heathman TR, Nienow AW, McCall MJ, Coopman K, Kara B, Hewitt CJ, The translation of cell-based therapies: Clinical landscape and manufacturing challenges, Vol. 10, Future Medicine Ltd., 2015, pp. 49–64. [DOI] [PubMed] [Google Scholar]
- [245].Cervera-Carrascon V, Havunen R, Hemminki A, Expert Opin. Biol. Ther 2019, 19, 443. [DOI] [PubMed] [Google Scholar]
- [246].Hewitt SL, Bai A, Bailey D, Ichikawa K, Zielinski J, Karp R, Apte A, Arnold K, Zacharek SJ, Iliou MS, Bhatt K, Garnaas M, Musenge F, Davis A, Khatwani N, Su SV, MacLean G, Farlow SJ, Burke K, Frederick JP, Sci. Transl. Med 2019, 11. [DOI] [PubMed] [Google Scholar]
- [247].Bhatia S, Longino NV, Miller NJ, Kulikauskas R, Iyer JG, Ibrani D, Blom A, Byrd DR, Parvathaneni U, Twitty CG, Campbell JS, Le MH, Gargosky S, Pierce RH, Heller R, Daud AI, Nghiem P, Clin. Cancer Res 2020, 26, 598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Rehman H, Silk AW, Kane MP, Kaufman HL, J. Immunother. Cancer 2016, 4, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Goldberg EP, Hadba AR, Almond BA, Marotta JS, J. Pharm. Pharmacol 2002, 54, 159. [DOI] [PubMed] [Google Scholar]
- [250].Marabelle A, Andtbacka R, Harrington K, Melero I, Leidner R, De Baere T, Robert C, Ascierto PA, Baurain JF, Imperiale M, Rahimian S, Tersago D, Klumper E, Hendriks M, Kumar R, Stern M, Öhrling K, Massacesi C, Tchakov I, Tse A, Douillard JY, Tabernero J, Haanen J, Brody J, Ann. Oncol 2018, 29, 2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Kwong B, Liposome-Anchored Local Delivery of Immunomodulatory Agents for Tumor Therapy, Massachusetts Institute of Technology, 2012. [Google Scholar]
- [252].Franzé S, Selmin F, Samaritani E, Minghetti P, Cilurzo F, Pharmaceutics 2018, 10, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Lipiäinen T, Peltoniemi M, Sarkhel S, Yrjönen T, Vuorela H, Urtti A, Juppo A, J. Pharm. Sci 2015, 104, 307. [DOI] [PubMed] [Google Scholar]
- [254].Keyel ME, Reynolds CP, Spotlight on dinutuximab in the treatment of high-risk neuroblastoma: Development and place in therapy, Vol. 13, Dove Medical Press Ltd., 2019, pp. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Shaker MA, Younes HM, J. Pharm. Sci 2009, 98, 2268. [DOI] [PubMed] [Google Scholar]
- [256].Rothschilds AM, Wittrup KD, What, Why, Where, and When: Bringing Timing to Immuno-Oncology, Vol. 40, Elsevier Ltd, 2019, pp. 12–21. [DOI] [PubMed] [Google Scholar]
- [257].Rothschilds A, Tzeng A, Mehta NK, Moynihan KD, Irvine DJ, Wittrup KD, Oncoimmunology 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Liu J, Blake SJ, Yong MCR, Harjunpää H, Ngiow SF, Takeda K, Young A, O’Donnell JS, Allen S, Smyth MJ, Teng MWL, Cancer Discov. 2016, 6, 1382. [DOI] [PubMed] [Google Scholar]