

Abstract
Metastasis is an inefficient process in which the vast majority of cancer cells are fated to die, partly because they experience oxidative stress. Metastasizing cancer cells migrate through diverse environments that differ dramatically from their tumor of origin, leading to redox imbalances. The rare metastasizing cells that survive undergo reversible metabolic changes that confer oxidative stress resistance. We review the changes in redox regulation that cancer cells undergo during metastasis. By better understanding these mechanisms, it may be possible to develop pro-oxidant therapies that block disease progression by exacerbating oxidative stress in cancer cells.
Significance:
Oxidative stress often limits cancer cell survival during metastasis, raising the possibility of inhibiting cancer progression with pro-oxidant therapies. This is the opposite strategy of treating patients with antioxidants, an approach that worsened outcomes in large clinical trials.
Introduction
Metastasis is the leading cause of death in patients with cancer because disseminated disease is no longer curable by surgery and is often therapy-resistant (1). Metastasis requires cancer cells to delaminate from their tumor of origin, invade the surrounding tissue, then migrate through tissue, blood, and/or lymph to new sites, all while surviving diverse and changing environments (2). Very few cancer cells survive this process, and many that do are unable to proliferate or persist in metastatic sites (3–6).
Cancer cells must be plastic to survive metastasis (7, 8). Genetic heterogeneity increases with disease progression (9), contributing to therapy resistance (10). Whole-genome duplications, chromosomal rearrangements, and chromosomal instability contribute to the increase in genetic heterogeneity (9, 11, 12). The genetic changes do not appear to confer metastatic competence, but rather arise by chance within primary tumors and are positively or negatively selected during metastasis (8, 11, 13). For example, copy-number changes in MYC (14) or MAPK pathway components (15) can enhance survival during metastasis. Recurrent coding sequence mutations have generally not been observed to arise during metastasis (11, 15–17), suggesting that there are not specific metastasis suppressor mutations. Rather, cancer cells undergo epigenetic (18, 19), transcriptional (7, 20–22), and metabolic (23–25) changes during metastasis. These reversible sources of heterogeneity conspire with genetic heterogeneity to confer fitness upon rare cells to survive and grow in metastatic sites.
Multiple factors contribute to the death of cancer cells during metastasis, including immune-mediated destruction (26, 27), growth factor deprivation (28), and diverse metabolic stresses (29). Redox stress is one important metabolic stress that limits the survival of cancer cells (24, 30). We review the role of redox regulation in metastasis and the metabolic adaptations that confer oxidative stress resistance.
Metastasizing Cells Experience Oxidative Stress
Cancer cells experience oxidative stress during certain critical phases of their evolution and progression. The mechanisms that cause cancer cells to experience oxidative stress are poorly understood but likely include hyperactivation of anabolic pathways (31, 32), increased mitochondrial function (33), malfunction of the electron transport chain as a result of mitochondrial DNA mutations (34, 35), and oncogenic pathway activation (36–38). As a consequence, cancer cells are often more dependent than normal cells upon cellular antioxidants including glutathione (39, 40), thioredoxin (39), antioxidant enzymes (e.g., glutathione peroxidase, ref. 41; catalase, ref. 42; and superoxide dismutase, refs. 43, 44), and their transcriptional regulators, such as Nrf2 (45, 46) and BACH1 (ref. 47; Fig. 1A). Glutathione is an abundant redox buffer that is present mainly in the reduced form within cells. It opposes the development of oxidative stress by neutralizing (reducing) reactive oxygen species (ROS) including oxygen free radicals, peroxides, and lipid peroxides, as well as by glutathionylating thiol groups on proteins to protect them from oxidation (Fig. 1B). Glutathione can be regenerated from its oxidized form, glutathione disulfide, by glutathione reductase, using a reducing equivalent from NADPH. Consequently, metabolic pathways that generate NADPH from NADP+ are important sources of reducing equivalents for oxidative stress resistance (ref. 48; Fig. 1A).
Figure 1.
![Figure 1. Metabolic pathways that generate NADPH are important sources of reducing equivalents for oxidative stress resistance. A, Glutathione (GSH) and thioredoxin (TRXred) are redox buffers that are used by antioxidant enzymes such as superoxide dismutase (SOD), peroxiredoxin (PRDX), and glutathione peroxidase 4 (GPX4) to neutralize ROS, including O2−, H2O2, and lipid ROS. The reduced forms of GSH and TRX can then be regenerated from the oxidized forms [glutathione disulfide (GSSG); TRXox] by glutathione reductase (GR) or thioredoxin reductase (TRXR), which obtain reducing equivalents from NADPH. NADP+ is generated de novo from NAD+ by NAD kinase (NADK; ref. 167). NADP+ is then reduced to NADPH by the pentose phosphate pathway, the folate pathway, malic enzyme (ME1, 2, or 3), glutamate dehydrogenase (GDH1/2), or isocitrate dehydrogenase (IDH1/2; ref. 86). Other abbreviations in this panel include electron transport chain (ETC), glucose-6-phosphate dehydrogenase (G6PD), phosphogluconate dehydrogenase (PGD), dihydrofolate reductase (DHFR), methylenetetrahydrofolate dehydrogenase 1/2 (MTHFD1/2), NADPH oxidase (NOX), superoxide dismutase (SOD), and catalase (CAT). B, Schematic of reactions in which antioxidant enzymes transfer reducing equivalents between NADPH, GSH, and ROS.](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/79cb/9306307/c469072f6481/2682fig1.jpg)
Metabolic pathways that generate NADPH are important sources of reducing equivalents for oxidative stress resistance. A, Glutathione (GSH) and thioredoxin (TRXred) are redox buffers that are used by antioxidant enzymes such as superoxide dismutase (SOD), peroxiredoxin (PRDX), and glutathione peroxidase 4 (GPX4) to neutralize ROS, including O2−, H2O2, and lipid ROS. The reduced forms of GSH and TRX can then be regenerated from the oxidized forms [glutathione disulfide (GSSG); TRXox] by glutathione reductase (GR) or thioredoxin reductase (TRXR), which obtain reducing equivalents from NADPH. NADP+ is generated de novo from NAD+ by NAD kinase (NADK; ref. 167). NADP+ is then reduced to NADPH by the pentose phosphate pathway, the folate pathway, malic enzyme (ME1, 2, or 3), glutamate dehydrogenase (GDH1/2), or isocitrate dehydrogenase (IDH1/2; ref. 86). Other abbreviations in this panel include electron transport chain (ETC), glucose-6-phosphate dehydrogenase (G6PD), phosphogluconate dehydrogenase (PGD), dihydrofolate reductase (DHFR), methylenetetrahydrofolate dehydrogenase 1/2 (MTHFD1/2), NADPH oxidase (NOX), superoxide dismutase (SOD), and catalase (CAT). B, Schematic of reactions in which antioxidant enzymes transfer reducing equivalents between NADPH, GSH, and ROS.
Cancer cells that survive the oxidative stress they experience during transformation (39) are able to bring oxidative stress under control, allowing the activation of anabolic pathways to drive tumor growth (31). However, when cells in primary tumors detach from extracellular matrix during invasion, they experience changes in signaling pathway activation and metabolism that again increase oxidative stress (49–51). There is evidence that cancer cells either proliferate or invade surrounding tissues but generally do not do both at the same time (52, 53), raising the possibility that invasion requires cells to downregulate anabolic pathways.
Oxidative stress likely increases further when metastasizing cancer cells enter the blood (24, 54–57), which has among the highest levels of oxidants in the body, including oxygen and iron. Oxidative stress limits the survival of cancer cells during metastasis (24). Treatment of mice with antioxidants increases the frequency of circulating cancer cells in the blood and metastatic disease burden (24, 30, 47, 58, 59). This has been observed in multiple cancers, in patient-derived xenografts growing in immunocompromised mice as well as in mouse cancers growing in syngeneic immunocompetent mice. Consistent with this, cancer cells undergo metabolic changes during invasion and metastasis that would be expected to reduce the generation of ROS (60–65).
Nascent metastatic nodules continue to exhibit signs of oxidative stress, including increased ROS levels and low ratios of glutathione to oxidized glutathione and NADPH to NADP+ (24), although the degree of oxidative stress differs among sites of metastasis (66). Oxidative stress is likely to slow the ability of metastatic cells, at least in some sites, to fully reactivate the anabolic pathways required for tumor growth, even after they have extravasated from the blood. For example, lipogenesis requires reducing equivalents from NADPH; inhibiting acetyl-CoA carboxylase decreases NADPH consumption by fatty acid synthesis and preserves NADPH for other cellular processes (67). Cancer cells shut down anabolic pathways during metastasis to conserve reducing equivalents to manage oxidative stress. Indeed, it is conceivable that dormancy in metastatic cells is sometimes caused by a prolonged failure to fully bring oxidative stress under control, leading to prolonged quiescence. Nonetheless, once metastatic tumors have grown beyond a few millimeters in diameter, cancer cells likely have undergone the adaptations needed to control oxidative stress, allowing broad activation of anabolic pathways.
Dietary supplementation with antioxidants has been proposed to provide health benefits, including suppressing the development of cancer by reducing ROS levels (68). Consequently, many clinical trials have been performed to test whether dietary supplementation with antioxidants can suppress the development of cancer. However, dietary antioxidants have consistently failed to reduce cancer incidence or cancer-related deaths in human clinical trials (69). Consistent with the data from experimental models, dietary supplementation with antioxidants in humans tended to increase cancer incidence and cancer-related deaths (70–73). The data thus suggest that antioxidants generally promote the development and progression of cancer in both animal models and in humans.
Although oxidative stress commonly limits the survival of cancer cells during transformation and metastasis, ROS also promotes cancer progression in certain contexts (74). ROS can cause DNA damage, contributing to the formation of oncogenic mutations, and can serve as progrowth signaling molecules (33). Genetic changes that increase the generation of ROS can promote cancer progression, and treatment with antioxidants has sometimes been observed to inhibit metastasis (75–79). For example, inhibition of TIGAR, an enzyme that promotes the entry of glucose into the pentose phosphate pathway, increases ROS levels in pancreatic ductal adenocarcinoma, leading to increased migration, invasion, and metastasis (80). One possibility is that modest increases in ROS levels can promote the activation of signaling pathways that are adaptive for cancer cells (33), particularly in early-stage cancers, while the higher ROS levels observed in metastasizing cancer cells are toxic. Another possibility is that different types of ROS have different effects on cancer cells. For example, hydrogen peroxide created by mitochondrial ROS might promote metastasis (81), whereas lipid peroxides created by membrane lipid oxidation might undermine survival during metastasis (55).
There may also be differences among cancers or model systems, in which oxidative stress limits disease progression in certain cancers while promoting disease progression in others. It is conceivable that mouse models of cancer tend to have lower ROS levels than human cancers due to lower mutation burdens. Cancer cell lines may have been selected for the capacity to withstand oxidative stress as a result of being propagated in culture. These will be important possibilities to consider as the field dissects the role of ROS and oxidative stress in cancer progression.
Mechanisms of Oxidative Stress Resistance During Metastasis
There are heritable, stable, and cell-intrinsic differences among cancers in their metastatic potential based on metabolic and transcriptional differences, including those that confer oxidative stress resistance (54, 82, 83). There is also heterogeneity among cancer cells within the same tumor that influences metastatic potential (54, 84, 85). For example, melanoma cells within hypoxic regions of primary tumors express higher levels of the lactate transporter MCT1, and higher levels of MCT1 confer oxidative stress resistance that increases survival in the blood (54). MCT1 seems to promote oxidative stress resistance by increasing lactate uptake, which decreases intracellular pH and the NAD+/NADH ratio. This promotes pentose phosphate pathway function, a major source of NADPH for oxidative stress resistance (86). Consistent with this, hypoxic cells within primary tumors exhibit transcriptional changes that appear to confer an oxidative stress–resistant phenotype that promotes the survival of metastasizing cells in the blood, increasing their potential to form metastatic tumors (87). Increased MCT1 expression may be one element of this phenotype.
De novo serine synthesis (88) and serine degradation (89) both yield NADPH and are used by cancer cells to manage oxidative stress, particularly during hypoxia. Although cancer cells that metastasize through the blood would not be expected to be hypoxic, these pathways nonetheless promote metastasis, potentially by acting in cancer cells within hypoxic environments (e.g., in lymph or after extravasation into metastatic sites). Inhibition of either phosphoglycerate dehydrogenase, an enzyme involved in serine synthesis, or serine hydroxymethyltransferase, an enzyme involved in serine degradation, increases ROS levels and reduces the formation of metastatic tumors (88, 89). Serine biosynthesis also preferentially promotes the growth of metastatic tumors as compared with primary tumors by promoting mTORC1 signaling (90). It is not clear whether the change in mTORC1 signaling contributes to the change in ROS levels. ROS also induces the expression of β-globin, the oxygen-binding protein best known for its function in erythrocytes, in circulating breast cancer cells (57). This appears to protect the cancer cells from oxidative stress, perhaps by scavenging ROS.
There is genetic evidence that some cancers, including melanoma and lung cancer, give rise to polyclonal metastases (91, 92). There are likely multiple cellular mechanisms that contribute to the formation of polyclonal metastases, including metastasis-to-metastasis spread (93). Another mechanism that may contribute to polyclonal metastasis is that some circulating cancer cells move through the blood in clusters. Clustering can occur among cancer cells or between cancer cells and neutrophils. In both cases it increases cancer cell survival and their ability to form metastatic tumors as compared with single circulating cancer cells (94–96). Clustering may promote the survival of cancer cells in the blood partly by reducing their exposure to oxygen, reducing the production of mitochondrial ROS (97). E-cadherin expression by metastasizing cells also promotes survival by limiting oxidative stress (98). It is tempting to speculate that E-cadherin acts by promoting cell–cell interaction, although E-cadherin deletion does not reduce the fraction of cancer cells that are present in cell clusters.
Oxidative stress kills metastasizing cancer cells by inducing ferroptosis (55, 56), a form of cell death marked by lipid oxidation (Fig. 2A; ref. 99). During ferroptosis, polyunsaturated fatty acids (PUFA) in membrane phospholipids are oxidized by redox-active iron. The resulting lipid peroxides can be scavenged by dietary antioxidants such as vitamin E or by certain cellular antioxidant defenses, such as GPX4 (100–102); however, accumulation of the lipid peroxides can overwhelm the cellular antioxidant defenses, leading to the induction of ferroptosis. At least in melanoma, ferroptosis does not appear to limit the growth of primary cutaneous tumors, in which little oxidative stress is evident, but does limit the survival of metastasizing cells (55). Circulating melanoma cells attempt to manage lipid oxidation by increasing the transcription of transferrin, which reduces intracellular iron levels and lipid peroxidation (56), and by increasing the incorporation of monounsaturated fatty acids (MUFA) into membrane lipids to displace PUFAs (55). Ferroptosis sensitivity marks a therapy-resistant cell state that is observed across several cancers, including melanoma, and that involves the increased synthesis of PUFAs (103), including polyunsaturated ether phospholipids (104). This raises the possibility that many cancers may become more sensitive to ferroptosis during metastasis and that disease progression could be inhibited by interventions that increase lipid peroxidation (85, 103–105).
Figure 2.
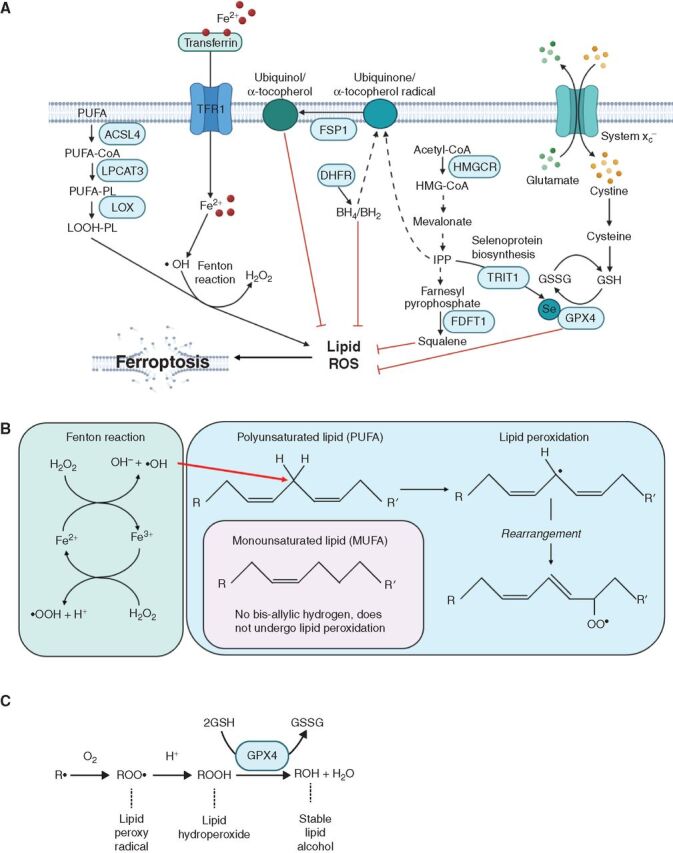
The regulation of ferroptosis. A, Lipid ROS, including lipid peroxides, arise as a result of the oxidation of polyunsaturated fatty acids (PUFA), driven by Fenton reactions in which redox active iron generates hydroxyl radicals (•OH). These PUFAs are present in membrane phospholipids (PL). Cells have multiple antioxidant defenses that oppose the accumulation of lipid ROS including the selenocystine (Se) enzyme, glutathione peroxidase 4 (GPX4), and the reducing agents squalene (100), tetrahydrobiopterin (BH4; ref. 105), and ubiquinol/α-tocopheral. Abbreviations include transferrin receptor protein 1 (TFR1), acyl-CoA synthetase long-chain family member 4 (ACSL4), lysophosphatidylcholine acyltransferase 3 (LPCAT3), lysyl oxidase (LOX), six-transmembrane epithelial antigen of prostate 3 (STEAP3), divalent metal transporter 1 (DMT1), ferroptosis suppressor protein 1 (FSP1; refs. 168, 169), dihydrofolate reductase (DHFR), 3-Hydroxy-3-Methylglutaryl-CoA Reductase (HMGCR), TRNA Isopentenyltransferase 1 (TRIT1), glutathione (GSH), glutathione disulfide (GSSG), farnesyl-diphosphate farnesyltransferase 1 (FDFT1). B, Schematic of reactions in which iron generates hydroxyl radicals (•OH) that react with bis-allylic hydrogens in PUFAs to generate lipid ROS (99). C, Generation of stable lipid alcohols from lipid ROS by GPX4.
The susceptibility of metastasizing cancer cells to ferroptosis appears to be influenced by both cell-autonomous lipid metabolism and by lipids taken up from the environment. Fatty acid transporters, including CD36, tend to be more highly expressed by cancer cells as compared with normal cells and promote metastasis or poor prognosis in multiple cancers (106, 107). Stearoyl-CoA desaturase (SCD1) is involved in the conversion of saturated to MUFAs in melanoma cells. Melanomas that are high for the Microphthalmia-associated transcription factor (MITF), which promotes aggressive proliferation but suppresses invasion (108), are dependent upon SCD1, perhaps to sustain membrane lipid biosynthesis (85, 109, 110). In contrast, melanomas that are low for MITF and less proliferative but more invasive are less dependent upon SCD1 (85). One possibility is that these MITFlo melanomas become more dependent upon MUFAs taken up from their environment during metastasis (55) because there is less SCD1-mediated production of MUFAs cell-intrinsically.
The literature on the effects of a high-fat diet on cancer is mixed (111). Some studies found that high-fat diets (112, 113) or dietary supplementation with palmitic acid, a saturated fatty acid (106), can promote metastasis. Other studies found that ketogenic high-fat diets can reduce metastatic disease burden, partly by increasing oxidative stress in cancer cells (114, 115). One possibility is that variability in outcomes among studies reflects differences in the PUFA or MUFA content of the diets that were administered. Many factors likely contribute to these differences in outcomes, including differences among high-fat diets in fatty acid, protein, and carbohydrate composition. In addition to the effects of fatty acids on redox homeostasis, fatty acids also play critical roles in membrane biosynthesis and energy metabolism that have effects on cancer progression independent of the effects on redox status (85, 116, 117).
Metastasis Through Lymphatics
Many cancers, including epithelial cancers and melanomas, form metastases in draining lymph nodes prior to forming metastases at distant sites (118–121). Genetic studies in human and mouse cancers have shown that regional lymph node metastases can give rise to distant metastases (91–93, 122). In mouse models, cancer cells in lymph nodes are capable of metastasizing to distant sites through the blood (123–125). However, some distant metastases arise from clones that differ from those in lymph nodes. In these instances, it is possible the metastatic cells entered the blood directly from primary tumors, or transited through lymphatics without forming lymph node tumors (92, 122). Obviously, it is also possible that they formed lymph node tumors that were neither detected nor sampled for analysis.
Lymphatics promote the migration and survival of cancer cells. Some cancers form more tumors after intralymphatic injection as compared with intravenous injection (55, 126). VEGFC and various chemokines promote the migration of cancer cells into lymphatic vessels, facilitating metastatic spread (127–131). When VEGFC is overexpressed in mouse lungs, it increases lymphatic vessel density, increasing the spread of cancer cells from the lung to other organs (131). The capacity to oxidize fatty acids promotes the survival of cancer cells in lymphatics (132) and their formation of metastatic tumors (106). Consistent with this, fatty acid oxidation promotes oxidative stress resistance and metastatic potential in colorectal cancer cells (133).
Melanoma cells that metastasize through lymph are metabolically different from cells that metastasize through blood (55). Melanoma cells in lymph experience less oxidative stress and form more metastases than melanoma cells in the blood (55). One of the ways in which lymph protects from ferroptosis is by having high levels of the MUFA oleic acid, which protects cells from lipid oxidation by reducing the abundance of PUFAs in membranes. PUFAs, but not MUFAs, are oxidized during ferroptosis due to the bis-allylic hydrogens they contain (Fig. 2B and C; ref. 99). Compared with the blood, lymph also contains lower concentrations of oxygen and iron, oxidants that contribute to ferroptosis (55). These observations suggest that melanoma cells tend to metastasize initially through lymphatics because lymph protects them from oxidative stress. Moreover, while in lymph, cancer cells increase MUFA incorporation into phospholipids, reducing their susceptibility to ferroptosis when they subsequently enter the blood.
Mitochondrial Function as a Determinant of Metastasis
Mitochondrial function has been studied only to a limited extent in cancer cells during metastasis, leaving many questions unanswered. One of the key impediments is that circulating cancer cells are rare, making it difficult to obtain enough cells for many assays. Nonetheless, mitochondria are a major source of ROS in cells and there is increasing evidence that mitochondrial function reduces the survival of metastasizing cancer cells, at least partly by increasing ROS levels (134). Mitochondrial mass and mitochondrial membrane potential decline in circulating melanoma cells in the blood as compared with the primary tumors from which they arise (24). One possibility is that these changes reflect decreased mitochondrial function in an effort to manage the production of mitochondrial ROS. However, flow cytometric measurements of mitochondrial membrane potential do not always correlate with mitochondrial or electron chain function (135). Lung cancer cell lines with metastatic potential have lower mitochondrial membrane potential and reduced mitochondrial function as compared with nonmetastatic lung cancer cell lines (136). PGC1α, a transcription factor that promotes mitochondrial biogenesis, seems to promote invasion and metastasis in some contexts (76) while inhibiting metastasis in others, including in melanoma (84, 137). Melanoma cells in primary tumors are heterogeneous for PGC1α expression, with PGC1αlo cells exhibiting increased metastatic potential, again consistent with the idea that reduced mitochondrial function promotes metastasis (84). However, there are many mechanisms downstream of PGC1α that appear to contribute to its effects on metastasis, including mechanisms independent of mitochondrial function (76, 84, 137). Additional studies of mitochondrial function during metastasis are required.
Metabolic pathways associated with mitochondrial function influence metastatic potential. For example, increased asparagine availability, either from the diet or from biosynthesis, promotes metastasis (138). Asparagine is synthesized from aspartate, and aspartate synthesis depends on electron transport chain function (139–141). This raises the possibility that asparagine is limiting in metastasizing cancer cells because mitochondrial function is limited in an effort to manage oxidative stress (136).
Pro-Oxidant Therapies
The studies above suggest that cancer progression might be inhibited with pro-oxidant therapies that exacerbate oxidative stress in cancer cells or block the metabolic adaptations that confer oxidative stress resistance (ref. 142; Fig. 3). The anticancer activity of radiation reflects, in part, the formation of hydroxyl radicals that attack DNA (143). Widely used chemotherapies, including procarbazine, paclitaxel, daunorubicin, and doxorubicin, kill cancer cells partly by promoting oxidative stress (144–146). Many small-molecule drugs with direct or indirect pro-oxidant effects have been tested in clinical trials for a wide range of cancers (147), and new strategies for developing prooxidant small molecules are being explored (148, 149). For example, Imexon is a small molecule that binds to thiols, depleting glutathione and increasing ROS levels, which has been tested for activity against non-Hodgkin lymphoma (150). Arsenic trioxide is used for the treatment of acute promyelocytic leukemia and may act partly by impairing electron transport chain function, leading to electron leakage and the generation of superoxide (151). These ROS-generating agents might damage mitochondrial DNA, which is more vulnerable to ROS than nuclear DNA (152), further increasing the generation of ROS as a result of defects in electron transport chain function (153). While a number of effective anticancer therapies have pro-oxidant effects, it is uncertain to what extent their anticancer activities reflect these pro-oxidant activities as compared with other activities independent of ROS.
Figure 3.

Potential pro-oxidant therapies. It may be possible to inhibit the metastasis or progression of some cancers using pro-oxidant therapies that exacerbate the oxidative stress experienced by cancer cells.
Ascorbate (vitamin C) is generally considered an antioxidant, but it exists in oxidized and reduced forms and when it is infused intravenously it selectively kills cancer cells by acting as a pro-oxidant (154). This is because the superphysiologic levels of ascorbate that can be achieved by intravenous infusion lead to the uptake of the fully oxidized form of ascorbate, dehydroascorbate, via the GLUT1 transporter, which is highly expressed in cancer cells with MAPK pathway activation. Once taken up by the cancer cells, dehydroascorbate is reduced back to ascorbate, inducing oxidative stress by consuming reducing equivalents. Ascorbate also alters the activity of epigenetic enzymes, such as TET2, which use ascorbate as a cofactor (155, 156). Building on the original studies by Linus Pauling that reported prolonged survival in patients with cancer administered high-dose intravenous ascorbate (157), the recent work demonstrating the pro-oxidant and epigenetic effects of high-dose ascorbate has led to a number of clinical trials testing activity against a wide range of cancers (158).
Dietary interventions could also have pro-oxidant effects. Ketogenic diets may suppress metastasis partly by increasing oxidative stress in cancer cells (114). Ketogenic diets are designed to minimize dietary carbohydrates, reducing blood glucose and insulin levels (159). However, ketogenic diets also increase dietary fat, commonly increasing PUFA levels. Increased incorporation of PUFAs into membrane phospholipids renders cancer cells more susceptible to the accumulation of lipid ROS and ferroptosis (160). This raises the possibility that ketogenic diets may exert anticancer effects partly by altering lipid metabolism (161) or by increasing PUFA levels in the membranes of cancer cells (162). Nonetheless, it remains to be tested whether a high PUFA diet or other approaches to promote PUFA incorporation into cancer cells could inhibit disease progression.
Future Directions
New technical approaches to study metastasis, including whole-body imaging of metastasis patterns (163), improved techniques for the isolation of circulating cancer cells from patients (164), screens to identify gene products that modulate metastasis (165, 166), and lineage tracing of bar-coded cancer cells to trace routes of metastasis (83), are accelerating progress.
In at least some cancers, metastasizing cells appear to experience unusually high levels of oxidative stress, raising the possibility that these cells might be particularly sensitive to pro-oxidant therapies. It is an open question whether such therapies could prevent disease progression in patients with high-risk primary or regionally metastatic lesions. Nonetheless, this merits deeper study in preclinical models. Beyond this big-picture question, there are a number of pressing biological questions central to understanding redox regulation during metastasis:
Does oxidative stress limit the survival of metastasizing cells from all cancers or only certain cancers?
What causes the oxidative stress experienced by metastasizing cells?
Are anabolic pathways downregulated in metastasizing cells to preserve reducing equivalents? Does this sometimes lead to dormancy in metastatic cells?
How is mitochondrial function modulated in metastasizing cancer cells as compared with the primary tumors from which they arise?
Do micrometastases continue to experience oxidative stress? For how long?
To what extent do interactions with immune and stromal cells influence oxidative stress in cancer cells?
Do differences in oxidative stress among distinct metastatic sites influence organotropism?
Acknowledgments
S.J. Morrison is a Howard Hughes Medical Institute Investigator, the Mary McDermott Cook Chair in Pediatric Genetics, the Kathryn and Gene Bishop Distinguished Chair in Pediatric Research, the director of the Hamon Laboratory for Stem Cells and Cancer, and a Cancer Prevention and Research Institute of Texas Scholar. This work was supported by the Cancer Prevention and Research Institute of Texas (RP170114 and RP180778) and by the NIH (U01 CA228608). A. Tasdogan was supported by the Leopoldina Fellowship (LPDS 2016-16) from the German National Academy of Sciences and the Fritz Thyssen Foundation. All figures were generated using BioRender (paid license).
Authors' Disclosures
S.J. Morrison reports personal fees from Kojin Therapeutics and other support from G1 Therapeutics outside the submitted work. No disclosures were reported by the other authors.
References
- 1. Lambert AW, Pattabiraman DR, Weinberg RA. Emerging biological principles of metastasis. Cell 2017;168:670–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vanharanta S, Massague J. Origins of metastatic traits. Cancer Cell 2013;24:410–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, et al. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol 1998;153:865–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cameron MD, Schmidt EE, Kerkvliet N, Nadkarni KV, Morris VL, Groom AC, et al. Temporal progression of metastasis in lung: cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res 2000;60:2541–6. [PubMed] [Google Scholar]
- 5. Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WE, Goldbrunner R, Herms J, et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med 2010;16:116–22. [DOI] [PubMed] [Google Scholar]
- 6. Sela Y, Li J, Kuri P, Merrell AJ, Li N, Lengner C, et al. Dissecting phenotypic transitions in metastatic disease via photoconversion-based isolation. Elife 2021;10:e63270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marjanovic ND, Hofree M, Chan JE, Canner D, Wu K, Trakala M, et al. Emergence of a high-plasticity cell state during lung cancer evolution. Cancer Cell 2020;38:229–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ganesh K, Massague J. Targeting metastatic cancer. Nat Med 2021;27:34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bailey C, Black JRM, Reading JL, Litchfield K, Turajlic S, McGranahan N, et al. Tracking cancer evolution through the disease course. Cancer Discov 2021;11:916–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Salgueiro L, Buccitelli C, Rowald K, Somogyi K, Kandala S, Korbel JO, et al. Acquisition of chromosome instability is a mechanism to evade oncogene addiction. EMBO Mol Med 2020;12:e10941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Priestley P, Baber J, Lolkema MP, Steeghs N, de Bruijn E, Shale C, et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019;575:210–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Watkins TBK, Lim EL, Petkovic M, Elizalde S, Birkbak NJ, Wilson GA, et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature 2020;587:126–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jacob LS, Vanharanta S, Obenauf AC, Pirun M, Viale A, Socci ND, et al. Metastatic competence can emerge with selection of preexisting oncogenic alleles without a need of new mutations. Cancer Res 2015;75:3713–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shih DJH, Nayyar N, Bihun I, Dagogo-Jack I, Gill CM, Aquilanti E, et al. Genomic characterization of human brain metastases identifies drivers of metastatic lung adenocarcinoma. Nat Genet 2020;52:371–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shain AH, Joseph NM, Yu R, Benhamida J, Liu S, Prow T, et al. Genomic and transcriptomic analysis reveals incremental disruption of key signaling pathways during melanoma evolution. Cancer Cell 2018;34:45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hu Z, Li Z, Ma Z, Curtis C. Multi-cancer analysis of clonality and the timing of systemic spread in paired primary tumors and metastases. Nat Genet 2020;52:701–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reiter JG, Makohon-Moore AP, Gerold JM, Heyde A, Attiyeh MA, Kohutek ZA, et al. Minimal functional driver gene heterogeneity among untreated metastases. Science 2018;361:1033–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McDonald OG, Li X, Saunders T, Tryggvadottir R, Mentch SJ, Warmoes MO, et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet 2017;49:367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vanharanta S, Shu W, Brenet F, Hakimi AA, Heguy A, Viale A, et al. Epigenetic expansion of VHL-HIF signal output drives multiorgan metastasis in renal cancer. Nat Med 2013;19:50–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roe JS, Hwang CI, Somerville TDD, Milazzo JP, Lee EJ, Da Silva B, et al. Enhancer reprogramming promotes pancreatic cancer metastasis. Cell 2017;170:875–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Whittle MC, Izeradjene K, Rani PG, Feng L, Carlson MA, DelGiorno KE, et al. RUNX3 controls a metastatic switch in pancreatic ductal adenocarcinoma. Cell 2015;161:1345–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Denny SK, Yang D, Chuang CH, Brady JJ, Lim JS, Gruner BM, et al. Nfib promotes metastasis through a widespread increase in chromatin accessibility. Cell 2016;166:328–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shi X, Tasdogan A, Huang F, Hu Z, Morrison SJ, DeBerardinis RJ. The abundance of metabolites related to protein methylation correlates with the metastatic capacity of human melanoma xenografts. Sci Adv 2017;3:eaao5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015;527:186–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lehuede C, Dupuy F, Rabinovitch R, Jones RG, Siegel PM. Metabolic plasticity as a determinant of tumor growth and metastasis. Cancer Res 2016;76:5201–8. [DOI] [PubMed] [Google Scholar]
- 26. Laughney AM, Hu J, Campbell NR, Bakhoum SF, Setty M, Lavallee VP, et al. Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat Med 2020;26:259–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garner H, de Visser KE. Immune crosstalk in cancer progression and metastatic spread: a complex conversation. Nat Rev Immunol 2020;20:483–97. [DOI] [PubMed] [Google Scholar]
- 28. Lowery FJ, Yu D. Growth factor signaling in metastasis: current understanding and future opportunities. Cancer Metastasis Rev 2012;31:479–91. [DOI] [PubMed] [Google Scholar]
- 29. Bergers G, Fendt SM. The metabolism of cancer cells during metastasis. Nat Rev Cancer 2021;21:162–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Le Gal K, Ibrahim MX, Wiel C, Sayin VI, Akula MK, Karlsson C, et al. Antioxidants can increase melanoma metastasis in mice. Sci Transl Med 2015;7:308re8. [DOI] [PubMed] [Google Scholar]
- 31. Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012;22:66–79. [DOI] [PubMed] [Google Scholar]
- 32. Ju HQ, Lin JF, Tian T, Xie D, Xu RH. NADPH homeostasis in cancer: functions, mechanisms and therapeutic implications. Signal Transduct Target Ther 2020;5:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sullivan LB, Chandel NS. Mitochondrial reactive oxygen species and cancer. Cancer Metab 2014;2:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Carew JS, Zhou Y, Albitar M, Carew JD, Keating MJ, Huang P. Mitochondrial DNA mutations in primary leukemia cells after chemotherapy: clinical significance and therapeutic implications. Leukemia 2003;17:1437–47. [DOI] [PubMed] [Google Scholar]
- 35. Hahn A, Zuryn S. Mitochondrial genome (mtDNA) mutations that generate reactive oxygen species. Antioxidants 2019;8:392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, et al. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem 1997;272:217–21. [PubMed] [Google Scholar]
- 37. Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, et al. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 1997;275:1649–52. [DOI] [PubMed] [Google Scholar]
- 38. Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, et al. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell 2002;9:1031–44. [DOI] [PubMed] [Google Scholar]
- 39. Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015;27:211–22. [DOI] [PubMed] [Google Scholar]
- 40. Cramer SL, Saha A, Liu J, Tadi S, Tiziani S, Yan W, et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat Med 2017;23:120–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Barrett CW, Ning W, Chen X, Smith JJ, Washington MK, Hill KE, et al. Tumor suppressor function of the plasma glutathione peroxidase gpx3 in colitis-associated carcinoma. Cancer Res 2013;73:1245–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Davison CA, Durbin SM, Thau MR, Zellmer VR, Chapman SE, Diener J, et al. Antioxidant enzymes mediate survival of breast cancer cells deprived of extracellular matrix. Cancer Res 2013;73:3704–15. [DOI] [PubMed] [Google Scholar]
- 43. Gomez ML, Shah N, Kenny TC, Jenkins EC Jr, Germain D. SOD1 is essential for oncogene-driven mammary tumor formation but dispensable for normal development and proliferation. Oncogene 2019;38:5751–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang X, Zhang H, Sapio R, Yang J, Wong J, Zhang X, et al. SOD1 regulates ribosome biogenesis in KRAS mutant non-small cell lung cancer. Nat Commun 2021;12:2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011;475:106–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lignitto L, LeBoeuf SE, Homer H, Jiang S, Askenazi M, Karakousi TR, et al. Nrf2 activation promotes lung cancer metastasis by inhibiting the degradation of Bach1. Cell 2019;178:316–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wiel C, Le Gal K, Ibrahim MX, Jahangir CA, Kashif M, Yao H, et al. BACH1 stabilization by antioxidants stimulates lung cancer metastasis. Cell 2019;178:330–45. [DOI] [PubMed] [Google Scholar]
- 48. Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014;510:298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009;461:109–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hawk MA, Schafer ZT. Mechanisms of redox metabolism and cancer cell survival during extracellular matrix detachment. J Biol Chem 2018;293:7531–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jiang L, Shestov AA, Swain P, Yang C, Parker SJ, Wang QA, et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 2016;532:255–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kohrman AQ, Matus DQ. Divide or conquer: cell cycle regulation of invasive behavior. Trends Cell Biol 2017;27:12–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Matus DQ, Lohmer LL, Kelley LC, Schindler AJ, Kohrman AQ, Barkoulas M, et al. Invasive cell fate requires G1 cell-cycle arrest and histone deacetylase-mediated changes in gene expression. Dev Cell 2015;35:162–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tasdogan A, Faubert B, Ramesh V, Ubellacker JM, Shen B, Solmonson A, et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 2020;577:115–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ubellacker JM, Tasdogan A, Ramesh V, Shen B, Mitchell EC, Martin-Sandoval MS, et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 2020;585:113–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hong X, Roh W, Sullivan RJ, Wong KHK, Wittner BS, Guo H, et al. The lipogenic regulator SREBP2 induces transferrin in circulating melanoma cells and suppresses ferroptosis. Cancer Discov 2021;11:678–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zheng Y, Miyamoto DT, Wittner BS, Sullivan JP, Aceto N, Jordan NV, et al. Expression of beta-globin by cancer cells promotes cell survival during blood-borne dissemination. Nat Commun 2017;8:14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, Bergo MO. Antioxidants accelerate lung cancer progression in mice. Sci Transl Med 2014;6:221ra15. [DOI] [PubMed] [Google Scholar]
- 59. Wang H, Liu X, Long M, Huang Y, Zhang L, Zhang R, et al. NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Sci Transl Med 2016;8:334ra51. [DOI] [PubMed] [Google Scholar]
- 60. Dey S, Sayers CM, Verginadis II, Lehman SL, Cheng Y, Cerniglia GJ, et al. ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J Clin Invest 2015;125:2592–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013;23:316–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kamarajugadda S, Cai Q, Chen H, Nayak S, Zhu J, He M, et al. Manganese superoxide dismutase promotes anoikis resistance and tumor metastasis. Cell Death Dis 2013;4:e504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Qu Y, Wang J, Ray PS, Guo H, Huang J, Shin-Sim M, et al. Thioredoxin-like 2 regulates human cancer cell growth and metastasis via redox homeostasis and NF-kappaB signaling. J Clin Invest 2011;121:212–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chen EI, Hewel J, Krueger JS, Tiraby C, Weber MR, Kralli A, et al. Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res 2007;67:1472–86. [DOI] [PubMed] [Google Scholar]
- 65. Lu X, Bennet B, Mu E, Rabinowitz J, Kang Y. Metabolomic changes accompanying transformation and acquisition of metastatic potential in a syngeneic mouse mammary tumor model. J Biol Chem 2010;285:9317–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Basnet H, Tian L, Ganesh K, Huang YH, Macalinao DG, Brogi E, et al. Flura-seq identifies organ-specific metabolic adaptations during early metastatic colonization. Elife 2019;8:e43627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012;485:661–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Goodman M, Bostick RM, Kucuk O, Jones DP. Clinical trials of antioxidants as cancer prevention agents: past, present, and future. Free Radic Biol Med 2011;51:1068–84. [DOI] [PubMed] [Google Scholar]
- 69. Chandel NS, Tuveson DA. The promise and perils of antioxidants for cancer patients. N Engl J Med 2014;371:177–8. [DOI] [PubMed] [Google Scholar]
- 70. Alpha-Tocopherol BCCPSG. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N Engl J Med 1994;330:1029–35. [DOI] [PubMed] [Google Scholar]
- 71. Klein EA, Thompson IM Jr, Tangen CM, Crowley JJ, Lucia MS, Goodman PJ, et al. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011;306:1549–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Goodman GE, Thornquist MD, Balmes J, Cullen MR, Meyskens FL Jr, Omenn GS, et al. The Beta-Carotene and Retinol Efficacy Trial: incidence of lung cancer and cardiovascular disease mortality during 6-year follow-up after stopping beta-carotene and retinol supplements. J Natl Cancer Inst 2004;96:1743–50. [DOI] [PubMed] [Google Scholar]
- 73. Ebbing M, Bonaa KH, Nygard O, Arnesen E, Ueland PM, Nordrehaug JE, et al. Cancer incidence and mortality after treatment with folic acid and vitamin B12. JAMA 2009;302:2119–26. [DOI] [PubMed] [Google Scholar]
- 74. Chio IIC, Tuveson DA. ROS in cancer: the burning question. Trends Mol Med 2017;23:411–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Porporato PE, Payen VL, Perez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, et al. A mitochondrial switch promotes tumor metastasis. Cell Rep 2014;8:754–66. [DOI] [PubMed] [Google Scholar]
- 76. LeBleu VS, O'Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol 2014;16:992–1003, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008;320:661–4. [DOI] [PubMed] [Google Scholar]
- 78. Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A 2010;107:8788–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Cheung EC, Lee P, Ceteci F, Nixon C, Blyth K, Sansom OJ, et al. Opposing effects of TIGAR- and RAC1-derived ROS on Wnt-driven proliferation in the mouse intestine. Genes Dev 2016;30:52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cheung EC, DeNicola GM, Nixon C, Blyth K, Labuschagne CF, Tuveson DA, et al. Dynamic ROS control by TIGAR regulates the initiation and progression of pancreatic cancer. Cancer Cell 2020;37:168–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Goh J, Enns L, Fatemie S, Hopkins H, Morton J, Pettan-Brewer C, et al. Mitochondrial targeted catalase suppresses invasive breast cancer in mice. BMC Cancer 2011;11:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Quintana E, Piskounova E, Shackleton M, Weinberg D, Eskiocak U, Fullen DR, et al. Human melanoma metastasis in NSG mice correlates with clinical outcome in patients. Sci Transl Med 2012;4:159ra49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Quinn JJ, Jones MG, Okimoto RA, Nanjo S, Chan MM, Yosef N, et al. Single-cell lineages reveal the rates, routes, and drivers of metastasis in cancer xenografts. Science 2021;371:eabc1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Luo C, Lim JH, Lee Y, Granter SR, Thomas A, Vazquez F, et al. A PGC1alpha-mediated transcriptional axis suppresses melanoma metastasis. Nature 2016;537:422–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Vivas-Garcia Y, Falletta P, Liebing J, Louphrasitthiphol P, Feng Y, Chauhan J, et al. Lineage-restricted regulation of SCD and fatty acid saturation by MITF controls melanoma phenotypic plasticity. Mol Cell 2020;77:120–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Chen L, Zhang Z, Hoshino A, Zheng HD, Morley M, Arany Z, et al. NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nat Metab 2019;1:404–15. [PMC free article] [PubMed] [Google Scholar]
- 87. Godet I, Shin YJ, Ju JA, Ye IC, Wang G, Gilkes DM. Fate-mapping post-hypoxic tumor cells reveals a ROS-resistant phenotype that promotes metastasis. Nat Commun 2019;10:4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Samanta D, Park Y, Andrabi SA, Shelton LM, Gilkes DM, Semenza GL. PHGDH expression is required for mitochondrial redox homeostasis, breast cancer stem cell maintenance, and lung metastasis. Cancer Res 2016;76:4430–42. [DOI] [PubMed] [Google Scholar]
- 89. Ye J, Fan J, Venneti S, Wan YW, Pawel BR, Zhang J, et al. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov 2014;4:1406–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Rinaldi G, Pranzini E, Van Elsen J, Broekaert D, Funk CM, Planque M, et al. In vivo evidence for serine biosynthesis-defined sensitivity of lung metastasis, but not of primary breast tumors, to mTORC1 inhibition. Mol Cell 2021;81:386–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. McFadden DG, Papagiannakopoulos T, Taylor-Weiner A, Stewart C, Carter SL, Cibulskis K, et al. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell 2014;156:1298–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sanborn JZ, Chung J, Purdom E, Wang NJ, Kakavand H, Wilmott JS, et al. Phylogenetic analyses of melanoma reveal complex patterns of metastatic dissemination. Proc Natl Acad Sci U S A 2015;112:10995–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMC, Papaemmanuil E, et al. The evolutionary history of lethal metastatic prostate cancer. Nature 2015;520:353–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014;158:1110–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Szczerba BM, Castro-Giner F, Vetter M, Krol I, Gkountela S, Landin J, et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019;566:553–7. [DOI] [PubMed] [Google Scholar]
- 96. Donato C, Kunz L, Castro-Giner F, Paasinen-Sohns A, Strittmatter K, Szczerba BM, et al. Hypoxia triggers the intravasation of clustered circulating tumor cells. Cell Rep 2020;32:108105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Labuschagne CF, Cheung EC, Blagih J, Domart MC, Vousden KH. Cell clustering promotes a metabolic switch that supports metastatic colonization. Cell Metab 2019;30:720–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Padmanaban V, Krol I, Suhail Y, Szczerba BM, Aceto N, Bader JS, et al. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 2019;573:439–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dixon SJ, Stockwell BR. The hallmarks of ferroptosis. Annu Rev Cancer Biol 2019;3:35–54. [Google Scholar]
- 100. Garcia-Bermudez J, Baudrier L, Bayraktar EC, Shen Y, La K, Guarecuco R, et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature 2019;567:118–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Bieri JG. An effect of selenium and cystine on lipide peroxidation in tissues deficient in vitamin E. Nature 1959;184:1148–9. [DOI] [PubMed] [Google Scholar]
- 102. Maiorino M, Conrad M, Ursini F. GPx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues. Antioxid Redox Signal 2018;29:61–74. [DOI] [PubMed] [Google Scholar]
- 103. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017;547:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 2020;585:603–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol 2020;16:1351–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017;541:41–5. [DOI] [PubMed] [Google Scholar]
- 107. Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer 2020;122:4–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Carreira S, Goodall J, Denat L, Rodriguez M, Nuciforo P, Hoek KS, et al. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev 2006;20:3426–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Carreira S, Goodall J, Aksan I, La Rocca SA, Galibert MD, Denat L, et al. Mitf cooperates with Rb1 and activates p21Cip1 expression to regulate cell cycle progression. Nature 2005;433:764–9. [DOI] [PubMed] [Google Scholar]
- 110. Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005;436:117–22. [DOI] [PubMed] [Google Scholar]
- 111. Ludwig DS, Willett WC, Volek JS, Neuhouser ML. Dietary fat: From foe to friend? Science 2018;362:764–70. [DOI] [PubMed] [Google Scholar]
- 112. Chen M, Zhang J, Sampieri K, Clohessy JG, Mendez L, Gonzalez-Billalabeitia E, et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat Genet 2018;50:206–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Pandey V, Vijayakumar MV, Ajay AK, Malvi P, Bhat MK. Diet-induced obesity increases melanoma progression: involvement of Cav-1 and FASN. Int J Cancer 2012;130:497–508. [DOI] [PubMed] [Google Scholar]
- 114. Allen BG, Bhatia SK, Buatti JM, Brandt KE, Lindholm KE, Button AM, et al. Ketogenic diets enhance oxidative stress and radio-chemo-therapy responses in lung cancer xenografts. Clin Cancer Res 2013;19:3905–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Poff AM, Ari C, Seyfried TN, D'Agostino DP. The ketogenic diet and hyperbaric oxygen therapy prolong survival in mice with systemic metastatic cancer. PLoS One 2013;8:e65522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Triki M, Rinaldi G, Planque M, Broekaert D, Winkelkotte AM, Maier CR, et al. mTOR signaling and SREBP activity increase FADS2 expression and can activate sapienate biosynthesis. Cell Rep 2020;31:107806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Vriens K, Christen S, Parik S, Broekaert D, Yoshinaga K, Talebi A, et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature 2019;566:403–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Sleeman J, Schmid A, Thiele W. Tumor lymphatics. Semin Cancer Biol 2009;19:285–97. [DOI] [PubMed] [Google Scholar]
- 119. Alitalo A, Detmar M. Interaction of tumor cells and lymphatic vessels in cancer progression. Oncogene 2012;31:4499–508. [DOI] [PubMed] [Google Scholar]
- 120. Leong SP, Gershenwald JE, Soong SJ, Schadendorf D, Tarhini AA, Agarwala S, et al. Cutaneous melanoma: a model to study cancer metastasis. J Surg Oncol 2011;103:538–49. [DOI] [PubMed] [Google Scholar]
- 121. Willis RA. The spread of tumours in the human body. London, United Kingdom: Butterworths; 1973;xi:p. 417. [Google Scholar]
- 122. Naxerova K, Reiter JG, Brachtel E, Lennerz JK, van de Wetering M, Rowan A, et al. Origins of lymphatic and distant metastases in human colorectal cancer. Science 2017;357:55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Pereira ER, Kedrin D, Seano G, Gautier O, Meijer EFJ, Jones D, et al. Lymph node metastases can invade local blood vessels, exit the node, and colonize distant organs in mice. Science 2018;359:1403–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Brown M, Assen FP, Leithner A, Abe J, Schachner H, Asfour G, et al. Lymph node blood vessels provide exit routes for metastatic tumor cell dissemination in mice. Science 2018;359:1408–11. [DOI] [PubMed] [Google Scholar]
- 125. Crile G Jr, Isbister W, Deodhar SD. Demonstration that large metastases in lymph nodes disseminate cancer cells to blood and lungs. Cancer 1971;28:657. [DOI] [PubMed] [Google Scholar]
- 126. Wallace AC, Hollenberg NK. The transplantability of tumours by intravenous and intralymphatic routes. Br J Cancer 1965;19:338–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Issa A, Le TX, Shoushtari AN, Shields JD, Swartz MA. Vascular endothelial growth factor-C and C-C chemokine receptor 7 in tumor cell-lymphatic cross-talk promote invasive phenotype. Cancer Res 2009;69:349–57. [DOI] [PubMed] [Google Scholar]
- 128. Burton JB, Priceman SJ, Sung JL, Brakenhielm E, An DS, Pytowski B, et al. Suppression of prostate cancer nodal and systemic metastasis by blockade of the lymphangiogenic axis. Cancer Res 2008;68:7828–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Hoshida T, Isaka N, Hagendoorn J, di Tomaso E, Chen YL, Pytowski B, et al. Imaging steps of lymphatic metastasis reveals that vascular endothelial growth factor-C increases metastasis by increasing delivery of cancer cells to lymph nodes: therapeutic implications. Cancer Res 2006;66:8065–75. [DOI] [PubMed] [Google Scholar]
- 130. Das S, Sarrou E, Podgrabinska S, Cassella M, Mungamuri SK, Feirt N, et al. Tumor cell entry into the lymph node is controlled by CCL1 chemokine expressed by lymph node lymphatic sinuses. J Exp Med 2013;210:1509–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Ma Q, Dieterich LC, Ikenberg K, Bachmann SB, Mangana J, Proulx ST, et al. Unexpected contribution of lymphatic vessels to promotion of distant metastatic tumor spread. Sci Adv 2018;4:eaat4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Lee CK, Jeong SH, Jang C, Bae H, Kim YH, Park I, et al. Tumor metastasis to lymph nodes requires YAP-dependent metabolic adaptation. Science 2019;363:644–9. [DOI] [PubMed] [Google Scholar]
- 133. Wang YN, Zeng ZL, Lu J, Wang Y, Liu ZX, He MM, et al. CPT1A-mediated fatty acid oxidation promotes colorectal cancer cell metastasis by inhibiting anoikis. Oncogene 2018;37:6025–40. [DOI] [PubMed] [Google Scholar]
- 134. Dupuy F, Tabaries S, Andrzejewski S, Dong Z, Blagih J, Annis MG, et al. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab 2015;22:577–89. [DOI] [PubMed] [Google Scholar]
- 135. Perry SW, Norman JP, Barbieri J, Brown EB, Gelbard HA. Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. BioTechniques 2011;50:98–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Chuang CH, Dorsch M, Dujardin P, Silas S, Ueffing K, Holken JM, et al. Altered mitochondria functionality defines a metastatic cell state in lung cancer and creates an exploitable vulnerability. Cancer Res 2021;81:567–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Torrano V, Valcarcel-Jimenez L, Cortazar AR, Liu X, Urosevic J, Castillo-Martin M, et al. The metabolic co-regulator PGC1alpha suppresses prostate cancer metastasis. Nat Cell Biol 2016;18:645–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Knott SRV, Wagenblast E, Khan S, Kim SY, Soto M, Wagner M, et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 2018;554:378–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 2015;162:540–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 2015;162:552–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Krall AS, Mullen PJ, Surjono F, Momcilovic M, Schmid EW, Halbrook CJ, et al. Asparagine couples mitochondrial respiration to ATF4 activity and tumor growth. Cell Metab 2021;33:1013–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Fry FH, Jacob C. Sensor/effector drug design with potential relevance to cancer. Curr Pharm Des 2006;12:4479–99. [DOI] [PubMed] [Google Scholar]
- 143. Ward JF. Biochemistry of DNA lesions. Radiat Res Suppl 1985;8:S103–11. [PubMed] [Google Scholar]
- 144. Cummings J, Anderson L, Willmott N, Smyth JF. The molecular pharmacology of doxorubicin in vivo. Eur J Cancer 1991;27:532–5. [DOI] [PubMed] [Google Scholar]
- 145. Ramanathan B, Jan KY, Chen CH, Hour TC, Yu HJ, Pu YS. Resistance to paclitaxel is proportional to cellular total antioxidant capacity. Cancer Res 2005;65:8455–60. [DOI] [PubMed] [Google Scholar]
- 146. Renschler MF. The emerging role of reactive oxygen species in cancer therapy. Eur J Cancer 2004;40:1934–40. [DOI] [PubMed] [Google Scholar]
- 147. Wang Y, Qi H, Liu Y, Duan C, Liu X, Xia T, et al. The double-edged roles of ROS in cancer prevention and therapy. Theranostics 2021;11:4839–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Shimada K, Reznik E, Stokes ME, Krishnamoorthy L, Bos PH, Song Y, et al. Copper-binding small molecule induces oxidative stress and cell-cycle arrest in glioblastoma-patient-derived cells. Cell Chem Biol 2018;25:585–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Adams DJ, Boskovic ZV, Theriault JR, Wang AJ, Stern AM, Wagner BK, et al. Discovery of small-molecule enhancers of reactive oxygen species that are nontoxic or cause genotype-selective cell death. ACS Chem Biol 2013;8:923–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Barr PM, Miller TP, Friedberg JW, Peterson DR, Baran AM, Herr M, et al. Phase 2 study of imexon, a prooxidant molecule, in relapsed and refractory B-cell non-Hodgkin lymphoma. Blood 2014;124:1259–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Pelicano H, Feng L, Zhou Y, Carew JS, Hileman EO, Plunkett W, et al. Inhibition of mitochondrial respiration: a novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J Biol Chem 2003;278:37832–9. [DOI] [PubMed] [Google Scholar]
- 152. Alexeyev M, Shokolenko I, Wilson G, LeDoux S. The maintenance of mitochondrial DNA integrity–critical analysis and update. Cold Spring Harb Perspect Biol 2013;5:a012641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat 2004;7:97–110. [DOI] [PubMed] [Google Scholar]
- 154. Yun J, Mullarky E, Lu C, Bosch KN, Kavalier A, Rivera K, et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015;350:1391–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Agathocleous M, Meacham CE, Burgess RJ, Piskounova E, Zhao Z, Crane GM, et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 2017;549:476–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, et al. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell 2017;170:1079–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Cameron E, Pauling L. Supplemental ascorbate in the supportive treatment of cancer: prolongation of survival times in terminal human cancer. Proc Natl Acad Sci U S A 1976;73:3685–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Ngo B, Van Riper JM, Cantley LC, Yun J. Targeting cancer vulnerabilities with high-dose vitamin C. Nat Rev Cancer 2019;19:271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Hopkins BD, Pauli C, Du X, Wang DG, Li X, Wu D, et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018;560:499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 2017;13:91–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Lien EC, Westermark AM, Li Z, Sapp KM, Heiden MGV. Caloric restriction alters lipid metabolism to contribute to tumor growth inhibition. bioRxiv 2021. [Google Scholar]
- 162. Perez MA, Magtanong L, Dixon SJ, Watts JL. Dietary lipids induce ferroptosis in caenorhabditiselegans and human cancer cells. Dev Cell 2020;54:447–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Olmeda D, Cerezo-Wallis D, Riveiro-Falkenbach E, Pennacchi PC, Contreras-Alcalde M, Ibarz N, et al. Whole-body imaging of lymphovascular niches identifies pre-metastatic roles of midkine. Nature 2017;546:676–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Girotti MR, Gremel G, Lee R, Galvani E, Rothwell D, Viros A, et al. Application of sequencing, liquid biopsies, and patient-derived xenografts for personalized medicine in melanoma. Cancer Discov 2016;6:286–99. [DOI] [PubMed] [Google Scholar]
- 165. Chen S, Sanjana NE, Zheng K, Shalem O, Lee K, Shi X, et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 2015;160:1246–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. van der Weyden L, Arends MJ, Campbell AD, Bald T, Wardle-Jones H, Griggs N, et al. Genome-wide in vivo screen identifies novel host regulators of metastatic colonization. Nature 2017;541:233–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Hoxhaj G, Ben-Sahra I, Lockwood SE, Timson RC, Byles V, Henning GT, et al. Direct stimulation of NADP(+) synthesis through Akt-mediated phosphorylation of NAD kinase. Science 2019;363:1088–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019;575:693–8. [DOI] [PubMed] [Google Scholar]
- 169. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019;575:688–92. [DOI] [PMC free article] [PubMed] [Google Scholar]