

Abstract
Exposure to environmental chemicals can produce effects on the endocrine system through epigenetic mechanisms. These can considerably decrease or increase the sensitivity of multiple hormones depending on the dose, route, or time of exposure. The exposure of endocrine disrupting chemicals (EDCs) during the in utero period could be a critical window, altering the epigenome profile. Recently, several researchers suggest a role of EDCs in the obesity epidemic. In this brief review, we focused on how four EDCs (bisphenol A, dichlorodiphenyltrichloroethane, di-(2-ethylhexyl) phthalate and tributyltin) may underlay transgenerational epigenetic effects. We also discuss the adipogenesis signaling pathway and the impact of exposure to individual or mixtures of EDCs on the developing endocrine system. Understanding the molecular determinants of epigenetic memory across generations will provide essential insight into how environmental exposure can affect the health of individuals, as well as subsequent generations.
Keywords: Endocrine Disrupting Chemicals, In Utero, Obesity, Epigenetics, Obesogen
Introduction
The prevalence of obesity and related non-communicable diseases has grown significantly, imposing a significant burden on human health at the individual and public health levels [1,2]. Over 36% of the adult U.S. population and 13% of the population worldwide is clinically obese (body mass index [BMI] ≥ 30) [3,4]. Furthermore, the proportion of overweight adults in the U.S. (BMI ≥ 25) is predicted to increase from 68% to 86% by 2020 [3]. While BMI measurements do not differentiate between subcutaneous adiposity, visceral adiposity, and muscularity, the trend of increasing BMI rates has been significantly associated with an increase in visceral adiposity [5]. Visceral adiposity describes the abdominal fat linked to cardiovascular disease, metabolic syndrome, and diabetes [6]. Furthermore, this epidemic has spread to children with current childhood obesity rates around 17% for children among 2- to- 19 years old [4]. The increasing obesity rate is expected to worsen health outcomes, shorten life expectancy, and increase health-related costs in the coming years [7–9].
In recent decades, researchers have obtained persuasive evidence for the role of several epigenetics mechanisms in the obesity epidemic due to in utero exposure(s) to adverse maternal environments affected by nutrition, smoking, stress, alcohol, various industrial chemicals, etc [10–13]. This review briefly focuses on whether exposure to four endocrine disruptor chemicals (EDCs)-bisphenol A, dichlorodiphenyltrichloroethane, di-(2-ethylhexyl) phthalate, and tributyltin-may induce transgenerational epigenetic inheritance of obesity. These EDCs were selected for this study since they are commonly detected in the environment, and in human tissues. Based upon the data from the epidemiological and data mining literature noted below, they are also linked with metabolic diseases such as diabetes and obesity. We also examine the adverse effects of exposure to individual or mixtures of EDCs during the development of different organ systems.
Epigenetic regulation of gene expression
Epigenetics refers to the study of heritable changes that are mitotically and/or meiotically stable and affect gene expression without altering the underlying DNA sequence [14,15]. The principals epigenetic mechanisms are DNA methylation, histone modifications, and noncoding RNAs [14]. All these mechanisms can alter the folding of DNA into a three-dimensional structure which may promote or inhibit gene expression [16]. Furthermore, epigenetic marks are tissue-specific and can change during our lifetime [14].
The most studied method in humans, DNA methylation, consists of the addition of a methyl group at the carbon-5 position of cytosine (C) neighboring by guanine (G) nucleotides, in the context of the CpG dinucleotides, and generally acts to repress transcription through the modification of chromatin structure to regulate the binding of proteins in the DNA major groove; these hyper methylated genomic regions generally decreased gene expression [17,18]. DNA methylation at CpG sites has been shown to be relatively stable, resulting in early-life DNA methylation to persist throughout an individual’s life [15].
In contrast to the stability of DNA methylation, histone modifications are a more dynamic epigenetic mechanism that tighten or loosen the packing of DNA around histone protein complexes [13]. Tightly packaged chromatin inhibits the binding of transcription factors, leading to reduced transcriptional activity; loosely packaged chromatin is more active since transcriptional protein complexes can better access the chromatin.
There are several post-translational modifications to histone proteins, including acetylation, methylation, phosphorylation, ubiquitination, and sumoylation, which induce changes of the chromatin structure that ultimately affect gene expression [13]. For instance, histone acetylation involves the transfer of acetyl groups onto lysines on the N-terminal tails of histones by histone acetyl transferases (HATs), removing the positive charge of lysine. This acetylation is associated with transcriptional activation of specific chromosomal regions as the elimination of the positive charge relaxes the chromatin to facilitate access of transcription factors to promoter sequences. In contrast, histone methylation can lead to either the activation or repression of chromatin transcription depending on the location where a methyl group is transferred to the ε-amino group of lysine and arginine often on the histone 3 and histone 4 tails [19]. For an extensive review on the other histone modifications [19,20].
MicroRNA (miRNA) is another important epigenetic mechanism that contributes to the regulation of the epigenome. miRNAs are small noncoding RNA molecules that are usually 20 to 30 nucleotides long [14]. They can regulate gene expression at the post-transcriptional level by imperfect complementarity with a target mRNA, usually resulting in their silencing via translational repression or target degradation [1,21–26]. These epigenetic mechanisms promote heritable multi- and trans-generational effects that affect subsequent phenotypic expression.
Transgenerational inheritance of epigenetic changes
DNA methylation and histone modifications are epigenetic mechanisms that develop predominantly during early stages of mammalian embryonic development and throughout life [27]. During embryonic development, prior DNA methylation modifications are erased from the genome and are restoring the methylation patterns in later developmental stages to maintain accurate imprint reprogramming through this method of non-Mendelian inheritance [28,29]. During the process of demethylation, the genome is more sensitive to environmental factors that may induce de novo methylation changes, altering the imprinting pattern and, subsequently, changing the expression of particular genes [30].
A potential mechanism in which epigenetic changes induce transgenerational phenotypic alterations is suggested through the association between DNA methylation and transgenerational gene expression [31]. Laboratory rodent exposures to various conditions-chemicals and maternal nutrition-can alter the cytosine methylation pattern at metastable epialleles-alleles known to be particularly vulnerable to environmental influences [32] (i.e. Agouti yellow allele)-to induce certain phenotypes, like obesity [16]. Evidence in laboratory rodents is possibly relevant to human fetus exposures, representing the potential for similar changes in the epigenome. Furthermore, studies of transgenerational inheritance of diseases and particular phenotype variations have found DNA sequence motifs, such as zinc finger binding regions and guanine quadruplex sequences, in differential DNA methylation regions in sperm [33,34]. The interaction of these sequences with molecular factors could alter chromatin structure and accessibility of proteins with DNA methyltransferases-enzymes that facilitate DNA methylation-altering de novo DNA methylation patterns [32,33].
Another assumed transgenerationally inherited epimutation (heritable change in gene activity that is not associated with a DNA mutation but rather with gain or loss of DNA methylation or other heritable modifications of chromatin) is the 5’ hydroxymethylation of cytosines (5-hmeC) [35]. Accumulating evidence associates 5-hmeC with stem cell differentiation, especially with regard to controlling development [17,31,36]. Furthermore, 5-hmeC is associated with demethylation, suggesting its critical role in DNA methylation [37]. The similar roles of 5-hmeC epimutations and DNA methylation in stem cells suggests a central role in gene expression and heritable characteristics in stem cell differentiation [37]. These hypotheses continue to gain support but evidence remains scarce.
New approach on obesity epidemic
While significant evidence supports the thermodynamic model of fat accumulation balancing caloric intake and caloric expenditure, researchers are now investigating other influences [38]; diet and physical activity alone do not appear to explain the rapid worldwide increase in the prevalence of adult overweight and obesity reaching values around 29% in 1980 to 37% in 2013 [39]. Several studies pointed out an important genetic component contributing to the risk of developing obesity [40]. However, until now, all genetic loci identified can only explain part of obesity heritability, accounting for around 2 – 3% of the total genetic variance in BMI. This value is far from the BMI heritability estimates around 40 – 70% [40]. As such, a mechanism beyond the thermodynamic model and genetics must be implicated in the growing obesity epidemic.
It is now generally accepted that gene expression is altered by epigenetic factors in response to environmental exposures throughout the lifetime. Furthermore, it is also true that human diet suffered profound changes in the last century, marked by innovations in food technology, novel ingredients, and bioactive molecules altering the human diet which can interfere in epigenetic changes [14]. With the advances in the field of epigenetics, many scholars suggest that epigenetic factors may play a greater role than genetics in the development of obesity due in part to our new lifestyle [14,40]. Furthermore, these different kinds of epigenetic modifications were suggested to be one possible source for the non-genetic heritability that still needs to be explained in obesity.
For instance, animals living in proximity to humans-pets or laboratory animals-demonstrated considerable higher rates of obesity when comparing to their counterparts in the wild [41]. The study examined twenty-four animal populations from eight different species showing an increase in weight over the past several decades, suggesting that a change in environmental exposures may lead to obesity in both humans and animals [41]. The authors had suggested that, sheer chance likely could not explain this trend in weight gain, nor can a possible change in the treatment of animals since experimental design strictly controls for potential confounding factors, such as physical activity and feeding. Thus, a recent hypothesis suggests that this increasing trend of obesity is rooted in the prenatal environment, which may predispose infants to increased fat accumulation throughout their lives [42].
Endocrine disrupting chemicals (EDCs) and obesity
In the past several decades, the increasing obesity epidemic has correlated with the increased use of industrial chemicals [43]. Inevitably, these chemicals subsequently leach into the environment, including food and water supplies. A considerable portion of these chemicals are known as endocrine disrupting chemicals (EDCs) [43]. The World Health Organization (WHO) and the United Nations Environment Programme (UNEP) define an EDC as “an exogenous substance or mixture that alters function(s) of the endocrine system and consequently causes adverse health effects in an intact organism, or its progeny, or (sub)populations” [44]. In short, EDCs are chemicals that disrupt normal hormone function [45]. An increasing number of studies are linking EDCs with the obesity epidemic [33,46,47]. These substances are being termed “obesogens” or molecules that induce adverse effects on lipid metabolism and adipogenesis, which can lead to obesity [45,48–50].
The obesogen hypothesis
Blumberg and colleagues [42] proposed the obesogen hypothesis in 2006, and defined obesogens as chemicals that increase the number of fat cells and/or the storage of fat into existing adipocytes to directly stimulate obesity. Obesogens can also promote obesity through altering basal metabolic rate and hormonal control of appetite and satiety [42,51,52], increasing the proportion of calorie storage, and inducing food storage via gut microbiota [53]. Recent studies have identified several obesogenic EDCs, including dichlorodiphenyltrichloroethane (DDT) [54,55], bisphenol A (BPA) [56,57], tributyltin (TBT) [58,59], diethylstilbestrol (DES) [60], perfluorooctanoic acid (PFOA) [61], and phthalates [62]. Recent literature has extensively compiled the evidence for the obesogen hypothesis and will not be further addressed in this review (for further information [50]).
One of the most compelling components to the obesogen hypothesis is the developmental origins of health and disease hypothesis, which suggests that humans develop predispositions to various diseases, such as obesity, when exposed to a particular in utero environment during a critical window of fetal development [2,21,63,64]. While this review focuses on in utero EDC exposures that promote obesity, other factors that alter the in utero environment are critical in understanding the etiology of obesity. For instance, several studies have examined the association between maternal malnutrition and the onset of obesity through the up-regulation of the glucocorticoid receptor [65,66]. However, this review will not address the non-EDC in utero obesogens. For future studies, researchers may consider these other risk factors for obesity determined by the in utero environment when performing and analyzing their experiments around developmental EDC exposures.
A recent concern around obesogens stems from evidence of the long-lasting effects of in utero exposure to EDCs and the subsequent onset of childhood or later-life obesity [67]. Because early development requires precise timing of hormonal action to support proper coordination of tissue and organ growth, alterations in hormonal activity induced by EDCs can hinder normal endogenous activities and induce various endocrine-related disorders in humans [21]. Focusing obesogenic research on in utero exposures will target one of the most vulnerable stages in a human’s life, which can help curb certain predispositions towards obesity [52].
Overview of adipogenesis
Adipogenesis describes the differentiation of mesenchymal stem cells (MSCs) and their more lineage-restricted derivatives into adipocytes during both development and adulthood to produce and maintain fat cell numbers [68,69]. MSCs inhabit the perivascular region of many organs [70] and stimulate the development of many cells types (i.e. adipose, bone, and muscle) following treatment by specific differentiation-stimulating mixtures in vitro. Further research is required to determine whether differing tissue localization of MSCs in vivo will demonstrate the same lineage potential as these in vitro results, or whether tissue localization restricts lineage potential [71].
Although there is a strong understanding of how other cells that have committed to the adipocyte lineage differentiate into mature adipocytes, little is known about the mechanisms through which MSCs commit to the adipocyte lineage and what environmental influences affect this commitment [72]. Recent studies have identified important signals in the adipocyte commitment lineage, such as Wnt (proteins used for cell-cell communication), bone morphogenic proteins (BMPs), and PI3K/Akt signaling, that are likely regulated by the expression of certain genes, including TCF7-like 1 [68], Zfp423 [73], Zfp521 [74], and S6K1 [75]. Mediation of PPARγ-the master regulator of adipogenesis [76,77]-controls the mutually exclusive cell commitment into either adipocyte or osteoblast lineages [78]; up-regulation of PPARγ induces commitment to the adipogenic lineage, while down-regulation of PPARγ through Wnt signaling stimulates MSC commitment to the osteogenic lineage [68]. Thus, active BMP/TGF-β [79] and PI3K/Akt signaling [75] coupled with the repression of Wnt-3a/10b [80–82] and Wnt-5a [82] signaling induces MSC adipogenesis. Increasingly, non-coding RNAs are also being associated with the promotion of adipogenesis [83]. Future research will be critical to understanding the commitment of MSCs to adipogenesis and how various EDCs may affect the manifestation of adipocytes in the development of excess fat, and, possibly, obesity.
EDCs and reprogramming MSCs
Despite the multiple signaling pathways influencing adipogenesis in MSCs that EDCs could potentially disrupt, only a few studies have examined the effect of EDCs on the programming of MSCs. One study found that in vitro treatment of 3T3-L1 preadipocytes with the EDC obesogen TBT and pharmaceutical obesogen rosiglitazone (ROSI) induced adipocyte differentiation [64]. In the same study, prenatal exposure of pregnant mice to TBT or ROSI induced higher fat deposition at birth. The authors concluded that prenatal TBT or ROSI exposure led to adipogenesis in a PPARγ-dependent manner, significantly shifting MSC commitment from osteogenic to adipogenic [63]. The in vitro reprogramming of cell lineage commitment is evident in vivo as well, wherein treatment with TBT or ROSI in rats evidenced increased expression of adipogenic markers and decreased expression of osteogenic markers, as well as larger adipocytes and adipose depots where fat is stored [58].
Recent studies have suggested transgenerational effects of various EDC exposures following MSC programming [24,54,55]. Transgenerational effects describe genomic changes observed in the F3 or later generations that have not been directly exposed to a particular chemical. Multigenerational effects, in contrast, describe the F1 and F2 genomes following direct exposure to the chemicals in utero [24,54,55]. Therefore, observed genomic alterations in F3 generations stemming from F1 in utero obesogen exposure are due to genetic, or more likely, aforementioned epigenetic, modifications [24,54,55]. Prenatal TBT exposure in pregnant F0 animals increased fat depot size and MSC adipogenesis through the F3 generation [58]. Thus, prenatal TBT exposure likely causes heritable alterations in the germ cell genome of the directly exposed F1 fetuses, increasing the commitment of MSCs to adipocytes rather than the osteoblast lineage. This finding implies an in utero programming event that causes permanent MSC adipogenesis, subsequently manifesting as observable adult phenotypes; although, currently there are no studies that explain the heritable effects of obesogen exposures on MSC differentiation [69].
Beyond the transgenerational effects discussed, further work has shown that BPA, diethylhexyl phthalate (DEHP), and dibutyl phthalate (DBP) [62], and the widespread pesticide, DDT, induce transgenerational effects in rats [54,55]. Section 2.4 contains a deeper discussion of these studies, which are summarized in table 1. Several groups, including Blumberg and Skinner, are actively investigating the EDCs influence on programming MSC adipogenic fate, as well as the underlying epigenetic mechanisms of multi- and trans-generational effects [33,69].
Table 1:
Studies conducting epigenetic analyses of EDC-induced obesogenic effects.
| Chemical | Dose | Species | Treatment period | Obesity measure | F1 observed epigenetic effect (direct exposure) | F3 observed epigenetic effect (transgenerational exposure) | Suggested mechanism | Gene expression changes | Reference |
|---|---|---|---|---|---|---|---|---|---|
| BPA | 50 μg/kg/day | Wistar rats | GD0-weaning | Insulin and glucose; DNA methylation | Week 3: No significant changes; Week 21: Serum insulin and HOMA-IR increased; insulin sensitivity index and hepatic glycogen storage decreased | N/A | Hypermethylation of Gck | Decreased Gck expression; Alteration in DNA methylation occurring in early development persists as the animals grow older | [47] |
| BPA | 40 μg/kg/day | SD rats | GD0-PND21 | Insulin and glucose | Hypermethylation of Gck | F0 exposure during gestation and lactation can affect glucose and insulin tolerance in the F2 offspring | Hypermethylation of Gck | Decreased Gck expression as mechanism underlying glucose and insulin intolerance in F2 BPA-treated offspring; a permanent epigenetic change in male germline that transmits multigenerational epigenetic alterations | [46] |
| p,p’-DDT | 25 or 50 mg/kg/day | SD rats | GD8–14 | Abdominal adiposity and body weight; TUNEL assay | No observed change | LD males and females: 50% obese; ND males: 75% obese | Females: male/both parental germline transmission; Males: female germline transmission | Tubb3, Slc4a4, Carm1 epimutations in F3 sperm to increase visceral adiposity | [55] |
| DEHP | 1, 10, or 100 mg/kg/day | Wistar rats | GD9–21 | Glucose and insulin | F1 predisposed to glucometabolic dysfunction at adulthood by down-regulation of Glut4 | N/A | Glut4 epigenetic alterations via DNA methylation of CpG islands; Glut4 as primary GLUT in adipose and muscle tissues | Hypermethylation of Glut4 promoter at MYOD-binding sites with decreased GLUT4 expression; negative correlation between Glut4 expression and CpG island methylation | [84] |
| TBT | 5.42, 54.2, or 542 nM |
Mice | 7 days before mating; GD0-weaning | WAT and BAT weight; adipocyte size and number | Males: Increased perirenal and interscapular WAT depot weight and adipocyte size/number; Females: Increased WAT depot weight and adipocyte size. Minimal weight gain in male and female but decreased BAT | Males: Increased all WAT depot weights and adipocyte size/number; Females: Adipocyte number/size increased in all WAT depots besides interscapular and ovarian. LD exposure led to increase in AT for ≥ 3 generations | Transgenerational reprogramming of MSCs to favor the adipocyte lineage | Sharp increase in expression of all adipogenic markers and decreased expression of Pref-1 (adipocyte differentiation inhibitor) | [58] |
| Mixture (BPA, DEHP, DBP) | 50 (BPA), 750 (DEHP), 66 (DBP) mg/kg/day | SD rats | GD8–14 | Body weight and abdominal adiposity | No observed change | Increased obesity in F3 females and F3 LD males | Gdnf and Esrra DNA methylation | Hypermethylation to decrease expression of Gdnf and Esrra | [62] |
Abbreviations: GD: Gestation Day; PN: Post-Natal; MeDIP: methylated DNA Fragments; QPCR, MeDIP: Quantitative Polymerase Chain Reaction; LD: Low Dose; ND: Normal Dose; SD: Sprague Dawley; N/A: Non-Available; BPA: Bisphenol A; DDT: Dichlorodiphenyltrichloroethane; DEHP: Di-(2-ethylhexyl) Phthalate; TBT: Tributylin.
Underlying mechanisms of multi- and trans-generational epigenetic effects of obesogenic EDC exposure
Several studies have identified potential mechanisms in which in utero exposures to various obesogenic EDCs produce multi- and trans-generational epigenetic effects in vivo [46,47,54,55,58,62,84]. Table 1 summarizes these critical findings and proposed mechanisms, while a discussion is provided in the subsequent subsections for each individual chemical. A short discussion of chemical mixtures will follow, although more research is needed to elucidate the epigenetic effects associated with mixtures of multiple obesogenic EDCs. Studies on EDC mixtures will better represent the complex reality, where humans are exposed to hundreds of chemicals every day-not a single chemical in isolation [16,85,86]. The articles cited in this literature review were obtained from a comprehensive PubMed search, and subsequently filtered using the method outlined in figure 1. The studies in table 1 were selected as the papers offering evidence for strictly obesity and obesity-related phenotypes (i.e. type II diabetes) inherited through potential epigenetic mechanisms following in utero EDC exposure. The rest of the articles obtained from this literature search were excluded from the specific study, but used to supplement and contextualize these studies. The final excluded full texts provided current information on EDC epigenetic regulation of other diseases and/or EDC-related heritable phenotypes. This review excluded the human epidemiological studies and experimental animal studies associating these EDCs with obesity since they have already undergone exhaustive review [87].
Figure 1:
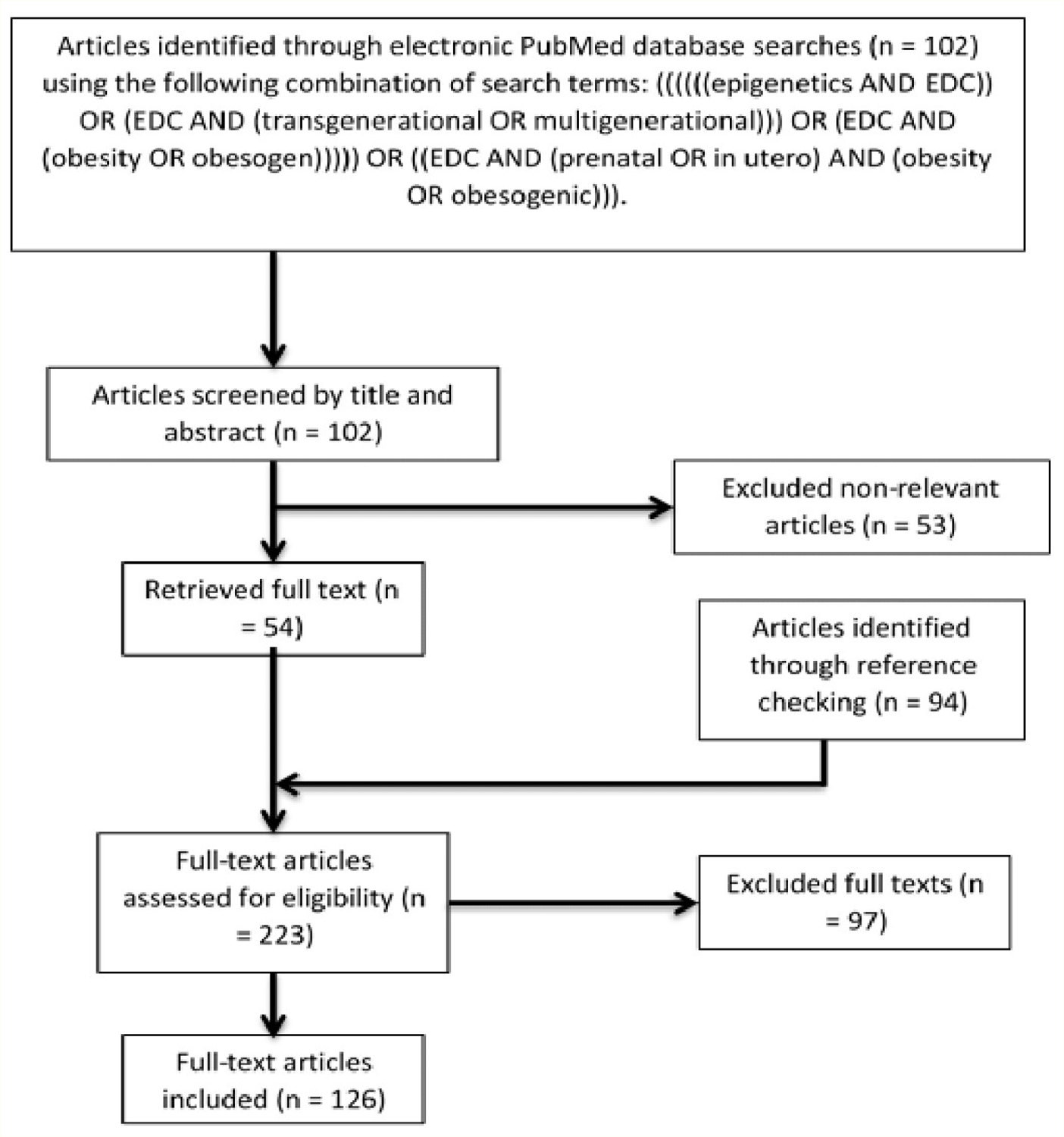
Flowchart of study selection. One reviewer to determine the eligibility and relevance of the literature to the literature review screened articles. Articles were deemed relevant if they were full-text and discussed any experiments or mechanisms involving epigenetics, obesity, and obesogenic chemicals.
For each EDC, this literature review examined the epigenetic studies in animals, including the physiological changes associated with obesity, proposed epigenetic mechanisms, and potential multi- or trans-generational epigenetic alterations of the EDC. These studies differed based on whether the affected generation had direct exposure to the original environmental chemical or metabolite exposure. When a pregnant mother (F0) is exposed to an EDC, her child (F1) could be affected by the same EDC throughout gestation exposure. Furthermore, because the germ cells of the F1 offspring are developing throughout gestation, the grandchildren (F2) are also directly exposed. Effects observed in the F2 generation would be considered multigenerational. In contrast, effects observed in the F3 generation that had no direct exposure to the original EDC would be transgenerational. When the EDCs’ exposure occurs through the F0 father, the transgenerational effects are observed in the F2 generation, as the only other generation directly exposed to the original exposure is the future F1 offspring, which is exposed as a germ cell (Figure 2).
Figure 2:
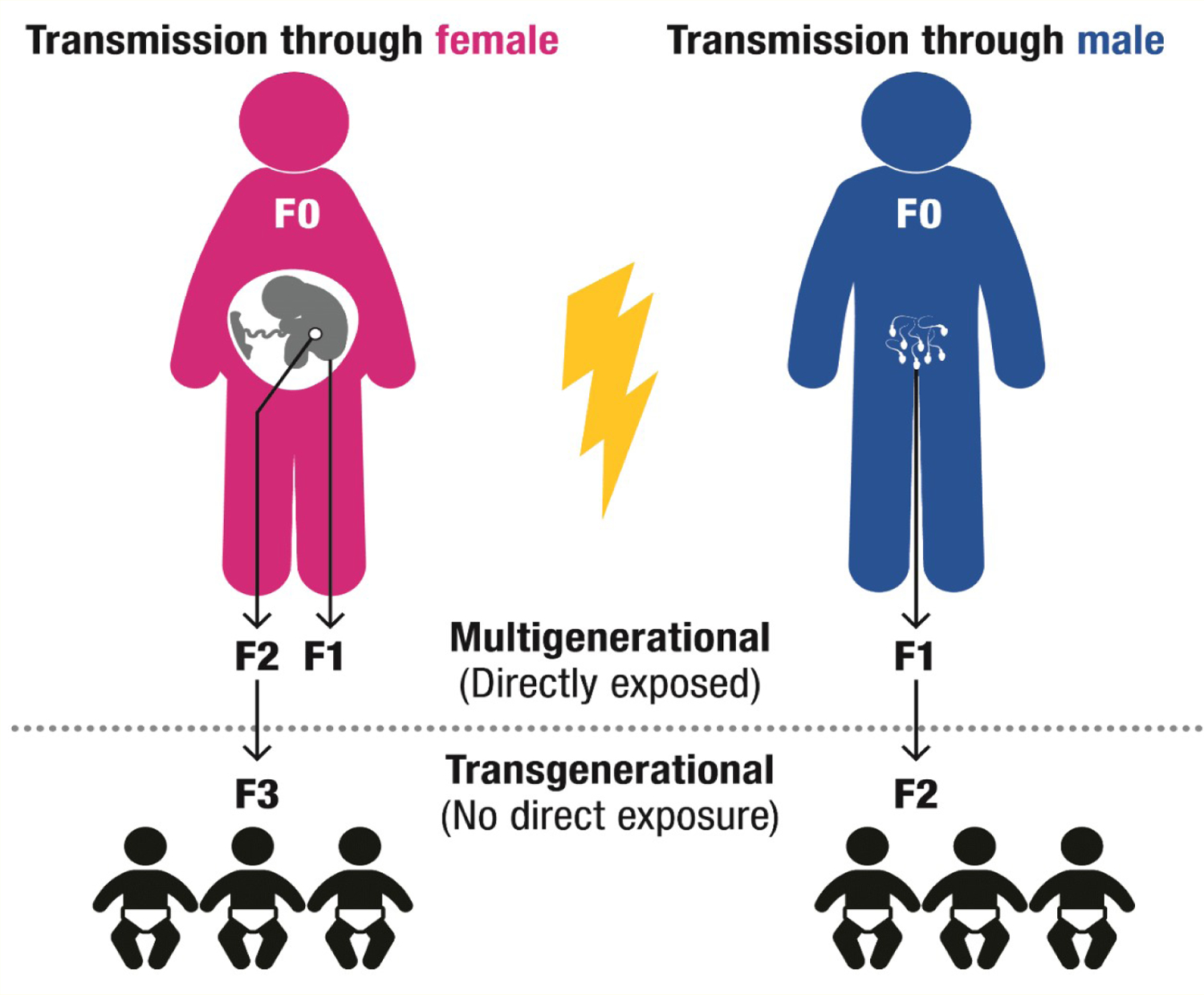
Potential multi- or trans-generational epigenetic alterations.
Bisphenol A
Bisphenol A (BPA) is a monomer used in the manufacturing of polycarbonate plastics and epoxy resins produce a multitude of consumer products, food and drink containers, and medical devices [88]. BPA has demonstrated the ability to be transferred both transplacentally and lactationally into the fetus or infant from the mother [89,90]. Mechanistically, BPA mimics estrogen both in vitro and in vivo, binding to the estrogen receptors ERα and ERβ, although with relatively weak binding affinities [91].
In utero exposure to “safe” or lower than safe doses of BPA (as defined by the current EPA standard of 50 ug/kg/day) has been associated with increased DNA methylation levels, resulting in impaired regulation of insulin and glucose control [46]. Following direct F0 rat exposure to BPA during gestation and lactation, the F1 offspring demonstrated no significant changes in glucose/insulin tolerance by week 3 beyond birth; but by week 21, rat pups showed increased serum insulin and HOMA-IR levels (Homeostatic Model Assessment of Insulin Resistance) as evidenced by analysis of hepatic tissue (Table 1). Additionally, insulin sensitivity and hepatic glycogen storage decreased [47]. In rat pups, significant promoter hypermethylation of Gck gene was observed in the liver of F1 at weeks 3 and 21 when compared with the control [47]. Gck is a key regulator of glucose use in hepatocytes as Gck initiates glucose metabolism through glucose phosphorylation [92]. Previously, decreased Gck activity was associated with type 2 diabetes mellitus [93], and mutations in the Gck gene have been linked to maturity-onset diabetes of the young (MODY)-a form of diabetes diagnosed in late childhood [94]. Thus, Gck is considered a diabetes susceptibility gene. Furthermore, this alteration in DNA methylation during early development was shown to persist throughout animal maturation [92].
A subsequent study supported that in utero BPA exposure induced the hypermethylation of Gck, and further identified the multigenerational inheritance of these epigenetic mutations into the F2 offspring (Table 1) [46]. F0 exposure during gestation and lactation affected glucose and insulin tolerance in the F2 offspring, predisposing the offspring of each generation to diabetes. This decreased Gck expression provides a possible mechanism underlying glucose and insulin intolerance in subsequent BPA-treated generations since global DNA hypomethylation in liver was previously associated with diabetes in rats [95]. These examples demonstrate multigenerational inheritance of epigenetic alterations that cause diabetes-a primary risk factor for obesity-but further research is needed to study the potential transgenerational inheritance of these epimutations. One important avenue of research is studying germline mutations, such as the methylation status of the Gck promoter region in sperm following BPA exposure, as a vehicle for transgenerational inheritance.
Dichlorodiphenyltrichloroethane
Dichlorodiphenyltrichloroethane (DDT) is historically one of the most commonly used insecticides [96]. While DDT has been banned in the U.S. since 1972 and by the UN for most uses since 2001, many countries have continued spraying DDT for malaria control [96–98]. Subsequently, due to the globalization of food supplies, DDT contaminated food is circumventing the U.S. ban and reaching U.S. consumers [96]. Estimates of global DDT use for disease vector control fluctuate between 4,000 to 5,000 metric tons per year [99]. Furthermore, evidence is accumulating for the association between exposure to DDT and its metabolite dichlorodiphenyldichloroethylene (DDE), diabetes and obesity [100–103]. While transgenerational phenomena have been demonstrated in humans [104], rodents [105], worms [106], flies [107], and plants [108] for DDT-induced diseases, only one study today has examined underlying epigenetic mechanisms of DDT-exposures contributing to the obesity epidemic [54,55].
Ancestral DDT exposure during a critical window of germline development promoted epigenetic transgenerational inheritance of obesity into the F3 rat generation (Table 1) [54,55]. Broadly, the study found some differential DNA methylation regions (DMR) in TUBB3, SLC4A4, and CARM1 genes in F3 sperm that resulted in increased visceral adiposity following both lower dose (25 mg/kg/day) and higher dose (50 mg/kg/day) F0 exposures during gestational development. Both the lower and higher doses of DDT used were consistent with wildlife and human environmental exposure levels [109]. The F2 generation (those directly exposed in utero) were not found to develop obesity, but developed kidney, prostate, and ovarian disease and tumor development as adults. In the F3 generation, 50% of lower dose males and females developed obesity and 75% of the higher dose males (although not females) developed obesity [54,55], suggesting transgenerational transmission of disease through both the female and male germlines-the egg and sperm, respectively. In contrast, the F1 obesity pathology was due to direct exposure of fetal somatic cells and was distinct from the germline exposure mechanism of the F3 generation [110,111]. Lower density CpG regions, also called “CpG deserts”, were identified within DMRs [110]. The CpG desert possesses less methylation capabilities due to a lack of cytosines available to accept methyl groups, and it was suggested to be involved in transgenerational inheritance [110]. However, further studies are needed to determine the functional significance of CpG deserts in epigenetics.
Di-(2-ethylhexyl) phthalate
Di-(2-ethylhexyl) phthalate (DEHP) is widely used as a plasticizer in everyday consumer products. Estimated annual DEHP production is approximately 2 million tons [112]. As a ubiquitous environmental pollutant, DEHP has been found in human amniotic fluid, umbilical cord blood, milk, semen and saliva [112]. Exposures to DEHP have been recently associated with energy imbalance and metabolic disorders [113] making DEHP a concern in the obesity epidemic. DEHP reduces blood glucose utilization and hepatic glycogenesis and glycogenolysis in rats [114]. DEHP-exposed rats also have reduced muscle glucose and lactate transport, reductions in muscle hexokinase and hepatic glucokinase, and glycogen synthesis [115], as well as disrupted pancreas and whole-body glucose homeostasis [116]. Furthermore, recent epidemiological studies identified a positive correlation between increased urinary phthalate metabolites, abdominal obesity and insulin resistance in both adolescent and adult males [117–119]. To date, only one study has examined the epigenetic effects of in utero DEHP exposures on obesity and obesity-related disorders, primarily type 2 diabetes [84]. However, no studies have yet focused their attention on transgenerational epigenetic alterations associated with these diseases.
One group studied the effects of gestational DEHP exposure on insulin signaling molecules and glucose transporter 4 (Glut4 (SLC2A4)) and its epigenome in the gastrocnemius muscle of F1 rat offspring (Table 1) [84]. They found that DEHP-exposed F1 offspring were predisposed to glucometabolic dysfunction at adulthood due to down-regulated Glut4 as a result of hypermethylation of the Glut4 gene promoter. They found a negative correlation between Glut4 expression and methylation level of the CpG islands. These Glut4 epigenetic alterations suggest a mechanism for multigenerational inheritance of obesity and obesity-related phenotypes since Glut4 is the primary GLUT-family gene in adipose and muscle tissues that acts as a major transporter protein for insulin-mediated whole-body glucose uptake [120]. In short, DEHP-induced epigenetic alterations in Glut4 gene appear to be a key component of the observed disposition towards metabolic abnormality, but more studies are needed to identify the potential transgenerational effects and underlying mechanisms leading to obesity.
Tributyltin
Tributyltin (TBT), a marine paint additive, is an obesogenic EDC banned by many international agencies (i.e. International Maritime Organization in 2008) but persists in our water and soil today [121]. TBT directly mediates adipogenesis through the RXR/PPARγ pathway [59,122]. In utero exposures to TBT have been observed to predispose MSCs to the adipocyte lineage at the expense of the osteogenic lineage, through epigenetic imprinting, both in vitro and in vivo [63]. To date, only one group to has studied the transgenerational epigenetic effects of TBT in promoting obesity (Table 1) [58]. They exposed pregnant mice to three different concentrations of TBT in drinking water to deliver approximately 50-fold lower (5.42 nM), 5-fold lower (54.2 nM), and 2-fold higher (542 nM) doses compared with the established no observable adverse effect level (NOAEL) of 25 µg/kg/day for mice [123]. This threshold is comparable to the established human tolerable daily intake of 250 ng/kg/day, derived by applying a 100-fold safety factor to the mouse NOAEL [124,125].
In these experiments, F2 and F3 mice exhibited increased adipose depot (fat storage) weight, had larger adipocyte size, increased adipocyte number, and greater differentiation of MSCs toward the adipocyte lineage [58]. These obesogenic effects occurred despite regulated caloric consumption, suggesting that these results were not attributable to a high caloric diet. Specifically, F1 males demonstrated increased perirenal and interscapular white adipose tissue (WAT) depot weight and adipocyte size and number. F1 females demonstrated increased WAT depot weight and adipocyte size. Both F1 males and females demonstrated minimal weight gain likely due to decreased brown adipose tissue (BAT-associated with high-energy metabolism). In the F3 generation, males showed increased WAT depot weights and adipocyte size and number, and females demonstrated increased adipocyte number and size in all WAT depots except interscapular and ovarian. In all 3 concentrations, prenatal exposure led to increased adipose tissue for at least 3 generations, indicating permanent, transgenerational obesogenic effects through germline alterations [58]. Further research is needed to better understand the mechanisms of TBT-induced adipocyte differentiation.
Mixtures
While the aforementioned studies focus on the epigenetic mechanisms underlying the obesogenic effects of developmental EDC exposures, those studies examine only single chemicals. Arguably, the most significant results will come from experimental studies on the adverse health effects of mixtures, which would more realistically represent daily human exposure to the large number of EDCs in the environment. To date, only one group has studied the epigenetic transgenerational inheritance of obesity via exposure to a chemical mixture (Table 1) [62]. They studied the mixture of BPA, DEHP, and dibutyl phthalate (DBP)-another common plasticizer used in latex adhesives, cosmetics, cellulose plastics, and as a solvent for dyes [126]. These three chemicals have been found in humans [127,128], and are all derived from various plastic products [129]. In this study, rats were exposed to BPA, DEHP, and DBP during gestational development at similar concentrations to single component EDC studies (Table 1). Increased obesity was observed in both F3 females and males, without obesogenic effects in the F1 offspring [62]. The observation that obesity was not seen in the directly exposed F1 generation and only the indirectly exposed F3 generation implies the involvement of a transgenerational mechanism. Analysis of DNA methylation in the altered sperm epigenome identified several epimutations on Gdnf and Esrra genes promoters that have also been previously associated with obesity [130,131]. Further studies are needed to confirm that BPA, DEHP, and DBP exposure will influence Gdnf and Esrra obesogenic effects in adipocytes. Furthermore, studies investigating a number of other EDC mixtures are needed to determine the combined epigenetic mechanisms fueling obesogenic effects relevant to our daily exposures. For instance, recent work integrated environmental, epidemiologic, genomic, and bioinformatics approaches to better apply laboratory data to the general population and identify the common environmental and molecular risk factors between breast cancer and endometriosis [132]. More integrated approaches as outlined by this methodological framework could be used to more effectively apply laboratory studies to a more realistic population burden of EDC exposures.
The present review aims to stimulate experimental laboratory research by identifying important biological pathways and EDCs candidates for investigation. Specific avenues of laboratory research might include in vitro and in vivo studies that should be conducted using exposure to selected endocrine-disrupting chemicals either individually or as mixtures. Specific receptors or pathway nodes of interest identified using an integrated or combined in vitro, in vivo, bioinformatics, computational systems biology/chemistry/or toxicology approaches could be technically evaluated by application of genomic, proteomic, or metabolomics methods. Thus, researchers could examine potential linkages for combined exposures to specific EDCs, cellular pathway alterations, and metabolic disturbances related to the development of important clinical outcomes [133].
For example, DEHP is converted after ingestion to mono-(2-ethylhexyl)phthalate (active metabolite of DEHP) which directly activates PPAR-gamma, induces its co-repressor N-CoR release and promotes recruitment of coactivators TRIP2 and PPARGC1 (PGC1-alpha). Mono (2-ethylhexyl) phthalate-induced fat cell differentiation may contribute to the development of obesity and associated metabolic disorders [113]. It is known that Tributyltin induces fat cell differentiation by direct activation of PPAR-gamma and, possibly, RXR-alpha and promotes recruitment of the coactivators NCOA2 (GRIP1/TIF2) [63]. Activated PPAR-gamma, RXR-alpha and/or PPAR-gamma/RXR-alpha dimers stimulate transcription of early adipogenic markers A-FABP [59,63], PPAR-gamma and the late marker Leptin. In addition, Tributyltin inhibits expression of adipogenesis inhibitor DLK via an unknown pathway [63].
Mono (2-ethylhexyl) phthalate and tributyltin-dependent expression of PPAR-gamma and RXR-alpha targets leads to fat cell differentiation [63,113]. DEHP and tributyltin are considered obesogens, which carry out their action via stimulation of PPAR-gamma (Figure 3). By integrating available information and bridging the gap between toxicology, epidemiology, computational methods, bioinformatics and chemistry within the world of disease mechanisms, scientific community can look further beyond the primary target of the individual or combined chemicals by several steps down the relevant pathway [133].
Figure 3:
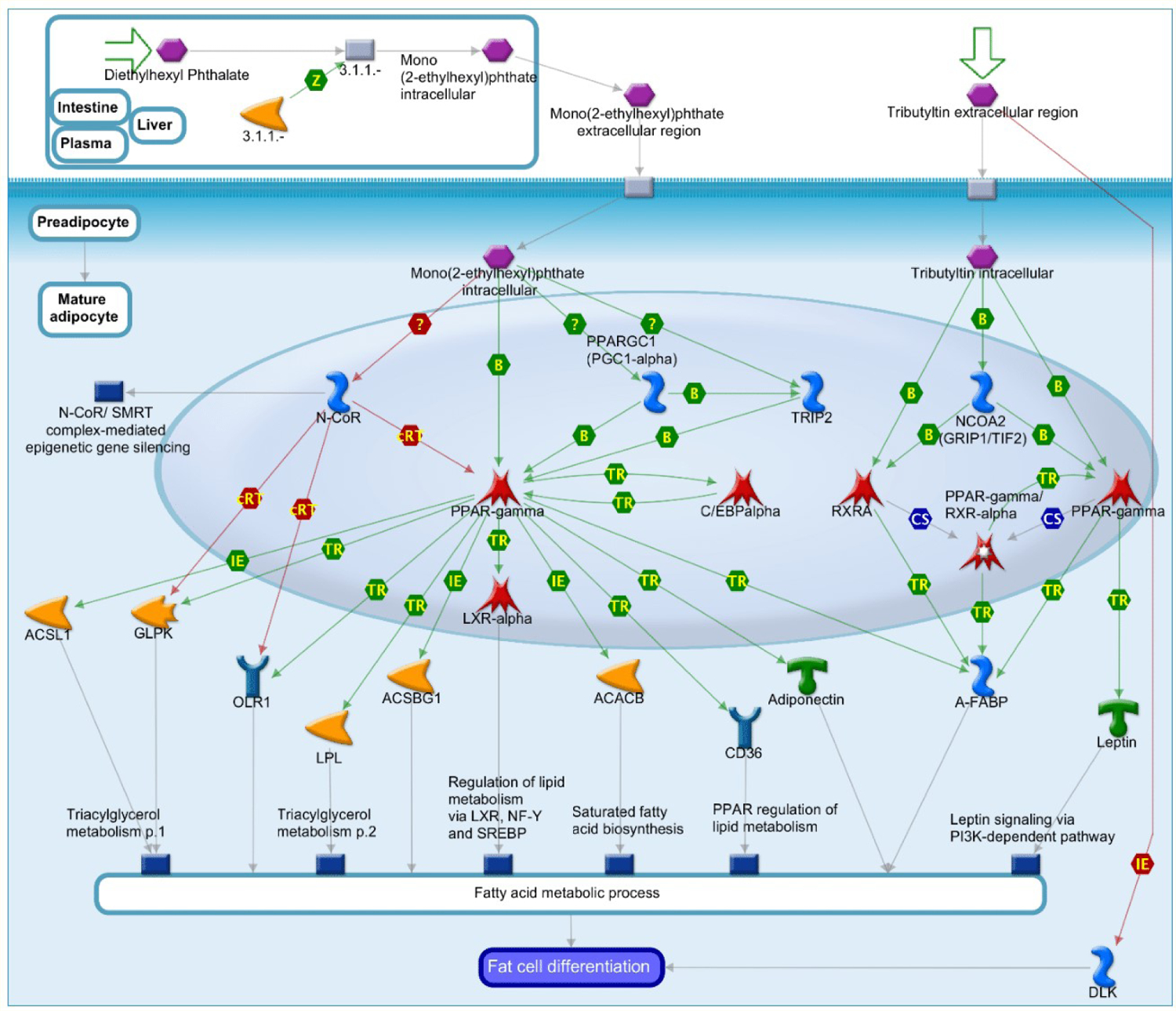
Role of Diethylhexyl Phthalate and Tributyltin in fat cell differentiation. Green arrows = activating interactions; red arrows = inhibiting interactions. Catalytic factors = yellow; transcription factors = red; cytokines and lipoproteins = green; receptors and adaptor proteins = blue.
Future Work and Conclusions
Determining the potential impact of prenatal and postnatal EDCs exposure compared to the risk of childhood and adult onset diseases is a current challenge to regulatory government agencies. The increasing evidence that EDCs exposures may produce epigenetics marks associated with epigenetic phenotypes that can extend beyond a current population exposed to EDCs to future generations is persuasive. This potential transgenerational transmission of obesity by EDCs is important and highlights a major public health concern. While several studies have begun to reveal the epigenetic mechanisms underlying transgenerational obesogenic effects following developmental EDC exposures (F3 generation for F0 exposure during pregnancy), these can exert a transgenerational epigenetic effect on phenotypes [46,47,54,55,58,62,84]. However, it is important to mention that not all studies have reported similar results. These incongruences may be related to differences in the administration route of EDC exposure or experimental protocols, which may influence the transgenerational epigenetic effect. More research needs to be completed in an integrated approach to disease etiology, especially around realistic concentrations of environmentally relevant EDCs or EDC mixtures in humans and animals. The initial integrated studies are those that examine mixtures relevant to actual human exposures, as opposed to single EDCs in isolation. Subsequent studies need to incorporate environmental, epidemiologic, genomic, and bioinformatics studies into conclusions about the effects of EDC mixture exposures on obesity. Certainly, determination of environmentally relevant EDC concentrations that humans may be exposed to daily would be a helpful next step in these EDC or EDCs mixture studies, as they will be key to illustrating the distinction between multigenerational versus transgenerational epigenetic inheritance.
Understanding the molecular determinants of established epigenetic memory across generations would provide essential insight into how environmental changes can affect the health of exposed individuals, as well as subsequent generations. Studying the epigenetic inheritance of EDC-induced obesity and other diseases may reveal findings that might help curb the health effects associated with EDCs exposure. A better understanding of the network and biological pathway mechanisms underlying how EDC exposures affect adipogenesis, transgenerational inherence, and obesity are still needed. Acceptance, implementation, as well as new methods and new requirements for deep sequencing-based expression analysis, will show major advances in robustness and inter-lab transferability over microarray platforms for gene expression analysis. Chemical-gene-disease interaction information can also be applied for designing animal and cell-based laboratory experiments that can continue to investigate the established transgenerational obesity hypotheses more effectively. Furthermore, there is a need to understand the windows of susceptibility of different vulnerable populations’ exposure to EDCs and the associated effects. Very little is known about associations between dietary intakes and potential obesogenic-EDCs exposure. Future research needs to focus on investigating the comparative contributions of genetic and epigenetic changes to transgenerational effects of EDCs.
Innovative methods of analysis such as deep sequence, microarrays, machine learning, bioinformatics, and computational tools are available and can be used to identify specific outcome pathways from complex data. The use of these methods or integration of these methods may help identify particular and sensitive biomarkers. Cluster identification of biomarkers as signatures of individual or combined exposure may help advance the development of EDC mixtures risk assessment methods. Biomonitoring and epidemiological studies need to assess important markers related to metabolic diseases such as obesity. The sensitivity and specificity of these available biomarkers that are influenced by a range of modifying factors (chemical mixture components, diet, genetics, age, sex, ethnicity, diseases, gut biota, etc.) can be studied using multiple sophisticated techniques. Innovative biomarkers could be developed for their use in human population studies, disease prevention and clinical use to detect individual or multiple chemical exposures.
The body burden of multiple chemicals, particularly those EDCs considered obesogenic chemicals should be considered within the larger framework for obesity and other chronic diseases prevention. Further investigations carried out to study the influence of factors such as transgenerational effects, multiple chemical exposures, nutrition, age, gender, ethnicity and genetic variations will help develop personalized specific treatment protocols for these complex diseases.
Acknowledgements
The findings and conclusions in this report are those of the author(s) and do not necessarily represent the official position of the Centers for Disease Control and Prevention/the Agency for Toxic Substances and Disease Registry. Mention of trade names is not an endorsement of any commercial product. The authors declare they have no actual or potential financial interests. David Albuquerque is recipient of a post-doctoral grant (SFRH/BPD/109043/2015) from Fundação para a Ciência e a Tecnologia (FCT).
Disclaimer
The findings and conclusions in this report are those of the author(s) and do not necessarily represent the official position of the Centers for Disease Control and Prevention/the Agency for Toxic Substances and Disease Registry.
Bibliography
- 1.Koletzko B, et al. “Early Nutrition Programming of Long-Term Health”. Proceedings of the Nutrition Society 71.3 (2012): 371–378. [DOI] [PubMed] [Google Scholar]
- 2.Zama AM and Uzumcu M. “Epigenetic Effects of Endocrine-Disrupting Chemicals on Female Reproduction: An Ovarian Perspective”. Frontiers in Neuroendocrinology 31.4 (2010): 420–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flegal KM., et al. “Prevalence of Obesity and Trends in the Distribution of Body Mass Index among Us Adults, 1999–2010”. JAMA 307.5 (2012): 491–497. [DOI] [PubMed] [Google Scholar]
- 4.Ogden CL., et al. “Prevalence of Childhood and Adult Obesity in the United States, 2011–2012”. JAMA 311.8 (2014): 806–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lustig RH. “Childhood Obesity: Behavioral Aberration or Biochemical Drive? Reinterpreting the First Law of Thermodynamics”. Nature Clinical Practice Endocrinology and Metabolism 2.8 (2006): 447–458. [DOI] [PubMed] [Google Scholar]
- 6.Freedland ES “Role of a Critical Visceral Adipose Tissue Threshold (Cvatt) in Metabolic Syndrome: Implications for Controlling Dietary Carbohydrates: A Review”. Nutrition & Metabolism (Lond) 1.1 (2004): 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swinburn BA., et al. “The Global Obesity Pandemic: Shaped by Global Drivers and Local Environments”. Lancet 378.9793 (2011): 804–814. [DOI] [PubMed] [Google Scholar]
- 8.Finucane MM., et al. “National, Regional, and Global Trends in Body-Mass Index since 1980: Systematic Analysis of Health Examination Surveys and Epidemiological Studies with 960 Country-Years and 9.1 Million Participants”. Lancet 377.9765 (2011): 557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.NCD-RisC. “Trends in Adult Body-Mass Index in 200 Countries from 1975 to 2014: A Pooled Analysis of 1698 Population-Based Measurement Studies with 19.2 Million Participants”. Lancet 387.10026 (2016): 1377–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hill JO and Peters JC. “Environmental Contributions to the Obesity Epidemic”. Science 280.5368 (1998): 1371–1374. [DOI] [PubMed] [Google Scholar]
- 11.Power C and Jefferis BJ. “Fetal Environment and Subsequent Obesity: A Study of Maternal Smoking”. International Journal of Epidemiology 31.2 (2002): 413–419. [PubMed] [Google Scholar]
- 12.Braithwaite EC., et al. “Maternal Prenatal Depressive Symptoms Predict Infant Nr3c1 1f and Bdnf Iv DNA Methylation”. Epigenetics 10.5 (2015): 408–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frias AE and Grove KL. “Obesity: A Transgenerational Problem Linked to Nutrition During Pregnancy”. Seminars in Reproductive Medicine 30.6 (2012): 472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Albuquerque D, et al. “Epigenetics of Human Obesity: A Link between Genetics and Nutrition”. In: Molecular Mechanisms Underpinning the Development of Obesity. Eds. Nóbrega Clévio and Rodriguez-López Raquel. Cham: Springer International Publishing; (2014): 101–127. [Google Scholar]
- 15.Gary Felsenfeld. “A Brief History of Epigenetics”. Cold Spring Harbor Perspectives in Biology 6.1 (2014): a018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stel J and Legler J. “The Role of Epigenetics in the Latent Effects of Early Life Exposure to Obesogenic Endocrine Disrupting Chemicals”. Endocrinology 156.10 (2015): 3466–4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bernal AJ and Jirtle RL. “Epigenomic Disruption: The Effects of Early Developmental Exposures”. Birth Defects Research Part A: Clinical and Molecular Teratology 88.10 (2010): 938–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones PA and Takai D. “The Role of DNA Methylation in Mammalian Epigenetics”. Science 293.5532 (2001): 1068–1070. [DOI] [PubMed] [Google Scholar]
- 19.Sadakierska-Chudy A and Filip M. “A Comprehensive View of the Epigenetic Landscape. Part Ii: Histone Post-Translational Modification, Nucleosome Level, and Chromatin Regulation by Ncrnas”. Neurotoxicity Research 27.2 (2015): 172–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sadakierska-Chudy A, et al. “A Comprehensive View of the Epigenetic Landscape Part I: DNA Methylation, Passive and Active DNA Demethylation Pathways and Histone Variants”. Neurotoxicity Research 27 (2015): 84–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xin F, et al. “Multigenerational and Transgenerational Effects of Endocrine Disrupting Chemicals: A Role for Altered Epigenetic Regulation?” Seminars in Cell and Developmental Biology 43 (2015): 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castel SE and Martienssen RA. “Rna Interference in the Nucleus: Roles for Small Rnas in Transcription, Epigenetics and Beyond”. Nature Reviews Genetics 14.2 (2013): 100–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mercer Tim R and Mattick John S. “Structure and Function of Long Noncoding Rnas in Epigenetic Regulation”. Nature Structural and Molecular Biology 20.3 (2013): 300–307. [DOI] [PubMed] [Google Scholar]
- 24.Remely M, et al. “Obesity: Epigenetic Regulation - Recent Observations”. BioMolecular Concepts 6.3 (2015): 163–175. [DOI] [PubMed] [Google Scholar]
- 25.Bhandari RK., et al. “Transgenerational Effects from Early Developmental Exposures to Bisphenol a or 17alpha-Ethinylestradiol in Medaka, Oryzias Latipes”. Science Reports 5 (2015): 9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.LaRocca J, et al. “First-Trimester Urine Concentrations of Phthalate Metabolites and Phenols and Placenta Mirna Expression in a Cohort of U.S. Women”. Environmental Health Perspectives (2015). [DOI] [PMC free article] [PubMed]
- 27.Reik W, et al. “Epigenetic Reprogramming in Mammalian Development”. Science 293.5532 (2001): 1089–1093. [DOI] [PubMed] [Google Scholar]
- 28.Liu FL., et al. “Epigenetic Regulation and Related Diseases During Placental Development”. Yi Chuan 39.4 (2017): 263–275. [DOI] [PubMed] [Google Scholar]
- 29.Maccani MA and Marsit CJ. “Epigenetics in the Placenta”. American Journal of Reproductive Immunology 62.2 (2009): 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaenisch R and Bird A. “Epigenetic Regulation of Gene Expression: How the Genome Integrates Intrinsic and Environmental Signals”. Nature Genetics 33 (2003): 245–254. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X and Ho SM. “Epigenetics Meets Endocrinology”. Journal of Molecular Endocrinology 46.1 (2011): R11–R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dolinoy DC., et al. “Metastable Epialleles, Imprinting, and the Fetal Origins of Adult Diseases”. Pediatric Research 61.5 (2007): 30R–37R. [DOI] [PubMed] [Google Scholar]
- 33.Skinner MK., et al. “Environmentally Induced Epigenetic Transgenerational Inheritance of Sperm Epimutations Promote Genetic Mutations”. Epigenetics 10.8 (2015): 762–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guerrero-Bosagna C., et al. “Identification of Genomic Features in Environmentally Induced Epigenetic Transgenerational Inherited Sperm Epimutations”. PLoS One 9.6 (2014): e100194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chamorro-Garcia R and Blumberg B. “Transgenerational Effects of Obesogens and the Obesity Epidemic”. Current Opinion in Pharmacology 19 (2014): 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nilsson EE and Skinner MK. “Environmentally Induced Epigenetic Transgenerational Inheritance of Disease Susceptibility”. Tranlational Research 165.1 (2015): 12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guibert S and Weber M. “Functions of DNA Methylation and Hydroxymethylation in Mammalian Development”. Current Topics in Developmental Biology 104 (2013): 47–83. [DOI] [PubMed] [Google Scholar]
- 38.Hall KD., et al. “Energy Balance and Its Components: Implications for Body Weight Regulation”. The American Journal of Clinical Nutrition 95.4 (2012): 989–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ng M, et al. “Global, Regional, and National Prevalence of Overweight and Obesity in Children and Adults During 1980–2013: A Systematic Analysis for the Global Burden of Disease Study 2013”. Lancet 384.9945 (2014): 766–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Albuquerque D, et al. “Current Review of Genetics of Human Obesity: From Molecular Mechanisms to an Evolutionary Perspective”. Molecular Genetics and Genomics 290.4 (2015): 1191–221. [DOI] [PubMed] [Google Scholar]
- 41.Klimentidis YC, et al. “Canaries in the Coal Mine: A Cross-Species Analysis of the Plurality of Obesity Epidemics”. Proceedings Biological Sciences 278.1712 (2011): 1626–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Janesick A and Blumberg B. “Endocrine Disrupting Chemicals and the Developmental Programming of Adipogenesis and Obesity”. Birth Defects Research part C Embryo Today 93.1 (2011): 34–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baillie-Hamilton PF. “Chemical Toxins: A Hypothesis to Explain the Global Obesity Epidemic”. Journal of Alternative and Complementary Medicine 8.2 (2002): 185–192. [DOI] [PubMed] [Google Scholar]
- 44.WHO/UNEP. “State of the Science of Endocrine Disrupting Chemicals -- 2012” (2013). [DOI] [PubMed]
- 45.Diamanti-Kandarakis E, et al. “Endocrine-Disrupting Chemicals: An Endocrine Society Scientific Statement”. Endocrine Reviews 30.4 (2009): 293–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li G, et al. “F0 Maternal Bpa Exposure Induced Glucose Intolerance of F2 Generation through DNA Methylation Change in Gck”. Toxicology Letters 228.3 (2014): 192–199. [DOI] [PubMed] [Google Scholar]
- 47.Ma Y, et al. “Hepatic DNA Methylation Modifications in Early Development of Rats Resulting from Perinatal Bpa Exposure Contribute to Insulin Resistance in Adulthood”. Diabetologia 56.9 (2013): 2059–2067. [DOI] [PubMed] [Google Scholar]
- 48.Kelishadi., et al. “Role of Environmental Chemicals in Obesity: A Systematic Review on the Current Evidence”. International Journal of Environmental Research and Public Health 2013 (2013): 896789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Elobeid MA., et al. “Endocrine Disruptors and Obesity: An Examination of Selected Persistent Organic Pollutants in the Nhanes 1999–2002 Data”. International Journal of Environmental Research and Public Health 7.7 (2010): 2988–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Cock M and van de Bor M. “Obesogenic Effects of Endocrine Disruptors, What Do We Know from Animal and Human Studies?” Environment International 70 (2014): 15–24. [DOI] [PubMed] [Google Scholar]
- 51.La Merrill M and Birnbaum LS. “Childhood Obesity and Environmental Chemicals”. Mount Sinai Journal of Medicine 78.1 (2011): 22–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Janesick and Blumberg. “Obesogens, Stem Cells and the Developmental Programming of Obesity”. International Journal of Andrology 35.3 (2012): 437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Snedeker SM and Hay AG. “Do Interactions between Gut Ecology and Environmental Chemicals Contribute to Obesity and Diabetes?” Environmental Health Perspectives 120.3 (2012): 332–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Skinner MK., et al. “Epigenetic Transgenerational Actions of Environmental Factors in Disease Etiology”. Trends in Endocrinology and Metabolism 21.4 (2010): 214–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Skinner MK., et al. “Ancestral Dichlorodiphenyltrichloroethane (Ddt) Exposure Promotes Epigenetic Transgenerational Inheritance of Obesity”. BMC Medicine 11 (2013): 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rubin BS. “Bisphenol A: An Endocrine Disruptor with Widespread Exposure and Multiple Effects”. Journal of Steroid Biochemistry and Molecular Biology 127.1–2 (2011): 27–34. [DOI] [PubMed] [Google Scholar]
- 57.Rubin BS., et al. “Perinatal Exposure to Low Doses of Bisphenol a Affects Body Weight, Patterns of Estrous Cyclicity, and Plasma Lh Levels”. Environmental Health Perspectives 109.7 (2001): 675–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chamorro-Garcia R, et al. “Transgenerational Inheritance of Increased Fat Depot Size, Stem Cell Reprogramming, and Hepatic Steatosis Elicited by Prenatal Exposure to the Obesogen Tributyltin in Mice”. Environmental Health Sciences 121.3 (2013): 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grun F, et al. “Endocrine-Disrupting Organotin Compounds Are Potent Inducers of Adipogenesis in Vertebrates”. Molecular Endocrinology 20.9 (2006): 2141–2155. [DOI] [PubMed] [Google Scholar]
- 60.Newbold RR., et al. “Perinatal Exposure to Environmental Estrogens and the Development of Obesity”. Molecular Nutrition nd Food Research 51.7 (2007): 912–917. [DOI] [PubMed] [Google Scholar]
- 61.Hines EP., et al. “Phenotypic Dichotomy Following Developmental Exposure to Perfluorooctanoic Acid (Pfoa) in Female Cd-1 Mice: Low Doses Induce Elevated Serum Leptin and Insulin, and Overweight in Mid-Life”. Molecular Cellular Endocrinology 304.1–2 (2009): 97–105. [DOI] [PubMed] [Google Scholar]
- 62.Manikkam M, et al. “Plastics Derived Endocrine Disruptors (Bpa, Dehp and Dbp) Induce Epigenetic Transgenerational Inheritance of Obesity, Reproductive Disease and Sperm Epimutations”. PLOS One 8.1 (2013): e55387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kirchner S, et al. “Prenatal Exposure to the Environmental Obesogen Tributyltin Predisposes Multipotent Stem Cells to Become Adipocytes”. Molecular Endocrinology 24.3 (2010): 526–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thayer KA., et al. “Role of Environmental Chemicals in Diabetes and Obesity: A National Toxicology Program Workshop Review”. Environmental Health Perspectives 120.6 (2012): 779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Budge H, et al. “Maternal Nutritional Programming of Fetal Adipose Tissue Development: Long-Term Consequences for Later Obesity”. Birth Defects Research Part C: Embryo Today 75.3 (2005): 193–199. [DOI] [PubMed] [Google Scholar]
- 66.Correia-Branco AE., et al. “Maternal Undernutrition and Fetal Developmental Programming of Obesity: The Glucocorticoid Connection”. Reproductive Sciences 22.2 (2015): 138–145. [DOI] [PubMed] [Google Scholar]
- 67.Boekelheide K, et al. “Predicting Later-Life Outcomes of Early-Life Exposures”. Environmental Health Sciences 120.10 (2012): 1353–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cristancho Ana G and Lazar Mitchell A. “Forming Functional Fat: A Growing Understanding of Adipocyte Differentiation”. Nature Reviews Molecular Cell Biology 12.11 (2011): 722–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Janesick AS., et al. “Transgenerational Inheritance of Prenatal Obesogen Exposure”. Molecular and Cellular Endocrinology 398.1–2 (2014): 31–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Crisan M, et al. “A Perivascular Origin for Mesenchymal Stem Cells in Multiple Human Organs”. Cell Stem Cell 3.3 (2008): 301–313. [DOI] [PubMed] [Google Scholar]
- 71.Bianco P “Back to the Future: Moving Beyond “Mesenchymal Stem Cells”. Journal of Cellular Biochemistry 112.7 (2011): 1713–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rosen ED and Spiegelman BM. “What We Talk About When We Talk About Fat”. Cell 156.1–2 (2014): 20–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gupta RK., et al. “Zfp423 Expression Identifies Committed Preadipocytes and Localizes to Adipose Endothelial and Perivascular Cells”. Cell Metabolism 15.2 (2012): 230–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kang S, et al. “Regulation of Early Adipose Commitment by Zfp521”. PLOS Biology 10.11 (2012): e1001433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Carnevalli LS., et al. “S6k1 Plays a Critical Role in Early Adipocyte Differentiation”. Developmental Cell 18.5 (2010): 763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Remillard RB and Bunce NJ. “Linking Dioxins to Diabetes: Epidemiology and Biologic Plausibility”. Environmental Health Perspectives 110.9 (2002): 853–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kersten S “Peroxisome Proliferator Activated Receptors and Obesity”. European Journal of Pharmacology 440.2–3 (2002): 223–234. [DOI] [PubMed] [Google Scholar]
- 78.Shockley KR., et al. “Ppargamma2 Regulates a Molecular Signature of Marrow Mesenchymal Stem Cells”. PPAR Research 2007 (2007): 81219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zamani N and Brown CW. “Emerging Roles for the Transforming Growth Factor-{Beta} Superfamily in Regulating Adiposity and Energy Expenditure”. Endocrine Reviews 32.3 (2011): 387–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kang S, et al. “Wnt Signaling Stimulates Osteoblastogenesis of Mesenchymal Precursors by Suppressing Ccaat/Enhancer-Binding Protein Alpha and Peroxisome Proliferator-Activated Receptor Gamma”. Journal of Biological Chemistry 282.19 (2007): 14515–14524. [DOI] [PubMed] [Google Scholar]
- 81.Kawai M, et al. “Wnt/Lrp/Beta-Catenin Signaling Suppresses Adipogenesis by Inhibiting Mutual Activation of Ppargamma and C/Ebpalpha”. Biochemical and Biophysical Research Communications 363.2 (2007): 276–282. [DOI] [PubMed] [Google Scholar]
- 82.Takada I, et al. “Suppression of Ppar Transactivation Switches Cell Fate of Bone Marrow Stem Cells from Adipocytes into Osteoblasts”. Annals of the New York Academy of Sciences 1116 (2007): 182–195. [DOI] [PubMed] [Google Scholar]
- 83.Sun L, et al. “Long Noncoding Rnas Regulate Adipogenesis”. Proceedings of the National Academy of Sciences of the United States of America 110.9 (2013): 3387–3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rajesh P and Balasubramanian K. “Phthalate Exposure in Utero Causes Epigenetic Changes and Impairs Insulin Signalling”. Journal of Endocrinology 223.1 (2014): 47–66. [DOI] [PubMed] [Google Scholar]
- 85.Biemann RB., et al. “Adipogenic Effects of a Combination of the Endocrine-Disrupting Compounds Bisphenol a, Diethylhexylphthalate, and Tributyltin”. Obesity Facts 7.1 (2014): 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lubrano C, et al. “ Obesity and Metabolic Comorbidities: Environmental Diseases?” Oxidative Medicine and Cellular Longevity 2013 (2013): 640673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gore AC, et al. “Edc-2: The Endocrine Society’s Second Scientific Statement on Endocrine-Disrupting Chemicals”. Endocrine Reviews 36.6 (2015): E1–E150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chianese R, et al. “Bisphenol a in Reproduction: Epigenetic Effects”. Current Medicinal Chemistry (2017). [DOI] [PubMed]
- 89.vom Saal FS., et al. “Chapel Hill Bisphenol a Expert Panel Consensus Statement: Integration of Mechanisms, Effects in Animals and Potential to Impact Human Health at Current Levels of Exposure”. Reproductive Toxicology 24.2 (2007): 131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vandenberg LN., et al. “Bisphenol-a and the Great Divide: A Review of Controversies in the Field of Endocrine Disruption”. Endocrine Reviews 30.1 (2009): 75–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Calafat AM, et al. “Exposure of the U.S. Population to Bisphenol a and 4-Tertiary-Octylphenol: 2003–2004”. Environmental Health Perspectives 116.1 (2008): 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Walker DG and Rao S. “The Role of Glucokinase in the Phosphorylation of Glucose by Rat Liver”. Biochemical Journal 90.2 (1964): 360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Caro JF., et al. “Liver Glucokinase: Decreased Activity in Patients with Type Ii Diabetes”. Hormone and Metabolic Research 27.1 (1995): 19–22. [DOI] [PubMed] [Google Scholar]
- 94.Flack JR., et al. “Gck Monogenic Diabetes and Gestational Diabetes: Possible Diagnosis on Clinical Grounds”. Diabetic Medicine 32.12 (2015): 1596–601. [DOI] [PubMed] [Google Scholar]
- 95.Williams KT., et al. “Type I Diabetes Leads to Tissue-Specific DNA Hypomethylation in Male Rats”. Journal of Nutrition 138.11 (2008): 2064–2069. [DOI] [PubMed] [Google Scholar]
- 96.Sadasivaiah S, et al. “Dichlorodiphenyltrichloroethane (Ddt) for Indoor Residual Spraying in Africa: How Can It Be Used for Malaria Control?” American Journal of Tropical Medicine and Hygiene 77.6 (2007): 249–263. [PubMed] [Google Scholar]
- 97.Packard RM. “The Making of a Tropical Disease: A Short History of Malaria.” Johns Hopkins University Press; (2007). [Google Scholar]
- 98.Guimaraes RM., et al. “Ddt Reintroduction for Malaria Control: The Cost-Benefit Debate for Public Health”. Cadernos de Saúde Pública 23.12 (2007): 2835–2844. [DOI] [PubMed] [Google Scholar]
- 99.Kabasenche WP and Skinner MK. “Ddt, Epigenetic Harm, and Transgenerational Environmental Justice”. Environmental Health 13 (2014): 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Valvi D, et al. “Prenatal Concentrations of Polychlorinated Biphenyls, Dde, and Ddt and Overweight in Children: A Prospective Birth Cohort Study”. Environmental Health Perspectives 120.3 (2012): 451–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Heggeseth B, et al. “Detecting Associations between Early-Life Ddt Exposures and Childhood Growth Patterns: A Novel Statistical Approach”. PLoS One 10.6 (2015): e0131443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Warner M, et al. “In Utero Ddt and Dde Exposure and Obesity Status of 7-Year-Old Mexican-American Children in the Chamacos Cohort”. Environmental Health Perspectives 121.5 (2013): 631–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Warner M, et al. “Prenatal Exposure to Dichlorodiphenyltrichloroethane and Obesity at 9 Years of Age in the Chamacos Study Cohort”. American Journal of Epidemiology 179.11 (2014): 1312–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pembrey ME. “Male-Line Transgenerational Responses in Humans”. Human Fertility 13.4 (2010): 268–271. [DOI] [PubMed] [Google Scholar]
- 105.Anway MD., et al. “Epigenetic Transgenerational Actions of Endocrine Disruptors and Male Fertility”. Science 308.5727 (2005): 1466–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Greer EL., et al. “Transgenerational Epigenetic Inheritance of Longevity in Caenorhabditis Elegans”. Nature 479.7373 (2011): 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ruden DM and Lu X. “Hsp90 Affecting Chromatin Remodeling Might Explain Transgenerational Epigenetic Inheritance in Drosophila”. Current Genomics 9.7 (2008): 500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hauser MT., et al. “Transgenerational Epigenetic Inheritance in Plants”. Biochimica et Biophysica Acta 1809.8 (2011): 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.ATSDR. “Toxicological Profile for 4,4′-Ddt, 4,4′-Dde, 4, 4′-Ddd (Update)”. ATSDR: Agency for Toxic Substances and Diseases Registry (ATSDR)/US Public Health Service; 1994 (2015). [Google Scholar]
- 110.Skinner MK and Guerrero-Bosagna C. “Role of Cpg Deserts in the Epigenetic Transgenerational Inheritance of Differential DNA Methylation Regions”. BMC Genomics 15 (2014): 692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jirtle RL and Skinner M. “Environmental Epigenomics and Disease Susceptibility”. Nature Reviews Genetics 8.4 (2007): 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moosa Fabiband., et al. “Human Biological Monitoring of Suspected Endocrine-Disrupting Compounds”. Asian Journal of Andrology 16.1 (2014): 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Desvergne B, et al. “Ppar-Mediated Activity of Phthalates: A Link to the Obesity Epidemic?” Molecular and Cellular Endocrinology 304.1–2 (2009): 43–48. [DOI] [PubMed] [Google Scholar]
- 114.Mushtaq M, et al. “Effect of Di-2-Ethylhexyl Phthalate (Dehp) on Glycogen Metabolism in Rat Liver”. Toxicology 16.2 (1980): 153–161. [DOI] [PubMed] [Google Scholar]
- 115.Martinelli MI., et al. “Dietary Di(2-Ethylhexyl)Phthalate-Impaired Glucose Metabolism in Experimental Animals”. Human and Experimental Toxicology 25.9 (2006): 531–538. [DOI] [PubMed] [Google Scholar]
- 116.Lin Y, et al. “Developmental Exposure to Di(2-Ethylhexyl) Phthalate Impairs Endocrine Pancreas and Leads to Long-Term Adverse Effects on Glucose Homeostasis in the Rat”. American Journal of Physiology Endocrinology and Metabolism 301.3 (2011): E527–E538. [DOI] [PubMed] [Google Scholar]
- 117.Stahlhut Richard W, et al. “Concentrations of Urinary Phthalate Metabolites Are Associated with Increased Waist Circumference and Insulin Resistance in Adult U.S. Males”. Environmental Health Perspectives 115.6 (2007): 876–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hatch EE., et al. “Association of Urinary Phthalate Metabolite Concentrations with Body Mass Index and Waist Circumference: A Cross-Sectional Study of Nhanes Data, 1999–2002”. Environmental Health 7 (2008): 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Trasande L, et al. “Urinary Phthalates and Increased Insulin Resistance in Adolescents”. Pediatrics 132.3 (2013): e646–e655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yokomori N, et al. “DNA Demethylation During the Differentiation of 3t3-L1 Cells Affects the Expression of the Mouse Glut4 Gene”. Diabetes 48.4 (1999): 685–690. [DOI] [PubMed] [Google Scholar]
- 121.Antizar-Ladislao Blanca. “Environmental Levels, Toxicity and Human Exposure to Tributyltin (Tbt)-Contaminated Marine Environment. A Review”. Environment International 34.2 (2008): 292–308. [DOI] [PubMed] [Google Scholar]
- 122.Li XJ Ycaza and Blumberg B. “The Environmental Obesogen Tributyltin Chloride Acts Via Peroxisome Proliferator Activated Receptor Gamma to Induce Adipogenesis in Murine 3t3-L1 Preadipocytes”. The Journal of Steroid Biochemistry and Molecular Biology 127.1–2 (2011): 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Vos JG., et al. “Immunotoxicity of Bis(Tri-N-Butyltin)Oxide in the Rat: Effects on Thymus-Dependent Immunity and on Nonspecific Resistance Following Long-Term Exposure in Young Versus Aged Rats”. Toxicology and Applied Pharmacology 105.1 (1990): 144–155. [DOI] [PubMed] [Google Scholar]
- 124.Airaksinen R, et al. “Organotin Intake through Fish Consumption in Finland”. Environmental Research 110.6 (2010): 544–547. [DOI] [PubMed] [Google Scholar]
- 125.Fristachi A, et al. “Using Probabilistic Modeling to Evaluate Human Exposure to Organotin in Drinking Water Transported by Polyvinyl Chloride Pipe”. Risk Analysis 29.11 (2009): 1615–1628. [DOI] [PubMed] [Google Scholar]
- 126.Heudorf U, et al. “Phthalates: Toxicology and Exposure”. International Journal of Hygiene and Environmental Health 210.5 (2007): 623–634. [DOI] [PubMed] [Google Scholar]
- 127.Talsness CE., et al. “Components of Plastic: Experimental Studies in Animals and Relevance for Human Health”. Philosophical Transactions of the Royal Society B: Biological Sciences 364.1526 (2009): 2079–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Maffini MV., et al. “Endocrine Disruptors and Reproductive Health: The Case of Bisphenol-A”. Molecular and Cellular Endocrinology 254–255 (2006): 179–186. [DOI] [PubMed] [Google Scholar]
- 129.Simoneau C, et al. “Identification and Quantification of the Migration of Chemicals from Plastic Baby Bottles Used as Substitutes for Polycarbonate”. Food Additives and Contaminants: Part A: Chemistry, Analysis, Control, Exposure and Risk Assessment 29.3 (2012): 469–480. [DOI] [PubMed] [Google Scholar]
- 130.Baudry C, et al. “Diet-Induced Obesity Has Neuroprotective Effects in Murine Gastric Enteric Nervous System: Involvement of Leptin and Glial Cell Line-Derived Neurotrophic Factor”. Journal of Physiology 590.3 (2012): 533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Slocum N, et al. “Responses of Brown Adipose Tissue to Diet-Induced Obesity, Exercise, Dietary Restriction and Ephedrine Treatment”. Experimental and Toxicologic Pathology 65.5 (2013): 549–557. [DOI] [PubMed] [Google Scholar]
- 132.Roy D, et al. “Integrated Bioinformatics, Environmental Epidemiologic and Genomic Approaches to Identify Environmental and Molecular Links between Endometriosis and Breast Cancer”. International Journal of Molecular Sciences 16.10 (2015): 25285–25322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ruiz P, et al. “A Systems Biology Approach Reveals Converging Molecular Mechanisms That Link Different Pops to Common Metabolic Diseases”. Environmental Health Perspectives 124.7 (2016): 1034–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]